

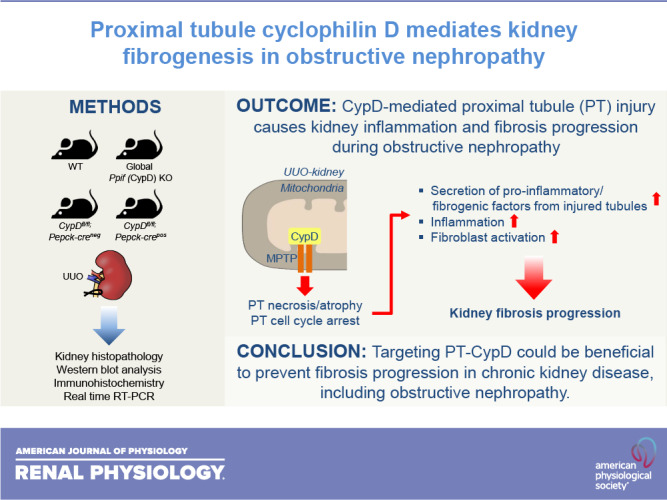
Keywords: chronic kidney disease, cyclophilin D, fibrosis, obstructive nephropathy, proximal tubule
Abstract
The proximal tubule (PT) is highly vulnerable to acute injury, including ischemic insult and nephrotoxins, and chronic kidney injury. It has been established that PT injury is a primary cause of the development of chronic kidney disease, but the underlying molecular mechanism remains to be defined. Here, we tested whether PT cyclophilin D (CypD), a mitochondrial matrix protein, is a critical factor to cause kidney fibrosis progression. To define the role of CypD in kidney fibrosis, we used an established mouse model for kidney fibrosis: the unilateral ureteral obstruction (UUO) model in global and PT-specific CypD knockout (KO). Global CypD KO blunted kidney fibrosis progression with inhibition of myofibroblast activation and fibrosis. UUO-induced tubular atrophy was suppressed in kidneys of global CypD KO but not tubular dilation or apoptotic cell death. PT cell cycle arrest was highly increased in wild-type UUO kidneys but was markedly attenuated in global CypD KO UUO kidneys. The number of macrophages and neutrophils was less in UUO kidneys of global CypD KO than those of wild-type kidneys. Proinflammatory and profibrotic factors were all inhibited in global CypD KO. In line with those of global CypD KO, PT-specific CypD KO also blunted kidney fibrosis progression, along with less tubular atrophy, renal parenchymal loss, cell cycle arrest in PT, and inflammation, indicating a critical role for PT CypD in fibrogenesis. Collectively, our data demonstrate that CypD in the PT is a critical factor contributing to kidney fibrosis in UUO, providing a new paradigm for mitochondria-targeted therapeutics of fibrotic diseases.
NEW & NOTEWORTHY It has been established that renal proximal tubule (PT) injury is a primary cause of the development of chronic kidney disease, but the underlying molecular mechanism remains to be defined. Here, we show that cyclophilin D, a mitochondrial matrix protein, in the PT causes kidney fibrogenesis in obstructive nephropathy. Our data suggest that targeting PT cyclophilin D could be beneficial to prevent fibrosis progression.
INTRODUCTION
Tissue fibrosis is an initial response for recovering parenchymal cell injury in diverse tissues, including the kidney, which results in the accumulation of the extracellular matrix in interstitial areas (1, 2). However, a maladaptive or excessive fibrotic response causes kidney dysfunction (3–5). Recent experimental and clinical data have suggested that the severity, frequency, and duration of initial injury in renal epithelial cells determine the dynamics of fibrosis progression (6–8). It has been recognized that renal tubular epithelial cell injury serves as a primary cause of kidney fibrosis. Of note, injury in the proximal tubule (PT), which is highly vulnerable to both acute and chronic injury, has been shown to play a critical role in kidney fibrosis, as injury or deletion of the PT initiates and perpetuates kidney fibrosis (8–13). However, the molecular mechanism by which PT cell injury causes kidney fibrogenesis remains to be fully uncovered.
Mitochondrial disturbance, including structural and functional injury, has been shown to manifest chronic tubular damage leading to fibrosis progression (14–16). The mitochondrial matrix protein cyclophilin D (CypD), with cis-trans peptidyl prolyl isomerase activity, is characterized as the regulatory component of mitochondrial permeability transition pore (MPTP) that regulates MPTP opening and MPTP-dependent necrotic cell death (17–20). Many reports have shown that targeting CypD is effective to prevent acute injury in diverse organs, including the kidney (21–27). However, the role of CypD in regulating kidney fibrosis has not been investigated. In the present study, we investigated whether CypD causes kidney fibrosis progression and whether its inhibition can mitigate chronic kidney disease (CKD) progression following ureteral obstruction.
MATERIALS AND METHODS
Mice and Surgical Preparation
Eight- to 12-wk-old male mice were cared for before and during the experimental procedures in accordance with policies of the Institutional Animal Care and Use Committee of University of Nebraska Medical Center and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All protocols had received prior approval from the Institutional Animal Care and Use Committee of University of Nebraska Medical Center. Global Ppif (CypD) knockout (KO) and Ppiffl/fl (CypDfl/fl; Jackson Lab, Bar Harbor, ME) and PEPCK Cre recombinase-expressing mice (28) (a kind gift from Dr. Volker Haase, Vanderbilt University) were used. Floxed mice of CypD and PEPCK Cre recombinase-expressing mice were mated to generate mice with PT-specific KO of CypD. Global CypD KO, PT-CypD KO, and their respective control mice were anesthetized by intraperitoneal injection of a cocktail containing ketamine (200 mg/kg body wt) and xylazine (16 mg/kg body wt). The right ureter was obstructed completely near the renal pelvis using 5-0 silk, as previously described (11, 29, 30). Sham-operated mice underwent the same surgical procedures except for the ureter ligation. After 7 days of unilateral ureteral obstruction (UUO) or sham, kidneys were either fixed in 4% formaldehyde for histological experiments or snap frozen in liquid nitrogen for biochemical experiments. Periodic acid-Schiff-stained sections were used for histological damage scores.
Collagen Deposition
Collagen deposition was assessed by Sirius red staining and hydroxyproline assay as previously described (11, 31). Sirius red-positive areas were expressed as the ratio of Sirius red-positive area to total area in five randomly chosen fields per kidney in a blinded manner.
Histology and evaluation of tubular injury.
Periodic acid-Schiff-stained sections were used for evaluating tubular injury as we have previously described (11). Tubular atrophy and dilatation were expressed as a ratio of the number of atrophied or dilated tubules to the number of total tubules in five randomly chosen fields per kidney in a blinded manner. To visualize the PT and evaluate PT injury, Lotus tetragonolobus lectin (LTL) or Dolichos biflorus agglutinin (DBA) was used. The ratio of the Lotus tetragonolobus lectin-positive area (or non-DBA-positive area) to total area was evaluated using ImageJ software (NIH, Bethesda, MD).
Apoptotic Cell Death
The TUNEL assay on kidney sections to evaluate apoptotic cells was carried out using the In Situ Cell Death Detection Kit (Fluorescein, Roche, Mannheim, Germany), as we have previously described (11).
Immunohistochemistry and Immunofluorescent Staining
Immunohistochemical staining of the kidneys was performed on paraffin-embedded sections as previously described (32, 33). Briefly, 4% paraformaldehyde-fixed kidney sections were rehydrated and labeled with antibodies against α-smooth muscle actin (α-SMA; Sigma, St. Louis, MO), polymorphonuclear leukocytes (PMN; Accurate, Westbury, NY), F4/80 (Proteintech, Chicago, IL), phosphorylated (p-)histone H3 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Smad3 (p-Smad3; Abcam, Cambridge, MA), Ki-67 (Novus Biologicals, Littleton, CO), and collagen type IV (Southernbiotech, Birmingham, AL). Sections were then incubated with peroxidase- or FITC-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA). DAPI (Sigma) or Meyer’s hematoxylin (Electron Microscopy Sciences, Hatfield, PA) was used. The α-SMA-positive area was measured in five randomly chosen fields per kidney in a blinded manner using ImageJ software (NIH). The respective numbers or positive areas were counted in five randomly chosen fields per kidney.
Western Blot Analysis
Western blot analysis was conducted as previously described (34) using various antibodies against the following proteins: p53, ICAM-1, transforming growth factor-β1 (TGF-β1), NLR family pyrin domain containing 3, GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA), α-SMA (Sigma), p-Smad3, IL-1β, poly(ADP-ribose) polymerase (PARP-1; Abcam, Cambridge, MA), cyclin B1, and cyclin D1 (Cell signaling, Danvers, MA). GAPDH immunoblot analysis was used as a loading control on stripped membranes. Band intensities were analyzed by ImageJ software (NIH).
RNA Extraction
Total RNA was extracted from kidneys using TRIzol reagent (Invitrogen) and cleaned with an RNeasy mini kit (Qiagen, Hilden, Germany) as previously described (26). First-strand cDNA was prepared by reverse transcription using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). To determine the conditions for the logarithmic phase during PCR amplification with target mRNA, aliquots (1 µg) were amplified using different numbers of cycles. A linear relationship between PCR product band visibility and the number of amplification cycles was observed for target mRNAs.
