

Abstract
Mechanical ventilation is a life-sustaining therapy for patients with respiratory failure but can cause further lung damage known as ventilator-induced lung injury (VILI). However, the intrinsic molecular mechanisms underlying recovery of VILI remain unknown. Phagocytosis of apoptotic cells (also known as efferocytosis) is a key mechanism orchestrating successful resolution of inflammation. Here we show the positive regulation of macrophage Toll-like receptor (TLR) 4 in efferocytosis and resolution of VILI. Mice were depleted of alveolar macrophages and then subjected to injurious ventilation (tidal volume, 20 mL/kg) for 4 h. On day 1 after mechanical ventilation, Tlr4+/+ or Tlr4−/− bone marrow-derived macrophages (BMDMs) were intratracheally administered to alveolar macrophage-depleted mice. We observed that mice depleted of alveolar macrophages exhibited defective resolution of neutrophilic inflammation, exuded protein, lung edema, and lung tissue injury after ventilation, whereas these delayed responses were reversed by administration of Tlr4+/+ BMDMs. Importantly, these proresolving effects by Tlr4+/+ BMDMs were abolished in mice receiving Tlr4−/− BMDMs. The number of macrophages containing apoptotic cells or bodies in bronchoalveolar lavage fluid was much less in mice receiving Tlr4−/− BMDMs than that in those receiving Tlr4+/+ BMDMs. Macrophage TLR4 deletion facilitated a disintegrin and metalloprotease 17 maturation and enhanced Mer cleavage in response to mechanical ventilation. Heat shock protein 70 dramatically increased Mer tyrosine kinase surface expression, phagocytosis of apoptotic neutrophils, and rescued the inflammatory phenotype in alveolar macrophage-depleted mice receiving Tlr4+/+ BMDMs, but not Tlr4−/− BMDMs. Our results suggest that macrophage TLR4 promotes resolution of VILI via modulation of Mer-mediated efferocytosis.
Keywords: efferocytosis, inflammation, macrophage, TLR4, ventilator-induced lung injury
INTRODUCTION
Mechanical ventilation is an indispensable therapy for patients with acute respiratory failure/acute respiratory distress syndrome (ARDS), but accumulating evidence from experimental and clinical studies indicates that it can also damage lung structure and compromise lung function, termed ventilator-induced lung injury (VILI) (1). Mechanical stress and strain can trigger an inflammatory response in the lung, characterized by proinflammatory cytokine upregulation and neutrophil infiltration (1). Studies from our laboratory (2) and others (3) have shown that alveolar macrophages are the primary source of inflammatory cytokines in response to mechanical stimuli and contribute to the pathogenesis of VILI.
The pathophysiology of VILI has been well documented, although the endogenous resolution mechanisms behind mechanical ventilation-induced lung inflammation and injury are still relatively poorly understood. In a rat model of injurious ventilation, the inflammatory response and histological lesions in the lung start to recover 24 h after VILI, reaching normality after 72 h (4, 5). During repair following VILI, collagen and matrix metalloproteinases-2 and -9 increase and persist at higher levels in lung tissues and bronchoalveolar lavage (BAL) fluid. Matrix metalloproteinase inhibition delays lung repair in a mouse model of mechanical ventilation-induced lung injury with high pressures (6, 7). Mechanical stretch also activates a transient inflammatory and fibroproliferative repair response which contributes to recovery of disrupted lung architecture (4). During the resolution phase of acute neutrophilic inflammation, recruited neutrophils rapidly undergo apoptosis or other forms of cell death at inflamed sites and are subsequently eliminated through phagocytosis by macrophages and dendritic cells in a process known as efferocytosis, thus avoiding further cellular disruption and release of proinflammatory mediators such as TNF-α and IL-6 and favoring the production of anti-inflammatory cytokines such as TGF-β and IL-10 (8). Although successful resolution of lung inflammation is dependent on efficient efferocytosis (9), the connection between efferocytosis by alveolar macrophages and recovery from VILI has not been established thus far.
Several studies including ours have reported that Toll-like receptor 4 (TLR4) is actively involved in the development of lung inflammatory injury during mechanical ventilation (10, 11). Mechanical ventilation activates the TLR4-NF-κB signaling pathway which upregulates the expression of proinflammatory mediators (12). During VILI, the inflammatory response in the lung is also regulated by a variety of endogenous TLR4 ligands such as low molecular weight hyaluronic acid, uric acid, heparin sulfate, tenascin-C, and high mobility group box protein 1 and heat shock protein (Hsp) 70 (13). Activation of TLR4 not only induces inflammation but also contributes to the resolution of inflammation by eradicating bacteria through increased phagocytosis (13, 14). In an in vitro experiment, activation of TLR4 by LPS increased the phagocytosis of apoptotic thymocytes by macrophages (15). However, the ability of TLR4 to directly modulate macrophage efferocytosis and subsequent resolution of VILI remains unclear, and no molecular mechanism for such a process has been described.
In the current study, we identified that macrophage TLR4 is required for clearance of apoptotic neutrophils and subsequent recovery from VILI. TLR4 activation by Hsp70 after mechanical ventilation facilitates macrophage efferocytosis by blocking a disintegrin and metalloprotease 17 (ADAM17)-Mer tyrosine kinase signaling. Our results reveal a novel function for macrophage TLR4 in regulating efferocytosis and suggest a new mechanism by which TLR4 promotes resolution and recovery of VILI.
MATERIALS AND METHODS
Mice
C57BL/6J (Tlr4+/+) and Tlr4−/− mice were acquired from Jackson Laboratory (Bar Harbor, ME), housed in a specific pathogen-free barrier facility. Age-matched (12–16 wk old) male and female mice (ratio 1:1) were used in all experiments. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago. All studies were performed under anesthesia using either 1%–3% isoflurane (inhalation) or ketamine-xylazine (100/10 mg/kg ip). A simple randomization procedure by means of drawing lots was used for assigning experimental groups.
Isolation and Culture of Bone Marrow-Derived Macrophages
Murine bone marrow-derived macrophages (BMDMs) were isolated and cultured as described previously (16). Briefly, both tibias and femur were aseptically taken from euthanized mice. Bone marrow was collected from the bones using differentiation medium (RPMI 1640 medium supplemented with 12.5% FBS, 20% L929-cell conditioned medium, 10 mM l-glutamine, 100 IU/mL penicillin, and 100 mg/mL streptomycin). Bone marrow cells (4 × 105) were plated in a Petri dish in 10 mL macrophage complete medium and incubated at 37°C and 5% CO2; 5-mL macrophage complete medium was added to each dish on day 3. On day 7 in culture, after nonadherent cells were removed, adherent cells were >95% pure macrophages, as determined by the expression of cell-surface markers HLA-DR, CD11b, and CD206, and these BMDMs were used for further experiments.
Isolation of Alveolar Macrophages
Alveolar macrophages were isolated from BAL fluid as described previously (2). At the end of experiments, mice were euthanized by exsanguination. The lungs were then excised and lavaged more than 10 times by slowly instilling and withdrawing 1 mL of warm (37°C) Ca2+/Mg2+-free HBSS (pH 7.4) containing EDTA (0.6 mM). BAL fluid was recovered and then centrifuged at 400 g for 10 min at 4°C. BAL cells were plated in 100-mm sterilized polystyrene Petri dishes and incubated for 2 h at 37°C. Dishes were washed three times with media to remove any nonadherent cells, and the adherent macrophages were collected for further experimental use. The purity of isolated alveolar macrophages was >95% as determined using fluorescently labeled antibodies that specifically recognize proteins expressed by mice macrophages (surface antigens F4/80 and CD11b). The viability was >98% as evaluated by trypan blue exclusion.
Isolation of Neutrophils and Induction of Apoptosis
Polymorphonuclear neutrophils were isolated from mouse peripheral blood by density-gradient Ficoll-Histopaque as described previously (17). Apoptosis was induced by exposure of neutrophils to UV irradiation (254 nm, UVS-26, 6-W bulb, 0.02 J/s/cm2) for 15 min and then incubated for 3 h in a 5% CO2 incubator at 37°C. Over 90% of neutrophils were apoptotic as verified by annexin V+ cells using flow cytometric analysis.
