

Summary
The Linear Ubiquitin Chain Assembly Complex (LUBAC), composed of HOIP, HOIL-1L, and SHARPIN, promotes tumor necrosis factor (TNF)-dependent NF-κB signaling in diverse cell types. HOIL-1L contains an Npl4 Zinc Finger (NZF) domain that specifically recognizes linear ubiquitin chains, but its physiological role in vivo has remained unclear. Here, we demonstrate that the HOIL-1L NZF domain has important regulatory functions in inflammation and immune responses in mice. We generated knockin mice (Hoil-1lT201A;R208A/T201A;R208A) expressing a HOIL-1L NZF mutant and observed attenuated responses to TNF- and LPS-induced shock, including prolonged survival, stabilized body temperature, reduced cytokine production, and liver damage markers. Cells derived from Hoil-1lT201A;R208A/T201A;R208A mice show reduced TNF-dependent NF-κB activation and incomplete recruitment of HOIL-1L into TNF Receptor (TNFR) Complex I. We further show that HOIL-1L NZF cooperates with SHARPIN to prevent TNFR-dependent skin inflammation. Collectively, our data suggest that linear ubiquitin-chain binding by HOIL-1L regulates immune responses and inflammation in vivo.
Subject areas: Biological sciences, Molecular biology, Immune response
Graphical abstract
Highlights
-
•
An RBR-type E3 ligase HOIL-1L decodes linear ubiquitin chains via the NZF domain
-
•
HOIL-1L NZF is essential for proper responses to LPS and TNF-induced shock in mice
-
•
Intact HOIL-1L NZF is required for activating the TNF-induced NF-kB pathway
-
•
HOIL-1L NZF cooperates with SHARPIN to control inflammation in mice
Biological sciences; Molecular biology; Immune response
Introduction
Tumor necrosis factor (TNF) is a pleiotropic cytokine involved in the maintenance and regulation of homeostasis and in the response to bacterial and viral infections (Webster and Vucic, 2020; Karki et al., 2021). TNF binding to TNF Receptor 1 (TNFR1) leads to the formation of TNFR Complex I and, in turn, nuclear factor kappa B (NF-κB) signaling. This signaling activates cell survival by inducing the transcription of pro-inflammatory and anti-apoptotic genes (Gómez-Díaz and Ikeda, 2019; Peltzer and Walczak, 2019). TNFR Complex I includes, among other proteins, Receptor Interacting serine/threonine Protein Kinase 1 (RIPK1), TNF Receptor type 1 Associated DEATH Domain (TRADD), TNF Receptor Associated Factor 2 (TRAF2), and cellular Inhibitor of Apoptosis Protein (cIAP)1/2 (Figure S1A) (Peltzer and Walczak, 2019).
Post-translational modifications, including ubiquitination and phosphorylation, regulate signal transduction downstream of TNFR Complex I. For example, the E3 ubiquitin ligases cIAP1/2 ubiquitinate themselves and RIPK1, leading to the recruitment of the Linear UBiquitin Chain Assembly Complex (LUBAC) (Haas et al., 2009) and a kinase complex IKK. LUBAC is the only known mammalian E3 ubiquitin ligase, which generates linear (Met1-linked) ubiquitin chains and stabilizes TNFR Complex I by ubiquitinating various substrates, such as NF-κB Essential Modifier (NEMO) and RIPK1 (Justus and Ting, 2015; Gómez-Díaz and Ikeda, 2019; Peltzer and Walczak, 2019). Thus, LUBAC promotes NF-κB signaling and cell survival via TNFR Complex I (Gómez-Díaz and Ikeda, 2019).
Sustained or excessive TNF stimulation leads to dissociation of RIPK1 from TNFR Complex I and its association with TNFR Complex II (Justus and Ting, 2015; Witt and Vucic, 2017). TNFR Complex II induces either apoptosis, by activating caspase-8, followed by caspase-3 (Witt and Vucic, 2017), or necroptosis by activating RIPK3 and Mixed Lineage Kinase domain Like pseudo kinase (MLKL) (Witt and Vucic, 2017). LUBAC-mediated linear ubiquitination negatively regulates TNFR Complex II-dependent apoptosis (Asaoka and Ikeda, 2015; Sasaki and Iwai, 2015; Peltzer and Walczak, 2019).
LUBAC consists of three proteins: two RING-in between-RING (RBR)-E3 ubiquitin ligases, the Heme-Oxidized IRP2 ubiquitin Ligase 1 (HOIL-1L) and the HOIL-1L Interacting Protein (HOIP) and the adaptor the Shank-Associated RH Domain-Interacting Protein (SHARPIN) (Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011). Genetic deletion of HOIP or HOIL-1L in mice leads to embryonic lethality due to aberrant endothelial cell death, namely, apoptosis and necroptosis in the vasculature (Peltzer et al., 2014, 2018). SHARPIN-deficient mice (Sharpincpdm/cpdm) are viable and display an inflammatory phenotype characterized by chronic proliferative dermatitis (Seymour et al., 2007), which is largely resolved by loss of TNF or TNFR (Gerlach et al., 2011; Kumari et al., 2014; Rickard et al., 2014). Mutations in genes encoding HOIL-1L and HOIP have been associated with autoimmune disorders, implicating LUBAC in the regulation of immune responses in humans (Boisson et al., 2012, 2015).
