

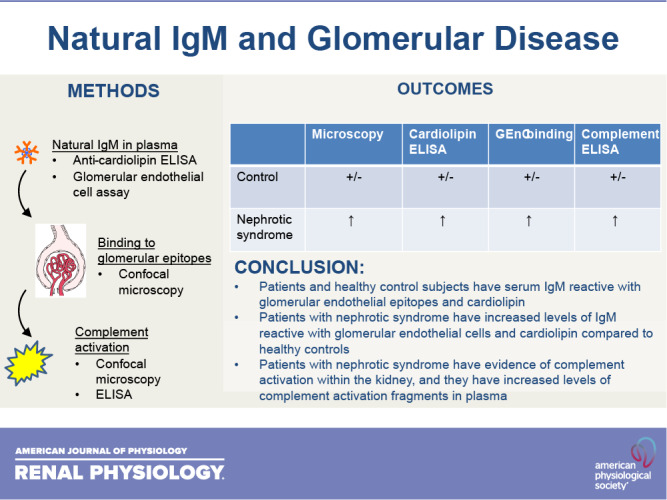
Keywords: complement, focal segmental glomerulosclerosis, IgM, minimal change disease, nephrotic syndrome
Abstract
Focal segmental glomerulosclerosis (FSGS) and minimal change disease (MCD) are common forms of idiopathic nephrotic syndrome. The causes of these diseases are incompletely understood, but the response of patients to immunosuppressive therapies suggests that their pathogenesis is at least in part immune mediated. Preclinical and clinical research indicates that activation of the classical pathway of complement contributes to glomerular injury in FSGS. Glomerular IgM deposits are also prominent in some patients, raising the possibility that IgM is a trigger of classical pathway activation. In the present study, we examined the pattern of complement activation in the glomeruli and plasma of patients with nephrotic syndrome. We also tested whether patients with FSGS and MCD have elevated levels of natural IgM reactive with epitopes on glomerular endothelial cells and cardiolipin. We found evidence of classical pathway activation in patients with idiopathic nephrotic syndrome compared with healthy control subjects. We also detected higher levels of self-reactive IgM to both targets. Based on these results, IgM and classical pathway activation may contribute to disease pathogenesis in some patients with FSGS and MCD.
NEW & NOTEWORTHY IgM is detected in biopsies from some patients with nephrotic syndrome, although this has been attributed to passive trapping of the protein. We found, however, that IgM colocalizes with complement activation fragments in some glomeruli. We also found that affected patients had higher levels of IgM reactive to glomerular endothelial cell epitopes. Thus, IgM activates the complement system in the glomeruli of some patients with nephrotic syndrome and may contribute to injury.
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is the most common cause of primary nephrotic syndrome in adults, and the incidence is increasing in children and adults (1, 2). Minimal change disease (MCD) is a related form of nephrotic syndrome that is also associated with various systemic conditions but usually presents as a primary glomerular disease (3). FSGS and MCD can overlap clinically, and many experts believe that they may represent two entities on a spectrum that share pathophysiological mechanisms. The molecular causes of these diseases are incompletely understood, however, hindering our ability to identify patients at risk for the diseases or to monitor their response to treatment.
There is mounting evidence that FSGS and MCD are immune-mediated diseases. First, many patients with these conditions respond to treatment with glucocorticoids or calcineurin inhibitors (4). In addition, C3 and IgM are deposited in the glomeruli of a subset of patients with both diseases (5), and several clinical studies have found that these deposits are associated with worse clinical outcomes. In one study, for example, patients with both glomerular IgM and C3 had a worse prognosis than patients who had no detectable IgM deposits or who had isolated IgM deposits without concomitant C3 (6). Lower plasma C3 levels are also associated with more rapid disease progression, further implicating complement activation in disease pathogenesis (7). Although renal deposition of IgM and C3 has been attributed to trapping of these large proteins in areas of scar, studies in animal models of FSGS have shown that the deposition of complement proteins in injured glomeruli is due to activation of the complement cascade, not simply passive trapping of the proteins (8, 9). Furthermore, complement deficiency is protective in these models.
IgM is a potent activator of the classical pathway of complement, and several studies have provided evidence that the classical pathway is activated in FSGS. Several years ago, we reported that C4a (a marker of classical pathway activation) was higher in plasma from patients with FSGS compared with control subjects (10), and a study recently reported that C4d levels are similarly elevated (11). Another study showed that both C4d and C1q deposits were detectable in the majority of FSGS biopsies and that the proteins colocalized within glomeruli (12). Importantly, C4d deposition was seen in nonsclerotic glomeruli, and it preceded the development of segmental scars in kidney allografts with recurrent disease. These observations argue against passive trapping and point to a pathogenic role for complement proteins.
“Natural” antibody is germline-encoded Ig that frequently binds to self-epitopes, including modified phospholipids displayed on the surface of injured cells (13, 14). Natural antibody facilitates the clearance of injured cells, but it can also activate complement within damaged tissues, exacerbating injury (15, 16). Although the identity of the glomerular epitopes bound by IgM in FSGS are not known, we previously generated a monoclonal natural IgM antibody that bound to murine endothelial cells in vitro and to injured glomeruli when injected in vivo (17). The natural IgM antibody that bound renal tissue was screened against a number of phospholipids and was found to bind to cardiolipin (15), raising the possibility that this is a target antigen for natural IgM in FSGS. Injury to glomerular cells may expose cardiolipin or other phospholipid epitopes on the surface of injured cells. Natural antibody could then bind to these epitopes and activate complement, causing immune-mediated injury to the glomerulus.
The level of specific natural antibodies can vary among individuals (18, 19). In the setting of acquired glomerular injury, patients who have higher levels of natural IgM reactive to glomerular endothelial cells (GEnCs) or cardiolipin might be more prone to secondary activation of the classical pathway in the glomerulus. Levels of natural IgM and complement activation fragments could, therefore, contribute to the pathogenesis of idiopathic nephrotic syndrome and predict disease progression. To explore this hypothesis, we analyzed biopsy tissue, plasma, and urine samples from patients with nephrotic syndrome. We compared the results with those obtained in healthy and disease controls.
METHODS
Study Population
Biopsy samples.
We analyzed leftover kidney biopsy tissue from 10 patients with FSGS (Table 1). These samples were chosen based on the availability of tissue and included six cases diagnosed as primary FSGS and four cases diagnosed as secondary disease. As negative controls, we included 10 biopsies with thin glomerular basement membrane disease (TBM) and peritumoral tissue from two patients who underwent nephrectomy for renal cell carcinoma.
