

Keywords: bladder, cystitis, inflammation, obstruction, proresolution
Abstract
Bladder outlet obstruction (BOO) is ultimately experienced by ≈90% of men, most commonly secondary to benign prostatic hyperplasia. Inflammation is a critical driver of BOO pathology in the bladder and can be divided into two critical steps: initiation and resolution. Although great strides have been made toward understanding the initiation of inflammation in the bladder [through the NLR family pyrin domain containing 3 (NLRP3) inflammasome], no studies have examined resolution. Resolution is controlled by five classes of compounds known as specialized proresolving mediators (SPMs), all of which bind to one or more of the seven different receptors. Using immunocytochemistry, we showed the presence of six of the known SPM receptors in the bladder of control and BOO rats; the seventh SPM receptor has no rodent homolog. Expression was predominantly localized to urothelia, often with some expression in smooth muscle, but little to none in interstitial cells. We next examined the therapeutic potential of the annexin-A1 resolution system, also present in control and BOO bladders. Using the peptide mimetic Ac2-26, we blocked inflammation-initiating pathways (NLRP3 activation), diminished BOO-induced inflammation (Evans blue dye extravasation), and normalized bladder dysfunction (urodynamics). Excitingly, Ac2-26 also promoted faster and more complete functional recovery after surgical deobstruction. Together, the results demonstrate that the bladder expresses a wide variety of potential proresolving pathways and that modulation of just one of these pathways can alleviate many detrimental aspects of BOO and speed recovery after deobstruction. This work establishes a precedent for future studies evaluating SPM effectiveness in resolving the many conditions associated with bladder inflammation.
NEW & NOTEWORTHY To our knowledge, this is the first study of proinflammation-resolving pathways in the bladder, which is the basis of a new pharmacological genus-dubbed “resolution pharmacology” aimed at reducing inflammation without creating an immunocompromised state. Inflammation plays a causative or exacerbating role in numerous bladder maladies. We documented proresolution receptors in the rat bladder and the effectiveness of a specialized proresolving mediator, annexin-A1, in alleviating detrimental aspects of bladder outlet obstruction and speeding recovery after deobstruction.
INTRODUCTION
Inflammation is a major driving force of the symptoms, dysfunction, and damage to the bladder in numerous benign urologic maladies such as bladder outlet obstruction (BOO), diabetic bladder dysfunction, urinary tract infections, interstitial cystitis/bladder pain syndrome, and even aging. Despite the relevance to so many urological diseases, no inflammation-based therapeutics are available for any of these conditions. Clearly, there is a significant unmet need to understand and control bladder inflammation with the ultimate goal of alleviating the morbidity of all of these disorders.
In general, inflammation is a complex and tightly regulated response to diverse sterile and infectious stimuli. It plays an essential role in host defense through an orchestrated series of events marked by local vascular changes, production of soluble mediators, and infiltration of inflammatory cells into damaged tissue (1). There are two critical stages of inflammation: initiation and resolution (2, 3). Initiation is triggered by antigens from invading pathogens (pathogen-associated molecular patterns that cause infectious inflammation) or endogenous antigens released by cellular damage (damage-associated molecular patterns that cause sterile inflammation) (4, 5). There has recently been a major breakthrough in the mechanics of the initiation phase with the discovery of multimeric structures called inflammasomes. Relatively few inflammasomes recognize the vast panoply of damage-associated molecular patterns and pathogen-associated molecular patterns and activate the same underlying proinflammatory cascade. However, we have shown a critical role for one of these, the NLR family pyrin domain-containing (NLRP)3 inflammasome, in triggering inflammation in the bladder in numerous benign disorders, including BOO (6, 7). Most efforts to control inflammation have been centered almost exclusively on inhibiting the activation phase of the inflammatory response. Although these efforts have been somewhat effective, there are weaknesses to this approach. Most notably, diminishing the responsive capacity of the innate immune system can leave the host vulnerable to opportunistic infection (8). With regard to many of the inflammatory urological maladies, there is the additional problem that symptoms that cause a patient to present to their physician only become apparent after inflammation has been present for a considerable amount of time. For example, in the case of BOO secondary to prostatic enlargement in men, longitudinal studies have suggested that inflammation and mild obstructive symptoms are often present for decades before they become severe enough for the patient to seek medical attention (9). In this common situation, strategies that prevent the initiation and propagation of inflammation may no longer have a significant role in arresting the advanced process, much less reversing the damage that has already occurred. Thus, strategies aimed at reducing, or resolving, current inflammation in the context of chronic disease will be critical to alleviating the sufferings of real-world patients (10).
The second critical phase in the natural course of inflammation is resolution (2, 3). In 1984, Serhan et al. (11, 12) discovered lipoxins, metabolites of arachidonic acid that could specifically promote the resolution of inflammation. This was quickly broadened by the discovery of multiple classes of functionally similar compounds that were given the umbrella term specialized proresolving mediators (SPMs) (13–15). Thus, the state of an inflammatory reaction is a delicate balance between initiating factors and these resolution factors (16). Moreover, it is currently thought that chronic inflammation, a defining characteristic of many bladder diseases, is more likely to result from a failure in proresolving pathways rather than an overstimulation of initiating pathways (8, 17–19). Finally, treatments aimed at ramping up the resolution phase are far less likely to produce immunocompromised patients than those which block the initiation phase (8).
Since their initial discovery, the number of SPMs has expanded to include at least 26 mediators in five classes (20). One of these classes is composed of a single polypeptide, annexin-A1. Although the annexin family has 12 members with diverse functions (21), annexin-A1 is the only identified SPM. The other classes of SPMs are lipids such as lipoxins, resolvins, protectins, and maresins along with their peptide-conjugated and n-3 docosapentaenoic acid-derived counterparts (20). Somewhat surprisingly, only seven independent receptors have been identified to mediate SPM activity, and there appears to be a significant ligand polypharmacology (single SPMs binding to more than one receptor) and receptor pleiotropy (a single receptor activated by multiple ligands) (22–24). Despite 35 years passing since the discovery of SPMs, to our knowledge, no studies have examined a potential role for them in the bladder, although one study did examine the expression of annexin family members (25). In that study, Monastyrskaya et al. (25) found that annexin-A1 was localized to the urothelium and that expression was decreased in biopsies taken from patients with interstitial cystitis/bladder pain syndrome. It is tempting to speculate from these limited data that a diminished capacity for inflammatory resolution in these patients, through reduced production of annexin-A1, could be responsible for the persistent inflammation, pain, and urinary symptoms of this disease, although this remains to be thoroughly investigated.
In the present study, we assessed the presence of six of the SPM receptors in the rat bladder by immunocytochemistry (the seventh SPM receptor has no rodent homolog) and alterations in their expression profiles during BOO. We further explored the annexin-A1 resolution system by performing immunocytochemistry for annexin-A1 in control and BOO bladders. In addition, we used a mimetic of annexin-A1 (Ac2-26) to test whether this SPM can block NLRP3 activation in urothelial cells and inhibit the sterile inflammation that occurs during BOO, as these are central to defining annexin-A1 as an SPM in the bladder. Most importantly, we administered Ac2-26 to rats that had been surgically obstructed and those that were deobstructed (after a period of obstruction) to evaluate the ability of Ac2-26 to heal the bladder and reverse the dysfunction and tissue damage that occurred.
MATERIALS AND METHODS
Animals
All protocols adhere to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Duke University Medical Center. Naive female Sprague-Dawley rats (≈200 g, 40−50-days-old, Envigo, Indianapolis, IN) were used in all experiments.
BOO Surgery
Females are the standard for rodent BOO studies (26–28), even though BOO predominates in males. The reasons for this are plentiful but mostly stem from the tortuosity of the male urethra along with various ducts entering into the urethra. Thus, inserting a catheter into a male is considerably more likely to cause injury that will elicit inflammation and confound results, particularly for experiments aimed at studying inflammation.