HOIL-1L is required for proper inflammatory responses and embryonic development (Peltzer et al., 2018; Kelsall et al., 2019; Fuseya et al., 2020). It consists of multiple domains including the Npl4 Zinc Finger (NZF), the ubiquitin-like domain (UBL), and the RBR domains (Figures 1A and S1B). Some of the biochemical functions of each domain are known: NZF binds a linear ubiquitin chain (Sato et al., 2011; Fennell et al., 2018), UBL binds other LUBAC components, and RBR catalyzes ester-bond linkage of ubiquitination (Kelsall et al., 2019; Carvajal, 2021). Based on the crystal structural study of the isolated NZF region of HOIL-1L, Threonine 203 and Arginine 210 within human HOIL-NZF were identified to be essential to establish the interaction with a linear di-ubiquitin chain (Sato et al., 2011). Mutation of these residues results in decreased LUBAC-induced NF-κB reporter activation in over-expression-based luciferase assays (Sato et al., 2011). However, it remains unclear whether the HOIL-1L NZF domain directly regulates the ubiquitin ligase activity of LUBAC and to what extent it controls immunity and inflammation in vivo.
Figure 1.

Mutations in the HOIL-1L NZF domain (Thr 203 Ala/Arg 210 Ala) reduce the NF-κB reporter activity without blocking LUBAC catalytic activity
(A) Schematic representation of human HOIL-1L with the annotated domains and the T203A/R210A double point mutation in the HOIL-1L NZF domain (human HOIL-1L TA/RA).
(B) GST-pull-down assay examining interaction between linear di-ubiquitin and HOIL-1L (WT or TA/RA). Total cell lysates of HEK293T cells transfected with indicated plasmids were incubated with GST-empty control or GST-tagged linear di-ubiquitin (GST-Lin Ub2). Total cell lysates (Input) and pull-down samples were examined by immunoblotting using an α-HOIL-1L antibody. Input amount of total cell lysates and GST-fusion proteins was visualized by Ponceau S staining.
(C) Luciferase-based NF-κB reporter assay using lysates of HEK293T cells co-transfected with an NF-κB reporter plasmid, Renilla-luciferase expression plasmid (internal control), with or without Myc-HOIP (WT or catalytic inactive C885A mutant), Flag-SHARPIN (WT), and HOIL-1L-HA (WT or TA/RA) plasmids.
(D) Immunoblots of co-immunoprecipitation samples examining interaction between Myc-HOIP and HOIL-1L-HA (WT or T203A/R210A) or GFP-SHARPIN. Total cell lysates of HEK293T cells transiently expressing tagged LUBAC components as indicated, were immunoprecipitated by using an α-Myc antibody. Total cell lysates (Input) and immunoprecipitates (IP: α-Myc) were subjected to SDS-PAGE followed by immunoblotting using the indicated antibodies.
(E) In vitro ubiquitination assays using the recombinant proteins of ubiquitin (Ub), mouse Ube1 (E1), E2 (UbcH7), HOIP, HOIL-1L (WT, TA/RA or a mutant of ubiquitin loading site C460A), SHARPIN, and NEMO. Samples were subjected to SDS-PAGE and linear ubiquitin chain formation and modifications of NEMO, HOIP, HOIL-1L, and SHARPIN were detected by immunoblotting with the indicated antibodies.
(F) In vitro ubiquitination assays using the recombinant proteins of ubiquitin (Ub), Ube1 (E1), E2 (UbcH7), HOIP, HOIL-1L (WT, TA/RA, or a mutant of ubiquitin loading site C460A), and NEMO. Linear ubiquitin chain formation and modifications of NEMO, HOIP, and HOIL-1L were detected by immunoblotting with the indicated antibodies.
(G) LUBAC-induced ubiquitination of NEMO and linear ubiquitin chain formation in HEK293T cells determined by immunoblotting. Total cell lysates transiently expressing Flag-NEMO, Myc-HOIP (WT or C885A mutant), GFP-SHARPIN, HOIL-1L-HA (WT or T203A/R210A mutant) were subjected to SDS-PAGE followed by immunoblot with the indicated antibodies. An α-Vinculin antibody was used to monitor loading amount. Data are representative of at least three independent experiments. (C) Data are represented as mean ± SD, ANOVA, n = 4, ∗p value ≤0.05, ∗∗∗∗p value ≤0.0001.
Results
HOIL-1L NZF mutations block linear ubiquitin chain binding and NF-κB activation without disrupting LUBAC activity
The NZF domain of HOIL-1L (aa 192–250) specifically recognizes linear ubiquitin chains via two key residues, Threonine 203 and Arginine 210 (Figure 1A) (Sato et al., 2011). To confirm the importance of these residues for binding, we expressed HOIL-1L wild type (WT) or HOIL-1L T203A/R210A in HEK293T cells and performed pull-down assays with GST-linear-di-ubiquitin (GST-Lin Ub2). Similar to the previous observations using recombinant proteins (Sato et al., 2011), WT HOIL-1L but not HOIL-1L T203A/R210A expressed in mammalian cells interacted with GST-Lin Ub2 (Figure 1B).
