

Abstract
The regulation of intracellular calcium (Ca2+) homeostasis is fundamental to maintain normal functions in many cell types. The ryanodine receptor (RyR), the largest intracellular calcium release channel located on the sarco/endoplasmic reticulum (SR/ER), plays a key role in the intracellular Ca2+ handling. Abnormal type 2 ryanodine receptor (RyR2) function, associated to mutations (ryanopathies) or pathological remodeling, has been reported, not only in cardiac diseases, but also in neuronal and pancreatic disorders. While animal models and in vitro studies provided valuable contributions to our knowledge on RyR2 dysfunctions, the human cell models derived from patients’ cells offer new hope for improving our understanding of human clinical diseases and enrich the development of great medical advances. We here discuss the current knowledge on RyR2 dysfunctions associated with mutations and post-translational remodeling. We then reviewed the novel human cellular technologies allowing the correlation of patient’s genome with their cellular environment and providing approaches for personalized RyR-targeted therapeutics.
Subject terms: Ventricular tachycardia, Ventricular tachycardia
Facts
RyR2 dysfunctions are due to mutations and pathological post-translational modifications.
Ryanopathies are not limited to cardiac diseases but rather cause a multiple-organ dysfunction.
The hiPSC technology brings new tools to investigate ryanopathies in multiple organs in patient-specific molecular and cellular environment.
Introduction
The ryanodine receptor is the largest membrane protein we know. It is a huge intracellular macromolecular complex formed by a homotetramer embedded in the endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR). Among three isoforms, the type 2 is mostly expressed in the heart, brain, and pancreas [1]. In cardiomyocytes (CMs), the opening of the L-type calcium voltage-dependent channels causes a first entry of the extracellular Ca2+ that triggers the binding of Ca2+ to cardiac ryanodine receptor/calcium (Ca2+) release channel (RyR2) and allows a further release of Ca2+ from the SR to reach the cardiac contraction. This mechanism involving RyR2 is known as calcium-induced-calcium-release (CICR) brought by Fabiato [2]. In neurons, intracellular Ca2+ cycling involves the RyR and inositol [1, 3, 4]-trisphosphate receptors (IP3R). RyR2 expression is distributed through the central nervous system including hippocampus. RyR2 Ca2+ release contributes to translate synaptic activity into neuronal function [5] and stimulate learning and memory [3]. RyR2 mutations are associated to inherited disorders. The first mutations found in the RyR2, were associated to catecholaminergic polymorphic ventricular tachycardia (CPVT) [4, 6]. Today, numerous mutations of the RyR2, defined as ryanopathies, are related to some arrhythmogenic disorders including CPVT, arrhythmogenic right ventricular cardiomyopathies (ARVC) under stress conditions or short-coupled polymorphic ventricular tachycardia (VT) when at rest- [7–10]. While novel RyR2 mutations have been identified in patients, our understanding of the link between RyR2 mutations and arrhythmias has been driven by experimental models, mostly through transgenic mice and heterologous recombinant RyR2 expression systems. However, the main issue with such models remains the further transposition of identifiable mechanisms in human [11, 12]. Hence, the electrophysiological kinetic properties of a mouse heart are different from the human ones in terms of both, action potential (AP) and electrocardiogram (ECG) [13]. The murine resting heart rate is about ten times as fast as that of human. In rodents, 90–92% of the reuptake of Ca2+ is ensured by SR Ca²+-ATPase (SERCA) compared to only 76% in human. The Ca2+ left is extruded out of the cell via the sodium-calcium exchanger (NCX). It has been shown that the stimulation frequency can affect the relative contribution of SERCA and NCX in human but not in rat [14]. Consequently, the murine model is sensitive to evaluate the SR-dependent arrhythmias [15]. Murine CMs therefore present electrical properties that are divergent when compared to that of their human counterparts [16].
The investigation of the recombinant RyR2 proteins in vitro has been very fruitful to provide for molecular patterns. However, the cellular context and some key RyR2 partners were missing. In fact, the lab animal and in vitro recombinant models tend to oversimplify the pathological mechanisms and phenotypes observed in patients. Human models are continuously needed to improve our understanding of the RyR2 physiological and pathophysiological roles [13].
Investigating arrhythmogenic disorders by obtaining fresh ventricular cardiac biopsies from patients and healthy control subjects is both ethically and technically difficult. This is where the human stem cell technologies provide for new possibilities to resolve these limits, particularly the human-induced pluripotent stem cells (hiPSCs). The hiPSCs are self-renewable cells that are theoretically capable to differentiate into many somatic cell types [17]. They have very limited ethical issues when compared to human embryonic induced pluripotent stem cells (hESC). The hiPSC harbor the genetic background of the patients. Thus, patient-specific hiPSC-derived cardiomyocytes (hiPSC-CMs) represent a novel complementary alternative model to investigate inherited arrhythmogenic disorders associated with ryanopathies and RyR2 dysfunctions.
The present review focuses on the ryanopathies and pathological RyR2 remodeling responsible for cardiac, neuronal, and pancreatic dysfunctions as a multi-organ syndrome. We also discuss the interest of exploiting hiPSCs as a tool to model the ryanopathies in a patient-specific context to further study the genome/phenotype relationship.
The RyR2 dysfunction and cardiac pathophysiological conditions
Ryanopathies inducing catecholaminergic polymorphic ventricular tachycardia (CPVT)
The CPVT is an inherited cardiac syndrome that is characterized by exercise-induced VT leading to some episodic syncope. CPVT is responsible for sudden cardiac death (SCD) during exercise, acute stress and/or emotions. CPVT patients have no structural cardiac abnormalities [18–20].
CPVT syndromes have been associated with several mutated proteins in the following manner:
-
(i)
In more than 70% of the cases, with dominant cardiac ryanodine receptor (RYR2) mutations (denoted CPVT1) [4, 8].
-
(ii)
In less than 5%, with autosomal recessive or dominant calsequestrin2 (CASQ2) mutations (denoted CPVT2) [21]. They affect Ca2+ buffering in the SR and lower its binding to RyR2 [22].
-
(iii)
Some homozygous mutations of the Trans-2,3-Enoyl-CoA Reductase Like gene (TECRL) are associated with both the CPVT syndrome (denoted CPVT3) and the long-QT syndrome (LQTS). They lead to an increase of the diastolic Ca2+ concentration and reduce the SR Ca2+ load, triggering then the delayed-after-depolarizations (DADs), that are responsible for these arrhythmogenic disorders [23].
-
(iv)
Some autosomal dominant mutations of calmodulin (CALM1, CALM2, and CALM3) (denoted CPVT4). They perturb the CaM affinity for Ca2+ and RyR2 at low Ca2+ concentration levels, leading to diastolic SR Ca2+ leak [24].
-
(v)
Some autosomal triadin gene (TRDN) recessive mutations (denoted as CPVT5). These provoke a total absence of this protein, further disrupting the FKBP12.6 and RyR2 interaction or either causing a reduced calsequestrin expression; calcium homeostasis in the heart is then disturbed [25, 26].
-
(vi)
Other genes are implicated in the pathophysiological mechanism of CPVT but in far lesser extent such as ankyrin 2 (ANK2) [27, 28], sodium voltage-gated channel alpha subunit 5 (SCN5A) [29] and potassium voltage-gated Channel subfamily J member 2 (KCNJ2) [30–32]. However, the diagnosis of CPVT syndrome remains questionable for these cases [33].
Nowadays, over 150 RyR2 variants are associated with CPVT (see Table 1). The majority of these RyR2 variants are in four domains called hot-spot domains, well conserved between human, dog, rat, mouse, and pig.
Table 1.
List of the RyR2 mutations associated with the CPVT syndrome.
| Localization | Mutations | Findings | Ref. | |
|---|---|---|---|---|
| Functional characterization of the RyR2 mutants | N-terminal domain | E189D | The RyR2-E189D mutation increased the propensity for SOICR, without altering the FKBP12.6 affinity to bind to the channel. | [1] |
| G230C | This novel CPVT mutation enhances RyR2 cytosolic Ca2+ sensitivity which leads to diastolic SR Ca2+ leak under stress conditions. RyR2 leak was associated with a depletion of the stabilizing FKBP12.6 protein, which eventually provoked arrhythmias. | [2] | ||
| ΔExon 3 | The RYR2 exon 3 deletion causes a NTD alteration and results in a Ca2+ release properties adjustment. Although this deletion is rescued by the β strand switching, it affects interfaces with other RYR2 domains. This suggests some N-terminal domain and channel pore coupling. | [3] | ||
| G357S | The RyR2-G357S mutation reduced the expression of the RyR2 protein and increased the arrhythmogenic SOICR in HEK293 cells, which might be responsible for the CPVT syndrome. | [4] | ||
| A165D | The RyR2-A165D mutation was first identified in a CPVT patient. When using a knock-in mice model, the A165D mutation induced SR Ca2+ release triggering DADs. The A165D mutation was located in the conformational stability loop, which explained the occurrence of some diastolic leak that is responsible for arrhythmias. | [5] | ||
| Helical domain 1 | S2246L | Increase of Ca2+ release in HL-1 cardiomyocytes expressing mutant hRyR2, after caffeine and β-adrenergic activation. | [6] | |
| P2328S | This mutation decreases FKBP12.6 binding to the RyR2. Sensitivity increases with cytosolic Ca2+ allowing a higher open probability of RyR2 channels at low diastolic levels, causing SR Ca2+ leaks in the CPVT1 syndrome. The JTV519 Rycal molecule rescued a normal RyR2 function. | [7] | ||
| R2401H | RyR2-R2401H mutation is located in the FKBP12.6 RyR2 binding region, which could affect the CICR and the ECC resulting in a CPVT. | [8] | ||
| S2246L, R2474S | RyR2 mutations increased both store-overload-induced Ca2+ release (SOICR) activity and sensitivity towards luminal calcium, without affecting the channel affinity for the FKBP12.6 in CPVT. | [9] | ||
| N23861 | The RyR2-N23861 mutation induced some sensitivity impairment towards Ca2+ dependent channel inhibition. | [10] | ||
| R2267H | A novel mutation was identified in sudden infant death syndrome cases. When using some heterologous system expression, this mutation was leaky under beta-adrenergic stimulation, leading to a PKA-phosphorylation that triggers cardiac arrhythmias. Interestingly, another study demonstrated a lack of pathogenicity of this variant. Thus, the in vitro functional findings were not translated to human phenotype. | [11, 12] | ||
| R2474S | The RyR2-R2474S mutation perturbed the interdomain conformational changes, which destabilized the closed state of the RyR2 and lead to a leaky channel. | [13, 14] | ||
| Central domain | N4104K | See findings of the S2246L mutation. | [6] | |
| Q4201R | See findings of the P2328S mutation. | [7] | ||
| Q4201R | See findings of the S2246L and R2474S mutations. | [9] | ||
| S4153R | This novel RyR2 heterozygous mutation was first described in a 25-year-old CPVT syndrome female patient. This mutation is characterized by some RyR2 gain-of-function that is induced by the SOICR threshold reduction and some propensity increase for spontaneous calcium release. | [15, 16] | ||
| Channel domain | R4497C | See findings of the S2246L mutation. | [6] | |
| V4653F | See findings of the P2328S mutation. | [7] | ||
| I4867M, | See findings of the S2246L and R2474S mutations. | [9] | ||
| A4860G | When using mice models and HEK293 cells, the RyR2-A4860G mutation reduced the channel activity by inhibiting Ca2+ release during the diastole and by overloading the SR with Ca2+. Consequently, it prolonged Ca2+ release and corresponding AP, leading to the activation of the NCX exchanger. The ITi current triggers the early afterdepolarizations (EADs) that are responsible for CPVT pathogenesis. | [17, 18] | ||
| S4565R | Two novel mutations were identified in sudden infant death syndrome cases. When using some heterologous system expression, these 2 mutations were leaky under beta-adrenergic stimulation, leading to a PKA-phosphorylation that triggers cardiac arrhythmias. | [11] | ||
