

Abstract
Pulmonary hypertension is characterized by sustained vasoconstriction and remodelling of the small pulmonary arteries, which is associated with persistent depolarization of the resting membrane potential (Em) of pulmonary arterial smooth muscle cells (PASMCs). It is well-known that the underlying mechanism of this depolarization includes inhibition of K+ channels; however, whether other ion channels contribute to this depolarization is unknown. We previously reported that acid-sensing ion channel 1 (ASIC1), a non-selective cation channel that conducts both Na+ and Ca2+, is present in PASMCs and contributes to the development of chronic hypoxia (CH)-induced pulmonary hypertension. Therefore, we tested the hypothesis that ASIC1-mediated Na+ influx contributes to PASMC Em regulation following CH-induced pulmonary hypertension. Using sharp electrode intracellular recordings in isolated, pressurized small pulmonary arteries from rats and mice, we show exposure to CH leads to PASMC membrane depolarization compared to control animals, and this is independent of intraluminal pressure-induced depolarization. In addition to a decrease in PASMC whole-cell K+ currents following CH, we demonstrate whole-cell non-selective cation channel (NSCC) currents are increased and essential to the persistent CH-induced Em depolarization in PASMCs. Both the specific inhibitor of ASIC1, psalmotoxin 1, and global knockout of ASIC1 (Asic1−/−) prevents CH-induced Em depolarization and largely inhibits whole-cell NSCC currents, without affecting whole-cell K+ currents. Our results show a combination of factors, including inhibition of K+ efflux and augmented Na+ influx, mediate CH-induced PASMC depolarization. Furthermore, this study demonstrates a novel role for ASIC1 in the regulation of Em in PASMCs during CH-induced pulmonary hypertension.
Keywords: membrane potential, intraluminal pressure, whole-cell patch-clamp, sharp electrode, psalmotoxin 1, amiloride
Graphical Abstract
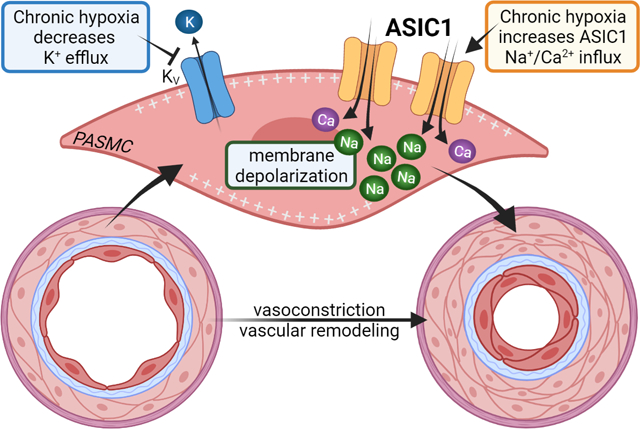
INTRODUCTION
Pulmonary hypertension is a multifactorial and progressive pulmonary vascular disease leading to increased pulmonary vascular resistance, elevated pulmonary artery pressure, and right ventricular dysfunction/failure. New guidelines define pulmonary hypertension as a mean pulmonary arterial pressure of >20 mmHg, pulmonary arterial wedge pressure of ≤15 mmHg, and pulmonary vascular resistance of ≥3 Wood Units (Simonneau et al., 2019). It is well characterized that the pulmonary vascular disease associated with pulmonary hypertension results from sustained pulmonary vasoconstriction and vascular remodelling primarily due to smooth muscle hyperreactivity, endothelial dysfunction, excessive cell proliferation, impaired apoptosis, and cellular trans-differentiation (i.e. endothelial-mesenchymal and smooth muscle transformations) (Voelkel & Tuder, 2000; Shimoda & Laurie, 2013; Gao et al., 2016). Although the pathogenesis of pulmonary hypertension is complex, clear correlations have been identified between pulmonary vascular disease and abnormalities in membrane potential, transmembrane flux of ions, and homeostasis of intracellular Ca2+ levels ([Ca2+]i).
Resting membrane potential (Em) in PASMCs of isolated pulmonary arteries is −50 to −65 mV when measured by intracellular sharp electrode (Suzuki & Twarog, 1982a; Naik et al., 2005) and −40 to −50 mV in cultured and freshly dissociated PASMCs when using the current-clamp technique (Yuan et al., 1993; Archer et al., 1998). Resting Em is primarily reliant on K+ permeability, but PASMC Em is more positive than the calculated equilibrium potential for K+ (−85 mV based on the Nernst equation), indicating other cations (e.g., Na+, Ca2+, H+, Mg2+) and anions (e.g., Cl−, HCO3−) contribute to the regulation of Em in PASMCs.
In pulmonary hypertensive patients and animal models of pulmonary hypertension, the resting Em in PASMCs is persistently depolarized and this correlates with reduced voltage-gated K+ (KV) channel expression and activity (Suzuki & Twarog, 1982b; Smirnov et al., 1994; Wang et al., 1997; Priest et al., 1998; Yuan et al., 1998; Platoshyn et al., 2000; Hong et al., 2004; Naik et al., 2005; Fike et al., 2006). The subsequent activation of voltage-gated calcium channels (VGCCs) is thought to be the major cause of the elevated [Ca2+]i and resultant sustained pulmonary vasoconstriction and PASMC proliferation (Muramatsu et al., 1997; Luke et al., 2012; Wan et al., 2013). However, in distal pulmonary arteries, increases in [Ca2+]i and vasoconstriction were, at most, partially inhibited by VGCC blockers (Shimoda et al., 2000; Lin et al., 2004; Jernigan et al., 2012). Instead, several studies demonstrate an essential role for non-selective cation channels (NSCCs) in mediating elevated [Ca2+]i, augmented vasoconstriction, and pulmonary vascular remodelling (Lin et al., 2004; Wang et al., 2006; Liu et al., 2011; Jernigan et al., 2012). It is possible that the enhanced activation of NSCCs also contributes to chronic Em depolarization of PASMCs in pulmonary hypertension.
Our laboratory has previously demonstrated that the NSCC, acid-sensing ion channel 1 (ASIC1), plays an important role in the development of chronic hypoxia (CH)-induced pulmonary hypertension by contributing to the enhanced PASMC Ca2+ influx, agonist-induced vasoconstriction, and vascular remodelling (Jernigan et al., 2012; Nitta et al., 2014). ASICs are voltage-insensitive and highly selective for Na+ over other ions with a permeability ratio PNa/PK of 13 (Waldmann et al., 1997; Yang & Palmer, 2014). Unlike other ASIC subtypes, ASIC1 is unique in that it is also permeable to Ca2+ (Xiong et al., 2004; Yermolaieva et al., 2004). In neurons, the influx of Na+ and Ca2+ following activation of ASIC1 contributes to membrane depolarization and generation of action potentials, activation of Ca2+/calmodulin-dependent mechanisms, and other second-messenger pathways signifying the diverse role these channels play in intracellular signalling and excitability (Chu & Xiong, 2013; Wemmie et al., 2013). Based on the known roles of ASIC1 to regulate Em, we tested the hypothesis that ASIC1 contributes to PASMC Em regulation following chronic hypoxia (CH)-induced pulmonary hypertension.
METHODS
Ethical Approval
All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine (Protocol #19-200899-HSC) and abide by the National Institutes of Health guidelines for animal use. All animals were anesthetized with an overdose of pentobarbital sodium (200 mg/kg, i.p.) and immediately euthanized by exsanguination after the loss of consciousness. The study complies with the ethical principles under which The Journal of Physiology operates.
Animals
Studies were completed in adult male Wistar rats (200–250 g body wt, Envigo) or ASIC1 [B6.129-Asic1tm1Wsh/J; Jackson Laboratory Stock #013733; (Wemmie et al., 2002)] knockout (Asic1−/−) mice and age-matched C57BL/6J (Jackson Laboratory Stock # 000664) wildtype controls (Asic1+/+). Homozygotes and/or heterozygotes were bred and disruption of ASIC1 was confirmed by PCR and agarose gel electrophoresis using a three-primer system to detect both wildtype and disrupted alleles: 5’-CAT GTC ACC AAG CTC GAC GAG GTG-3’ (Asic1+/+ forward primer), 5’-TGG ATG TGG AAT GTG TGC GA-3’ (Asic1−/− forward primer), 5’-CCG CCT TGA GCGGCA GGT TTA AAG G-3’ (Asic1+/+and Asic1−/− reverse primer). Animals were housed 1–5 per cage in a specific pathogen-free (SPF) animal care facility and maintained on a 12:12-h light-dark cycle. Standard chow (Teklad soy protein-free diet #2920, Envigo) and water were provided ad libitum. Animals were randomly allocated to experimental groups and when possible, the treatment assignments were blinded to investigators. To obtain sufficient numbers of knockout mice and reduce the number of animals being bred, this study equally used both male and female mice (14–20 weeks old). Our previous experiments showed no significant interaction between sex and the development of pulmonary hypertension (Nitta et al., 2014).
Exposure to Chronic Hypoxia.
Chronic hypoxia (CH) is a common complication of chronic lung diseases and a key stimulus in the development of pulmonary hypertension. Animals designated for exposure to CH were housed in a clear-plexiglass hypobaric chamber (~0.5 m3) with barometric pressure maintained at ~380 mmHg for 4 wk. The hypobaric chamber was partially evacuated with a vacuum pump allowing for continuous airflow of 30 L/min through the chamber. The chamber was opened 3 times/week to change bedding and provide fresh water and food. Age-matched control animals were housed at ambient barometric pressure (~630 mmHg in Albuquerque, NM). We have previously demonstrated that this rat and mouse model mimics many of the cardiopulmonary changes observed in human pulmonary hypertension including increased right ventricular systolic pressure, right ventricular hypertrophy, enhanced vasoconstriction, and arterial remodelling (Nitta et al., 2014; Jernigan et al., 2017).