Plasma Membrane Extraction
The plasma membrane fraction was extracted using a membrane protein extraction kit (Thermo Fisher) according to the manufacturer’s instructions. Alveolar macrophages isolated from mouse lungs were incubated with 0.75-mL permeabilization buffer for 10 min at 4°C and then centrifuged at 16,000 g for 15 min. The resulting pellet was incubated with 0.5 mL of solubilization buffer for 30 min at 4°C and centrifuged at 16,000 g for 15 min at 4°C. The supernatant was harvested for analysis of solubilized membrane and membrane-associated proteins.
In Vitro Macrophage Efferocytosis Assay
An established protocol was performed to examine macrophage efferocytosis in vitro (17). Briefly, both mouse BMDMs and apoptotic neutrophils (2–3 × 107/mL) were separately labeled by incubating cells with CellTracker Green and CellTracker Red at 37°C for 15 min, respectively. The labeled apoptotic cells were added to macrophage cultures at a ratio of 10 to 1 for 2 h of incubation. The extracellular fluorescence of membrane-bound but nonengulfed apoptotic cells or bodies was quenched with trypan blue (0.04% in PBS). The phagocytosis of apoptotic neutrophils by BMDMs was determined using fluorescence microscopy. Phagocytic index was expressed as the percentage of macrophages containing at least one engulfed neutrophil.
In Vivo Macrophage Efferocytosis Assay
Alveolar macrophage efferocytosis following mechanical ventilation was assessed by counting macrophages ingesting apoptotic cells and apoptotic bodies in BAL fluid (17). At the end of experiments, mice were euthanized by exsanguination. The trachea was surgically exposed, and the lungs underwent lavage with 1 mL prewarmed PBS for three times. After the collected BAL fluid was centrifuged, cell pellets were suspended in PBS. Single cell suspensions were spun onto a microscope slide by use of a cytocentrifuge (Shandon). Slides were stained with Diff-Quik dye (Dade Behring) and examined at magnifications of ×20 and ×40 by light microscopy. A minimum of 300 macrophages were counted for each sample. Efferocytosis was calculated as the percentage of alveolar macrophages containing apoptotic cells and apoptotic bodies.
Western Blotting Analysis
Alveolar macrophages and BMDMs were washed twice with ice-cold PBS, collected, and lysed using radioimmunoprecipitation assay buffer (Boston Bio Products, BP-115) supplemented with 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Sigma), and 1 mM Na3VO4. Protein concentrations of the samples were determined with DC Protein Assay Reagent (Bio-Rad). Equal amounts of protein were loaded for electrophoresis on 8%–14% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes (Millipore, 0.45 µm, IPVH00010). Membranes were blocked in 5% nonfat skim milk with Tris-buffered saline/0.1% Tween-20 for 1 h, incubated with primary antibodies (Abs) overnight at 4°C, and then with horseradish peroxidase (HRP)-conjugated secondary Abs (1:3,000–5,000) for 1 h at room temperature. The membranes were incubated in the chemiluminescent substrate solution and the protein bands shown with Odyssey Fc Imager (LI-COR Biosciences). Western blots were quantified from scanned films using ImageJ Software (NIH, Bethesda, MD) (16, 17).
Detection of Apoptosis by TUNEL Assay
Apoptotic cells in the BAL fluid were evaluated using terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling (TUNEL) kit (Boehringer Mannheim, Indianapolis, IN), according to the manufacturer’s protocols. Briefly, BAL cells were spun onto a microscope slide by use of a cytocentrifuge (Shandon) and fixed in 4% paraformaldehyde at room temperature for 5 min. The slides were incubated with the buffer provided and then immersed in terminal deoxynucleotidyl transferase buffer. Terminal deoxynucleotidyl transferase and fluorescein‐dUTP were added and allowed to incubate for 60 min at 37°C. Finally, the slides were washed with PBS, counterstained with propidium iodide, and examined under a fluorescence microscope.
Depletion of Alveolar Macrophages in Mice
Depletion of alveolar macrophages was performed as described previously (17). The clodronate liposome (Encapsula NanoSciences) was intratracheally delivered to the anesthetized mice with ketamine-xylazine (100/10 mg/kg ip). Lavageable alveolar macrophage count was reduced by 95% at 2 days and 92% at 5 days following instillation of clodronate liposome.
Animal Protocols
For induction of ventilator-induced lung injury, mice were anesthetized with ketamine-xylazine (100/10 mg/kg) intraperitoneally, intubated orally with a 20-gauge IV catheter, and then ventilated with a Harvard Apparatus ventilator (MiniVent, Harvard Biosciences) for 4 h. Mice were randomly assigned to the different treatment groups with blinding. Ventilated animals received high VT (20 mL/kg injurious ventilation) with a respiratory rate 60 breaths/min, room air, and zero end-expiratory pressure. Arterial partial pressure of carbon dioxide () was maintained between 35 and 45 Torr (4.7–6.0 kPa) by adding ∼5 mL of dead space to the ventilator circuit. During ventilation, a heating pad was provided to maintain body temperature between 37°C and 38°C. After mechanical ventilation, 0.25 mL of 0.9% prewarm normal saline was administered intraperitoneally every 2 h to ensure adequate blood volume. Additional doses of ketamine-xylazine (0.5 mg/0.025 mg) were administered intraperitoneally to maintain adequate anesthesia throughout the experiment. For some experiments, at day 1 after mechanical ventilation, PBS or BMDMs isolated from Tlr4+/+ or Tlr4−/− mice (2 × 106 cells, 200 μL of total volume each) were intratracheally instilled to alveolar macrophage-depleted mice via as described previously (17, 18). At different time points after mechanical ventilation, mice were euthanized with a lethal dose of the anesthetic agent, and lung tissues and BAL fluid were collected for the assessment of lung inflammation and injury.
Extracellular Hsp70 Level Detection
The concentrations of extracellular Hsp70 in BAL fluid of mice were measured using an Hsp70 ELISA detection kit (DuoSet IC; R&D Systems, Inc.) following the manufacturer’s instructions. Visualization was carried out using substrate solution, and the enzyme-substrate reaction was terminated by the addition of 10% sulfuric acid solution. Absorbance was read on an ELISA plate reader at 450/620 nm.
Determination of Neutrophil Counts in BAL Fluid
At the end of the experiments, mice were euthanized by exsanguination. BAL was performed by intratracheal injection of 1 mL of PBS and repeated three times. Recovered BAL fluid was centrifuged at 800 rpm for 5 min at 4°C. BAL cells were resuspended and counted and adjusted to 1 × 105 cells/mL. Then 200 µL of cells were cytospun onto slides at 300 rpm for 5 min with a cytocentrifuge (Shandon). Slides were immediately fixed, stained using Diff-Quick dye (Siemens, B4132-1A), and examined by microscopy. Three hundred cells in randomly selected fields were counted and the percentage of neutrophils was calculated.
Cytokine Concentration in BAL Fluid
The concentrations of TNF-α, IL-6, IL-10, and TGF-β in BAL fluid were measured with ELISA kits (BioLegend) or CBA kits (eBioscience) according to the manufacturers’ instructions.
Determination of Lung Edema Formation and Vascular Permeability
Total protein concentration in BAL fluid was measured using Bradford protein assay (Bio-Rad) to determine the permeability of the alveolar-capillary barriers (17). The results were converted to μg/μL using values based on a standard curve generated with serial dilutions of bovine serum albumin (BSA). At the end of the experiments, lungs were excised, weighed, dried at 60°C for 48 h, and then reweighed. Wet-to-dry lung weight ratio was calculated and used as an index of lung water content and edema.