| R4496C (human: R4497C) | The RyR2-R4496C mutation induced an increase of the SR Ca2+ load responsible for Ca2+ waves and arrhythmias in CPVT murine model. | [19, 20] | ||
| K4750Q | The RyR2-K4750Q mutation mediated-CPVT induced diastolic SR Ca2+ leak caused by an enhance of propensity to activation of cytosolic and luminal Ca2+ and by the loss of cytosolic Ca2+/Mg2+-mediated inactivation. | [21] | ||
| I4855M | The RyR2-I4855M mutation was present in 2 members of a CPVT-affected family. The RyR2-I4855M shows with some loss-of-function and is characterized by some CICR inhibition of the HEK293 cells. The I4855A may interfere with Ca2+ permeation and may affect interactions between the RyR2 pore subunits. | [22] | ||
| Case reports and genotyping studies of patient cohorts | N-terminal domain | R414L, I419F, P164S | Novel RyR2 mutations were associated with the CPVT1 syndrome in a swimming-triggered arrhythmia syndrome using direct DNA sequencing and denaturing high-performance liquid chromatography. The 388 unrelated patients were chosen according to family or personal history of drowning or swimming related cardiac events. However, considering the large number of the cohort they did not specify the cardiac phenotype of each patient. | [23] |
| ΔExon 3, A77V |
In a 17-year-old boy postmortem study, the RyR2-A77V mutation was associated with both an arrhythmogenic right ventricular cardiomyopathy and a CPVT syndrome, in the same family. This 17-year-old boy presented right ventricular fibrofatty and fatty myocardium replacement and calcium phosphate deposits in right ventricular cardiomyocytes that were mostly restrained into mitochondria. His mother and his sister presented normal right and left ventricles volume and no kinetic alterations. The exercise treadmill stress test revealed polymorphic ventricular tachycardia that were successfully abolished with β-blocker (Acebutolol) treatment. The same RyR2-A77V mutation led to distinct diseases in the same family members. This reflect the complexity of clinical diagnosis and the variable phenotype that can be present even among family members of the same family. De novo RYR2 exon 3 deletion was reported in a severe CPVT case. This patient also developed some left ventricular non-compaction (LVNC), which exacerbates the arrhythmia. This patient showed no sign of endomyocardial inflammation and displayed normal heart structure. Multiform of premature ventricular contractions, ectopic atrial rhythm and ventricular triplet were observed during exercise. She experienced ventricular fibrillation and underwent ICD implantation together with the administration of Metoprolol and then Satolol treatment. Due the severity of her phenotype, she started Flecainide and Nadolol treatment and underwent bilateral sympathectomy. The interaction between RyR2- ΔExon 3 and LVNC that may represent a predictive clinical marker for a more severe CPVT phenotype remains unclear. |
[24, 25] | ||
| R414C | The molecular autopsy revealed novel mediated CPVT syndrome RyR2 mutations in 2 unexplained drowning cases. This patient carrying the RyR2-R414C variant experienced syncope and seizure-like symptoms. Unexceptional and unremarkable EEG and physical examination were found. She was first diagnosed with acute seizure activity secondary to trauma. Due to the nature of the sudden death, direct DNA sequencing and polymerase chain reaction, denaturing high-performance liquid chromatography were performed which revealed this missense novel RyR2 mutation. As this patient presented normal structural heart and absence of fatty infiltration, she was considered as a CPVT patient. | [26] | ||
| V186M, P164S | Four patients (3 males) out of 8 patients, were presented RyR2 mutations associated with some CPVT syndrome. Each patient presented specific symptoms which reflect the heterogeneity of CPVT phenotypes. Some patients had palpitations and seizure-like activity others cardiac arrest with ventricular fibrillation. Unfortunately, they did not match each RyR2-variant with its specific phenotype. | [27] | ||
| R169Q | One RyR2 novel heterozygous mutation in exon 8 was screened in an 18 years old female patient presenting a CPVT syndrome. This patient presented sudden collapse due to exercise and had bidirectional ventricular tachycardia during exercise stress test. She had a good response to the β-blocker treatment. This same mutation was found recently in three unrelated females. Interestingly, all of these patients presented left ventricular non-compaction cardiomyopathy and two of them survived sudden cardiac arrest. In vitro functional analysis of this mutation revealed an increase of the Ca2+ fractional release from the SR and a decreased threshold for overload-induced Ca2+ release. It was suggested that this RyR2-R169Q mutation leads to local structural abnormalities within or near the hot-spot regions which in turn leads to functional perturbations. It leads to allosteric dysregulation by reducing the side chains size and diminishing the positive charge and stacking interaction of the RyR2 protein. | [28–30] | ||
| L62F, M81L, P164S, E243K, F329L, R332W, V377M, G357S, T415R, R420Q, V507I, A549V, S616L, H240R | A cohort of CPVT patients was screened to investigate RYR2 gene mutations. 34 novel mutations were identified. They did not specify the clinical phenotype of the 155 unrelated patients examined in this study. Interestingly, they proposed a novel targeted genetic testing for CPVT syndrome. They emphasized also genotype/phenotype relationship as the majority of these mutations were localized in the so-called hot-spot regions. | [31] | ||
| D242V, E243K | The long-term follow-up of 101 CPVT patients showed high cardiac events, despite some β-blockers treatment in 21% of patients with 13% of fatal or near-fatal events. Some of these patients survived cardiac arrest and presented palpitations and syncope accompanied or not with seizures. 80% of these patients were treated with β-blockers (mostly with Nadolol but also with Propranolol, Bisoprolol, Acebutolol and Pindolol). ICD implantation and Verapamil were added to some patients after the 1st cardiac event. Even though β-blockers lower the cardiac events rate they are not sufficient alone to prevent arrhythmias. | [32] | ||
| R169L | This mutation was identified in an 8 years old boy with CPVT and Left Ventricular Hypertrophy. This boy presented with two episodes of emotion-triggered syncope and could not survive the third one that led to sudden cardiac death. This patient carried two other mutations the G1339 variant in ATP-binding cassette, subfamily C member 9 (ABCC9) and R52H variant in Potassium Inwardly Rectifying Channel Subfamily J Member 5 (KCNJ5). These 2 variants have unknown significance. The combination between CPVT and Left Ventricular Hypertrophy might lead to a more severe fatal phenotype. However, more studies are needed to elucidate the pathophysiological mechanism underlying the structural alterations of this RyR2 mutation. This same mutation was also reported in another 9 years old girl who experienced syncopal episode. The ECG findings were not reported. | [33, 34] | ||
| SPRY1 | R739H | See findings of the L62F mutation. | [31] | |
| P1 | R1013Q, R1051P | See findings of the L62F mutation. | [31] | |
| SPRY2 | A1136V, T1107M, | See findings of the L62F mutation. | [31] | |
| Handle domain | E1724K | Independently of the localization of the RyR2 mutations, all CPVT patients presented some bradycardia and responded to the β-blockers (Nadolol, Propranolol and Metoprolol) treatment. These patients presented mono or polymorphic premature ventricular beats (MPVB/PPVB) that trigger bidirectional ventricular tachycardia and polymorphic ventricular tachycardia (PMVT) salvos. 9-year-old was the median age of symptoms onset. The proband carrying the RyR2-E1724K mutation presented monomorphic bigeminy (BG) and PMVT upon exercise stress test. | [35] | |
| E1837K, E2045G | See findings of the L62F mutation. | [31] | ||
| V1810L | A novel CPVT syndrome associated RyR2 mutation was identified during the screening of 35 Kazakhstani patients. This low-penetrance variant was found in a 42-year-old Korean proband. Initially, this patient was diagnosed with idiopathic arrhythmia characterized by unstable paroxysms of ventricular tachycardia. He presented bigeminy with a sinus rate of 83 bpm and reached 220 bpm during VT which was monomorphic. | [36] | ||
| Helical domain 1 | S2246L, R2474S | Priori’s group was the first who reported a direct relationship between RyR2 missense variants and CPVT syndrome. 4 missense mutations have been identified including 3 de novo. The RyR2-S2246L variant was identified in an 8-year-old boy who presented spontaneous onset of bidirectional VT upon isoproterenol infusion. Nadolol and ICD implantation proved effective for this proband. The RyR2-R2474S variant was also found in an 8-year-old boy who presented non-sustained bidirectional VT upon exercise stress test. He was treated with Atenolol. | [37, 38] | |
| P2328S | Missense RyR2 gene mutation was identified in CPVT patients, which could affect the myocardial calcium signaling. | [39] | ||
| R2311D, E2311D | The arrhythmogenic events occurred in young RyR2 mutations-affected patients, when compared to ungenotyped CPVT patients, with a higher risk of syncope for males. | [40] | ||
| V2306I, P2328S | Novel mutations were found to be associated with the CPVT syndrome in 12 Finnish probands. | [41] | ||
| A2387P | Novel RyR2 mutation was screened and identified using the DHPLC approach. | [42] | ||
| A2403T | See findings of the R414L mutation. | [23] | ||
| L2487I | RyR2 mutation was detected in 6% of unrelated genotype negative and atypical LQTS, that was considered as CPVT patients. | [43] | ||
| A2254V, A2394G | Independently of the localization of the RyR2 mutations, all CPVT patients presented some bradycardia and responded to the β-blockers treatment. 9-year-old was the median age of symptoms onset. The proband carrying the RyR2-A2254V mutation survived cardiac arrest (CA) and presented BG and polymorphic couplets (PC) upon exercise stress test. Whereas, the patient carrying the RyR2-A2394G mutation presented with seizures during the syncopal events and survived CA. Her exercise stress test revealed MPVB and PMVT. | [35] | ||
| V2475F | The molecular autopsy revealed novel mediated CPVT syndrome RyR2 mutations in 2 unexplained drowning cases. The boy had negative toxicology screen results and no sign for trauma and structural cardiovascular abnormalities. Direct DNA sequencing revealed the presence of this novel RyR2-V2475F variant. | [26] | ||
| R2359Q | Novel RyR2 mutations was identified in 2 CPVT families. The ECG performed for 3 patients from these families, revealed U-wave alterations | [44] | ||
| L2534V | A 13-year-old boy case study, with some novel RyR2 heterozygous mutation. An implantable recording loop was used to diagnose arrhythmogenic disorders. | [45] | ||
| R2404T | Some RyR2 novel heterozygous mutations were showed to be associated with a CPVT syndrome, in a family exhibiting some long QT-syndrome. | [46] | ||
| F2307L | Genetic screening for long QT and CPVT syndrome patients in Norway. | [47] | ||
| V2113M, Y2156C, H2168Q, E2183V, D2216V, E2296Q, F2307L, V2321M, R2404T, R2420W, M2389L | See findings of the L62F mutation. | [31] | ||
| H2217Y, C2402Y | See findings of the D242V mutation. | [32] | ||
| G2337V | The β-blockers treatment suppressed severe arrhythmias in stress-induced CPVT related RyR2 mutations, though it did not prevent the less severe ones. | [48] | ||
| L2527W | Determination of a novel RyR2 heterozygous mutation in a 9-year-old Chinese boy, misdiagnosed with epilepsy and CPVT syndrome. The β-blocker (Metoprolol) treatment proved unfavorable. | [49] | ||
| E2296K | This RyR2-E2296K mutation was identified in a 5-year-old Chinese boy with CPVT using the whole exome sequencing. This mutation might reduce the protein stability. However, further investigations are needed to prove its causality. | [50] | ||
| V2193L | The RyR2-V2193L mutation was identified in a 9-year-old Chinese boy who presented with both epilepsy and CPVT syndrome. The exercise stress test revealed frequent PPVB and PMVT with the presence of R on T. His electroencephalogram (EEG) showed frequent epileptiform discharges during stage II, stage III and REM sleep. He was successfully treated with Metoprolol and Levetiracetam. | [51] | ||
