

Keywords: astrocytes, gap junctions, patch-clamp, potassium channels
Abstract
Predominant expression of leak-type K+ channels provides astrocytes a high membrane permeability to K+ ions and a hyperpolarized membrane potential that are crucial for astrocyte function in brain homeostasis. In functionally mature astrocytes, the expression of leak K+ channels creates a unique membrane K+ conductance that lacks voltage-dependent rectification. Accordingly, the conductance is named ohmic or passive K+ conductance. Several inwardly rectifying and two-pore domain K+ channels have been investigated for their contributions to passive conductance. Meanwhile, gap junctional coupling has been postulated to underlie the passive behavior of membrane conductance. It is now clear that the intrinsic properties of K+ channels and gap junctional coupling can each act alone or together to bring about a passive behavior of astrocyte conductance. Additionally, while the passive conductance can generally be viewed as a K+ conductance, the actual representation of this conductance is a combined expression of multiple known and unknown K+ channels, which has been further modified by the intricate morphology of individual astrocytes and syncytial gap junctional coupling. The expression of the inwardly rectifying K+ channels explains the inward-going component of passive conductance disobeying Goldman–Hodgkin–Katz constant field outward rectification. However, the K+ channels encoding the outward-going passive currents remain to be determined in the future. Here, we review our current understanding of ion channels and biophysical mechanisms engaged in the passive astrocyte K+ conductance, propose new studies to resolve this long-standing puzzle in astrocyte physiology, and discuss the functional implication(s) of passive behavior of K+ conductance on astrocyte physiology.
INTRODUCTION
Astrocytes are a major glial subtype in the brain. Anatomically, these star-shaped glial cells tile the entire brain and establish a syncytium through gap junctional coupling. From a functional standpoint, homeostatic support is a commonly recognized function of astrocytes in the brain (1–4), and the expression of a large K+ conductance is a prerequisite for this crucial astrocyte function. First, this conductance renders astrocytes with a high membrane permeability to K+ ions so that K+ can be freely redistributed across the membrane following the electrochemical gradient. This feature is crucial for maintaining a physiological level of extracellular K+ (∼3 mM) whereby neuronal electrical impulses can be regenerated for signaling transduction (5, 6). Secondly, to support neuronal synaptic transmission, astrocytes are equipped with various transporter systems by which neurotransmitters, such as glutamate, GABA, and glycine, can be timely removed from the extracellular spaces. These uptake transporter systems are driven by the inward Na+ gradient across membrane; hence, the uptake efficacy increases along with the hyperpolarizing magnitude of the membrane potential (7–9). In other words, the closer the membrane potential is to the K+ equilibrium potential (approximately −90 mV), the higher the efficiency of the transporter systems in astrocytes. The abundant expression of K+ channels serves well for these astrocyte functions.
Given the significance of this conductance, it is important to understand the details (species, quantity, etc.) regarding K+ channels that make for this passive conductance. In this review, we will begin by recounting the history of the evolving understanding of astrocyte K+ conductance, focusing on the initial discovery of an ohmic behavior of K+ conductance from neuroglia, moving to the confirmation of a universal link of this conductance to functionally mature astrocytes. This will be followed by the discussion of the biophysical/physio-anatomical interpretation of this conductance, and an assessment of our current understanding of the ion channels, gap junctional coupling, and biophysical mechanisms engaged in the passive behavior of astrocyte K+ conductance. We will then summarize the known and unknown K+ channel species involved in astrocyte passive conductance. Finally, we will outline critical future studies that may help to resolve several remaining issues, such as the molecular identities of those unknown K+ channels and the function of passive conductance in astrocyte physiology and pathology.
ELECTRICAL PASSIVITY OF ASTROCYTE K+ CONDUCTANCE
Stephen Kuffler (10) is credited for being the first to report the basic electrophysiological properties of astrocytes in the mid-1960s. He and his associates examined the electrophysiological properties of astrocytes located at the optic nerve of an aquatic species of salamander, mudpuppies, using an elegant triple-sharp-electrode recording mode. Despite the use of “neuroglia” in their report, the cells under investigation were actually white matter fibrous astrocytes. Notably, until now, Kuffler’s findings are still the steadfast cornerstone for the contemporary study of astrocyte physiology (2, 4). With respect to their physiology, astrocytes are electrically nonexcitable cells, and the cell membrane exhibits a selective permeability to K+ ions compared with Na+ and Cl−. This K+ dominant permeability establishes a membrane conductance that is characterized by ohmic membrane voltage responses to the injected currents, or ohmic behavioral conductance (Fig. 1, C1 and D1). In the early 1990s, the application of the patch clamp to live brain slices confirmed the existence of the same glial subtype in the gray and white matter of the juvenile rodent brain (12–14) (Fig. 1, C2 and D2). These subtype of glial cells were then termed as “passive astrocytes'', and it was also speculated that the passive behavior indicates the functional maturation of astrocytes (14). This speculation was later confirmed by tracing this glial phenotype in the developing rodent brain and their correlation with astrocyte markers glial fibrillary acidic proteins (GFAP) and glutamate aspartate transporter (GLAST) (15, 16).
Figure 1.

Electrical passivity of astrocyte membrane K+ conductance. A: differential interference contrast (DIC) image of an astrocytic soma (left, arrow) during whole cell recording from a hippocampal slice prepared from ALDH1L1-eGFP astrocyte reporter mouse. B1, C1, and D1: current command (Ic; B1)-induced ohmic voltage response (C1 and D1) from an astrocyte. B2, C2, and D2: voltage command (Vc; B2)-induced linear (passive) membrane current (C2 and D2) from the same astrocyte. E: recording of a single freshly dissociated astrocyte with K+-free/Na+-containing electrode solution ([Na+]p) in 3.5 mM artificial cerebral spinal fluid (aCSF) solution. The absence of K+-conducting ion inside the astrocyte completely eliminated the outward-going K+ conductance (left). Removal of 3.5 mM K+ from aCSF further eliminated the inward-going passive conductance in recorded astrocyte (middle), and the inward-going currents could be fully restored by reemerging of 3.5 mM K+ in aCSF bath solution (right), indicating a high selectivity and permeability of astrocyte membrane to K+ ions. E is reproduced from Ref. 11 by permission of John Wiley and Sons. Im, membrane current; Vm, membrane potential.
After decades of investigation, a ubiquitous link of passive K+ conductance to the diverse subtypes of astrocytes in the adult brain has been firmly established in several species (from low to high), including human astrocytes (10, 15–21).
Currently, the consensus is that, in the early postnatal brain, the voltage-gated outwardly transient (IKa), delayed rectifying (IKd), and a low-density of Kir4.1 channels are the major K+ channels expressed by newborn/immature astrocytes (15, 16, 22). Additionally, beginning very early in the neonatal brain, the lack of depolarization-induced inward voltage-gated Na+ and Ca2+ conductance appears to be a defining feature of astrocytes (22). During postnatal development, the early expression of IKa and IKd gradually disappears from the whole cell conductance; it is not yet known whether these rectifying channels would simply be overshadowed or substituted by the increasing expression of leak K+ channel conductances. Nevertheless, the disappearance of rectifying channel components ultimately gives rise to a linear current-voltage passive conductance. In rodents, protoplasmic astrocytes uniformly adopt a passive conductance at the end of the third postnatal week (15, 16), and this passive conductance appears throughout the morphologically diverse subtypes of astrocytes within the adult brain (17–19, 21, 23–28). Therefore, passive conductance is a distinctive and defining electrophysiological feature of astrocytes in the adult brain.
THE BIOPHYSICAL ASPECTS OF ELECTRICAL PASSIVITY OF ASTROCYTE K+ CONDUCTANCE
The term ohmic behavior reminds one of Ohm’s law. Indeed, this happens to be a perfect example in which a physics law operates in the biological system to generate a key electrophysiological feature of astrocytes. In Stephen Kuffler’s seminal study (10), the K+ conductance exhibited a linear relationship of membrane voltage response to the injected currents, which closely followed the prediction of Ohm’s law for the current-voltage relationship (Fig. 1, C1 and D1). In the whole cell voltage-clamp recording, passive behavior is indicated by a linear membrane current response to the applied command voltages. However, strictly speaking, the amplitudes of inward-going currents induced by the hyperpolarizing command voltages are slightly larger than that of the outward-going currents induced by depolarizing voltages (see Fig. 1, C2 and D2) (29, 30). Therefore, a weakly inward rectifying K+ conductance is a more accurate term for describing this astrocyte K+ conductance. Nevertheless, to follow up on the convention, we use “ohmic” or “passive” conductance throughout this discussion.
To understand this electrical phenomenon, we will first introduce the concept of cells as electrical equivalent circuits: a combination of resistors and a capacitor (31) (Fig. 2A). While the double lipid bilayer membranes are high resistance insulators (∼1015 Ω/cm), the presence of various ion channels span the membrane creating pathways for charged ions to pass through (31). This reduces the otherwise high membrane resistance (Rm), a readout that is inversely proportional to the quantity of leak-type channels opening at a cell’s resting condition, or voltage-gated ion channels that are active upon the depolarization of membrane potential (Vm) to their activation thresholds. The high Rm is also able to separate opposing charges inside and outside the cells, thus acting as a membrane capacitance (Cm). Because Rm and Cm occur across the cell membrane, this creates a circuit of parallel Rm and Cm known as a RC circuit (Fig. 2A).
Figure 2.

Characteristics of astrocyte leak K+ conductance. A: Rm and Cm circuit representation of a cell. In voltage-clamp recording, a command voltage (Vc) is applied to the cell through an electrode with an access resistance (Ra) of 5 MΩ in series with membrane resistance (Rm) in the circuit. The Cm is the capacitance of the cell in parallel with Rm. B: in a model neuron, Rm =100 MΩ, a Δ10-mV Vc from resting membrane potential (Vm) −70 mV (B1) induces rapid charge/discharge of the capacitance (Icap, blue) and only small charges through the leak-type K+ channels (IK,leak, red). Iout is the total charge output in response to Vc. B2: Vc-induced output membrane potential (Vout) that is 95% of the Vc. C: in an astrocyte, Rm = 6.4 MΩ, a Δ10-mV Vc from resting Vm −80 mV (C2) induces Icap (blue) and IK,leak (red) that are superimposed (C1). The induced Vout is only 56% of the Vc. D and E: real recordings from a neuron and astrocyte as indicated. In a neuron, at the steady-state baseline marked by *, the neuron is voltage-clamped by the applied Vc. At an Rm=100 MΩ, the Ra,eff is close to the actual Ra, i.e., Ra,eff = RaRm/(Ra + Rm) hence, the Ra can be confidently resolved from Ra = (Vc − Vr)/Ipeak · Cm can be resolved from τ = Cm · Ra and the Rm can be resolved from Rm= Rtotal − Ra· E: applying a Δ10-mV Vc to a low Rm (6.4 MΩ) astrocyte with a low Ra,eff, the actual Ipeak cannot be measured for Ra calculation. An immediate merging of Icap with IK,leak makes it impossible to resolve the τ. F: in a high Rm neuron, the voltage clamping was readily achieved at the beginning of a +Δ50 mV Vc (marked by *), and that was followed by sequential activation of the voltage-gated inward INa, outward IKa, and IKd.