Pressure Myography in Small Pulmonary Arteries
Small intrapulmonary arteries were cannulated and pressurized for sharp electrode membrane potential recordings as previously described (Naik et al., 2005; Broughton et al., 2010). Briefly, animals were anesthetized with pentobarbital sodium (200 mg/kg, i.p.). The left lung was removed, and small intrapulmonary arteries (4th – 5th order, ~100–150 μm passive inner diameter) were dissected free of bronchioles and lung parenchyma, transferred to a vessel chamber (CH-1, Living Systems Instrumentation), and secured to tapered glass pipettes with a single strand of silk ligature. After cannulation, the artery was pressurized with a servo-controlled peristaltic pump (PS-200-P/S, Living Systems Instrumentation) to 12 or 35 mmHg. These pressures are estimates of in vivo pressures in conscious rats from each group, as determined previously (Resta et al., 1999). Any artery that failed to maintain pressure was discarded. The vessel chamber was superfused (5 ml/min at 37°C) with HEPES-based saline buffer containing in mM: 130 NaCl, 4 KCl, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 1.8 CaCl2, and 6 glucose; pH adjusted to 7.4 with NaOH (all reagents from Sigma-Aldrich).
Measurement of PASMC Em.
PASMC Em values were recorded using intracellular sharp electrodes in isolated pressurized, small pulmonary arteries from control and CH rats and Asic1+/+ and Asic1−/− mice as previously described (Naik et al., 2005). PASMCs were impaled with sharp electrodes (60–80 MΩ) filled with 3 M KCl. A Neuroprobe amplifier (model 1600, A-M Systems) was used to record Em. Analog output from the amplifier was low-pass filtered at 1 kHz and routed to an oscilloscope (Gould, 420) and a digitizer acquisition system (MiniDigi 1A, Axon Instruments) and recorded using WinDaq software (Dataq Instruments). Criteria for acceptance of Em recordings were as follows: 1) an abrupt negative deflection in potential as the microelectrode is advanced into the cell, 2) stable Em for ≥1 min, and 3) an abrupt change in potential to ~0 mV after the electrode is retracted from the cell. When possible, recordings at 12 and 35 mmHg were made in the same pulmonary artery. Em was recorded in separate arteries for the following conditions: vehicle, replace extracellular NaCl with N-methyl-D-glucamine (NMDG, 140 mM, Sigma-Aldrich M2004), LaCl3 (30 μM; Sigma-Aldrich L4131), amiloride (30 μM, Enzo ALX-550–212-G001), or psalmotoxin 1 (PcTX1; 20 nM, Phoenix Peptides 063–22).
Isolation of Pulmonary Arterial Smooth Muscle Cells
Freshly dispersed PASMCs were used for patch-clamp electrophysiology. Briefly, animals were anesthetized with pentobarbital sodium (200 mg/kg body weight, i.p.), and the heart and lungs were removed by midline thoracotomy. Intrapulmonary arteries (~2nd–5th order) were dissected from surrounding lung parenchyma and enzymatically digested by incubating in 100 nM Ca2+-containing Hank’s Balanced Salt Solution (HBSS; Lonza 10-547F) containing papain (9.5 U/ml, Sigma-Aldrich P4762), type-I collagenase (2 mg/ml, Sigma-Aldrich C-0130), dithiothreitol (1 mg/ml, Sigma-Aldrich D0632), and BSA (2 mg/ml, Sigma-Aldrich A9647) at 37°C for 20 min. Single smooth muscle cells were dispersed by gentle trituration with a fire-polished pipette in Ca2+-free HBSS.
Patch Clamp Electrophysiology.
K+ and NSCC current recordings were performed in freshly dispersed PASMCs by using conventional whole-cell patch-clamp configuration in voltage-clamp mode with the following biophysical criteria: seal resistance > 1 GΩ and series resistance < 25 MΩ. These criteria were confirmed after membrane rupture and monitored through the course of the experiment. Freshly isolated PASMCs were superfused in extracellular solution under constant flow (2 ml/min) at room temperature (~23°C). Whole-cell current data were generated with an Axopatch 200B amplifier (Axon Instruments) using 4- to 6-MΩ patch electrodes controlled by a motorized micromanipulator (MP-225, Sutter Instruments). Electrodes were pulled from 1.5-mm borosilicate filamented-glass (Sutter Instruments) with a micropipette puller (P-87, Sutter Instruments) and fire-polished with a microforge (MF-830, Narishige). Currents were filtered with a lowpass Bessel filter at 1 kHz and digitized (1440A Digitizer, Molecular Devices) and recorded using pClamp software (Molecular Devices). Currents were normalized to cell capacitance to obtain current density (pA/pF), and current-voltage (I–V) relationships were generated from the last 50 ms of each stimulus.
K+ current recordings.
The extracellular solution contained (in mM) 140 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 10 glucose and buffered to pH 7.4 with NaOH. The pipette was filled with an intracellular solution containing (in mM) 140 KCl, 1 MgCl2, 5 MgATP, 10 HEPES, and 1 EGTA and adjusted to pH 7.2 with KOH. The liquid junction potential is 4.943 mV and calculated according to the stationary Nernst-Planck equation using LCPcalc (Harden, 2020). The osmolarity of all solutions was adjusted to 290–300 mOsm/L with sucrose. After a 5-min dialysis period, whole-cell K+ currents were measured in response to voltage steps applied from −60 to +150 mV (rats) or −80 to +120 mV (mice) in 10-mV increments from a holding potential of −50 mV.
NSCC current recordings.
The extracellular solution contained (in mM): 135 NaCl, 1 MgSO4, 4 NaHCO3, 1 NaH2PO4, 3 EGTA, 10 HEPES, and 6 Glucose and buffered to pH 7.4 with NaOH. To determine the contribution of Na+ to NSCC currents, NaCl was replaced with 140 mM NMDG to remove the chemical driving force of Na+ across the membrane. The extracellular Na+ concentration was approximately 5 mM and buffered to pH 7.4 with HCl. The pipette was filled with intracellular solution containing (in mM) 138 CsOH, 138 L-aspartic acid, 1.15 EGTA, 1 CaCl2, 2 MgATP, 10 HEPES and adjusted to pH 7.2 with CsOH. CaCl2 was added to yield a free Ca2+ concentration of 150 nM, as calculated using Maxchelator software (Bers et al., 1994). The osmolarity of all solutions was adjusted to 290–300 mOsm/L with sucrose. After a 5-min dialysis period, whole-cell Na+ currents were measured in response to voltage steps applied from −80 to +80 mV in 10-mV increments from a holding potential of 0 mV immediately before and between each voltage step to inactivate voltage-gated channels (otherwise, the holding potential was −50 mV). The liquid junction potential is 17.661 mV and calculated according to the stationary Nernst-Planck equation using LCPcalc (Harden, 2020). NSCC currents were recorded under the following conditions: vehicle, 5-nitro-2-(3-phenylpropylamino) benzoic acid (5-NPPB, 100 μM; Sigma-Aldrich N4779) and N-phenylanthranilic acid (NPA, 1 mM; Sigma-Aldrich 78150), amiloride (30 μM), or psalmotoxin 1 (PcTX1; 20 nM).
Statistics
All data are expressed as means (SD). For experiments involving measurements from multiple cells from an individual animal, data were averaged for each animal and considered as a single observation. Statistical comparisons were made using Prism 8 (GraphPad Software). The statistical tests used and exact p values are reported either in the figure/legend or the supplemental Statistical Summary Document. A probability of <0.05 with a power level of 0.80 was accepted as statistically significant for all comparisons.
RESULTS
CH-induced PASMC Em depolarization is independent of intraluminal pressure.
Isolated small pulmonary arteries from control and CH rats were pressurized to 12 mmHg or 35 mmHg to approximate in vivo pressures as previously determined in conscious control and pulmonary hypertensive rats (Resta et al., 1999). The Em of PASMCs from CH rats was depolarized ~16–18 mV relative to Em from control rats at each pressure, respectively (Figure 1A–B). Consistent with our previous reports (Naik et al., 2005), we found that increasing pressure from 12 to 35 mmHg resulted in a similar depolarization (p = 0.386) of Em in both control [Δ of 11.5 (2.5) mV] and CH [Δ of 13.6 (5.1) mV] PASMCs. These data demonstrate the persistent PASMC depolarization following CH is independent of pressure/stretch-induced Em depolarization mediated by higher intraluminal pressures following CH-induced pulmonary hypertension.
Figure 1. CH-induced PASMC Em depolarization is independent of intraluminal pressure.