We performed BOO surgery as previously described (6, 7, 29, 30). Female Sprague-Dawley rats (≈200 g) were anesthetized with an intraperitoneal injection of ketamine hydrochloride (90 mg/kg) and xylazine (10 mg/kg). Rats were given subcutaneous injections of amikacin (12.5 mg/kg) for antibiotic prophylaxis and carprofen (5 mg/kg) for pain relief. For BOO and sham-operated (sham) rats, a P50 catheter of 1-mm outer diameter was inserted transurethrally until urine was expressed, indicating its placement within the bladder, and then hermetically sealed. A posterior, vertical abdominal incision was made, and the bladder was externalized. The urethra was isolated, and a 5-0 silk suture was passed dorsally. The suture was tied tightly for BOO and loosely for sham. The muscular layer and the skin layer of the abdomen were then closed separately using a 5-0 polyglycolic acid suture. All rats were then maintained for 12 days. Animals in the deobstruction experiments were then anesthetized, and the abdomen was reopened. The suture around the urethra was then removed, and the abdomen was sutured closed.
Immunohistochemistry
Bladders were removed, fixed overnight in 10% neutral-buffered formalin (4°C), and stored in 70% ethanol at 4°C until processed for histology by our core facility in the Department of Surgery at Duke University Medical Center. Samples were dehydrated and paraffin embedded, and transverse sections (5 µm) were cut from the caudal third of the bladder and mounted onto slides. Slides were then rehydrated and subjected to immunohistochemistry using standard techniques. The primary antibodies used in this study are shown in Table 1. Staining was developed using a Vectastain ABC peroxidase staining kit (Cat. No. PK-4000, Vector Laboratories, Burlingame, CA) and the provided anti-rabbit secondary antibody. Micrographs were captured on a ZEISS Axio Imager Microscope. For the specific micrographs presented in the figures, all slides were stained on the same day and all parameters (incubation times, horseradish peroxidase product development times, etc.) were identical between all slides. In addition, all slides were photographed on the same day and all microscope and photographic parameters (magnification, lamp intensity, exposure times, etc.) were identical. Antibody dilutions (reported in the figures) were a product of the specific antibody, but identical dilutions were used between controls and BOO samples. All staining was repeated a minimum of three times to ensure consistency.
Table 1.
Primary antibodies used in the present study
| Antigen | Host | Company | Catalog Number | Reference(s) |
|---|---|---|---|---|
| IgG isotype control | Rabbit | Novus Biologicals | NBP2-36463 | |
| FPR-2 | Rabbit | Novus Biologicals | NLS1878 | 31–33 |
| BLT-1 | Rabbit | LifeSpan Biosciences | LS-A1494 | |
| ChemR23 | Rabbit | LifeSpan Biosciences | LS-B12924 | |
| GPR37 | Rabbit | Abcam | ab21834 | |
| LGR6 | Rabbit | Abcam | ab126747 | 34–36 |
| GPR18 | Rabbit | Abcam | ab150618 | 37 |
| Annexin-A1 | Rabbit | LifeSpan Biosciences | LS+B6711 | 38 |
BLT-1, leukotriene B4 receptor 2; ChemR23, chemerin receptor 23; FPR-2, formyl-peptide receptor-2; GPR, G protein-coupled receptor; LGR6, leucine-rich repeat-containing G protein-coupled receptor 6.
Caspase-1 Assays
Urothelial cells were isolated, and caspase-1 assays were performed as previously described (39, 40). Briefly, rats were euthanized, and their bladders were harvested and placed in sterile PBS. Using an 18-gauge blunt-tip needle, bladders were inverted and inflated with PBS. A purse-string suture was used to tie off the bladder. Inflated bladders were then submerged in collagenase P (1 mg/mL in complete media) and shaken for 1 h at 37°C. Cells were then passed through a 40-µm nylon mesh, pelleted, and resuspended in complete media [F-12K media (HyClone Laboratories, Logan, UT) supplemented with 10% low-endotoxin, dialyzed FBS (HyClone Laboratories), 10-µM nonessential amino acids (HyClone Laboratories), 1.0 µg/mL hydrocortisone (Sigma Aldrich, St. Louis, MO), 10 µg/mL insulin (Gibco Laboratories, Gaithersburg, MD), 5 µg/mL transferrin (Gibco Laboratories), 6.7 ng/mL selenium (Gibco Laboratories), 100 U/mL penicillin (Gibco Laboratories), and 100 µg/mL streptomycin (Gibco Laboratories)]. Cells were counted and plated in a black-walled 96-well plate at 50,000 cells/well in 90 µL complete media. Cells were placed in an incubator (water saturated, 37°C, 95% air, and 5% CO2) for 24 h. Media were then removed and replaced with 90 µL of serum-free media. Cells were incubated for 30 min to allow them to acclimate to the new media. Ac2-26 was prepared as a stock concentration of 10 µM in PBS and then serially diluted 1:2 in serum-free media; 10 µL of the appropriate dilution of Ac2-26 (or serum-free media) were then added to the cells and incubated for 1 h. ATP (an activator of NLRP3) was then added at an EC50 value of 0.625 mM, and cells were incubated for an additional hour. One hour before the end of incubation, untreated control wells were treated with 1.25 mM ATP (a maximal dose) to serve as a standard for the maximal caspase response. The caspase-1 assay was then performed as previously reported (39, 41). Briefly, media were removed and 50 µL of lysis buffer (10 mM MgCl2 and 0.25% Igepal CA-630) was added for 5 min followed by 50 µL of storage buffer [40 mM HEPES (pH 7.4), 20 mM NaCl, 2 mM EDTA, and 20% glycerol]. The plates were then frozen at −80°C until use (>30 min). For analysis, the plates were thawed and 50 µL of 50 mM HEPES with 10% sucrose, 0.1% CHAPS, 10 µL dithiothreitol (final concentration of 5.5 mM), and 20 µL Z-YVAD-AFC substrate (final concentration of 110 µM) were added to each well. The plates were incubated in the dark for 1 h at 37°C with mild shaking. The fluorescence was then measured (excitation = 400 nm and emission = 505 nm). The measured fluorescence in untreated wells (no. Ac2-26 or ATP) was subtracted from all wells, and the results were normalized to the maximal ATP response. Results are reported as percentages of the maximal ATP response.
In Vivo Treatment Paradigms
Two treatment paradigms were studied: BOO and BOO + deobstruction. For the BOO experiments, rats were given intraperitoneal injections of Ac2-26 (1 mg/kg in ≈0.2 mL) or vehicle 1 h before BOO surgery and then daily thereafter at approximately the same time of day. On day 12, animals were analyzed 4 h after injections. For the deobstruction experiments, animals were given BOO surgery and 12 days later they were deobstructed. They received no treatments during this period. At the time of deobstruction, animals were administered Ac2-26 (1 mg/kg in ≈0.2 mL) or vehicle and again on postoperative days 1 and 2. Rats were analyzed 4 h after the last treatment.
Evans Blue Assay
For the Evans blue extravasation assay, rats were injected with 25 mg/kg Evans blue dye (from a 5 mg/mL stock) via a tail vein injection. One hour later, the bladders were harvested, weighed, and placed in 1 mL of formamide. Bladders were incubated overnight at 56°C with shaking. The following day, the absorbance of formamide was measured (620 nm), and the results were calculated from a standard curve.