To verify that the HOIL-1L NZF domain promotes NF-κB activation via LUBAC, we employed luciferase-based NF-κB reporter assays in HEK293T cells co-expressing HOIP. HOIL-1L WT activated the NF-κB reporter with or without SHARPIN, as previously shown (Tokunaga et al., 2009; Gerlach et al., 2011; Ikeda et al., 2011) (Figures 11C, and S1B–S1D). In contrast, NF-κB reporter activity was decreased in cells expressing either HOIL-1L T203A/R210A or HOIL-1L-ΔNZF (Figures 1C, and S1B–S1E), even though HOIL-1L T203A/R210A supported LUBAC formation (Figure 1D). We also examined a HOIL-1L-RBR-NZF mutant and the catalytic inactive mutant HOIL-1L C460A and observed that NF-κB reporter activity was decreased with HOIL-1L- RBR-NZF but increased with HOIL-1L C460A (Figures S1D, S1F and S1G).
To assess the catalytic activity of LUBAC containing HOIL-1L T203A/R210A, we examined the formation of unanchored linear ubiquitin chains as well as the ubiquitination of known substrates by recombinant proteins in vitro (Figures 1E, 1F, and S1H). Similar to HOIL-1L WT, LUBAC containing HOIL-1L T203A/R210A supported the formation of unanchored linear ubiquitin chains and the ubiquitination of NEMO, HOIP, HOIL-1L, and SHARPIN (Figure 1E). In addition, LUBAC containing HOIL-1L T203A/R210A supported NEMO ubiquitination in HEK293T cells (Figure 1G). Collectively, these results suggest that loss of the HOIL-1L NZF domain impairs LUBAC-dependent activation of the NF-κB pathway without severely compromising LUBAC ligase activity.
Of note, in the absence of SHARPIN, unanchored and anchored ubiquitin chain formation was clearly delayed with HOIL-1L T203A/R210A compared with HOIL-1L WT (Figure 1F), suggesting that SHARPIN and the HOIL-1L NZF domain have a collaborative role in promoting the E3 ligase functions of LUBAC.
In summary, our data suggest that the reduction of NF-κB reporter activity by the HOIL-1L NZF mutations are not due to loss of LUBAC activity.
The HOIL-1L NZF domain is required for endogenous TNF-induced NF-κB signaling
To determine if the HOIL-1L NZF domain and binding linear ubiquitin regulates immune signaling cascades in vivo, we generated Hoil-1lT201A;R208A/T201A;R208A knockin mice (mutations at the equivalent residues to T203 and R210 in human HOIL-1L) using CRISPR-Cas9 technology (Figures S2A–S2C). Hoil-1lT201A;R208A/T201A;R208A (referred as Hoil-1lnzf∗/nzf∗) mice were born at nearly the expected ratio and showed no obvious developmental defects (Figures 2A and 2B). Histological analysis of various tissues (spleen, liver, intestine, and skin) from 11-week-old male mice displayed no clear differences between Hoil-1l+/+ (wild type) and Hoil-1lnzf∗/nzf∗ further supporting that these mice develop normally under basal conditions (Figure 2C).
Figure 2.

Hoil-1lnzf∗/nzf∗ knockin mice display no developmental phenotype yet TNF-induced NF-κB activation is reduced in Hoil-1lnzf∗/nzf∗cells
(A) Numbers of weaned and expected pups of the indicated genotypes from Hoil-1l+/nzf∗crosses.
(B) Gross appearance image of 11-week-old Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ mice. Scale bar: 10 mm.
(C) H&E staining of the indicated tissues from 11-week-old Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ mice. Scale bar: 200 μm.
(D) Immunoblotting to detect phosphorylation of IKK-α/β in Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ MEFs treated with mouse TNF (20 ng/ml) for the indicated time. An α-Vinculin antibody was used to monitor loading amount. ∗ Indicates modified HOIL-1L.
(E) mRNA levels of NF-κB targets (TNF, IκB-α, A20, and ICAM) determined by qRT-PCR in Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ BMDMs treated with mouse TNF (20 ng/mL) for the indicated time. Normalization was done to β-actin.
(F) Immunoblotting to detect TNFR Complex I formation in Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ MEFs. Total cell lysates (Input) of cells treated with or without human TNF (100 ng/mL), and immunoprecipitates (IP: α-FLAG) were subjected to SDS-PAGE. Recruitment of HOIP, SHARPIN, and HOIL-1L was monitored by immunoblotting. An α-HOIL-1L antibody (Atlas Antibody, HPA 024185) was used for detecting HOIL-1L in TNFR Complex I and input. α-HOIL-1L (Merck Millipore, MABC576) was additionally used to detect HOIL-1L in input. α-Vinculin antibody was used to monitor loading amount. Data are representative of at least three independent experiments. (E) Data are represented as mean ± SD. ANOVA, n = 3, ∗∗p value ≤0.01, ∗∗∗p value ≤0.001, ∗∗∗∗p value ≤0.0001.
To assess the physiological role of the HOIL-1L NZF domain in NF-κB signaling, we established immortalized MEFs from Hoil-1lnzf∗/nzf∗ mice and exposed them to TNF. Hoil-1lnzf∗/nzf∗ MEFs displayed similar levels of unmodified HOIL-1L but lower levels of modified HOIL-1L (presumably mono-ubiquitination) (Kelsall et al., 2019) compared with wild-type MEFs (Figure 2D), suggesting that the HOIL-1L NZF domain and binding linear ubiquitin chains contribute to HOIL-1L mono-ubiquitination in cells. Of importance, TNF-induced phosphorylation of the NF-κB signaling component IKKα/β and degradation of IκB-α were reduced in Hoil-1lnzf∗/nzf∗ MEFs compared with wild-type MEFs (Figures 2D, and S2D). Similar results were observed in Hoil-1lnzf∗/nzf∗ bone marrow-derived macrophages (BMDMs) (Figure S2E). Moreover, the TNF-induced expression of four NF-κB-target genes was reduced in both Hoil-1lnzf∗/nzf∗ MEFs (Figure S2F) and Hoil-1lnzf∗/nzf∗ BMDMs (Figure 2E). These data indicate that loss of the HOIL-1L NZF domain function inhibits TNF-induced NF-κB signaling in non-immune and immune cells.