Ohmic Behavior Indicates a Constant Nonrectifying Membrane Resistance
An ohmic current-voltage relationship indicates a constant Rm in this RC circuit (Fig. 2A) (31). From a biological context, a constant Rm indicates a dominant expression of leak-type K+ channels with negligible copresence of voltage-dependent channel conductances. It should be noted that the interastrocytic gap junction channels also behave as leak channels and therefore contribute (in part) to the measured passive conductance and Rm in whole cell voltage-clamp recordings (22, 27, 32–36). The contribution of gap junctional coupling to the passive behavior will be elaborated further in Passive Behavior as a Result of Gap Junctional Coupling.
Electrical Passivity Signifies the Inability in Voltage-Clamp Control of a Recorded Astrocyte
To understand the nature of electrical passivity, we should briefly review the rationale of voltage-clamp recording. To identify voltage- and time-dependent ion/anion channels (rectifying conductance), a command voltage (Vc) is delivered to experimentally alter the Vm of a recorded cell from the resting condition to the desired level (Fig. 2, B and F). Successful attainment of the command voltage (Vc) is then indicated by a decline of the capacitive charge from the peak to the baseline (* in Fig. 2, B and F), a time point wherein the resting Vm is reset to Vc in the recorded cell (Fig. 2, B, D, and F). Using a neuronal recording as an example (Fig. 2F), the attainment of the Vc to the activation threshold of voltage-gated Na+ and K+ channels is followed by sequential activation of the following channels: the voltage-gated inward Na+ (INa), the outward transient (IKa), and the delayed rectifying (IKd) K+ conductances (Fig. 2F).
While this electrophysiological method serves well for the high Rm cells, such as neurons (100 MΩ to 200 MΩ) (37), it is of limited value for low Rm astrocytes (38). In an astrocyte, the low Rm (6.4 MΩ) creates a scenario wherein a large amount of injected current, instead of driving the resting Vm to Vc, instantaneously and continuously flows out of the recorded cell through the leak K+ channels (Ik,leak, Fig. 2, B and C) (39). A visible consequence is the lack of separation of capacitive charge/discharge (Icap, Fig. 2, C and E) from Ik,leak in astrocyte whole cell current profile (Fig. 2C) (40). In other words, the Vc-induced Icap and Ik,leak are temporally superimposed (Fig. 2, C and E), so that the Vc could never be attained. There is a theoretically estimated and experimentally detected >50% deficiency in Vc during astrocyte recording (38, 41) (Fig. 3, A–D). Thus the passive behavior indicates a severe Vc deficiency in voltage-clamp studies of astrocytes (33, 38, 41).
Figure 3.

Voltage deficiency in voltage-clamp recording of astrocyte and space constant. A: dual patch recording from an astrocyte in situ. B1: command voltages (Vc), −200 mV to +40 mV, 40-mV increment, delivered through an electrode to the astrocyte (① in A). B2: Vc-induced changes of astrocyte membrane potential (Vm) recorded by the second electrode (② in A). C: Vc-induced membrane current (Im) recorded by the first electrode (① in A). D: the plots of Im-Vc and Im-Vm revealed an >50% of voltage deficiency in voltage-clamping study of astrocytes. Note, a Goldman–Hodgkin–Katz (GHK) fit is superimposed to show the disobedience of astrocyte K+ conductance to a classic view of leak-type K channels with constant field outward rectification. E and F: calculation of space (length) constant (λ) from dual patch single astrocyte recording data. The fallout of the applied Vc from electrode ① (Vo) along the distance (d) to electrode ② in an astrocyte soma (E), yielding a λ of 3.6 µm according the equation shown in F. The λ here is calculated based on the data published in Ref. 41.
One can better understand the above discussion quantitatively using mathematical modeling. For the RC circuit shown in Fig. 2A, the membrane potential (Vm) satisfies the differential equation
where Ip represents the time-dependent injected current, Vr is the resting potential, , is the effective access resistance, and is the Vc-induced actual voltage drop on the cell. Solutions of this equation satisfy Vm(t) → Vout with time constant τ = CmRa,eff.
For neurons, Ra << Rm and, therefore, Vout ≈ Vc. For example, suppose that Rm =100 MΩ, Ra = 5 MΩ, Vc = −60 mV, and Vr = −70 mV. Then, Vout = −60.48 mV so that (Vout – Vr) = 0.952(Vc – Vr) (Fig. 2B). For astrocytes Ra and Rm are comparable. Suppose that Rm = 6.4 MΩ, Ra = 5 MΩ, Vc = −70 mV, and Vr = −80 mV. Then, Vout = −74.39 mV, so that (Vout – Vr) = 0.561(Vc – Vr) (Fig. 2C). Therefore, in response to an identical amplitude of Vc, there is 95% of the applied voltage distributed on a neuron versus 56% on an astrocyte.
Passive Conductance Disobeys the Goldman–Hodgkin–Katz’s Prediction for Classic Leak-Type K+ Channels
Another distinctive feature of passive conductance is the disobedience of the current activation kinetics to the prediction of Goldman–Hodgkin–Katz’s (GHK’s) current equation for the leak-type K+ channels (42, 43) (Fig. 3D). The leak K+ conductance is expected to show a constant field outward rectification in physiological recording solutions; in the recording made with a K+ gradient of intracellular/extracellular K+ concentration ([K+]i:[K+]o) = 140 mM:3.5 mM, the theoretical GHK fit is shown in Fig. 3D However, in actuality, the amplitude of the inward and outward passive conductance is rather symmetric (Fig. 1). This puzzle, i.e., the presence of a large inward-going passive conductance, can be partially explained by the presence of several inwardly rectifying K+ channels, such as Kir4.1, Kir5.1, and Kir6.1 (44–52) (Fig. 4). Due to the tendency to conduct greater inward currents (relative to outward going K+ currents against GHK prediction) inward rectifiers were initially considered as “anomalous” leak K+ channels (53). Thus the functional expression of inwardly rectifying K+ channels provides some sensible explanation for the inward component of passive conductance.
Figure 4.

Characteristics of Kir4.1 activation kinetics in astrocytes. A: inwardly rectifying currents were recorded from a neonatal astrocyte after inhibition of gap junctional coupling by 100 µM meclofenamic acid (MFA) (Icontrol). The command voltage (Vc) is shown in the inset. B: 100 µM Ba2+ inhibited a large portion of the inward component of currents but left the outward currents intact (IBa). C: the Ba2+ -sensitive Kir4.1 currents (IBa-sensitive) isolated by subtraction of traces in B from traces in A. D: shown the typical Kir4.1 currents (blue) that have a reversal potential (Vrev) of −80 mV, exhibit a large absence of depolarization-induced outward-going currents due to plugging of Mg2+ ions to Kir4.1 channel pores, and inhibition of inward-going currents at the Vc more hyperpolarizing than −140 mV (*) due to competitive Na+ binding to Kir4.1 channel pores with K+ ions. Modified from Ref. 22.
Passive Conductance Confers a Short Space Constant for Efficient Buffering of Large Electrical Inputs
The space (or length) constant (λ) is a measure of the steady-state decay of experimentally introduced voltage with distance in a cell. Due to the low Rm, there is a 74% reduction in the applied voltage within a short 5-µm distance in an astrocyte soma (41). Such a large and rapid voltage fallout along an astrocyte soma corresponds to a rather short space constant of 3.6 µm, indicating a high absorbance capacity of the astrocyte membrane to the electrical inputs, such as high K+-induced regional depolarization. Although this is an estimate from the astrocytic soma, the leak K+ channels, such as Kir4.1, show a more enriched expression in the terminal processes surrounding the synapses and blood vessels (54, 55). Hence this biophysical feature is likely also preserved within the astrocytic processes, allowing them to have a similar capacity to buffer the electrical disturbances from the extracellular space. At the syncytial levels, such a buffering capacity is further enhanced by syncytial isopotentiality to minimize the fluctuation of astrocyte Vm induced from neuronal depolarization inputs (56, 57). Interestingly, in a voltage-sensitive dye (VSD) study, the activation of astrocytic glutamate transporter was shown to generate a brief VSD transient (<5 ms) (58). Nevertheless, despite rapid progress in VSD techniques, lack of cellular resolution remains a technical barrier to determine if the neuronal activity would be able to induce significant sub-regional Vm depolarization in astrocytes (59).
THE PHYSIOANATOMICAL ASPECTS OF ELECTRICAL PASSIVITY OF ASTROCYTE K+ CONDUCTANCE
The voltage-clamp patch clamp recording mode was designed for identifying rectifying ion channels in a cell under investigation. Specifically, to achieve adequate voltage-clamping control, it is necessary to have a >90% change in Vm toward Vc (Fig. 2B). However, the presence of a low astrocytic membrane resistance (Rm, 6.4 MΩ) in series with a comparable value of electrode access resistance (Ra, i.e., 5 MΩ) substantially shifts the distribution of Vc from Rm to Ra. The resultant >50% voltage error makes it next to impossible to reach the activation threshold of voltage-gated K+ channels (41) (Fig. 3).
In addition to the low Rm, the coupling of astrocytes into a syncytium further exacerbates the voltage-clamp deficiency. A patch-clamp amplifier controls the voltage of a recorded astrocyte through a narrow electrical pathway, i.e., an electrode with a diameter <1 µm and typically 5 MΩ in resistance. However, the recorded astrocyte directly couples to seven to nine nearest neighbors through an even lower inter-astrocytic resistance of 4.2 MΩ in a parallel electrical circuit (11, 19). This creates a scenario wherein a patch-clamp amplifier is forced to compete with seven to nine directly coupled astrocytes as well as their extended neighbors to achieve a Vc. Meanwhile, the entire astrocyte network is sturdily voltage clamped by the syncytial isopotentiality at the resting physiological level (11). Together, these two factors cause several issues for the electrophysiological study of astrocytes.
How to Interpret the Whole Cell Current from the Voltage-Clamp Study?
The assumption that the passive conductance is K+ conductance was first deduced from the reversal potential of the conductance that is close to the K+ equilibrium potential (EK = −90 mV) and absence of member permeability to Na+ and Cl− (10, 14, 60) (Fig. 1D2), and later directly demonstrated by experiments where substitution of K+ ions in the recording solutions abolished the passive conductance (11) (Fig. 1E). Thus it is safe to claim that the passive conductance under investigation is largely mediated by K+ channels. However, it remains unfeasible to correlate passive conductance to any specific K+ channel conductance for the following reasons.