Quantitative RT-PCR
Quantitative real-time PCR was performed with 1 µL of the cDNA template added to 10 µL of 2× SYBR Premix Ex Taq (Applied Biosystems, Foster City, CA) and specific primers (10 pM each) as previously described (26). Real-time PCR (Bio-Rad) was carried out for 40 cycles of denaturation at 95°C for 15 s, annealing at 58°C for 15 s, and extension at 72°C for 15 s. Target gene expression was quantified relative to that of an internal control gene (GAPDH) based on the comparison of the threshold cycle (CT) at constant fluorescence intensity. The amount of transcript was inversely related to the observed CT, and the CT was expected to increase by 1 for every twofold dilution of the transcript. Relative expression (R) was calculated using the equation R = 2 − (ΔCT sample − ΔCT control). All data were normalized relative to GAPDH and the respective controls. Primer sequences are provided in the Supplemental Data (all Supplemental Material is available at http://doi.org/10.6084/m9.figshare.14462037).
Statistical Analyses
Data are expressed as means ± SD. Differences between two groups were assessed by a two-tailed unpaired Student’s t test. For multiple group comparisons, one-way ANOVA with Bonferroni analysis was applied (GraphPrism 5.0 software). A P value of <0.05 was considered statistically significant.
RESULTS
Global Inhibition of CypD Reduces UUO-Induced Kidney Tubular Injury and Fibrosis
To investigate whether CypD contributes to fibrosis progression, we performed UUO in wild-type (WT) and global CypD KO mice. First, we checked collagen deposition by Sirius red stain and hydroxyproline analysis and fibroblast activation using expression levels of α-SMA, a marker of myofibroblasts. UUO highly increased myofibroblast activation, as shown by increased α-SMA, and collagen deposition in WT mouse kidneys (Fig. 1), resulting in kidney fibrosis. However, kidney fibrosis was significantly suppressed in global CypD KO mouse kidneys with downregulated expression of α-SMA and collagen (Fig. 1), indicating the protective effect of CypD KO in UUO-induced kidney fibrosis. Next, since tubular injury can initiate kidney fibrosis (9), we examined tubular cell injury and cell fate, including cell cycle arrest, in UUO kidneys. UUO increased tubular atrophy, dilation, and apoptosis in WT kidneys post-UUO (Fig. 2, A–D). CypD KO suppressed UUO-induced tubular atrophy, which is caused by inappropriate and failed tubular recovery (5), but not tubular dilation and apoptosis (Fig. 2, A–D). Kidney tubular injury-associated factors (PARP-1 and p53) were increased in WT kidneys during UUO (Fig. 2, E–H), a pattern similar to that of increased tubular injury score. Genetic deletion of CypD prevented upregulation of PARP-1 and p53 (Fig. 2, E–H), suggesting that CypD worsens tubular injury, particularly necrotic cell death, leading to kidney fibrosis during UUO.
Figure 1.
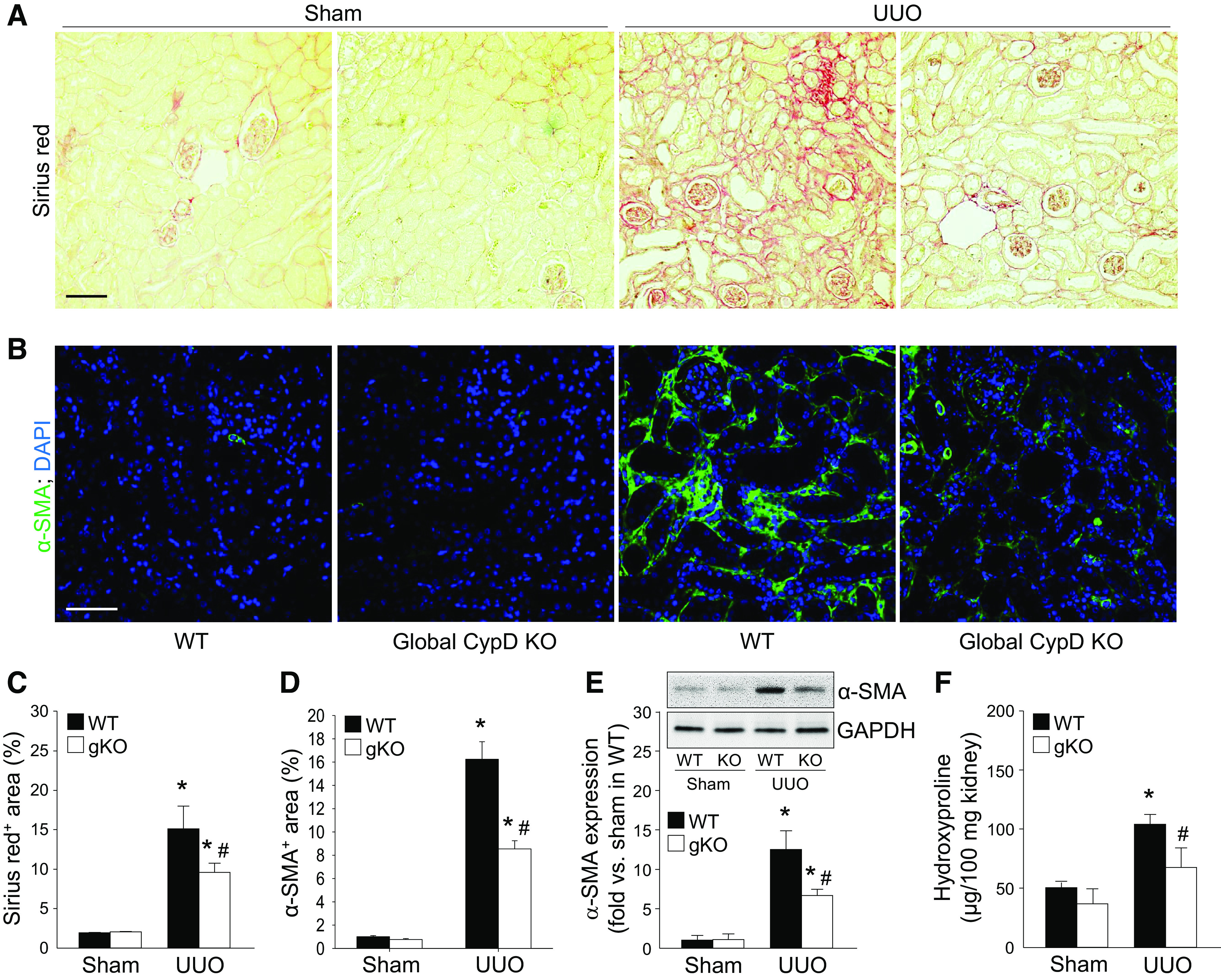
Prevention of unilateral ureteral obstruction (UUO)-induced kidney fibrosis by genetic deletion of cyclophilin D (CypD). Wild-type (WT) and global CypD knockout (KO) (gKO) mice were subjected to UUO or sham operation for 7 days. A and B: paraffin-embedded kidney sections were used to carry out Sirius red stain for collagen deposition (A) and immunofluorescent stain for evaluating α-smooth muscle actin (α-SMA), a marker of myofibroblasts (n = 5 or 6) (B). C and D: data for Sirius red-positive collagen deposition (red color; C) and α-SMA-positive area (green color; D) were quantified from five randomly chosen fields per kidney (n = 5 or 6). DAPI was used for counterstaining. Expression levels were evaluated using Image J software. E: expression of α-SMA was examined by Western blot analysis using specific antibodies. The Western blot bands were used as representative (n = 5). Anti-GAPDH antibody was used as a loading control. Expression levels were evaluated using ImageJ software. F: total collagen levels were evaluated by hydroxyproline assays in kidney lysates. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
Figure 2.
