

Abstract
Natural killer (NK) cells are innate lymphocytes that recognize and clear infected and transformed cells. The importance of NK cells in tumor surveillance underlies the development of NK cell therapy as cancer treatment. The NK-92 cell line has been successfully modified to express high-affinity CD16 receptor for antibody-dependent cellular cytotoxicity and/or chimeric antigen receptors (CARs) that can recognize antigens expressed on tumor cells and mediate NK cell activation. Since there is no need for human leukocyte antigen matching or prior exposure to the tumor antigens, NK-92 provides an opportunity for the development of next-generation off-the-shelf cell therapy platforms. CAR-engineered NK-92 cells have demonstrated robust antitumor activity in in vitro and in vivo preclinical studies, propelling the clinical development of CAR NK-92 cells. Preliminary phase 1 data indicate that CAR NK-92 can be safely administered in the clinic. In this review, we provide an overview of recent advances in the research and clinical application of this novel cell immunotherapy.
Keywords: natural killer cells, adoptive cell therapy, NK-92, high-affinity NK, haNK, CAR NK, targeted high-affinity NK, t-haNK
Graphical abstract
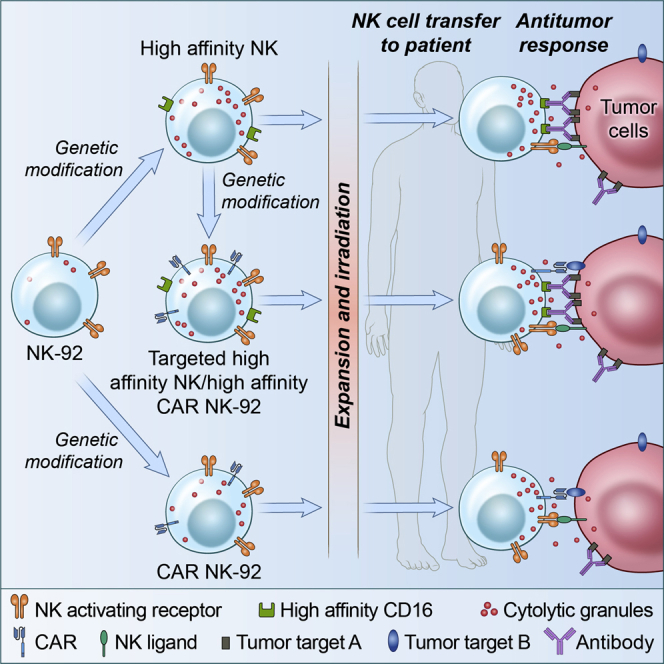
NK-92 cells engineered to express high-affinity CD16 and/or chimeric antigen receptors are potential allogeneic off-the-shelf cell therapy products that show a favorable safety profile based on preliminary results of phase 1 studies.
Introduction
Natural killer (NK) cells are innate effector lymphocytes that play an important role in clearing infected and transformed cells. In humans, NK cells are typically defined as CD3−CD56+ cells and represent 5%–20% of the circulating lymphocytes. Human NK cells can be subclassified into immature NK cells (CD56brightCD16dim), which are potent inflammatory cytokine producers that play a role in immunomodulation, and mature NK cells (CD56dimCD16bright), which mediate the cytolytic function of the human NK cells. In mice, NK cells are phenotypically CD3− cells that express NK1.1, NCR1, and/or CD49b, depending on mouse background.1
In contrast to T cells and B cells, NK cell recognition is not dependent on antigen specificity and does not require prior sensitization. Instead, the activity of NK cells is controlled by the integration of signals from various NK cell surface inhibitory and activating receptors. Most of the inhibitory receptors bind major histocompatibility class (MHC) class I molecules and can detect missing self-markers on target cells that downregulated MHC class I to evade T cells. In humans, examples of inhibitory NK receptors include killer cell immunoglobulin-like receptors (KIRs), leukocyte immunoglobulin-like receptors (LILRs), and CD94-NKG2A receptors. Non-human leukocyte antigen (HLA)-specific inhibitory receptors such as PD-1, TIGIT, and CD96 can also be upregulated on the NK cell surface in pathological conditions to limit exacerbated immune responses during viral infection; conversely, they may enable tumor escape. Activating receptors discriminate between healthy cells and abnormal cells by detecting pathogen or cell stress-induced cell ligands. Examples of NK cell activating receptors are Fc gamma receptor (FcγR)IIA (also known as CD16), NKG2D, and members of the natural cytotoxicity receptors (NCRs) such as NKp46, NKp30, and NKp44. Except for CD16, activating receptors must be simultaneously crosslinked in pairs or combinations to elicit synergistic activation signals for NK cell function.2, 3, 4
NK cells can directly kill infected and transformed cells through several processes. One NK cytotoxic response involves the targeted release of cytolytic granules that contain perforin and granzymes. NK cells also mediate killing of target cells via engagement of death receptors. Death ligands on NK cells, such as Fas ligand (FasL) and TRAIL, bind to their cognate receptors on target cells and result in apoptosis of target cells.1,5 NK cells are also able to induce antibody-dependent cell-mediated cytotoxicity (ADCC) through the CD16 receptors that detect target cells that are opsonized with immunoglobulin (Ig)G antibodies. The ADCC function of NK cells is considered to contribute to the clinical activity of cancer therapeutic antibodies.6,7 In addition to direct killing, NK cells are also potent producers of cytokines such as interferon-γ (IFNγ), tumor necrosis factor (TNF)-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF) as well as chemokines (e.g., CCL3, CCL4, CCL5, XCL1, and CXCL8) that can modulate the function of other immune mediators.1
The ability of NK cells to differentiate normal cells from those that have undergone malignant transformation makes them a key player in tumor immunosurveillance. Tumor-infiltrating NK cells correlate with improved patient prognosis and survival in colorectal carcinoma,8 gastric carcinoma,9 pulmonary adenocarcinoma,10 and squamous cell lung carcinoma.11 In renal cell carcinoma, NK cell infiltration of the lung metastases is associated with improved survival.12 Inversely, an 11-year follow-up prospective cohort study from Japan suggested that low NK cell cytotoxicity is associated with increased cancer risk.13 Low NK activity was also correlated with cancer recurrence following surgical tumor resection of colorectal cancer14 and incidence of metastasis in head and neck15 and pharyngeal16 cancer.
In an actively developing area of investigation, harnessing NK cells is proving to be an attractive strategy to target cancer, either by activating endogenous NK cells or adoptive cell transfer. The clinical application of NK cells in the cancer setting was first explored in the 1980s when high infusions of interleukin (IL)-2 and lymphokine-activated killer (LAK) cells were tested in renal cell carcinoma and melanoma.17 LAK cells were generated from freshly isolated peripheral blood mononuclear cells (PBMCs) that were cultured with IL-2, which activates NK cytotoxicity and supports NK and T cell proliferation. The NK cell population was primarily responsible for the anti-tumor cytotoxicity of LAK cells;18 however, due to the minimal benefit of LAK cells and the toxicities linked to high-dose IL-2, LAK cell-based therapies were not developed further.17,19
Since then, our understanding of NK biology has advanced, resulting in the development of improved NK cell therapies with a better safety profile and efficacy. Furthermore, the clinical success of chimeric antigen receptor (CAR)-engineered T cells in hematological malignancies propelled the development of CAR-engineered NK cells.19 NK cells for adoptive cell therapy can be derived or generated from peripheral blood, umbilical cord blood, and hematopoietic stem cells/induced pluripotent stem cells. The advantages and limitations of isolating, expanding, and engineering NK cells from these sources were reviewed in Daher et al.20 NK cells can also be sourced from immortalized NK cell lines such as NKG, KHYG-1, NK-YS, YT, YTS, NK3.3, NKL, and NK-92.21 All of these cell lines proliferate easily in culture and can potentially provide a steady supply of off-the-shelf “pure” NK cells; however, only NK-92 has demonstrated consistent cytotoxic activity against cancer targets.22 Compared to peripheral blood NK cells, NK-92 can easily be manipulated to express receptors or ligands that enhance tumor targeting.22 In this review, we highlight recent developments in CAR-engineered NK-92 cells as off-the-shelf targeted cancer immunotherapy. Other advances in NK-based immunotherapy were expertly reviewed in Franks et al.21
NK-92 cells
NK-92 is an established NK cell line derived from the peripheral blood of a 50-year-old male non-Hodgkin’s lymphoma patient.23 NK-92 cells express CD56bright, CD2, CD7, CD11a, CD28, CD45, and CD54 and are negative for the cell surface markers CD3, CD4, CD8, CD16, CD1, CD5, CD10, CD14, CD19, CD20, CD23, CD34, and HLA-DR.24 Despite being CD56brightCD16−, which conventionally characterizes cytokine-producing NK cells,1 NK-92 cells express high levels of granzyme B and perforin, making them highly cytotoxic.23,24
NK-92 cells express a relatively large number of activating receptors and very few inhibitory receptors. NKG2D, NKp30, NKp46, and 2B4 have all been detected on NK-92 cells and are important in the function of the cell line.24 Target cells that express the NKG2D ligands MHC class I chain-related genes (MIC)A/B are susceptible to NK-92 killing, while those that do not are resistant to NK-92-mediated lysis.25 NK-92 cells express the inhibitory receptors CD94/NKG2A and LIR-1 but lack most of the receptors in the KIR family, except for KIR2DL4.24 KIR2DL4 is an unusual KIR that has an inhibitory potential due to the immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic tail, but it functionally activates NK cytotoxicity.26 The presence of a variety of activating receptors and the relative absence of inhibitory receptors contribute to the superior cytotoxicity of NK-92 cells.