For some experiments, the Evans blue dye technique was applied to assess lung microvascular permeability as previously described (19). Briefly, Evans blue dye (30 mg/kg) was intravenously injected into mice through the tail vein 35 min before lung collection. Lung was then excised, homogenized in ice-cold PBS, incubated with formamide for 18 h at 60°C, and centrifuged at 13,000 g for 20 min. Evans blue content was measured at absorbances of A620 and A740, and calculated as follows: A620 (corrected) = A620–(1.1649 × A740 + 0.004). The Evans blue index was expressed as the amount of dye in the lung relative to the weight of lung tissue (µg Evans blue/g lung/min).
Lung Histology and Lung Injury Scoring
Lungs were instilled, inflated at 15 cmH2O, and preserved for 24–48 h in 10% buffered formalin. Fixed lungs were washed with PBS and dehydrated in 70% ethanol followed by paraffin embedding. Slices at 4–5 µm thickness were subsequently stained with hematoxylin and eosin and visualized under light microscopy. Lung injury scores were quantified by an investigator blinded to the treatment groups as described previously (20). In brief, the following four pathological changes were scored on a scale of 0–4: 1) alveolar congestion, 2) hemorrhage, 3) infiltration of inflammatory cells or aggregation of neutrophils in air space or the vessel wall, and 4) alveolar wall thickness. The criteria are as follows: 0 = normal lungs; 1 = mild, <25% lung involvement; 2 = moderate, 25%–50% lung involvement; 3 = severe, 50%–75% lung involvement; and 4 = very severe, >75% lung involvement. Twenty random high-power fields (×40 magnification) were scored per animal. The overall VILI score was obtained by adding all the averaged scores.
Antibody Validation
The specificity of all antibodies used in this study has been tested and reported in previous studies. Na+/K+-ATPase (No. 3010) and GAPDH (No. 2118) Abs were from Cell Signaling (Danvers, MA) (17). Anti-ADAM17 (ab57484, ab39163) Abs were ordered from Abcam (Cambridge, MA) (17). Anti-TLR4 (M300) (13) and anti-Mer (sc-66399) (18) Abs were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Statistics
Data were analyzed using SPSS statistics software (version 23.0, IBM). Two-sample t test was used for comparisons between two groups, and one-way ANOVA with post hoc test (Bonferroni test) for three or more groups. All data are expressed as means ± SD. All tests were two-tailed, and differences were considered significant when P < 0.05.
RESULTS
Resolution of Ventilator-Induced Inflammation and Injury Is Impaired in Alveolar Macrophage-Depleted Mice
To determine the role of alveolar macrophages in the resolution of ventilator-induced lung inflammatory injury, C57BL/6J mice were depleted of alveolar macrophages with clodronate liposome and subjected to mechanical ventilation at a high tidal volume of 20 mL/kg (Fig. 1A). In comparison with control mice, alveolar macrophage-depleted mice displayed less lung inflammation and injury as reflected by lower lung wet/dry weight ratio (Fig. 1B), protein concentration (Fig. 1C), and neutrophil count in BAL fluid (Fig. 1D) at 4 h, day 1, and day 2 following ventilation. These injury parameters peaked on day 2 in both groups and were significantly higher in control mice than those in alveolar macrophage-depleted mice (Fig. 1, B–D). Lung wet/dry weight ratio (Fig. 1B) and protein concentration in BAL fluid (Fig. 1C) almost completely recovered by day 7 in control mice but remained elevated in alveolar macrophage-depleted mice. Neutrophil count in BAL fluid gradually decreased in control mice but remained elevated in alveolar macrophage-depleted mice (Fig. 1D) on day 7 after mechanical ventilation. Lung histology showed severe histological changes, including alveolar congestion, exudates, and infiltration of inflammatory cells at day 2 following mechanical ventilation in both control and alveolar macrophage-depleted mice (Fig. 1E, left). These histopathological alterations in lung tissues were significantly ameliorated by day 7 following mechanical ventilation, as noted by the significantly reduced lung injury score in control mice, but not in alveolar macrophage-depleted mice (Fig. 1E, right). These results indicate that alveolar macrophage ablation results in less lung inflammatory injury but delays the resolution of lung inflammation and injury following mechanical ventilation.
Figure 1.

Alveolar macrophage depletion impairs resolution of ventilator-induced lung injury. A: flow diagram indicating timelines for experimental interventions. Following depletion of alveolar macrophages with clodronate liposome (CLOD), mice were mechanically ventilated with a high tidal volume (20 mL/kg) (MV) for 4 h. Resolution of lung inflammatory injury was assessed on days 1, 2, 4, and 7 after ventilation. B: wet-to-dry lung weight ratio. C: protein concentration in bronchoalveolar lavage (BAL) fluid. D: polymorphonuclear neutrophils (PMN) counts in the BAL fluid. E: hematoxylin and eosin staining of sections of lungs. Left: representative lung histology. Magnification, ×20; inset, ×40. Right: quantification of histopathological lung injury scores. All images for control and treatment groups were collected at the same time under the same conditions. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05 and **P < 0.01 vs. control group (PBS), one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD.
Macrophage TLR4 Promotes the Recovery of VILI
To assess the role of TLR4 expression in the proresolving effect of alveolar macrophages following mechanical ventilation-induced lung inflammation and injury, we used an alveolar macrophage-depleted mouse model of VILI to evaluate the effect of instilled Tlr4+/+ or Tlr4−/− BMDMs. Our previous studies demonstrated that intratracheal instillation of BMDMs resulted in uniform distribution of macrophages in alveolar macrophage-depleted lungs (17, 18). Following depletion of alveolar macrophages, mice were ventilated with a high tidal volume (20 mL/kg) for 4 h. On day 1 after mechanical ventilation, BMDMs were intratracheally administered to alveolar macrophage-depleted mice. Lung inflammatory injury was assessed on day 4 after ventilation (Fig. 2A). TLR4 deletion in BMDMs was verified by Western blot analysis (Fig. 2B). Alveolar macrophage-depleted mice receiving Tlr4+/+ BMDMs showed a significant decrease in wet-to-dry lung weight ratio (Fig. 2C), protein level in BAL fluid (Fig. 2D), lung histological injury (Fig. 2, E and F), and neutrophil counts in BAL fluid (Fig. 2G) compared with control mice (PBS group). However, alveolar macrophage-depleted mice receiving Tlr4−/− BMDMs exhibited no improvement in lung inflammation and injury (Fig. 2, C–G). To decipher the mechanisms responsible for impaired resolution of lung inflammation by Tlr4−/− macrophages, we examined the levels of key inflammatory mediators in BAL fluid and found that the levels of proinflammatory cytokines TNF-α (Fig. 3A) and IL-6 (Fig. 3B) in the BAL fluid were elevated in alveolar macrophage-depleted lungs on day 4 after mechanical ventilation. Alveolar macrophage-depleted mice receiving Tlr4+/+ BMDMs showed lower levels of TNF-α (Fig. 3A) and IL-6 (Fig. 3B), whereas those mice receiving Tlr4−/− BMDMs exhibited higher levels of TNF-α (Fig. 3A) and IL-6 (Fig. 3B). In contrast, alveolar macrophage-depleted mice receiving Tlr4−/− BMDMs had a remarkably decreased level of anti-inflammatory cytokines IL-10 (Fig. 3C) and TGF-β (Fig. 3D) compared with those receiving Tlr4+/+ BMDMs. These results suggest that TLR4 expression in alveolar macrophage may play an important role in resolving lung inflammation during repair of VILI.
Figure 2.