Urodynamics
For rats undergoing urodynamics, we also implanted a suprapubic catheter at the time of obstruction. Following the placement of the suture around the urethra, a purse-string suture (5-0 silk with a tapered needle) was placed in the dome of the bladder, a hole was cut through the bladder wall in the middle of the purse string, and a piece of PE-50 tubing was inserted. The PE-50 tubing had a flared intravesicular end with a 3-mm to 4-mm piece of silicon tubing under the flare that served as a spacer between the flare and bladder wall to prevent overgrowth of urothelia. The purse string was tied, and the catheter was tunneled to the back of the neck and secured to interscapular tissue using a 5-0 silk suture. It was then cut 3−4 cm from the body and cauterized. Following the closure of the abdomen, the skin around the exit site of the catheter was closed with a 5-0 polyglycolic acid suture. Rats were then singly housed to avoid damage to the catheter by other rats.
Urodynamic experiments were performed similar to our previous study (6, 42). Briefly, on postoperative day 12 for BOO rats and 2 days after deobstruction for deobstructed rats, rats were placed into a restrainer (modified from Item No. HLD-RM, Kent Scientific, Torrington, CT) and put inside of a small animal cystometry laboratory station (Catamount, St. Albans, VT). Sterile degassed PBS was infused at a rate of 80 µL/min, whereas an in-line pressure transducer measured intravesical pressure. The rat was situated above an analytical scale that captured and recorded voided volumes. Rats underwent urodynamic testing for 1−2 h, whereas vesicular pressure and scale readings were recorded every 0.25 s using Med-CMG software (Catamount). After the last void, postvoid residual volume (PVR) was measured by drawing back on the suprapubic catheter with a syringe.
Quantitation of the urodynamics tracings was performed with CMG Analysis software (v. 1.06, Catamount). Typically, 5−10 micturitions were analyzed once the tracing stabilized (20−30 min). A micturition cycle was defined as the time intravesicular pressure returned to baseline after a previous void until it returned to baseline following the next void. Peak intravesical pressure for a given void was recorded as the voiding pressure, and the change in the analytical scale concurrent with the pressure peak was noted as the void volume. This void volume was divided by the length of time it took for the volume change to be fully registered on the scale to calculate flow rate. The time between successive peaks in voiding pressure was recorded as the intercontraction interval (ICI) and used to calculate frequency (3,600/ICI = number of micturitions/h). For each parameter, the average of the recorded values was used in statistical analysis for that given rat and was considered as n = 1.
Statistical Analysis
Statistical analysis was performed by one-way ANOVA followed by Student-Newman-Keuls post hoc analysis using GraphPad InStat software (v. 3.1, La Jolla, CA).
RESULTS
SPM Receptors Are Present in the Bladder, and Some Alter the Location of Expression in Response to BOO
To understand the extent of the potential repertoire of SPM systems active in the bladder, we stained for the known SPM receptors in bladders from control and BOO rats. The exception was G protein-coupled receptor (GPR)32, which has no rodent homolog (43) and thus was not examined. As shown in Fig. 1, formyl-peptide receptor (FPR)-2, block lipid transport-1 (BLT-1), chemerin receptor 23 (ChemR23), GPR37, leucine-rich repeat-containing G protein-coupled receptor 6 (LGR6), and GPR18 were all expressed in control bladders. FPR-2 (Fig. 1, A–E), BLT-1 (Fig. 1, F–J), and ChemR23 (Fig. 1, K–O) were expressed mostly, but not exclusively, in the luminal layer of the urothelia (the umbrella cells, best seen in the higher-magnification micrographs) with limited expression in the detrusor and no apparent expression in the interstitium. During BOO, however, expression was much more evenly distributed across the entire urothelial layer (which had also undergone the expected hyperplasia). Subjective observation of the intensity of staining suggested increased urothelial expression of at least BLT-1 and ChemR23 in response to BOO, although proof of this requires quantitative analysis. These micrographs also showed expression of FRP-2, BLT-1, and ChemR23 in the detrusor in response to BOO, whereas expression in the interstitium remained restricted. Smooth muscle expression of FPR-2 (44), BLT-1 (45), and ChemR23 (46) have been previously documented in other tissues.
Figure 1.
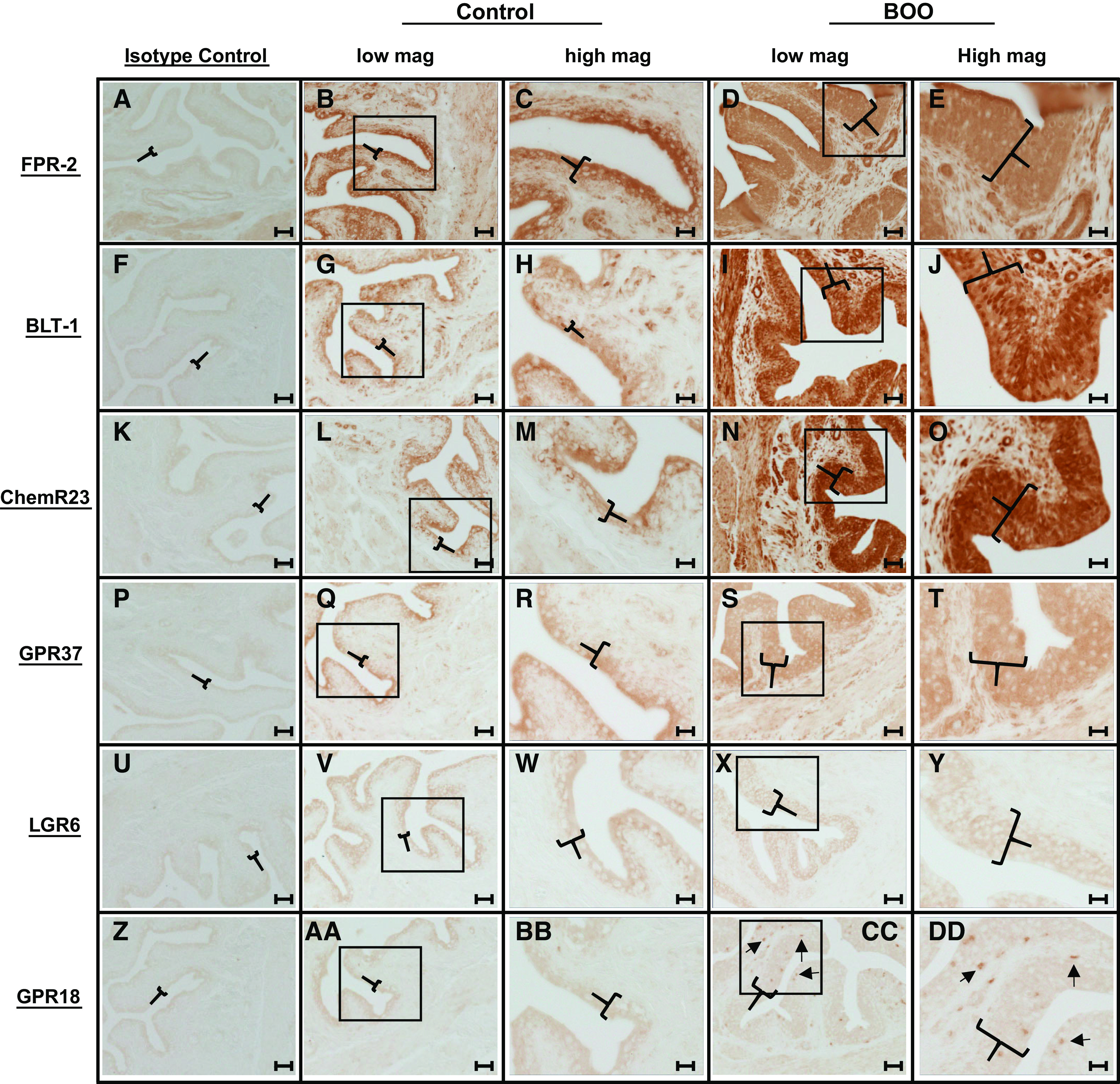
Several specialized proresolving mediator receptors are expressed in the rat bladder, and the tissue expression patterns of some change during bladder outlet obstruction (BOO). All images show immunostaining of bladder sections (5 µm) using rabbit primary antibodies. The first column shows sections from control rats stained with a rabbit isotype control antibody. The second column shows sections from control bladders but stained with antibodies to the indicated receptors (low mag). The third column shows higher magnitude (high mag) micrographs from the area indicated by the box in the appropriate image from the second column. The fourth column shows sections from bladders taken from 12-day BOO rats and stained for the indicated receptors. The fifth column shows higher magnitude micrographs from the area indicated by the box in the appropriate picture from the fourth column. Brackets indicate the urothelium. In CC and DD, arrows indicate individual cells positive for G protein-coupled receptor (GPR)18. Slides were developed using a Vectastain ABC-horseradish peroxidase (HRP) [HRP_ kit (PK-4001), Vector Laboratories]. All parameters for the slides used in this figure (incubation times, HRP product development times, etc.) were identical between all samples and for each antibody. All slides specifically used in this figure were stained on the same day, and identical antibody dilutions were used between control and BOO samples. Micrographs were taken with identical settings (exposure times, light intensity, etc.). Scale bar = 50 µm for the first, second, and fourth columns (low mag) and 20 µm for the third and fifth columns (high mag). All staining was repeated a minimum of three times to ensure consistency. The primary antibodies were as follows: isotype control (NBP2-36463; Novus, 1:100; A, F, K, P, U, and Z), formyl-peptide receptor-2 (FPR-2; NLS1878, Novus, 1:100; B–E), leukotriene B4 receptor 2 (BLT-1; LS-A1494, LS-Bio, 1:100; G–H), chemerin receptor 23 (ChemR23; LS-B12924, LS-Bio, 1:100; L–O), GPR37 (ab218134, Abcam, 1:200; Q–T), leucine-rich repeat-containing G protein-coupled receptor 6 (LGR6; ab126747, Abcam, 1:100; V–Y), and GPR18 (ab150618, Abcam, 1:250; AA–DD).