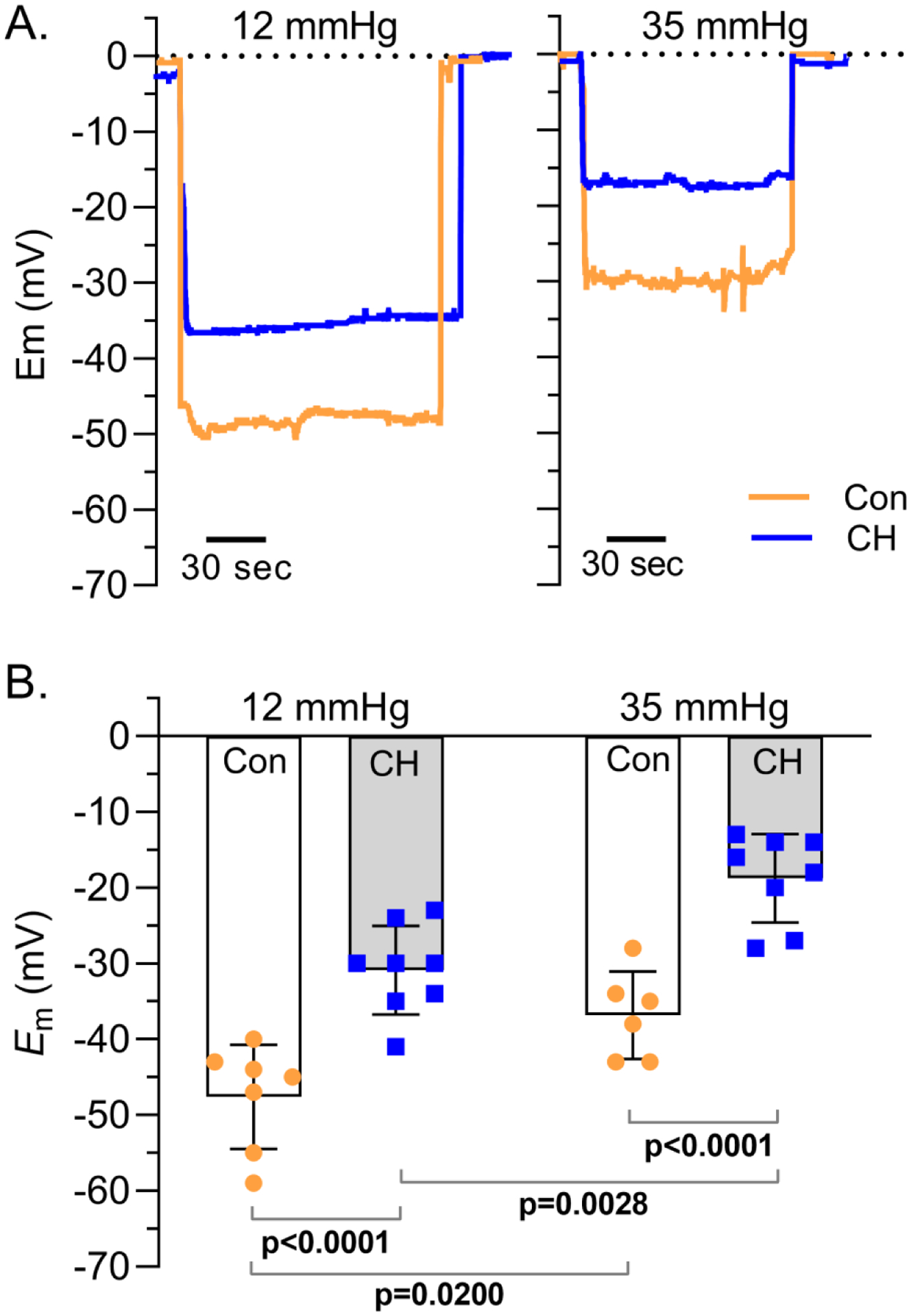
A) Representative traces and B) summary data showing PASMC membrane potential (Em; mV) in isolated small pulmonary arteries from control (circles) and CH (squares) rats pressurized to 12 (left) or 35 (right) mmHg. Values are means (SD); dots indicate n as # of animals/group; analyzed by two-way ANOVA, individual groups compared using Tukey’s multiple comparisons test.
PASMCs from CH rats have decreased K+ and increased NSCC conductance.
The recorded outward K+ currents (IK) in PASMCs were mostly slowly-activating and non-inactivating as shown in Figure 2A. Consistent with previous reports (Suzuki & Twarog, 1982b; Smirnov et al., 1994; Platoshyn et al., 2001), IK was significantly less in PASMCs from CH rats at all voltages from −50 to 150 mV. On average, the cell membrane capacitance (Cm) was greater (p = 0.0001) in PASMCs from CH [9.3 (4.1) pF, n = 43] compared to control rats [6.5 (2.4) pF, n = 46]. Because Cm is proportional to cell surface area, these data are indicative of hypertrophy of PASMCs from CH-induced pulmonary hypertensive rats. Since Cm was greater in PASMCs from CH rats, currents were normalized to Cm to obtain current density (pA/pF), which was also significantly reduced in PASMCs following CH (Figure 2C). NSCC currents (INSCC) (Figure 2D and 2E) and current densities (Figure 2F) were linear and significantly greater in PASMCs from CH rats at all voltages from −80 to 80 mV (except 0 mV). The inward component of INSCC was nearly abolished by the replacement of extracellular Na+ with NMDG in PASMCs from both control and CH rats (Figure 3; p < 0.05 from −80 to −10 mV in controls and −80 to 80 mV, except 0 mV in CH). The outward current was not affected by NMDG in PASMC from control animals, but NMDG blunted outward current in PASMC from CH rats (Figure 3B). Although the effect of NMDG to blunt outward currents in PASMC from CH rats is unclear, it may be due to a direct effect of NMDG to inhibit outward currents, as demonstrated by others (Thuma & Hooper, 2018). On the other hand, INSCC was minimally affected by the Cl− channel blockers, 5-nitro-2-(3-phenylpropylamino) benzoic acid (5-NPPB, 100 μM) and N-phenylanthranilic acid (NPA, 1 mM) added to bath and pipette (Figure 3; p < 0.05 from 30 to 80 mV in PASMCs from CH rats). Since K+ and Ca2+ were omitted from the extracellular solution, these data indicate Na+ influx through NSCCs is increased following CH.
Figure 2. PASMC from CH rats have decreased K+ and increased NSCC conductance.
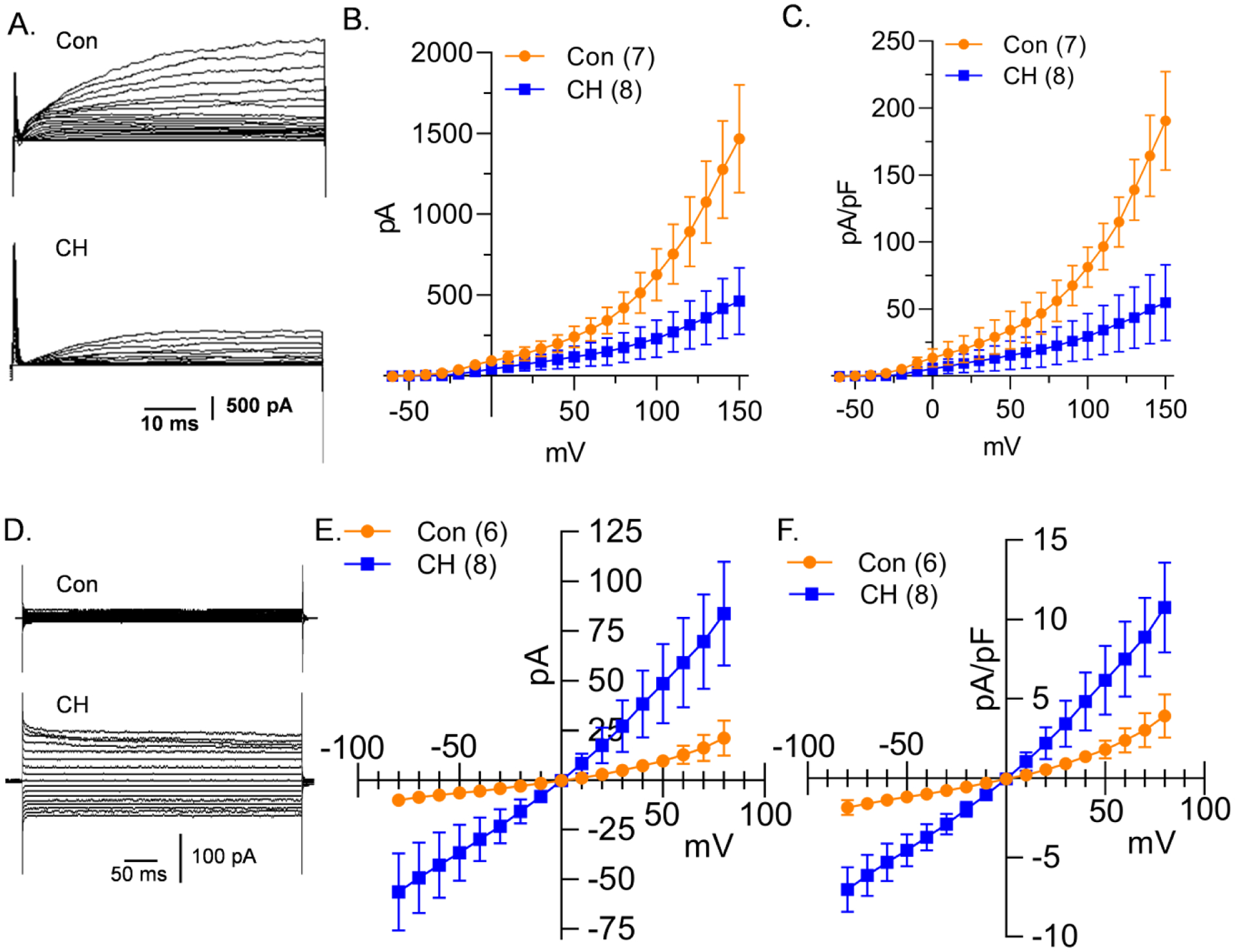
A) Representative K+ current traces and average B) current (pA) and C) current density (pA/pF) plotted as a function of voltage (mV) recorded in PASMC from control (circles) and CH (squares) rats. D) Representative NSCC current traces and average E) current (pA) and F) current density (pA/pF) plotted as a function of voltage (mV) recorded in PASMC from control (circles) and CH (squares) rats. Values are means (SD); n indicated in parentheses as the number of rats/group. Each voltage increment was analyzed individually using multiple unpaired t-tests, without assuming a consistent standard deviation using the Holm-Šidák method (see Statistical Summary Document for P-values at each voltage).
Figure 3. Na+ is the major influx ion of the NSCC current.