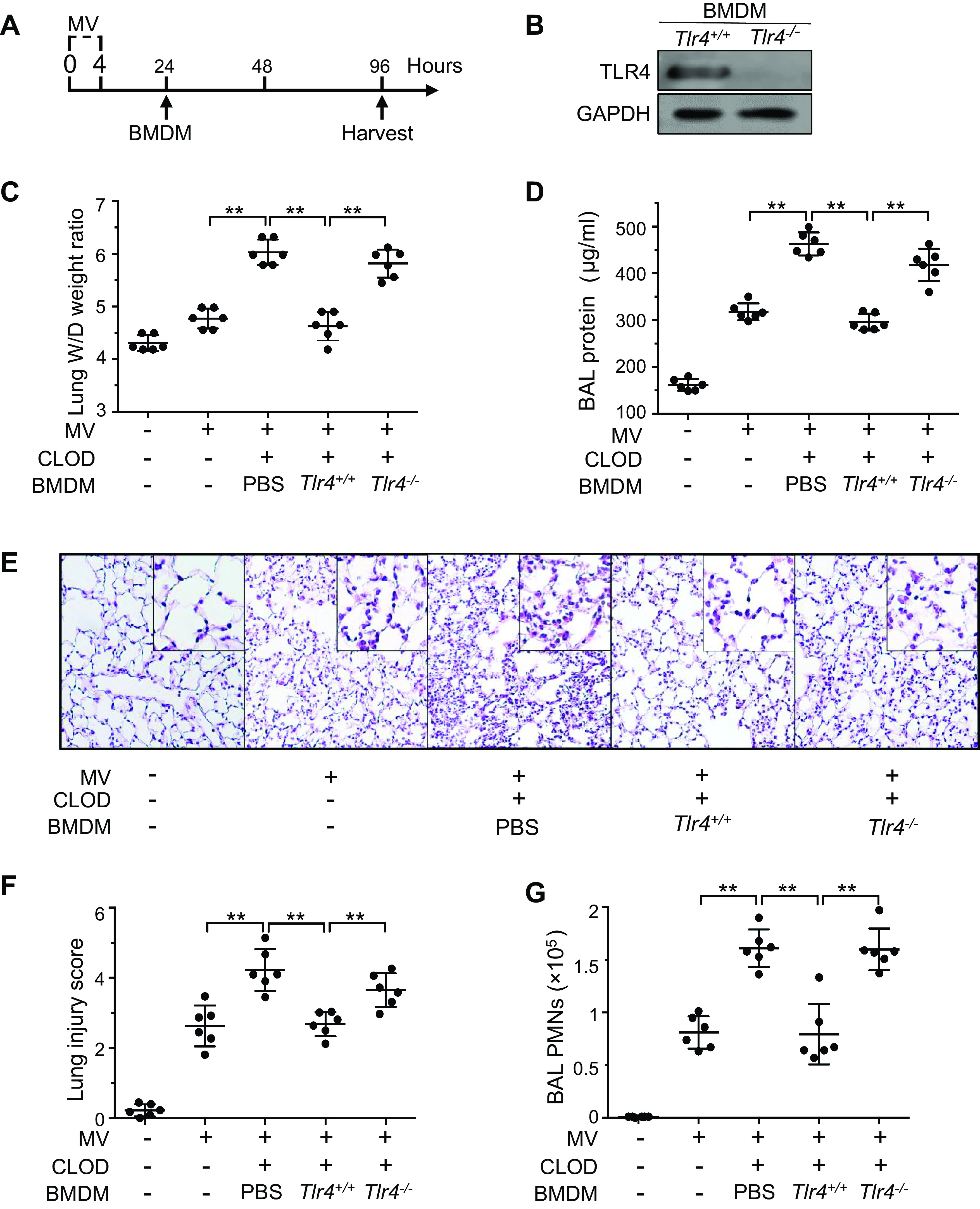
Effects of lung transplantation of bone marrow-derived macrophages (BMDMs) on resolution of VILI. A: flow diagram indicating timelines for experimental interventions. Following depletion of alveolar macrophages with clodronate liposome (CLOD), mice were mechanically ventilated with a high tidal volume (20 mL/kg) (MV) for 4 h. On day 1 after mechanical ventilation, PBS or Tlr4+/+ or TLR4−/− bone marrow-derived macrophages (BMDMs) were intratracheally administered to alveolar macrophage-depleted mice. Lung inflammatory injury was assessed on day 4 after ventilation. B: TLR4 protein expression in BMDMs. C: wet-to-dry (W/D) lung weight ratio. D: protein concentrations in the bronchoalveolar lavage (BAL) fluid. E: representative lung histology. Hematoxylin and eosin staining of sections of lungs. Magnification, ×20; inset, ×40. All images for control and treatment groups were collected at the same time under the same conditions. F: quantification of histopathological lung injury scores. G: polymorphonuclear neutrophils (PMN) counts in the BAL fluid. n = 6 lungs (from 3 male and 3 female mice). **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. TLR4, Toll-like receptor 4; VILI, ventilator-induced lung injury.
Figure 3.

Effect of lung transplantation of BMDMs on release of cytokines during ventilator-induced lung injury. Following depletion of alveolar macrophages with clodronate liposome (CLOD), mice were mechanically ventilated with a high tidal volume (20 mL/kg) (MV) for 4 h. On day 1 after mechanical ventilation, Tlr4+/+ or Tlr4−/− bone marrow-derived macrophages (BMDMs) were intratracheally administered to alveolar macrophage-depleted mice. Lung inflammatory injury was assessed on day 4 after ventilation. A: TNFα in the bronchoalveolar lavage (BAL) fluid. B: IL-6 in BAL fluid. C: IL-10 in BAL fluid. D: TGF-β in BAL fluid. n = 6 lungs (from 3 male and 3 female mice). **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD.
TLR4 Accelerates Clearance of Apoptotic Neutrophils by Alveolar Macrophages during VILI
Alveolar macrophages that phagocytose apoptotic neutrophils polarize into a proresolving phenotype that allows them to secrete TGF-β and IL-10 as opposed to proinflammatory cytokines such as TNF-α and IL-6 (17, 18, 21). We thus examined the role of TLR4 expression in alveolar macrophages in clearing apoptotic neutrophils in vivo. We observed that the number of macrophages containing apoptotic bodies or cells in BAL fluid was much lower in alveolar macrophage-depleted lungs receiving Tlr4−/− BMDMs than in those receiving Tlr4+/+ BMDMs (Fig. 4A). As further support of a role for TLR4 expression in efferocytosis by alveolar macrophages, using the TUNEL assay, we determined the relative abundance of apoptotic neutrophils in the lung. In alveolar macrophage-depleted lungs, the BAL cells were predominantly neutrophils with some free (nonengulfed) apoptotic cells as indicated by bright fluorescent TUNEL staining (Fig. 4B). In contrast, Tlr4+/+ BMDM-treated mice showed fewer free apoptotic cells in BAL fluid (Fig. 4B). Consistent with the earlier finding, we observed a great number of free apoptotic cells in BAL fluid from Tlr4−/− BMDM-treated mice compared with Tlr4+/+BMDM-treated mice (Fig. 4B). These data suggest that macrophage TLR4 regulates resolution of ventilator-induced lung inflammation and injury by modulating efferocytosis.
Figure 4.

Depletion of macrophage TLR4 reduces the clearance of apoptotic neutrophils by alveolar macrophages during VILI. Mice were depleted of alveolar macrophages by intratracheal administration of clodronate (CLOD) liposomes, followed by mechanical ventilation with a high tidal volume (20 mL/kg) (MV) for 4 h. On day 1 after ventilation, Tlr4+/+ or Tlr4−/− bone marrow-derived macrophages (BMDMs) were intratracheally administered to alveolar macrophage-depleted mice. A: effects of TLR4 on efferocytosis. Left: representative photomicrographs of cytospin preparations of the bronchoalveolar lavage (BAL) cells 2 days after transplantation of BMDMs. Arrows indicate apoptotic cells or bodies. Original magnification, ×40. Right: quantification of macrophages (MФ) containing apoptotic cells or bodies in the BAL fluid. B: effects of TLR4 on the number of apoptotic neutrophils. Left: representative photomicrographs of cytospin preparations showing apoptotic neutrophils. Cytospin preparations of the BAL cells were processed for terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) assay using fluorescein-labeled dUTP (green signal) and counterstained with the nuclear stain (DAPI). Right: quantification of apoptotic neutrophils in the BAL fluid. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. All images for control and treatment groups were collected at the same time under the same conditions. TLR4, Toll-like receptor 4; VILI, ventilator-induced lung injury.