In addition to FPR-2, BLT-1, and ChemR23, GPR37 (Fig. 1, P–T) was expressed mostly on the luminal surface in control urothelia, although BOO changed the expression to include the entire urothelial layer with some expression now seen in the detrusor and perhaps the interstitium. LGR6 (Fig. 1, U–Y) was similarly expressed in the luminal cell layer in controls and the entire urothelia during BOO but without any detrusor or interstitium expression. GPR18 (Fig. 1, Z–DD) had a weak expression in the luminal layer in controls, not unlike GPR37, but with BOO bladders we saw a clear and distinct staining of individual cells in the urothelial layer (as opposed to the entire urothelia or specific layers) and interstitial layer. Overall, the data clearly show the presence of multiple proresolving receptors, and thus presumably intact pathways, in control urothelia and that the location of expression in specific tissues changes for some receptors in response to the inflammatory stimulus of BOO.
Annexin-A1 Is Expressed in the Bladder
To determine if SPMs are likely to play important roles in the bladder, we first evaluated the annexin-A1 resolution system whose proresolution effects are mediated by FPR-2. Figure 2 shows immunocytochemical staining of annexin-A1 in the bladder from control and BOO rats. As shown in Fig. 2, A–C, annexin-A1 was expressed in the control bladder and localized primarily to the bladder urothelium, although there was clear expression in the detrusor with a small amount in interstitial cells. Smooth muscle expression has been previously documented in other tissues (44). The expression pattern did not demonstrably change in response to BOO (Fig. 2, D and E), although the amount of urothelia increased due to the well-known hyperplasia associated with this disorder.
Figure 2.
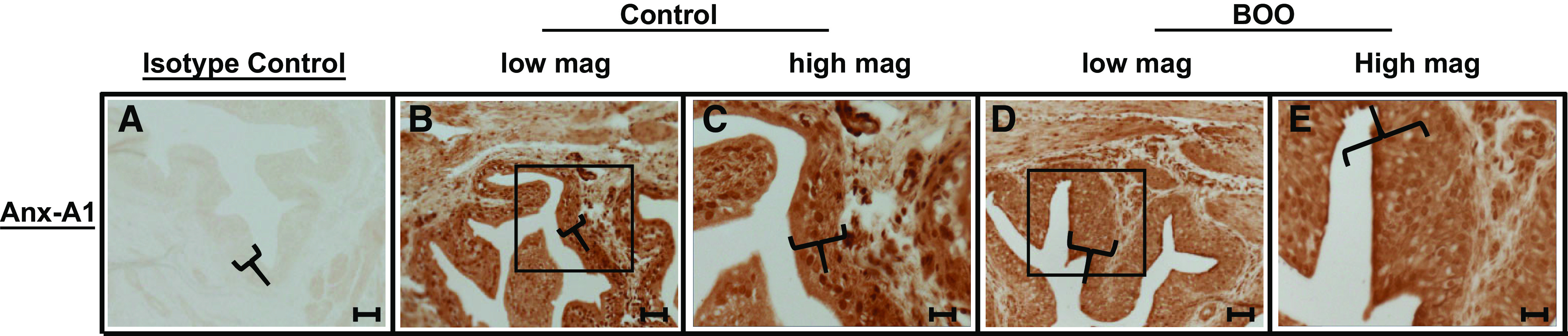
Annexin-A1 (Anx-A1) is expressed in the urothelium and detrusor from control bladders and bladders with bladder outlet obstruction (BOO). Sections (5 µm) of bladders from control rats (A−C) or 12-day BOO rats (D and E) were stained for annexin-A1 or with isotype control serum, as described in materials and methods. Primary antibody sources are shown in Table 1. Scale bar = 50 µm for A, B, and D and 20 µm for C and E.
Ac2-26 Inhibits Inflammation-Initiating Pathways (NLRP3) in Urothelia
A key characteristic of all SPMs is the ability to suppress the initiation of inflammation (47, 48). One well-known initiator of sterile inflammation is the NLRP3 inflammasome, and we have identified a key role for NLRP3 during BOO and in other inflammatory bladder conditions (6, 39, 40, 49, 50). Thus, we sought to determine if annexin-A1 might prevent NLRP3 activation in urothelia. For this, primary urothelia were placed in culture for 24 h and pretreated with the annexin-A1 mimetic Ac2-26 for 1 h, and NLRP3 was then activated with ATP (an activator of NLRP3) for 1 h. Ac2-26 is a peptide consisting of amino acids 2−26 of intact annexin-A1 that retains full activity (51, 52) while having better stability and lower immunogenicity than full-length annexin-A1 (53). While it binds to both FPR-1 and FPR-2, its anti-inflammatory activity is thought to be mediated entirely through FPR-2. As a readout for NLRP3 activation, caspase-1 was assessed, which is the enzymatic product of the activated inflammasome. As shown in Fig. 3, Ac2-26 dose-dependently suppressed NLRP3 activation with an IC50 value of ≈0.25 µM.
Figure 3.

Ac2-26 inhibits NLR family pyrin domain containing 3 (NLRP3) activation. Primary urothelial cells were incubated in 96-well plates overnight, washed, and given Ac2-26 for 1 h at the indicated doses. NLRP3 was then activated with 0.625 mM ATP for 1 h. Caspase-1 activity was assessed by the cleavage of DEVD-AFC as described in materials and methods. Results were normalized to ATP-only controls and are presented as means ± SE; n = 8, 8, 7, 8, 7, 8, 8, 7, 8, 7, 8, and 8, respectively. **P < 0.01 and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test.
Ac2-26 Suppresses the Increase in Bladder Weight During BOO and Enhances Its Resolution After Deobstruction
To assess the ability of annexin-A1 to prevent inflammatory changes during BOO, we administered the annexin-A1 mimetic Ac2-26 (1 mg/kg/day ip) starting 1 h before BOO surgery until 12 days postsurgery when various endpoints were measured. We first determined bladder weights because increases in bladder weight represent edematous inflammation with a component from smooth muscle hypertrophy. As shown in Fig. 4, bladder weights were increased by BOO and this increase was significantly blunted with Ac2-26.