The “leaky” membrane and syncytial coupling together impose a severe difficulty in the identification of the channels that mediate passive conductance. We now use two examples to illustrate this problem. First, a commonly used paradigm in the voltage-clamp study is to step an astrocyte from resting potential (−80 mV) to a series of voltage steps in the range of ±Δ100 mV. In a high Rm neuron, or a neonatal and decoupled astrocyte, the IKa and IKd can be readily identified at their activation threshold of −50 mV and −40 mV, respectively (Fig. 2F and Fig. 7, B2 and B3). However, with a voltage error > 74% directly measured from the astrocyte recording (41), the maximum depolarization induced by a Δ +100 mV Vc can only shift the membrane voltage from the resting −80 mV to −55 mV, which is still far below the activation thresholds for IKa and IKd (61–63). Second, the Ba2+-sensitive Kir4.1 currents inactivate at Vm that are more hyperpolarized than −130 mV to −140 mV (22, 64, 65) (Fig. 4). This is caused by the competitive binding of Kir4.1 channel pores by Na+ at negative membrane potentials (66, 67). However, the actuality is that a Δ − 100-mV Vc drives only a change of the Vm from resting potential −80 mV to −105 mV. Hence, the distinct Na+-mediated Kir4.1 inactivation kinetics do not appear in astrocyte passive conductance (Fig. 5). Likewise, the isolated Ba2+-sensitive currents also do not exhibit the characteristics of Kir4.1 reported from neonatal or isolated astrocytes (Fig. 4). It is worth noting that in single isolated mature astrocytes [>postnatal (P) day 21], the Na+-mediated Kir4.1 inactivation is still invisible (11, 39) (Fig. 5), indicating an insufficiently improved voltage-clamp quality even in recordings made from single isolated astrocytes and the existence of other leak-type K+ channels contributing to the inward-going passive conductance.
Figure 7.

Gap junctional coupling is sufficient to bring about a passive behavior of membrane conductance. A: recording of the passive membrane conductance from a neonatal astrocyte in the hippocampal CA1 region (A1). Inhibition of gap junctional coupling with 100 µM meclofenamic acid (MFA) for 15 min resulted in a conversion of passive (A1) to rectifying current profile (A2) and substantial reduction of inward-going conductance (A3). B: the rectifying conductances in A2 comprised of inwardly rectifying Kir4.1 (B1), voltage-gated outward IKa (B2), and delayed rectifying IKd (B3). C–E: treatment of brain slices with MFA converted all the rectifying astrocytes to passive astrocytes (C), increased input resistance (Rin) from 50 MΩ to > 200 MΩ (D), and altered the Ioutward/Iinward ratio (E) from a linear, i.e.,∼ 1, to a strong outward rectification, 6.8. Modified from Ref. 22.
Figure 5.

Absence of the characteristics of Kir4.1 activation kinetics in mature astrocytes. A: 100 μM Ba2+ brought about a 2.5 mV and 14 mV depolarization of membrane potential (VM) in a syncytial coupled (black trace) and a freshly dissociated astrocyte (gray trace), respectively. B and C: in a syncytial coupled astrocyte, the Ba2+-sensitive currents remained a linear current-voltage (I-V) relationship, exhibiting neither an Mg2+ nor Na+ inhibition of outward- and inward-going currents noted in Fig. 4. D and E: in a single freshly dissociated astrocyte, 100 μM Ba2+ were able to substantially inhibit the passive conductance but failed to isolate a current component fitting in the activation kinetics of Kir4.1 shown in Fig. 4, indicating a mixed expression of multiple Ba2+-sensitive currents other than Kir4.1. VC, command voltage. Modified from Ref. 29.
Second, the low Rm also makes the quantitative analysis of passive conductance less reliable. Due to a drop of a large portion of Vc on Ra, instead of Rm, just a minor variation of Ra can introduce significant artifacts into the recorded currents. For instance, suppose that the astrocyte Rm be a constant (6.4 MΩ). The minor variations of Ra, e.g., changing from the initial 5 MΩ to 7.5 MΩ and 10 MΩ, will respectively reduce the whole-cell current by 18.0% and 43.9%. Thus, in addition to the fact that the Kir4.1 cannot be discerned from the passive conductance, the pharmacological analysis, such as 100 µM Ba2+-induced Kir4.1 inhibition, cannot be quantitatively analyzed with confidence (38) (Fig. 5). Furthermore, 100 µM Ba2+ typically induce a 3- to 5-mV depolarization in brain slice studies (29, 68), whereas the depolarization induced by the same concentration of Ba2+ was at least threefold greater in single freshly isolated astrocytes (Fig. 5A), suggesting syncytial coupled astrocytes, particularly those astrocytes residing deeper inside the brain slices, have an uneven exposure to the bath applied Ba2+ and therefore variably contributed to Vm depolarization in the recorded astrocyte (69).
In summary, although the passive conductance can be generally viewed as a K+ conductance, the conductance actually represents a compound expression of multiple known and unknown K+ channels (see known and unknown ion channels involved in passive conductance), and the recorded passive conductance is a result that has been further modified by the intricate morphology of individual astrocytes and syncytial coupling. The interrelated issues described above create a formidable barrier for reliable identification and quantitative analysis of the K+ channels expressed by astrocytes.
The Impact of Low Rm on the Measurement of Membrane Passive Electrical Properties
As a result of low Rm and the passivity of astrocyte membrane conductance, another commonly neglected problem is the inability to resolve the membrane passive electrical properties of astrocytes, i.e., access resistance (Ra), membrane resistance (Rm), and membrane capacitance (Cm).
Following a step in Vc, there is a sharp rise in the injected current Ip to a peak Ipeak = (Vc – Vr)/Ra (Fig. 2, B and D). This is followed by an exponential decay with time constant τ = CmRa,eff, where as we defined earlier. For neurons, Ra << Rm and, therefore, Ra,eff ≈ Ra. One can, therefore, resolve Ra, Cm, and Rm: , Cm = τ/Ra, Rm = Rt − Ra, where Rt is the total membrane resistance.
Unlike voltage-gated channels, the leak K+ conductance is altered instantaneously at the onset of a command voltage (Vc) that immediately merges into the upraise peak and decay phases of capacitive charge/discharge (Fig. 2, C and E). This “contamination” makes it impossible to know the actual peak amplitude of capacitive currents’ charge and, therefore, resolve Ra from (Fig. 2C). Because Ra,eff ≅ Rm in astrocytes, the Ra measured from “Membrane test” (pClamp9.0) is thus erroneous (38). Furthermore, the absence of the decay of the capacitive discharge makes the computation of membrane time constant (τ) erroneous, as is the membrane capacitance (Cm) that is resolved from Cm = τ/Ra (38).
Because Cm cannot be measured from astrocytes, it should not therefore, be used as a readout of astrocyte surface area for current density analysis. Without a reliable readout for Cm and Ra, the electronic Ra/Cm compensation, a designed feature of a patch-clamp amplifier, cannot be implemented during astrocyte whole cell recording. Because Rm also cannot be resolved from total membrane resistance (Rt, Rm = Rt − Ra), the membrane input resistance (Rin) becomes a useful measurement and has been used by several research groups to quantify the passive K+ conductance (68, 70–72).
Impact of the Low Rm on the Measurement of Membrane Potential
In the whole cell recording, the K+ gradient in the recording solutions is experimentally created, and as such, the ionic concentrations do not precisely mimic those of an actual cell. Thus, this method is unable to inform us of the actual Vm of astrocytes. However, when comparing the Vm values from different studies, we are reminded of how crucial it is to know the K+ gradients used in each experiment. Further, because of a low Na+ to K+ relative permeability (PNa/PK ≅ 0.018) and absence of resting Cl− permeability (51, 73, 74), the astrocyte Vm is highly sensitive to even a minor change in the extracellular K+ concentration. Because of this, the Vm of astrocytes is famously known as a “K+ electrode” which reports dynamic changes in extracellular K+ concentrations (75–77). For example, a change in extracellular K+ from 3.5 mM to 2.5 mM would theoretically hyperpolarize the Vm by −5 mV according to the GHK prediction (31). In published studies, K+ concentrations varied from 2.5 mM, 3 mM, and 3.5 mM in bath artificial cerebral spinal fluid (aCSF) solutions from different research groups. Indeed, in studies using 2.5 mM K+ in bath solution, the reported resting Vm is typically more hyperpolarizing than −82 mV, whereas resting Vm around −76 mV is reported in studies using 3.5 mM K+ in aCSF solutions.
Turning back to our original point, we ask the question: does a low Rm also affect the measurement of resting Vm from astrocytes? The answer is most certainly, yes! Based on the voltage divider equation below (74),
a lower Rm makes the recorded Vm measurement closer to the cell’s actual membrane potential, Em. Under an identical glass-membrane high gigaohm seal resistance (Rseal) made between different types of cells, such as astrocytes and neurons, the Vm in low Rm astrocytes is closer to the Em than that of the high Rm neurons.
To explain this further with the actual numbers, we can assume a 2 GΩ Rseal to be achieved in both a 6.4 MΩ Rm astrocyte and a 100 MΩ Rm neuron that respectively have an Em of −90 mV and −70 mV. Based on the voltage divider equation, the recorded Vm from this astrocyte and neuron would be −89.7 mV and −66.7 mV, respectively. However, the Vm in the low Rm astrocyte deviates only 0.33% from the actual Em compared with a large 4.7% deviation of Vm from Em in neuron. Thus the Vm measurement reflects more closely the actual battery potential (Em) of astrocytes. Another reminder one can take from this analysis is that the recorded neuronal Vm is somewhat less than the actual negative battery potentials (Em); this error should also be considered and can be corrected based on the Rm of the neurons under investigation.
Furthermore, the low Rm also creates a technical barrier to ensure high-quality whole-cell recording from astrocytes. As we discussed above, a high gigaohm seal is a prerequisite for reliable measurement of Vm, and whole-cell study of membrane conductance. Once a high gigaohm (>2 GΩ) is established, a leak K+ conductance immediately steps into the “membrane seal test” currents (Fig. 2C). This masks the initially established Rseal, or, in other words, the Rseal becomes “invisible.” This makes it difficult to ensure whether a high gigaohm seal remains during the whole cell recording, and consequently, the extent to which the measured Vm would still be reliable. The powerful influence of the leak K+ conductance on the “visibility” of the Rseal was demonstrated in a report where the cell-attached recording mode was used to record astrocyte Vm (69). In cell-attached mode, a recording mode able to gain an electrical access to the interior of a cell without rupturing the membrane, there was a spontaneous decline of membrane input resistance (Rin) and a progressive hyperpolarizing shift in Vm that ultimately reached a resting Vm value comparable to that of the whole cell recording at approximately −76 mV (69). This study showed that, because of the low Rm in astrocytes, even a high gigaohm seal remains during the recording, and the Rseal becomes invisible to confirm the quality of the recorded Vm. Therefore, additional criteria should be applied to warrant the quality of astrocyte whole cell recording. One of the empirical practices is the requirement of an initial Rseal to reach at least 2 GΩ high at the initial phase of cell-attached mode. Another practice following the attainment of a gigaohm seal is to disregard those recordings that had suffered from a depolarizing Vm shift during the rupturing of the membrane into the whole cell mode (69), a sign of transient or permanent deterioration of the initially established Rseal.