| C2277R | The RyR2-C2277R variant, located in the calstabin-binding domain, was identified in 8 members of the same family. The proband and her other family members presented ventricular extrasystoles (VE), bigeminy and/or trigeminy, doublets and non-sustained VT upon exercise stress test and adrenaline test. These patients showed similar response but different ventricular arrhythmias complexity degrees. The proband was treated with the combination of ICD implantation, Flecainide and Nadolol. The other family members were treated either with Atenolol, or Nadolol or with the combination of Nadolol and Flecainide or Atenolol and Flecainide which proved effective. | [52] | ||
| G3037D | Identification of a novel RyR2 heterozygous mutation in a 2 years old patient exhibiting some CPVT syndrome. | [53] | ||
| Helical domain 2 | N4104K | See findings of the mutation S2246L. The RyR2-N4104K variant was identified in a 14-year-old boy who presented non-sustained bidirectional VT upon exercise stress test. This proband was efficiently treated with Atenolol. | [37, 38] | |
| Central domain | Q4201R | Missense RyR2 gene mutation was identified in CPVT patients, which could affect the myocardial calcium signaling. | [39] | |
| L3778F, G3946S | See findings of the R2311D mutation. | [40, 54] | ||
| N4097S, E4146K, T4158P | In a postmortem genetic testing model, 3 novel mutations were identified in 7 cases of sudden unexplained death, that might potentially cause CPVT. | [55] | ||
| F4020L, E4076K, N4104I, H4108N, H4108Q | Independently of the localization of the RyR2 mutations, all CPVT patients presented some bradycardia and responded to the β-blockers treatment. 9-year-old was the median age of symptoms onset. The proband carrying the RyR2-F4020L mutation presented with seizures during the syncopal events. His exercise stress test revealed BG, PC, and PMVT. Unfortunately, he died suddenly at the age of 20. The proband carrying the RyR2-E4076K mutation presented BG and PMVT upon exercise stress test. The patient carrying the RyR2-N4104I mutation presented with seizures during the syncopal events. His exercise stress test revealed PPVB and sustained PMVT. The proband carrying the RyR2-H4108N mutation survived CA and presented BG, PC, and PMVT upon exercise stress test. Whereas, the patient carrying the RyR2-H4108Q mutation presented MPVB and PMVT upon exercise stress test. The symptoms of these patients reflect the complexity and the variability of the clinical phenotype of CPVT patients which allowed the assessment of a genotype-phenotype correlation. | [35] | ||
| S3938R, T4196A, | See findings of the V186M mutation. | [27] | ||
| L4105F | Novel mutation of the RyR2 mediated CPVT syndrome in 21 years old male. A β-blocker (Metoprolol) and calcium channel blocker (Verapamil) treatment, combined with the successful placement of a dual-chamber implantable cardioverter defibrillator proved effective. | [56] | ||
| R4144C | See findings of the F2307L mutation. | [47] | ||
| L3879P, Q3925E, G3946A, S3959L, M3972I, D3973H, L3974Q, K3997E, S4124G, Y4149s, R4157Q, Q4159P, N4178S, E4187Q | See findings of the L62F mutation. | [31] | ||
| S3799P, G3946D, D3977Y, A4091V, A4091T | See findings of the D242V mutation. | [32] | ||
| F4174L | A novel heterozygous mutation of the RYR2 gene associated CPVT syndrome was identified in a 17-year-old Caucasian boy. Interestingly, arrhythmias had occurred both at rest and under sympatho-adrenergic stimulation conditions. | [57] | ||
| A4282V, R4307C, G4315E | See findings of the L62F mutation. | [31] | ||
| Unspecified domain | K4392R | Case report of an athlete woman harboring some gain-of-function RyR2-K4392R mutation associated CPVT syndrome. | [58] | |
| R4497C | See findings of the mutation S2246L. The RyR2-R4497C variant was identified in a 30-year-old female who presented non-sustained bidirectional polymorphic VT upon exercise stress test. Two of her sisters died suddenly at the age of 14 and 16 respectively. Variable age-related manifestation of the disease has been thus suggested. This proband was treated with ICD implantation. | [37, 38] | ||
| Channel domain | V4653F | Missense RyR2 gene mutation was identified in CPVT patients, which could affect the myocardial calcium signaling. | [39] | |
| V4771I, A4860G, I4867M, N4895D, E4950K | See findings of the R2311D mutation. | [40] | ||
| P4902L, R4959Q | Three novel mutations were found to be associated with the CPVT syndrome in 12 Finnish probands. | [41] | ||
| N4504I, A4608P, V4880A, M4504I, A4607P | Four novel RyR2 mutations were screened and identified using the DHPLC approach. | [42] | ||
| F4499C, A4510T, G4671R, I4848V | See findings of the R414L mutation. | [23] | ||
| A4556T, 4657–4658EYinsertion, G4671R | RyR2 mutations were detected in 6% of unrelated genotype negative and atypical LQTS, that were considered as CPVT patients. | [43] | ||
| G4662S, H4762P, P4902S | Independently of the localization of the RyR2 mutations, all CPVT patients presented some bradycardia and responded to the β-blockers treatment. 9-year-old was the median age of symptoms onset. The probands carrying the RyR2-G4662S and RyR2-H4762P mutations presented BG and PMVT upon exercise stress test. The patient carrying the RyR2-P4902S presented PPVB and PMVT upon exercise stress test. | [35] | ||
| R4959Q | This mutation was identified in 11 patients of the same family. Four patients were diagnosed with bidirectional tachycardia. Five patients presented monomorphic ventricular tachycardia. Two patients died suddenly while asleep. | [59] | ||
| F4851C, N4895D | Two novel RyR2 mutations were identified in 2 CPVT families. The ECG performed for 3 patients from these families, revealed U-wave alterations | [44] | ||
| F4511L | See findings of the R2404T mutation. | [46] | ||
| E4431K, E4611K | Genetic screening for long QT and CPVT syndrome patients in Norwegia. | [47] | ||
| S4565R, E4611K, W4645R, K4650E, N4736 Del, R4790Q, K4805R, R4822H, G4936R | See findings of the L62F mutation. | [31] | ||
| F4851L | See findings of the D242V mutation. | [32] | ||
| G4671V | See findings of the G2337V mutation. | [48] | ||
| D4631V | A novel CPVT syndrome associated RyR2 mutation was identified during the screening of 35 Kazakhstani patients. This de-novo missense variant was identified in a 23-year-old female Kazakh. 13-year-old was the age of symptom onset. She experienced syncopal episodes and MPVB/PPVB that trigger bidirectional ventricular tachycardia and PMVT salvos. Since childhood this patient suffered from dizziness, frequent respiratory infections, scoliosis, palpitation, and chronic pyelonephritis along with the CPVT syndrome. She underwent ICD implantation together with the administration of β-blockers treatment. | [36] |
Thanks to the available 3D RyR1 and 2 structures, we now know that the majority of the RyR2 related CPVT mutations are located in domains involved in channel activation and gating including the pore, pseudo-voltage sensor and central domains (Fig. 1) [1, 34, 35]. A potential link between mutation localization and phenotype severity has been emphasized [36], in particular mutations in the C-terminal hot-spot domain being of greater risk to patients. Mutations in the bridging solenoid and the N-terminal domains, which form the RyR2 interprotomer contact domain, influence the cytoplasmic shell dynamics: they affect the outward shell movement that is necessary for the channel pore opening. An altered RyR2 channel gating mechanism via pore opening facilitation has been suggested for elevated frequency of spontaneous Ca2+ sparks under-stress [1]. Mutations in the C-terminal domain directly promote the activation of the channel either by facilitating the binding of Ca2+ and ATP or restraining the channel inactivation. It should be noted that FKBP12.6 binds to the bridging solenoid, stabilizing the interaction between the cytoplasmic region and the pore of the channel.
Fig. 1. Representative 3D structure of RyR2.

A Illustration of the 3D structure of RyR2 viewed from the cytoplasmic side and adapted from Peng et al. [35]. B Domain organization of the RyR2. NTD N-terminal domain, SPRY splA kinase and RyR domain, P1/P2 RyR repeat domain rich in phosphorylation sites, HD1/HD2 Helical domain.
The FKBP12.6 binding has not been always shown affected by CPVT-linked ryanopathies. However, ryanopathies trigger Ca2+ leak in any case [37, 38]. The so-called store-overload-induced calcium release (SOICR) hypothesis suggests that the CPVT-linked RyR2 mutations increase both, the sensitivity of the channel to luminal calcium and SOICR activity resulting in SR Ca2+ leak, DADs and arrhythmias, without affecting the affinity of the channel to FKBP12.6 [39]. Another group has demonstrated that the mitochondrial ROS induced by the Ca2+ leak exacerbate the RyR2 activity in a proarrhythmic feedback cycle in CPVT mouse model [40].
RyR2 homozygous multi-exon duplication was identified in 2 large Amish family with unexplained sudden deaths. Highly penetrant RYR2 homozygous duplication, exons 1 through 4 of RYR2 and RYR2’s 5′UTR/promoter region, was the main cause of death of these young subjects. This RYR2 homozygous duplication might result in a loss of function of the channel however the pathophysiological mechanism related to the SCD has not been elucidated so far [41].
Phenotypic relationship in CPVT individuals by classifying and correlating the RYR2 missense variants has been evaluated. The authors found that CPVT-associated RYR2 variants predominantly occur at more conserved amino-acid positions located between amino-acid positions 3949–4332 and 4867–4967. These variants were also located in RyR and IP3R homology-associated and ion transport domains. However, the RYR2 variants associated with sudden death during sleep were exclusively located in the C-terminal domain [42].
Finally, ryanopathies lead, in the majority of the cases, to gain of function (GOF) of the channel and in a lesser extent to a loss of function (LOF) (Fig. 2).
Fig. 2. Representative diagram illustrating the consequences of RyR2 gain-of-function or loss-of-function heterozygous mutations.

A During diastole, the PKA- or the CaMKII-phosphorylation of the RyR2 gain-of-function mutant channels induces a SR Ca2+ leak that increases the diastolic Ca2+ concentration and activates the NCX exchanger. This leads to ITi current which generates DADs and arrhythmias. B During AP, the ICaL activates the phosphorylated RyR2-WT channels that generate a normal calcium transient with a lower amplitude which in consequences, induces an accumulation of the residual Ca2+ in the SR which gradually increases. When the SR Ca2+ load achieves the threshold of stimulation of the phosphorylated RyR2 loss-of-function mutant channels, it releases Ca2+. This in turn activates the neighboring RyR2-WT enhancing thus the CICR mechanism. Hence, the Ca2+ release through the RyR2 loss-of-function mutant channels at the end of the Ca2+ transient induces a second phase of Ca2+ release. It activates the NCX exchanger leading to ITi current which generates EADs responsible of the pathogenesis of the CPVT syndrome.
Ryanopathies and short-coupled polymorphic ventricular tachycardia (SC-PMVT)
The Torsade de Pointes (TdP), also called ‘twisting peaks’, are cardiac rhythm disorders. These sometimes revert back into sinus rhythm or either degenerate into ventricular fibrillations (VF), syncope, and SCDs [10]. The ECG performed shows the typical TdP initiating-mode with the presence of a short-long-short sequence pattern [43, 44]. Short-coupled TdP (SC-TdP) and polymorphic ventricular tachycardia (PMVT) occur in patients with normal structural heart, in the absence of QT prolongation, myocardial ischemia and stress-induced arrhythmia exercise tests [45, 46]. They entail, in 8% of cases, the SCD of patients [10].
We first reported the existing link between a novel heterozygous mutation, RyR2-H29D, with a clinical phenotype of short-coupled PMVT at rest [10]. A RyR2-H29D mutation-harboring by mother and daughter, suffered a syncope and a short-coupled PMVT at rest. We used a heterologous recombinant system and compared the functional and molecular RyR2-H29D properties to control channels in presence of the FKBP12.6. RyR2-H29D leads to leaky RyR2 channel, at low diastolic Ca2+ levels, under non-stressful conditions and along with FKBP12.6 depletion. These results were challenged by Chen’s group by introducing the same variant into the RyR2 mouse sequence [47]. They could not evidence any changes in RyR2 open probability, [3H] ryanodine RyR2 binding, caffeine sensitivity and spontaneous Ca2+ release. The controversial conclusions of both studies are yet unexplained but might be related to some experimental conditions.