Figure 4.

Ac2-26 (Ac) suppresses the increase in bladder weight during bladder outlet obstruction (BOO) and reduces weight after deobstruction (Deobs). At the end of the described treatment regimens, the bladders were removed and weighed. Data are presented as means ± SE; n = 8, 15, 38, 11, 16, and 20, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test. Con, control.
Since annexin-A1 drives the resolution of inflammation, we examined its efficacy in speeding recovery following deobstruction surgery, analogous to transurethral resection of the prostate surgery often performed on men with advanced BOO. For these experiments, rats were not given Ac2-26 before or after BOO surgery. After 12 days, the urethral ligature was cut and removed, and the rats were then given Ac2-26 on that day and on days 1 and 2 postoperatively. Endpoints were assessed 4 h after the last dose. As shown in Fig. 4, bladder weights from deobstructed rats were still increased from nonsurgical controls and were not significantly different from what they were after 12 days of BOO (i.e., before deobstruction). However, Ac2-26 administration during this period was able to significantly reduce bladder weights during these 2 days, albeit not back to control or sham levels.
Ac2-26 Suppresses Bladder Inflammation During BOO and Hastens Its Resolution After Deobstruction
We next assessed inflammation in the bladder of BOO and BOO + deobstructed rats using the Evans blue dye extravasation method (54). As shown in Fig. 5, the results were very similar to those seen with bladder weights. Evans blue in the tissue was increased in response to BOO but maintained at control levels when Ac2-26 was administered during the entire course of BOO. Not surprisingly, deobstruction reduced inflammation slightly, but significantly, after 2 days. However, Ac2-26 administration during those 2 days resolved inflammation back to control and sham levels.
Figure 5.

Ac2-26 (Ac) suppresses bladder inflammation during bladder outlet obstruction (BOO) and enhances resolution after deobstruction (Deobs). Inflammation was assessed by Evan’s blue dye extravasation. Dye (25 mg/kg) was injected intravenously, and 1 h later bladders were removed, weighed, and placed in 1 mL formamide overnight (56°C). Absorbance (620 nm) was measured, and the results were calculated from a standard curve. Data are presented as means ± SE; n = 7, 7, 27, 9, 14, and 21, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test. Con, control.
Ac2-26 Normalizes Important Indexes of Bladder Function During Obstruction and After Deobstruction
To assess the effects of Ac2-26 on the deterioration of bladder function during BOO, urodynamics was performed. Ac2-26 was administered daily for 12 days after BOO, followed by cystometry. Representative pressure and scale tracings are shown in Fig. 6. Urodynamically, obstruction is defined as high-pressure voiding with decreased flow rate. Figures 6E and 7A show high-pressure voiding, whereas Figs. 6F and 7B show low flow rates in obstructed rats, as expected. Ac2-26 has no effect on these parameters (Figs. 6, G and H, and 7) since this drug will not affect physical outflow resistance. Deobstruction also normalized these parameters (Figs. 6, I and J, and 7, A and B), whereas Ac2-26 had no effect (Figs. 6, K and L, and 7, A and B), again as expected.
Figure 6.

Representative urodynamics pressure and scale tracings from the various experimental groups described in materials and methods. A and B: control untreated rats. C and D: sham-operated (sham) rats. E and F: rats with bladder outlet obstruction (BOO). G and H: BOO rats treated with Ac2-26. I and J: rats obstructed for 12 days and then deobstructed for 2 days (Deobs). K and L: rats obstructed for 12 days and then deobstructed and treated with Ac2-26 for 2 days. A, C, E, G, I, and K: intravesicular pressure tracings for each group through several micturition cycles. The tracing is oriented so the first peaks of voiding pressure align for ease of comparison. The tracings reveal voiding pressure and intercontraction interval, the latter of which was used to calculate frequency. B, D, F, H, J, and L: continuous tracings from the scale showing a single void recorded during urodynamics for each group. The beginnings of a void are all aligned for ease of comparison. The tracings reveal voided volume and flow rate.
Figure 7.

Bladder outlet obstruction (BOO) causes high-pressure voiding and low flow rates that are not affected by Ac2-26 (Ac). Deobstruction (Deobs) restored normal voiding pressures and flow rates with no effect from Ac2-26. Animals were subjected to the treatment regimens described elsewhere and subjected to urodynamics. Recordings were analyzed using Med-CMG software (Catamount, St. Albans, VT). Typically, 5−10 individual micturition cycles were quantitated per animal and averaged for n = 1. A: voiding pressure = maximum pressure associated with the void. B: voiding flow rate = volume of a void divided by the duration of flow. Data are presented as means ± SE; n = 15, 13, 8, 7, 9, and 7 for both, respectively. **P < 0.01 and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test. Con, control.
We have established that overactive bladder-like symptoms occur by 12 days (6) in this BOO model. Overactive bladder is defined clinically as an increase in urinary frequency and urgency, but urgency cannot be measured in rats. In rats, overactive bladder-like symptoms are defined cystometrically as an increase in urinary frequency with a decrease in void volume. As shown in Figs. 6, E and F, and 8, A, and B, BOO rats demonstrated a large increase in frequency and a significant decrease in voided volume. Excitingly, the development of overactive bladder-like parameters was prevented by daily Ac2-26 administration (Figs. 6, G and H, and 8, A and B). In deobstructed rats, frequency returned to normal 2 days after deobstruction (Figs. 6I and 8A). However, void volume was significantly increased (Figs. 6J and 8B), a result likely due to an increased voiding efficiency after a decrease in outflow resistance. Importantly, Ac2-26 normalized this parameter (Figs. 6L and 8B). BOO rats also retained a significant PVR (Fig. 9A), and, markedly, Ac2-26 did not improve this parameter. Figure 9B shows that this PVR resulted from a decrease in voiding efficiency for both groups. Deobstructed rats demonstrated a measurable, but reduced, PVR after 2 days (Fig. 9A). This resulted in an increase in efficiency (Fig. 9B), which may help explain the higher voiding volume in deobstructed rats as shown in Fig. 8B. Ac2-26 did not affect PVR or voiding efficiency (Fig. 9).
Figure 8.

Ac2-26 (Ac) normalizes urinary frequency and void volume in response to bladder outlet obstruction (BOO) and after deobstruction (Deobs). Animals were subjected to the treatment regimens described elsewhere and subjected to urodynamics. Recordings were analyzed using Med-CMG software (Catamount, St. Albans, VT). Typically, 5−10 individual micturition cycles were quantitated per animal and averaged for n = 1. A: voiding frequency = number of voids divided by total time for those cycles. B: void volume = total volume voided. Data are presented as means ± SE; n = 15, 13, 8, 7, 9, and 7 for all, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test. Con, control.
Figure 9.

Ac2-26 (Ac) does not affect postvoid residual volume (PVR) or voiding efficiency in response to bladder outlet obstruction (BOO) or after deobstruction (Deobs). Animals were subjected to the treatment regimens described elsewhere and subjected to urodynamics. Recordings were analyzed using Med-CMG software (Catamount, St. Albans, VT). Typically, 5−10 individual micturition cycles were quantitated per animal and averaged for n = 1. A: PVR = total amount remaining after voiding (measured by drawing back on the catheter immediately after the last void and measuring any volume recovered). B: voiding efficiency = voided volume divided by (void volume + PVR). Data are presented as means ± SE; n = 15, 13, 8, 7, 9, and 7 for all, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 by ANOVA and a Student-Newman-Keuls test. Con, control.