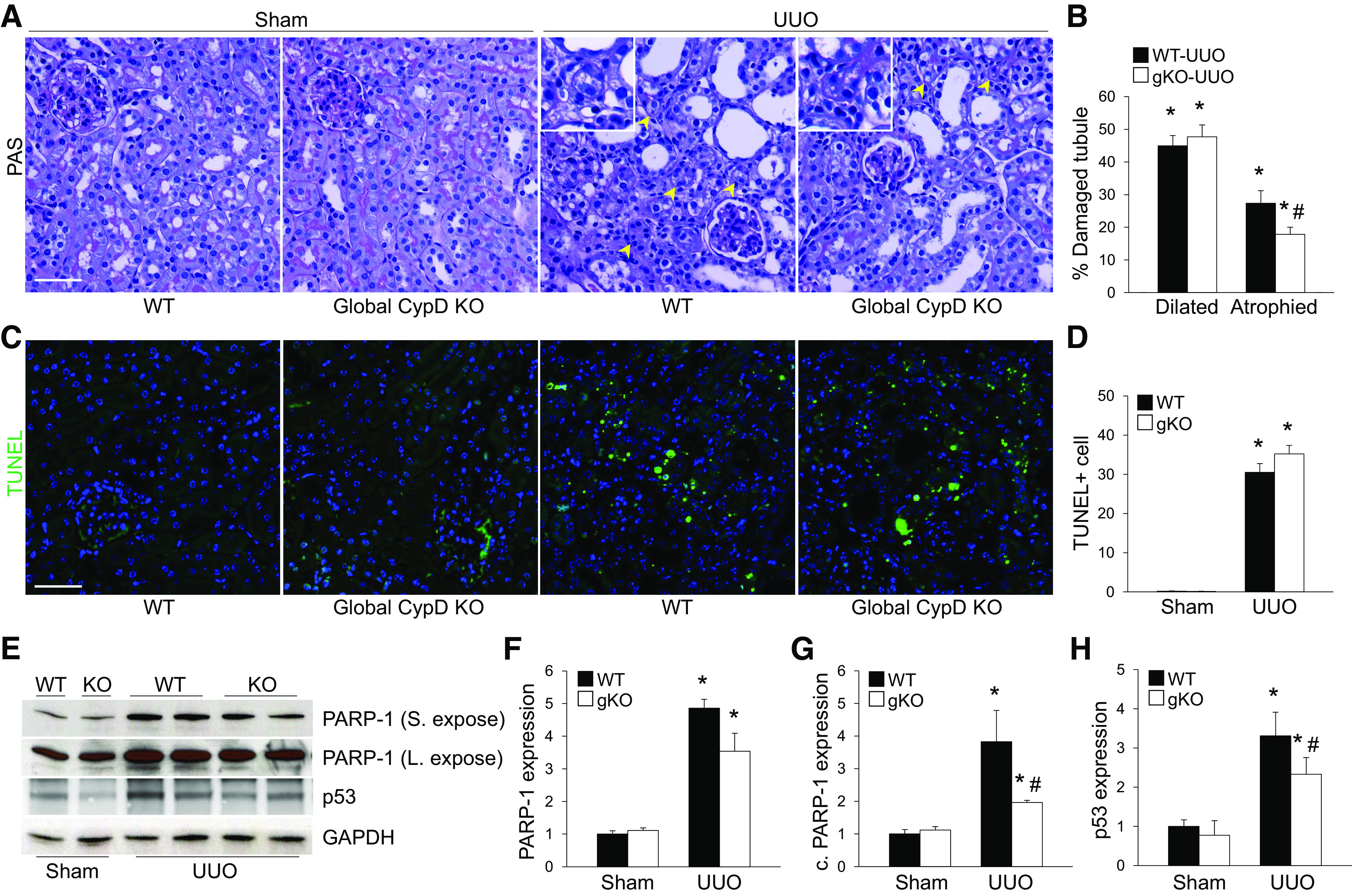
Global cyclophilin D (CypD) knockout (KO) (gKO) inhibits kidney tubular injury and atrophy during unilateral ureteral obstruction (UUO). Wild-type (WT) and global CypD KO mice were subjected to UUO or sham operation for 7 days. A and C: paraffin-embedded kidney sections were used to carry out periodic acid-Schiff (PAS) stain (A) for evaluating histopathology and TUNEL stain (C) for evaluating tubular apoptotic cell death (n = 5 or 6). Yellow arrowheads indicate atrophied tubules. B: percent ratios of atrophied and dilated tubules to total tubules were calculated from 100 tubules in 5 randomly chosen fields per kidney (n = 5 or 6). D: numbers of TUNEL-positive apoptotic cells (green) were counted from 5 randomly chosen fields per kidney (n = 5 or 6). DAPI (blue) was used for counterstaining. E: expression levels of poly(ADP-ribose) polymerase (PARP-1) and p53 were examined by Western blot analysis (n = 5), and representative Western blots are shown. F−H: quantification for PARP-1 and p53 was performed. Levels of PARP-1 were expressed by PARP-1 and cleaved PARP-1 (c. PARP-1) separately. Anti-GAPDH antibody was used as a loading control. Expression levels were evaluated using ImageJ software. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO. S. expose, short exposure; L. expose, long exposure.
Genetic Deletion of CypD Attenuates Cell Cycle Arrest in the PT During UUO
To examine the role of CypD in tubular cell fate during UUO, we checked cell proliferation and cycle arrest in UUO kidneys. Tubular Ki-67-positive proliferating cells were increased in both LTL-positive and LTL-negative PTs, although to a lesser degree in LTL-negative non-PTs, in WT UUO kidneys compared with those of sham kidneys (Fig. 3A). Genetic deletion of CypD did not change UUO-induced tubular cell proliferation (Fig. 3A). Interstitial cell proliferation was highly increased in UUO kidneys (Fig. 3A), indicating an expansion of interstitial cells, fibroblasts, and inflammatory cells, which play major roles in the progression of kidney fibrosis (32, 35). Interstitial cell proliferation was significantly inhibited in UUO kidneys of CypD KO mice (Fig. 3A), indicating a role for CypD in interstitial cell expansion.
Figure 3.
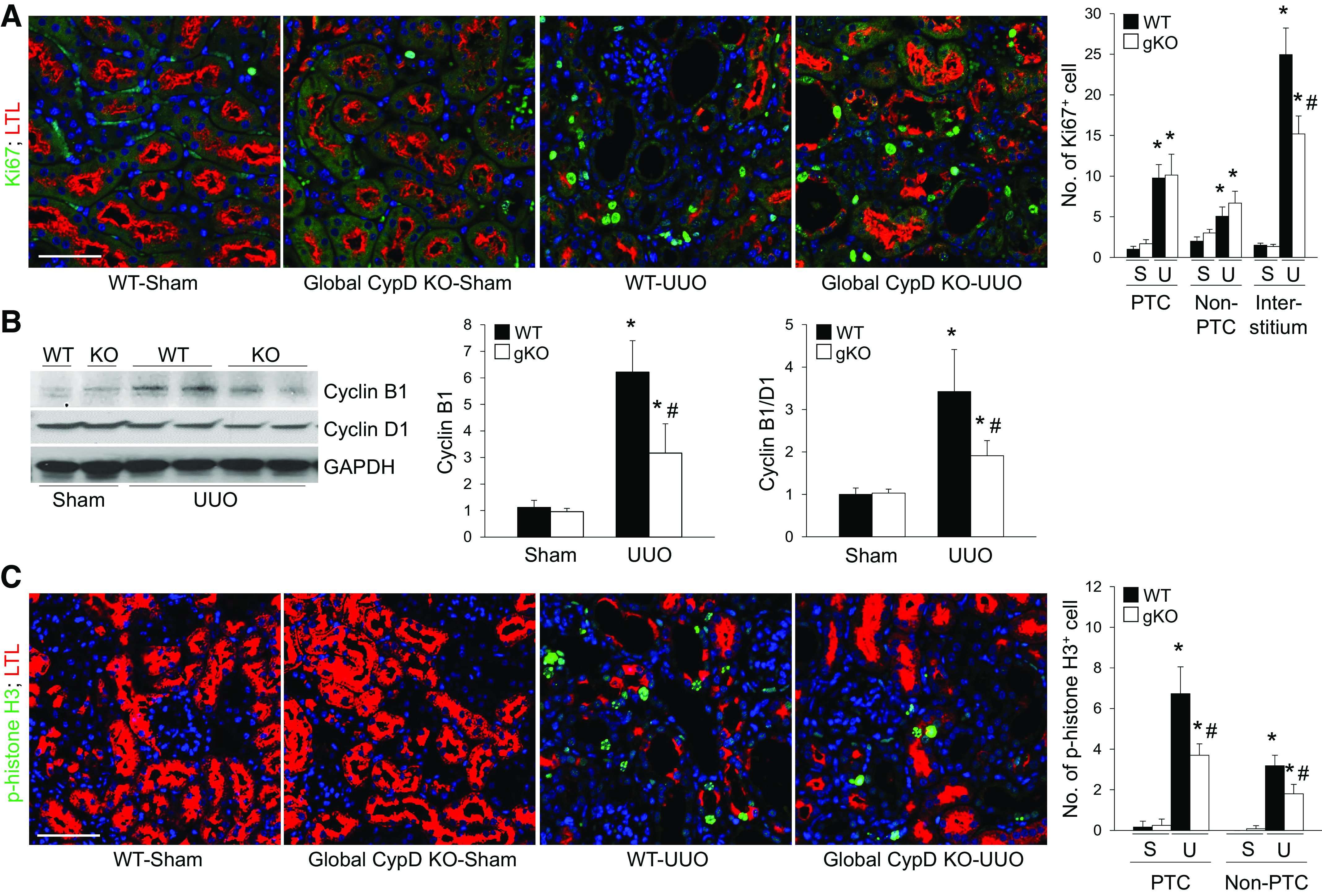
Global cyclophilin D (CypD) knockout (KO) (gKO) suppresses proximal tubule cell cycle arrest during unilateral ureteral obstruction (UUO). Wild-type (WT) and global CypD KO mice were subjected to UUO or sham operation for 7 days. A: paraffin-embedded kidney sections were used to carry out immunofluorescent staining for evaluating the amount of Ki-67-positive cell proliferation (green) (n = 5 or 6). Lotus tetragonolobus lectin (LTL; red) was used for a marker of proximal tubule. Ki-67-positive proliferating cells and phospho-histone H3 (p-histone H3)-positive cell cycle-arrested cells were evaluated from 5 randomly chosen fields per kidney (n = 5 or 6). DAPI (blue) was used for counterstaining. B: expression levels of cyclin B1 and D1 were examined by Western blot analysis (n = 5), and representative Western blots are shown. Anti-GAPDH antibody was used as a loading control. Expression levels were evaluated using ImageJ software. C: number of phospho-histone H3 (green)-positive cells as a marker of G2/M arrest were counted in the same method as that of Ki-67. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham. #P < 0.05 vs. WT UUO.