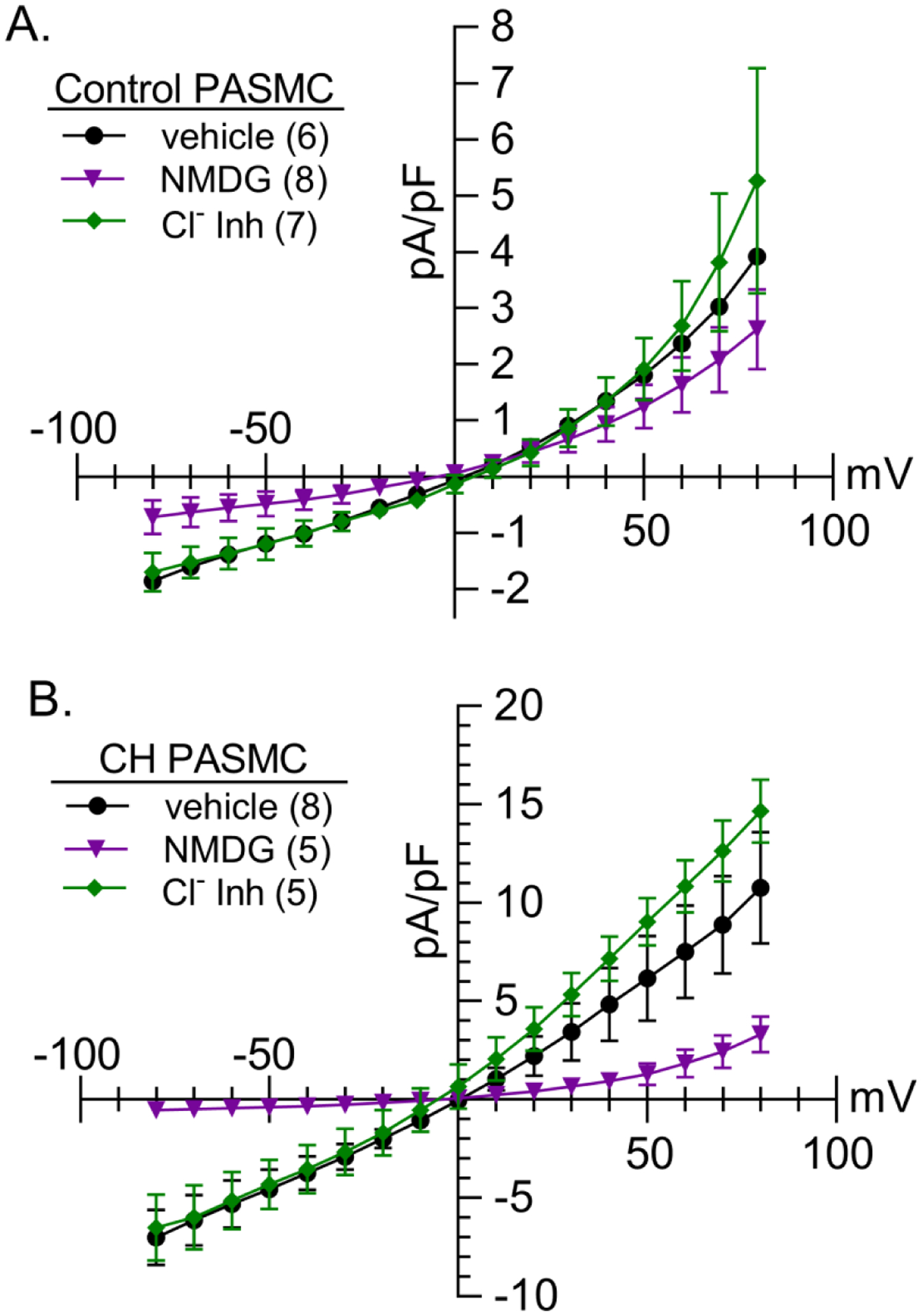
NSCC current density (pA/pF)-voltage (mV) relationship in PASMCs from A) control and B) CH rats under vehicle conditions (circles), following replacement of extracellular NaCl with N-methyl-D-glucamine (NMDG, 140 mM; triangles), or in the presence of the Cl− channel inhibitors (Cl− Inh, diamonds), 5-nitro-2-(3-phenylpropylamino)benzoic acid (5-NPPB, 100 μM) and N-phenylanthranilic acid (NPA, 1 mM). Con and CH vehicles are the same as Fig 2F. Values are means (SD); n indicated in parentheses as the number of rats/group; analyzed by two-way ANOVA, individual groups compared using Šidák multiple comparisons test (see Statistical Summary Document for P-values at each voltage).
Na+ influx contributes to CH-induced Em depolarization.
To determine the importance of Na+ influx to Em regulation we replaced extracellular Na+ with NMDG. The CH-induced Em depolarization in pulmonary arteries pressurized to 12 and 35 mmHg was absent, however, the pressure-induced depolarization remained in both groups (Figure 4A). These data establish the importance of Na+ influx to CH-induced PASMC depolarization but suggest other ions contribute to the pressure-induced depolarization.
Figure 4. NSCC influx contributes to CH-induced Em depolarization.
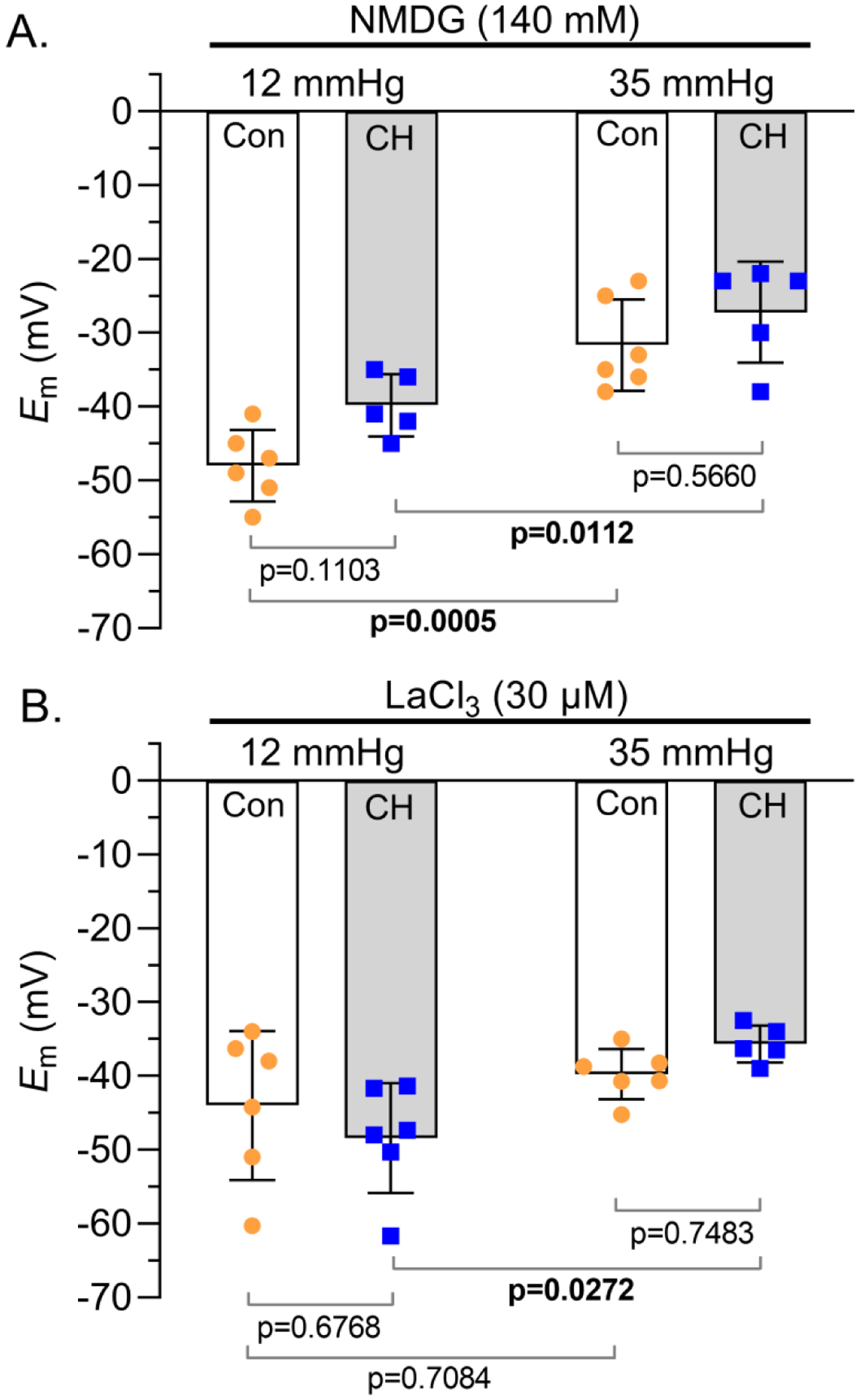
Summary membrane potential (Em; mV) recordings at intraluminal pressures of 12 and 35 mmHg in isolated, pressurized small pulmonary arteries from control (circles) and CH (squares) rats B) following replacement of extracellular NaCl with NMDG (140 mM) or C) in the presence of LaCl3 (30 μM). Values are means (SD); dots indicate n as # of rats/group; analyzed by two-way ANOVA and individual groups compared using Tukey’s multiple comparisons test.
Na+ is mainly transported across PASMC membranes by NSCCs, as well as various transporters and exchangers. To determine the involvement of non-selective cation channels, we conducted Em recordings in the presence of LaCl3. LaCl3 prevented the CH-induced depolarization at both 12 and 35 mmHg (Figure 4B), implicating an important role for NSCCs to mediate CH-induced depolarization. Increasing pressure from 12 to 35 mmHg in the presence of LaCl3 did not induce a depolarization in PASMCs from control arteries but did cause depolarization in PASMCs from CH animals, suggesting different mechanisms contribute to pressure-dependent depolarization in PASMCs from control and CH animals.
ASIC1 contributes to INSCC and CH-induced Em depolarization
The pan-ASIC blocker, amiloride, had no effect on NSCC current density in PASMCs from control rats, but significantly inhibited NSCC current density in PASMCs from CH rats (Figure 5A), without affecting K+ conductance (Figure 5B) in PASMCs from either group. In the presence of amiloride, neither the CH-induced depolarization nor pressure-induced depolarization was present (Figure 5C). These data demonstrate that even with reduced K+ conductance, inhibition of NSCC flux normalizes Em in PASMCs from CH rats to that of controls.
Figure 5. Amiloride prevents NSCC influx and CH- and pressure-induced Em depolarization.