Table 1.
Biopsy sample patient characteristics
| Thin Basement Membrane Biopsy Samples |
FSGS Biopsy Samples |
|
|---|---|---|
| n | 10 | 10 |
| Age, yr | 47.5 (36.4, 49.7)* | 37 (31.75, 50.25)* |
| Sex (male/female) | 2/8 | 8/2 |
| Race | ||
| European American | 10 | 3 |
| African American | 7 | |
| Hispanic | ||
| Asian | ||
| FSGS subtype | ||
| Primary | 6 | |
| Secondary | 4 | |
| Serum creatinine, mg/dL | 0.80 (0.72, 0.85)* | 2.0 (1.2, 5.98)* |
| Urine protein excretion, g/day | 0.08 mg/g creatinine (0.07, 0.14)* | 4.85 g/day (2.4, 11.1)* |
FSGS, focal segmental glomerulosclerosis; IQR, interquartile range. *Median (IQR).
Plasma and urine samples.
We analyzed plasma, serum, and urine samples from 95 patients with FSGS or MCD enrolled in the Nephrotic Syndrome Study Network (NEPTUNE), a long-term observational study of patients with proteinuric kidney diseases (Table 2) (20). Baseline samples and samples collected up to 36 mo after study entry were analyzed. To validate the results from the NEPTUNE samples, we prospectively collected samples from 15 patients with nephrotic syndrome collected at baseline and 3–10 mo after starting tacrolimus. As controls for these studies, we analyzed plasma, serum, and urine samples from 30 healthy subjects. Anti-cardiolipin antibodies were also measured in samples from 30 patients with nonproteinuric chronic kidney disease (CKD) (21). Patient characteristics are shown in Table 2. All of the samples were collected with the approval of the Institutional Review Board and the Human Subjects Committee at the institution where they were obtained, and all of the subjects consented to the use of these samples for research.
Table 2.
Plasma and urine sample patient characteristics
| Healthy Control Plasma |
NEPTUNE Cohort Plasma |
Validation Cohort Plasma |
|
|---|---|---|---|
| n | 30 | 95 | 15 |
| Age, yr | 38 ± 13 (20−65 yr) | 37 ± 18 | 12 ± 5 |
| Sex (male/female) | 6/24 | 59/36 | 9/6 |
| Race | |||
| European American | 27 (90%) | 55 | 6 |
| African American | 18 | 1 | |
| Hispanic | 23 | 2 | |
| Asian | 13 | 6 | |
| Disease | |||
| Focal segmental glomerulosclerosis | 45 | 8 | |
| Minimal change disease | 30 | 7 | |
| Systolic blood pressure, mmHg | 116 ± 10 | 121 ± 15 | 111 ± 12 |
| Diastolic blood pressure, mmHg | 72 ± 8 | 75 ± 11 | 65 ± 7 |
| Estimated glomerular filtration rate, mL/min/1.73 m2 | 82 ± 17 | 71 ± 32 | 142 ± 100 |
| Urine protein:creatinine, mg/g | 0.08 ± 0.09 | 2.56 ± 4.21 | 1.43 ± 1.85 |
| Immunosuppression | |||
| Glucocorticoids | 29 | 6 | |
| Calcineurin inhibitors | 3 | 14 | |
| Mycophenolate mofetil | 1 | 2 | |
| Other | 3 | ||
| Renin-angiotensin-aldosterone system inhibition | 38 (40%) | 3 (20%) |
NEPTUNE, Nephrotic Syndrome Study Network Consortium.
Antibodies
Antibodies used for tissue staining included mouse anti-human C4d (Quidel, San Diego, CA), DyLight 650 anti-human IgM, μ-chain (Abcam, Cambridge, UK), a DyLight 550-labeled antibody that reacts with human iC3b/C3d (22), a mouse monoclonal antibody to a human C9 neoepitope (C9neo) generated when the terminal complement complex forms in cell membranes (23), Alexa 488 goat anti-mouse IgG (Invitrogen, Waltham, MA), Cy3 goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), Alexa 647 goat IgG (Southern Biotechnology, Birmingham, AL), mouse IgG1, clone MOPC-21 (BioLegend, San Diego, CA), and DyLight 650-mouse IgG2a isotype (created and validated in house). Binding of IgM to endothelial cells was detected using a Cy3-conjugated goat anti-mouse IgM µ-chain antibody (Jackson ImmunoResearch Laboratories). The control IgM antibody used for this experiment was a murine anti-lipopolysaccharide IgM (Southern Biotech). Deposition of C3c on endothelial cells was detected using a FITC-conjugated rabbit anti-human C3c antibody (Aligent/Dako). C2 is a natural IgM κ hybridoma generated from a peritoneal B1 cell of a C57BL/6 mouse (15). The antibody was purified from hybridoma supernatant by affinity chromatography with anti-mouse IgM (µ-chain specific)-agarose (Sigma-Aldrich). Previous work has shown that the C2 antibody binds to cardiolipin and to endothelial cells (15, 17).
Complement and IgM ELISAs
Complement fragments Ba, C4a, and sC5b-9 were detected in patient plasma and urine using MicroVue enzyme immunoassay kits (Quidel). Plasma was diluted 1:40 (C4a), 1:10 (sC5b-9), and 1:1,000 (Ba). Urine was diluted 1:5 (C4a and sC5b-9) and 1:200 (Ba). Serum IgM levels were measured using Human IgM ELISA Kits (Abcam). Samples were diluted 1:60,000 and run in triplicate. Assays were performed, and analyte concentrations were determined according to manufacturer specifications.