Other studies carried out identified novel RyR2 mutations entailing the occurrence of the SC-TdP. The RyR2-M995V variant, located in the cytoplasmic loop, outside the hot-spot region primary amino-acid sequence, was identified in a SC-TdP patient who experienced many syncopes [48]. This particular patient had a family history of sudden death and both her brother and her father carry the same RyR2-M995V variant. Despite the lack of functional characterization in this study, the co-segregation of the variant in the family suggests its involvement in the SC-TdP observed in the patient. Another study reported non-cited RyR2 variant, but its pathogenicity was studied in case of idiopathic VF case. The proband survived a SCD episode and recovered from a short-coupled extrasystole degenerating into TdP [49]. Moreover, the RYR2 gene sequencing in 7 SC-TdP patients revealed three novel heterozygous mutations (RyR2-S4938F, -V1024I, and -A2673V) and 1 polymorphism (RyR2-N1551S) that were all located inside the cytoplasmic domain. In this study, researchers found that the RyR2-S4938F exhibited a loss of function associated with higher ER Ca2+ signals, [3H] ryanodine binding reduction to the channel and Ca2+ release decrease. In contrast, the RyR2-N1551S, -V1024I, and -A2673V variants demonstrated some gain-of-function properties [9]. These results suggest that, similarly to CPVT, PMVT-related RyR2 mutations are associated with both some gain- and loss-of-function. Interestingly, a novel reported heterozygous mutation RyR2-F4174I in a 17 years old Caucasian patient caused polymorphic VT both at rest and under stress conditions. This mutation may cause arrhythmia independently of the sympatho-adrenergic stimulation via unclear pathophysiological mechanism [50].
Two LOF mutations were recently identified. The RyR2-E4146K and -G4935R were associated with Ca2+ release deficiency syndrome and IVF. Using recombinant expression in HEK cells, Zhong et al. found that the RyR2-G4935R suppresses completely the [3H] RyR2 binding and the RyR2 caffeine activation quasi-similar to the RyR2-E4146K. In addition, the cytosolic Ca2+ activation, SOICR activity and RyR2 sensitivity to luminal Ca2+ were abolished in RyR2-E4146K. To compare with CPVT RyR2 GOF mutations, RyR2-E4146K and -G4935R mutations induce cardiac arrhythmias under non-stressful conditions and are silent under exercise stress testing. Therefore, RyR2 LOF mutations are associated with a distinct phenotype than CPVT and constitute a new entity of cardiac arrythmias [51].
It is noteworthy that the overexpression of the DPP6 gene enhances the Ito current in Purkinje fibers, resulting in arrhythmias in IVF patients [52]. Other groups identified GOF mutations of KCNE5 which enhances the Ito current leading to AP shortening of Purkinje cells in IVF [53]. Moreover, loss of function mutations of the His-Purkinje system transcription factor IRX3 were identified to induce arrhythmias in IVF mouse model [54].
Ryanopathies and arrhythmogenic right ventricular cardiomyopathy (ARVC)
Arrhythmogenic right ventricular dysplasia is also known as the arrhythmogenic right ventricular cardiomyopathy (ARVD/ARVC). This is an inherited cardiac muscle disease characterized by the replacement of the CM by fatty or fibro-fatty tissues that further means its degeneration [55]. ARVD occurs most especially in the right ventricle and provokes some abnormal contraction and ventricle dilatation with posterior ventricular arrhythmias and SCD [56]. In fifty percent of cases, the ARVD is caused by some autosomal dominant mutation in gene encoding desmosomal proteins like desmoplakin (DSP), plakoglobin (JUP), plakophilin-2 (PKP2), desmoglein-2 (DSG2) and desmocollin-2 (DSC2) [57]. In fact, the desmosome plays a key role in maintaining the integrity of the cell membranes of the heart. Desmosomes are intercellular cell-to-cell junctions that provide a strong adhesion of the myocardium cells, enabling them to resist the mechanical stress induced during contraction itself [58].
Recessive mutations were also reported in other studies but were mainly associated with skin disorders. Other ARVD forms are often associated with mutations of the transforming beta-3 growth factor regulator (TGFβ3) inducing ARVD1 [59], transmembrane protein 43 (TMEM43) [60], desmin [61], lamin A/C [62], striatin [63], and titin [64].
The arrhythmogenic right ventricular cardiomyopathy type 2 (ARVC/D2) differs from other ARVD forms because it is a stress-induced disorder responsible for the occurrence of VT [65]. Interestingly, dominant mutations of the RyR2 were reported in the ARVD2 pathophysiological mechanism but in a far lesser extent [66]. In 2001, Tiso et al. identified four missense RyR2 mutations: RyR2-R176Q, -L433P, -N2386I, and -T2504M. These were reported to be in a critical domain for maintaining normal Ca2+ channel regulations and were associated with ARVD2 in Italians patients. RyR2-N2386I and -T2504M are located in the FKBP12.6 interaction domain. RyR2-R176Q and -L433P are situated in the cytosolic domain. Under β-adrenergic stimulation, these mutations induce decrease affinity of the FKBP12.6 for RyR2, some hypersensitivity of the RyR2 channel leading to a SR Ca2+ leak, that triggers, not only arrhythmias that are prevented with the use of β-blockers, but also some apoptosis and/or cell necrosis, which are responsible for the degenerative pathways observed in ARVD2 [67–69]. It is noteworthy that the RyR2-L433P mutation is the only one to induce a LOF resulting in a decrease of response to the caffeine activation of RyR2. The dantrolene treatment proved effective. One explanation given by the authors to this is that dantrolene restores the inter-subunit interaction of the RyR2 N-terminal domain and normal channel functions [70]. Other studies demonstrated the implication of RyR2 in ARVD2 pathogenesis, and highlighted other RyR2 mutations like RyR2-R420W, -Y2392C, and -A77V in Italian patients treated with β-blockers, that seemed to prevent the Ca2+ homeostasis in-balance [71]. Moreover, a German group identified 2 single nucleotide RyR2 polymorphisms, RyR2-G1885E and -G1886S in a cohort of 85 ARVC patients. When present in a trans configuration, they induced an abnormal calcium homeostasis and SR Ca2+ leak responsible for the arrhythmogenic disorders observed in these patients [72]. In a canine model (Boxer dogs) of ARVC2, lower RyR2 proteins and mRNA levels were reported in all heart chambers, with the lowest expression in the right ventricle. In the same model, other group demonstrated a depletion of the FKBP12.6 that led eventually to SR Ca2+ leak, which together could explain the pathophysiological mechanisms lying behind the ARVC2 syndrome [73]. When screening the RYR2 gene in a cohort of 64 ARVC/D patients without mutation in desmosomes genes, Roux-Buisson et al. identified 6 missense rare variants: RyR2- P1583S, -A2213S, -G2367R, -Y2932H, -V3219M, and -L4670V- that were associated with a conventional phenotype of ARVC/D [74]. Interestingly, it was reported that the RyR2-T1107M mutation is, not only associated with CPVT syndrome, but also with hypertrophic cardiomyopathy (HCM) in a patient who suffered severe left ventricular outflow tract obstruction and VF [75]. HEK293 cells expressing the corresponding murine mutant, RyR2-A1107M, exhibited diminished fractional Ca2+ release and increased luminal Ca2+ threshold release termination which could reflect the HCM pathophysiological mechanism [76].
An isoform-specific effect on RyR structure was revealed by evaluating the effect of the RyR2-R176Q mutant involved in CPVT and ARVD2, and its homologous the skeletal muscle RyR1-R163C, implicated in malignant hyperthermia (MH) and central core disease. Both mutations are located in the N-terminal domain. Despite the equivalent position in 3D structure and sequence of the positive charge loss, the RyR1-R163C mutant exhibited a preactivated altered conformation and displayed salt bridge network distortion, Ca2+ binding site alteration, rotation of a cytoplasmic domain induced by a molecular latch and partial progression to channel open state [77].
Ryanopathies as a multiple-organ dysfunction syndrome
Ryanopathies and neuronal disorders
The brain regionally expresses the three RyR isoforms. The RyR2 is predominant in the cerebral cortex and in dental gyrus of the hippocampus [78]. The role of RyR2 in neuronal function and behavior has been investigated in the last decade. It has been shown that nicotine induces RyR2 upregulation via activation of the transcription factor CREB in the cortex and ventral midbrain. This upregulation induces itself a phosphorylation of CREB leading to positive feedback signaling loop [79]. Downregulation of RyR2 induces spatial memory defects while the brain-derived neurotrophic factor induces RyR2 upregulation through ROS generation which is crucial for structural plasticity [80].
Neuronal consequences of ryanopathies have been reported; Lehnart et al. showed a link between CPVT syndrome and independent tonic-clonic seizures in the CPVT RyR2-R2474S transgenic murine model. This mutation induced a Ca2+ leak through the RyR2, that was responsible for both arrhythmia and epilepsy episodes under stress. Both disorders were prevented by stabilizing the RyR2 closed state conformation using the Rycal S107 compound [81]. A study also evaluated the RyR2 involvement in epilepsy for CPVT patients [82]. Another recent study showed the existing link between the RyR2-A77T mutation and a generalized epilepsy in a 32 years old woman presenting no anterior cardiac manifestations. However, her brother carrying the same mutation, was diagnosed with a CPVT syndrome and survived a cardiac arrest [83].
Furthermore, the role of RyR2 in cerebral ischemia pathogenesis was reported. Increased RyR2 S-glutathionylation proved to actually enhance the CICR, resulting in an amplification of the Ca2+ entry and leading to further cortical neuronal death [84]. RyR2 was also reported to be involved in memory impairment. In a mouse model of stress-induced cognitive dysfunction, neuronal RyR2 was shown to be remodeled by oxidation, S-nitrosylation, PKA-phosphorylation, and depletion of FKBP12.6 and leaky. Ca2+ fixing with the administration of Rycal compounds or prevention of PKA phosphorylation with genetic ablation of serine 2808, rescued the cognitive dysfunction [85]. Our group also attested the existing connection between neuronal RyR2 dysfunctions and neurodegenerative disorders, notably the occurrence of the Alzheimer disease (AD). In sporadic-AD-human-patient-brain samples and in two familial AD murine models, the “biochemical signature” of the leaky RyR2 with PKA-phosphorylation -FKBP12.6 depletion was associated with an ER Ca2+ leak, through increased RyR2 open probability and a loss of memory [86]. Preventing the RyR2 PKA-phosphorylation on the Ser2808 site by crossing AD mice with S2808A+/+ mice improved the cognitive behavior, while S2808D+/+ mice, which harbor constitutively some PKA-phosphorylated RyR2, exhibit early cognitive and synaptic dysfunctions. These results strongly support the role played by the RyR2 PKA phosphorylation and the β-adrenergic receptor signaling cascade in AD patterns. An interplay between the β-adrenergic signaling pathway, the amyloid β (Aβ) and the altered Ca2+ homeostasis through the leaky RyR2 channels in AD, was also observed [87, 88]. Altered Ca2+ signaling via the RyR2 channels in the pathogenesis of AD was highlighted in the literature by other groups too [89–91]. Recently, Chen’s group demonstrated a hyperactivity-directed strategy to counter the progression of AD despite the continuous β-amyloid accumulation. Genetically limiting the open time of the RyR2 through the RyR2-E4872Q mutation prohibits the learning and memory alteration, intrinsic membrane hyperexcitability, neuronal hyperactivity and neuronal cell death in AD mouse model. It enhances as well the A-type K+ current that drive neuronal excitability. These results were confirmed with the use of a pharmacological RyR2 open time limiting, the R-carvedilol enantiomer [92]. Moreover, loss of RyR2 induces dendritic spine structural plasticity impairment during memory acquisition. Dendritic sparsification, loss of excitatory synapses and over compensatory excitability were also observed. These alterations suggest a crucial role of RyR2 in neurodegenerative diseases [93].