To decipher how the HOIL-1L NZF domain regulates the TNF-induced NF-κB pathway, we stimulated MEFs with Flag-TNF and performed pull-down assays to analyze TNFR Complex I formation. Wild-type and Hoil-1lnzf∗/nzf∗ MEFs expressed similar levels of the individual LUBAC components (Figure 2F, input) and showed comparable TNF-induced recruitment of IKKα and RIPK1 in the TNFR Complex I (Figures S2G and S2H). We also observed a similar pattern of the modification of RIPK1 in the TNFR Complex I in wild-type and Hoil-1lnzf∗/nzf∗ MEFs (Figure S2H). In contrast, recruitment of HOIL-1L into the TNFR Complex I was mildly reduced at the 15-min time point in Hoil-1lnzf∗/nzf∗ MEFs (Figure 2F, indicated with arrows). These data suggest that more transient recruitment of HOIL-1L to TNFR Complex I could underlie the reduced NF-κB activation in Hoil-1lnzf∗/nzf∗ MEFs.
LUBAC negatively regulates TNF-dependent apoptosis (Asaoka and Ikeda, 2015; Sasaki and Iwai, 2015; Peltzer and Walczak, 2019). Thus, we next examined whether the HOIL-1L NZF domain participates in the regulation of TNF-dependent apoptosis. Wild-type and Hoil-1lnzf∗/nzf∗ MEFs treated with TNF and cycloheximide showed similar caspase-8 activity (Figure S2I), and similar levels of cleaved caspase-3 and cleaved poly ADP ribose polymerase (PARP) (Figure S2J), suggesting that the HOIL-1L NZF domain is not required for LUBAC to inhibit TNF-dependent apoptosis induction in these cells.
Collectively, these results suggest that the HOIL-1L NZF domain is dispensable for inhibition of TNF-induced apoptosis by LUBAC, but promotes TNF-induced NF-κB signaling, likely by maintaining LUBAC components in the TNFR Complex I.
HOIL-1L NZF mutations attenuate TNF-induced shock and LPS-induced septic shock in mice
Having determined that the HOIL-1L NZF domain regulates the TNF-induced NF-κB pathway in cells, we sought to address its contribution in vivo during TNF-induced shock. To this end, mouse TNF (mTNF) was intravenously injected into wild-type and Hoil-1lnzf∗/nzf∗ mice and their responses were monitored for up to 12 hours. Wild-type mice displayed a drop in body surface temperature starting at 6 hours post TNF injection, as expected, whereas Hoil-1lnzf∗/nzf∗ mice were more resistant (Figure 3A). We investigated the TNF-induced immune responses in these mice by measuring cytokine levels in the serum. The TNF-induced pro-inflammatory cytokines interleukin (IL-6), IL-12 (p70), and granulocyte colony stimulating factor (G-CSF), which are all NF-κB targets (Pahl, 1999), were lower in serum from Hoil-1lnzf∗/nzf∗ mice than in serum from wild-type mice (Figures 3B–3D). Overall, these data suggest that Hoil-1lnzf∗/nzf∗ mice have lower TNF-induced NF-κB signaling and are thus more resistant to TNF-induced shock.
Figure 3.

Hoil-1lnzf∗/nzf∗ mice show reduced responses to TNF-induced shock
(A) Temperature of Hoil-1l+/+ (n = 5) and Hoil-1lnzf∗/nzf∗ (n = 4) upon intravenous TNF injection (450 μg/kg) monitored over time.
(B–D) IL-6 (B), IL-12 (C), and G-CSF (D) levels in the serum of unstimulated (UNST) and TNF-injected mice. After 12 h of TNF injection, serum levels of indicted cytokines were measured by ELISA. UNST: Hoil-1l+/+ (n = 6), Hoil-1lnzf∗/nzf∗ (n = 6), TNF: Hoil-1l+/+ (n = 5), Hoil-1lnzf∗/nzf∗ (n = 4). Data are represented as mean ± SD. ANOVA, ∗p value ≤0.05, ∗∗∗p value ≤0.001.
To further address the role of the HOIL-1L NZF domain in immune responses, we examined LPS-induced septic shock, which is largely dependent on the TNF signaling pathway (Mandal et al., 2018; Vandewalle et al., 2019). Intriguingly, Hoil-1lnzf∗/nzf∗ mice survived longer than wild-type littermates after LPS-induced septic shock (Figure 4A), despite similarly sharp drops in temperature (Figure 4B), similar levels of infiltrating myeloid cells in the peritoneal cavity (Figures S3A–S3D), and similar serum levels of the pro-inflammatory cytokines, IL-6, IL-27, and G-CSF (Figures 4D–4F). LPS-dependent induction of four NF-κB target genes was also comparable in wild-type and Hoil-1lnzf∗/nzf∗ BMDMs, suggesting that HOIL-1L NZF is not directly involved in the LPS-TLR4-NF-κB pathway at least in BMDMs (Figure 4C). However, the serum levels of TNF, IL-1α, and IL-1β were decreased in Hoil-1lnzf∗/nzf∗mice compared with wild-type mice (Figures 4G–4I), suggesting that these cytokines could underlie the distinct LPS-induced immune responses. Furthermore, Hoil-1lnzf∗/nzf∗ mice displayed decreased serum levels of the liver enzyme aspartate aminotransferase (AST) compared with the wild type after LPS injection, indicating reduced liver damage markers in Hoil-1lnzf∗/nzf∗ mice (Figures 4J and 4K). These results indicate that the HOIL-1L NZF domain promotes LPS-induced septic shock, accompanied by selected cytokine induction and increased levels of liver damage markers.