Immunofluorescent Confocal Microscopy
Kidney biopsy sections were warmed to room temperature, fixed in absolute acetone for 10 min, and washed twice with cold PBS. Nonspecific binding was blocked with 1% BSA, 5% heat-inactivated goat serum, and 2.5% heat-inactivated FBS in PBS for 1 h at room temperature. Primary and secondary antibodies were diluted in 2% heat-inactivated FBS and 1% BSA in PBS. Tissues were incubated for 14 h at 4°C with primary antibodies to C4d (16.7 μg/mL), IgM (0.5 μg/mL), iC3b/C3d (6 μg/mL), and/or C9neo (14.5 μg/mL). The tissues were then washed three times with PBS and incubated with Alexa 488 goat anti-mouse secondary antibody (2.5 μg/mL) for 1 h at room temperature. Tissues were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride to detect nuclei, washed three times in cold PBS, and mounted with a 1:1 solution of PBS and glycerol. Slides were then sealed and imaged in a blinded fashion with an Olympus FV1000 FCS inverted confocal microscope (Olympus Life Science, Tokyo, Japan) at ×100, ×200, or ×600 original magnification. A minimum of 10 fields of view were captured per sample. Images were converted from the binary data format and analyzed with Olympus FV-10ASW software (version 04.02.02.09). Quantification was performed on images collected at ×100 magnification by region of interest selection and measurement of fluor intensity in each specific channel following subtraction of background autofluorescence. Every glomerulus was analyzed, and 5–20 regions of interest in the tubulointerstitium were analyzed depending on the size of the biopsy.
IgM Cell Binding Assays
To detect binding of IgM in normal human serum to endothelial cells grown in vitro, human microvascular endothelial cells (American Type Culture Collection, Manassas, VA) were exposed to various concentrations of H2O2 for 10 min. Cells were washed three times and then exposed to 25% normal human serum (Complement Technology, Tyler, TX) for 60 min at 37°C. Cells were stained for bound IgM or deposited C3C and imaged with a BioTek Cytation1 imaging system (BioTek, Winooski, VT). Image analysis was performed using Gen5 3.05 software.
To measure the level of IgM in patient samples that is reactive with epitopes displayed on whole cells, we incubated conditionally immortalized human GEnCs with serum samples and measured bound IgM by flow cytometry (24). Cells were grown in tissue culture flasks. After the cells were differentiated, they were detached from the flasks with Accutase (Innovative Cell Technologies, San Diego, CA) and washed in PBS. Then, 0.5 × 106 cells were suspended in 50-µL PBS containing 5% patient serum. The mixture was incubated on ice for 1 h and then gently vortexed every 20 min to keep the cells in suspension. Cells were then washed with PBS and incubated for 1 h at 4°C in 50-µL DyLight 650 anti-human IgM, μ-chain, diluted 1:200. Cells were again washed, and fluorescence was then measured using a FACScalibur flow cytometer. Approximately 300,000 events were collected for each sample. The results were analyzed using FlowJo software, and cells stained with aliquots of a pooled sample of normal human serum were used to normalize the results between assays.
Anti-Cardiolipin Antibody Assays
Detection of autoantibodies (IgG and IgM) reactive with cardiolipin was based on previously published methods (25). Briefly, a nontreated polystyrene microtiter plate was coated with 50 μg/mL cardiolipin from the bovine heart (Sigma-Aldrich) diluted in absolute ethanol. The plate was incubated uncovered for 16–18 h at 4°C with gentle rocking. It was then washed twice with PBS (pH 7.4), gently blotted dry, and then blocked with 10% active FBS (Thermo Fisher, Grand Island, NY) in PBS for 2 h at room temperature. Human anti-cardiolipin IgG and IgM standards were purchased from Louisville APL Diagnostics and prepared according to the manufacturer’s instructions. Human serum samples were diluted in cold blocking buffer, added to the ELISA plate, and incubated for 1 h at 4°C. Plates were then washed three times. Horseradish peroxidase-conjugated goat anti-human antibodies (EMD Millipore, Darmstadt, Germany) were then used to detect IgM and IgG (diluted 1:6,000 and 1:3,000, respectively). The plates were then developed with 3,3′,5,5′-tetramethylbenzidine, and the reaction was terminated with 0.2 M sulfuric acid. The color change was measured at 450 nm. A log-log calibration curve was generated using background adjusted mean values of human standards and was used to determine the levels of anti-cardiolipin IgM and IgG (CL-M and CL-G, respectively) in each sample.
Statistical Analysis
Descriptive statistics are reported for all variables as n (%) for categorical variables and means (SD) for continuous variables. Comparisons between two groups were performed using a two-tailed t test. A paired t test and correlation analysis were performed if appropriate. Comparisons between multiple groups were performed using ANOVA with Tukey’s test for pairwise multiple comparisons. Analysis of linear regression was performed to assess the cross-sectional association between baseline clinical parameters and the experimental measurements, with and without adjustment for age, sex, race, and cohort. A linear mixed effects model with random intercept using longitudinal data was performed to investigate how treatment with steroids impacted the experimental measurements, with and without adjustment for covariates. The above analyses were performed using SAS 9.4 (SAS Institute, Cary, NC). Pearson’s r value calculations and the corresponding correlation matrixes for fluorescence values were generated through multiple variable analysis in GraphPad Prism. A two-sided significance level of 0.05 was used to determine significance of all results.
RESULTS
IgM and Complement Activation Fragments Are Deposited in Biopsies of Patients with FSGS
It has long been known that complement proteins are deposited in the glomeruli of patients with FSGS (5, 6), but the presence of complement proteins has been attributed to the passive trapping of plasma proteins. To address this question, we stained kidney biopsy tissue from patients with FSGS using antibodies specific for fragments generated during complement activation. We stained tissues for IgM, C4d, iC3b/C3d fragments (22) and an epitope on C9 that is generated during the formation of the C5b-9 complex (C9neo), and the slides were imaged by confocal microscopy. Isotype control staining was performed to confirm the specificity of the staining (Supplemental Fig. S1, https://doi.org/10.6084/m9.figshare.14690733), and biopsies with TBM were used as a disease control. Deposits of the four immune proteins were detected within some glomeruli in all 10 of the FSGS biopsies examined, although the abundance of the deposits varied among different glomeruli, even within the same biopsy (Fig. 1A). All of the immune proteins were deposited within some glomeruli (Fig. 1B), supportive of the idea that IgM can bind to glomerular epitopes and activate the classical pathway. We detected low levels of complement proteins in TBM samples (Fig. 1C), similar to what has been previously reported (26). There was heterogeneity in the pattern of the deposits among glomeruli within each kidney biopsy and among the kidney biopsies for both FSGS (Fig. 1 and Supplemental Fig. S2, https://doi.org/10.6084/m9.figshare.14690760) and TBM samples (Fig. 1 and Supplemental Fig. S3, https://doi.org/10.6084/m9.figshare.14690781). In some glomeruli, there were regions of IgM and C4d deposition, but little iC3b/C3d or C9neo were seen. Furthermore, iC3b/C3d and C9neo were more prominent in areas of glomerular injury (Supplemental Fig. S2, https://doi.org/10.6084/m9.figshare.14690760). Nevertheless, the positive staining with anti-iC3b/C3d and C9 neo antibodies indicated that these fragments are due to activation of the complement cascade and are not simply due to trapping of intact C3 and C9 proteins.