The ability to expand NK-92 cells easily and reproducibly in good manufacturing practice (GMP) conditions allows for the development of off-the-shelf cell therapy products using this cell line as a backbone.27 NK-92 cells can be continuously grown in culture with a doubling time of 24–36 h.27 NK-92 cell growth is dependent on IL-2, and withdrawal of IL-2 causes a decline in cytotoxicity after 24 h.23,28 IL-7 can support short-term proliferation of NK-92 but cannot support sustained growth of the cells.23 IL-15 was also reported to augment NK-92 cell proliferation and activity.29,30
NK-92 cells are effective against a broad spectrum of tumor targets. Early studies demonstrated the cytolytic activity of NK-92 against malignant cells of hematologic origin in vitro.23,28,31 Preclinical studies in severe combined immunodeficient (SCID) mice have shown the antitumor activity of NK-92 in T acute lymphoblastic leukemia (T-ALL),31 acute myelogenous leukemia (AML),31 myeloma,32 and melanoma33 xenograft models. Meanwhile, targeting of nonmalignant allogeneic cells, such as hematopoietic cells derived from normal donors, was not reported.31 Furthermore, NK-92 cells were not tumorigenic in immunocompromised SCID mice.22 These studies demonstrated that in the preclinical setting, NK-92 has antitumor activity with minimal side effects.
Clinical trials with NK-92 cells
Several phase 1 clinical studies have been completed to establish the safety of administering NK-92 cells as allogeneic cell therapy in hematologic and solid cancers. Four clinical trials included patients with solid tumors such as renal cell cancer, lung cancer, melanoma,34,35 and blood-related malignancies including AML, multiple myeloma (MM), and other relapsed/refractory hematological cancers.35, 36, 37 In these studies, one treatment course consisted of two to three infusions of escalating doses of NK-92 cells given 24–48 h apart. The NK-92 cells were irradiated at 10 Gy prior to infusion to prevent proliferation in vivo and eliminate the tumorigenic potential.28
These clinical studies demonstrated that NK-92 cell infusion is generally well tolerated and is correlated with some clinical responses. In a phase 1 study performed in Chicago, 12 patients with renal cell cancer or metastatic melanoma were enrolled and received NK-92 cell dose levels of 1 × 108, 3 × 108, 1 × 109, or 3 × 109 cells/m2. Most of the NK-92 infusion-related toxicities were mild except for one grade 3 fever and one grade 4 hypoglycemic episode in the cohort that received the highest dose level. The melanoma patient exhibited a minor response during the study period, while one renal cell cancer patient had a mixed response. A minor response was defined as the regression of a target tumor lesion by 10%–30% without the formation of new lesions and without the progression of non-target lesions, while a mixed response was the regression of some lesions but simultaneous progression of others. One patient was alive with disease at 4 years post-treatment.34 In the study performed in Frankfurt, no NK-92 cell infusion-related toxicities were observed even at the highest dose level tested, which was 1 × 1010 cells/m2. One patient, however, had to discontinue the second infusion due to back pain that was likely related to abdominal distension caused by the fluid bolus given before and during the infusion. Three of the four patients with advanced lung cancer had some antitumor response. Two of the lung cancer patients had metastatic lesions that disappeared after two infusions of NK-92, while one patient had stable disease for approximately 2 years.35 In the QUILT-3.018 study (ClinicalTrials.gov: NCT00900809), seven patients with AML received a total of 20 infusions of NK-92 at 1 × 109 or 3 × 109 cells/m2. No patient experienced dose-limiting toxicities during infusion or within the 21 days of the post-infusion observation period. In addition, no grade 3–4 toxicities related to the infusion were observed. In one patient the blast percentage was reduced, and in two patients the blast percentage remained stable.36 In another study, 12 patients with lymphoma or MM who relapsed after autologous hematopoietic cell transplantation (AHCT) for relapsed disease were enrolled and the highest NK-92 dose level tested was 3 × 109 cells/m2 (ClinicalTrials.gov: NCT00990717). Minor acute infusion-related toxicities were observed but no grade 3 or 4 infusion-related toxicities, no delayed toxicity, no graft-versus-host disease, and no cytokine release syndrome were noted. Complete response was achieved in one patient with Hodgkin’s lymphoma (alive 10 years after therapy) and one patient with MM. A mixed response was observed in two patients, one with Hodgkin’s lymphoma and one with diffuse large B cell lymphoma (DLBCL). One patient with chronic lymphocytic leukemia (CLL) had clinical improvement in the trial.37 Several phase 1 and 2 trials are underway to evaluate the safety and efficacy of NK-92 in combination with other anticancer agents in stage II or IV Merkel cell carcinoma (ClincialTrials.gov: NCT02465957), hematological cancers (ClinicalTrials.gov: NCT02727803), and pancreatic cancer (ClinicalTrials.gov: NCT03136406) (Table 1).
Table 1.
Clinical studies with NK-92 and haNK cells
| Trial identifier: ClinicalTrials.gov: | Disease condition | NK cell | Combination agent | Phase | Status |
|---|---|---|---|---|---|
| NCT02465957 | stage IIIB and IV Merkel cell carcinoma | NK-92 | phase 2 | active, not recruiting | |
| NCT02727803 | myelodysplastic syndrome, leukemia, lymphoma, MM | NK-92 | biological: anti-thymocyte globulin, rituximab | phase 2 | active, recruiting |
| drug: busulfan, clofarabine, cyclophosphamide, fludarabine phosphate, melphalan | |||||
| radiation: total-body irradiation | |||||
| procedure: umbilical cord blood transplantation | |||||
| NCT03136406 | pancreatic cancer | NK-92 | drug: cyclophosphamide, oxaliplatin, capecitabine, 5-fluorouracil, leucovorin, nab-paclitaxel | phase 1/2 | active, not recruiting |
| biological: bevacizumab, avelumab, N-803, Ad-CEA, RAS-yeast | |||||
| NCT03387085 | TNBC | haNK | drug: aldoxorubicin HCl, capecitabine, cisplatin, cyclophosphamide, 5-fluorouracil, leucovorin, nab-paclitaxel | phase 1/2 | unknown |
| biological: N-803, Ad-CEA, Ad-brachyury, Ad-MUC1, RAS-yeast, CEA-yeast, brachyury-yeast, avelumab, bevacizumab | |||||
| procedure: SBRT | |||||
| NCT03563157 | metastatic CRC | haNK | drug: aldoxorubicin HCl, capecitabine, cisplatin, cyclophosphamide, 5-fluorouracil, leucovorin, nab-paclitaxel | phase 1/2 | active, not recruiting |
| biological: N-803, Ad-CEA, Ad-brachyury, Ad-MUC1, yeast-RAS, yeast-CEA, yeast-brachyury, avelumab, bevacizumab | |||||
| procedure: SBRT | |||||
| NCT03387111 | squamous cell carcinoma | haNK | drug: aldoxorubicin HCl, capecitabine, cisplatin, cyclophosphamide, 5-fluorouracil, leucovorin, nab-paclitaxel | phase 1/2 | active, not recruiting |
| biological: N-803, Ad-CEA, Ad-brachyury, Ad-MUC1, yeast-RAS, yeast-CEA, yeast-brachyury, avelumab, bevacizumab, necitumumab | |||||
| procedure: SBRT | |||||
| NCT03329248 | pancreatic cancer | haNK | biological: N-803, Ad-CEA, RAS-yeast, avelumab, bevacizumab | phase 1/2 | active, not recruiting |
| drug: capecitabine, cyclophosphamide, 5-fluorouracil, leucovorin, nab-paclitaxel, Lovaza, oxaliplatin | |||||
| procedure: SBRT | |||||
| NCT03853317 | Merkel cell carcinoma | haNK | biological: avelumab, N-803 | phase 2 | active, recruiting |
MM, multiple myeloma; Ad, adenovirus; CEA, carcinoembryonic antigen; RAS, rat sarcoma virus; TNBC, triple-negative breast cancer; haNK, high-affinity natural killer; SBRT, stereotactic body radiation therapy; CRC, colorectal cancer; MUC1, mucin 1.