These reports clearly demonstrated that similar ryanopathies can affect the heart and brain organs independently. However, these results also underlined the complex interpretation of such disorders, with the same family mutation triggering different effects: CPVT and/or epilepsy. As an 11 years old boy carrying the RyR2-R2401H mutation was long-term diagnosed with epilepsy instead of CPVT [94]. This misdiagnosis was common for CPVT syndrome [95]. Thereby, this ambivalence appears to be patient-specific and therefore, the use of hiPSC biotechnology constitutes a novel tool to model these neuro-cardiac disorders associated with ryanopathies.
Ryanopathies and metabolic syndromes
Diabetes mellitus is a chronic disease characterized by high blood sugar levels, called hyperglycemia. The glucose intolerance in these patients is due to: (i) abnormal secretion of insulin from the Langerhans cell islet located in the pancreas and induced by an autoimmune destruction of β-pancreatic cells that causes a lack of the insulin secretion usually called type I diabetes, (ii) resistance of target cells to the action of insulin inducing resistance, denoted as type II diabetes, or (iii) combination of both mechanisms [96, 97].
Insulin secretion is stimulated by glucose level elevation. Basically, glucose incorporating the β-pancreatic cells causes an increase in ATP production and induces the depolarization of the cellular membrane by inactivating the ATP-sensitive K+ channels. This is followed by the calcium entry through the Ca2+ voltage-dependent channels that further triggers a huge release of Ca2+ from the ER. In this mechanism, the precise role of the RyR2 in regulating the insulin secretion from the β-pancreatic cells is not fully understood. Dixit et al. demonstrated that the RyR2 GOF by constitutive CaMKII hyperphosphorylation at Ser2814 contributes to basal RyR2 ER leak, glucose intolerance, impaired glucose-stimulated insulin secretion, which constitute pre-diabetes features [98]. Is the RyR2 GOF causing glucose intolerance and promoting diabetes?
Santulli et al. brought an answer to that question by investigating the β-pancreatic cells of CPVT patients harboring RyR2 mutations. They found that CPVT RyR2 mutated patients as well as transgenic CPVT mice exhibited glucose intolerance and impaired glucose homeostasis. They also found that CPVT RyR2 channels in the pancreas were oxidized and S-nitrosylated, which activated an ER stress response, some mitochondrial dysfunction and decreased fuel-stimulated insulin release [99]. By preventing the ER Ca2+ leak pharmacologically, the closed conformation of RyR2 prevents these metabolic abnormalities in β-pancreatic cells.
Other groups have also investigated the role of RyR2 in insulin secretion patterns in diabetes mellitus [100, 101]. Our group demonstrated an altered Ca2+ homeostasis that included increased frequency of Ca2+ sparks, reduced amplitude of Ca2+ transients and depressed SR Ca2+ load in diabetic rats [102]. These modifications were associated with RyR2 PKA-hyperphosphorylation and a depletion of the FKBP12.6 protein. The possibility of differentiating hiPSC in insulin-producing ß-like cells [103] also opens interesting perspectives in this field of research.
Ryanopathies modeled through human stem cell technologies
The knowledge acquired in cardiac physiology and physiopathology has been significant thanks to the animal models used [104]. However, the animal models do not always recapitulate the phenotypes observed in patients. Investigating stress consequences in murine CMs, with electrical pacing is practically impossible, because 10 Hz pacing is too fast and leads to cell death. Actually, imposing some 1 or 2 Hz electrical pacing in mouse or rat CMs is so far away from the usual physiological rodent heart rate, that it does not account for such intended stress conditions. From a genomic point-of-view, the mouse response differs from the human one in several diseases investigated [105]. The use of complementary tools and models, beside that of the mouse, in the cardiovascular research field is therefore quite necessary.
The hiPSCs were first introduced in 2007, when Yamanaka’s group succeeded at reprogramming somatic cells- the human dermal fibroblasts-, into hiPSCs, by overexpressing four defined growth factors (Oct3/4, Sox2, c-Myc, and KLF4). These are now referred as Yamanaka’s reprogramming factors [106]. These generated hiPSCs actually exhibit the main properties of hESC, including their proliferation ability, morphology, pluripotency markers, telomerase activity, epigenetic and gene expressions. They show an incredible potential to differentiate into the three germ layers and also abolish all ethical issues arising with the use of hESC [17, 106]. After these results, several groups were able to reprogram other somatic cell types into hiPSCs, including peripheral blood cells, keratinocytes, dental pulp, and urine-derived cells [107–112].
With the hiPSCs, researchers use patient-specific cells for their potential for self-renewal; these can be theoretically differentiated into all somatic cell types. The generation of hiPSC-CMs is therefore of growing interest because of its multiple applications. It can be used, first of all, to elucidate important underlying cardiac disease-driving molecular mechanisms and test novel therapies [17, 113, 114]. Secondly, the access to an in vitro human development model enhances the study of human heart patterns that would not otherwise be possible. Thirdly, the stem cell derived CMs are used as a human cardiac model to research on diverse basic questions ranging from cellular electrophysiology to protein biochemistry. Furthermore, the ability to generate hiPSCs, from patients with inherited cardiac disease, provides unprecedented opportunities to study the disease in human CMs.
The hiPSC-CMs technique provides multiple advantages, including an unlimited supply of human CMs. Obtaining patient-specific hiPSC-CMs allows for a more personalized medicine and assessment of the genotype-phenotype association, by characterizing the effect of a single-point mutation in a patient, while conserving all its genetic defects and genetic background [17]. The cardiac differentiation of hiPSC into CMs is induced by applying in vitro a specific growth factor, to mimic the signaling pathway involved in the cardiomyogenesis during the early phases of embryogenesis. This leads to the formation of either pacemaker, ventricular or atrial-like CMs, depending on the application of various cytokines concentration combinations and on time-dependent schemes. The cardiac cell fate seems to be controlled in the hiPSC, as it is in embryos.
Albeit these great advantages, the hiPSC-CMs face many limitations. The most challenging one is that represented by the degree of maturity. The ventricular-like CMs differentiated from the hiPSC have a neonatal phenotype that includes some kind of metabolic, structural, functional, and electrophysiological immaturity. The lack of T-tubules and of a normal ultrastructure affect the distribution of the RyR2 and influence calcium homeostasis, which is slower in the hiPSC-CMs as compared to that of adult CMs [115].
Studies in the field develop efforts to improve this maturity. In an attempt to overcome these obstacles, several groups have engineered methods to further mature the hiPSC-CMs by using techniques which include some electrical and mechanical approaches. These are known as environmental manipulations, three-dimensional approaches and biochemical approaches [116].
The environmental manipulations consist of targeting the electrical and mechanical properties of the hiPSC-CMs. The electrical stimulation improves the calcium handling, increases the ultrastructure organization of the myofibrils and conduction velocity [117]. A specific substrate with grooves promotes the cell alignment, sarcomere orientation and bipolar localizations of the gap junctions to a level close to the in vivo arrangement [118]. Cyclic stretch in combination with electrical pacing lead to a more mature phenotype by increasing cell alignment, contractility, and cell size [119]. However, the mechanism by which the electrical and the mechanical stimulation induce hiPSC-CMs maturation is not fully understood. Interestingly, a recent study has subjected the early differentiated hiPSC-CMs (12 days old hiPSC-CMs, following immediately the first spontaneous cardiac contractions) to electromechanical conditioning. At 28 days of culture, the hiPSC-CMs exhibit adult-like phenotype including the cardiac gene expressions, the mitochondria density, the sarcomere length, the presence of the T-tubules, well organized ultrastructure, oxidative metabolism and positive force/frequency relationship [120, 121]. The combination of cell differentiation and electrical field conditioning results in atrial and ventricular tissues with specific drug responses and gene expression [122].
The three-dimensional (3D) approaches including 3D culture systems, known as organoids, improve the transcriptomic and the metabolic maturation and offer a more accurate model for disease modeling and drug testing by representing the in vivo morphology of the heart [123]. Engineered heart tissue displays greater maturation compared to 2D cultures, as shown by the expression of cardiac markers, better alignment of the cells and developed T-tubules [124]. Despite their great interest, the maintenance of a pure CM population and the provision for the adequate exposure to oxygen and nutrients in the 3D approaches, are challenging. In addition, the 3D approaches are subjected to many disadvantages. For example, disease modeling requires the use of single cells to characterize the diseased phenotype. These single cells are required due to their potential clinical use. Finally, the dose used for drug testing may not be accurate in the organoids since the cell numbers and the size of aggregates are not optimized yet.
Biochemical approaches involve the addition of growth factors, a change in culture medium and in hiPSC-CMs culture duration. For example, the Matrigel mattress improves the contractility in a comparable state of the adult rabbit ventricular CMs and increases both the expression of cardiac markers and sarcomere lengths [125]. Specific cell culture differentiation protocols, such as the differentiation in serum-albumin-free basal medium or simple serum with minimal signaling pathway factors, improve both the structural and physiological maturation properties. Their co-culture with mesenchymal stem cells, which secrete key soluble factors, enhances the differentiation and maturation by increasing the gap junctions, the energy production as well as the structural alignment of hiPSC-CMs [126]. Maturating by a medium enriched in T3 (triiodothyronine) hormone, which is crucial for normal cardiac development through the regulation of isoform-switching at the perinatal period, leads to the extension of the T-tubules network, increases the sarcomere length and the cell size, to further improve the contractility [127]. The combination of T3 and/or dexamethasone with the Matrigel mattress (extracellular matrix with physiological stiffness), induces the ventricle-like ECC characterized by T-tubules development, a uniform Ca2+ release, a promoted RyR2 structural organization and enhanced CICR mechanism [128]. The long-term culture (of up to 360 days) induces the formation of the sarcomeric M-band, a hallmark of structural sarcomeric maturation, yet, it remains as a quite unpractical method [129]. Some studies even applied a culture media supplied with lactose and deprived of glucose to reach a more mature metabolic activity towards the oxidative metabolism, which also improved the electrophysiological parameters and increased the sarcomere length [130]. Recently, we demonstrated that the low oxygen exposure during the first stage of 3D-cardiac differentiation is essential to enhance the contractile force and to promote a better Ca2+ handling properties [131]. These approaches improved the maturity of hiPSC-CMs. However, since they did not reach the adult-like state; and despite as described some methods for inducing more T-tubule maturation, most laboratories struggle and fail to obtain a physiological control of calcium physiology in hiPSC-CMs; the improvement of maturation techniques is still required.
Cardiac ryanopathies harbored by hiPSC-CMs
CPVT syndrome and hiPSC-CMs
Investigating the basic underlying pathological mechanisms leading to CPVT has faced difficulties and controversies due to the limited models available. Fatima et al. were the first to model the CPVT syndrome, using patient-specific hiPSC-CMs. In this work, they characterized a novel RyR2 heterozygous mutation, the p.F2483I-, associated with the CPVT syndrome. This mutation induced spontaneous Ca2+ events that led eventually to DAD and arrhythmias after catecholaminergic stimulations [132].