Figure 1.

IgM and complement deposits in focal segmental glomerulosclerosis (FSGS). Biopsy samples from patients with FSGS or thin glomerular basement membrane disease (TBM) were stained for IgM, C4d, iC3b/C3d, and C9neo. A: representative images from a single primary FSGS biopsy containing three glomeruli. Individual glomeruli are indicated with a circle, rectangle, and hexagon. iC3b/C3d (red) and C9neo (shown in turquoise in the right image) were seen in only a subset of the glomeruli that were positive for IgM (blue) and C4d (green). Original magnification: ×200. Scale bars = 100 µm. B: high-powered views of a single glomerulus from a FSGS biopsy showing that IgM (blue), C4d (green), and iC3b/C3d (red) were deposited in a similar pattern within the glomerulus. Original magnification: ×600. Scale bars = 20 µm. C: high-powered views of a glomerulus from a TBM biopsy showing that all of the immune proteins could be detected, although to a lesser degree than in FSGS. Original magnification: ×600.
Complement Activation Fragments Are Elevated in Plasma from Patients with Nephrotic Syndrome
We measured complement fragments in plasma samples from patients with nephrotic syndrome who were part of the NEPTUNE cohort and also from a second cohort of 15 pediatric patients with idiopathic nephrotic syndrome (“validation” group). C4a levels in both groups of patients with nephrotic syndrome were higher than in healthy controls, indicating that there was classical pathway activation in these patients (Fig. 2A). We did not see a difference in C4a levels between patients with MCD and FSGS or between patients who did or did not achieve a complete or partial remission during follow-up. sC5b-9 levels were higher in both cohorts compared with healthy controls, although the increase in the validation cohort did not reach statistical significance (Fig. 2B). There was a trend toward lower Ba levels in nephrotic syndrome cohorts compared with healthy controls. Ba levels were higher in patients with FSGS compared with MCD, and they were also higher in those patients who did not have a complete or partial remission during follow-up compared with those who did (Fig. 2C). However, Ba levels were inversely proportional to estimated glomerular filtration rate (eGFR; Supplemental Fig. S4, https://doi.org/10.6084/m9.figshare.14690799; Supplemental Fig. S5, https://doi.org/10.6084/m9.figshare.14690871.v1). Therefore, changes in Ba are more likely to be due to a reduction in GFR than to the presence and/or etiology of nephrotic syndrome. Levels of all three complement fragments were stable over time in the NEPTUNE cohort (Fig. 3).
Figure 2.

Complement activation fragments in plasma from patients with nephrotic syndrome. We measured C4a, Ba, and sC5b-9 in baseline plasma from patients with nephrotic syndrome and healthy controls. A: C4a levels were higher in both cohorts of nephrotic patients compared with healthy controls. B: sC5b-9 levels were higher in samples from the Nephrotic Syndrome Study Network Consortium (NEPTUNE) cohort of patients with nephrotic syndrome compared with healthy controls. C: Ba levels did not significantly differ between patients with nephrotic syndrome or control subjects, but they were significantly higher in patient with focal segmental glomerulosclerosis (FSGS) compared with patients with minimal change disease (MCD). Ba levels were also higher in those who did not achieve a complete or partial remission during follow-up (no PR) compared with those who did (CR/PR). n = 29 for the healthy controls, 90 for the NEPTUNE samples, and 15 for the validation cohort. The red bars indicate medians for each group. **P < 0.01; ***P < 0.001.
Figure 3.

Changes in complement activation fragments over time and after immunosuppression. We measured C4a, Ba, and sC5b-9 in plasma collected in 83 patients with nephrotic syndrome from the Nephrotic Syndrome Study Network Consortium cohort at baseline and during follow-up visits. No significant changes in any of the complement fragments were seen over time. Graphs show the medians for each time point; bars indicate interquartile ranges.
Urine C4a levels were higher in samples from both groups of patients with nephrotic syndrome compared with control samples (Fig. 4). Urine sC5b-9 levels were higher in the NEPTUNE cohort and trended higher (P = 0.05) in the validation cohort. It is worth noting that increases in urine complement fragments can be a nonspecific result of passage of plasma complement proteins into the urinary space and might not reflect activation within the glomerulus (27).
Figure 4.

Complement activation fragments in urine from patients with nephrotic syndrome. We measured C4a, Ba, and sC5b-9 in baseline urine from patients with nephrotic syndrome and healthy controls. C4a levels were higher in both cohorts of patients compared with healthy controls. Ba levels were not significantly elevated in either of the patient cohorts. sC5b-9 levels were higher in samples from the Nephrotic Syndrome Study Network Consortium (NEPTUNE) cohort of patients with nephrotic syndrome compared with healthy controls. n = 29 for the healthy controls, 90 for the NEPTUNE samples, and 15 for the validation cohort. The red bars indicate medians and interquartile ranges. ***P < 0.001.
IgM with Reactivity to GEnCs Was Elevated in Plasma of Patients with Nephrotic Syndrome Compared with Healthy Controls
The natural IgM clone C2 did not react with unmanipulated microvascular endothelial cells grown in culture, but C2 did bind to cells after they were exposed to H2O2 (Supplemental Fig. S6, https://doi.org/10.6084/m9.figshare.14691000). This was not simply due to nonspecific binding, as control monoclonal IgM did not bind to the cells under either condition. IgM in normal serum also bound to injured endothelial cells and was associated with complement activation on the cell surface (Supplemental Fig. S6, B and C). This indicates that normal subjects have IgM that reacts with epitopes on injured endothelial cells. C2 and IgM in normal human serum also bound to GEnCs after they were enzymatically detached from tissue culture plates, suggesting that this process induces mild injury in the cells (Fig. 5A). Natural antibody clones reacted to varying degrees with injured human GEnCs in vitro, indicating different affinities of the various clones for the cells (Fig. 5A).
Figure 5.