To clarify whether CypD plays a critical role in tubular cell cycle arrest, we analyzed tubular cell cycle arrest-associated factors in UUO kidneys as a function of CypD. The ratio of cyclin B1 to cyclin D1 was significantly upregulated in WT UUO kidneys, suggesting G2/M arrest, whereas it was reduced in kidneys from CypD KO mice (Fig. 3B). Next, to find out which tubular cells underwent cell cycle arrest, we performed immunohistochemistry for p-histone H3, a marker of G2/M arrest, in UUO kidneys. Expression of p-histone H3 was increased in LTL-positive PTs, and to a lesser degree in LTL-negative non-PTs, in UUO kidneys compared with sham kidneys (Fig. 3C). Genetic KO of CypD prevented these increases in both PTs and non-PTs, although the effect was greater in PTs than in non-PTs (Fig. 3C). These data suggest that CypD-mediated pathways can cause tubular cell cycle arrest in predominantly PTs during UUO, which may lead to the development of kidney fibrosis.
CypD Inhibition Mitigates Inflammation and Fibrotic Signaling During UUO
To check out the contribution of proinflammatory and profibrotic signaling in CypD-mediated kidney fibrosis during UUO, we evaluated inflammatory cell number and expression levels of proinflammatory/profibrotic signaling molecules. UUO induced increases of F4/80-positive macrophages and PMN-positive neutrophils in WT mice, but CypD KO suppressed the increase of these inflammatory cells (Fig. 4, A–C). Similar to the number of inflammatory cells, expression of ICAM-1, IL-1β, and NLR family pyrin domain containing 3, which are all well-known contributors to kidney fibrosis and tubular injury, was highly upregulated in WT UUO kidneys (Fig. 4, D–G). However, expression of these factors was mitigated in CypD KO kidneys with UUO (Fig. 4, D–G). Immunohistochemical and quantitative analyses for the profibrotic signaling molecule p-Smad3 showed that UUO significantly increased its expression in WT UUO kidneys (Fig. 5, A–C). CypD deficiency resulted in suppressed expression of p-Smad3 in kidney sections and whole kidney lysates (Fig. 5, A–C). Expression of TGF-β was similar to that of Smad3 (Fig. 5, B and D), suggesting that CypD upregulates proinflammatory/profibrotic signaling molecules to induce kidney fibrosis during UUO.
Figure 4.
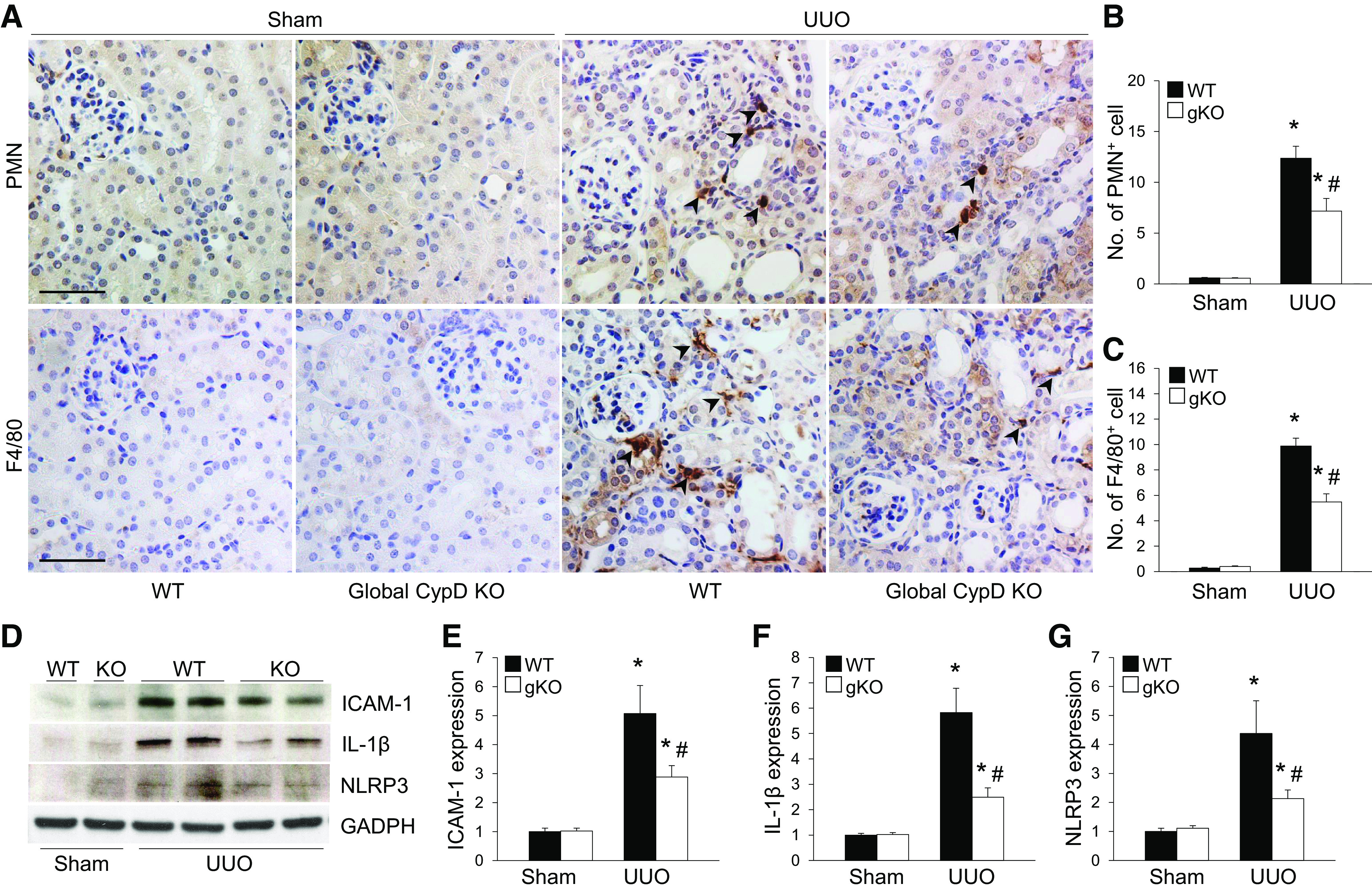
Cyclophilin D (CypD) knockout (KO) prevents kidney inflammation during unilateral ureteral obstruction (UUO). Wild-type (WT) and global CypD KO mice (gKO) were subjected to UUO or sham operation for 7 days. A: paraffin-embedded kidney sections were used to carry out immunohistochemistry for evaluating the number of polymorphonuclear leukocyte (PMN)- and F4/80-positive cells (n = 5 or 6). Hematoxylin stain (blue color) was used for counterstaining. Arrowheads indicate PMN- or F4/80-positive cells. B and C: numbers or areas of PMN-positive neutrophils (B; brown color; arrowheads) and F/80-positive macrophages (C; brown color; arrowheads) were counted from 5 randomly chosen fields per kidney (n = 5 or 6). Expression levels were evaluated using ImageJ software. D: expression of ICAM-1, IL-1β, and NLR family pyrin domain containing 3 (NLRP3) was examined by Western blot analysis (n = 5), and representative Western blots are shown. Anti-GAPDH antibody was used as a loading control. E−G: expression levels were evaluated using ImageJ software. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
Figure 5.