Other studies have used hiPSC-CMs to characterize novel heterozygous mutations associated with the CPVT syndrome. These are summarized in Table 2. The use of the hiPSC model as a tool for drug screening was emphasized in 2012 by Itzaki and coworkers. They found that the Thapsigargin treatment,—a specific inhibitor of the SERCA2A—, and that the Flecainide,—a sodium channel blocker—, were effective to abolish the abnormal release of Ca2+ in patient-specific CPVT hiPSC-CMs [133]. Another group reported, for the first time, some electrophysiological defects characterized by the occurrence of early and delayed afterdepolarizations,—a key marker of the pathogenesis of the CPVT syndrome—. An abnormal intracellular Ca2+ cycling, reduced SR Ca2+ load and SR Ca2+ leak was also reported in RyR2-P2328S hiPSC-CMs [134]. A group showed abnormal Ca2+ transients and DADs responsible for the arrhythmic phenotype in 3D beating CPVT hiPSC-CMs clusters, under both stress and resting conditions. The CaMKII inhibition rescued the cardiac function by reducing the DADs in CPVT hiPSC-CMs [135]. Using specific compounds and genome editing, the critical roles of CaMKII-dependent re-entry and CaMKII-dependent phosphorylation at Ser2814 in RyR2 were shown to be deleterious for the first time in the two human engineered tissue models of CPVT, harboring the RyR2-D358N and RyR2-R4651I mutations, respectively. Adrenergic stimulation and quick electrical stimulation, but not resting condition, caused re-entrant rhythms in the CPVT tissues [136]. We demonstrated that the RyR2-D3638A mutant channels cause, not only a SR Ca2+ leak and some impaired contractile properties under stress conditions, but also some post-translational modifications including the FKBP12.6 (FKBP12.6) depletion from the RyR2 macromolecular complex in patient-specific hiPSC-CMs [137]. This particular patient was resistant to the β-blocker Metoprolol treatment but not to Flecainide. The Metoprolol resistance observed in the proband was also revealed in the patient-specific hiPSC-CMs, which highlights the interest in hiPSC-CMs for channelopathy modeling and personalized medicine [137]. Table 2 summarizes the RyR2 mutations associated with CPVT1 syndrome that were investigated using hiPSC-CMs. The localization of the mutations on the RyR2 structural domains is displayed.
Table 2.
List of CPVT1 syndromes modeled using the hiPSC-CMs.
| Localization | Mutations | Findings | Ref. |
|---|---|---|---|
| D358N | CPVT tissues display re-entrant rhythms under stress that are prevented by CaMKII inhibition. | [60] | |
| N-terminal domain | S406L | The β-adrenergic stimulation by isoproterenol induced DADs and diastolic Ca2+ leak, that were reduced with the Dantrolene treatment. | [61] |
| E2311D/Q231D | Increased spontaneous calcium sparks and DADs, that were normalized by a CaMKII inhibition. | [62] | |
| R420Q | Non-ionotropic and lusitropic effects, increased arrhythmias and intracellular Ca2+ associated with immature ultrastructural features. | [63] | |
| ΔExon 3 | Dantrolene treatment reduced the premature ventricular complexes and the abnormal Ca2+ release in 4 CPVT patients and CPVT hiPSC-CMs. However, Dantrolene was not effective to treat patients carrying mutations in or near the transmembrane domain of the RyR2. | [64] | |
| Helical domain 1 | F2483I | The reduction of Ca2+ stores induced by a higher CICR mechanism led to an abnormal Ca2+ homeostasis. These abnormalities were verified in 2018 in gene-edited CPVT hiPSC-CMs generated by the CRISPR/Cas9 technology. | [65–67] |
| P2328S | The abnormal calcium homeostasis and the reduction of the SR Ca2+ load led to EADs and DADs at baseline and under isoproterenol stimulation. Another study found that the CPVT hiPSC-CMs exhibit increased non-alternating variability of Ca2+ transients and slow depolarization under isoproterenol stimulation. | [68, 69] | |
| P2328S, T2538R | See findings of the ΔExon 3 mutation. | [64] | |
| Y2476D | Arrhythmic events and impairment of the calcium handling and beating properties of CPVT hiPSC-CMs. These abnormalities were more pronounced under β-adrenergic stress. | [70] | |
| Central domain | M4109R | The β-adrenergic stimulation induces DADs and irregular Ca2+ transients that were abolished with the Flecainide and Thapsigargin treatments. | [71] |
| L4115F, Q4201R | See findings of the ΔExon 3 mutation. | [64] | |
| L3741P | The Flecainide treatment abolished the DADs and the spontaneous calcium sparks. | [72] | |
| D3638A | The RyR2 macromolecular complex remodeling, including FKBP12.6 depletion, SR Ca2+ leak and impaired contractile properties were observed in RyR2-D3638A hiPSC-CMs under stress conditions. Abnormal release of Ca2+ were prevented with the Flecainide and S107 treatments but not with the Metoprolol. | [73] | |
| R4651I | CPVT tissues display re-entrant rhythms under stress that are prevented by CaMKII inhibition. | [60] | |
| Channel domain | V4653F | See findings of the P2328S mutation. | [64] |
| I4587V | DADs and abnormal diastolic Ca2+ release were observed under β-adrenergic stress. The S107 treatment reduced the occurrence of DADs. | [74] | |
| R4959Q | See findings of the Y2476D mutation. | [70] |
In all these studies, the CPVT hiPSC-CMs were generated from patients harboring RyR2 mutations. Modeling of monogenic arrhythmias through hiPSC-CMs are a pertinent tool to provide insights for risk prediction, pathophysiological mechanisms and the discovery of potential new drugs.
Moreover, the hiPSC model was used to characterize a mutation in the TERCL gene leading to the CPVT syndrome. Higher diastolic Ca2+ concentration, lower SR Ca2+ load due to decreased SERCA and NCX activity, smaller Ca2+ transient amplitude and prolonged APs were observed in homozygous TERCL-hiPSC-CMs [138]. Prolonged APs were verified in TERCL knockdown hESC-CMs. However, the Flecainide treatment was effective to reduce the triggering activity of the TERCL-hiPSC-CMs.
The CRISPR/Cas9 technology was used to introduce 3 different CPVT1 mutations, the RyR2-R420Q, the RyR2-Q4201R and the RyR2-F2484I in hiPSC-CMs. These mutant hiPSC-CMs exhibited aberrant Ca2+ releases and irregular Ca2+ sparks. In contrast to RyR2-R420Q, the RyR2-Q4201R and the RyR2-F2484I hiPSC-CMs exhibited large SR Ca2+ leak and decreased SR Ca2+ content. These abnormalities were effectively supressed by the Rycal compound, the JTV519, which was more effective than Flecainide and dantrolene. These results suggested mutation site-specific effect on drug responsiveness and Ca2+ signaling abnormalities [139].
It should be noted that immature hiPSC-CMs that lack T-tubules are not a very appropriate model for CPVT and the use of these cells to model arrhythmogenic disorders is quite challenging. However, by using patient-derived CMs and the exciting CRISPR/Cas9 technology allowing genome editing, we are now able to generate isogenic CMs that differ only at the single RyR2 variant. The phenotypes for the mutant patient-derived hiPSC-CMs is perhaps the best evidence yet for a causal link for a RyR2 variant and inherited cardiac arrhythmias. Thus, such combined innovative biotechnologies definitely support the hiPSC for disease modeling.
SC-PMVT syndrome and hiPSC-CMs
The consequences of RyR2-associated mutation with the SC-PMVT at rest are unclear. Our group was the first who revealed the effect of RyR2-H29D mutation associated with PMVT syndrome, a CPVT-like phenotype, at rest using a human cardiac model [140]. The RyR2-H29D mutation causes a diastolic SR Ca2+ leak, abnormal mechanical and electrical properties such as AP shortening and DADs and post-translational modification of the RyR2 macromolecular complex including PKA-phosphorylation, S-nitrosylation, oxidation and depletion of FKBP12.6 under non stress conditions. These abnormalities were completely abolished when the RyR2-H29D mutation was reverted using CRISPR/Cas9 technology [140].
Conclusions
To sum up, the RyR2 obviously plays a major part in the pathogenesis of different diseases related to cardiac, neurodegenerative, and metabolic disorders. Calcium homeostasis regulated by the RyR2 is crucial for cell metabolism and it also helps signaling some physiological and pathophysiological conditions. Ryanopathies and RyR2 post-translational modifications lead to a SR Ca2+ leak that is responsible for various pathogenic patterns. Finding new tools to model human ryanopathies in the dish and provide for some patient-specific methods is therefore of obvious interest for researchers of the field (Fig. 3).
Fig. 3. Potential applications of patient-specific induced pluripotent stem cells for patients harboring ryanopathies.
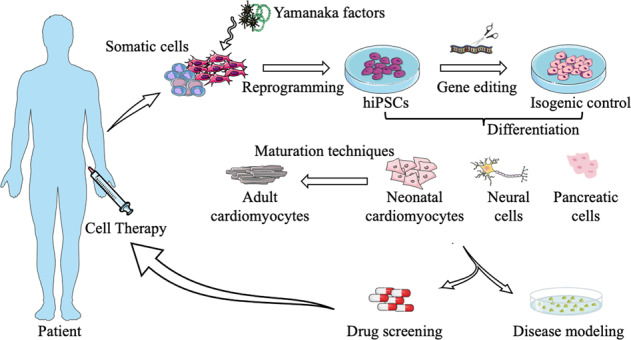
Recapitulative scheme illustrating the potential use of hiPSC generated from a patient blood sample or somatic cells and carrying RyR2 mutation. Some isogenic control hiPSC could be generated by correcting the single-RyR2-point mutation using CRISPR/Cas9 technology. The hiPSC could then be differentiated into cardiomyocytes, neural and pancreatic cells. These generated cells could be used for disease modeling and drug screening approaches for a potential patient-specific cell therapy.
Author contributions
YS and ACM wrote the review. AL and ACM supervised the work.
Funding
YS was supported by a post-doctoral grant of the Fonds Marion Elisabeth Brancher. This work was supported by grants of the French Muscular Dystrophy Association (AFM; project 16073, MNM2 2012 and 20225), the “Fondation Coeur et Recherche” and the “Institut National pour la Santé et la Recherche Médicale” (INSERM).
Data availability
Correspondence and requests for materials should be addressed to ACM.
Competing interests
The authors declare no competing interests.
Footnotes
Edited by Dr Daniele Bano
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
11/30/2022
A Correction to this paper has been published: 10.1038/s41419-022-05468-3
References
- 1.Santulli G, Lewis D, des Georges A, Marks AR, Frank J. Ryanodine receptor structure and function in health and disease. Subcell Biochem. 2018;87:329–52.. doi: 10.1007/978-981-10-7757-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–95. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lobos P, Córdova A, Vega-Vásquez I, Ramírez OA, Adasme T, Toledo J, et al. RyR-mediated Ca<sup>2 + </sup> release elicited by neuronal activity induces nuclear Ca<sup>2 + </sup> signals, CREB phosphorylation, and Npas4/RyR2 expression. Proc Natl Acad Sci. 2021;118:e2102265118. doi: 10.1073/pnas.2102265118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.CIR.103.2.196. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Yun H-M, Baik J-H, Chung KC, Nah S-Y, Rhim H. Functional Interaction of Neuronal Cav1.3 L-type Calcium Channel with Ryanodine Receptor Type 2 in the Rat Hippocampus*. J Biol Chem. 2007;282:32877–89. doi: 10.1074/jbc.M701418200. [DOI] [PubMed] [Google Scholar]
- 6.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90. doi: 10.1161/01.CIR.103.4.485. [DOI] [PubMed] [Google Scholar]
- 7.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–94. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 8.Meli AC, Refaat MM, Dura M, Reiken S, Wronska A, Wojciak J, et al. A novel ryanodine receptor mutation linked to sudden death increases sensitivity to cytosolic calcium. Circ Res. 2011;109:281–90. doi: 10.1161/CIRCRESAHA.111.244970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujii Y, Itoh H, Ohno S, Murayama T, Kurebayashi N, Aoki H, et al. A type 2 ryanodine receptor variant associated with reduced Ca(2+) release and short-coupled torsades de pointes ventricular arrhythmia. Heart Rhythm. 2017;14:98–107. doi: 10.1016/j.hrthm.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 10.Cheung JW, Meli AC, Xie W, Mittal S, Reiken S, Wronska A, et al. Short-coupled polymorphic ventricular tachycardia at rest linked to a novel ryanodine receptor (RyR2) mutation: leaky RyR2 channels under non-stress conditions. Int J Cardiol. 2015;180:228–36. doi: 10.1016/j.ijcard.2014.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandao KO, Tabel VA, Atsma DE, Mummery CL, Davis RP. Human pluripotent stem cell models of cardiac disease: from mechanisms to therapies. Dis Model Mech. 2017;10:1039–59.. doi: 10.1242/dmm.030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol Med. 2011;17:475–84. doi: 10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–8. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milani-Nejad N, Janssen PM. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharm Ther. 2014;141:235–49. doi: 10.1016/j.pharmthera.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards AG, Louch WE. Species-dependent mechanisms of cardiac arrhythmia: a cellular focus. Clin Med Insights Cardiol. 2017;11:1179546816686061. doi: 10.1177/1179546816686061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP. The need for speed: mice, men, and myocardial kinetic reserve. Circ Res. 2016;119:418–21. doi: 10.1161/CIRCRESAHA.116.309126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acimovic I, Vilotic A, Pesl M, Lacampagne A, Dvorak P, Rotrekl V, et al. Human pluripotent stem cell-derived cardiomyocytes as research and therapeutic tools. Biomed Res Int. 2014;2014:512831. doi: 10.1155/2014/512831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–9. doi: 10.1161/01.CIR.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 19.Leenhardt A, Maison-Blanche P, Denjoy I, Cauchemez B, Joubert JP, Coumel P. [Mechanism of spontaneous occurrence of tachycardia] Arch Mal Coeur Vaiss. 1999;1:17–22. [PubMed] [Google Scholar]
- 20.Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6:1652–9. doi: 10.1016/j.hrthm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 21.Napolitano C, Priori SG, Bloise R. Catecholaminergic Polymorphic Ventricular Tachycardia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)): Seattle (WA); 1993.