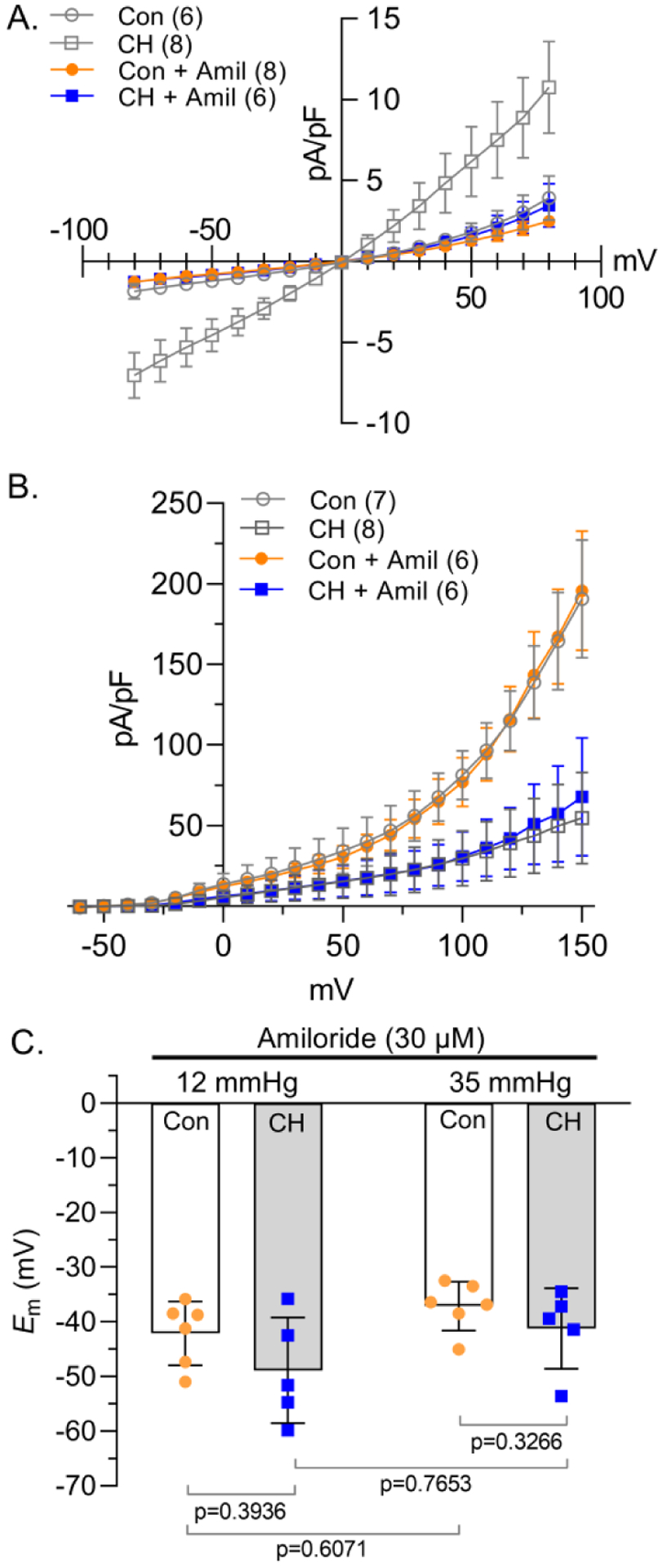
A) NSCC and B) K+ current density (pA/pF)-voltage (mV) relationship in PASMCs from control (circles) and CH (squares) rats in the presence of amiloride (Amil; 30 μM). Con and CH vehicles (open grey symbols) are the same as Fig 2C and 2F. C) Summary membrane potential (Em; mV) recordings at intraluminal pressures of 12 and 35 mmHg in isolated, pressurized small pulmonary arteries from control (circles) and CH (squares) rats in the presence of amiloride (30 μM). Values are means (SD); n indicated in parentheses or dots and represents # of animals/group; each voltage was analyzed by two-way ANOVA and individual groups compared using Tukey’s multiple comparisons test (see Statistical Summary Document for P-values).
The specific ASIC1 inhibitor, psalmotoxin 1 (PcTX1), had similar effects as amiloride to reduce NSCC current density in PASMCs from CH, but not control rats (Figure 6A). In addition, PcTX1 eliminated the CH-induced depolarization (Figure 6B) and similar to LaCl3, prevented the pressure-induced depolarization in arteries from control, but not CH animals.
Figure 6. ASIC1 contributes to enhanced NSCC influx and CH-induced Em depolarization.
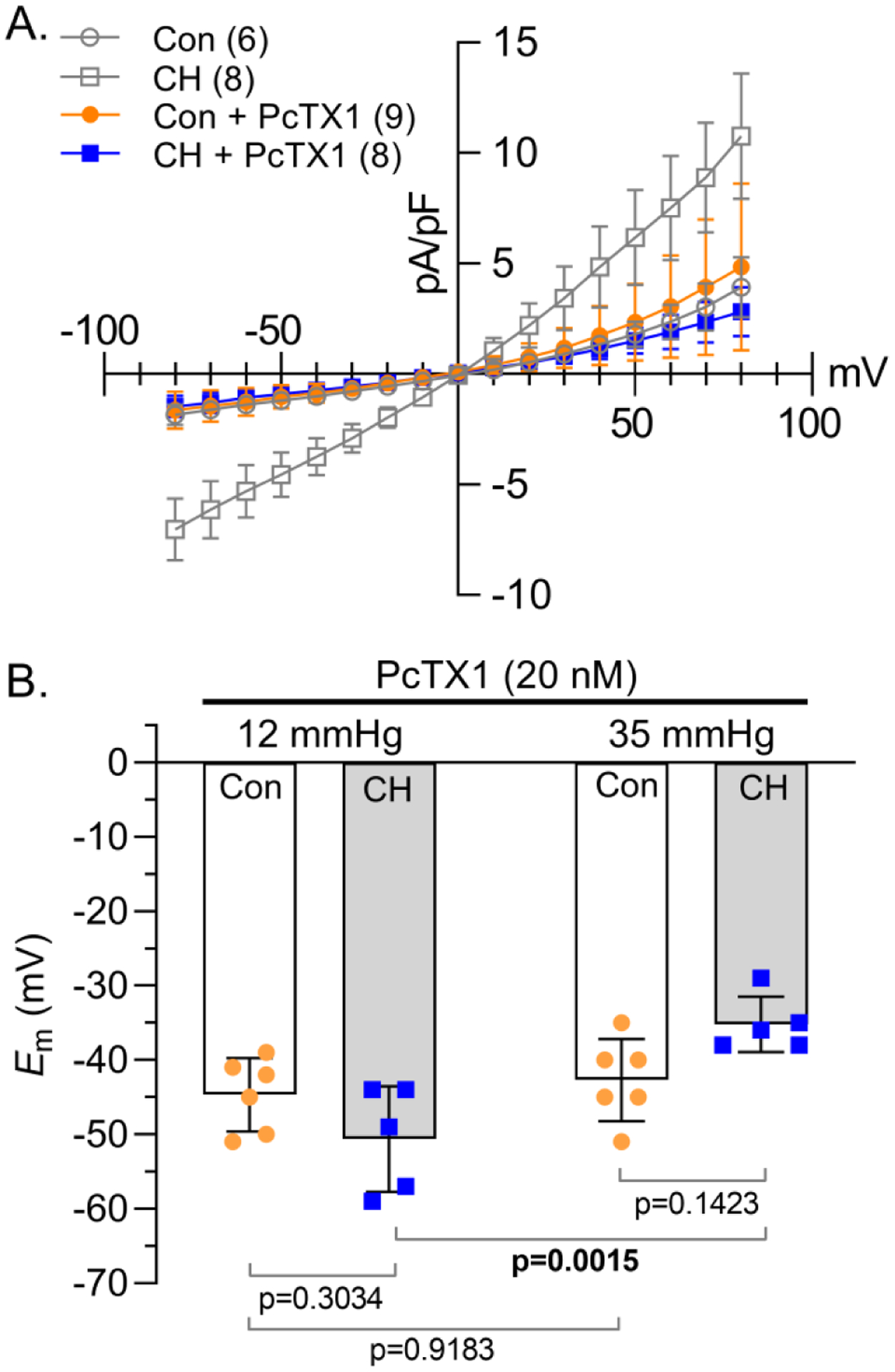
A) NSCC current density (pA/pF)-voltage (mV) relationship in PASMCs from control (circles) and CH (squares) rats in the presence of selective ASIC1 antagonist psalmotoxin 1 (PcTX1; 20 nM). Con and CH vehicles (open grey symbols) are the same as Fig 2F. B) Summary membrane potential (Em; mV) recordings at intraluminal pressures of 12 and 35 mmHg in isolated, pressurized small pulmonary arteries from control (circles) and CH (squares) rats in the presence PcTX1 (20 nM). Values are means (SD); n indicated in parentheses or dots and represents # of animals/group. Each voltage increment was analyzed individually using A) multiple unpaired t-tests, without assuming a consistent standard deviation using the Holm-Šidák method or B–C) analyzed by two-way ANOVA and individual groups compared using Tukey’s multiple comparisons test (see Statistical Summary Document for P-values).
To verify the involvement of ASIC1, we examined NSCC and K+ current density and Em in PASMCs from Asic1+/+ and Asic1−/− mice. NSCC current density was greater in PASMCs from Asic1+/+ mice exposed to CH relative to controls and the inward INSCC was largely abolished when extracellular Na+ was replaced with NMDG in both groups (Figure 7A). Under control conditions, PASMC NSCC current density was relatively small and not different between Asic1+/+ and Asic1−/− mice, suggesting there is very little ASIC1 activity under control conditions (Figure 7A–B). Exposing Asic1−/− mice to CH did not augment PASMC NSCC conductance (Figure 7B), indicating the importance of ASIC1 in mediating increased NSCC conductance in PASMCs following CH. Similar to rats, PASMC Cm was significantly larger in Asic1+/+ mice following exposure to CH (Figure 7C). PASMC Cm was unaltered by CH in PASMCs from Asic1−/− mice (Figure 7C) indicating ASIC1 may be necessary for CH-induced PASMC hypertrophy. Exposure to CH similarly reduced K+ current density in PASMCs from both Asic1+/+ and Asic1−/− mice (Figure 7D), suggesting knockout of ASIC1 does not interfere with CH regulation of K+ conductance.
Figure 7. ASIC1 mediates enhanced CH-induced NSCC.