TLR4 Suppresses Shedding of Mer Receptor Tyrosine Kinase by Inactivating ADAM17 Signaling
We next set out to understand the mechanism whereby TLR4 expression enhances the ability of macrophages to phagocytose apoptotic neutrophils. Clearance of apoptotic cells requires the surface expression of receptors on macrophages that recognize apoptotic cell-associated ligands such as phosphatidylserine (22). We found that the surface expression of Mer receptor tyrosine kinase on alveolar macrophages isolated from Tlr4+/+ lungs was downregulated after mechanical ventilation at high tidal volume, and this response was further exaggerated in alveolar macrophages isolated from ventilated Tlr4−/− lungs (Fig. 5A). The level of surface Mer expression on macrophages is known to be regulated by ADAM17. Proteolytic cleavage of the extracellular domain of transmembrane Mer by ADAM17 leads to the production of the soluble form of Mer protein (sMer) (18, 23). Consistent with decreased surface Mer expression on macrophages, there was a corresponding increase in sMer in BAL fluid from Tlr4−/− lungs (Fig. 5A). To ascertain whether increased cleavage of Mer on Tlr4−/− macrophages is related to activation of ADAM17, the premature (p) versus mature (m) forms of ADAM17 were analyzed in alveolar macrophages isolated from ventilated lungs. Mechanical ventilation at high tidal volume increased maturation of ADAM17 in Tlr4+/+ lungs, whereas depletion of TLR4 further enhanced ventilation-induced ADAM17 maturation (Fig. 5B, left), and band densitometry analysis showed a significant increase in the ratio of mADAM17 and GAPDH (Fig. 5B, right). Taken together, these findings indicate that TLR4 stabilizes Mer surface expression on macrophages by inactivating ADAM17 during mechanical ventilation.
Figure 5.

TLR4 regulates surface expression of Mer on alveolar macrophages. Tlr4+/+ and Tlr4−/− mice were mechanically ventilated with a high (20 mL/kg) tidal volume for 4 h. Bronchoalveolar lavage (BAL) fluid was collected, and alveolar macrophages were isolated from ventilated lungs on day 2. The plasma membrane fraction was extracted, and protein expression determined by Western blot analysis. A: effects of TLR4 on the surface expression of Mer on alveolar macrophages and the release of soluble Mer (sMer) in the BAL fluid. Top: representative blots showing the surface expression of Mer on alveolar macrophages and the level of sMer in the BAL fluid. Bottom: protein quantification (normalized to Na+/K+-ATPase) by densitometry. B: effects of TLR4 on immature proform a disintegrin and metalloprotease 17 (pADAM17) and mature ADAM17 (mADAM17) in alveolar macrophages. Left: representative blots showing protein expression of pADAM17 and mADAM17. Right: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. TLR4, Toll-like receptor 4.
TLR4 Interaction with Hsp70 Promotes Macrophage Efferocytosis
TLR4 is primarily activated by interaction with its ligands to induce a signaling cascade. During mechanical ventilation, numerous endogenous TLR4 ligands including Hsp70 are released from damaged lung tissues or cells. Hsp70 in BAL fluid has been shown to increase after high-tidal-volume ventilation in fetal sheep (24). Given the importance of Hsp70 in anti-inflammatory responses (25), we examined the potential role of TLR4-Hsp70 interaction in regulating macrophage efferocytosis during resolution of VILI. We first monitored the dynamics of extracellular Hsp70 levels in mouse lungs at various time intervals following mechanical ventilation. The level of Hsp70 in BAL fluid was significantly increased at 24 h, reached a peak at 48 h after mechanical ventilation, and then decreased at days 4 and 7 (Fig. 6A). Next, we determined the effect of Hsp70 on efferocytosis by Tlr4+/+ or Tlr4−/− macrophages. We observed that incubation of Tlr4+/+ BMDMs with Hsp70 markedly increased the phagocytosis of apoptotic neutrophils. However, treatment of Tlr4−/− BMDMs with Hsp70 was unable to stimulate the internalization of apoptotic cells (Fig. 6B). These results demonstrate that TLR4-Hsp70 interaction is essential for phagocytosis of apoptotic neutrophils during the repair of VILI.
Figure 6.

TLR4 interaction with Hsp70 promotes efferocytosis by macrophages. A: changes of heat shock protein 70 (Hsp70) in the lung during ventilator-induced lung injury. Mice were mechanically ventilated with a high tidal volume (20 mL/kg) (MV) for the indicated time. The concentration of Hsp70 in the bronchoalveolar lavage (BAL) fluid was measured on days 1, 2, 4, and 7 after ventilation. n = 6 lungs (from 3 male and 3 female mice) each time point. *P < 0.05 and **P < 0.01 vs. control unventilated group (0 h), one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. B: TLR4 is required for Hsp70-mediated efferocytosis by bone marrow-derived macrophages (BMDMs). BMDMs were treated with Hsp70 (40 μg/mL) or vehicle. Macrophages were labeled with CellTracker Green and incubated with CellTracker Red-labeled apoptotic polymorphonuclear neutrophils (PMN) at a 1:10 ratio for 2 h. The cells were then mounted on a slide and analyzed by fluorescence microscopy. Top: representative fluorescent images showing macrophages engulfing apoptotic PMNs. Scale bars, 10 μm. Bottom: phagocytic index based on the fluorescent images. n = 6 mice (from 3 male and 3 female mice). **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. All images for control and treatment groups were collected at the same time under the same conditions. TLR4, Toll-like receptor 4.
TLR4 Interaction with Hsp70 Suppresses ADAM17-Mediated Mer Cleavage
On the basis of our observations that TLR4 in the presence of Hsp70 stabilized surface expression of macrophage Mer, we explored the possible role of Hsp70 in regulating surface expression of Mer receptor tyrosine kinase and activation of ADAM17. As shown in Fig. 7A, compared with the control group, treatment of Tlr4+/+ macrophages with Hsp70 significantly upregulated surface expression of Mer, and this was not observed on Tlr4−/− macrophages in the absence or presence of Hsp70. Consistently, treatment of Tlr4+/+ macrophages with Hsp70 dramatically reduced sMer secretion, whereas TLR4 depletion reversed Hsp70-induced decrease in sMer level in the medium (Fig. 7A). Similarly, Hsp70 treatment markedly downregulated the level of mature ADAM17 in Tlr4+/+ macrophages and this response was reversed in Tlr4−/− macrophages (Fig. 7B). These data suggest that Hsp70 binding to TLR4 suppresses ADAM17 activation leading to upregulation of Mer expression on the macrophage surface and inhibition of sMer generation in the lung.
Figure 7.

Effects of Hsp70 on surface expression of Mer and ADAM17 maturation in alveolar macrophages following mechanical ventilation. Tlr4+/+ and Tlr4−/− mice were injected intravenously with 10 μg recombinant human Hsp70 or vehicle and then mechanically ventilated with a high tidal volume (20 mL/kg) for 4 h. Bronchoalveolar lavage (BAL) fluid was collected, and alveolar macrophages were isolated from ventilated lungs on day 2. The plasma membrane fraction was extracted, and protein expression was determined by Western blot analysis. A: effects of Hsp70 on surface expression of Mer on alveolar macrophages and soluble Mer (sMer) in BAL fluid after ventilation. Top: representative blots showing surface expression of Mer on alveolar macrophages and soluble Mer (sMer) in BAL fluid. Bottom: protein quantification (normalized to Na+/K+-ATPase) by densitometry. B: effects of Hsp70 on immature proform a disintegrin and metalloprotease 17 (pADAM17) and mature ADAM17 (mADAM17) in alveolar macrophages. Left: representative blots showing protein expression of pADAM17 and mADAM17. Right: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. TLR4, Toll-like receptor 4.