High-affinity NK cells
The parental NK-92 cell line is dependent on exogenous IL-2 for cytotoxicity23,28 and is devoid of the ADCC-mediating receptor CD16.6,7,24 Hence, a novel NK-92 cell line, called high-affinity NK (haNK), has been engineered to express to high-affinity 158V FcγRIIIa (CD16) receptor as well as endoplasmic reticulum-retained IL-2.38,39 Injection of haNK cells into athymic mice did not result in tumor formation, but to further ensure safety, haNK cells were irradiated to decrease the tumorigenic potential. Irradiated haNK cells continued to produce IL-2, IFNγ, and IL-8 for at least 48 h and maintained cytotoxicity against various cancer cell lines in vitro. haNK cells have high levels of granzyme and are more cytotoxic against tumor targets compared to healthy human donor NK cells.39
Several studies have shown that patients with the 158V/V FcγRIIIa (CD16) polymorphism experience a better antitumor response and increased survival upon treatment with monoclonal antibodies (mAbs) such as anti-CD20 rituximab (Rituxan) for lymphoma,40,41 anti-ErbB2/HER2 trastuzumab (Herceptin) for breast cancer,42 and anti-epithelial growth factor receptor (EGFR) cetuximab (Erbitux) for colorectal cancer43,44 and squamous cell head and neck cancer.45 However, only ∼10% of the population are homozygous for the high-affinity 158V allele.46 In addition, the role of ADCC in cancer therapy remains controversial since ADCC has yet to be directly observed.47 Nevertheless, the findings in the abovementioned retrospective studies provide a strong rationale for the combination treatment of haNK cells with tumor-targeting IgG1 antibodies. Improved haNK cell-mediated targeting of cervical, ovarian, breast, and lung cancer cell lines was observed in the presence of cetuximab, trastuzumab, or anti-HER2 pertuzumab (Perjeta) mAbs. Blocking the CD16 receptor diminished tumor cell lysis, indicating that ADCC plays a role in the antitumor activity of haNK cells in combination with tumor-associated antigen (TAA)-targeted antibodies.39 Furthermore, in a CD38+ NCI-H929 multiple myeloma xenograft model in NOD-scid IL2Rγnull (NSG) mice, combination treatment with haNK cells and anti-CD38 mAb daratumumab (Darzalex) resulted in improved survival compared to tumor-bearing mice that received haNK cells with isotype control.38
Clinical trials with haNK cells
A phase 1 3+3 dose escalation study, with a starting dose of 2 × 109 haNK cells per infusion, has been designed to determine the safety of haNK cell infusion in patients with metastatic or locally advanced solid tumors (ClinicalTrials.gov: NCT03027128). This study has completed enrollment but has yet to post results. An ongoing phase 2 study aims to evaluate the therapeutic effect of haNK cells with anti-programmed death ligand 1 (PD-L1) avelumab (Bavencio) and the IL-15 superagonist N-803 in Merkel cell carcinoma patients who have progressed on or within 6 months of completing treatment with avelumab or anti-programmed cell death 1 (PD-1) pembrolizumab (Keytruda) by objective response rate (ORR) using response evaluation criteria in solid tumors version 1.1 (RECIST 1.1) (ClinicalTrials.gov: NCT03853317). Another ongoing phase 1b trial (ClinicalTrials.gov: NCT03387085) evaluates the safety and efficacy of haNK cell therapy in combination with immune checkpoint inhibition, IL-15 superagonist (N-803) administration, cancer vaccines, and chemoradiation in patients with refractory, metastatic, or unresectable triple-negative breast cancer (TNBC). Preliminary results indicate that the combination is safe and tolerable with a disease control rate of 78%, overall response rate of 67%, and complete response of 22%.48 Similar phase 1/2 trials evaluate the safety and efficacy of haNK cells in combination with immunotherapy and chemoradiation in metastatic colorectal cancer (ClinicalTrials.gov: NCT03563157), squamous cell carcinoma (ClinicalTrials.gov: NCT03387111), and pancreatic cancer (ClinicalTrials.gov: NCT03329248) (Table 1).
CAR-engineered NK-92 and haNK cells
Advantages of CAR NK over CAR T cells
CAR T cells represent a cutting-edge immunotherapeutic approach that has revolutionized cancer treatment. Currently, the U.S. Food and Drug Administration (FDA) has approved four autologous CD19-directed CAR T cell therapies for the treatment of relapsed or refractory diffuse large B cell lymphoma, primary mediastinal B cell lymphoma, high-grade B cell lymphoma, and transformed follicular lymphoma,49 as well as one autologous B cell maturation antigen (BCMA)-directed CAR T cell therapy for the treatment of relapsed/refractory MM.50 Despite the successes of CAR T cell therapy in the clinic, several challenges remain unaddressed. While CAR T cells have been effective in hematological cancers, they currently provide minimal therapeutic benefits in solid tumor settings.51 Preparation of CAR T cells also requires an autologous source since allogeneic T cells cause graft-versus-host disease (GVHD).52 Moreover, CAR T cell therapy may induce cytokine release syndrome and neurologic toxicities that can be life-threatening.53,54
NK cells provide a CAR-engineering platform that is safer and more advantageous compared to T cells.55 With the absence of GVHD after allogeneic NK cell infusion34, 35, 36, 37,56,57 and the availability of immortalized NK cell lines such as NK-92,21 CAR NK cells not only are a safer alternative but also one that potentially has a broader off-the-shelf clinical application not limited by individualized preparation.58 Another safety advantage of CAR NK cells over CAR T cells is the type of cytokines that NK cells produce. The cytokine release syndrome induced by CAR T cells is associated with elevated levels of pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1.54,55,59,60 CAR NK-92 cells, alternatively, have a cytokine profile that is less likely to induce cytokine release syndrome. CAR NK-92 cells were reported to secrete high levels of IFNγ, macrophage inflammatory protein (MIP)-1α (CCL3), GM-CSF, and moderate levels of TNF-α.55,61 Furthermore, persistent CAR T cells that can attack normal cells cause on-target/off-tumor effects. CD19-targeting CAR T cells can persist as memory CAR T cells and target normal B cells, which may lead to prolonged B cell deficiency.58,62 In one colorectal cancer patient, HER-2-targeting CAR T cells possibly recognized low levels of HER2/neu on lung epithelial cells, resulting in acute respiratory failure.63 Meanwhile, irradiated parental NK-92 cells have a limited lifespan in vivo and do not develop memory, minimizing the risk of these side effects.28 However, as a consequence, repeated and frequent infusions of irradiated NK-92 and NK-92-based cells would be required to maintain in vivo cell numbers.64 Lastly, CAR T cells are mostly dependent on the artificial receptor for tumor targeting. The heterogenous nature of most tumors entails that a population of the malignant cells will not be recognized and attacked by the CAR T cells.65 CAR NK-92 cells retain the expression of activating receptors, allowing these effector cells to detect even the tumor cells that do not express the CAR target.55 In addition to natural cytotoxicity, CAR-engineered haNK cells may also have the potential to mediate ADCC in the presence of TAA-specific mAbs.38,39