In summary, the low Rm (on one hand) provides a high fidelity for Em reporting for astrocytes, but on the other hand, it makes the quality control of whole-cell astrocyte recording difficult. Heterogeneity of astrocytes has attracted increasingly attention. However, with careful consideration of all the factors listed above, a wealth of in vivo and in situ results favor a view that homogeneity in resting Vm is a unique feature of astrocytes (19, 24, 25, 34, 51, 75, 78–84).
MECHANISMS UNDERLYING THE PASSIVE BEHAVIOR OF ASTROCYTE K+ CONDUCTANCE
Two potential mechanisms have been put forth to explain the ohmic behavior of astrocyte K+ conductance (85). In the first theory, the intrinsic properties of K+ channels are speculated to be the cause of passive behavior, i.e., the activation of multiple leak and rectifying K+ channels act in synergy to generate the inward and outward portions of passive conductance. The second hypothesis is that the gap junctional coupling causes the passive behavior of astrocyte membrane conductance.
Passive Behavior as a Result of the Intrinsic Properties of Ion Channels
The first evidence favoring this hypothesis came from the study of excised outside-out patches from hippocampal astrocytes in which the passive behavior remained in the majority of excised astrocyte membrane patches (86). However, to directly test this hypothesis, it is necessary to avoid syncytial coupling by using of freshly dissociated astrocytes (87). It is also necessary to examine this question using astrocytes isolated from >P21 animals when the passive conductance becomes a uniform feature of astrocytes (16). Furthermore, after the third postnatal week, the gene expression, cellular morphology, and functional properties of astrocytes all reach maturity (33, 44, 63, 65, 88). By electrophysiologically examining isolated mature astrocytes, this hypothesis was definitively proved by two independent studies, indicating that the passivity reflects the intrinsic properties of astrocyte K+ channels (25, 39) (Figs. 6 and 8). This notion was further corroborated by studies where astrocytic connexin43 (Cx43) and connexin30 (Cx30) were genetically deleted. In Cx43/Cx30 double gene knockout mice, the passive behavior K+ conductance remained intact (34, 83). Thus, the intrinsic properties of K+ channels are sufficient to create a passive behavior of astrocyte membrane conductance. It is worth noting that the input resistance (Rin) in single isolated astrocytes remains at a comparably lower level than that of their counterparts in situ, i.e., 6.4 MΩ (39) (Fig. 6), indicating that the voltage-clamp control is still limited to a rather small somatic area around the patch electrode in a single astrocyte recording. Hence, the passive behavior most likely reflects the ion channel properties of the soma, whereas the precise contribution of the distal astrocyte processes to the passive conductance remains unknown (85).
Figure 6.

The intrinsic properties of ion channels are sufficient to bring about a passive behavior of membrane conductance in astrocytes. A and B: the passive current profile appeared in astrocytes either recorded in situ (left) or from a single astrocyte freshly dissociated from the syncytial coupling (right). C: plots of the current-voltage relationship of the 2 astrocyte recordings shown in B. D: no difference of input resistance (R-input) between astrocytes in situ and dissociated single astrocytes. Astrocytes recordings were taken from mice that were at ages older than postnatal day (P)21. DIC, differential interference contrast; Vc, command voltage. Modified from Ref. 39.
Figure 8.

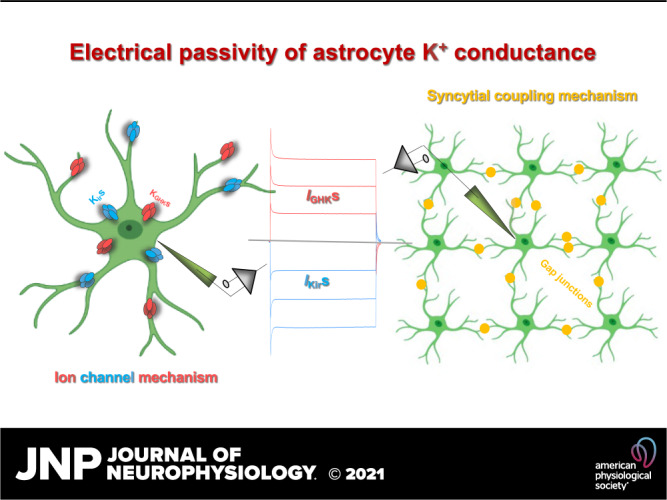
Mechanisms underlying the electrical passivity of astrocyte passive K+ conductance. The combined expression of inward-going inwardly rectifiers (Kirs) and yet unknown outward-going Goldman–Hodgkin–Katz rectifiers (KGHKs) is sufficient to give rise to a passive behavior K+ conductance (top: ion channel mechanism). Syncytial coupling alone is also able to generate a passive behavior of membrane conductance (IKirs and IGHKs; bottom: syncytial coupling mechanism). The two mechanisms work in concert to underlie the electrical passivity of astrocyte K+ conductance in functionally mature astrocytes.
Passive Behavior as a Result of Gap Junctional Coupling
Olfactory bulb ensheathing cells (OECs) are gap junctional coupled and are electrically passive in the whole cell current profile. Interestingly, pharmacological uncoupling of OECs converted the membrane current from a passive to a rectifying profile (89). A similar observation was reported from the pharmacological uncoupling of gap junctional coupled GFAP-expressing cells from the postnatal subventricular zone (90). These observations imply that electrical coupling alone would be sufficient to create a passive behavior of membrane conductance.
This hypothesis was demonstrated in a study using hippocampal slices prepared from early postnatal ages (<P10) where inhibition of gap junctional coupling resulted in substantial suppression of passive conductance, and that, in turn, activated rectifying K+ conductance (65). A more definitive evidence in support of this hypothesis was provided by a study of neonatal astrocytes in situ (22). Neonatal astrocytes predominantly express voltage-gated IKa and IKd, and to a much lesser extent, the Kir4.1. However, 8% of neonatal astrocytes have already adopted a passive whole cell current profile. Interestingly, following pharmacological inhibition of gap junctional coupling (100 µM meclofenamic acid), IKa and IKd appeared to be the dominant K+ conductance that transformed the passive to rectifying current profile (Fig. 7A). Therefore, gap junctional coupling is sufficient to mask or suppress the activation of rectifying K+ conductance. Thus, cell-to-cell coupling alone is sufficient to bring about a passive behavior of membrane conductance (Fig. 8).
Interestingly, a large portion of inward passive conductance appears to be the gap junctional conductance in low Kir4.1-expressing neonatal astrocytes. Under an uncoupled condition, all neonatal astrocytes express a rather low density of Ba2+-sensitive Kir4.1 (Fig. 7A2) and high density of IKa and IKd (Fig. 7, B2 and B3), whereas syncytial coupling creates a symmetric passive conductance. More specifically, syncytial coupling enhances the inward passive conductance by 5.5- to 9.4-fold (Fig. 7A3), in contrast to only a 1.6-fold increase in the outward IKd after uncoupling (Fig. 7A3) (22). Because IKa and IKd are predominant K+ conductances in neonatal astrocytes, this observation suggests a “filter effect” of gap junctions; preferring the passage of leak K+ conductance versus the rectifying K+ conductances. Indeed, a syncytial coupling facilitates the distribution of leak K+ conductance, and that in turn prevents the Vm deviation of individual astrocytes and cardiomyocytes from syncytial isopotentiality (69, 91). Although the biophysical mechanisms engaged in this behavior are unknown, gap junctional coupling appears to allow aggregation of inward passive conductance from neighboring astrocytes into a recorded astrocyte, which then “amplifies” the inward portion of passive conductance (Fig. 7A).
How Does Gap Junction Coupling Alter the Behavior of Membrane Conductance?
In neonatal astrocytes, the magic swing of membrane conductance between rectifying and passive states appears to be the result of three coordinated acts (22). First, coupling lowers the input resistance (Rin) of individual astrocytes and that in turn masks the activation of rectifying IKa and IKd. Evidently, uncoupling of the recorded neonatal astrocytes increases the membrane Rin from 50 MΩ to ∼200 MΩ (Fig. 7D), a Rin that is comparable to neurons. This enables the activation of IKa and IKd in neonatal astrocytes. In short, gap junctional coupling suppresses the activation of voltage-gated channels and facilitates the intercellular transfer of leak K+ conductance in an astrocyte syncytium. Second, the cell-cell coupling prevents the activation of the rectifying component of inward passive conductance. The Kir4.1 currents show a characteristic inactivation at the command voltages more negative than −130 mV to −140 mV (Fig. 4D) (22, 63–65, 92). Under coupled conditions, this rectifying component disappears due to the voltage deficiency (see the biophysical aspects of electrical passivity of astrocyte k+ conductance). Third, coupling recruits junctional conductance from neighboring astrocytes into the recorded passive conductance. Note that the so-called junctional conductance is also K+ conductance-generated from K+ channels of neighboring astrocytes. As a result of gap junctional coupling, these together generate a linear passive conductance (Fig. 1).
In summary, gap junctional coupling alone is sufficient to bring about a passive behavior conductance. In the neonatal and early postnatal stages, this is the sole mechanism accounting for the passivity of neonatal immature astrocytes. Nevertheless, a prerequisite to achieve this is the presence of a low number of leak K+ channels. Second, in the adult brain, the intrinsic property of the expressed K+ channels alone is sufficient to underlie passive behavior. However, in functionally mature astrocytes, gap junctional coupling and the expression of various leak K+ channels work in concert to account for the passive membrane conductance (Fig. 8).
KNOWN AND UNKNOWN ION CHANNELS INVOLVED IN PASSIVE CONDUCTANCE
We know now that the intrinsic properties of astrocyte K+ channels are sufficient to give rise to a passive conductance. Additionally, the expression of inwardly rectifying K+ channels provides an explanation for the inward-going component of passive conductance. However, the K+ channels encoding the outward-going passive conductance are still largely unknown. Hence, gaining a better understanding of those leak K+ channels that follow the GHK constant field outward rectification should be a central focus of future studies.
Now three categories of leak K+ channels have been identified: 1) the inwardly rectifying K+ channels that preferentially conduct K+ ions in the inward direction; 2) two-pore domain K+ channels that are constitutively leak and sensitive to various physiochemical stimuli; and 3) the Ca2+-activated K+ channels that are sensitive to the change of intracellular Ca2+ concentrations. In each of these leak channel families, several K+ channel isoforms and some of their auxiliary units are expressed in astrocytes at the molecular and functional levels (44, 45).