TLR4 Favors the Recovery of VILI through Its Interaction with Hsp70
By using two well-characterized in vivo models, we addressed whether Hsp70 binding to TLR4 facilitates resolution of ventilator-induced lung inflammation and injury. The effect of Hsp70 treatment on lung inflammatory injury in ventilated Tlr4+/+ mice was first examined. Alternatively, alveolar macrophage-depleted mice were ventilated with a high tidal volume (20 mL/kg) for 4 h and then intratracheally instilled with Hsp70-pretreated Tlr4+/+ or Tlr4−/− BMDMs on day 1 post mechanical ventilation (17, 18). We found that both treatment of Tlr4+/+ mice with Hsp70 and transplantation of Hsp70-pretreated Tlr4+/+ BMDMs remarkedly reduced Evans blue dye accumulation (Fig. 8A), lung injury score (Fig. 8B), and neutrophil count in BAL fluid (Fig. 8C). However, transplantation of Hsp70-pretreated Tlr4−/− BMDMs failed to reduce Evans blue dye accumulation (Fig. 8A), lung injury score (Fig. 8B), and neutrophil count in BAL fluid (Fig. 8C) in alveolar macrophage depleted and ventilated mice. Mechanistically, the number of macrophages containing apoptotic bodies or cells (Fig. 8D) and the levels of anti-inflammatory cytokine IL-10 (Fig. 8E) in BAL fluid were much higher in both Hsp70-treated Tlr4+/+ mice and alveolar macrophage deficient mice transplanted with Hsp70 pretreated Tlr4+/+ BMDMs following mechanical ventilation. These effects were not seen in alveolar macrophage deficient mice transplanted with Hsp70-pretreated Tlr4−/− BMDMs (Fig. 8, D and E). These results suggest that TLR4 mediates the recovery and resolution of VILI through its interaction with Hsp70.
Figure 8.

Hsp70 promotes the recovery and resolution of lung inflammatory injury in Tlr4+/+ BMDM-treated mice, but not in Tlr4−/− BMDM-treated mice. Mice were depleted of alveolar macrophages by intratracheal administration of clodronate (CLOD) liposomes, followed by mechanical ventilation with a high tidal volume (20 mL/kg) for 4 h. On day 1 after ventilation, these mice were injected intravenously with 10 μg recombinant human HSP70 or vehicle and Tlr4+/+or Tlr4−/− bone marrow-derived macrophages (BMDMs) were then intratracheally administered. Lung inflammatory injury was assessed on day 4 after ventilation. A: pulmonary vascular permeability measured by lung Evans blue dye (EBD) extravasation. Top: representative lung appearance after Evans blue dye administration. Bottom: quantitative analysis of Evans blue dye extravasation. n = 6/group. *P < 0.05, **P < 0.01, one-way ANOVA. B: hematoxylin and eosin staining of sections of lungs. Top: representative lung histology. Magnification, ×20; inset, ×40. Bottom: quantification of histopathological lung injury scores. n = 6/group. *P < 0.05, **P < 0.01, one-way ANOVA. C: polymorphonuclear neutrophils (PMN) counts in the bronchoalveolar lavage (BAL) fluid. *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. D: clearance of apoptotic neutrophils by alveolar macrophages. Top: representative photomicrographs of cytospin preparations of BAL cells 2 days after transplantation of BMDMs. Arrows indicate apoptotic bodies. Original magnification, ×40. Bottom: quantification of BAL fluid macrophages (MФ) containing apoptotic bodies. E: IL-10 in BAL fluid. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. All images for control and treatment groups were collected at the same time under the same conditions.
DISCUSSION
As a type of pattern recognition receptors, TLR4 senses invading pathogens or endogenous damage signals and regulates both innate and adaptive immune responses throughout the course of infection (26). TLR4 also functions in noninfectious lung inflammation induced by alveolar overdistention during mechanical ventilation (11, 27). In this study, we identified the proresolution function of macrophage TLR4 which was attributed to effective clearance of apoptotic cells during recovery of VILI. TLR4 activation with Hsp70 enhanced the ability of macrophages to engulf apoptotic cells by modulating ADAM17-Mer receptor tyrosine kinase signaling. Such an enhanced ability to clear apoptotic cells could be a self-resolving mechanism by which hosts are able to constrain the spreading of inflammation and accelerate repair of VILI. Alveolar macrophages constitute the predominant immune cell in the lung in the steady state and play a crucial role in both initiation and resolution of lung inflammation in response to noxious stimuli (28). Emerging evidence suggests that alveolar macrophages contribute to the lung inflammatory response and alveolar barrier dysfunction in ventilator-induced lung injury (2, 3). In the clodronate model of alveolar macrophage depletion, we confirmed the proinflammatory role of alveolar macrophages in the development of VILI. Although depletion of alveolar macrophages reduced lung inflammation, the extent of recovery following VILI was also significantly impaired. Importantly, repletion of macrophages in the lung after mechanical ventilation markedly promoted the resolution of lung inflammatory injury. These findings demonstrate a critical role for alveolar macrophages in both the proinflammatory phase and resolving phase of VILI, rendering them a potential therapeutic target.
TLR4 has been shown to play a major role in the resolution of lung inflammation and repair of lung injury in several pathological conditions (29–31). TLR4 expression is important for type 2 alveolar epithelial cell renewal and repair of bleomycin-induced lung injury (29). Interaction of hyaluronan fragments with TLR4 maintains lung epithelial cell integrity and promotes repair and recovery from acute lung injury (30). In the mouse models of bleomycin- or silica-induced acute or chronic lung injury, basal TLR4 activity is crucial for facilitating resolution of acute or chronic inflammation and fibrogenesis after acute or chronic lung injury (31). This study describes what we believe is a novel role for macrophage TLR4 in resolution of mechanical ventilation-induced lung inflammatory injury. In alveolar macrophage-depleted mice, loss of macrophage TLR4 impaired the ability of transplanted macrophages to resolve lung inflammation following injurious mechanical ventilation, demonstrating the critical role of TLR4 expression on alveolar macrophages in repairing VILI.
Efficient efferocytosis represents a critical step in tissue remodeling, modulation of immune responses, and resolution of inflammation. The impairment of efferocytosis is directly correlated with increased morbidity and mortality after lung inflammatory injury (32, 33). Decreased efferocytosis delayed the recovery of acute and chronic pulmonary inflammatory diseases (32, 34), whereas enhanced alveolar macrophage-mediated efferocytosis accelerated resolution of lung inflammation in the Escherichia coli peritonitis-associated acute lung injury model (35). In this study, deletion of TLR4 in BMDMs dramatically reduced phagocytosis of apoptotic neutrophils. Importantly, pulmonary transplantation of TLR4 deficient BMDMs into alveolar macrophage-depleted mice following injurious ventilation decreased the number of macrophages containing apoptotic bodies in BAL fluid, whereas reconstitution with Tlr4+/+ BMDMs increased efferocytosis by macrophages in the lung. These findings couple with the resolution of lung inflammation and recovery of lung injury following injurious ventilation. Therefore, this study raises the possibility that TLR4 expression on alveolar macrophages promotes the repair of VILI by modulating the clearance of apoptotic neutrophils, the hallmarks of proresolving mechanisms.
Efferocytosis is a tightly orchestrated process by which macrophages are recruited to sites of apoptotic activity, and then recognize, engulf, and finally clear apoptotic cells. The efficiency of efferocytosis is determined by three signaling cascades: find-me, eat-me, and postengulfment signaling. Eat-me signals are the key to recognition of apoptotic cells and initiation of the phagocytosis process by activating receptors on macrophages (22). The TAM (Tyro3, Axl, Mer) tyrosine kinases are a family of efferocytosis receptors that recognize phosphatidylserine on apoptotic cells through the binding to protein S and growth arrest-specific 6 (Gas6) (36). Mer is a type I transmembrane protein that is highly expressed in both human (37) and mouse (38) alveolar macrophages, where it is thought to guide phagocytic clearance of apoptotic cells and lung tissue repair. Proteolytic ectodomain shedding of Mer by ADAM17 leads to low Mer surface expression, and generation of soluble Mer (23), which then acts as a decoy receptor and inhibits efferocytosis (39). Upon LPS challenge, TLR4 signaling has been shown to accelerate activation of ADAM17 and subsequent cleavage of Mer (23) and CD163 (40). In this report, we describe the opposite role for TLR4 in macrophages following mechanical ventilation, where the receptor suppresses ADAM17 activation and stabilizes the surface expression of Mer. Our results showed that TLR4 deletion dramatically reduced Mer surface expression on macrophages and markedly increased the level of sMer in the lung following mechanical ventilation. Furthermore, TLR4 ablation significantly enhanced mechanical ventilation-induced increase in ADAM17 activity. These findings strongly support the concept that TLR4 promotes macrophage efferocytosis after mechanical ventilation by preventing ADAM17-mediated cleavage of Mer. In contrast to LPS-induced TLR4 inflammatory signaling, it is likely that during the resolution phase, mechanical stress stimulates lung cells to generate endogenous ligands which activate the TLR4 anti-inflammatory signaling to inhibit the ADAM17-mediated Mer cleavage.