Alternatively, the repeated infusion of an allogeneic cell product such as CAR NK-92 may trigger the patient’s immune response and possibly limit the effect of NK-92 cell-based therapies. Hence, the development of humoral and T cell responses has to be monitored. In the phase 1 clinical trials, the formation of HLA antibodies against NK-92 cells occurred in less than half of the recipients.34,35 Furthermore, mixed lymphocyte reactions using the patients’ lymphocytes and irradiated NK-92 cells showed that NK-92 cells are only mild stimulators.22 Whether the differences in the immune status of the patients and prior blood transfusion events contribute to the variability in host responses against NK-92 cell therapy is yet to be determined. Nevertheless, patients who develop HLA antibodies may have to avoid retreatment with NK-92 cell products beyond a 7-day window in order to avert an anamnestic response.34
Generation of CAR NK-92 cells
CARs are based on the T cell receptor (TCR) and are composed of an extracellular antibody single-chain variable fragment (scFv) that recognizes specific surface antigens on the tumor, a hinge, a transmembrane domain, and an intracellular signaling domain.66,67 Most CAR NK-92 studies utilize first-generation CARs, which contain a single signaling domain composed of CD3ζ, FcεR1γ, or DAP-12.61,68,69 CAR-modified NK-92 cells were found to lyse tumor cells more effectively than CD16-engineered NK-92 cells acting through ADCC in vitro,64,70 which indicates that even first-generation CARs are more potent than ADCC-mediated tumor killing. Second- and third-generation CARs employ one or two costimulatory domains in conjunction with CD3ζ to improve cytotoxic activity. The co-activating proteins are usually based on the CD28 family (CD28 or ICOS), the TNF receptor family (4-1BB, OX40, or CD27), or the signaling lymphocytic activation molecule (SLAM)-related receptor family (2B4).67,71 A study comparing ErbB2-targeted NK cells expressing CD3ζ alone, CD28/CD3ζ, or CD137/CD3ζ found that the second-generation constructs displayed increased cytotoxicity compared to the first-generation CAR.72
Sustained CAR expression on NK cells requires stable gene transfer. The main gene modification methods utilized to generate CAR NK cells are viral transduction and transfection.58,71 Retrovirus and lentivirus-based vectors have been widely applied in the production of CAR NK cells due to stable gene integration and high transduction rates, especially in blood-derived NK cells. Transduction levels of up to 60% were achieved in peripheral blood NK cells using retroviral vectors,73 while a 73% transduction efficacy was achieved using lentiviral vectors in NK cells derived from cord blood.74 Viral transduction, however, carries the risk of insertional mutations that can result in oncogenesis and other adverse events. Non-viral transfection methods with either naked plasmid DNA, transposase DNA-mediated integration, or mRNA by electroporation are inexpensive and low immunogenicity alternatives to viral transduction.58 In fact, electroporation with mRNA was found to result in high transfection efficiencies in NK-92 cells. This method results only in transient gene transfer and, hence, short-term CAR expression.22,74,75
Preclinical studies with CAR NK-92 cells
The first CAR NK-92 reported was engineered using a first-generation CAR transgene that consisted of a ErbB2 (HER2/neu)-specific scFv, a CD8 hinge, and a CD3ζ signaling domain that was delivered via a retroviral vector.76 The value of targeting ErbB2 has been demonstrated by the positive preclinical and clinical observations with ErbB2-specific antibodies and CAR T cells.77,78 ErbB2-targeting CAR NK-92 cells demonstrated specific cytotoxicity toward ErbB2-expressing breast, ovarian, and squamous cell carcinoma cells in vitro. In vivo, cell therapy with ErbB2-CAR-NK-92 suppressed the tumor growth of human-ErbB2+ NIH 3T3 fibroblasts in CD-1 nude mice.76 ErbB2-CAR-NK-92 cells were also demonstrated to migrate and accumulate in ErbB2+ tumors, further illustrating the specificity of the modified cells.79, 80, 81 Second-generation ErbB2-CAR-NK-92 cells that included the costimulatory CD28 domain in addition to CD3ζ displayed increased cytotoxicity compared to the first-generation CAR72 and exhibited potent antitumor activity in glioblastoma82 and breast cancer models.83
Other CAR NK-92 cells have been developed against different TAA targets and have shown in vivo activity in preclinical tumor models. CAR NK-92 cells specific for B cell differentiation antigens CD19 and CD20 in CLL, B cell ALL (B-ALL), and lymphoma,84, 85, 86, 87 CD138 and CS1 in MM,88,89 and CD3, CD5, and CD7 in T-ALL90, 91, 92, 93 inhibited the tumor progression of these hematological cancers in corresponding xenograft models in NSG mice. CAR NK-92 cells for various surface antigens expressed by solid tumors have also been studied. Targeting EGFR/EGFRvIII in glioblastoma,94,95 epithelial cell adhesion molecule (EPCAM) in colorectal and renal cell carcinoma,96,97 GD2 in neuroblastoma,98 GPC3 in hepatocellular carcinoma,99 mesothelin in ovarian cancer,100 and prostate-specific membrane antigen (PSMA) in prostate cancer101 using CAR NK-92 cells decreased disease burden and/or prolonged the survival of tumor-bearing mice. In addition to TAAs, NK-92 cells engineered with scFv that recognizes TCR peptide epitopes complexed with HLA-A2, such as gp100/HLA-A2 in melanoma and WT1/HLA-A2 in leukemia, were shown to be effective in targeting the epitope-presenting tumors.102,103 Immune checkpoint molecules, which are expressed by multiple tumor types and are essential in immune evasion,104 represent another optimal target for CAR NK-92. For instance, B7-H3-targeted CAR NK-92 suppressed the tumor growth of non-small cell lung cancer and improved the survival of the tumor-bearing mice.105 PD-L1 targeted haNK (PD-L1 t-haNK) cell therapy is highly cytotoxic against a broad spectrum of tumor cell lines in vitro and induces potent anti-cancer effects in murine xenograft models of TNBC, bladder cancer, and lung cancer.64,106 Overall, these pre-clinical data demonstrate the potential therapeutic benefit of CAR NK-92 cell therapy and, therefore, provide a strong rationale for the application of this novel cell therapy in the clinic.
Clinical trials with CAR NK-92 cells
The first-in-human clinical trial using an off-the-shelf CAR NK-92 targeted CD33 in patients with relapse or refractory AML (ClinicalTrials.gov: NCT02944162).107 This CD33 CAR NK-92 cell line was generated via lentiviral transfection and utilized a third-generation CAR involving costimulatory molecules CD28 and CD137. The highest dose administered was 5 × 109 cells and even at this level, no grade 3–4 adverse events were observed. However, all three patients suffered from infusion-related fever that abated after a day or two. In a similar phase 1 study using CD33-targeted CAR T cells for treatment of relapsed or refractory AML (ClinicalTrials.gov: NCT03126864), grade 3 cytokine release syndrome and grade 2 neurotoxicity syndrome were observed in one of the three patients.108 The CD33 CAR NK-92 cells were detected 1 week post-infusion in all three patients, suggesting that irradiated CAR NK-92 cells can persist, albeit in the short term, in vivo.107 Although no obvious clinical efficacy was observed, this phase 1 study showed that CAR NK-92 cell therapy may be used safely in patients and may provide an off-the-shelf treatment option for AML and/or other cancers.107 Other CAR NK-92 cell products being clinically tested for safety in hematological cancers target CD7 (ClinicalTrials.gov: NCT02742727) and CD19 (ClinicalTrials.gov: NCT02892695) in leukemia and lymphoma, and BCMA (ClinicalTrials.gov: NCT03940833) for MM (Table 2).