Inwardly Rectifying K+ Channels
The inwardly rectifying Kir4.1 is the most extensively studied leak K+ channel in astrocytes. In cultured and freshly dissociated astrocytes, Kir4.1 shows a moderate to strong inward rectification, resulting from the blockage of the channel pore by Mg2+ and polyamine (Fig. 4). In native astrocytes, Kir4.1 is highly sensitive to micromolar concentrations of barium (Ba) (IC50 = 7.1 µM) (65). Hence the currents can be effectively inhibited by barium at 100 µM concentration (63–65, 92–95). Therefore, pharmacological isolation of Kir4.1 by 100 µM Ba is a commonly accepted approach to study this leak K+ channel. The functional expression of Kir4.1 in astrocytes was documented in studies carried out in the early postnatal brain in situ and freshly dissociated single astrocytes (22, 63, 65, 92, 94). The activation kinetics and the current sensitivity to micromolar concentrations of barium are consistent with Kir4.1 being expressed in cultured astrocytes (64). Another powerful way to study the contribution of Kir4.1 to passive conductance is animal genetic manipulation, conditional knockout (cKO) of Kir4.1 from astrocytes. Indeed, Kir4.1 deletion resulted in a substantial membrane potential depolarization (68). Nevertheless, Kir4.1 expression is up-regulated during development (44, 65, 96), whereas only a few Kir4.1 cKO animals in this study survived to adulthood (68). These results/observations call into question the actual contribution of Kir4.1 to the passive conductance. An additional concern regarding the contribution of Kir4.1 to the passive conductance is the lack of barium sensitivity. In isolated single postnatal astrocytes, 100 µM barium can completely inhibit Kir4.1 currents (65). However, in brain slice recordings, barium (at the same concentration) depolarized astrocytes by only 3–5 mV with insignificant reduction in the passive conductance (29, 38, 68, 97) (Fig. 5F). A more precise dual-patch single astrocyte analysis showed that the Ba2+-sensitive Kir4.1 accounts for 48% of passive conductance (38). Consequently, these results imply that Kir4.1, perhaps together with other channels in the inwardly rectifying K+ channel family, is a major class of K+ channels contributing to the passive conductance. However, additional unknown leak K+ channels may also contribute significantly to the passive conductance, especially the outward-going component of passive conductance, such as GHK rectifiers (KGHKs) (Fig. 8).
Two-Pore Domain K+ Channels
The lack of barium sensitivity of passive conductance and the disobedient nature of activation kinetics of passive conductance to GHK constant field rectification indicated the involvement of other (perhaps equally important K+ channels) to the passive conductance, such as two-pore domain K+ channels (K2Ps) (41). Among 15 members in the K2P family, TAKS-1, TREK-1, TREK2, and TWIK-1 showed expression in astrocytes (41, 65, 98–102), and the expression of TWIK-1 and TREK-1 are particularly high at both transcriptomic and protein levels (29, 41, 65, 103). Furthermore, a TWIK-1 mutant (TWIK-1.K274E) can produce a conductance very close to astrocytic passive conductance (104, 105), and TREK-1 is a GHK rectifier (106). These studies suggested that the combined expression of TWIK-1 and TREK-1 could sensibly explain the passive conductance. However, the opposite results were generated in follow-up studies using a variety of approaches (i.e., gene knockdown and TWIK-1/TREK-1 double gene knockout mice) (22, 29, 30, 97, 103, 107). It should be noted that both genetic approaches are inherently subject to different limitations. For example, the germline gene knockout method may potentially alter the expression of other genes, including K+ channels, and shRNA-mediated gene knockdown has potential off-target effects as well (108). Thus an additional and more definitive approach, such as Cre/Lox conditional knockout system, is necessary to clarify the actual contribution of TWIK-1 and TREK-1 to astrocyte passive conductance.
Ca2+-Activated K+ Channels
Astrocytes also show the expression of another class of leak K+ channels - calcium-activated K+ channels (KCa). There are three subclasses of KCa: large-conductance KCa (BK, KCa1.1), intermediate-conductance KCa2+ (IK, KCa3.1), and small-conductance KCa (SK, KCa2.1, KCa2.2, and KCa 2.3) (109). BK expression is enriched in astrocytic processes surrounding vasculature (110). Evidence shows that oscillations in astrocyte intracellular Ca2+ ([Ca2+]i) induces dilation/constriction of blood vessels via efflux of K+ ions, and this process is mediated by astrocytic BK channels. The release of K+ ions through astrocytic BK channels can sequentially activate the Kir and voltage-gated Ca2+ channels in the smooth muscles enwrapping vascular lumen. The Ca2+ influx through voltage-gated Ca2+ channels then initiates vessel dilation/constriction to regulate blood flow (111, 112). Additionally, IK and SK are also expressed in astrocytic processes and have been indicated to regulate the tone of the vasculature (113, 114). Given the preferential perivascular location of KCa, the study of astrocytic KCa has mainly been in the context of their role in neurovascular coupling. Thus the extent to which KCa contributes to the passive conductance remains unknown.
Voltage-Gated K+ Channels
It is necessary to note that despite the fact that IKd and IKa are outward K+ conductance channels, they are not leak-type K+ channels. Due to a significant voltage deficiency, they also likely have minimal contribution to the outward-going passive conductance. Otherwise, these voltage-dependent conductances can be temporally separated from the initial capacitive discharge with distinctive outward transient and delayed rectifying characteristics (Fig. 2). Additionally, given a rather depolarized activation threshold, the contribution of these rectifying K+ channels to the passive conductance should be minimal.
Another unknown question is whether the rectifying IKa and IKd seen in the neonatal and postnatal developmental stage continue to be present in mature astrocytes. If so, it would be intriguing to know where these channels are located. Furthermore, it may be interesting to investigate the conditions that activate these channels and how their activation influences astrocyte physiology and pathophysiology. Future studies that examine these ideas are of high merit.
CAN THE PATCH-CLAMP REGAIN THE POWER FOR PHYSIOLOGICAL STUDIES OF ASTROCYTES?
Independent of how cells can be categorized into excitable or nonexcitable subclasses, the electrical properties always inform the important function of a living cell under investigation, and this has not been an exception in astrocyte studies over the past several decades. In this regard, the electrophysiological approach remains an irreplaceable and indispensable means for studies of astrocyte physiology and pathology. Additionally, there has been a broad extension of the traditional use of the electrophysiological methods in conjunction with others, such as imaging studies of Na+ and glucose dynamics and topology of astrocyte networks, and the innovative use of the same method with computational modeling for analyzing the electrical coupling of the syncytial networks (33, 115–120).
Although progress has been made with respect to our understanding of the nature of astrocyte passive conductance, a major question yet to be determined is the entire repertoire of K+ channels involved in astrocyte membrane conductance. An answer to this question would allow the patch-clamp method to regain momentum for astrocyte physiology studies. To illustrate this possibility, we present two examples in this discussion: one conducted in Müller glia (121) and another from freshly dissociated hippocampal astrocytes (65). In these studies, the channel blockers for Kir4.1, IKa, IKd, and two-pore domain K+ channels showed an additive suppression of the membrane passive conductance and stepwise depolarization of Vm in Müller glia and isolated postnatal hippocampal astrocytes (65, 121). These studies were designed, in part, based on the pharmacology for the known Kir4.1 and “fishing expedition” for other speculated unknown K+ channels in Müller glia and astrocytes. These experimental results demonstrated that once we have a full knowledge of the molecular identities of K+ channels, a similar strategy can be used to study a given K+ channel of interest upon suppression of the remainder of K+ channels in astrocytes either pharmacologically or through the use of genetically modified mice lacking a defined set of K+ channels.
Several single-astrocyte RNA-seq databases have now been made publicly available. For example, the Khakh laboratory (45) has identified nine highly expressed K+ channels and two auxiliary subunits in their publication and database generated from isolated adult striatum, hippocampus, and cortical astrocytes. In addition to the high expression of Kir4.1, other leak K+ channels in the two-pore domain and Ca2+-activated channel family also show high expression at the transcriptomic level. Interestingly, several K+ channels encoding IKa (Kcnd2) and IKd (Kcna2, Kcna6, and Kanq3) also exhibit a moderately high expression in two RNA-seq databases, i.e., Brain RNA-seq (Barres Lab) (http://www.brainrnaseq.org/) (122). Data sources that are more easily accessible are valuable for cross-examination and validation of those candidate genes before delving into large-scale functional studies.
For astrocytes, this approach can be conducted in the presence of gap junction inhibitors, such as meclofenamic acid, which can effectively inhibit astrocyte gap junctional coupling by >99% (11, 35, 36). Furthermore, the cell-cell coupling can be completely circumvented by the use of freshly dissociated astrocytes (39, 63, 65, 116, 123, 124).
FUNCTIONAL SIGNIFICANCE OF THE PASSIVE BEHAVIOR OF ASTROCYTE K+ CONDUCTANCE?
Perhaps the most basic question to ask is the biological significance of passivity of astrocyte K+ conductance. To begin this discussion, we must first be aware of several key facts. First, each astrocyte exhibits an intricate spongiform morphology (36, 125–128). Second, numerous cellular units are then aggregated into a syncytial network (1, 56, 127, 129–131). Additionally, different isoforms of K+ channels, such as Kir4.1, are distributed at varying subcellular compartments in astrocytes (54, 55). However, in electrophysiological studies, the passive behavior is an electrical phenomenon extracted from astrocyte somatic recordings; hence, the anatomical/functional representation of this electrical feature to individual astrocytes and to astrocyte networks must first be understood.
What Does Passive Behavior Mean for Astrocyte Cellular Functionality?
In single isolated astrocytes, we’ve learned that both inward and outward passive conductances are the conductances of leak K+ channels (11) (Fig. 1E). Additionally, several known and unknown K+ channels work in concert to make up a passive conductance (63, 65). A key function of passive K+ conductance is offering astrocytes the ability to redistribute K+ ions across the membrane at an equal efficacy in both inward and outward directions (Fig. 1E).
At the individual cellular level, inwardly rectifying K+ channels should primarily contribute to the inward-going component of passive conductance. The tendency of inwardly rectifiers for inward transfer of K+ ion makes them ideal for K+ uptake. An unresolved issue is the identities of other leak K+ channels encode the outward passive conductance.
Another poorly understood issue is the subcellular location of leak K+ channels. The currently available evidence favors a nonuniform distribution of these channels. In retina Müller glia, the Kir4.1 has a preferential association with the end-feet adjacent to vitreous humor, which is believed to allow for the release of spatially transferred K+ ions from the inner plexiform (28, 136). Kir4.1 has also been shown to have a preferential association in fine astrocytic processes abutting synapses (54) and blood vessels (55).
What Does Passive Behavior Mean for Astrocyte Functionality at Syncytial Levels?
We now know that gap junctional coupling alone is sufficient to generate a passive behavior of a membrane conductance (22, 89, 90). However, in the context of an astrocyte syncytium, the coupling-mediated membrane passivity must be established upon the intrinsic properties of membrane K+ channels. First, gap junctional coupling may create a passive behavior of membrane conductance, but it cannot render astrocyte membrane with a high selectivity and permeability to K+ ions. Second, the ability to generate a symmetric conductance also depends on the intrinsic properties of expressed K+ channels in astrocytes. Connexin 43 and connexin 30, the two major gap junction channels in astrocytes, do not show voltage-dependent gating rectification (132–135).