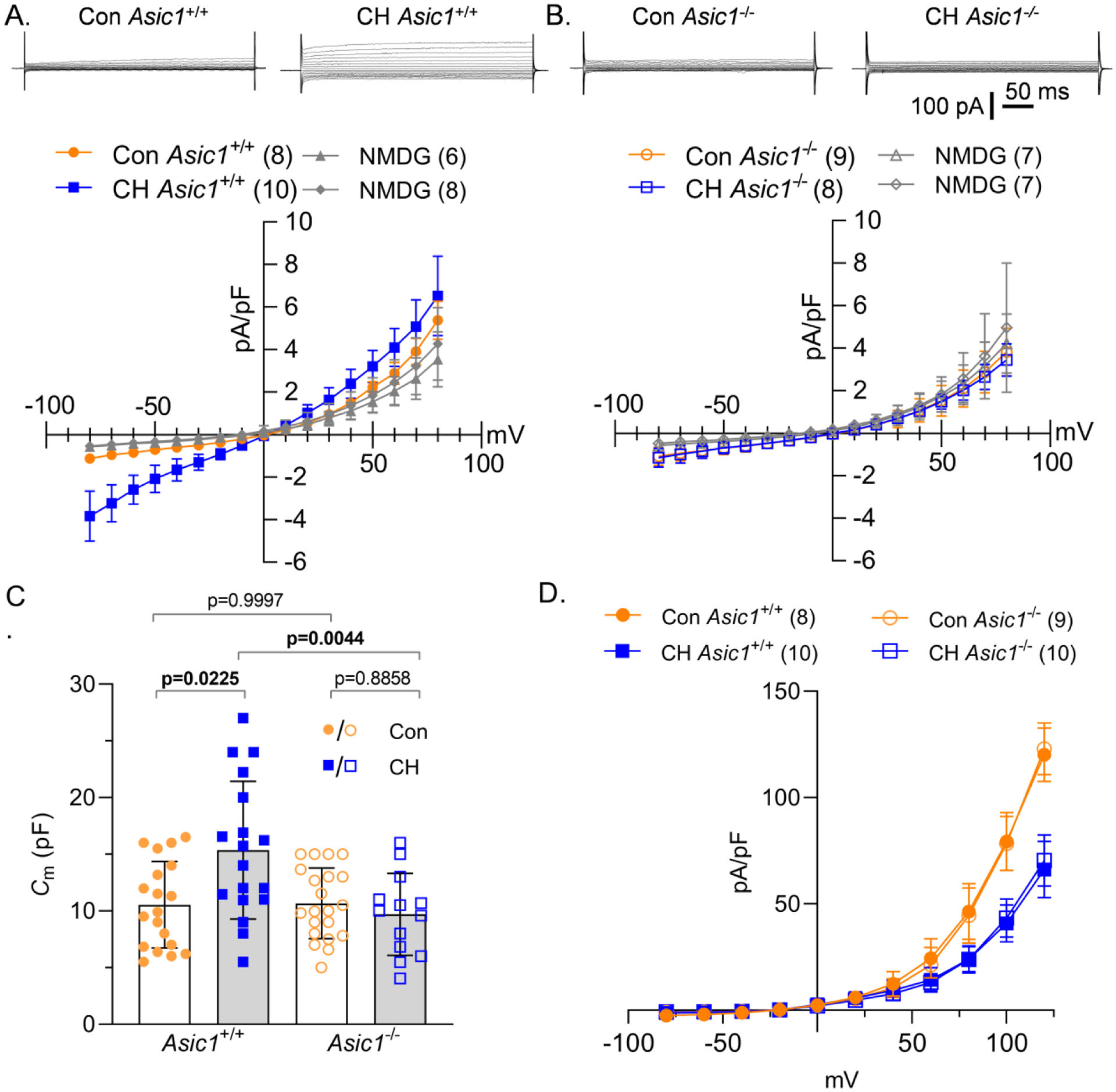
Representative traces and average NSCC current density (pA/pF)-voltage (mV) relationship in PASMCs from control (circles) and CH (squares) A) Asic1+/+ (filled symbols) or B) Asic1−/− (open symbols) mice under vehicle conditions or following replacement of extracellular NaCl with NMDG (140 mM, Con: triangles, CH: diamonds). C) Membrane capacitance (Cm; pF) and D) K+ current density (pA/pF)-voltage (mV) relationship in PASMCs from each group. Values are means (SD); n indicated in parentheses or dots as the number of mice/group. Each voltage increment was analyzed individually by two-way ANOVA and individual groups were compared using Tukey’s multiple comparisons test (see Statistical Summary Document for P-values).
We then examined Em in isolated small pulmonary arteries from control and CH, Asic1+/+ and Asic1−/− mice pressurized to 12 mmHg or 35 mmHg. Analogous to our findings in rats, Em was depolarized in PASMCs from CH Asic1+/+ mice compared to control Asic1+/+ mice at both pressures (Figure 8B). Exposing Asic1−/− mice to CH did not result in CH-induced Em depolarization. However, increasing pressure from 12 to 35 mmHg caused Em depolarization in all groups. Together, these data suggest ASIC1 does not regulate Em under control conditions and does not mediate pressure-induced depolarization. However, activation of ASIC1 following CH leads to increased Na+ influx which contributes to CH-induced Em depolarization.
Figure 8. ASIC1 mediates CH-, but not pressure-induced Em depolarization.
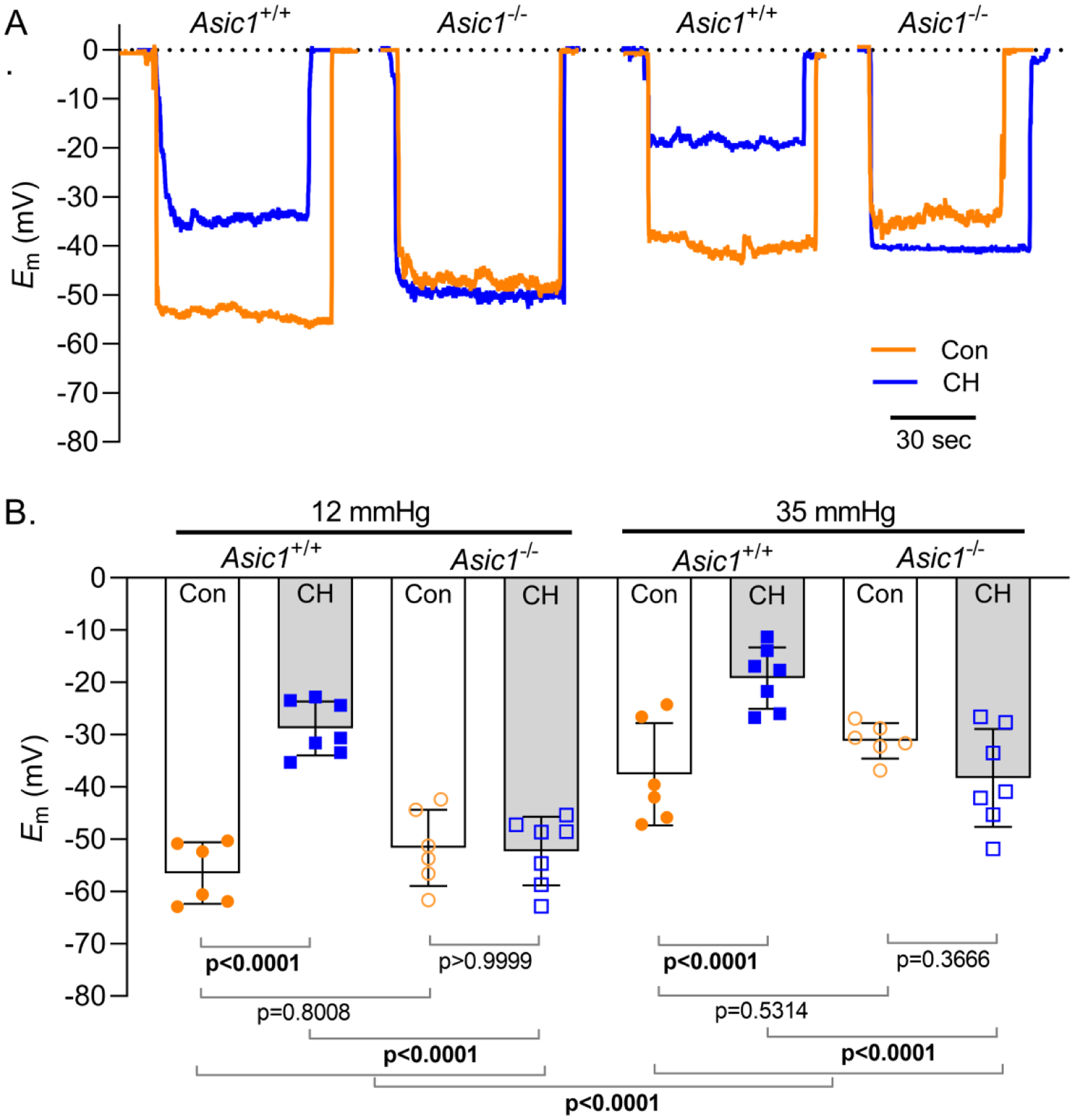
Summary membrane potential (Em; mV) recordings at intraluminal pressures of 12 and 35 mmHg in isolated, pressurized small pulmonary arteries from control (circles) and CH (squares) Asic1+/+ (filled symbols) or Asic1−/− (open symbols) mice. Values are means (SD); dots indicate n as # of animals/group; analyzed by two-way ANOVA and individual groups compared using Tukey’s multiple comparisons test.
DISCUSSION
The objective of the current study is to determine the contribution of ASIC1-mediated Na+ influx to PASMC Em regulation in control and CH-induced pulmonary hypertensive animals. In addition to the well-established decrease in PASMC K+ conductance following CH, the current study demonstrates NSCC conductance is increased and essential to the persistent Em depolarization in PASMCs following CH-induced pulmonary hypertension. Pharmacologic and genetic inhibition of ASIC1 prevented CH-induced Em depolarization of PASMCs. Although the mechanisms underlying this depolarization in PASMCs are likely complex, our results suggest a combination of factors, including inhibition of K+ efflux and augmented Na+ influx, are involved. Furthermore, this study demonstrates a novel role of ASIC1 to contribute to the regulation of Em in PASMCs and the pathogenesis of pulmonary hypertension.