IgM in the serum of patients with nephrotic syndrome binds to glomerular endothelial cells (GenCs). A: we tested binding of various natural IgM clones to human GenCs and confirmed that the natural antibody clone C2 reacted with human cells (dark black line). The isotype control is shown as a shaded gray peak. The other curves represent distinct natural antibody clones, illustrating the variable ability to bind GEnCs. B: serum from healthy controls (n = 78) and from the Nephrotic Syndrome Study Network Consortium (NEPTUNE) cohort of patients (n = 115) with nephrotic syndrome were incubated with GEnCs in vitro, and binding of IgM was analyzed by flow cytometry (GEnC IgM). Greater binding of IgM to GEnCs was seen in patients with nephrotic syndrome. *P < 0.05. C: GEnC IgM was inversely correlated with estimated glomerular filtration rate (eGFR). D: GEnC IgM levels were in patients who had a complete or partial remission (CR/PR) during follow-up compared with patients who did not have either a complete or partial remission (no PR). E: total serum IgM levels were lower in serum samples from the NEPTUNE cohort of patients (n = 30) compared with healthy controls (n = 10). **P < 0.01. RFU, relative fluorescence units.
To test the reactivity of IgM in the serum of patients with nephrotic syndrome against epitopes displayed on GEnCs, we incubated cells with serum from patients in the NEPTUNE cohort and healthy controls. We then measured the amount of IgM that bound to the cells by flow cytometry (GEnC IgM). A greater amount of IgM bound to GEnCs after incubation with serum from patients with nephrotic syndrome compared with control samples, although it was only about a 10% increase and there was a large degree of overlap in the groups (Fig. 5B). When we correlated GEnC IgM values with clinical data, there was an inverse relationship of GEnC IgM with eGFR (Fig. 5C). Average GEnC IgM levels were also higher in those who failed to achieve either a complete or partial remission over the course of follow-up than in those who did have a remission (Fig. 5D). No correlation between GEnC IgM levels and proteinuria was seen, and levels were not significantly different between the subset of patients with FSGS compared with the subset of patients with MCD (data not shown). It has been previously reported that total IgM levels increase in patients with nephrotic syndrome (28). We measured total IgM levels in the serum of a subset of the NEPTUNE and control samples and found that IgM levels were decreased in patients with nephrotic syndrome (Fig. 5E). This indicates that the elevated levels of GEnC IgM are not simply due to a nonspecific increase in IgM in these patients.
Circulating IgM Reactive to Cardiolipin Was Elevated in Patients with Nephrotic Syndrome Compared with Healthy Controls
To explore whether levels of Ig reactive with cardiolipin are elevated in patients with nephrotic syndrome, we measured titers of CL-M and CL-G in serum from patients with nephrotic syndrome, healthy controls, and patients with nonproteinuric CKD. The level of CL-M was approximately sixfold higher in both cohorts of patients with nephrotic syndrome compared with healthy subjects and patients with CKD (P < 0.05 by ANOVA; Fig. 6A). CL-G levels were not different between the groups (Fig. 6B). CL-M levels were relatively stable in patients from the NEPTUNE cohort over time (Fig. 6C). CL-M levels were approximately threefold higher in samples from patients with FSGS compared with patients with MCD, although the difference was not statistically significant (data not shown). Although there appeared to be a weak correlation between CL-M levels and GEnC IgM levels in individual patients, it was not statistically significant (Fig. 6E). CL-M levels were similar in patients who had a complete or partial remission over the course of follow-up compared with those who did not have a remission (data not shown).
Figure 6.

IgM in the serum of patients with nephrotic syndrome binds to cardiolipin. We measured levels of Ig that reacted with cardiolipin in serum from healthy controls, patients with chronic kidney disease (CKD), the Nephrotic Syndrome Study Network Consortium (NEPTUNE) cohort of patients with nephrotic syndrome, and a second cohort of patients with nephrotic syndrome (validation cohort). A: anti-cardiolipin IgM (CL-M) levels were significantly higher in the NEPTUNE and validation cohorts than in healthy subjects or patients with CKD. *P < 0.05. B: anti-cardiolipin IgG (CL-G) levels were not significantly different among the groups. C: CL-M levels were measured in serially collected samples from the NEPTUNE cohort of patients with nephrotic syndrome (n = 94). Plotted data are means for each time point; error bars show SDs.
We did not detect an interaction between CL-M titers with kidney function or proteinuria (data not shown). We also did not see any correlations between IgM GEnC or CL-M levels with complement fragments (Supplemental Fig. S7, https://doi.org/10.6084/m9.figshare.14691021.v1).
Effects of Immunosuppression on Levels of IgM Reactive to Cardiolipin and Complement Activation Fragments
We next analyzed the effects of immunosuppressive treatment on complement fragments. When the effect of treatment with glucocorticoids was examined in samples from the NEPTUNE cohort, an interaction was seen between the duration of treatment and the CL-M titer (Fig. 7A). Similarly, when patients in the NEPTUNE cohort started on prednisone during the observation period, CL-M levels were lower at the subsequent study visit (Fig. 7B). We did not see an interaction between treatment with prednisone and levels of C4a and sC5b-9 over time, and in patients who were started on prednisone during the study, the C4a level did not change at the subsequent visit (data not shown). Ba levels, however, were lower at the next visit after treatment with prednisone was started (Fig. 7C). All of the patients in the validation cohort were treated with tacrolimus. In this group, there was a reduction in systemic C4a levels following the initiation of treatment (Fig. 7D). However, no changes in CL-M, Ba, or sC5b-9 levels were observed (data not shown).
Figure 7.

Effects of immunosuppression on levels of anti-cardiolipin IgM (CL-M) and complement fragments. A: in the subset of Nephrotic Syndrome Study Network Consortium (NEPTUNE) patients treated with glucocorticoids (n = 47), a significant interaction between treatment with glucocorticoids and CL-M levels was seen, and the level of CL-M decreased over time in this group. No interaction was seen between CL-M levels and age, sex, or race (not shown). In NEPTUNE patients started on prednisone during the study, CL-M (B) and plasma Ba (C) were lower at the subsequent visit. All of the patients in the validation cohort were treated with tacrolimus. D: the level of C4a was significantly decreased at the subsequent visit after tacrolimus was started. *P < 0.05.