Figure 4.

Hoil-1lnzf∗/nzf∗mice are more resistant to LPS-induced septic shock than wild-type mice
(A) Survival curve of Hoil-1l+/+ (n = 9) and Hoil-1lnzf∗/nzf∗ (n = 8) mice upon LPS injection. Hoil-1l+/+ (n = 9) and Hoil-1lnzf∗/nzf∗ (n = 8) mice were injected with 30 mg/kg of LPS, and the survival was monitored up to 80 h.
(B) Body temperature of Hoil-1l+/+ (n = 5) and Hoil-1lnzf∗/nzf∗ (n = 5) mice upon LPS injection (30 mg/kg) monitored over time.
(C) qRT-PCR to monitor mRNA transcript levels of TNF, IL-6, IκB-α, IL-1β determined in Hoil-1l+/+ and Hoil-1lnzf∗/nzf∗ BMDMs treated with LPS (10 ng/mL) for the indicated time. Normalization was done to β-actin.
(D–I) ELISA to monitor IL-27, G-CSF, IL-6, TNF, IL-1α, and IL-1β levels in the serum of control and LPS-injected (30 mg/kg) mice. Samples for LPS-injected mice were collected after 12 h of LPS injection. Unstimulated (UNST): Hoil-1l+/+ (n = 6) and Hoil-1lnzf∗/nzf∗ (n = 6), and LPS-injected (LPS): Hoil-1l+/+ (n = 6) and Hoil-1lnzf∗/nzf∗ (n = 7).
(J and K) ALT and AST levels in the sera of Hoil-1l+/+ (n = 7) and Hoil-1lnzf∗/nzf∗ (n = 7) mice 12 h post LPS injection (30 mg/kg). (A) Log-rank (Mantel-Cox test) ∗∗p value = 0.0018. (B-I) Data are represented as mean ± SD, ANOVA. (C) Representative data from three independent experiments, n = 3, ∗p value ≤0.05. (D-I) ∗p value ≤0.05, ∗∗∗p value ≤0.001. (J-K) Data are presented as mean ± SD, Student’s t, ∗p value ≤0.05.
Collectively, we infer that mutations in the HOIL-1L NZF domain reduce TNF-induced NF-κB signaling and render mice resistant to TNF- and LPS-induced shock.
HOIL-1L NZF mutations exacerbate systemic inflammation in Sharpincpdm/cpdm mice
Given the partially redundant biochemical properties of HOIL-1L and SHARPIN in vitro, we investigated redundancy in vivo by generating Hoil-1lnzf∗/nzf∗ mice in a SHARPIN-deficient background (Sharpincpdm/cpdm). Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice were viable and born nearly at the Mendelian ratio (Figure 5A) but presented visible skin inflammation already at 2 weeks after birth, appeared smaller, and had reduced body weight compared with all other littermates, including Sharpincpdm/cpdm mice (Figures 5B and 5C). At 4 weeks after birth, Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice displayed systemic inflammation characterized by inflammatory cell infiltration in the liver, intestines, and lung (Figure S4A).
Figure 5.

Hoil-1lnzf∗/nzf; Sharpincpdm/cpdm mice show systemic inflammation and a distinctive immune cell composition
(A) Numbers of weaned and expected pups of the indicated genotypes from Hoil-1l+/nzf∗; Sharpin+/cpdm crosses.
(B) Representative image of the gross appearance from 2-week-old mice of the indicated genotypes. Scale bar: 10 mm.
(C) Body weight of 4-week-old mice from the indicated genotypes.
(D) Representative images of the spleen of indicated genotype. Scale bar: 10 mm.
(E) Spleen weight of 4-week-old mice from the indicated genotypes.
(F) H&E staining, CD45R and CD3 immunostaining of spleen sections from 4-week-old mice of the indicated genotypes. Scale bar: 50 μm.
(G) Percentage of viable myeloid cells (CD11b+), normalized to the frequency of viable CD45+ cells, in the spleen from 4-week-old mice of the indicated genotypes.
(H) Percentage of neutrophils (CD11bHi Ly6G+) out of viable CD45+ cells in the spleen from 4-week-old mice of the indicated genotypes.
(I and J) Percentage of inflammatory monocytes (CD11c- CD11b+ Ly6G− Ly6C+) (I) and conventional monocytes (CD11c− CD11b+ Ly6G− Ly6C−) (J), out of viable CD45+ CD11b+ cells, in the spleen from 4-week-old mice of the indicated genotypes.