The increase in pulmonary arterial pressure associated with CH could induce a pressure (or stretch)-induced depolarization of PASMCs. Although both an increase in intraluminal pressure and CH resulted in PASMC depolarization, our data indicate that pressure- and CH-induced depolarization are likely unrelated mechanisms. While CH-induced depolarization is prevented by removal of extracellular Na+, NSCC inhibition with LaCl3, and pharmacological and genetic ASIC1 inhibition; only amiloride effectively blocked pressure-induced depolarization in both groups. Replacement of extracellular Na+ with NMDG did not affect pressure-dependent depolarization in either group, indicating other ions contribute to this response. LaCl3 prevented pressure-induced depolarization in PASMC from control, but not CH animals. This suggests involvement of NSCCs under control conditions, likely mechanosensitive- or stretch-activated channels. However, following CH, pressure/stretch likely activates other ion channels, receptors, transporters, or transmembrane structures involved in mechanical integrity, mechanosensing, and signal transduction. Although PcTX1 had a similar effect as LaCl3 to prevent pressure-induced depolarization in PASMCs from control but not CH rats, this was not corroborated in the Asic1−/− mice where there was no effect of ASIC1 knockout on pressure-dependent depolarization in either group. PcTX1 is a potent and selective blocker of homomeric ASIC1a. ASIC1a/2a heteromeric channels, which may represent a significant fraction of ASICs in vivo, can also be inhibited by PcTX1 (Liu et al., 2018). ASIC2 is a well-known mechanosensitive ion channel (Ruan et al., 2021) and therefore may mediate pressure-dependent depolarization inhibited by PcTX1 in controls. Amiloride, which is routinely used to inhibit ASICs (ASIC IC50 = 1–10 μM), effectively inhibited pressure-induced depolarization in both groups. However, amiloride has been shown to inhibit several other ion channels, membrane transporters, membrane receptors, and various protein kinases and enzymes. Most notably, amiloride inhibits α1-adrenergic receptors (IC50 = 33 μM), muscarinic receptors (IC50 = 40 μM), the Na+/H+ exchanger (IC50 = 84 μM), voltage-gated Ca2+ (IC50 = 90 μM) and Na+ (IC50 = 600 μM) channels, and at much higher concentrations the Na+/Ca2+ exchanger (IC50 = ~1mM), and Na+/K+-ATPase (IC50 = >3 mM) (Kleyman & Cragoe, 1988). These off-target effects of amiloride may account for the inhibition of pressure-dependent depolarization following CH.
It is well known that in pulmonary hypertensive patients and animal models of pulmonary hypertension, the persistent depolarization of resting Em in PASMCs correlates with reduced KV channel expression and activity (Fike et al., 2006; Hong et al., 2004; Naik et al., 2005; Platoshyn et al., 2001; Smirnov et al., 1994; Suzuki and Twarog, 1982b; Wang et al., 1997; Yuan et al., 1998). Under these conditions, the relative K+ permeability (PK) is decreased, which would be expected to lead to a greater contribution of PNa and/or PCl to draw resting Em to more positive potentials. Indeed, pulmonary hypertension has been associated with increased Cl− currents due to upregulation and increased activity of Ca2+-activated Cl− channels (Sun et al., 2012; Forrest et al., 2012). In the current study, we show NSCC currents are augmented in PASMCs and more importantly, contribute to Em regulation following CH exposure. Inhibition of ASIC1 following CH normalized Em, yet K+ current density remained blunted in PASMC from CH Asic1−/− mice or in the presence of amiloride. These studies demonstrate that inhibition of ASIC1 does not indirectly affect K+ currents and further supports the requirement for increased PNa in mediating CH-induced depolarization. Our findings are in line with studies in neurons showing that in addition to reduced KV channel activity, a large component of the hypoxia-induced depolarization is due to influx of Na+ (Müller & Somjen, 2000; Raley-Susman et al., 2001; Sheldon et al., 2004) and demonstrate a unique inverse association between K+/Na+ conductance in PASMCs from animals exposed to CH.
Na+ is mainly transported across PASMC membranes through non-selective cation channels, as well as various transporters and exchangers. Although cultured human PASMCs have been shown to express tetrodotoxin-sensitive voltage-gated Na+ channels, the physiological function of these channels remains unresolved since inhibition of these voltage-gated Na+ channels does not alter Em, intracellular Ca2+ release, or proliferation in human PASMCs, nor contraction in rat isolated pulmonary arteries (Platoshyn et al., 2005). Although we cannot rule out the possible role of the Na+/H+ or Na+/Ca2+ exchanger, the effect of La3+ to prevent CH-induced depolarization suggests the involvement of NSCCs. Even though the ASIC1-mediated Na+ currents were small and did not contribute to Em in control PASMCs, the CH-induced depolarization was prevented following ASIC1 inhibition. This is consistent with the known role of ASICs to induce membrane depolarization and generate action potentials in neurons (Vukicevic & Kellenberger, 2004; Poirot et al., 2006) and provides novel evidence that ASIC1-mediated Na+ influx induces membrane depolarization and regulates Em in PASMCs following CH exposure.
Our data further show that CH-induced depolarization can be acutely reversed by pharmacological inhibition of ASIC1, suggesting the channel is constitutively active following CH. Unlike many other ion channels/proteins involved in pulmonary hypertension, the requirement for ASIC1 in the development of pulmonary hypertension is not dependent on an increase in PASMC ASIC1 protein expression. Rather, through a cell-surface biotinylation assay, we have demonstrated that CH promotes a RhoA-dependent localization of ASIC1 to the plasma membrane (Herbert et al., 2018). In addition to enhanced plasma membrane trafficking leading to greater activity of the channel, it is known that the properties of ASICs can be modulated by signalling molecules or biochemical changes associated with pathological conditions that can dramatically augment and/or prolong the activity of these channels (Chu & Xiong, 2013). This activation of ASIC1 specifically under pathological conditions makes it an ideal therapeutic target.
ASIC1 is well-known to be activated by extracellular H+ and exhibits a fast desensitizing current with pH values below 7.0. By contrast, several non-proton ligands can trigger sustained ASIC currents at physiological pH (Yu et al., 2010; Gautschi et al., 2017). Although we have not fully elucidated the role of acidosis in the activation of ASIC1 following CH, our previous studies have described a distinct role for ASIC1 in regulating PASMC Ca2+ influx in both physiological and pathophysiological settings, thus contributing to pulmonary vasoconstriction elicited by various Gq-coupled receptor agonists (UTP, endothelin-1, U-46619), store-operated Ca2+ entry, and alveolar hypoxia (Jernigan et al., 2009, 2012; Nitta et al., 2014). The effects of CH to augment receptor-mediated vasoconstriction and store-operated Ca2+ entry in pulmonary arteries from Asic1+/+ mice were not observed after CH in Asic1−/− mice (Nitta et al., 2014). In contrast, depolarization-induced pulmonary arterial constriction in response to increasing concentrations of KCl is similar between Asic1+/+ and Asic1−/− mice, indicating that the decreased vasoconstrictor responsiveness in pulmonary arteries from Asic1−/− mice is specific to receptor-mediated responses and not a result of a generalized decrease in vasoreactivity. Furthermore, Asic1−/− mice exhibit diminished right ventricular systolic pressure and lack evidence of right ventricular hypertrophy or arterial remodelling in response to CH (Nitta et al., 2014). Taken together, these data demonstrate that conduction of both Na+ and Ca2+ through ASIC1 is a central component to the active vasoconstriction and vascular remodelling associated with the development of hypoxic pulmonary hypertension and right ventricular hypertrophy.
In addition to regulating vascular tone, Em depolarization can affect a wide range of cellular functions, including cell proliferation, migration, and differentiation. Indeed, dysfunction of Kv channels has been linked to inhibition of apoptosis and medial hypertrophy in PASMCs from hypoxic animals and patients with pulmonary arterial hypertension (Michelakis et al., 2002; Pozeg et al., 2003; Bonnet et al., 2007). Our previous evidence that ASIC1 contributes to medial arterial remodelling following CH (Nitta et al., 2014) is supported by the current data showing the CH-induced increase in PASMC Cm is absent in PASMCs from Asic1−/− mice, demonstrating a contribution of ASIC1 to PASMC hypertrophy. Further investigation is necessary to determine the exact mechanism by which ASIC1 channel activity and depolarization modify PASMC growth, proliferation, and migration.
In summary, the current study provides novel evidence that ASIC1-mediated Na+ influx induces membrane depolarization and regulates Em in PASMCs following CH exposure. Together with previous studies from our laboratory demonstrating the involvement of ASIC1 in PASMC Ca2+ influx, enhanced agonist-induced pulmonary vasoconstriction, pulmonary arterial remodelling, elevated right ventricular systolic pressure, and right ventricular hypertrophy these results indicate an essential role for ASIC1 in mediating hypoxic pulmonary hypertension (Jernigan et al., 2012; Nitta et al., 2014). Since ASIC1 is activated by acidosis, ischemia, and inflammation (Xu & Chen, 2020), modulation of ASICs by these mediators can lead to dramatically enhanced or prolonged activity of these channels, thus promoting their pathological functions which may differ from their physiological function. As there is increasing evidence indicating the involvement of these channels in pathological processes such as neurodegenerative diseases, ischemic brain injury, and a variety of cancers (Chu & Xiong, 2013), a better understanding of how ASICs behave in pathological conditions may help define novel therapeutic interventions for a variety of disease states.
KEY POINT SUMMARY.
In pulmonary hypertensive patients and animal models of pulmonary hypertension, the resting membrane potential (Em) of pulmonary arterial smooth muscle cells (PASMCs) is persistently depolarized.
In addition to the well-established reduction of K+ conductance, we show non-selective cation channel (NSCC) currents are increased and essential to the persistent Em depolarization in PASMCs following chronic hypoxia (CH)-induced pulmonary hypertension.
The current study provides novel evidence that acid-sensing ion channel 1 (ASIC1)-mediated Na+ influx induces membrane depolarization and regulates Em in PASMCs following CH exposure.
Although fairly quiescent under control conditions, our findings demonstrate a pathological function of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension.
ACKNOWLEDGEMENTS
The authors would like to thank Lindsay Herbert for her assistance with genotyping the Asic1+/+ and Asic1−/− animals.
FUNDING
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL-111084 (to N.L. Jernigan) and R01 HL-132883 (to T.C. Resta) and American Heart Association grant 18TPA34110281 (to N.L. Jernigan).