DISCUSSION
Bladder inflammation is the root cause, or an exacerbating factor, in many different benign urological disorders. Recent work suggests that chronic inflammation likely results from a failure in proresolving pathways (8) and that manipulation of proresolving pathways has the potential for both halting the inflammatory process and reversing tissue damage (55, 56). This study is the first to explore the repertoire of resolving pathways in the bladder. We also assessed the therapeutic potential of one SPM, annexin-A1, to alleviate inflammation and bladder dysfunction in a rat model of BOO.
There are at least 26 SPMs, all of which bind to one or more of only seven GPRs (20, 22–24). Thus, to assess the potential resolving pathways present, we performed immunocytochemistry for six SPM receptors on bladders from control and BOO rats. The seventh receptor, GPR32, does not have an analog in rodents (43) and hence it was not examined. We found that, in fact, all of the receptors were present and localized primarily to the urothelia, although there was a minor expression for some in smooth muscle. It is interesting that we found a change in the expression pattern in a subset of receptors in response to BOO, mostly toward a more even expression in the entire urothelia instead of being concentrated in the luminal layer. In previous work, we have shown that NLRP3 was also expressed in the entire urothelia (6, 41). Colocalization of the proinflammatory NLRP3 inflammasome and SPM receptors is unsurprising considering that they form a tight feedback loop to regulate inflammation (for reviews, see Refs. 3 and 57). It is noteworthy that two other NLRs that have been implicated in triggering resolution pathways in other tissues, NLRP6 (58) and NLRP12 (59, 60), have also been found by our laboratory to be expressed in the bladder urothelium (41), although their functions have yet to be explored. One SPM receptor, GPR18, had a unique change in expression pattern with numerous individual positive cells appearing in both the urothelial and interstitial layers during BOO. It is unclear at this time if these GPR18+ cells are a subclass of individual urothelial cells that have turned on expression or immune cells that have migrated into the bladder in response to BOO. Overall, the data clearly show the presence of multiple proresolving receptors, with presumably intact proresolution pathways, in control urothelia and that the location of expression in specific tissues changes in response to BOO for some of these receptors.
We chose to focus on annexin-A1 in this study since it had previously been localized to the bladder and associated with a proinflammatory disease state (25). Moreover, it is well known to exert proresolution effects through a single receptor (FPR-2), and our survey clearly showed expression of FPR-2 in the bladder. Given the receptor pleiotropy of many of the other SPMs, the single-receptor association of annexin-A1 allowed for a clearer interpretation of the response. Immunohistochemical staining of annexin-A1 showed its basal presence predominantly in the urothelium with lesser amounts found in the detrusor and interstitial cells. We did not see a change in expression distribution in response to BOO, but the number of urothelial cells increased due to the well-known hyperplastic response.
Inflammation-initiating pathways often upregulate SPMs, which work to downregulate the expression or activity of the initiating pathways, forming a negative feedback loop (3, 57). To test the ability of Ac2-26 to act as an SPM on urothelial cells, we assessed its ability to prevent NLRP3 inflammasome activation by ATP in vitro (47, 48). We found a typical inhibition curve with an EC50 value of ≈0.25 µM and maximal inhibition at ≈0.5 µM, suggesting that Ac2-26 could indeed function as an SPM on urothelia. However, the interaction of annexin-A1 with NLRP3 is more complex than a simple inhibitor. In neutrophils and macrophages, endogenous annexin-A1 is actually needed for inflammasome priming (61) (upregulation of pro-IL-1β expression), possibly by binding directly to NLRP3 (61). This priming involves cytoplasmic annexin-A1 and is independent of FPR-2 (61). Since we have seen both NLRP3 (6, 41) and annexin-A1 (this study) localized to the urothelia, it is possible that this priming activity is also active in the bladder. In contrast, exogenous annexin-A1, conceivably derived from paracrine sources, can work through FPR-2 to negatively regulate NLRP3 (62), as we have also seen in this study. Therefore, in the bladder, annexin-A1 could be proinflammatory or proresolution depending on whether it is acting on its receptor from the extracellular space or directly on the inflammasome within the cytoplasm. Future studies will explore this possibility.
Having established the presence of annexin-A1 in the urothelium and its ability to act as an SPM, we sought to determine if it might be effective in vivo to alleviate the inflammation and bladder dysfunction associated with BOO and to enhance the recovery of function following deobstruction surgery. Consistent with our prior findings, rats with BOO demonstrated an increase in bladder weight and inflammation (Evans blue extravasation) (6). Concomitant administration of Ac2-26 diminished the bladder weight gain and inflammation, demonstrating that it is an effective SPM for bladder inflammation. From a functional standpoint, obstructed animals developed overactive bladder-like symptoms with increased urinary frequency and smaller voiding volumes, an effect we have shown is dependent on NLRP3-induced inflammation (6). Voiding efficiency was also decreased in those animals as PVR increased and bladders entered the decompensated phase of BOO. Interestingly, rats that were given Ac2-26 did not develop overactive bladder-like voiding, but they also were not protected from the decrease in voiding efficiency. It is clear that Ac2-26 is capable of suppressing the appearance of overactive bladder-like symptoms in this surgical rat model of BOO and conferring some degree of protection. Although the mechanism of this effect remains to be explored, we anticipate that it is a consequence of its effects on inflammation mediated by FPR-2. Inflammation during BOO is well established to cause overactive bladder symptoms, and we have shown that this is mediated through NLRP3 (6). Thus, direct inhibition of NLRP3 by Ac2-26 (as shown in this study) is one mechanism by which Ac2-26 may promote the return to normalcy from overactivity. Ac2-26 also promotes MAPK-dependent production of anti-inflammatory cytokines such as IL-10, which would further limit inflammation. These, and many other possible mechanisms by which Ac2-26 may affect overactive symptoms in the bladder, will provide much fodder for future mechanistic studies.
Deobstruction of the BOO model is ideal for studying the resolution of inflammation because elimination of the inciting inflammatory stimuli is as simple as cutting the restrictive suture around the urethra. In untreated animals, deobstruction leads to a resolution of inflammation and should lead to a decrease in bladder weight back to normal if enough time is allowed to pass. In our study, which only followed the animals for 2 days after deobstruction, there was not enough recovery time for bladder weight to return to baseline, although there was a significant reduction in inflammation (as measured by the Evans blue assay). Administration of Ac2-26 after deobstruction hastened the resolution of inflammation back to baseline and promoted a significant reduction of bladder weight. Functional recovery was not as impressive, with most urodynamic parameters essentially equal in deobstructed animals regardless of treatment. It is likely that the short period for the return of function (2 days) precludes observation of a therapeutic effect of annexin-A1, and these studies may also benefit from a chronic model that better captures the decompensated phase (7). Regardless, the data from these experiments establish the potential of an SPM, annexin-A1, to promote recovery from BOO after deobstruction.
In conclusion, we have found that the six known rodent SPM receptors are all present in the rat bladder along with the only protein SPM, annexin-A1. Furthermore, we found that an active biomimetic of annexin-A1, Ac2-26, is capable of antagonizing NLRP3 activation in cultured urothelial cells and prevents the initiation of inflammation in a surgical rat model of BOO. Excitingly, it significantly diminished BOO-induced bladder dysfunction and quickened the recovery of tissue health, and possibly return to function, after deobstruction.
GRANTS
This work was supported by a Poindexter Fellowship (to S.N.H), Duke University School of Medicine (to J.T.P.), National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK103534 and R01DK117890 (to J.T.P.), and the Urology Care Foundation (to S.N.H.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.M.H.Jr., S.N.H., and J.T.P. conceived and designed research; F.M.H.Jr., S.N.H., B.D.N., A.A., M.T.Z., and H.J. performed experiments; F.M.H.Jr., S.N.H., and B.D.N. analyzed data; F.M.H.Jr., S.N.H., B.D.N., and J.T.P. interpreted results of experiments; F.M.H.Jr. prepared figures; F.M.H.Jr. and S.N.H. drafted manuscript; F.M.H.Jr., S.N.H., B.D.N., A.A., and J.T.P. edited and revised manuscript; F.M.H.Jr. and J.T.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Julie Fuller and the Substrate Services Core and Research Support Services in the Department of Surgery for the help with histological embedding and sectioning. The authors also thank the Light Microscopy Core Facility and Yasheng Gao for the help in obtaining images.