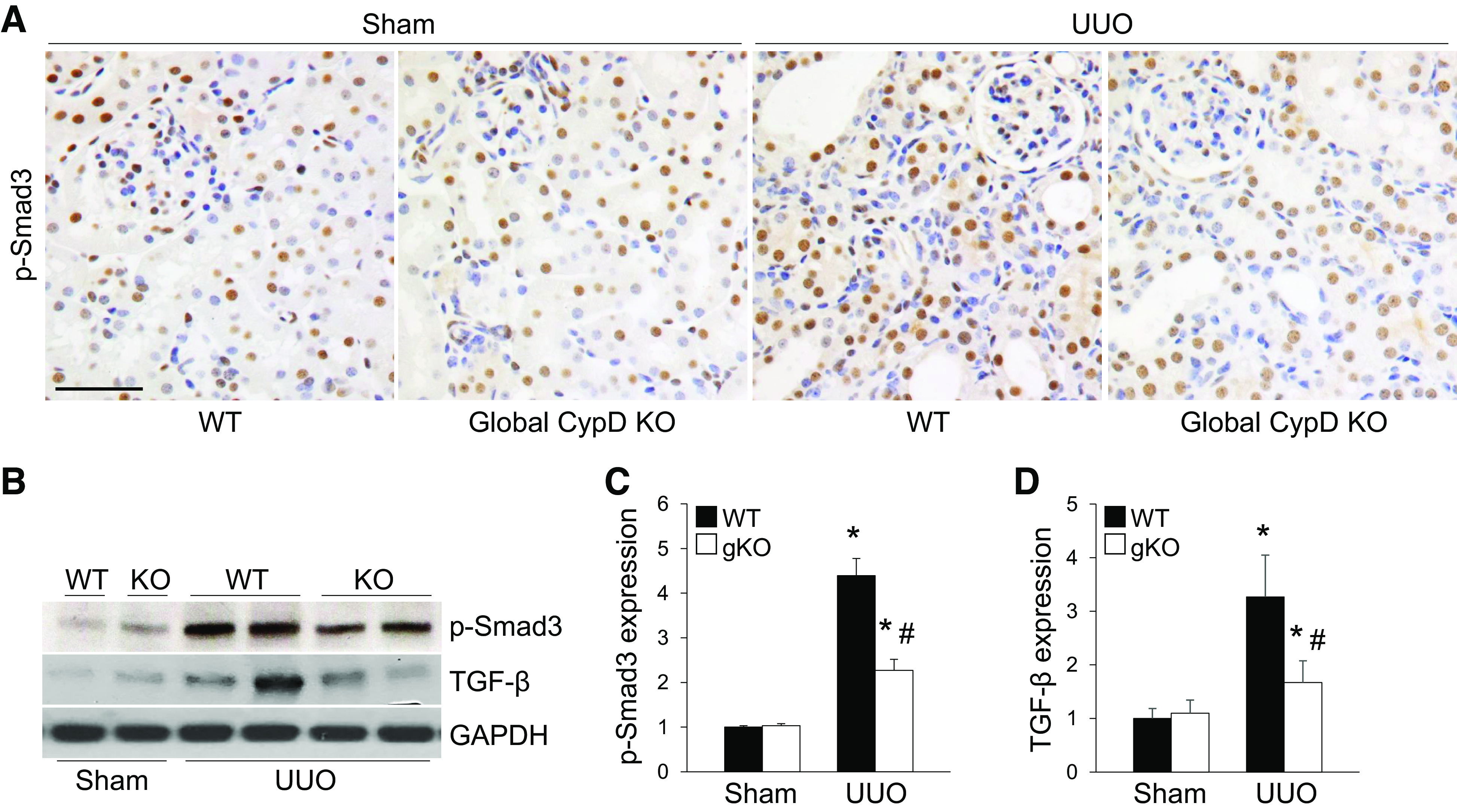
Cyclophilin D (CypD) knockout (KO) downregulates fibrogenic signaling molecules in kidneys with unilateral ureteral obstruction (UUO). Wild-type (WT) and global CypD KO mice (gKO) were subjected to UUO or sham operation for 7 days. A: paraffin-embedded kidney sections were used to carry out immunohistochemistry for evaluating the expression level of phospho-Smad3 (p-Smad3) (n = 5 or 6). Hematoxylin stain (blue color) was used for counterstaining. B: expression of p-Smad3 and transforming growth factor-β (TGF-β) was examined by Western blot analysis (n = 5), and representative Western blots are shown. Anti-GAPDH antibody was used as a loading control. C and D: expression levels were evaluated using ImageJ software. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
PT-Specific CypD KO Parallels the Effects of Global CypD KO During UUO
To clarify the role of PT CypD in CypD-mediated kidney fibrosis progression during UUO, we generated PT-specific CypD KO (PT-CypD KO) mice by crossing CypD floxed mice with PEPCK Cre recombinase-expressing mice (26). Mice were then subjected to UUO. We confirmed whether PT-CypD KO suppresses fibrosis progression evaluated by collagen deposition and expression of α-SMA. The Sirius red-positive area of collagen and expression levels of collagen type IV and α-SMA were inhibited in PT-CypD KO mice during UUO compared with those of WT controls (Cre-negative mice; Fig. 6, A–D). PT-CypD KO also suppressed UUO-induced increase of mRNA expression levels of collagen type I-α1 (Col1A1), α-SMA (Acta2), and fibrogenic cytokines [connective tissue growth factor (Ctgf) and Tgf-β)] (Fig. 6E). PT-specific CypD deficiency significantly prevented kidney tubular cell injury during UUO along with more preserved LTL-positive PT cells (Fig. 7, A–C). Similar to injury levels, PT cell cycle arrest, evaluated by immunostaining for p-histone H3, was mitigated in PT-CypD KO kidneys during UUO (Fig. 7, A and D), indicating that PT-specific CypD has a critical role in CypD-mediated kidney tubular injury and kidney fibrosis during UUO. To determine whether mitochondrial damage affects CypD-mediated fibrosis progression in obstructive nephropathy, we analyzed expression levels of mitochondrial electron transporter chain complexes. UUO resulted in downregulation of electron transport chain complexes, except complex IV, but the levels were not different between WT and PT-CypD KO mice (Supplemental Fig. S1), suggesting a mitochondrial damage-independent pathway of CypD-mediated kidney fibrosis progression. Inflammation and interstitial cell proliferation showed a similar pattern with that of PT injury and its consequence, kidney fibrosis in WT controls (Cre-negative mice) (Fig. 8 and Supplemental Fig. S2). PT-CypD KO suppressed increases of macrophage and neutrophil infiltration and Ki-67-positive interstitial cells (Fig. 8 and Supplemental Fig. S2). These data demonstrate that CypD in the PT plays a critical role in UUO-induced kidney inflammation and fibrosis progression.
Figure 6.
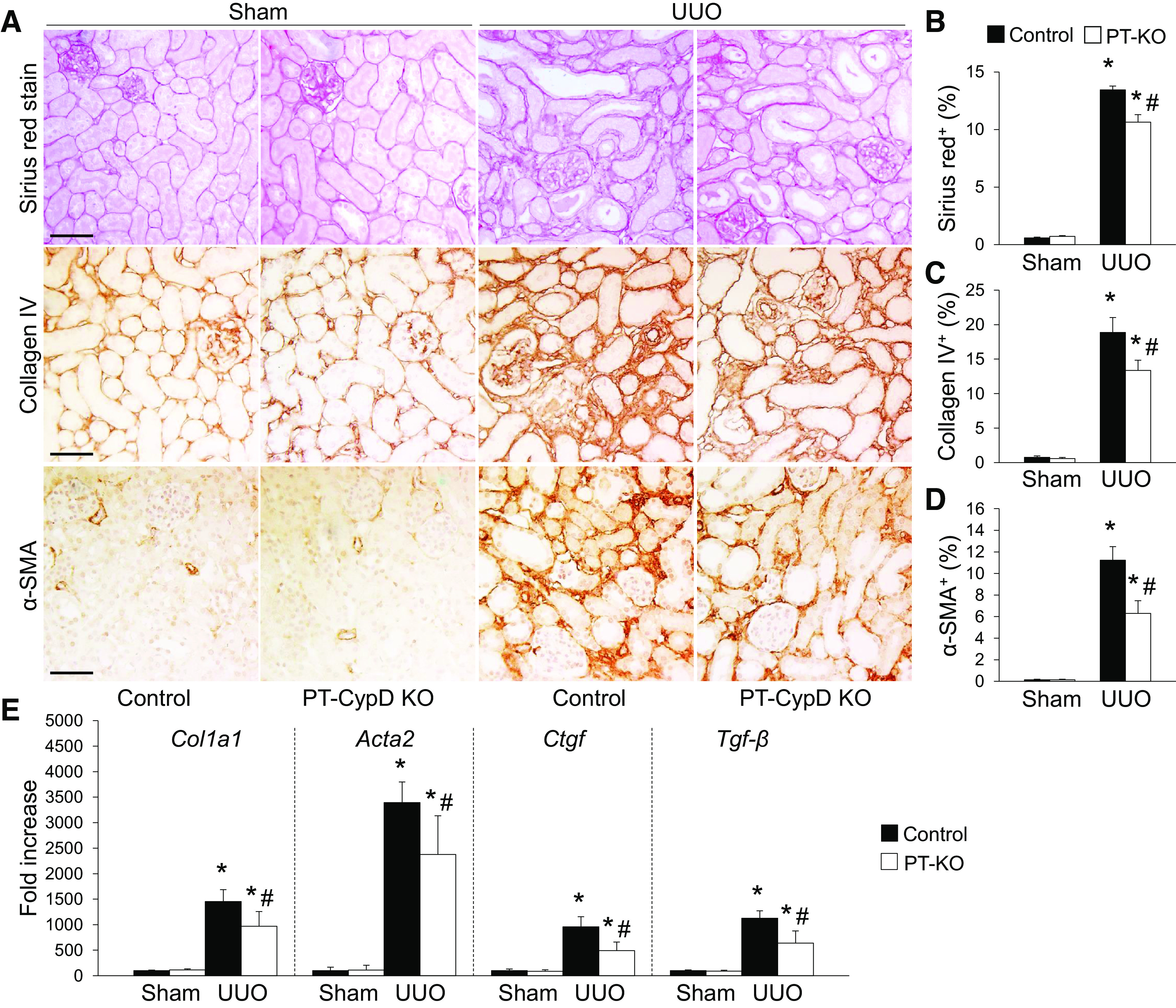
Proximal tubule-specific cyclophilin D knockout (PT-CypD KO) mitigates unilateral ureteral obstruction (UUO)-induced kidney fibrosis. Wild-type (WT) control and PT-CypD KO (PT-KO) mice were subjected to UUO or sham operation for 7 days. A: paraffin-embedded kidney sections were used to carry out Sirius red stain or immunohistochemistry for evaluating Sirius red-, collagen type IV, and α-smooth muscle actin (α-SMA)-positive areas (n = 5 or 6). Hematoxylin stain (blue color) was used for counterstaining. B−D: Sirius red (red color)-, collagen type IV (brown color)-, and α-SMA (brown color)-positive areas were counted from 5 randomly chosen fields per kidney (n = 5 or 6). Expression levels were evaluated using ImageJ software. E: collagen type I-α1 (Col1a1), α-SMA (Acta2), connective tissue growth factor (Ctgf), and transforming growth factor-β (Tgf-β) mRNA levels were evaluated using quantitative RT-PCR and calculated using a formula described in materials and methods (n = 5 or 6). Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
Figure 7.