As a result of syncytial coupling-mediated passivity, the K+ conductance generated from individual astrocytes becomes a shared characteristic of a syncytial network. This idea is supported by a study where the intracellular K+ ions, conducting ions for outward K+ currents, were eliminated by dialysis of the cell with K+-free/Na+-containing electrode solution (11). In theory and actuality, the removal of intracellular K+ content abolishes the outward passive conductance (Fig. 1E). However, outward passive conductance remained in the recording resulting from transjunctional sharing of outward K+ conductance from neighboring astrocytes (11), indicating that part of the passive conductance in a syncytial coupled astrocyte is derived from neighboring astrocytes. In other words, the currents recorded in an astrocyte soma do have a shared representation of the K+ conductance from the peripheral processes of the same cell and neighboring astrocytes.
In summary, the abundant expression of leak K+ channels, the biophysical features of gap junction channels, and the low interastrocytic resistance create a powerful isopotential reticular system (130). The electrical passivity of this system is the functional basis for homeostatic regulation of brain function.
Does Passive Behavior Suppress the Activation of Rectifying K+ Conductance?
Currently, the function of rectifying K+ channels that are present throughout the transcriptome and appear function in neonatal and freshly isolated astrocytes remains a mystery (44, 45, 63, 65, 124). Given the expression of several voltage-gated K+ channels in transcriptomic databases, these channels appear to be a continuing feature of mature astrocytes (44, 45, 122). However, the conditions that enable the activation of these rectifying channels are still poorly understood.
In neonatal astrocytes, despite abundant expression, cell-to-cell coupling can effectively mask the activation of rectifying K+ channels (22). In the mature brain, a stronger syncytial isopotentiality can further obscure the activation of these channels in the somatic whole cell recording. Nevertheless, because syncytial isopotentiality is a functional readout from astrocyte soma recordings, it is possible that some of the astrocytic processes, especially those that do not form astrocyte-astrocyte pathways, could be weakly controlled by syncytial isopotentiality (127). Accordingly, environmental depolarization inputs may be sufficient to activate these rectifying K+ channels. If so, it would be interesting to follow-up by examining the functional consequence of rectifying K+ channel activation. Activation of IKa and IKd causes efflux of K+ ions. If these K+ channels do have a preferential expression on astrocytic end-feet, they may regulate the tone of vascular smooth muscles, and consequently, regional cerebral blood flow.
To answer this question (and others), we must first precisely pinpoint the location of these rectifying channels. From there, we are better positioned to understand their functions within the brain. For example, one may postulate that if these channels are “strategically” located in astrocytic processes that enwrap bundles of axons, as revealed in a recent EM study (127), then these rectifying K+ channels may be activated upon neuronal firing. Hence, the resulting release of K+ ions may vary extracellular K+ concentration, which subsequently influences the frequency and pattern of neuronal firing. One could also imagine that the perisynaptic location of these channels may largely escape the control of syncytial isopotentiality, and the resultant channel activation may attenuate depolarization inputs during synaptic transmission. Overall, how leak K+ channels, rectifying K+ channels, and syncytial isopotentiality work in concert in astrocyte function remains an open question for future study.
CONCLUDING REMARKS
Although recent studies have led to significant progress toward our knowledge of the electrical passivity of astrocyte membrane conductance, our understanding of the physiological implication(s) of this unique feature of astrocytes is still in its infancy. At the very least, we know now that the passivity is a complex outcome of the interplay between multiple factors such as intricate cellular morphology, membrane ion channels, intercellular gap junctional coupling, and syncytial isopotentiality (Fig. 8). Because of the complex nature of this passivity, one, unfortunately, cannot directly extrapolate the needed information, such as changes in ion channel or gap junctional coupling, simply based on an apparent change in the passive conductance. Thus experimental conditions should be carefully created to study these functional properties of astrocytes. For example, to study the functional expression of membrane K+ channels, uncoupling astrocytes from their network, either pharmacologically or genetically, would be crucial. Furthermore, if one is interested in a specific ion channel, inhibition of other major astrocytic leak K+ channels is necessary. Likewise, to study the actual strength of electrical coupling, the use of pairs of freshly dissociated astrocytes is desirable, and the elimination of membrane leak K+ conductance is also necessary. Finally, accumulating evidence has demonstrated that the electrical passivity of membrane behavior coupled with a rather hyperpolarizing resting membrane potential is a gold standard for the differentiation of astrocytes in healthy conditions from those that undergo pathological changes in disease states.
GRANTS
This work is sponsored by a grant from the National Institute of Neurological Disorder and Stroke Grant RO1NS116059 (to M. Z.) and an Alumni Grant for Graduate Research and Scholarship, Ohio State University (to S.A.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.Z., Y.D., S.A., and D.T. prepared figures; M.Z., Y.D., S.A., and D.T. drafted manuscript; M.Z., Y.D., S.A., and D.T. edited and revised manuscript; M.Z., Y.D., S.A., and D.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Harold K. Kimelberg (1941–2021), a pioneer in the field of astrocyte biology, for discussions on a broad range of issues in glial research, including the question in this review article. We thank Dr. Serguei Skatchkov for providing information on the early study of neuroglia and astrocyte passive K+ conductance with Dr. Richard Orkand and the provision of related literature.
Present address of S. Aten: Dept. of Neurology, Division of Sleep Medicine, and Program in Neuroscience, Harvard Medical School, Beth Israel Deaconess Medical Center, Boston, MA 02215.
REFERENCES
- 1.Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11: 87–99, 2010. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- 2.Kimelberg HK. Functions of mature mammalian astrocytes: a current view. Neuroscientist 16: 79–106, 2010. doi: 10.1177/1073858409342593. [DOI] [PubMed] [Google Scholar]
- 3.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 119: 7–35, 2010. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev 98: 239–389, 2018. doi: 10.1152/physrev.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36: 291–300, 2000. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 6.Walz W. Role of glial cells in the regulation of the brain ion microenvironment. Prog Neurobiol 33: 309–333, 1989. doi: 10.1016/0301-0082(89)90005-1. [DOI] [PubMed] [Google Scholar]
- 7.Danbolt NC. Glutamate uptake. Prog Neurobiol 65: 1–105, 2001. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Corcuera B, Benito-Munoz C, Aragon C. Glycine transporters in glia cells: structural studies. Adv Neurobiol 16: 13–32, 2017. doi: 10.1007/978-3-319-55769-4_2. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Danbolt NC. GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 4: 165, 2013. doi: 10.3389/fendo.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol 29: 768–787, 1966. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- 11.Ma B, Buckalew R, Du Y, Kiyoshi CM, Alford CC, Wang W, McTigue DM, Enyeart JJ, Terman D, Zhou M. Gap junction coupling confers isopotentiality on astrocyte syncytium. Glia 64: 214–226, 2016. doi: 10.1002/glia.22924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger T, Schnitzer J, Kettenmann H. Developmental changes in the membrane current pattern, K+ buffer capacity, and morphology of glial cells in the corpus callosum slice. J Neurosci 11: 3008–3024, 1991. doi: 10.1523/JNEUROSCI.11-10-03008.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chvatal A, Pastor A, Mauch M, Sykova E, Kettenmann H. Distinct populations of identified glial cells in the developing rat spinal cord slice: ion channel properties and cell morphology. Eur J Neurosci 7: 129–142, 1995. doi: 10.1111/j.1460-9568.1995.tb01027.x. [DOI] [PubMed] [Google Scholar]
- 14.Steinhauser C, Berger T, Frotscher M, Kettenmann H. Heterogeneity in the membrane current pattern of identified glial cells in the hippocampal slice. Eur J Neurosci 4: 472–484, 1992. doi: 10.1111/j.1460-9568.1992.tb00897.x. [DOI] [PubMed] [Google Scholar]
- 15.Kafitz KW, Meier SD, Stephan J, Rose CR. Developmental profile and properties of sulforhodamine 101-labeled glial cells in acute brain slices of rat hippocampus. J Neurosci Methods 169: 84–92, 2008. doi: 10.1016/j.jneumeth.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 16.Zhou M, Schools GP, Kimelberg HK. Development of GLAST(+) astrocytes and NG2(+) glia in rat hippocampus CA1: mature astrocytes are electrophysiologically passive. J Neurophysiol 95: 134–143, 2006. doi: 10.1152/jn.00570.2005. [DOI] [PubMed] [Google Scholar]
- 17.Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, Silva AJ, Takano T, Goldman SA, Nedergaard M. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell stem cell 12: 342–353, 2013. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirischuk S, Moller T, Voitenko N, Kettenmann H, Verkhratsky A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosci 15: 7861–7871, 1995. doi: 10.1523/JNEUROSCI.15-12-07861.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiyoshi CM, Du Y, Zhong S, Wang W, Taylor AT, Xiong B, Ma B, Terman D, Zhou M. Syncytial isopotentiality: a system-wide electrical feature of astrocytic networks in the brain. Glia 66: 2756–2769, 2018. doi: 10.1002/glia.23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuffler SW, Potter DD. Glia in the leech central nervous system: physiological properties and neuron-glia relationship. J Neurophysiol 27: 290–320, 1964. doi: 10.1152/jn.1964.27.2.290. [DOI] [PubMed] [Google Scholar]