Numerous endogenous TLR4 ligands are released from cellular compartments that can activate TLR4 signaling following mechanical ventilation (41). Among all endogenous TLR4 ligands, Hsp70 has been shown to exert a protective effect on ventilator-induced lung injury by blocking the inflammatory response (42). Anti-inflammatory and protective effects of Hsp70 have been demonstrated in various mouse models of inflammation (25). The mechanisms by which Hsp70 confers protection are not fully understood, but most likely may relate to its ability to stabilize damaged intracellular proteins and suppress NF-κB signaling-mediated production of proinflammatory mediators such as TNF-α and IL-1β (25, 43). The protective effect of Hsp70-TLR4-Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF) signaling in endothelial cells was also reported in oxidant-induced lung injury (44). In this report, several lines of compelling evidence argue that Hsp70 engagement with TLR4 plays a crucial role in TLR4-mediated efferocytosis and resolution of ventilator-induced lung injury as observed in our model system. First, extracellular Hsp70 in the lung was elevated during the repair phase of VILI (days 2–7). Second, Hsp70 treatment increased the phagocytosis of neutrophils by Tlr4+/+ BMDMs, but not by Tlr4−/− BMDMs in vitro. Third, Hsp70 engagement with TLR4 increased Mer surface expression and decreased the level of sMer in the lung through suppression of ADAM17-mediated Mer cleavage. Finally, administration of exogenous Hsp70 promoted the resolution of inflammatory injury in alveolar macrophage-depleted mice receiving Tlr4+/+ BMDMs but failed to do so in mice receiving Tlr4−/− BMDMs. These results clearly support the notion that TLR4 interaction with Hsp70 enhances phagocytosis of apoptotic neutrophils by blocking proteolytic cleavage of surface Mer receptor on macrophages, leading to resolution and recovery of lung inflammation and injury following mechanical ventilation.
It is worth noting that there exist phenotypic and functional differences between BMDMs and resident lung macrophages. However, in our model, intratracheal administration of wild type BMDMs following mechanical ventilation completely rescued delayed recovery of lung inflammatory injury induced by alveolar macrophage depletion, demonstrating the comparable role of instilled BMDMs in the alveolar space in lung inflammation. Therefore, we believe that this study strongly supports the proresolving role for the Hsp70-TLR4 signaling in lung macrophages in VILI.
Based on our findings, targeting the Hsp70-TLR4 signaling axis may be a therapeutic strategy to accelerate recovery of VILI. Because specifically overexpressing TLR4 on alveolar macrophages is challenging to achieve in vivo, it remains a poor choice for therapeutic intervention. Our data suggest that Hsp70 is a critical member of this signaling pathway for clearance of apoptotic neutrophils during resolution of VILI. Hsp70 could be targeted by developing pharmacological inducers of Hsp70 or direct delivery of purified Hsp70 protein, such as recombinant Hsp70 (45). Taken together, our findings reveal the ability of TLR4 to activate efferocytosis signaling following mechanical ventilation. Leveraging this mechanism may sensitize alveolar macrophages to damage signals in the prorepair microenvironment and provide a unique therapeutic opportunity to promote the resolution of lung inflammation.
The activation of TLR4 is required for host defense against invading microbes and mechanical ventilation-induced lung inflammation. This is the first report to our knowledge that TLR4 expression on macrophages is essential for phagocytosis of apoptotic neutrophils and recovery of ventilator-induced lung injury. Following mechanical ventilation, endogenous TLR4 ligand Hsp70 is secreted, directly binds TLR4, and thus induces ADAM17 inactivation which prevents Mer cleavage, leading to increased Mer surface expression on macrophages and decreased sMer. These changes to Mer mediated by TLR4 ultimately result in increased efferocytosis and resolution of lung inflammation and injury (Fig. 9). These results, combined with our previous findings (15), demonstrate that TLR4 expression in macrophages plays pivotal roles in both host defense and resolution of lung inflammation following mechanical ventilation. This study provides newfound knowledge of the dual role of TLR4 in regulating lung inflammation in different microenvironmental cues in response to mechanical stress. In conclusion, the data presented here have revealed important insights into the intrinsic mechanisms underlying the resolution and repair of lung inflammation and injury induced by mechanical ventilation.
Figure 9.

Model of TLR4-mediated recovery of ventilator-induced lung injury. Hsp70 engagement with TLR4 inhibits the generation of mature a disintegrin and metalloprotease 17 (mADAM17) from immature proform ADAM17 (pADAM17), leading to the stabilization of Mer surface expression on macrophages which facilitates phagocytosis of apoptotic cells and subsequent resolution of ventilator-induced lung inflammation and injury. TLR4 deletion in macrophages promotes ADAM17 maturation which causes Mer shedding and the release of the soluble Mer (sMer), and thus impairs phagocytosis of apoptotic cells. TLR4, Toll-like receptor 4.
GRANTS
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number R01HL152696 (to G. Hu), R01HL104092 (to G. Hu) and by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R21AI152249 (to G. Hu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.S., L.B., C.J., Y.-Y.Z., R.D.M., and G.H. conceived and designed research; K.S., L.B., and C.J. performed experiments; K.S., L.B., and G.H. analyzed data; C.J., X.D., Y.-Y.Z., R.D.M., and G.H. interpreted results of experiments; K.S. and L.B. prepared figures; L.B. and G.H. drafted manuscript; K.S., X.D., Y.-Y.Z., R.D.M., and G.H. edited and revised manuscript; K.S., L.B., C.J., X.D., Y.-Y.Z., R.D.M., and G.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Maricela Castellon (Department of Anesthesiology, University of Illinois College of Medicine) for technical assistance.