Biography

Author Profile/Biography: Nikki Jernigan is a cardiovascular physiologist. She received her Ph.D. from the University of New Mexico School of Medicine in Albuquerque, New Mexico and did her postdoctoral training at the Cardiovascular-Renal Research Center at the University of Mississippi Medical Center in Jackson, Mississippi. She returned to the University of New Mexico as faculty of the Department of Cell Biology and Physiology and has built a strong research program that focuses on altered vascular signalling mechanisms associated with cardiovascular disease.
Footnotes
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request: njernigan@salud.unm.edu
COMPETING INTERESTS
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- Archer S, Souil E, Dinh-Xuan A, Schremmer B, Mercier J, El Yaagoubi A, Nguyen-Huu L, Reeve H & Hampl V (1998). Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101, 2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Patton CW & Nuccitelli R (1994). A practical guide to the preparation of Ca2+ buffers. Methods in Cell Biology 40, 3–29. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN & Michelakis ED (2007). The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. PNAS 104, 11418–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton BR, Jernigan NL, Norton CE, Walker BR & Resta TC (2010). Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298, L232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X-P & Xiong Z-G (2013). Acid-Sensing Ion Channels in Pathological Conditions. Ad Exp Med Biol. 961, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fike CD, Kaplowitz MR, Zhang Y & Madden JA (2006). Voltage-gated K+ channels at an early stage of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 291, L1169–76. [DOI] [PubMed] [Google Scholar]
- Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA & Leblanc N (2012). Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303, C1229–C1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Chen T & Raj JU (2016). Endothelial and Smooth Muscle Cell Interactions in the Pathobiology of Pulmonary Hypertension. Am J Respir Cell Mol Biol 54, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi I, Bemmelen MX van & Schild L (2017). Proton and non-proton activation of ASIC channels. PLOS ONE 12, e0175293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden SW and Brogioli D (2020). LJPcalc [Online]. Available: https://swharden.com/software/LJPcalc
- Herbert LM, Resta TC & Jernigan NL (2018). RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am J Physiol Cell Physiol 314, C166–C176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Weir EK, Nelson DP & Olschewski A (2004). Subacute hypoxia decreases voltage-activated potassium channel expression and function in pulmonary artery myocytes. Am J Respir Cell Mol Biol 31, 337–343. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, Herbert LM, Walker BR & Resta TC (2012). Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol 302, C931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan NL, Naik JS, Weise-Cross L, Detweiler ND, Herbert LM, Yellowhair TR & Resta TC (2017). Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS ONE 12, e0180455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan NL, Paffett ML, Walker BR & Resta TC (2009). ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca(2+) entry. Am J Physiol Lung Cell Mol Physiol 297, L271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyman T & Cragoe EJr (1988). Amiloride and its analogs as tools in the study of ion transport. J Membr Biol 105, 1–21. [DOI] [PubMed] [Google Scholar]
- Lin M-J, Leung GPH, Zhang W-M, Yang X-R, Yip K-P, Tse C-M & Sham JSK (2004). Chronic Hypoxia-Induced Upregulation of Store-Operated and Receptor-Operated Ca2+ Channels in Pulmonary Arterial Smooth Muscle Cells: A Novel Mechanism of Hypoxic Pulmonary Hypertension. Circ Res 95, 496–505. [DOI] [PubMed] [Google Scholar]
- Liu X-R, Zhang M-F, Yang N, Liu Q, Wang R-X, Cao Y-N, Yang X-R, Sham JSK & Lin M-J (2011). Enhanced Store-Operated Ca2+ Entry and TRPC channel Expression in Pulmonary Arteries of Monocrotaline-Induced Pulmonary Hypertensive Rats. Am J Physiol Cell Physiol 302, C77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hagan R & Schoellerman J (2018). Dual actions of Psalmotoxin at ASIC1a and ASIC2a heteromeric channels (ASIC1a/2a). Sci Rep 8, 7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke T, Maylor J, Undem C, Sylvester JT & Shimoda LA (2012). Kinase-dependent activation of voltage-gated Ca2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 302, L1128–L1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R & Archer SL (2002). Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105, 244–250. [DOI] [PubMed] [Google Scholar]
- Müller M & Somjen GG (2000). Na+ Dependence and the Role of Glutamate Receptors and Na+ Channels in Ion Fluxes During Hypoxia of Rat Hippocampal Slices. J Neurophysiol 84, 1869–1880. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Tyler RC, Rodman DM & McMurtry IF (1997). Possible role of T-type Ca2+ channels in L-NNA vasoconstriction of hypertensive rat lungs. Am J Physiol Heart Circ Physiol 272, H2616–H2621. [DOI] [PubMed] [Google Scholar]
- Naik JS, Earley S, Resta TC & Walker BR (2005). Pressure-induced smooth muscle cell depolarization in pulmonary arteries from control and chronically hypoxic rats does not cause myogenic vasoconstriction. J Appl Physiol 98, 1119–1124. [DOI] [PubMed] [Google Scholar]
- Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR & Jernigan NL (2014). Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306, H41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ & Yuan JX-J (2000). Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279, C1540–C1549. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Remillard CV, Fantozzi I, Sison T & Yuan JXJ (2005). Identification of functional voltage-gated Na(+) channels in cultured human pulmonary artery smooth muscle cells. Pflugers Arch 451, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ & Yuan JX (2001). Chronic hypoxia decreases K(V) channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 280, L801–12. [DOI] [PubMed] [Google Scholar]
- Poirot O, Berta T, Decosterd I & Kellenberger S (2006). Distinct ASIC currents are expressed in rat putative nociceptors and are modulated by nerve injury. J Physiol 576, 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozeg ZI, Michelakis ED, McMurtry MS, Thébaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A & Archer SL (2003). In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 107, 2037–2044. [DOI] [PubMed] [Google Scholar]
- Priest RM, Robertson TP, Leach RM & Ward JP (1998). Membrane potential-dependent and - independent vasodilation in small pulmonary arteries from chronically hypoxic rats. J Pharmacol Exp Ther 285, 975–982. [PubMed] [Google Scholar]
- Raley-Susman KM, Kass IS, Cottrell JE, Newman RB, Chambers G & Wang J (2001). Sodium Influx Blockade and Hypoxic Damage to CA1 Pyramidal Neurons in Rat Hippocampal Slices. J Neurophysiol 86, 2715–2726. [DOI] [PubMed] [Google Scholar]
- Resta TC, Chicoine LG, Omdahl JL & Walker BR (1999). Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol 276, H699–708. [DOI] [PubMed] [Google Scholar]
- Ruan N, Tribble J, Peterson AM, Jiang Q, Wang JQ & Chu X-P (2021). Acid-Sensing Ion Channels and Mechanosensation. Int J Mol Sci 22, 4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon C, Diarra A, Cheng YM & Church J (2004). Sodium Influx Pathways during and after Anoxia in Rat Hippocampal Neurons. J Neurosci 24, 11057–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA & Laurie SS (2013). Vascular remodeling in pulmonary hypertension. J Mol Med (Berl) 91, 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA, Sham JS, Shimoda TH & Sylvester JT (2000). L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279, L884–94. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG & Souza R (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53, 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov S, Robertson T, Ward J & Aaronson P (1994). Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol 266, H365–70. [DOI] [PubMed] [Google Scholar]
- Sun H, Xia Y, Paudel O, Yang X-R & Sham JSK (2012). Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 590, 3507–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H & Twarog BM (1982a). Membrane properties of smooth muscle cells in pulmonary arteries of the rat. Am J Physiol 242, H900–H906. [DOI] [PubMed] [Google Scholar]
- Suzuki H & Twarog BM (1982b). Membrane properties of smooth muscle cells in pulmonary hypertensive rats. Am J Physiol 242, H907–15. [DOI] [PubMed] [Google Scholar]
- Thuma JB & Hooper SL (2018). Choline and NMDG directly reduce outward currents: reduced outward current when these substances replace Na+ is alone not evidence of Na+-activated K+ currents. Journal of Neurophysiology 120, 3217–3233. [DOI] [PubMed] [Google Scholar]
- Voelkel NF & Tuder RM (2000). Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest 106, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukicevic M & Kellenberger S (2004). Modulatory effects of acid-sensing ion channels on action potential generation in hippocampal neurons. Am J Physiol Cell Physiol 287, C682–C690. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C & Lazdunski M (1997). A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177. [DOI] [PubMed] [Google Scholar]
- Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MSR, Ko EA, Wang J, Wang C, Makino A & Yuan JX-J (2013). Chronic Hypoxia Selectively Enhances L- and T-type Voltage-dependent Ca2+ Channel Activity in Pulmonary Artery by Upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 305, L154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Juhaszova M, Rubin LJ & Yuan XJ (1997). Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 100, 2347–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Weigand L, Lu W, Sylvester JT, L. Semenza G & Shimoda LA (2006). Hypoxia Inducible Factor 1 Mediates Hypoxia-Induced TRPC Expression and Elevated Intracellular Ca2+ in Pulmonary Arterial Smooth Muscle Cells. Circ Res 98, 1528–1537. [DOI] [PubMed] [Google Scholar]
- Wemmie J, Chen J, Askwith C, Hruska-Hageman A, Price M, Nolan B, Yoder P, Lamani E, Hoshi T, Freeman J & Welsh M (2002). The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Taugher RJ & Kreple CJ (2013). Acid-sensing ion channels in pain and disease. Nat Rev Neurosci 14, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ & Simon RP (2004). Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698. [DOI] [PubMed] [Google Scholar]
- Xu Y & Chen F (2020). Factors and Molecular Mechanisms Influencing the Protein Synthesis, Degradation and Membrane Trafficking of ASIC1a. Front Cell Dev Biol 8, 596304–596304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L & Palmer LG (2014). Ion conduction and selectivity in acid-sensing ion channel 1. J Gen Physiol 144, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM & Welsh MJ (2004). Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. PNAS 101, 6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H & Xu TL (2010). A nonproton ligand sensor in the acid-sensing ion channel. Neuron 68, 61–72. [DOI] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB & Rubin LJ (1998). Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98, 1400–1406. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Goldman WF, Tod ML, Rubin LJ & Blaustein MP (1993). Ionic currents in rat pulmonary and mesenteric arterial myocytes in primary culture and subculture. Am J Physiol 264, L107–15. [DOI] [PubMed] [Google Scholar]