(K) Percentage of plasma cells (CD138+ CD28+), out of viable B220+ Lin− cells, in the mesenteric lymph node (mLN) from 4-week-old mice of the indicated genotypes. The lineage (Lin) cocktail consists of TCRβ, DX5, CD11b, and CD23. Data are represented as mean ± SD. ANOVA, ∗∗p value≤0.01, ∗∗∗p value≤0.001, ∗∗∗∗p value ≤0.0001. (C and E) Hoil-1l+/+; Sharpin+/+; TnfrI+/+ (n = 13 for C, n = 15 for E), Hoil-1lnzf∗/nzf∗; Sharpin+/+;TnfrI+/+ (n = 10), Hoil-1l+/+; Sharpincpdm/cpdm ;TnfrI+/+ (n = 11 for C, n = 10 for E), Hoil-1l+/nzf∗; Sharpincpdm/cpdm;TnfrI+/+ (n = 25), Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm;TnfrI+/+ (n = 9 for C, n = 7 for E), Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm; TnfrI−/− (n = 5) (G-K) Hoil-1l+/+; Sharpin+/+ (n = 8 for G-J, n = 6 for K), Hoil-1lnzf∗/nzf∗; Sharpin+/+ (n = 6 for G-J, n = 7 for K), Hoil-1l+/+; Sharpincpdm/cpdm (n = 6), Hoil-1l+/nzf∗; Sharpincpdm/cpdm (n = 9 for G-J, n = 8 for K), Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm (n = 4).
Of note,Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice had smaller spleens compared with all other littermates (Figures 5D and 5E). However, histopathological analysis revealed that disruption of the HOIL-1L NZF did not exacerbate the disruption of the splenic structure in SHARPIN-deficient mice (Figure 5F, H&E). Mutation of the HOIL-1L NZF domain did not exacerbate the reduced frequency of CD45R+CD19+ B cells or the increased frequency of myeloid cells in Sharpincpdm/cpdm spleens (Figures S4B, 5G and S4E) (Gurung et al., 2016; Sharma et al., 2019). Although CD45R/B220+ B cells and CD3+ T cells were present in Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm spleens determined by histological analysis, their distributions were altered compared with spleens of littermates (Figure 5F). The frequency of mature B cells and plasma cells in the spleen were also similar in all analyzed genotypes (Figures S4B–S4D). Neutrophils and inflammatory monocytes were reduced in the spleens of Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice compared with Sharpincpdm/cpdm mice (Figures 5H, 5I, S4F, and S4G), whereas conventional monocytes were increased (Figures 5J and S4F). The percentage of total T cells in the spleen was similar across the genotypes analyzed (Figure S4H), as was the percentage of CD4+ and CD8+ T cells (Figures S4I and S4J). Collectively, our results show that mutation of the HOIL-1L NZF domain alters the composition of the myeloid cell compartment in the spleens of SHARPIN-deficient mice.
SHARPIN deficiency, irrespective of the HOIL-1L NZF domain, increased the frequency of myeloid cells (Figure S5A) and decreased the frequency of B cells (Figure S5B) in the mesenteric lymph node (mLN), without affecting the frequency of mature B cells (Figure S5C). Strikingly, only Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice displayed an increase in plasma cells in the mesenteric lymph nodes, revealing clear additive effects of HOIL-1L NZF and SHARPIN in plasma cell responses (Figures 5K and S5D).
Collectively, these results reveal profound differences in the immune cell composition of Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice compared with SHARPIN-deficient mice and suggest a different inflammatory process in these mice that could underlie their more severe phenotype.
TNFR1 knockout resolves the inflammatory skin phenotype of Hoil-1lnzf∗/nzf∗; Sharpin cpdm/cpdm mice
The main inflammatory phenotype in SHARPIN-deficient mice is chronic proliferative dermatitis accompanied by keratinocyte apoptosis (Gerlach et al., 2011; Ikeda et al., 2011; Kumari et al., 2014; Rickard et al., 2014). We analyzed skin sections derived from Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice and observed thickening of the keratin layer (H&E and Keratin 14 [KRT14]), apoptotic cells (cleaved caspase-3), and neutrophil infiltration (Lys6G) at 2 weeks of age (Figure 6A) and at 4 weeks of age (Figures 6B and 6C). These inflammatory characteristics were not observed in any of the other genotypes (Figures 6A–6C). At 4 weeks of age, dermatitis was observed even with a single HOIL-1L NZF mutant allele in the Sharpincpdm/cpdm background (Figures 6B and 6C). To test whether the increased apoptosis phenotype observed in the Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice was cell autonomous, we analyzed MEFs derived from Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm embryos and their respective controls (Figures S6A and S6B). As previously reported, the protein levels of HOIP and HOIL-1L in MEFs were reduced when SHARPIN was absent (Figure S6A). Against the apoptotic phenotype observed in the skin tissue, TNF-induced cleavage of caspase-3 and PARP was comparable between Hoil-1l+/+; Sharpincpdm/cpdm and Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm MEFs (Figure S6B), which was further confirmed by the caspase-8 activity assays (Figure S6C) and flow cytometry using a cell viability dye (Figure S6D). These data suggest that apoptosis observed in the Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm skin tissues and isolated MEFs have different mechanisms to control cell death. Loss of HOIP in mice converts the cell death-driven dermatitis in SHARPIN-deficient mice to T cell-predominant autoimmune lesions (Sasaki et al., 2019). Therefore, we investigated whether T cell infiltration in the skin of Sharpincpdm/cpdm mice was increased by the HOIL-1L NZF mutations. Indeed, the number of CD3-positive T cells in the skin was increased in Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice when compared with Sharpincpdm/cpdm mice (Figures 6D and 6E), suggesting that HOIL-1L NZF cooperates with SHARPIN to restrict inflammation displaying characteristics of autoimmunity-like processes.
Figure 6.