All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
REFERENCES
- 1.Kneyber MC, Zhang H, Slutsky AS. Ventilator-induced lung injury. Similarity and differences between children and adults. Am J Respir Crit Care Med 190: 258–265, 2014. doi: 10.1164/rccm.201401-0168CP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol 190: 3590–3599, 2013. doi: 10.4049/jimmunol.1200860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 291: L1191–L1198, 2006. doi: 10.1152/ajplung.00055.2006. [DOI] [PubMed] [Google Scholar]
- 4.Curley GF, Contreras M, Higgins B, O'Kane C, McAuley DF, O'Toole D, Laffey JG. Evolution of the inflammatory and fibroproliferative responses during resolution and repair after ventilator-induced lung injury in the rat. Anesthesiology 115: 1022–1032, 2011. doi: 10.1097/ALN.0b013e31823422c9. [DOI] [PubMed] [Google Scholar]
- 5.Nin N, Lorente JA, de Paula M, El Assar M, Vallejo S, Peñuelas O, Fernández-Segoviano P, Ferruelo A, Sánchez-Ferrer A, Esteban A. Rats surviving injurious mechanical ventilation show reversible pulmonary, vascular and inflammatory changes. Intensive Care Med 34: 948–956, 2008. doi: 10.1007/s00134-007-0959-6. [DOI] [PubMed] [Google Scholar]
- 6.Albaiceta GM, Gutiérrez-Fernández A, Parra D, Astudillo A, García-Prieto E, Taboada F, Fueyo A. Lack of matrix metalloproteinase-9 worsens ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 294: L535–L543, 2008. doi: 10.1152/ajplung.00334.2007. [DOI] [PubMed] [Google Scholar]
- 7.González-López A, Astudillo A, García-Prieto E, Fernández-García MS, López-Vázquez A, Batalla-Solís E, Taboada F, Fueyo A, Albaiceta GM. Inflammation and matrix remodeling during repair of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 301: L500–L509, 2011. doi: 10.1152/ajplung.00010.2011. [DOI] [PubMed] [Google Scholar]
- 8.Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 21: 398–414, 2020. doi: 10.1038/s41580-020-0232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest 143: 1750–1757, 2013. doi: 10.1378/chest.12-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuipers MT, van der Poll T, Schultz MJ, Wieland CW. Bench-to-bedside review: damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care 15: 235, 2011. doi: 10.1186/cc10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu G, Malik AB, Minshall RD. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 38: 194–201, 2010. doi: 10.1097/CCM.0b013e3181bc7c17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker JC. Mitochondrial damage pathways in ventilator induced lung injury (VILI): an update. J Lung Health Dis 2: 18–22, 2018. doi: 10.29245/2689-999X/2017/2.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain V, Halle A, Halmen KA, Lien E, Charrel-Dennis M, Ram S, Golenbock DT, Visintin A. Phagocytosis and intracellular killing of MD-2 opsonized gram-negative bacteria depend on TLR4 signaling. Blood 111: 4637–4645, 2008. doi: 10.1182/blood-2007-11-126862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Husebye H, Aune MH, Stenvik J, Samstad E, Skjeldal F, Halaas O, Nilsen NJ, Stenmark H, Latz E, Lien E, Mollnes TE, Bakke O, Espevik T. The Rab11a GTPase controls toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 33: 583–596, 2010. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Freitas A, Banerjee S, Xie N, Cui H, Davis KI, Friggeri A, Fu M, Abraham E, Liu G. Identification of TLT2 as an engulfment receptor for apoptotic cells. J Immunol 188: 6381–6388, 2012. doi: 10.4049/jimmunol.1200020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Sun D, Yan Z, Reynolds AB, Christman JW, Minshall RD, Malik AB, Zhang Y, Hu G. Differential role for p120-catenin in regulation of TLR4 signaling in macrophages. J Immunol 193: 1931–1941, 2014. doi: 10.4049/jimmunol.1302863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang C, Liu Z, Hu R, Bo L, Minshall RD, Malik AB, Hu G. Inactivation of Rab11a GTPase in macrophages facilitates phagocytosis of apoptotic neutrophils. J Immunol 198: 1660–1672, 2017. doi: 10.4049/jimmunol.1601495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du X, Jiang C, Lv Y, Dull RO, Zhao YY, Schwartz DE, Hu G. Isoflurane promotes phagocytosis of apoptotic neutrophils through AMPK-mediated ADAM17/Mer signaling. PLoS One 12: e0180213, 2017. doi: 10.1371/journal.pone.0180213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao YY, Hu G, Pan D, Malik AB, Hu G. YAP controls endothelial activation and vascular inflammation through TRAF6. Circ Res 123: 43–56, 2018. doi: 10.1161/CIRCRESAHA.118.313143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sue RD, Belperio JA, Burdick MD, Murray LA, Xue YY, Dy MC, Kwon JJ, Keane MP, Strieter RM. CXCR2 is critical to hyperoxia-induced lung injury. J Immunol 172: 3860–3868, 2004. doi: 10.4049/jimmunol.172.6.3860. [DOI] [PubMed] [Google Scholar]
- 21.Leitch AE, Duffin R, Haslett C, Rossi AG. Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol 1: 350–363, 2008. doi: 10.1038/mi.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol 20: 254–267, 2020. doi: 10.1038/s41577-019-0240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem 286: 33335–33344, 2011. doi: 10.1074/jbc.M111.263020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hillman NH, Moss TJ, Kallapur SG, Bachurski C, Pillow JJ, Polglase GR, Nitsos I, Kramer BW, Jobe AH. Brief, large tidal volume ventilation initiates lung injury and a systemic response in fetal sheep. Am J Respir Crit Care Med 176: 575–581, 2007. doi: 10.1164/rccm.200701-051OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borges TJ, Wieten L, van Herwijnen MJ, Broere F, van der Zee R, Bonorino C, van Eden W. The anti-inflammatory mechanisms of Hsp70. Front Immunol 3: 95, 2012. doi: 10.3389/fimmu.2012.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell 180: 1044–1066, 2020. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Su X, Yan X, Wasserloos K, Chao W, Kaynar AM, Liu ZQ, Leikauf GD, Pitt BR, Zhang LM. Toll-like receptor 4-myeloid differentiation factor 88 signaling contributes to ventilator-induced lung injury in mice. Anesthesiology 113: 619–629, 2010. doi: 10.1097/ALN.0b013e3181e89ab2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu G, Christman JW. Editorial: alveolar macrophages in lung inflammation and resolution. Front Immunol 10: 2275, 2019. doi: 10.3389/fimmu.2019.02275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, Noble PW. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med 22: 1285–1293, 2016. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med 11: 1173–1179, 2005. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 31.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, Yan J, Li Z, Liu H, Hua F, Lu W, Hu ZW. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol 180: 275–292, 2012. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 32.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol 76: 467–492, 2014. doi: 10.1146/annurev-physiol-021113-170408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medan D, Wang L, Yang X, Dokka S, Castranova V, Rojanasakul Y. Induction of neutrophil apoptosis and secondary necrosis during endotoxin-induced pulmonary inflammation in mice. J Cell Physiol 191: 320–326, 2002. doi: 10.1002/jcp.10105. [DOI] [PubMed] [Google Scholar]
- 34.Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol 14: 81–93, 2014. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 35.El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci USA 109: 14983–14988, 2012. doi: 10.1073/pnas.1206641109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer 18: 94, 2019. doi: 10.1186/s12943-019-1022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kazeros A, Harvey BG, Carolan BJ, Vanni H, Krause A, Crystal RG. Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am J Respir Cell Mol Biol 39: 747–757, 2008. doi: 10.1165/rcmb.2007-0306OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee YJ, Han JY, Byun J, Park HJ, Park EM, Chong YH, Cho MS, Kang JL. Inhibiting Mer receptor tyrosine kinase suppresses STAT1, SOCS1/3, and NF-κB activation and enhances inflammatory responses in lipopolysaccharide-induced acute lung injury. J Leukoc Biol 91: 921–932, 2012. doi: 10.1189/jlb.0611289. [DOI] [PubMed] [Google Scholar]
- 39.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 109: 1026–1033, 2007. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Z, Zhang H, Zhang X, He S, Dong W, Wang X, Chen Y, Liu X, Guo C. Lipopolysaccharide downregulates CD163 expression to inhibit PRRSV infection via TLR4-NF-κB pathway. Front Microbiol 11: 501, 2020. doi: 10.3389/fmicb.2020.00501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 109: 465–472, 2008. doi: 10.1097/ALN.0b013e318182aef1. [DOI] [PubMed] [Google Scholar]
- 42.Villar J, Méndez-Alvarez S. Heat shock proteins and ventilator-induced lung injury. Curr Opin Crit Care 9: 9–14, 2003. doi: 10.1097/00075198-200302000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Wheeler DS, Wong HR. Heat shock response and acute lung injury. Free Radic Biol Med 42: 1–14, 2007. doi: 10.1016/j.freeradbiomed.2006.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Zhang X, Shan P, Hunt CR, Pandita TK, Lee PJ. A protective Hsp70-TLR4 pathway in lethal oxidant lung injury. J Immunol 191: 1393–1403, 2013. doi: 10.4049/jimmunol.1300052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JY, Han Y, Lee JE, Yenari MA. The 70-kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opin Ther Targets 22: 191–199, 2018. doi: 10.1080/14728222.2018.1439477. [DOI] [PMC free article] [PubMed] [Google Scholar]