- 22.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA. 2003;100:11759–64. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhuiyan ZA, Hamdan MA, Shamsi ET, Postma AV, Mannens MM, Wilde AA, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18:1060–6. doi: 10.1111/j.1540-8167.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 24.Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–12. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–67. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cacheux M, Fauconnier J, Thireau J, Osseni A, Brocard J, Roux-Buisson N, et al. Interplay between Triadin and Calsequestrin in the Pathogenesis of CPVT in the Mouse. Mol Ther. 2019;28:171–9. [DOI] [PMC free article] [PubMed]
- 27.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 28.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci USA. 2004;101:9137–42. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swan H, Amarouch MY, Leinonen J, Marjamaa A, Kucera JP, Laitinen-Forsblom PJ, et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet. 2014;7:771–81. doi: 10.1161/CIRCGENETICS.114.000703. [DOI] [PubMed] [Google Scholar]
- 30.Kalscheur MM, Vaidyanathan R, Orland KM, Abozeid S, Fabry N, Maginot KR, et al. KCNJ2 mutation causes an adrenergic-dependent rectification abnormality with calcium sensitivity and ventricular arrhythmia. Heart Rhythm. 2014;11:885–94. doi: 10.1016/j.hrthm.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53. doi: 10.1161/CIRCGENETICS.111.962316. [DOI] [PubMed] [Google Scholar]
- 32.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511–9. doi: 10.1016/S0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 33.Sumitomo N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J Arrhythm. 2016;32:344–51.. doi: 10.1016/j.joa.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez A, Caputo C. Ryanodine interferes with charge movement repriming in amphibian skeletal muscle fibers. Biophysical J. 1996;70:376–82. doi: 10.1016/S0006-3495(96)79581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354:aah5324 [pii]. [DOI] [PubMed]
- 36.van der Werf C, Nederend I, Hofman N, van Geloven N, Ebink C, Frohn-Mulder IM, et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol. 2012;5:748–56. doi: 10.1161/CIRCEP.112.970517. [DOI] [PubMed] [Google Scholar]
- 37.Zissimopoulos S, Thomas NL, Jamaluddin WW, Lai FA. FKBP12.6 binding of ryanodine receptors carrying mutations associated with arrhythmogenic cardiac disease. Biochem J. 2009;419:273–8. doi: 10.1042/BJ20082324. [DOI] [PubMed] [Google Scholar]
- 38.Salvage SC, Gallant EM, Beard NA, Ahmad S, Valli H, Fraser JA, et al. Ion channel gating in cardiac ryanodine receptors from the arrhythmic RyR2-P2328S mouse. J Cell Sci. 2019;132:jcs229039. [DOI] [PMC free article] [PubMed]
- 39.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–81. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. 2020;115:38. doi: 10.1007/s00395-020-0797-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tester DJ, Bombei HM, Fitzgerald KK, Giudicessi JR, Pitel BA, Thorland EC, et al. Identification of a Novel Homozygous Multi-Exon Duplication in RYR2 Among Children With Exertion-Related Unexplained Sudden Deaths in the Amish Community. JAMA Cardiol. 2020;5:340–5. doi: 10.1001/jamacardio.2019.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olubando D, Hopton C, Eden J, Caswell R, Lowri Thomas N, Roberts SA, et al. Classification and correlation of RYR2 missense variants in individuals with catecholaminergic polymorphic ventricular tachycardia reveals phenotypic relationships. J Hum Genet. 2020;65:531–9. doi: 10.1038/s10038-020-0738-6. [DOI] [PubMed] [Google Scholar]
- 43.Kay GN, Plumb VJ, Arciniegas JG, Henthorn RW, Waldo AL. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J Am Coll Cardiol. 1983;2:806–17. doi: 10.1016/S0735-1097(83)80226-5. [DOI] [PubMed] [Google Scholar]
- 44.Noda T, Shimizu W, Satomi K, Suyama K, Kurita T, Aihara N, et al. Classification and mechanism of Torsade de Pointes initiation in patients with congenital long QT syndrome. Eur Heart J. 2004;25:2149–54. doi: 10.1016/j.ehj.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 45.Leenhardt A, Glaser E, Burguera M, Nurnberg M, Maison-Blanche P, Coumel P. Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation. 1994;89:206–15. doi: 10.1161/01.CIR.89.1.206. [DOI] [PubMed] [Google Scholar]
- 46.Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63:1300–8. doi: 10.1016/j.jacc.2013.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao Z, Guo W, Yuen SM, Wang R, Zhang L, Van Petegem F, et al. The H29D mutation does not enhance cytosolic Ca2+ activation of the cardiac ryanodine receptor. PLoS ONE. 2015;10:e0139058. doi: 10.1371/journal.pone.0139058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimura M, Fujisawa T, Aizawa Y, Matsuhashi N, Ito S, Nakajima K, et al. An RyR2 mutation found in a family with a short-coupled variant of torsade de pointes. Int J Cardiol. 2017;227:367–9. doi: 10.1016/j.ijcard.2016.11.052. [DOI] [PubMed] [Google Scholar]
- 49.Beach SR, Celano CM, Sugrue AM, Adams C, Ackerman MJ, Noseworthy PA, et al. QT Prolongation, Torsades de Pointes, and Psychotropic Medications: A 5-Year Update. Psychosomatics. 2018;59:105–22.. doi: 10.1016/j.psym.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Seidlmayer LK, Riediger F, Pagonas N, Nordbeck P, Ritter O, Sasko B. Description of a novel RyR2 mutation in a juvenile patient with symptomatic catecholaminergic polymorphic ventricular tachycardia in sleep and during exercise: a case report. J Med Case Rep. 2018;12:298. doi: 10.1186/s13256-018-1825-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong X, Guo W, Wei J, Tang Y, Liu Y, Zhang JZ, et al. Identification of loss-of-function RyR2 mutations associated with idiopathic ventricular fibrillation and sudden death. Biosci Rep. 2021;41:4. doi: 10.1042/BSR20210209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MW, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468–76.. doi: 10.1016/j.ajhg.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohno S, Zankov DP, Ding WG, Itoh H, Makiyama T, Doi T, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol. 2011;4:352–61.. doi: 10.1161/CIRCEP.110.959619. [DOI] [PubMed] [Google Scholar]
- 54.Koizumi A, Sasano T, Kimura W, Miyamoto Y, Aiba T, Ishikawa T, et al. Genetic defects in a His-Purkinje system transcription factor, IRX3, cause lethal cardiac arrhythmias. Eur Heart J. 2016;37:1469–75. doi: 10.1093/eurheartj/ehv449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–91.. doi: 10.1161/01.CIR.94.5.983. [DOI] [PubMed] [Google Scholar]
- 56.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–53. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 57.Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pr Cardiovasc Med. 2008;5:258–67. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–87. doi: 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 59.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–73. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 60.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–21. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19:4595–607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- 62.Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–36. doi: 10.1093/eurheartj/ehr451. [DOI] [PubMed] [Google Scholar]
- 63.Meurs KM, Mauceli E, Lahmers S, Acland GM, White SN, Lindblad-Toh K. Genome-wide association identifies a deletion in the 3’ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet. 2010;128:315–24. doi: 10.1007/s00439-010-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876–85. doi: 10.1161/CIRCULATIONAHA.110.005405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nava A, Martini B, Thiene G, Buja GF, Canciani B, Scognamiglio R, et al. [Arrhythmogenic right ventricular dysplasia. Study of a selected population]. G Ital Cardiol. 1988;18:2–9. [PubMed]
- 66.Rampazzo A, Nava A, Erne P, Eberhard M, Vian E, Slomp P, et al. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet. 1995;4:2151–4. doi: 10.1093/hmg/4.11.2151. [DOI] [PubMed] [Google Scholar]
- 67.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–94. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 68.Tiso N, Salamon M, Bagattin A, Danieli GA, Argenton F, Bortolussi M. The binding of the RyR2 calcium channel to its gating protein FKBP12.6 is oppositely affected by ARVD2 and VTSIP mutations. Biochem Biophys Res Commun. 2002;299:594–8. doi: 10.1016/S0006-291X(02)02689-X. [DOI] [PubMed] [Google Scholar]
- 69.Thomas NL, George CH, Lai FA. Functional heterogeneity of ryanodine receptor mutations associated with sudden cardiac death. Cardiovasc Res. 2004;64:52–60. doi: 10.1016/j.cardiores.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 70.Seidel M, Thomas NL, Williams AJ, Lai FA, Zissimopoulos S. Dantrolene rescues aberrant N-terminus intersubunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc Res. 2015;105:118–28. doi: 10.1093/cvr/cvu240. [DOI] [PubMed] [Google Scholar]
- 71.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, et al. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol. 2002;40:341–9. doi: 10.1016/S0735-1097(02)01946-0. [DOI] [PubMed] [Google Scholar]
- 72.Milting H, Lukas N, Klauke B, Korfer R, Perrot A, Osterziel KJ, et al. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Res. 2006;71:496–505. doi: 10.1016/j.cardiores.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 73.Meurs KM, Lacombe VA, Dryburgh K, Fox PR, Reiser PR, Kittleson MD. Differential expression of the cardiac ryanodine receptor in normal and arrhythmogenic right ventricular cardiomyopathy canine hearts. Hum Genet. 2006;120:111–8. doi: 10.1007/s00439-006-0193-2. [DOI] [PubMed] [Google Scholar]
- 74.Roux-Buisson N, Gandjbakhch E, Donal E, Probst V, Deharo JC, Chevalier P, et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: results of a systematic screening. Heart Rhythm. 2014;11:1999–2009. doi: 10.1016/j.hrthm.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 75.Fujino N, Ino H, Hayashi K, Uchiyama K, Nagata M, Konno T, et al. Abstract 915: A Novel Missense Mutation in Cardiac Ryanodine Receptor Gene as a Possible Cause of Hypertrophic Cardiomyopathy: Evidence From Familial Analysis. Circulation. 2006;114:II_165-II_.