Table 2.
Clinical studies with CAR-expressing NK-92 and haNK cells
| Trial identifier: ClinicalTrials.gov: | Antigen target | Disease condition | NK cell | Combination agent | Phase | Status |
|---|---|---|---|---|---|---|
| NCT02944162 | CD33 | AML | NK-92 | phase 1/2 | unknown | |
| NCT03940833 | BCMA | MM | NK-92 | phase 1/2 | active, recruiting | |
| NCT02742727 | CD7 | leukemia, lymphoma | NK-92 | phase 1/2 | unknown | |
| NCT02892695 | CD19 | leukemia, lymphoma | NK-92 | phase 1/2 | unknown | |
| NCT03383978 | ErbB2 | glioblastoma | NK-92 | phase 2 | active, recruiting | |
| NCT02839954 | MUC1 | hepatocellular carcinoma, pancreatic carcinoma, glioma, gastric carcinoma, NSCLC, TNBC, CRC | NK-92 | phase 1/2 | unknown | |
| NCT04050709 | PD-L1 | advanced solid tumor, metastatic tumor | haNK | phase 1 | active, not recruiting | |
| NCT03228667 | PD-L1 | NSCLC, SCLC, HNSCC, RCC, CRC, urothelial carcinoma, Merkel cell carcinoma, melanoma, gastric cancer, cervical cancer, hepatocellular carcinoma | haNK | biological: N-803, pembrolizumab, nivolumab, atezolizumab, avelumab, or durvalumab | phase 2 | active, not recruiting |
| NCT04847466 | PD-L1 | GEJ cancer, advanced HNSCC | haNK | biological: N-803, pembrolizumab | phase 2 | not yet recruiting |
| NCT04390399 | PD-L1 | pancreatic cancer | haNK | biological: N-803 | phase 2 | active, recruiting |
| drug: aldoxorubicin HCl, nab-paclitaxel, gemcitabine, cyclophosphamide, 5-fluorouracil, leucovorin, irinotecan liposome | ||||||
| procedure: SBRT | ||||||
| NCT04927884 | PD-L1 | TNBC | haNK | biological: N-803 | phase 1/2 | not yet recruiting |
| drug: sacituzumab govitecan-hziy, cyclophosphamide |
AML, acute myeloid leukemia; BCMA, B cell maturation antigen; MM, multiple myeloma; MUC1, mucin 1; NSCLC, non-small cell lung cancer; TNBC, triple-negative breast cancer; CRC, colorectal cancer; haNK, high-affinity natural killer; PD-L1, programmed death ligand 1; SCLC, small cell lung cancer; HNSCC, head and neck squamous cell carcinoma; RCC, renal cell carcinoma; GEJ, gastroesophageal junction.
A phase 1 3+3 dose escalation trial evaluating the safety, maximum tolerated dose (MTD), and response to a second-generation CAR NK-92 (NK-92/5.28.z) specific for ErbB2 in glioblastoma (ClinicalTrials.gov: NCT03383978) is underway (Table 2).109 In this study, patients with recurrent or refractory ErbB2-positive glioblastoma are administered with irradiated CAR NK-92 cells intracranially during relapse surgery. As of 2019, administration of the first two dose levels (1 × 107 and 3 × 107 cells) concluded and no dose-limiting toxicities were observed. The highest dose planned for this trial is 1 × 108 cells. Once dose escalation is completed, a planned expansion cohort would include patients who will receive up to 12 additional injections of the ErbB2-CAR-NK-92 cells into the resection cavity through an implanted catheter and reservoir. Peripheral blood and cerebrospinal fluid will be collected and analyzed for soluble factors and cells to determine the effects of the CAR NK on endogenous immune cells over the course of therapy.109
Another TAA being used as a CAR NK-92 target is mucin 1 (MUC1), an aberrantly glycosylated transmembrane glycoprotein overexpressed in a variety of epithelial cancers.110 A MUC1-CAR-NK-92 has been engineered by lentiviral gene transfer to express third-generation anti-MUC1 CAR with CD28/CD137 signaling moiety and has been shown to lyse MUC1+ tumor cells in vitro and in vivo.111 In a phase 1 clinical trial, 13 patients with MUC1+ expressing lung cancer, pancreatic cancer, colon cancer or ovarian cancer were enrolled and received 1 × 109 cells per infusion (ClinicalTrials.gov: NCT02839954; Table 2). No severe cytokine release syndrome and/or bone marrow suppression were reported, indicating that MUC1-CAR-NK-92 can be safely applied as therapy against different solid tumors. Furthermore, some minor clinical response was observed. Nine patients (69.2%) presented stable disease, and one patient had progressive disease.111
Based on preclinical data indicating the potency of PD-L1 t-haNK cells against a broad range of tumors,64,106 PD-L1 t-haNK cells have entered a phase I clinical trial to evaluate the safety and preliminary efficacy of these CAR-modified high affinity NK cells in locally advanced solid tumors and metastatic cancer (ClinicalTrials.gov: NCT04050709). This study will determine the maximum tolerated dose and designate the recommended dose for future phase 2 studies. PD-L1 t-haNK cells are also being studied in combination with other anticancer agents in several phase 1/2 trials. In a phase 2 trial, the efficacy of PD-L1 t-haNK with immune checkpoint inhibitor and N-803 is being determined in patients with solid tumors that have progressed and/or relapsed after checkpoint inhibitor therapy (ClinicalTrials.gov: NCT03228667). The primary and secondary outcomes will address objective response rate, survival, and incidence of adverse events. A similar combination with PD-L1 t-haNK, N-803, and anti-PD-1 (pembrolizumab) will be used to treat patients with advanced-form gastric or head and neck cancer in an upcoming phase 2 trial (ClinicalTrials.gov: NCT04847466). In patients with pancreatic cancer, the comparative efficacy and overall safety of PD-L1 t-haNK is being evaluated in combination with chemoradiation and N-803 (ClinicalTrials.gov: NCT04390399). Based on the positive initial results observed with haNK cells in combination with N-803 and low-dose chemotherapy in refractory TNBC,48 a phase 1B/2 study has been planned to evaluate the efficacy of sacituzumab govitecan-hziy (Trodelvy) with PD-L1 t-haNK cell therapy, cyclophosphamide, and N-803 in patients with TNBC after at least two prior regimens for metastatic disease (ClinicalTrials.gov: NCT04927884). Trodelvy is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate that is approved by the FDA for the treatment of TNBC patients with metastatic disease who have received at least two prior therapies.112 So far, no preliminary data have been posted on any of the PD-L1 t-haNK cell therapy studies (Table 2).
Future perspectives
Despite the tremendous potential of CAR NK-92 cells, several issues need to be addressed to inform better construction and application of this therapy. First, most of the CAR constructs being utilized in the generation of CAR NK-92 were designed for CAR T cells and may not be optimal for NK cells. For instance, NK cells do not naturally express CD28, and the function of this costimulatory molecule in CAR NK cells remains unclear.113 Furthermore, in YTS cells, CAR containing DAP12 outperformed a CAR that included CD28/CD3ζ, suggesting that DAP12 may be a better signaling moiety for NK cells.68 Second, the effects of an immunosuppressive environment on CAR NK-92 cells need to be further elucidated. In addition, methods to interfere with or redirect these negative signals present a scheme to enhance the antitumor capabilities of CAR NK-92 cells. In fact, a CAR NK-92 has been engineered to convert the immunosuppressive signal induced by TGF-β into an activating signal.114 Lastly, to increase the potency of CAR NK-92 cells especially against solid tumors, tumor infiltration must be promoted. Engineering chemokine receptors, such as CXCR4, into CAR NK-92 cells may enhance chemotaxis to the tumor.69
Conclusions
CAR NK-92 cells provide an off-the-shelf therapeutic platform that could be readily available and broadly applicable. Numerous preclinical studies suggest that CAR NK-92 cells have great promise as an effective cellular immunotherapy against a broad range of tumor types. The concluded NK-92 phase 1 studies and, importantly, initial reports on various CAR NK-92 cell therapies indicate that CAR NK-92 treatment strategy is safe for clinical use. Upcoming clinical data will illuminate the therapeutic potential of this CAR NK-92 and expound on the feasibility of combining this cell therapy with other anticancer agents. Advances in CAR NK-92 technology guarantee that the field will evolve and progress, potentially resulting in improved clinical outcomes for cancer patients, especially those with limited treatment options.