DISCUSSION
The mechanisms of progressive glomerular injury in FSGS and MCD are incompletely understood, but there is evidence that, at least in some cases, it is an immune-mediated process. In the current study, we present histological and serological evidence that IgM activates the classical pathway of complement in the glomeruli of some patients with these forms of nephrotic syndrome. IgM colocalized with the complement fragments C4d, iC3b/C3d, and C9neo in some glomeruli of patients with FSGS, consistent with a role for IgM as a trigger of the classical pathway. The antibodies used to detect iC3b/C3d and C9neo in these samples only detect activated fragments, not intact C3 and C9. Positive staining with these antibodies, therefore, indicates that the deposits were caused by enzymatic complement activation and are not simply due to trapping of intact C3 and C9 in areas of sclerosis (22). Although complement activation was detected in all of the FSGS biopsies, the pattern of complement activation was heterogeneous among glomeruli and within individual glomeruli. In some glomerular segments, activation was controlled before the level of C3 cleavage, for example, whereas in other regions IgM colocalized with C4d, iC3b/C3d, and C9neo.
Analysis of plasma samples from patients with FSGS and MCD revealed that C4a and sC5b-9 levels were elevated compared with controls. We also measured levels of IgM reactive with GEnC epitopes using a GEnC-based assay as well as an ELISA using purified cardiolipin as the target antigen. Patients with nephrotic syndrome had higher levels of reactive IgM compared with control subjects in both of these assays. Although the magnitude of the difference between nephrotic patients and healthy controls was greater using the cardiolipin ELISA, the results of the GEnC assay correlated better with clinical parameters. Anti-GEnC IgM levels were inversely proportional to eGFR, for example, and baseline levels of GEnC IgM were also higher in patients who did not have a partial or complete remission during follow-up than in patients who did have a remission. Given the shallow slope of the correlation between GEnC IgM and eGFR, however, the biological significance is unclear. It is possible that cardiolipin is just one of several epitopes expressed on injured GEnCs, so that the cell-based assay is a better reflection of the total pool of nephritogenic IgM.
Taken together, our results support a model in which natural IgM binds to epitopes expressed on injured GEnCs and activates the classical pathway. This could be a mechanism of glomerular disease progression in some patients, transforming self-limited injury of the glomerulus into a sustained pathological process. Complement activation on GEnCs could cause glomerular injury by several mechanisms. C5b-9 formation on GEnCs may directly injure these cells, for example, and the anaphylatoxins (C3a and C5a) could affect other nearby cells, including podocytes (29, 30). Nevertheless, the heterogeneity of complement activation in the glomeruli of patients with FSGS suggests that this process is modulated by several variables. One factor that is likely to be important is the expression of complement regulatory proteins within the glomerulus. For example, a recent study showed that the complement regulator CD55 is downregulated in experimental and human FSGS (31). Acquired defects in the ability to regulate complement could explain why complement activation is halted at the level of C4d deposition in some glomerular locations, whereas in other regions activation proceeds to the level of iC3b/C3d and C9neo deposition.
It is likely that multiple factors influence the overall degree of complement activation in the glomerulus, including the titer of nephritogenic IgM, expression of target epitope within the glomerulus, and local expression of complement regulatory proteins. Once complement is fully activated in the glomerulus, however, it could increase epitope expression and decrease expression of complement regulatory proteins, thereby perpetuating injury. Furthermore, natural IgM and complement may also contribute to other forms of progressive glomerular injury. These immune proteins are also frequently seen, for example, in biopsies of patients with diabetic nephropathy and hypertensive nephrosclerosis (32–34). Thus, this could represent a common final pathway of disease progression after a variety of glomerular insults.
The abundance of GEnC IgM that a person generates could affect their risk of glomerular disease progression, and it might also be a therapeutic target for immunosuppression. CL-M levels in nephrotic patients decreased in response to treatment with glucocorticoids. Interestingly, C4a levels did not decrease in patients treated with glucocorticoids. It is possible that IgM deposits persist in the glomeruli, continuing to activate complement even after serum levels of IgM decrease. In that regard, detection of complement activation fragments in kidney biopsy tissue and in the circulation may be more reflective of ongoing complement activation in the glomerulus than serum levels of nephritogenic IgM. Unfortunately, the biopsy tissue available for this study could not be paired with plasma samples from the same patient. This limits our ability to directly examine the correlation between plasma analytes and glomerular complement activation and prevents us from determining whether complement fragments in the plasma predict or associate with deposits in glomeruli. It is also noteworthy that C4a levels did decrease in patients started on tacrolimus, raising the possibility that calcineurin inhibitors are more effective than glucocorticoids for suppressing this immune process.
Interestingly, Ba levels were more closely associated with clinical parameters than C4a levels. Plasma Ba was significantly higher in patients with FSGS compared with those with MCD, and baseline levels were also higher in those who failed to have either a complete or partial remission at any point during follow-up than in patients who did have a remission. Ba levels also decreased after glucocorticoid treatment was begun. There was an inverse correlation between Ba and eGFR; however, and Ba levels of patients in the normal eGFR range were similar to levels in healthy controls. Furthermore, we have previously found that Ba levels are increased in nonproteinuric CKD (21). Thus, the alternative pathway appears to be activated as a consequence of a reduced GFR and is probably not specific for nephrotic syndrome or FSGS.
FSGS and MCD are considered to be diseases that primarily affect podocytes and can be caused by circulating factors, genetic variants, infections, toxins, and hemodynamic factors (35). Even if the initial insult is to the podocyte, however, other glomerular cells and structures are affected as the capillary collapses. In vitro, IgM only binds to GEnCs after the cells are injured. This suggests that natural IgM and complement do not initiate glomerular injury but rather they exacerbate injury caused by other factors and contribute to disease progression. This is consistent with the observation that there was overlap in the levels of IgM GEnC or CL-M between nephrotic patients and healthy subjects. Based on these findings, detection of the antibodies cannot be used to predict or diagnose the disease. Nevertheless, these findings do implicate natural antibodies and complement as potential mechanisms of disease progression. Potentially, detection of ongoing complement activation could be used to select patients for treatment with B cell-targeted or complement inhibitory drugs.
It is also worth noting that natural antibody IgM and the classical pathway can serve protective functions, helping to facilitate the removal of injured and apoptotic cells in a noninflammatory fashion (36–38). In fact, proteins such as C-reactive protein may help to suppress alternative pathway activation on apoptotic cells, allowing the protective functions of IgM and the classical pathway to proceed while inhibiting the inflammatory effects of alternative pathway activation (39). Although alternative pathway-deficient mice are protected in models of FSGS (8, 9), we are not aware of comparable studies using mice with targeted deletion of classical pathway proteins. Thus, it is possible that natural IgM and the classical pathway have protective functions but that impaired complement regulation in damaged glomeruli permits activation to proceed through the terminal complement proteins. If so, then a drug that selective blocks the alternative pathway may be more effective than a drug that blocks the classical pathway, even if activation is initiated through the classical pathway.