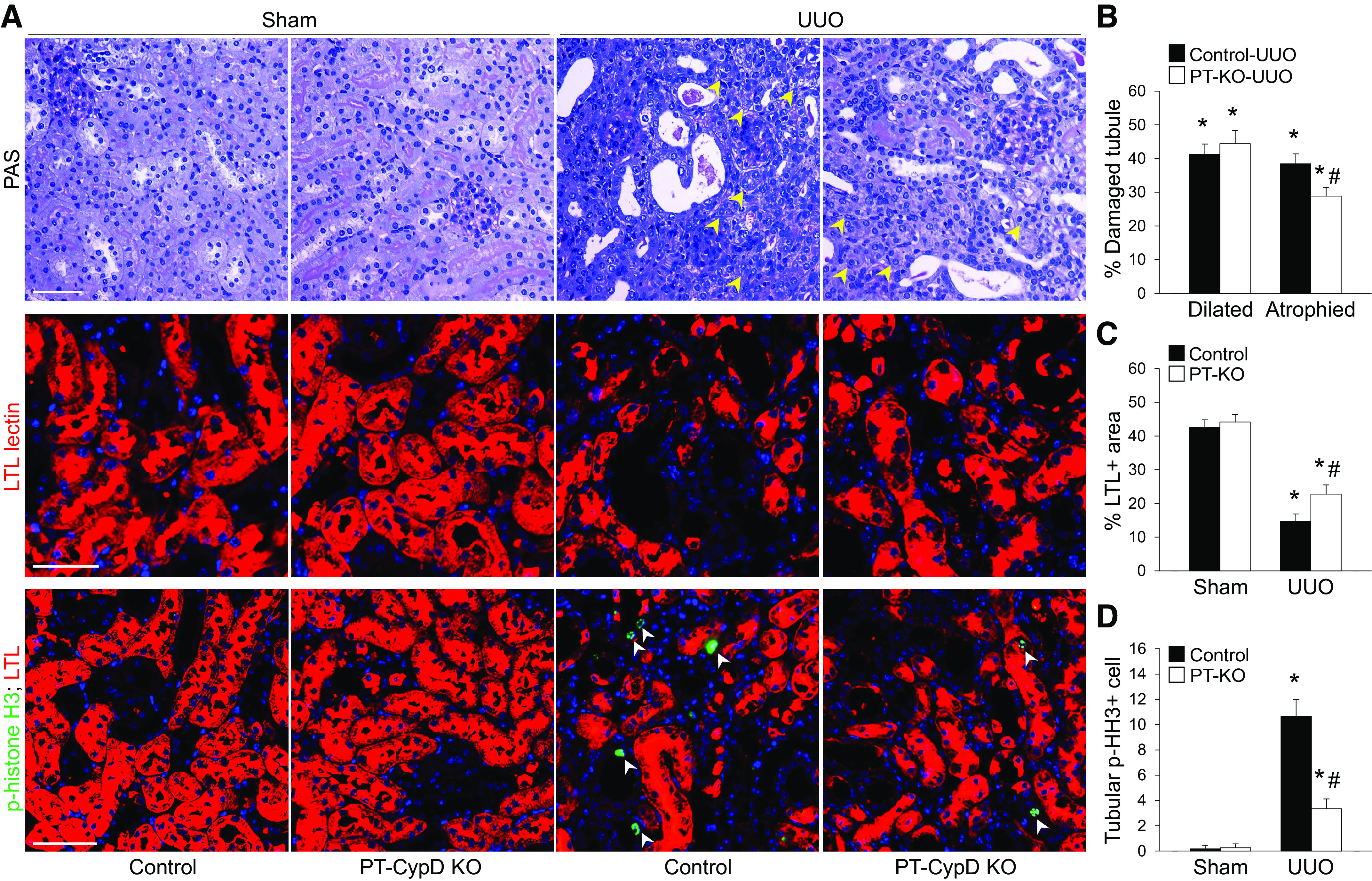
Proximal tubule-specific cyclophilin D knockout (PT-CypD KO) prevents unilateral ureteral obstruction (UUO)-induced tubule injury and loss and cell cycle arrest in proximal tubules during UUO. Wild-type (WT) control and PT-CypD KO (PT-KO) mice were subjected to UUO or sham operation for 7 days. A: paraffin-embedded kidney sections were used to carry out periodic acid-Schiff (PAS) and Lotus tetragonolobus lectin (LTL) staining (red) and immunohistochemistry for evaluating phospho-histone H3 (p-HH3; green) (n = 5 or 6). Hematoxylin and DAPI stain (both blue) were used for counterstaining. Yellow and white arrowheads indicate atrophied tubules and tubular p-HH3-positive cells, respectively. B: percent ratios of atrophied and dilated tubules to total tubules were calculated from 100 tubules in 5 randomly chosen fields per kidney (n = 5 or 6). C and D: LTL-positive areas and tubular p-HH3-positive cells were quantified from 5 randomly chosen fields per kidney (n = 5 or 6). Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
Figure 8.
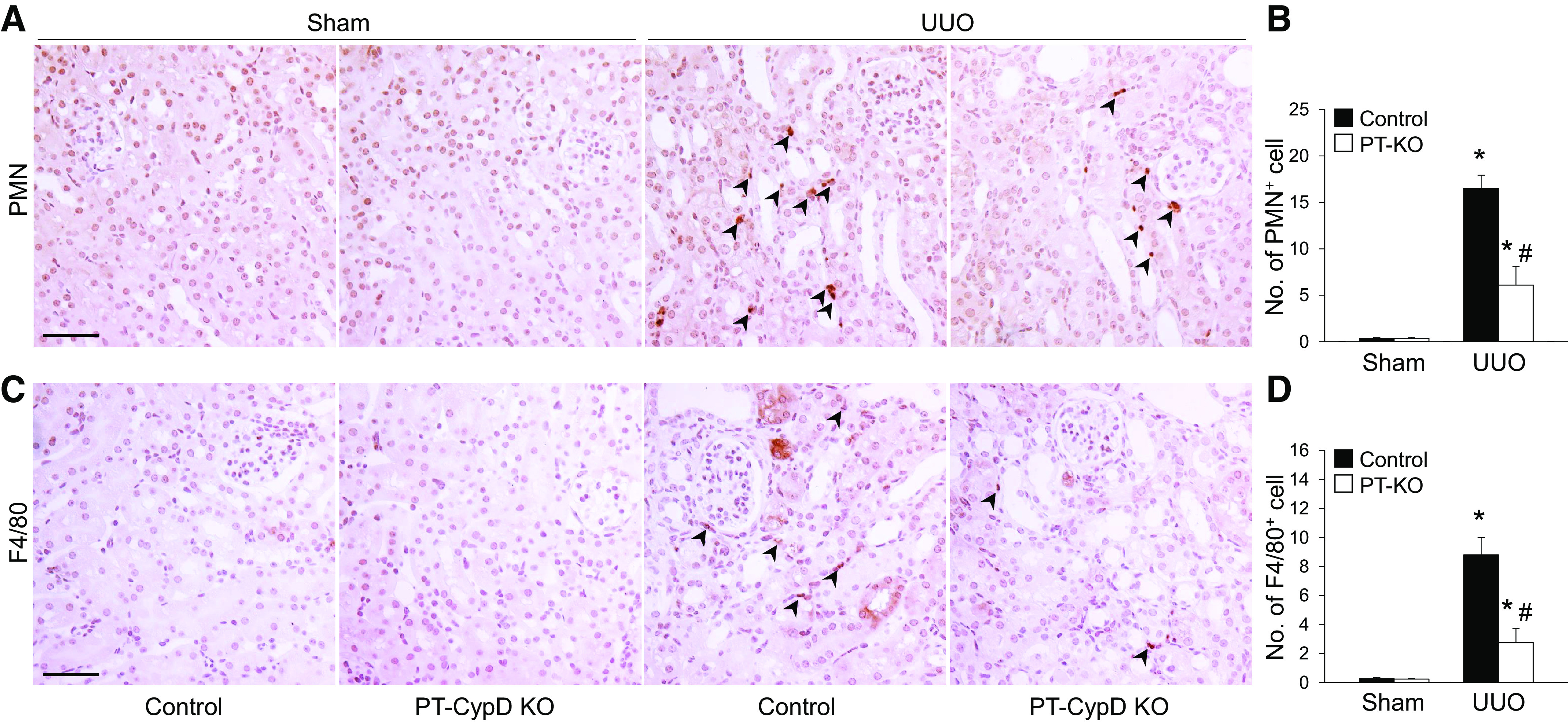
Proximal tubule-specific cyclophilin D knockout (PT-CypD KO) alleviates kidney inflammation during unilateral ureteral obstruction (UUO). Wild-type (WT) control and PT-CypD KO (PT-KO) mice were subjected to UUO or sham operation for 7 days. A and C: paraffin-embedded kidney sections were used to carry out immunohistochemistry to evaluate the number of polymorphonuclear leukocyte (PMN)- and F4/80-positive cells (n = 5 or 6). Hematoxylin stain (blue color) was used for counterstaining. Arrowheads indicate PMN- or F4/80-positive cells. B and D: numbers of PMN-positive neutrophils (B; brown color) and F/80-positive macrophages (D; brown color) were counted from 5 randomly chosen fields per kidney (n = 5 or 6). Expression levels were evaluated using ImageJ software. Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. sham; #P < 0.05 vs. WT UUO.