- 21.Zayas-Santiago A, Agte S, Rivera Y, Benedikt J, Ulbricht E, Karl A, Davila J, Savvinov A, Kucheryavykh Y, Inyushin M, Cubano LA, Pannicke T, Veh RW, Francke M, Verkhratsky A, Eaton MJ, Reichenbach A, Skatchkov SN. Unidirectional photoreceptor-to-Muller glia coupling and unique K+ channel expression in Caiman retina. PLoS One 9: e97155, 2014. doi: 10.1371/journal.pone.0097155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong S, Du Y, Kiyoshi CM, Ma B, Alford CC, Wang Q, Yang Y, Liu X, Zhou M. Electrophysiological behavior of neonatal astrocytes in hippocampal stratum radiatum. Mol Brain 9: 34, 2016. doi: 10.1186/s13041-016-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A 94: 14821–14825, 1997. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang AY, Woo J, Sardar D, Lozzi B, Bosquez Huerta NA, Lin CJ, Felice D, Jain A, Paulucci-Holthauzen A, Deneen B. Region-specific transcriptional control of astrocyte function oversees local circuit activities. Neuron 106: 992–1008, 2020. doi: 10.1016/j.neuron.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lalo U, Palygin O, Rasooli-Nejad S, Andrew J, Haydon PG, Pankratov Y. Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex. PLoS Biol 12: e1001747, 2014. [Erratum in PLoS Biol 12: e1001857, 2014]. doi: 10.1371/journal.pbio.1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci 26: 2673–2683, 2006. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller T, Moller T, Neuhaus J, Kettenmann H. Electrical coupling among Bergmann glial cells and its modulation by glutamate receptor activation. Glia 17: 274–284, 1996. doi: 10.1002/(SICI)1098-1136(199608)17:4<274::AID-GLIA2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Newman EA. Regional specialization of retinal glial cell membrane. Nature 309: 155–157, 1984. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Y, Kiyoshi CM, Wang Q, Wang W, Ma B, Alford CC, Zhong S, Wan Q, Chen H, Lloyd EE, Bryan RM, Zhou M. Genetic deletion of TREK-1 or TWIK-1/TREK-1 potassium channels does not alter the basic electrophysiological properties of mature hippocampal astrocytes in situ. Front Cell Neurosci 10: 13, 2016. doi: 10.3389/fncel.2016.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Kiyoshi CM, Du Y, Ma B, Alford CC, Chen H, Zhou M. mGluR3 activation recruits cytoplasmic TWIK-1 channels to membrane that enhances ammonium uptake in hippocampal astrocytes. Mol Neurobiol 53: 6169–6182, 2016. doi: 10.1007/s12035-015-9496-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hille B. Ion Channels of Excitable Cells. Sunderland, MA: Sinauer, 2001. [Google Scholar]
- 32.Dermietzel R, Hertberg EL, Kessler JA, Spray DC. Gap junctions between cultured astrocytes: immunocytochemical, molecular, and electrophysiological analysis. J Neurosci 11: 1421–1432, 1991. doi: 10.1523/JNEUROSCI.11-05-01421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephan J, Eitelmann S, Zhou M. Approaches to study gap junctional coupling. Front Cell Neurosci 15: 640406, 2021. doi: 10.3389/fncel.2021.640406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26: 5438–5447, 2006. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu G, Wang W, Kimelberg HK, Zhou M. Electrical coupling of astrocytes in rat hippocampal slices under physiological and simulated ischemic conditions. Glia 58: 481–493, 2010. doi: 10.1002/glia.20939. [DOI] [PubMed] [Google Scholar]
- 36.Xu G, Wang W, Zhou M. Spatial organization of NG2 glial cells and astrocytes in rat hippocampal CA1 region. Hippocampus 24: 383–395, 2014. doi: 10.1002/hipo.22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholls JG, Martin AB, Brown DA, Diamond ME, Weisblat DA, Fuchs PA. From Neuron to Brain (5th ed.). Sunderland, MA: Sinauer Associates. 2012. [Google Scholar]
- 38.Ma B, Xu G, Wang W, Enyeart JJ, Zhou M. Dual patch voltage clamp study of low membrane resistance astrocytes in situ. Mol Brain 7: 18, 2014. doi: 10.1186/1756-6606-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du Y, Ma B, Kiyoshi CM, Alford CC, Wang W, Zhou M. Freshly dissociated mature hippocampal astrocytes exhibit passive membrane conductance and low membrane resistance similarly to syncytial coupled astrocytes. J Neurophysiol 113: 3744–3750, 2015. doi: 10.1152/jn.00206.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enyedi P, Czirják G. background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605, 2010. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 41.Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, Chen H. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci 29: 8551–8564, 2009. doi: 10.1523/JNEUROSCI.5784-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldman DE. Potential, impedance, and rectification in membranes. J Gen Physiol 27: 37–60, 1943. doi: 10.1085/jgp.27.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol 108: 37–77, 1949. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28: 264–278, 2008. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, Coppola G, Khakh BS. Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95: 531–549.e539, 2017. doi: 10.1016/j.neuron.2017.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gotz S, Bribian A, Lopez-Mascaraque L, Gotz M, Grothe B, Kunz L. Heterogeneity of astrocytes: electrophysiological properties of juxtavascular astrocytes before and after brain injury. Glia 69: 346–361, 2021. doi: 10.1002/glia.23900. [DOI] [PubMed] [Google Scholar]
- 47.Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem 279: 44065–44073, 2004. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- 48.Mulkey DK, Wenker IC. Astrocyte chemoreceptors: mechanisms of H+ sensing by astrocytes in the retrotrapezoid nucleus and their possible contribution to respiratory drive. Exp Physiol 96: 400–406, 2011. doi: 10.1113/expphysiol.2010.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olsen ML, Khakh BS, Skatchkov SN, Zhou M, Lee CJ, Rouach N. New insights on astrocyte ion channels: critical for homeostasis and neuron-glia signaling. J Neurosci 35: 13827–13835, 2015. doi: 10.1523/JNEUROSCI.2603-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skatchkov SN, Thomzig A, Eaton MJ, Biedermann B, Eulitz D, Bringmann A, Pannicke T, Veh RW, Reichenbach A. Kir subfamily in frog retina: specific spatial distribution of Kir 6.1 in glial (Muller) cells. Neuroreport 12: 1437–1441, 2001. doi: 10.1097/00001756-200105250-00028. [DOI] [PubMed] [Google Scholar]
- 51.Stephan J, Haack N, Kafitz KW, Durry S, Koch D, Hochstrate P, Seifert G, Steinhauser C, Rose CR. Kir4.1 channels mediate a depolarization of hippocampal astrocytes under hyperammonemic conditions in situ. Glia 60: 965–978, 2012. doi: 10.1002/glia.22328. [DOI] [PubMed] [Google Scholar]
- 52.Thomzig A, Wenzel M, Karschin C, Eaton MJ, Skatchkov SN, Karschin A, Veh RW. Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol Cell Neurosci 18: 671–690, 2001. doi: 10.1006/mcne.2001.1048. [DOI] [PubMed] [Google Scholar]
- 53.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 54.Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol 281: C922–C931, 2001. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- 55.Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26: 47–54, 1999. doi: 10.1002/(SICI)1098-1136(199903)26:1<47::AID-GLIA5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Banaclocha M. Astroglial isopotentiality and calcium-associated biomagnetic field effects on cortical neuronal coupling. Cells 9: 439, 2020. doi: 10.3390/cells9020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terman D, Zhou M. Modeling the role of the astrocyte syncytium and K+ buffering in maintaining neuronal firing patterns. Oper Med Physiol 5: 7–16, 2019. doi: 10.20388/omp2019.001.0063. [DOI] [Google Scholar]
- 58.Pal I, Kardos J, Dobolyi A, Heja L. Appearance of fast astrocytic component in voltage-sensitive dye imaging of neural activity. Mol Brain 8: 35, 2015. doi: 10.1186/s13041-015-0127-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bando Y, Grimm C, Cornejo VH, Yuste R. Genetic voltage indicators. BMC Biol 17: 71, 2019. doi: 10.1186/s12915-019-0682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ransom BR, Goldring S. Slow depolarization in cells presumed to be glia in cerebral cortex of cat. J Neurophysiol 36: 869–878, 1973. doi: 10.1152/jn.1973.36.5.869. [DOI] [PubMed] [Google Scholar]
- 61.Sontheimer H, Black JA, Waxman SG. Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci 19: 325–331, 1996. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 62.Steinhauser C, Kressin K, Kuprijanova E, Weber M, Seifert G. Properties of voltage-activated Na+ and K+ currents in mouse hippocampal glial cells in situ and after acute isolation from tissue slices. Pflugers Arch 428: 610–620, 1994. doi: 10.1007/BF00374585. [DOI] [PubMed] [Google Scholar]
- 63.Zhou M, Kimelberg HK. Freshly isolated astrocytes from rat hippocampus show two distinct current patterns and different [K+]o uptake capabilities. J Neurophysiol 84: 2746–2757, 2000. doi: 10.1152/jn.2000.84.6.2746. [DOI] [PubMed] [Google Scholar]
- 64.Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol 73: 333–346, 1995. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- 65.Seifert G, Huttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhauser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 29: 7474–7488, 2009. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohmori H. Inactivation kinetics and steady-state current noise in the anomalous rectifier of tunicate egg cell membranes. J Physiol 281: 77–99, 1978. doi: 10.1113/jphysiol.1978.sp012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reichelt W, Pannicke T. Voltage-dependent K+ currents in guinea pig Muller (glia) cells show different sensitivities to blockade by Ba2+. Neurosci Lett 155: 15–18, 1993. doi: 10.1016/0304-3940(93)90663-6. [DOI] [PubMed] [Google Scholar]
- 68.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354–11365, 2007. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du Y, Wang W, Lutton AD, Kiyoshi CM, Ma B, Taylor AT, Olesik JW, McTigue DM, Askwith CC, Zhou M. Dissipation of transmembrane potassium gradient is the main cause of cerebral ischemia-induced depolarization in astrocytes and neurons. Exp Neurol 303: 1–11, 2018. doi: 10.1016/j.expneurol.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henneberger C, Rusakov DA. Monitoring local synaptic activity with astrocytic patch pipettes. Nat Protoc 7: 2171–2179, 2012. doi: 10.1038/nprot.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schnell C, Fresemann J, Hulsmann S. Determinants of functional coupling between astrocytes and respiratory neurons in the pre-Botzinger complex. PLoS One 6: e26309, 2011. doi: 10.1371/journal.pone.0026309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Q, Wang W, Aten S, Kiyoshi CM, Du Y, Zhou M. Epileptiform neuronal discharges impair astrocyte syncytial isopotentiality in acute hippocampal slices. Brain Sci 10: 208, 2020. doi: 10.3390/brainsci10040208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma BF, Xie MJ, Zhou M. Bicarbonate efflux via GABA(A) receptors depolarizes membrane potential and inhibits two-pore domain potassium channels of astrocytes in rat hippocampal slices. Glia 60: 1761–1772, 2012. doi: 10.1002/glia.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xie M, Lynch DT, Schools GP, Feustel PJ, Kimelberg HK, Zhou M. Sodium channel currents in rat hippocampal NG2 glia: characterization and contribution to resting membrane potential. Neuroscience 150: 853–862, 2007. doi: 10.1016/j.neuroscience.2007.09.057. [DOI] [PubMed] [Google Scholar]
- 75.Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci 22: 1042–1053, 2002. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol 29: 788–806, 1966. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 77.Ransom BR, Goldring S. Ionic determinants of membrane potential of cells presumed to be glia in cerebral cortex of cat. J Neurophysiol 36: 855–868, 1973. doi: 10.1152/jn.1973.36.5.855. [DOI] [PubMed] [Google Scholar]
- 78.Amzica F, Massimini M. Glial and neuronal interactions during slow wave and paroxysmal activities in the neocortex. Cereb Cortex 12: 1101–1113, 2002. doi: 10.1093/cercor/12.10.1101. [DOI] [PubMed] [Google Scholar]