REFERENCES
- 1.Sherwood ER, Toliver-Kinsky T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol 18: 385–405, 2004. doi: 10.1016/j.bpa.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, Teixeira MM. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther 139: 189–212, 2013. doi: 10.1016/j.pharmthera.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol 7: 160, 2016. doi: 10.3389/fimmu.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20: 95–112, 2020. doi: 10.1038/s41577-019-0215-7. [DOI] [PubMed] [Google Scholar]
- 5.Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 15: 493–518, 2020. doi: 10.1146/annurev-pathmechdis-012419-032847. [DOI] [PubMed] [Google Scholar]
- 6.Hughes FM , Jr., Hill HM, Wood CM, Edmondson AT, Dumas A, Foo WC, Oelsen JM, Rac G, Purves JT. The NLRP3 inflammasome mediates inflammation produced by bladder outlet obstruction. J Urol 195: 1598–1605, 2016. doi: 10.1016/j.juro.2015.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes FM , Jr., Sexton SJ, Ledig PD, Yun CE, Jin H, Purves JT. Bladder decompensation and reduction in nerve density in a rat model of chronic bladder outlet obstruction are attenuated with the NLRP3 inhibitor glyburide. Am J Physiol Renal Physiol 316: F113–F120, 2019. doi: 10.1152/ajprenal.00400.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnig C, Frossard N, Levy BD. Towards targeting resolution pathways of airway inflammation in asthma. Pharmacol Ther 186: 98–113, 2018. doi: 10.1016/j.pharmthera.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Fitzpatrick JM. The natural history of benign prostatic hyperplasia. BJU Int 97, Suppl 2: 3–6, 2006. doi: 10.1111/j.1464-410X.2006.06097.x. [DOI] [PubMed] [Google Scholar]
- 10.Purves JT, Hughes FM , Jr.. Inflammasomes in the urinary tract: a disease-based review. Am J Physiol Renal Physiol 311: F653–F662, 2016. doi: 10.1152/ajprenal.00607.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN, Hamberg M, Samuelsson B. Trihydroxytetraenes: a novel series of compounds formed from arachidonic acid in human leukocytes. Biochem Biophys Res Commun 118: 943–949, 1984. doi: 10.1016/0006-291x(84)91486-4. [DOI] [PubMed] [Google Scholar]
- 12.Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA 81: 5335–5339, 1984. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol 153, Suppl 1: S200–S215, 2008. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 510: 92–101, 2014. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serhan CN. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol Aspects Med 58: 1–11, 2017. doi: 10.1016/j.mam.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akasheh RT, Pang J, York JM, Fantuzzi G. New pathways to control inflammatory responses in adipose tissue. Curr Opin Pharmacol 13: 613–617, 2013. doi: 10.1016/j.coph.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaen RI, Sanchez-Garcia S, Fernandez-Velasco M, Bosca L, Prieto P. Resolution-based therapies: the potential of lipoxins to treat human diseases. Front Immunol 12: 658840, 2021. doi: 10.3389/fimmu.2021.658840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schett G, Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun 9: 3261, 2018. doi: 10.1038/s41467-018-05800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David S, Lopez-Vales R. Bioactive lipid mediators in the initiation and resolution of inflammation after spinal cord injury. Neuroscience 466: 273–297, 2021. doi: 10.1016/j.neuroscience.2021.04.026. [DOI] [PubMed] [Google Scholar]
- 20.Chiang N, Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem 64: 443–462, 2020. doi: 10.1042/EBC20200018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendez-Barbero N, Gutierrez-Munoz C, Blazquez-Serra R, Martin-Ventura JL, Blanco-Colio LM. Annexins: involvement in cholesterol homeostasis, inflammatory response and atherosclerosis. Clin Investig Arterioscler 33: 206–216, 2021. doi: 10.1016/j.arteri.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 22.Park J, Langmead CJ, Riddy DM. New advances in targeting the resolution of inflammation: implications for specialized pro-resolving mediator GPCR drug discovery. ACS Pharmacol Transl Sci 3: 88–106, 2020. doi: 10.1021/acsptsci.9b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panigrahy R, Singh B, Das SK. Diabetic uropathy and bladder dysfunctions. Diabetes Metab Syndr 11: 81–82, 2017. doi: 10.1016/j.dsx.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 24.Livshits G, Kalinkovich A. Receptors for pro-resolving mediators as a therapeutic tool for smooth muscle remodeling-associated disorders. Pharmacol Res 164: 105340, 2021. doi: 10.1016/j.phrs.2020.105340. [DOI] [PubMed] [Google Scholar]
- 25.Monastyrskaya K, Babiychuk EB, Draeger A, Burkhard FC. Down-regulation of annexin A1 in the urothelium decreases cell survival after bacterial toxin exposure. J Urol 190: 325–333, 2013. doi: 10.1016/j.juro.2013.01.088. [DOI] [PubMed] [Google Scholar]
- 26.Kitta T, Kakizaki H, Tanaka H, Sano H, Furuno T, Mitsui T, Moriya K, Nonomura K. An alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate glutamate-receptor antagonist can inhibit premicturition contractions in rats with bladder outlet obstruction. BJU Int 100: 181–186, 2007. doi: 10.1111/j.1464-410X.2007.06919.x. [DOI] [PubMed] [Google Scholar]
- 27.Kitta T, Kanno Y, Chiba H, Higuchi M, Ouchi M, Togo M, Moriya K, Shinohara N. Benefits and limitations of animal models in partial bladder outlet obstruction for translational research. Int J Urol 25: 36–44, 2018. doi: 10.1111/iju.13471. [DOI] [PubMed] [Google Scholar]
- 28.Reis LO, Sopena JM, Favaro WJ, Martin MC, Simao AF, Reis RB, Andrade MF, Domenech JD, Cardo CC. Anatomical features of the urethra and urinary bladder catheterization in female mice and rats. An essential translational tool. Acta Cir Bras 26 Suppl 2: 106–110, 2011. doi: 10.1590/s0102-86502011000800019. [DOI] [PubMed] [Google Scholar]
- 29.Hughes FM , Jr., Hirshman NA, Malick HA, White SW, Jin H, Harper SN, Purves JT. A possible mechanism underlying mood disorders associated with LUTS: chronic bladder outlet obstruction causes NLRP3-dependent inflammation in the hippocampus and depressive behavior in rats. Neurourol Urodyn 39: 1700–1707, 2020. doi: 10.1002/nau.24448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutolf R, Hughes FM , Jr., Inouye BM, Jin H, McMains JC, Pak ES, Hannan JL, Purves JT. NLRP3/IL-1β mediates denervation during bladder outlet obstruction in rats. Neurourol Urodyn 37: 952–959, 2018. doi: 10.1002/nau.23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelmoaty S, Wigerblad G, Bas DB, Codeluppi S, Fernandez-Zafra T, El-Awady e. S, Moustafa Y, Abdelhamid AD, Brodin E, Svensson CI. Spinal actions of lipoxin A4 and 17(R)-resolvin D1 attenuate inflammation-induced mechanical hypersensitivity and spinal TNF release. PLoS One 8: e75543, 2013. doi: 10.1371/journal.pone.0075543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen K, Liu M, Liu Y, Yoshimura T, Shen W, Le Y, Durum S, Gong W, Wang C, Gao JL, Murphy PM, Wang JM. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J Clin Invest 123: 1694–1704, 2013. doi: 10.1172/JCI65569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh D, Qi R, Jordan JL, San Mateo L, Kao CC. The human antimicrobial peptide LL-37, but not the mouse ortholog, mCRAMP, can stimulate signaling by poly(I:C) through a FPRL1-dependent pathway. J Biol Chem 288: 8258–8268, 2013. doi: 10.1074/jbc.M112.440883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jank BJ, Kadletz L, Dunkler D, Haas M, Schnoell J, Kenner L, Heiduschka G. Epithelial stem cell marker LGR6 expression identifies a low-risk subgroup in human papillomavirus positive oropharyngeal squamous cell carcinoma. Oral Oncol 105: 104657, 2020. doi: 10.1016/j.oraloncology.2020.104657. [DOI] [PubMed] [Google Scholar]