- 76.Tang Y, Tian X, Wang R, Fill M, Chen SR. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res. 2012;110:968–77. doi: 10.1161/CIRCRESAHA.111.256560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iyer KA, Hu Y, Nayak AR, Kurebayashi N, Murayama T, Samsó M. Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM. Sci Adv. 2020;6:eabb2964. doi: 10.1126/sciadv.abb2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ziviani E, Lippi G, Bano D, Munarriz E, Guiducci S, Zoli M, et al. Ryanodine receptor-2 upregulation and nicotine-mediated plasticity. EMBO J. 2011;30:194–204. doi: 10.1038/emboj.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.More JY, Bruna BA, Lobos PE, Galaz JL, Figueroa PL, Namias S, et al. Calcium release mediated by redox-sensitive RyR2 channels has a central role in hippocampal structural plasticity and spatial memory. Antioxid Redox Signal. 2018;29:1125–46.. doi: 10.1089/ars.2017.7277. [DOI] [PubMed] [Google Scholar]
- 81.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Investig. 2008;118:2230–45.. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nagrani T, Siyamwala M, Vahid G, Bekheit S. Ryanodine calcium channel: a novel channelopathy for seizures. Neurologist. 2011;17:91–4. doi: 10.1097/NRL.0b013e3181ef0d52. [DOI] [PubMed] [Google Scholar]
- 83.Yap SM, Smyth S. Ryanodine receptor 2 (RYR2) mutation: A potentially novel neurocardiac calcium channelopathy manifesting as primary generalised epilepsy. Seizure. 2019;67:11–4. doi: 10.1016/j.seizure.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 84.Bull R, Finkelstein JP, Galvez J, Sanchez G, Donoso P, Behrens MI, et al. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J Neurosci. 2008;28:9463–72. doi: 10.1523/JNEUROSCI.2286-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu X, Betzenhauser MJ, Reiken S, Meli AC, Xie W, Chen BX, et al. Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell. 2012;150:1055–67. doi: 10.1016/j.cell.2012.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017;134:749–67.. doi: 10.1007/s00401-017-1733-7. [DOI] [PubMed] [Google Scholar]
- 87.Bussiere R, Lacampagne A, Reiken S, Liu X, Scheuerman V, Zalk R, et al. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J Biol Chem. 2017;292:10153–68.. doi: 10.1074/jbc.M116.743070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meli AC. The impact of cardiovascular diseases and new gene variants in swaying Alzheimer’s disease. Cardiovasc Res. 2019;115:e102–e4.. doi: 10.1093/cvr/cvz196. [DOI] [PubMed] [Google Scholar]
- 89.Oules B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820–34. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–70. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yao J, Sun B, Institoris A, Zhan X, Guo W, Song Z, et al. Limiting RyR2 Open Time Prevents Alzheimer’s Disease-Related Neuronal Hyperactivity and Memory Loss but Not β-Amyloid Accumulation. Cell Rep. 2020;32:108169. [DOI] [PMC free article] [PubMed]
- 93.Bertan F, Wischhof L, Sosulina L, Mittag M, Dalügge D, Fornarelli A, et al. Loss of Ryanodine Receptor 2 impairs neuronal activity-dependent remodeling of dendritic spines and triggers compensatory neuronal hyperexcitability. Cell Death Differ. 2020;27:3354–73. doi: 10.1038/s41418-020-0584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu X, Li J, Shen Y, Hu J, He W, Zhou Q, et al. GW26-e4458 A long-term misdiagnosis of catecholaminergic polymorphic ventricular tachycardia as epilepsy caused by RyR2 mutation. J Am Coll Cardiol. 2015;66:C179. [Google Scholar]
- 95.Duan H, Lu Y, Yan S, Qiao L, Hua Y, Li Y, et al. A delayed diagnosis of catecholaminergic polymorphic ventricular tachycardia with a mutant of RYR2 at c.7580T>G for 6 years in a 9-year-old child. Medicine. 2018;97:e0368. doi: 10.1097/MD.0000000000010368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.You WP, Henneberg M. Type 1 diabetes prevalence increasing globally and regionally: the role of natural selection and life expectancy at birth. BMJ Open Diabetes Res Care. 2016;4:e000161. doi: 10.1136/bmjdrc-2015-000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Holman N, Young B, Gadsby R. Current prevalence of Type 1 and Type 2 diabetes in adults and children in the UK. Diabet Med. 2015;32:1119–20. doi: 10.1111/dme.12791. [DOI] [PubMed] [Google Scholar]
- 98.Dixit SS, Wang T, Manzano EJ, Yoo S, Lee J, Chiang DY, et al. Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PLoS One. 2013;8:e58655. doi: 10.1371/journal.pone.0058655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Investig. 2015;125:4316. doi: 10.1172/JCI84937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamamoto WR, Bone RN, Sohn P, Syed F, Reissaus CA, Mosley AL, et al. Endoplasmic reticulum stress alters ryanodine receptor function in the murine pancreatic beta cell. J Biol Chem. 2019;294:168–81. doi: 10.1074/jbc.RA118.005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luciani DS, Gwiazda KS, Yang T-LB, Kalynyak TB, Bychkivska Y, Frey MHZ, et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009;58:422–32. doi: 10.2337/db07-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yaras N, Ugur M, Ozdemir S, Gurdal H, Purali N, Lacampagne A, et al. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–8. doi: 10.2337/diabetes.54.11.3082. [DOI] [PubMed] [Google Scholar]
- 103.Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–72. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tamargo J, Caballero R, Nunez L, Gomez R, Vaquero M, Delpon E. Genetically engineered mice as a model for studying cardiac arrhythmias. Front Biosci. 2007;12:22–38. doi: 10.2741/2046. [DOI] [PubMed] [Google Scholar]
- 105.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 107.Xue Y, Cai X, Wang L, Liao B, Zhang H, Shan Y, et al. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells. PLoS One. 2013;8:e70573. doi: 10.1371/journal.pone.0070573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ohmine S, Squillace KA, Hartjes KA, Deeds MC, Armstrong AS, Thatava T, et al. Reprogrammed keratinocytes from elderly type 2 diabetes patients suppress senescence genes to acquire induced pluripotency. Aging. 2012;4:60–73. doi: 10.18632/aging.100428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Streckfuss-Bomeke K, Wolf F, Azizian A, Stauske M, Tiburcy M, Wagner S, et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur Heart J. 2013;34:2618–29. doi: 10.1093/eurheartj/ehs203. [DOI] [PubMed] [Google Scholar]
- 110.Aasen T, Izpisua Belmonte JC. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat Protoc. 2010;5:371–82. doi: 10.1038/nprot.2009.241. [DOI] [PubMed] [Google Scholar]
- 111.Nishizawa M, Chonabayashi K, Nomura M, Tanaka A, Nakamura M, Inagaki A, et al. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell. 2016;19:341–54. doi: 10.1016/j.stem.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 112.Pesl M, Acimovic I, Pribyl J, Hezova R, Vilotic A, Fauconnier J, et al. Forced aggregation and defined factors allow highly uniform-sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessels. 2013;29:834–46. [DOI] [PubMed]
- 113.Polonen RP, Swan H, Aalto-Setala K. Mutation-specific differences in arrhythmias and drug responses in CPVT patients: simultaneous patch clamp and video imaging of iPSC derived cardiomyocytes. Mol Biol Rep. 2019;47:1067–77. [DOI] [PubMed]
- 114.Maizels L, Huber I, Arbel G, Tijsen AJ, Gepstein A, Khoury A, et al. Patient-Specific Drug Screening Using a Human Induced Pluripotent Stem Cell Model of Catecholaminergic Polymorphic Ventricular Tachycardia Type 2. Circ Arrhythm Electrophysiol. 2017;10:e004725. [DOI] [PubMed]
- 115.Koivumaki JT, Naumenko N, Tuomainen T, Takalo J, Oksanen M, Puttonen KA, et al. Structural immaturity of human iPSC-derived cardiomyocytes: in silico investigation of effects on function and disease modeling. Front Physiol. 2018;9:80. doi: 10.3389/fphys.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Machiraju P, Greenway SC. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J stem cells. 2019;11:33–43. doi: 10.4252/wjsc.v11.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods. 2013;10:781–7. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim DH, Lipke EA, Kim P, Cheong R, Thompson S, Delannoy M, et al. Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc Natl Acad Sci USA. 2010;107:565–70. doi: 10.1073/pnas.0906504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ruan J-L, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, et al. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation. 2016;134:1557–67. doi: 10.1161/CIRCULATIONAHA.114.014998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ronaldson-Bouchard K, Yeager K, Teles D, Chen T, Ma S, Song L, et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nat Protoc. 2019;14:2781–817. doi: 10.1038/s41596-019-0189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–43. doi: 10.1038/s41586-018-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao Y, Rafatian N, Feric NT, Cox BJ, Aschar-Sobbi R, Wang EY, et al. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell. 2019;176:913–27 e18. doi: 10.1016/j.cell.2018.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feric NT, Radisic M. Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv Drug Deliv Rev. 2016;96:110–34. doi: 10.1016/j.addr.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stoehr A, Neuber C, Baldauf C, Vollert I, Friedrich FW, Flenner F, et al. Automated analysis of contractile force and Ca2+ transients in engineered heart tissue. Am J Physiol Heart Circulatory Physiol. 2014;306:H1353–H63. doi: 10.1152/ajpheart.00705.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, et al. Matrigel mattress: a method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2015;117:995–1000. doi: 10.1161/CIRCRESAHA.115.307580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yoshida S, Miyagawa S, Fukushima S, Kawamura T, Kashiyama N, Ohashi F, et al. Maturation of human induced pluripotent stem cell-derived cardiomyocytes by soluble factors from human mesenchymal stem cells. Mol Ther. 2018;26:2681–95. doi: 10.1016/j.ymthe.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ribeiro MC, Tertoolen LG, Guadix JA, Bellin M, Kosmidis G, D’Aniello C, et al. Functional maturation of human pluripotent stem cell derived cardiomyocytes in vitro-correlation between contraction force and electrophysiology. Biomaterials. 2015;51:138–50. doi: 10.1016/j.biomaterials.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 128.Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, et al. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2017;121:1323–30. doi: 10.1161/CIRCRESAHA.117.311920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J. 2013;77:1307–14. doi: 10.1253/circj.CJ-12-0987. [DOI] [PubMed] [Google Scholar]
- 130.Bhute VJ, Bao X, Dunn KK, Knutson KR, McCurry EC, Jin G, et al. Metabolomics identifies metabolic markers of maturation in human pluripotent stem cell-derived cardiomyocytes. Theranostics. 2017;7:2078–91. doi: 10.7150/thno.19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Souidi M, Sleiman Y, Acimovic I, Pribyl J, Charrabi A, Baecker V, et al. Oxygen Is an Ambivalent Factor for the Differentiation of Human Pluripotent Stem Cells in Cardiac 2D Monolayer and 3D Cardiac Spheroids. Int J Mol Sci. 2021;22:662. [DOI] [PMC free article] [PubMed]
- 132.Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnaiz-Cot JJ, et al. In vitro Modeling of Ryanodine Receptor 2 Dysfunction Using Human Induced Pluripotent Stem Cells. Cell Physiol Biochem. 2011;28:579–92. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000. doi: 10.1016/j.jacc.2012.02.066. [DOI] [PubMed] [Google Scholar]
- 134.Kujala K, Paavola J, Lahti A, Larsson K, Pekkanen-Mattila M, Viitasalo M, et al. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS One. 2012;7:e44660. doi: 10.1371/journal.pone.0044660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino-Cruz JE, Buonocore M, et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843. doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Park S-J, Zhang D, Qi Y, Li Y, Lee Keel Y, Bezzerides, Vassilios J, et al. Insights into the pathogenesis of catecholaminergic polymorphic ventricular tachycardia from engineered human heart tissue. Circulation. 2019;140:390–404. doi: 10.1161/CIRCULATIONAHA.119.039711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Acimovic I, Refaat MM, Moreau A, Salykin A, Reiken S, Sleiman Y, et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. J Clin Med. 2018;7:423. [DOI] [PMC free article] [PubMed]
- 138.Devalla HD, Gelinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016;8:1390–408. doi: 10.15252/emmm.201505719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang XH, Wei H, Xia Y, Morad M. Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R. Heart Rhythm. 2021;18:250–60. doi: 10.1016/j.hrthm.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sleiman Y, Souid M, Kumar R, Yang E, Jaffré F, Tiing Z, et al. Modeling polymorphic ventricular tachycardia at rest using patient-specific induced pluripotent stem cell-derived cardiomyocytes. EBioMedicine. 2020;60:103024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Correspondence and requests for materials should be addressed to ACM.