In conclusion, we have found that the classical pathway of complement is activated in the glomeruli of patients with FSGS and that patients with idiopathic nephrotic syndrome have higher levels of IgM reactive to endothelial cells and cardiolipin than healthy controls. Higher levels of Ig reactive with glomerular epitopes may be a mechanism of disease progression after a glomerular insult, and complement activation fragments in the plasma may be useful biomarkers of glomerular inflammation in these diseases. Many new therapeutic agents that target B cells and the complement cascade are in development, and these drugs may have a role for the treatment of FSGS and MCD. Our findings may provide noninvasive biomarkers for identifying the patients most likely to benefit from these treatments and for assessing the efficacy of these therapeutic agents.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14690733.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14690760.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.14690781.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.14690799.
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.14690871.v1.
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.14691000.
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.14691021.v1.
GRANTS
This work was supported by an ancillary study award from NephCure/Kidney International, National Institutes of Health Grants DK113586, DK076690, and CA225840, and Department of Defense Grant LR180050 (to J.M.T.). This work was also supported by a sponsored research agreement with Q32 Bio, Incorporated.
DISCLOSURES
J.M.T. and V.M.H. receive royalties from Alexion Pharmaceuticals, Inc., and are consultants for Q32 Bio, Inc., a company developing complement inhibitors. They also hold stock and will receive royalty income from Q32 Bio, Inc. S.L.K. and F.L. are employed by Q32 Bio, Inc. H.T. is a consultant to Otsuka (Chair, DSMB) and Chemocentryx (DSMB). He has consultancy agreements through NYU Grossman School of Medicine with Retrophin Inc. and Goldfinch Bio Inc. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
H.T., S.S., B.R., D.J., and J.M.T. conceived and designed research; C.C., L.K., F.L., S.L.K., V.M.H., D.J., J.L., C.L., B.R., A.F., S.P., S.E.P., W.Z., and P.C. performed experiments; H.T., Z.Y., S.S., B.R., S.L.K., D.J., J.M.T., J.L., and C.L. analyzed data; S.L.K., V.M.H., J.M.T., J.L., and C.L. interpreted results of experiments; J.M.T. prepared figures; B.R. and J.M.T. drafted manuscript; H.T., C.C., S.S., B.R., F.L., S.L.K., V.M.H., J.M.T., B.R., S.P., S.E.P., and W.Z. edited and revised manuscript; H.T., L.K., Z.Y., S.S., B.R., F.L., S.L.K., V.M.H., D.J., J.M.T., C.L., B.R., S.E.P., and P.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The Nephrotic Syndrome Study Network Consortium (NEPTUNE; U54DK083912) is a part of the National Institutes of Health Rare Disease Clinical Research Network, supported through a collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, and the National Institute of Diabetes, Digestive, and Kidney Diseases. Additional funding and/or programmatic support for this project was also provided by the University of Michigan, NephCure Kidney International, and the Halpin Foundation. The anti-C9neo antibody used in this study was a generous gift of Paul Morgan. We also thank Dr. Moin Saleem, who provided immortalized glomerular cells.
REFERENCES
- 1.Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 12: 502–517, 2017. [Erratum in Clin J Am Soc Nephrol 13: 1889, 2018]. doi: 10.2215/CJN.05960616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreoli SP. Racial and ethnic differences in the incidence and progression of focal segmental glomerulosclerosis in children. Adv Ren Replace Ther 11: 105–109, 2004. doi: 10.1053/j.arrt.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Waldman M, Crew RJ, Valeri A, Busch J, Stokes B, Markowitz G, D'Agati V, Appel G. Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol 2: 445–453, 2007. doi: 10.2215/CJN.03531006. [DOI] [PubMed] [Google Scholar]
- 4.Bose B, Cattran D; Toronto Glomerulonephritis Registry. Glomerular diseases: FSGS. Clin J Am Soc Nephrol 9: 626–632, 2014. doi: 10.2215/CJN.05810513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habib R, Girardin E, Gagnadoux MF, Hinglais N, Levy M, Broyer M. Immunopathological findings in idiopathic nephrosis: clinical significance of glomerular “immune deposits.” Pediatr Nephrol 2: 402–408, 1988. doi: 10.1007/BF00853431. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YM, Gu QH, Huang J, Qu Z, Wang X, Meng LQ, Wang F, Liu G, Cui Z, Zhao MH. Clinical significance of IgM and C3 glomerular deposition in primary focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 11: 1582–1589, 2016. doi: 10.2215/CJN.01190216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Xie J, Zhang X, Tong J, Hao X, Ren H, Wang W, Chen N. Serum C3 and renal outcome in patients with primary focal segmental glomerulosclerosis. Sci Rep 7: 4095, 2017. doi: 10.1038/s41598-017-03344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenderink AM, Liegel K, Ljubanović D, Coleman KE, Gilkeson GS, Holers VM, Thurman JM. The alternative pathway of complement is activated in the glomeruli and tubulointerstitium of mice with adriamycin nephropathy. Am J Physiol Renal Physiol 293: F555–F564, 2007. doi: 10.1152/ajprenal.00403.2006. [DOI] [PubMed] [Google Scholar]
- 9.Turnberg D, Lewis M, Moss J, Xu Y, Botto M, Cook HT. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol 177: 4094–4102, 2006. doi: 10.4049/jimmunol.177.6.4094. [DOI] [PubMed] [Google Scholar]
- 10.Thurman JM, Wong M, Renner B, Frazer-Abel A, Giclas PC, Joy MS, Jalal D, Radeva MK, Gassman J, Gipson DS, Kaskel F, Friedman A, Trachtman H. Complement activation in patients with focal segmental glomerulosclerosis. PloS One 10: e0136558, 2015. doi: 10.1371/journal.pone.0136558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Cui Z, Gu QH, Zhang YM, Qu Z, Wang X, Wang F, Cheng XY, Meng LQ, Liu G, Zhao MH. Complement activation profile of patients with primary focal segmental glomerulosclerosis. PloS One 15: e0234934, 2020. doi: 10.1371/journal.pone.0234934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van de Lest NA, Zandbergen M, Wolterbeek R, Kreutz R, Trouw LA, Dorresteijn EM, Bruijn JA, Bajema IM, Scharpfenecker M, Chua JS. Glomerular C4d deposition can precede the development of focal segmental glomerulosclerosis. Kidney Int 96: 738–749, 2019. doi: 10.1016/j.kint.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 13.Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol 10: 778–786, 2010. doi: 10.1038/nri2849. [DOI] [PubMed] [Google Scholar]