TNFR1 knockout resolves skin inflammation and keratinocyte apoptosis in Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice
(A and B) H&E staining, cleaved caspase-3, Keratin 14 (KRT14), and Ly6G immunostaining of ventral skin sections of 2-, 4- or 8-week-old mice from the indicated genotypes. Scale bar: 50 μm.
(C) Quantification of the total epidermal thickness from KRT14-immunostained ventral skin sections of 4-week-old mice from the indicated genotypes.
(D) CD3 staining of epidermis and dermis of ventral skin sections from 4-week-old mice of the indicated genotypes. Scale bar: 25 μm.
(E) Quantification of CD3 positive cells in the ventral skin (epidermis and dermis) from 4-week-old mice of the indicated genotypes.
(F) Representative image of the gross appearance from 8-week-old mice of the indicated genotypes. The red arrow indicates skin lesions. Data are represented as mean ± SD, ANOVA, ∗∗p value≤0.01, ∗∗∗p value≤0.001, ∗∗∗∗p value ≤0.0001. (C and E) Hoil-1l+/+; Sharpin+/+ (n = 5 for C, n = 8 for E), Hoil-1lnzf∗/nzf∗;Sharpin+/+ (n = 4 for C and E), Hoil-1l+/+;Sharpincpdm/cpdm (n = 4 for C and E), Hoil-1L+/nzf∗;Sharpincpdm/cpdm (n = 6 for C, n = 3 for E), Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm (n = 5 for C and E). (C) Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm;Tnfr1−/− (n = 3).
Because the Sharpincpdm/cpdm skin phenotype is dependent on TNFR1 signaling (Gerlach et al., 2011; Kumari et al., 2014; Rickard et al., 2014), we hypothesized that the increased skin inflammation and apoptosis observed in Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice are TNFR1 dependent. To address this, we generated TNFR1-deficient mice in the background of Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm and analyzed their phenotype. Consistent with our hypothesis, TNFR1 knockout mitigated the skin inflammation and apoptosis observed in the 4-week-old Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice (Figures 6B, 6C, and S6E). However, at a later time point, 8 weeks, these triple-mutant mice started to display mild signs of skin inflammation and apoptosis, suggesting the TNFR1-pathway is not the only pathway that regulates skin inflammation in these mice (Figures 6B and 6F). The TNFR1 knockout resolved additional phenotypes of Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice, including body weight, spleen weight, and spleen pathology (Figures 5C–5F), indicating an involvement of the TNFR signaling pathway for these phenotypes.
Collectively, the linear ubiquitin chain-binding domain of HOIL-1L cooperates with SHARPIN to restrict excessive skin inflammation and cell death, which is at least partially dependent on TNFR1. Furthermore, an imbalance in the population of immune cells could contribute to the inflammatory phenotype observed in Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm mice.
Discussion
HOIL-1L consists of multiple domains of which functions are not fully understood. A crystal structure of a HOIL-1L fragment revealed that the NZF domain has a special C-terminal stretch that enables specific binding of linear ubiquitin chains (Sato et al., 2011). Among seven known linear ubiquitin specific binders in different proteins (Fennell et al., 2018), HOIL-1L is the only one, which is a component of LUBAC. Yet the physiological function of this HOIL-1L activity in vivo had remained totally unknown. We previously showed that the linear ubiquitin chain-specific binding domain in NEMO (called UBAN) is required for NF-κB activation in cells (Rahighi et al., 2009). In this study, we uncovered that HOIL-1L NZF contributes to the TNF-induced signaling pathway by promoting proper recruitment of HOIL-1L in the TNFR Complex I, which is in line with previous observation using the HOIL-1L NZF-mutant reconstituted in HOIL-1L knockout MEFs (Peltzer et al., 2018). Furthermore, by generating HOIL-1L NZF mutant knockin mice by the CRISPR-Cas9 technology, we, for the first-time, revealed functions of the HOIL-1L NZF domain in immune responses in vivo.
In general, it is known that the TNF-dependent NF-κB signaling pathway contributes to LPS-induced septic shock by promoting pro-inflammatory cytokine induction (Mandal et al., 2018; Vandewalle et al., 2019). When the NF-κB pathway is blocked, target cytokine induction in vivo is reduced, immune responses are suppressed, and mice are more resistant to TNF- and LPS-induced shock (Sheehan et al., 1989; Marino et al., 1997; Tortola et al., 2016; Mandal et al., 2018; Vandewalle et al., 2019). In the case of Hoil-1lnzf∗/nzf∗ mice, LPS-induced and TNF-induced shock were both milder than in control mice, consistent with their reduced TNF-induced NF-κB target gene expression. In macrophages, we observed similar levels of NF-κB-dependent gene induction by the LSP-TLR4 pathway in wild type and Hoil-1lnzf∗/nzf∗; thus, we speculate that the resistance to LPS-induced septic shock is indirectly due to the diminished TNFR pathway (summarized in Figure 7). In addition to the NF-κB signaling, linear ubiquitin chain binding by the HOIL-1L NZF might be required for the inflammasome-dependent pathway induced by septic shock. Inflammasome formation leads to the maturation and release of IL-1β and IL-18 and to pyroptosis causing tissue damage (Kayagaki et al., 2011; Broz and Dixit, 2016). Indeed, HOIL-1L was reported as a positive regulator of inflammasome assembly in cells (Rodgers et al., 2014). In support of this alternative, LPS-induced septic shock in Hoil-1lnzf∗/nzf∗ mice resulted in lower serum levels of IL-1β than in similarly treated wild-type littermates. In the future, it would be important to decipher how HOIL-1L regulates inflammasome activation.