DISCUSSION
In the present study, we demonstrated that CypD mediates PT injury during obstructive nephropathy, leading to kidney fibrosis progression through increases of tubular cell death and atrophy and upregulation of proinflammatory and fibrogenic signaling molecules. Both global deletion and PT-targeted deletion of CypD resulted in a similar protective effect, suggesting that the PT is a major target for CypD-mediated tubular injury and inflammation and subsequent kidney fibrosis progression during obstructive nephropathy. Our findings highlight that targeting of the mitochondrial matrix protein CypD could be an effective strategy to prevent and treat kidney tubular injury, inflammation, and fibrosis progression in the UUO model.
The kidney PT is highly susceptible to various surgical and chemical stimuli due to its high energy demand for active reabsorption of glucose, ions, and nutrients (16, 36). Accumulating evidence suggests that the PT is a main target of kidney injury and its subsequent adverse outcomes during UUO (13). Ureteral obstruction causes acute and progressive PT injury along with persistent inflammation, tubular atrophy, and extracellular matrix accumulation, with consequent kidney fibrosis (37, 38). Cell injury/death-mediated tubular atrophy during UUO is one of the most significant features (39): blockade of cell death by apoptosis or necrosis halts or slows the progression of kidney fibrosis with suppressed tubular atrophy and proinflammatory and fibrogenic signaling (11, 30, 31, 40–42). Our present data show that CypD deficiency blunts kidney fibrosis with less tubular atrophy, but not tubular apoptosis, emphasizing the critical role of CypD in necrotic cell death during UUO (18, 24, 27).
Tubular G2/M cell cycle arrest has been recognized as a critical factor inducing maladaptive repair after acute kidney injury (3, 5, 43). The cell cycle-arrested PT upregulates profibrotic and proinflammatory factors to progress kidney fibrosis as a maladaptive repair mechanism (44). Our findings suggest that blockade of CypD prevents PT cell cycle arrest and may ultimately contribute to slow kidney fibrosis progression. Our results reiterate recent findings showing that limiting the secretion of proinflammatory and fibrotic signaling molecules from G2/M cell cycle-arrested PTs is critical to halt kidney fibrosis progression (45). Recent work has shown that cyclin G1, which controls cell cycle progression and is expressed in G2/M-arrested PT cells, and the related compartment, target of rapamycin-autophagy spatial coupling compartment (TASCC), instigates kidney fibrosis through PT cell cycle arrest and subsequent fibrotic secretion, but PT-specific deletion of TASCC suppresses these malevolent progression (46). On the contrary, appropriate recovery from acute kidney injury via facilitating cell cycle regulation seems to prevent kidney fibrosis progression. It has been reported that targeted blockade of ataxia telangiectasia and Rad3-related (ATR) in the PT worsens G2/M cell cycle arrest during UUO, resulting in augmented expression of TGF-β and connective tissue growth factor, contributing to kidney fibrosis progression (47). Recent reports have demonstrated that loss of mitochondrial function causes G1/S cell cycle arrest due to dysregulated cyclin E (48–50), suggesting a potential role for mitochondria in cell cycle regulation. Our study demonstrate that mitochondrial CypD could play a role in renal tubular cell cycle arrest during UUO.
CypD is likely associated with inflammatory signaling and tissue inflammation (51) as well as mitochondrial dysfunction (20). A recent study has demonstrated that CypD promotes inflammatory reprogramming and disease progression in the mouse liver during endotoxemia (52). RNA sequencing showed that CypD KO suppresses inflammatory signaling, including TNF receptor-mediated signaling, in septic liver injury. Similarly, genetic deletion of CypD attenuates the lipopolysaccharide-induced inflammatory response in mouse peritoneal macrophages by suppressing NF-κB signaling (53), showing a critical role of CypD in the inflammatory response of macrophages to acute stimuli. It has been shown that targeting CypD in aortic endothelial cells and in mice prevents angiotensin II- or calcium overload-mediated vascular inflammation (54, 55). In line with a recent report (56), our data clearly showed that both global and PT-CypD KO alleviate kidney inflammation with downregulated inflammatory cytokines and less accumulation of macrophages and neutrophils, suggesting a regulatory role of CypD in kidney inflammation, which may contribute to kidney fibrosis progression during obstructive nephropathy.
Accumulating evidence demonstrates that activation of CypD rather than its expression level is critical to both MPTP-dependent and MPTP-independent signaling (18). UUO did not change the expression level of CypD in the kidney (56, 57), suggesting that CypD-mediated kidney fibrosis progression would be associated with CypD activation. Posttranslational modifications, including phosphorylation, acetylation, and S-nitrosylation, have been shown to be associated with CypD activation in acute tissue injury (17, 58–60). CypD binding with mitochondrial or nonmitochondrial proteins, such as p53 and peroxisome proliferator-activated receptor-α, has also been suggested as a cause of CypD activation, which triggers cell death (26, 61). We have previously reported that CypD binds peroxisome proliferator-activated receptor-α in mitochondria to elicit impaired mitochondrial fatty acid oxidation, resulting in acute kidney injury (26). However, on the other hand, our data revealed that PT-CypD KO did not prevent mitochondrial damage, as evaluated by expression levels of electron transport chain complexes, in UUO kidneys. A previous report has shown that CypD in normal and cancer cells regulates cell proliferation and migration (62), suggesting extramitochondrial signaling of CypD. Future studies on determining whether CypD-mediated energy metabolism regulates kidney fibrosis progression and defining the role of CypD, independent of mitochondrial injury, in kidney fibrogenesis are warranted.
Taken together, our data emphasize PT injury as a driving force for the initiation and progression of tubular atrophy, inflammation, and kidney fibrosis during UUO. These data warrant further study for defining the precise mechanism by which CypD activation results in tubular atrophy and kidney fibrosis. Targeting PT CypD could be beneficial to prevent fibrosis progression in CKD, including obstructive nephropathy.
Perspectives and Significance
The mitochondrial matrix protein CypD is a component of the MPTP and a regulator of MPTP-dependent necrotic cell death. Blockade of CypD is well known to have a beneficial effect against acute tissue injury and death, but the effect in fibrotic diseases has not been defined. Here, we showed that PT CypD is critical to the progression of kidney fibrosis during obstructive nephropathy. This study provides new insights into a potential target to treat cell injury/death-mediated tissue remodeling and fibrogenesis. Further studies on determining whether and how a mitochondrial injury-independent role of CypD, such as regulation of glycolysis, fatty acid oxidation, cell cycle arrest, inflammation, and epithelial cell fate (62–65), affects kidney fibrosis progression would be of particular interest.
SUPPLEMENTAL DATA
All Supplemental Material: http://doi.org/10.6084/m9.figshare.14462037.
GRANTS
B.J.P. is supported by National Institutes of Health Grant Nos. DK116987, DK120533, and DK120846, and H.-S.J. is supported by American Heart Association Postdoctoral Fellowship Grant No. 15POST25130003.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.-S.J. and B.J.P. conceived and designed research; H.-S.J., M.R.N., L.H., and J.K. performed experiments; H.-S.J., M.R.N., and J.K. analyzed data; H.-S.J. and B.J.P. interpreted results of experiments; H.-S.J., M.R.N., and J.K. prepared figures; H.-S.J. drafted manuscript; H.-S.J. and B.J.P. edited and revised manuscript; H.-S.J., M.R.N., L.H., J.K., and B.J.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Volker H. Haase (Vanderbilt University) for providing the PEPCK-Cre transgenic mouse.