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. The authors thank Debra Weingarten for assistance in the preparation of this manuscript.
Author contributions
K.P.F. and J.W.H. wrote the manuscript and conducted the literature search. The authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Abel A.M., Yang C., Thakar M.S., Malarkannan S. Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 2018;9:1869. doi: 10.3389/fimmu.2018.01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanier L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quatrini L., Della Chiesa M., Sivori S., Mingari M.C., Pende D., Moretta L. Human NK cells, their receptors and function. Eur. J. Immunol. 2021;51:1566–1579. doi: 10.1002/eji.202049028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth M.J., Cretney E., Kelly J.M., Westwood J.A., Street S.E.A., Yagita H., Takeda K., van Dommelen S.L.H., Degli-Esposti M.A., Hayakawa Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005;42:501–510. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 6.Campbell K.S., Hasegawa J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013;132:536–544. doi: 10.1016/j.jaci.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo Nigro C., Macagno M., Sangiolo D., Bertolaccini L., Aglietta M., Merlano M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019;7:105. doi: 10.21037/atm.2019.01.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coca S., Perez-Piqueras J., Martinez D., Colmenarejo A., Saez M.A., Vallejo C., Martos J.A., Moreno M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–2328. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Ishigami S., Natsugoe S., Tokuda K., Nakajo A., Che X., Iwashige H., Aridome K., Hokita S., Aikou T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. 2000;88:577–583. [PubMed] [Google Scholar]
- 10.Takanami I., Takeuchi K., Giga M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J. Thorac. Cardiovasc. Surg. 2001;121:1058–1063. doi: 10.1067/mtc.2001.113026. [DOI] [PubMed] [Google Scholar]
- 11.Villegas F.R., Coca S., Villarrubia V.G., Jiménez R., Chillón M.J., Jareño J., Zuil M., Callol L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. 2002;35:23–28. doi: 10.1016/s0169-5002(01)00292-6. [DOI] [PubMed] [Google Scholar]
- 12.Remark R., Alifano M., Cremer I., Lupo A., Dieu-Nosjean M.-C., Riquet M., Crozet L., Ouakrim H., Goc J., Cazes A. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin. Cancer Res. 2013;19:4079–4091. doi: 10.1158/1078-0432.CCR-12-3847. [DOI] [PubMed] [Google Scholar]
- 13.Imai K., Matsuyama S., Miyake S., Suga K., Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356:1795–1799. doi: 10.1016/S0140-6736(00)03231-1. [DOI] [PubMed] [Google Scholar]
- 14.Tartter P.I., Steinberg B., Barron D.M., Martinelli G. The prognostic significance of natural killer cytotoxicity in patients with colorectal cancer. Arch. Surg. 1987;122:1264–1268. doi: 10.1001/archsurg.1987.01400230050009. [DOI] [PubMed] [Google Scholar]
- 15.Schantz S.P., Ordonez N.G. Quantitation of natural killer cell function and risk of metastatic poorly differentiated head and neck cancer. Nat. Immun. Cell Growth Regul. 1991;10:278–288. [PubMed] [Google Scholar]
- 16.Schantz S.P., Savage H.E., Racz T., Taylor D.L., Sacks P.G. Natural killer cells and metastases from pharyngeal carcinoma. Am. J. Surg. 1989;158:361–366. doi: 10.1016/0002-9610(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg S.A., Lotze M.T., Muul L.M., Chang A.E., Avis F.P., Leitman S., Linehan W.M., Robertson C.N., Lee R.E., Rubin J.T. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987;316:889–897. doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- 18.Liu S., Galat V., Galat Y., Lee Y.K.A., Wainwright D., Wu J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021;14:7. doi: 10.1186/s13045-020-01014-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendel P., Reindl L.M., Bexte T., Künnemeyer L., Särchen V., Albinger N., Mackensen A., Rettinger E., Bopp T., Ullrich E. Arming immune cells for battle: A brief journey through the advancements of T and NK cell immunotherapy. Cancers (Basel) 2021;13:1481. doi: 10.3390/cancers13061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daher M., Melo Garcia L., Li Y., Rezvani K. CAR-NK cells: The next wave of cellular therapy for cancer. Clin. Transl. Immunology. 2021;10:e1274. doi: 10.1002/cti2.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franks S.E., Wolfson B., Hodge J.W. Natural born killers: NK cells in cancer therapy. Cancers (Basel) 2020;12:E2131. doi: 10.3390/cancers12082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klingemann H., Boissel L., Toneguzzo F. Natural killer cells for immunotherapy—Advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong J.H., Maki G., Klingemann H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- 24.Maki G., Klingemann H.G., Martinson J.A., Tam Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001;10:369–383. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- 25.Romanski A., Bug G., Becker S., Kampfmann M., Seifried E., Hoelzer D., Ottmann O.G., Tonn T. Mechanisms of resistance to natural killer cell-mediated cytotoxicity in acute lymphoblastic leukemia. Exp. Hematol. 2005;33:344–352. doi: 10.1016/j.exphem.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Faure M., Long E.O. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J. Immunol. 2002;168:6208–6214. doi: 10.4049/jimmunol.168.12.6208. [DOI] [PubMed] [Google Scholar]
- 27.Tam Y.K., Martinson J.A., Doligosa K., Klingemann H.G. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy. 2003;5:259–272. doi: 10.1080/14653240310001523. [DOI] [PubMed] [Google Scholar]
- 28.Klingemann H.G., Wong E., Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol. Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 29.Zhang B., Zhang J., Tian Z. Comparison in the effects of IL-2, IL-12, IL-15 and IFNα on gene regulation of granzymes of human NK cell line NK-92. Int. Immunopharmacol. 2008;8:989–996. doi: 10.1016/j.intimp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Törnroos H., Hägerstrand H., Lindqvist C. Culturing the human natural killer cell line NK-92 in interleukin-2 and interleukin-15—Implications for clinical trials. Anticancer Res. 2019;39:107–112. doi: 10.21873/anticanres.13085. [DOI] [PubMed] [Google Scholar]
- 31.Yan Y., Steinherz P., Klingemann H.G., Dennig D., Childs B.H., McGuirk J., O’Reilly R.J. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res. 1998;4:2859–2868. [PubMed] [Google Scholar]
- 32.Swift B.E., Williams B.A., Kosaka Y., Wang X.H., Medin J.A., Viswanathan S., Martinez-Lopez J., Keating A. Natural killer cell lines preferentially kill clonogenic multiple myeloma cells and decrease myeloma engraftment in a bioluminescent xenograft mouse model. Haematologica. 2012;97:1020–1028. doi: 10.3324/haematol.2011.054254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tam Y.K., Miyagawa B., Ho V.C., Klingemann H.-G. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J. Hematother. 1999;8:281–290. doi: 10.1089/106161299320316. [DOI] [PubMed] [Google Scholar]
- 34.Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy. 2008;10:625–632. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 35.Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 36.Boyiadzis M., Agha M., Redner R.L., Sehgal A., Im A., Hou J.-Z., Farah R., Dorritie K.A., Raptis A., Lim S.H. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19:1225–1232. doi: 10.1016/j.jcyt.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Williams B.A., Law A.D., Routy B., denHollander N., Gupta V., Wang X.H., Chaboureau A., Viswanathan S., Keating A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8:89256–89268. doi: 10.18632/oncotarget.19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boissel L., Klingemann H., Campbell K., Nichols K., Toneguzzo F., Marcus P., Williams B., Keating A., Soon-Shiong P. An “off the shelf,” GMP-grade, IL-2-independent NK cell line expressing the high-affinity Fc-receptor to augment antibody therapeutics. Cancer Res. 2016;76(14, Suppl):2302. [Google Scholar]