- 79.Benedikt J, Inyushin M, Kucheryavykh YV, Rivera Y, Kucheryavykh LY, Nichols CG, Eaton MJ, Skatchkov SN. Intracellular polyamines enhance astrocytic coupling. Neuroreport 23: 1021–1025, 2012. doi: 10.1097/WNR.0b013e32835aa04b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19: 1297–1308, 1997. doi: 10.1016/S0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 81.Henneberger C, Rusakov DA. Synaptic plasticity and Ca2+ signalling in astrocytes. Neuron Glia Biol 6: 141–146, 2010. doi: 10.1017/S1740925X10000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mishima T, Hirase H. In vivo intracellular recording suggests that gray matter astrocytes in mature cerebral cortex and hippocampus are electrophysiologically homogeneous. J Neurosci 30: 3093–3100, 2010. doi: 10.1523/JNEUROSCI.5065-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A 108: 8467–8472, 2011. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17: 694–703, 2014. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dallerac G, Chever O, Rouach N. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front Cell Neurosci 7: 159, 2013. doi: 10.3389/fncel.2013.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schools GP, Zhou M, Kimelberg HK. Development of gap junctions in hippocampal astrocytes: evidence that whole cell electrophysiological phenotype is an intrinsic property of the individual cell. J Neurophysiol 96: 1383–1392, 2006. doi: 10.1152/jn.00449.2006. [DOI] [PubMed] [Google Scholar]
- 87.Kimelberg HK, Cai Z, Schools G, Zhou M. Acutely isolated astrocytes as models to probe astrocyte functions. Neurochem Int 36: 359–367, 2000. doi: 10.1016/S0197-0186(99)00144-8. [DOI] [PubMed] [Google Scholar]
- 88.Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22: 183–192, 2002. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rela L, Bordey A, Greer CA. Olfactory ensheathing cell membrane properties are shaped by connectivity. Glia 58: 665–678, 2010. doi: 10.1002/glia.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu X, Bolteus AJ, Balkin DM, Henschel O, Bordey A. GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54: 394–410, 2006. doi: 10.1002/glia.20392. [DOI] [PubMed] [Google Scholar]
- 91.Van de Sande DV, Kopljar I, Maaike A, Teisman A, Gallacher DJ, Bart L, Snyders DJ, Leybaert L, Lu HR, Labro AJ. The resting membrane potential of hSC-CM in a syncytium is more hyperpolarised than that of isolated cells. Channels (Austin) 15: 239–252, 2021. doi: 10.1080/19336950.2021.1871815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia 53: 516–528, 2006. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55: 274–281, 2007. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- 94.Olsen ML, Campbell SL, Sontheimer H. Differential distribution of Kir4.1 in spinal cord astrocytes suggests regional differences in K+ homeostasis. J Neurophysiol 98: 786–793, 2007. doi: 10.1152/jn.00340.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tang X, Taniguchi K, Kofuji P. Heterogeneity of Kir4.1 channel expression in glia revealed by mouse transgenesis. Glia 57: 1706–1715, 2009. doi: 10.1002/glia.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nwaobi SE, Lin E, Peramsetty SR, Olsen ML. DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia 62: 411–427, 2014. doi: 10.1002/glia.22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang W, Putra A, Schools GP, Ma B, Chen H, Kaczmarek LK, Barhanin J, Lesage F, Zhou M. The contribution of TWIK-1 channels to astrocyte K(+) current is limited by retention in intracellular compartments. Front Cell Neurosci 7: 246, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferroni S, Valente P, Caprini M, Nobile M, Schubert P, Rapisarda C. Arachidonic acid activates an open rectifier potassium channel in cultured rat cortical astrocytes. J Neurosci Res 72: 363–372, 2003. doi: 10.1002/jnr.10580. [DOI] [PubMed] [Google Scholar]
- 99.Gnatenco C, Han J, Snyder AK, Kim D. Functional expression of TREK-2 K+ channel in cultured rat brain astrocytes. Brain Res 931: 56–67, 2002. doi: 10.1016/S0006-8993(02)02261-8. [DOI] [PubMed] [Google Scholar]
- 100.Kindler CH, Pietruck C, Yost CS, Sampson ER, Gray AT. Localization of the tandem pore domain K+ channel TASK-1 in the rat central nervous system. Brain Res Mol Brain Res 80: 99–108, 2000. doi: 10.1016/s0169-328x(00)00136-4. [DOI] [PubMed] [Google Scholar]
- 101.Kucheryavykh LY, Kucheryavykh YV, Inyushin M, Shuba YM, Sanabria P, Cubano LA, Skatchkov SN, Eaton MJ. Ischemia increases TREK-2 channel expression in astrocytes: relevance to glutamate clearance. Open Neurosci J 3: 40–47, 2009. doi: 10.2174/1874082000903010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pruss H, Dewes M, Derst C, Fernandez-Klett F, Veh RW, Priller J. Potassium channel expression in adult murine neural progenitor cells. Neuroscience 180: 19–29, 2011. doi: 10.1016/j.neuroscience.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 103.Hwang EM, Kim E, Yarishkin O, Woo DH, Han KS, Park N, Bae Y, Woo J, Kim D, Park M, Lee CJ, Park JY. A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat Commun 5: 3227, 2014. doi: 10.1038/ncomms4227. [DOI] [PubMed] [Google Scholar]
- 104.Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15: 1004–1011, 1996. [PMC free article] [PubMed] [Google Scholar]
- 105.Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121: 37–47, 2005. [Erratum in Cell 141: 368, 2010]. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 106.Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854–6862, 1996. doi: 10.1002/j.1460-2075.1996.tb01077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang W, Kiyoshi CM, Du Y, Taylor AT, Sheehan ER, Wu X, Zhou M. TREK-1 null impairs neuronal excitability, synaptic plasticity, and cognitive function. Mol Neurobiol 57: 1332–1346, 2020. doi: 10.1007/s12035-019-01828-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Toro Cabrera G, Mueller C. Design of shRNA and miRNA for delivery to the CNS. Methods Mol Biol 1382: 67–80, 2016. doi: 10.1007/978-1-4939-3271-9_5. [DOI] [PubMed] [Google Scholar]
- 109.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321–329, 1998. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 110.Price DL, Ludwig JW, Mi H, Schwarz TL, Ellisman MH. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res 956: 183–193, 2002. doi: 10.1016/s0006-8993(02)03266-3. [DOI] [PubMed] [Google Scholar]
- 111.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9: 1397–1403, 2006. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 112.Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A 107: 3811–3816, 2010. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol Cell Physiol 299: C1068–C1078, 2010. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Longden TA, Dunn KM, Draheim HJ, Nelson MT, Weston AH, Edwards G. Intermediate-conductance calcium-activated potassium channels participate in neurovascular coupling. Br J Pharmacol 164: 922–933, 2011. doi: 10.1111/j.1476-5381.2011.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Augustin V, Bold C, Wadle SL, Langer J, Jabs R, Philippot C, Weingarten DJ, Rose CR, Steinhauser C, Stephan J. Functional anisotropic panglial networks in the lateral superior olive. Glia 64: 1892–1911, 2016. doi: 10.1002/glia.23031. [DOI] [PubMed] [Google Scholar]
- 116.Du Y, Kiyoshi CM, Terman D, Zhou M. Analysis of the functional states of an astrocyte syncytium. In: Basic Neurobiology Techniques Neuromethods, edited by Wright N. New York: Springer, 2020, vol 2, p. 285–313. [Google Scholar]
- 117.Eitelmann S, Hirtz JJ, Stephan J. A Vector-based method to analyze the topography of glial networks. Int J Mol Sci 20: 2821, 2019. doi: 10.3390/ijms20112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Felix L, Delekate A, Petzold GC, Rose CR. Sodium fluctuations in astroglia and their potential impact on astrocyte function. Front Physiol 11: 871, 2020. doi: 10.3389/fphys.2020.00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rose CR, Verkhratsky A. Principles of sodium homeostasis and sodium signalling in astroglia. Glia 64: 1611–1627, 2016. doi: 10.1002/glia.22964. [DOI] [PubMed] [Google Scholar]
- 120.Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322: 1551–1555, 2008. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- 121.Skatchkov SN, Eaton MJ, Shuba YM, Kucheryavykh YV, Derst C, Veh RW, Wurm A, Iandiev I, Pannicke T, Bringmann A, Reichenbach A. Tandem-pore domain potassium channels are functionally expressed in retinal (Muller) glial cells. Glia 53: 266–276, 2006. doi: 10.1002/glia.20280. [DOI] [PubMed] [Google Scholar]
- 122.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89: 37–53, 2016. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seifert G, Zhou M, Steinhauser C. Analysis of AMPA receptor properties during postnatal development of mouse hippocampal astrocytes. J Neurophysiol 78: 2916–2923, 1997. doi: 10.1152/jn.1997.78.6.2916. [DOI] [PubMed] [Google Scholar]
- 124.Tse FW, Fraser DD, Duffy S, MacVicar BA. Voltage-activated K+ currents in acutely isolated hippocampal astrocytes. J Neurosci 12: 1781–1788, 1992. doi: 10.1523/JNEUROSCI.12-05-01781.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bushong EA, Martone ME, Ellisman MH. Examination of the relationship between astrocyte morphology and laminar boundaries in the molecular layer of adult dentate gyrus. J Comp Neurol 462: 241–251, 2003. doi: 10.1002/cne.10728. [DOI] [PubMed] [Google Scholar]
- 126.Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27: 6473–6477, 2007. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kiyoshi CM, Aten S, Arzola EP, Patterson JA, Taylor AT, Du Y, Guiher AM, Philip M, Gerviacio Camacho E, Mediratta D, Collins K, Benson E, Kidd G, Terman D, Zhou M. Ultrastructural view of astrocyte-astrocyte and astrocyte-synapse contacts within the hippocampus (Preprint). BioRxiv 10.28.358200, 2020. doi: 10.1101/2020.10.28.358200. [DOI]
- 128.Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113: 221–233, 2002. doi: 10.1016/s0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 129.Gutnick MJ, Connors BW, Ransom BR. Dye-coupling between glial cells in the guinea pig neocortical slice. Brain Res 213: 486–492, 1981. doi: 10.1016/0006-8993(81)90259-6. [DOI] [PubMed] [Google Scholar]
- 130.Kiyoshi CM, Zhou M. Astrocyte syncytium: a functional reticular system in the brain. Neural Regen Res 14: 595–596, 2019. doi: 10.4103/1673-5374.247462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev 32: 29–44, 2000. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- 132.Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys 34: 325–472, 2001. [Erratum in Q Rev Biophys 35: 109, 2002]. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- 133.Moreno AP, Rook MB, Fishman GI, Spray DC. Gap junction channels: distinct voltage-sensitive and -insensitive conductance states. Biophysical J 67: 113–119, 1994. doi: 10.1016/S0006-3495(94)80460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Spray DC. Physiological properties of gap junction channels in the nervous system. In: Gap Junctions in the nervous system, edited by Spary DC, Dermietzel R.. Austin, TX: RG Landes Company, 1996, p. 35–59. [Google Scholar]
- 135.Valiunas V, Manthey D, Vogel R, Willecke K, Weingart R. Biophysical properties of mouse connexin30 gap junction channels studied in transfected human HeLa cells. J Physiol 519: 631–644, 1999. doi: 10.1111/j.1469-7793.1999.0631n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science 244: 578–580, 1989. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]