- 35.Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, Grimprel E, Landman-Parker J, Hentgen V, Marlin S, McElreavey K, Sarkisian T, Grateau G, Alnemri ES, Amselem S. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA 105: 1614–1619, 2008. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu V, Auchman M, Mollica PA, Sachs PC, Bruno RD. ALDH1A1 positive cells are a unique component of the tonsillar crypt niche and are lost along with NGFR positive stem cells during tumourigenesis. Pathology 50: 524–529, 2018. doi: 10.1016/j.pathol.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Baranowska-Kuczko M, Kozłowska H, Kloza M, Sadowska O, Kozłowski M, Kusaczuk M, Kasacka I, Malinowska B. Vasodilatory effects of cannabidiol in human pulmonary and rat small mesenteric arteries: modification by hypertension and the potential pharmacological opportunities. J Hypertens 38: 896–911, 2020. doi: 10.1097/HJH.0000000000002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seidel S, Neymeyer H, Kahl T, Roschel T, Mutig K, Flower R, Schnermann J, Bachmann S, Paliege A. Annexin A1 modulates macula densa function by inhibiting cyclooxygenase 2. Am J Physiol Renal Physiol 303: F845–F854, 2012. doi: 10.1152/ajprenal.00704.2011. [DOI] [PubMed] [Google Scholar]
- 39.Harper SN, Leidig PD, Hughes FM , Jr., Jin H, Purves JT. Calcium pyrophosphate and monosodium urate activate the NLRP3 inflammasome within bladder urothelium via reactive oxygen species and TXNIP. Res Rep Urol 11: 319–325, 2019. doi: 10.2147/RRU.S225767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes FM , Jr., Hirshman NA, Inouye BM, Jin H, Stanton EW, Yun CE, Davis LG, Routh JC, Purves JT. NLRP3 promotes diabetic bladder dysfunction and changes in symptom-specific bladder innervation. Diabetes 68: 430–440, 2019. doi: 10.2337/db18-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hughes FM , Jr., Turner DP, Purves JT. The potential repertoire of the innate immune system in the bladder: expression of pattern recognition receptors in the rat bladder and a rat urothelial cell line (MYP3 cells). Int Urol Nephrol 47: 1953–1964, 2015. doi: 10.1007/s11255-015-1126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider MP, Hughes FM , Jr., Engmann AK, Purves JT, Kasper H, Tedaldi M, Spruill LS, Gullo M, Schwab ME, Kessler TM. A novel urodynamic model for lower urinary tract assessment in awake rats. BJU Int 115, Suppl 6: 8–15, 2015. doi: 10.1111/bju.13039. [DOI] [PubMed] [Google Scholar]
- 43.Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR review 7. Br J Pharmacol 171: 3551–3574, 2014. doi: 10.1111/bph.12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petri MH, Laguna-Fernandez A, Gonzalez-Diez M, Paulsson-Berne G, Hansson GK, Back M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res 105: 65–74, 2015. doi: 10.1093/cvr/cvu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Back M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, Hansson GK. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci USA 102: 17501–17506, 2005. doi: 10.1073/pnas.0505845102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carracedo M, Artiach G, Witasp A, Claria J, Carlstrom M, Laguna-Fernandez A, Stenvinkel P, Back M. The G-protein coupled receptor ChemR23 determines smooth muscle cell phenotypic switching to enhance high phosphate-induced vascular calcification. Cardiovasc Res 115: 1557–1566, 2019. doi: 10.1093/cvr/cvy316. [DOI] [PubMed] [Google Scholar]
- 47.Duvall MG, Bruggemann TR, Levy BD. Bronchoprotective mechanisms for specialized pro-resolving mediators in the resolution of lung inflammation. Mol Aspects Med 58: 44–56, 2017. doi: 10.1016/j.mam.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RX, Colgan SP. Special pro-resolving mediator (SPM) actions in regulating gastro-intestinal inflammation and gut mucosal immune responses. Mol Aspects Med 58: 93–101, 2017. doi: 10.1016/j.mam.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hughes FM , Jr., Kennis JG, Youssef MN, Lowe DW, Shaner BE, Purves JT. The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome mediates inflammation and voiding dysfunction in a lipopolysaccharide-induced rat model of cystitis. J Clin Cell Immunol 7: 396, 2016. doi: 10.4172/2155-9899.1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes FM , Jr., Vivar NP, Kennis JG, Pratt-Thomas JD, Lowe DW, Shaner BE, Nietert PJ, Spruill LS, Purves JT. Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. Am J Physiol Renal Physiol 306: F299–F308, 2014. doi: 10.1152/ajprenal.00297.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cirino G, Cicala C, Sorrentino L, Ciliberto G, Arpaia G, Perretti M, Flower RJ. Anti-inflammatory actions of an N-terminal peptide from human lipocortin 1. Br J Pharmacol 108: 573–574, 1993. doi: 10.1111/j.1476-5381.1993.tb12843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheikh MH, Solito E. Annexin A1: uncovering the many talents of an old protein. Int J Mol Sci 19: 1045, 2018. doi: 10.3390/ijms19041045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin X, He L, Fan D, Liang W, Wang Q, Fang J. Targeting the resolution pathway of inflammation using Ac2–26 peptide-loaded PEGylated lipid nanoparticles for the remission of rheumatoid arthritis. Asian J Pharm Sci. In press. doi: 10.1016/j.ajps.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin Y, Avendaño C, Piedras MJ, Krzyzanowska A. Evaluation of Evans blue extravasation as a measure of peripheral inflammation. Protocol Exchange. In press. doi: 10.1038/protex.2010.209. [DOI] [Google Scholar]
- 55.Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterol 155: 282–302.e8, 2018. doi: 10.1053/j.gastro.2018.06.031. [DOI] [PubMed] [Google Scholar]
- 56.Musso G, Gambino R, Cassader M, Paschetta E, Sircana A. Specialized proresolving mediators: enhancing nonalcoholic steatohepatitis and fibrosis resolution. Trends Pharmacol Sci 39: 387–401, 2018. doi: 10.1016/j.tips.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 57.Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res 2016: 8239258, 2016. doi: 10.1155/2016/8239258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti TD. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488: 389–393, 2012. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, Ting JP. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol 178: 1256–1260, 2007. doi: 10.4049/jimmunol.178.3.1256. [DOI] [PubMed] [Google Scholar]
- 60.Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, Kanneganti TD. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20: 649–660, 2011. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galvao I, de Carvalho RVH, Vago JP, Silva ALN, Carvalho TG, Antunes MM, Ribeiro FM, Menezes GB, Zamboni DS, Sousa LP, Teixeira MM. The role of annexin A1 in the modulation of the NLRP3 inflammasome. Immunology 160: 78–89, 2020. doi: 10.1111/imm.13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanches JM, Correia-Silva RD, Duarte GHB, Fernandes A, Sanchez-Vinces S, Carvalho PO, Oliani SM, Bortoluci KR, Moreira V, Gil CD. Role of annexin A1 in NLRP3 inflammasome activation in murine neutrophils. Cells 10: 121, 2021. doi: 10.3390/cells10010121. [DOI] [PMC free article] [PubMed] [Google Scholar]