- 39.Jochems C., Hodge J.W., Fantini M., Fujii R., Morillon Y.M., 2nd, Greiner J.W., Padget M.R., Tritsch S.R., Tsang K.Y., Campbell K.S. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. 2016;7:86359–86373. doi: 10.18632/oncotarget.13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cartron G., Dacheux L., Salles G., Solal-Celigny P., Bardos P., Colombat P., Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 41.Weng W.K., Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Musolino A., Naldi N., Bortesi B., Pezzuolo D., Capelletti M., Missale G., Laccabue D., Zerbini A., Camisa R., Bisagni G. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008;26:1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 43.Zhang W., Gordon M., Schultheis A.M., Yang D.Y., Nagashima F., Azuma M., Chang H.-M., Borucka E., Lurje G., Sherrod A.E. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J. Clin. Oncol. 2007;25:3712–3718. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- 44.Bibeau F., Lopez-Crapez E., Di Fiore F., Thezenas S., Ychou M., Blanchard F., Lamy A., Penault-Llorca F., Frébourg T., Michel P. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009;27:1122–1129. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 45.Magnes T., Melchardt T., Hufnagl C., Weiss L., Mittermair C., Neureiter D., Klieser E., Rinnerthaler G., Roesch S., Gaggl A. The influence of FCGR2A and FCGR3A polymorphisms on the survival of patients with recurrent or metastatic squamous cell head and neck cancer treated with cetuximab. Pharmacogenomics J. 2018;18:474–479. doi: 10.1038/tpj.2017.37. [DOI] [PubMed] [Google Scholar]
- 46.Lehrnbecher T., Foster C.B., Zhu S., Leitman S.F., Goldin L.R., Huppi K., Chanock S.J. Variant genotypes of the low-affinity Fcγ receptors in two control populations and a review of low-affinity Fcγ receptor polymorphisms in control and disease populations. Blood. 1999;94:4220–4232. [PubMed] [Google Scholar]
- 47.Gómez Román V.R., Murray J.C., Weiner L.M. In: Antibody Fc: Linking Adaptive and Innate Immunity. Ackerman M.E., Nimmerjahn F., editors. Academic Press; 2014. Antibody-dependent cellular cytotoxicity (ADCC) pp. 1–27. [Google Scholar]
- 48.Kistler M., Nangia C., To C., Sender L., Lee J., Jones F., Jafari O., Seery T., Rabizadeh S., Niazi K., Rock A. Safety and efficacy from first-in-human immunotherapy combining NK and T cell activation with off-the-shelf high-affinity CD16 NK cell line (haNK) in patients with 2nd-line or greater metastatic triple-negative breast cancer (TNBC) Cancer Res. 2020;80(4, Suppl):P5-04-02. [Google Scholar]
- 49.Kanwal B. Relapsed/refractory non-Hodgkin lymphoma: engineering T-cells to express chimeric antigen receptors (CARs), a salvage? Cureus. 2021;13:e16307. doi: 10.7759/cureus.16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.George L.L., Deshpande S.R., Cortese M.J., Kendall E.K., Chattaraj A., Shah Z., Zhao J., Anwer F. Emerging targets and cellular therapy for relapsed refractory multiple myeloma: A systematic review. Clin. Lymphoma Myeloma Leuk. 2021 doi: 10.1016/j.clml.2021.06.003. Published online June 25, 2021. [DOI] [PubMed] [Google Scholar]
- 51.D’Aloia M.M., Zizzari I.G., Sacchetti B., Pierelli L., Alimandi M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018;9:282. doi: 10.1038/s41419-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ichiki Y., Bowlus C.L., Shimoda S., Ishibashi H., Vierling J.M., Gershwin M.E. T cell immunity and graft-versus-host disease (GVHD) Autoimmun. Rev. 2006;5:1–9. doi: 10.1016/j.autrev.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Gust J., Hay K.A., Hanafi L.-A., Li D., Myerson D., Gonzalez-Cuyar L.F., Yeung C., Liles W.C., Wurfel M., Lopez J.A. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klingemann H. Are natural killer cells superior CAR drivers? OncoImmunology. 2014;3:e28147. doi: 10.4161/onci.28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller J.S., Soignier Y., Panoskaltsis-Mortari A., McNearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 57.Shaffer B.C., Le Luduec J.-B., Forlenza C., Jakubowski A.A., Perales M.-A., Young J.W., Hsu K.C. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2016;22:705–709. doi: 10.1016/j.bbmt.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Y., Tian Z.G., Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol. Sin. 2018;39:167–176. doi: 10.1038/aps.2017.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teachey D.T., Lacey S.F., Shaw P.A., Melenhorst J.J., Maude S.L., Frey N., Pequignot E., Gonzalez V.E., Chen F., Finklestein J. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang C., Oberoi P., Oelsner S., Waldmann A., Lindner A., Tonn T., Wels W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017;8:533. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fabian K.P., Padget M.R., Donahue R.N., Solocinski K., Robbins Y., Allen C.T., Lee J.H., Rabizadeh S., Soon-Shiong P., Schlom J., Hodge J.W. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J. Immunother. Cancer. 2020;8:e000450. doi: 10.1136/jitc-2019-000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen N., Li X., Chintala N.K., Tano Z.E., Adusumilli P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018;51:103–110. doi: 10.1016/j.coi.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dotti G., Gottschalk S., Savoldo B., Brenner M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guedan S., Calderon H., Posey A.D., Jr., Maus M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2018;12:145–156. doi: 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Töpfer K., Cartellieri M., Michen S., Wiedemuth R., Müller N., Lindemann D., Bachmann M., Füssel M., Schackert G., Temme A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015;194:3201–3212. doi: 10.4049/jimmunol.1400330. [DOI] [PubMed] [Google Scholar]
- 69.Müller N., Michen S., Tietze S., Töpfer K., Schulte A., Lamszus K., Schmitz M., Schackert G., Pastan I., Temme A. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J. Immunother. 2015;38:197–210. doi: 10.1097/CJI.0000000000000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clémenceau B., Valsesia-Wittmann S., Jallas A.C., Vivien R., Rousseau R., Marabelle A., Caux C., Vié H. In vitro and in vivo comparison of lymphocytes transduced with a human CD16 or with a chimeric antigen receptor reveals potential off-target interactions due to the IgG2 CH2-CH3 CAR-spacer. J. Immunol. Res. 2015;2015:482089. doi: 10.1155/2015/482089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gong Y., Klein Wolterink R.G.J., Wang J., Bos G.M.J., Germeraad W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021;14:73. doi: 10.1186/s13045-021-01083-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schönfeld K., Sahm C., Zhang C., Naundorf S., Brendel C., Odendahl M., Nowakowska P., Bönig H., Köhl U., Kloess S. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015;23:330–338. doi: 10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suerth J.D., Morgan M.A., Kloess S., Heckl D., Neudörfl C., Falk C.S., Koehl U., Schambach A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J. Mol. Med. (Berl.) 2016;94:83–93. doi: 10.1007/s00109-015-1327-6. [DOI] [PubMed] [Google Scholar]
- 74.Boissel L., Betancur M., Lu W., Wels W.S., Marino T., Van Etten R.A., Klingemann H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk. Lymphoma. 2012;53:958–965. doi: 10.3109/10428194.2011.634048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boissel L., Betancur M., Wels W.S., Tuncer H., Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009;33:1255–1259. doi: 10.1016/j.leukres.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uherek C., Tonn T., Uherek B., Becker S., Schnierle B., Klingemann H.-G., Wels W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100:1265–1273. [PubMed] [Google Scholar]
- 77.Wong D.J., Hurvitz S.A. Recent advances in the development of anti-HER2 antibodies and antibody-drug conjugates. Ann. Transl. Med. 2014;2:122. doi: 10.3978/j.issn.2305-5839.2014.08.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu X., Zhang N., Shi H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget. 2017;8:62730–62741. doi: 10.18632/oncotarget.17528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daldrup-Link H.E., Meier R., Rudelius M., Piontek G., Piert M., Metz S., Settles M., Uherek C., Wels W., Schlegel J., Rummeny E.J. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radiol. 2005;15:4–13. doi: 10.1007/s00330-004-2526-7. [DOI] [PubMed] [Google Scholar]