- 14.Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Bäckhed F, Miller YI, Hörkkö S, Corr M, Witztum JL, Binder CJ. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest 119: 1335–1349, 2009. doi: 10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elvington A, Atkinson C, Kulik L, Zhu H, Yu J, Kindy MS, Holers VM, Tomlinson S. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J Immunol 188: 1460–1468, 2012. doi: 10.4049/jimmunol.1102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore FD, Carroll MC. Identification of the target self-antigens in reperfusion injury. J Exp Med 203: 141–152, 2006. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panzer SE, Laskowski J, Renner B, Kulik L, Ljubanovic D, Huber KM, Zhong W, Pickering MC, Holers VM, Thurman JM. IgM exacerbates glomerular disease progression in complement-induced glomerulopathy. Kidney Int 88: 528–537, 2015. doi: 10.1038/ki.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waffarn EE, Hastey CJ, Dixit N, Soo Choi Y, Cherry S, Kalinke U, Simon SI, Baumgarth N. Infection-induced type I interferons activate CD11b on B-1 cells for subsequent lymph node accumulation. Nat Commun 6: 8991, 2015. doi: 10.1038/ncomms9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binder CJ, Hörkkö S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9: 736–743, 2003. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 20.Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PX, Barisoni L, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 83: 749–756, 2013. doi: 10.1038/ki.2012.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jalal D, Renner B, Laskowski J, Stites E, Cooper J, Valente K, You Z, Perrenoud L, Le Quintrec M, Muhamed I, Christians U, Klawitter J, Lindorfer MA, Taylor RP, Holers VM, Thurman JM. Endothelial microparticles and systemic complement activation in patients with chronic kidney disease. J Am Heart Assoc 7: e007818, 2018. doi: 10.1161/JAHA.117.007818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thurman JM, Kulik L, Orth H, Wong M, Renner B, Sargsyan SA, Mitchell LM, Hourcade DE, Hannan JP, Kovacs JM, Coughlin B, Woodell AS, Pickering MC, Rohrer B, Holers VM. Detection of complement activation using monoclonal antibodies against C3d. J Clin Invest 123: 2218–2230, 2013. doi: 10.1172/JCI65861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kemp PA, Spragg JH, Brown JC, Morgan BP, Gunn CA, Taylor PW. Immunohistochemical determination of complement activation in joint tissues of patients with rheumatoid arthritis and osteoarthritis using neoantigen-specific monoclonal antibodies. J Clin Lab Immunol 37: 147–162, 1992. [PubMed] [Google Scholar]
- 24.Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O'Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int 69: 1633–1640, 2006. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 25.Pierangeli SS, Harris EN. A protocol for determination of anticardiolipin antibodies by ELISA. Nat Protoc 3: 840–848, 2008. doi: 10.1038/nprot.2008.48. [DOI] [PubMed] [Google Scholar]
- 26.Wilson HR, Medjeral-Thomas NR, Gilmore AC, Trivedi P, Seyb K, Farzaneh-Far R, Gunnarsson I, Zickert A, Cairns TD, Lightstone L, Cook HT, Pickering MC. Glomerular membrane attack complex is not a reliable marker of ongoing C5 activation in lupus nephritis. Kidney Int 95: 655–665, 2019. doi: 10.1016/j.kint.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol 11: 700–707, 2000. doi: 10.1681/ASN.V114700. [DOI] [PubMed] [Google Scholar]
- 28.Giangiacomo J, Cleary TG, Cole BR, Hoffsten P, Robson AM. Serum immunoglobulins in the nephrotic syndrome. A possible cause of minimal-change nephrotic syndrome. N Engl J Med 293: 8–12, 1975. doi: 10.1056/NEJM197507032930103. [DOI] [PubMed] [Google Scholar]
- 29.Uffing A, Pérez-Sáez MJ, Mazzali M, Manfro RC, Bauer AC, de Sottomaior Drumond F, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol 15: 247–256, 2020. doi: 10.2215/CJN.08970719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morigi M, Perico L, Corna D, Locatelli M, Cassis P, Carminati CE, Bolognini S, Zoja C, Remuzzi G, Benigni A, Buelli S. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 5: e131849, 2020. doi: 10.1172/jci.insight.131849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angeletti A, Cantarelli C, Petrosyan A, Andrighetto S, Budge K, D'Agati VD, Hartzell S, Malvi D, Donadei C, Thurman JM, Galešić-Ljubanović D, He JC, Xiao W, Campbell KN, Wong J, Fischman C, Manrique J, Zaza G, Fiaccadori E, Manna LG, Fribourg M, Leventhal J, Sacco DS, Perin L, Heeger PS, Cravedi P. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J Exp Med 217: e20191699, 2020. doi: 10.1084/jem.20191699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ainsworth SK, Hirsch HZ, Brackett NC Jr, Brissie RM, Williams AV Jr, Hennigar GR. Diabetic glomerulonephropathy: histopathologic, immunofluorescent, and ultrastructural studies of 16 cases. Hum Pathol 13: 470–478, 1982. doi: 10.1016/s0046-8177(82)80030-0. [DOI] [PubMed] [Google Scholar]
- 33.Gamble CN. The pathogenesis of hyaline arteriolosclerosis. Am J Pathol 122: 410–420, 1986. [PMC free article] [PubMed] [Google Scholar]
- 34.Mujais SK, Emmanouel DS, Kasinath BS, Spargo BH. Marked proteinuria in hypertensive nephrosclerosis. Am J Nephrol 5: 190–195, 1985. doi: 10.1159/000166931. [DOI] [PubMed] [Google Scholar]
- 35.Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol 2: 529–542, 2007. doi: 10.2215/CJN.04121206. [DOI] [PubMed] [Google Scholar]
- 36.Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 19: 56–59, 1998. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 37.Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med 188: 2313–2320, 1998. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaveri SV, Silverman GJ, Bayry J. Natural IgM in immune equilibrium and harnessing their therapeutic potential. J Immunol 188: 939–945, 2012. doi: 10.4049/jimmunol.1102107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med 192: 359–366, 2000. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]