REFERENCES
- 1.Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med 373: 95–96, 2015. doi: 10.1056/NEJMc1504848. [DOI] [PubMed] [Google Scholar]
- 2.Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest 128: 97–107, 2018. doi: 10.1172/JCI93563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C, Group AXW; ADQI XIII Work Group. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27: 687–697, 2016. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 26: 1765–1776, 2015. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82: 516–524, 2012. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 7.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol 27: 2393–2406, 2016. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphreys BD, Xu F, Sabbisetti V, Grgic I, Movahedi Naini S, Wang N, Chen G, Xiao S, Patel D, Henderson JM, Ichimura T, Mou S, Soeung S, McMahon AP, Kuchroo VK, Bonventre JV. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 123: 4023–4035, 2013. doi: 10.1172/JCI45361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang HS, Padanilam BJ. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 309: F540–F550, 2015. doi: 10.1152/ajprenal.00170.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68–69: 248–262, 2018. doi: 10.1016/j.matbio.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311: F145–F161, 2016. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 306: F367–F378, 2014. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 15.Szeto HH, Liu S, Soong Y, Seshan SV, Cohen-Gould L, Manichev V, Feldman LC, Gustafsson T. Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1β and IL-18 and arrests CKD. J Am Soc Nephrol 28: 1437–1449, 2017. doi: 10.1681/ASN.2016070761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang HS, Noh MR, Kim J, Padanilam BJ. Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front Med (Lausanne) 7: 65, 2020. doi: 10.3389/fmed.2020.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 77: 1111–1122, 2013. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol Life Sci 73: 2309–2324, 2016. doi: 10.1007/s00018-016-2202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Javadov S, Jang S, Parodi-Rullán R, Khuchua Z, Kuznetsov AV. Mitochondrial permeability transition in cardiac ischemia-reperfusion: whether cyclophilin D is a viable target for cardioprotection? Cell Mol Life Sci 74: 2795–2813, 2017. doi: 10.1007/s00018-017-2502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res 126: 280–293, 2020. doi: 10.1161/CIRCRESAHA.119.316306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 23.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010, 2005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 297: F749–F759, 2009. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 25.Park JS, Pasupulati R, Feldkamp T, Roeser NF, Weinberg JM. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol 301: F134–F150, 2011. doi: 10.1152/ajprenal.00033.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang HS, Noh MR, Jung EM, Kim WY, Southekal S, Guda C, Foster KW, Oupicky D, Ferrer FA, Padanilam BJ. Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int 97: 327–339, 2020. doi: 10.1016/j.kint.2019.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029, 2013. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 66: 2576–2583, 2006. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang HS, Kim JI, Noh M, Rhee MH, Park KM. Regulator of G protein signaling 2 (RGS2) deficiency accelerates the progression of kidney fibrosis. Biochim Biophys Acta 1842: 1733–1741, 2014. doi: 10.1016/j.bbadis.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 30.Noh MR, Woo CH, Park MJ, In Kim J, Park KM. Ablation of C/EBP homologous protein attenuates renal fibrosis after ureteral obstruction by reducing autophagy and microtubule disruption. Biochim Biophys Acta Mol Basis Dis 1864: 1634–1641, 2018. doi: 10.1016/j.bbadis.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Padanilam BJ. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am J Physiol Renal Physiol 301: F450–F459, 2011. doi: 10.1152/ajprenal.00059.2011. [DOI] [PubMed] [Google Scholar]
- 32.Jang HS, Kim JI, Han SJ, Park KM. Recruitment and subsequent proliferation of bone marrow-derived cells in the postischemic kidney are important to the progression of fibrosis. Am J Physiol Renal Physiol 306: F1451–F1461, 2014. doi: 10.1152/ajprenal.00017.2014. [DOI] [PubMed] [Google Scholar]
- 33.Noh MR, Kong MJ, Han SJ, Kim JI, Park KM. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol 34: 101548, 2020. doi: 10.1016/j.redox.2020.101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang HS, Kim JI, Kim J, Park JW, Park KM. Angiotensin II removes kidney resistance conferred by ischemic preconditioning. Biomed Res Int 2014: 602149, 2014. doi: 10.1155/2014/602149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang HS, Kim JI, Jung KJ, Kim J, Han KH, Park KM. Bone marrow-derived cells play a major role in kidney fibrosis via proliferation and differentiation in the infiltrated site. Biochim Biophys Acta 1832: 817–825, 2013. doi: 10.1016/j.bbadis.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Scholz H, Boivin FJ, Schmidt-Ott KM, Bachmann S, Eckardt KU, Scholl UI, Persson PB. Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat Rev Nephrol 17: 335–349, 2021. doi: 10.1038/s41581-021-00394-7. [DOI] [PubMed] [Google Scholar]
- 37.Forbes MS, Thornhill BA, Minor JJ, Gordon KA, Galarreta CI, Chevalier RL. Fight-or-flight: murine unilateral ureteral obstruction causes extensive proximal tubular degeneration, collecting duct dilatation, and minimal fibrosis. Am J Physiol Renal Physiol 303: F120–F129, 2012. doi: 10.1152/ajprenal.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176: 1767–1778, 2010. doi: 10.2353/ajpath.2010.090345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schelling JR. Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatr Nephrol 31: 693–706, 2016. doi: 10.1007/s00467-015-3169-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mei S, Li L, Wei Q, Hao J, Su Y, Mei C, Dong Z. Double knockout of Bax and Bak from kidney proximal tubules reduces unilateral urethral obstruction associated apoptosis and renal interstitial fibrosis. Sci Rep 7: 44892, 2017. doi: 10.1038/srep44892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying Y, Kim J, Westphal SN, Long KE, Padanilam BJ. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol 25: 2707–2716, 2014. doi: 10.1681/ASN.2013121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang H, Li R, Zhang L, Zhang S, Dong W, Chen Y, Wang W, Li C, Ye Z, Zhao X, Li Z, Wu Y, Zhang M, Liu S, Dong Z, Liang X. p53-cyclophilin D mediates renal tubular cell apoptosis in ischemia-reperfusion-induced acute kidney injury. Am J Physiol Renal Physiol 317: F1311–F1317, 2019. doi: 10.1152/ajprenal.00072.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee K, Gusella GL, He JC. Epithelial proliferation and cell cycle dysregulation in kidney injury and disease. Kidney Int 100: 67–78, 2021. doi: 10.1016/j.kint.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu SM, Bonventre JV. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr Opin Nephrol Hypertens 29: 310–318, 2020. doi: 10.1097/MNH.0000000000000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canaud G, Brooks CR, Kishi S, Taguchi K, Nishimura K, Magassa S, Scott A, Hsiao LL, Ichimura T, Terzi F, Yang L, Bonventre JV. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci Transl Med 11: eaav4754, 2019. doi: 10.1126/scitranslmed.aav4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishi S, Brooks CR, Taguchi K, Ichimura T, Mori Y, Akinfolarin A, Gupta N, Galichon P, Elias BC, Suzuki T, Wang Q, Gewin L, Morizane R, Bonventre JV. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J Clin Invest 129: 4797–4816, 2019. doi: 10.1172/JCI122313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1-S transition. Proc Natl Acad Sci USA 106: 11825–11826, 2009. doi: 10.1073/pnas.0906430106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell 9: 843–854, 2005. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106: 11960–11965, 2009. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naoumov NV. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol 61: 1166–1174, 2014. doi: 10.1016/j.jhep.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Veres B, Eros K, Antus C, Kalman N, Fonai F, Jakus PB, Boros E, Hegedus Z, Nagy I, Tretter L, Gallyas F Jr, Sumegi B. Cyclophilin D-dependent mitochondrial permeability transition amplifies inflammatory reprogramming in endotoxemia. FEBS Open Bio 11: 684–704, 2021. doi: 10.1002/2211-5463.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Priber J, Fonai F, Jakus PB, Racz B, Chinopoulos C, Tretter L, Gallyas F Jr, Sumegi B, Veres B. Cyclophilin D disruption attenuates lipopolysaccharide-induced inflammatory response in primary mouse macrophages. Biochem Cell Biol 93: 241–250, 2015. doi: 10.1139/bcb-2014-0120. [DOI] [PubMed] [Google Scholar]
- 54.Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, Dikalov SI. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension 67: 1218–1227, 2016. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Du H, Chen D, Yuan H, Chen W, Jia W, Wang X, Li X, Gao L. Cyclophilin D deficiency protects against the development of mitochondrial ROS and cellular inflammation in aorta. Biochem Biophys Res Commun 508: 1202–1208, 2019. doi: 10.1016/j.bbrc.2018.12.064. [DOI] [PubMed] [Google Scholar]
- 56.Hou W, Leong KG, Ozols E, Tesch GH, Nikolic-Paterson DJ, Ma FY. Cyclophilin D promotes tubular cell damage and the development of interstitial fibrosis in the obstructed kidney. Clin Exp Pharmacol Physiol 45: 250–260, 2018. doi: 10.1111/1440-1681.12881. [DOI] [PubMed] [Google Scholar]
- 57.Leong KG, Ozols E, Kanellis J, Badal SS, Liles JT, Nikolic-Paterson DJ, Ma FY. Cyclophilin inhibition protects against experimental acute kidney injury and renal interstitial fibrosis. Int J Mol Sci 22: 271, 2020. doi: 10.3390/ijms22010271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amanakis G, Sun J, Fergusson MM, McGinty S, Liu C, Molkentin JD, Murphy E. Cysteine 202 of cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc Res 117: 212–223, 2021. doi: 10.1093/cvr/cvaa053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hurst S, Gonnot F, Dia M, Crola Da Silva C, Gomez L, Sheu SS. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis 11: 661, 2020. doi: 10.1038/s41419-020-02864-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286: 40184–40192, 2011. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barreto-Torres G, Hernandez JS, Jang S, Rodríguez-Muñoz AR, Torres-Ramos CA, Basnakian AG, Javadov S. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARα-cyclophilin D interaction in cardiomyocytes. Am J Physiol Heart Circ Physiol 308: H749–H758, 2015. doi: 10.1152/ajpheart.00414.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tavecchio M, Lisanti S, Lam A, Ghosh JC, Martin NM, O'Connell M, Weeraratna AT, Kossenkov AV, Showe LC, Altieri DC. Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. J Biol Chem 288: 5553–5561, 2013. doi: 10.1074/jbc.M112.433045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120: 3680–3687, 2010. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klawitter J, Pennington A, Klawitter J, Thurman JM, Christians U. Mitochondrial cyclophilin D ablation is associated with the activation of Akt/p70S6K pathway in the mouse kidney. Sci Rep 7: 10540, 2017. doi: 10.1038/s41598-017-10076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tavecchio M, Lisanti S, Bennett MJ, Languino LR, Altieri DC. Deletion of cyclophilin D Impairs β-oxidation and promotes glucose metabolism. Sci Rep 5: 15981, 2015. doi: 10.1038/srep15981. [DOI] [PMC free article] [PubMed] [Google Scholar]