- 80.Meier R., Piert M., Piontek G., Rudelius M., Oostendorp R.A., Senekowitsch-Schmidtke R., Henning T.D., Wels W.S., Uherek C., Rummeny E.J., Daldrup-Link H.E. Tracking of [18F]FDG-labeled natural killer cells to HER2/neu-positive tumors. Nucl. Med. Biol. 2008;35:579–588. doi: 10.1016/j.nucmedbio.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 81.Alkins R., Burgess A., Ganguly M., Francia G., Kerbel R., Wels W.S., Hynynen K. Focused ultrasound delivers targeted immune cells to metastatic brain tumors. Cancer Res. 2013;73:1892–1899. doi: 10.1158/0008-5472.CAN-12-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C., Burger M.C., Jennewein L., Genßler S., Schönfeld K., Zeiner P., Hattingen E., Harter P.N., Mittelbronn M., Tonn T. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2015;108:djv375. doi: 10.1093/jnci/djv375. [DOI] [PubMed] [Google Scholar]
- 83.Liu H., Yang B., Sun T., Lin L., Hu Y., Deng M., Yang J., Liu T., Li J., Sun S., Jiao S. Specific growth inhibition of ErbB2-expressing human breast cancer cells by genetically modified NK-92 cells. Oncol. Rep. 2015;33:95–102. doi: 10.3892/or.2014.3548. [DOI] [PubMed] [Google Scholar]
- 84.Müller T., Uherek C., Maki G., Chow K.U., Schimpf A., Klingemann H.G., Tonn T., Wels W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008;57:411–423. doi: 10.1007/s00262-007-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boissel L., Betancur-Boissel M., Lu W., Krause D.S., Van Etten R.A., Wels W.S., Klingemann H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. OncoImmunology. 2013;2:e26527. doi: 10.4161/onci.26527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oelsner S., Friede M.E., Zhang C., Wagner J., Badura S., Bader P., Ullrich E., Ottmann O.G., Klingemann H., Tonn T., Wels W.S. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19:235–249. doi: 10.1016/j.jcyt.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 87.Liu Q., Xu Y., Mou J., Tang K., Fu X., Li Y., Xing Y., Rao Q., Xing H., Tian Z. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19+ malignancy in a mouse model. Cytotherapy. 2020;22:552–562. doi: 10.1016/j.jcyt.2020.06.003. [DOI] [PubMed] [Google Scholar]
- 88.Jiang H., Zhang W., Shang P., Zhang H., Fu W., Ye F., Zeng T., Huang H., Zhang X., Sun W. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014;8:297–310. doi: 10.1016/j.molonc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen K.H., Wada M., Firor A.E., Pinz K.G., Jares A., Liu H., Salman H., Golightly M., Lan F., Jiang X., Ma Y. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. 2016;7:56219–56232. doi: 10.18632/oncotarget.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen K.H., Wada M., Pinz K.G., Liu H., Lin K.W., Jares A., Firor A.E., Shuai X., Salman H., Golightly M. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31:2151–2160. doi: 10.1038/leu.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu Y., Liu Q., Zhong M., Wang Z., Chen Z., Zhang Y., Xing H., Tian Z., Tang K., Liao X. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019;12:49. doi: 10.1186/s13045-019-0732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.You F., Wang Y., Jiang L., Zhu X., Chen D., Yuan L., An G., Meng H., Yang L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019;9:64–78. [PMC free article] [PubMed] [Google Scholar]
- 94.Han J., Chu J., Keung Chan W., Zhang J., Wang Y., Cohen J.B., Victor A., Meisen W.H., Kim S.H., Grandi P. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015;5:11483. doi: 10.1038/srep11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Genßler S., Burger M.C., Zhang C., Oelsner S., Mildenberger I., Wagner M., Steinbach J.P., Wels W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology. 2015;5:e1119354. doi: 10.1080/2162402X.2015.1119354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang Q., Tian K., Xu J., Zhang H., Li L., Fu Q., Chai D., Li H., Zheng J. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J. Immunol. Res. 2017;2017:6915912. doi: 10.1155/2017/6915912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Q., Zhang H., Ding J., Liu H., Li H., Li H., Lu M., Miao Y., Li L., Zheng J. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J. Immunol. Res. 2018;2018:4263520. doi: 10.1155/2018/4263520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seidel D., Shibina A., Siebert N., Wels W.S., Reynolds C.P., Huebener N., Lode H.N. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 2015;64:621–634. doi: 10.1007/s00262-015-1669-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yu M., Luo H., Fan M., Wu X., Shi B., Di S., Liu Y., Pan Z., Jiang H., Li Z. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol. Ther. 2018;26:366–378. doi: 10.1016/j.ymthe.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao B., Liu M., Wang L., Liang B., Feng Y., Chen X., Shi Y., Zhang J., Ye X., Tian Y. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020;524:96–102. doi: 10.1016/j.bbrc.2020.01.053. [DOI] [PubMed] [Google Scholar]
- 101.Montagner I.M., Penna A., Fracasso G., Carpanese D., Dalla Pietà A., Barbieri V., Zuccolotto G., Rosato A. Anti-PSMA CAR-engineered NK-92 cells: An off-the-shelf cell therapy for prostate cancer. Cells. 2020;9:E1382. doi: 10.3390/cells9061382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang G., Liu R., Zhu X., Wang L., Ma J., Han H., Wang X., Zhang G., He W., Wang W. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013;91:615–624. doi: 10.1038/icb.2013.45. [DOI] [PubMed] [Google Scholar]
- 103.Zhao Q., Ahmed M., Tassev D.V., Hasan A., Kuo T.Y., Guo H.F., O’Reilly R.J., Cheung N.K.V. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015;29:2238–2247. doi: 10.1038/leu.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang S., Cao B., Zhou G., Zhu L., Wang L., Zhang L., Kwok H.F., Zhang Z., Zhao Q. Targeting B7-H3 immune checkpoint with chimeric antigen receptor-engineered natural killer cells exhibits potent cytotoxicity against non-small cell lung cancer. Front. Pharmacol. 2020;11:1089. doi: 10.3389/fphar.2020.01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robbins Y., Greene S., Friedman J., Clavijo P.E., Van Waes C., Fabian K.P., Padget M.R., Abdul Sater H., Lee J.H., Soon-Shiong P. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. eLife. 2020;9:e54854. doi: 10.7554/eLife.54854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 108.Tambaro F.P., Singh H., Jones E., Rytting M., Mahadeo K.M., Thompson P., Daver N., DiNardo C., Kadia T., Garcia-Manero G. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. 2021 doi: 10.1038/s41375-021-01232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Burger M.C., Zhang C., Harter P.N., Romanski A., Strassheimer F., Senft C., Tonn T., Steinbach J.P., Wels W.S. CAR-engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front. Immunol. 2019;10:2683. doi: 10.3389/fimmu.2019.02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nath S., Mukherjee P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014;20:332–342. doi: 10.1016/j.molmed.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Q., Wang Y., Lin M., Xia L., Bao Y., Sun X., Yang L. Phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunol. Res. 2019;7(2, Suppl):A014. [Google Scholar]
- 112.Adams E., Wildiers H., Neven P., Punie K. Sacituzumab govitecan and trastuzumab deruxtecan: Two new antibody-drug conjugates in the breast cancer treatment landscape. ESMO Open. 2021;6:100204. doi: 10.1016/j.esmoop.2021.100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hermanson D.L., Kaufman D.S. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front. Immunol. 2015;6:195. doi: 10.3389/fimmu.2015.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Z., Guo L., Song Y., Zhang Y., Lin D., Hu B., Mei Y., Sandikin D., Liu H. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-βR II and NKG2D. Cancer Immunol. Immunother. 2017;66:537–548. doi: 10.1007/s00262-017-1959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]