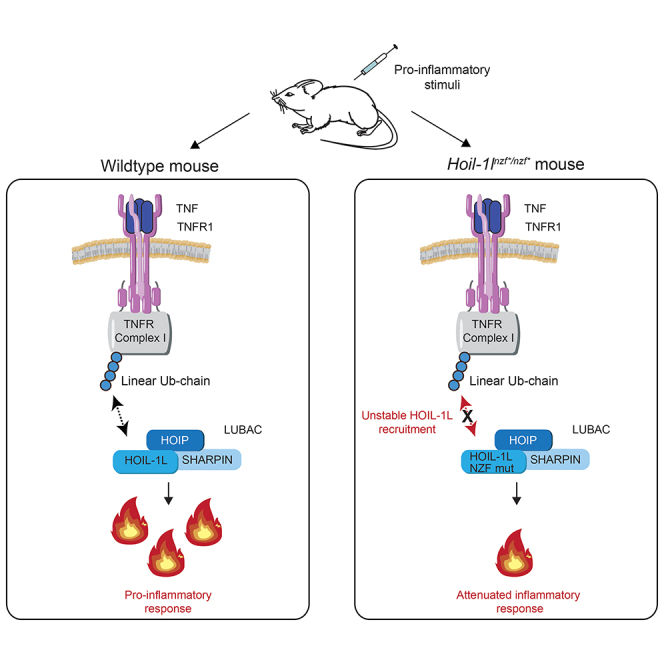
Figure 7.

Proposed model depicting the role of the linear ubiquitin chain recognition by HOIL-1L-NZF in the regulation of the LPS-induced responses via the TNFR pathway
In wild type mice (upper panel), LPS challenge leads to cytokine production, increased levels of liver damage markers, and death. At the cellular level, TNF-dependent NF-κB activation is mediated through LUBAC in which HOIL-1L-NZF binds to linear ubiquitin chains to form TNFR Complex I. Double point mutations in the HOIL-1L NZF domain, abrogating interaction with linear ubiquitin chains (lower panel), confers resistance to LPS-induced shock in mice, in which cytokine production is reduced. In cells from these mutant mice, recruitment of HOIL-1L to the TNFR Complex I is more transient than wild type thus leading to reduced NF-κB activation.
The responses to LPS- or TNF-induced shock vary in viable mice with LUBAC disruptions, including Sharpincpdm/cpdm (Nastase et al., 2016), HOIL-1L partial deletion (originally described as knockout) (Tokunaga et al., 2009), HOIL-1L ΔRING1 mice (Fuseya et al., 2020) and the HOIL-1L NZF mutant mice from this study. Sharpincpdm/cpdm mice have severe LPS-induced septic shock response, which is dependent on the caspase-1 pathway (Nastase et al., 2016). TNF-induced shock in HOIL-1L partial deletion mice leads to liver apoptosis (Tokunaga et al., 2009). HOIL-1L ΔRING1 mice are more resistant to LPS and D-GalN-induced shock with reduced apoptosis in the liver (Fuseya et al., 2020). These data further indicate that LUBAC is involved in multiple pathways that control immune responses in vivo.
Systemic inflammation of Sharpincpdm/cpdm mice can be resolved by different genetic modifications; for example, TNF KO (Gerlach et al., 2011), TNFR1 or TRADD KO (Kumari et al., 2014), and caspase-8 KO or MLKL KO (Rickard et al., 2014) largely resolve the inflammatory phenotype, indicating that TNF-induced cell death drives inflammation in Sharpincpdm/cpdm mice. Furthermore, caspase-1 KO (Nastase et al., 2016) and caspase-1/11 double KO (Douglas et al., 2015) also resolve the skin inflammation phenotype, suggesting that the caspase-1 pathway also plays an important role. More recently, HOIL-1L ΔRING1 mice were shown to resolve the Sharpincpdm/cpdm phenotype (Fuseya et al., 2020), whereas crosses with HOIL-1L ΔRBR mice (Shimizu et al., 2016) or HOIP mutant knockin mice of a specific ubiquitination site (Fennell et al., 2020) leads to embryonic lethality, suggesting that a fine balance of ubiquitination processes regulated by LUBAC contributes to the Sharpincpdm/cpdm phenotype. We observed that disruption of the HOIL-1L NZF domain exacerbated the skin inflammation phenotype of Sharpincpdm/cpdm mice, which was accompanied with keratinocyte apoptosis. Because TNFR1 KO largely resolved skin inflammation and apoptosis in these mice, we speculate that the HOIL-1L NZF cooperates with SHARPIN to prevent inflammation. Our results using isolated MEFs from Hoil-1lnzf∗/nzf∗;Sharpincpdm/cpdm and Hoil-1l+/+;Sharpincpdm/cpdm MEFs showed no clear differences in TNF-induced cell death, suggesting that the phenotype could be due to a cell type-specific effect. Alternatively, the increased inflammation and apoptosis in the skin tissues may be due to the dysregulated immune cell environment such as increased CD3-positive T cells. Indeed, the immune cell environment also in other tissues was altered by mutation of the HOIL-1L NZF domain in SHARPIN-deficient mice. Sharpincpdm/cpdm mice display splenomegaly and an increased percentage of neutrophils in the spleen, indicating that an imbalanced immune cell composition precedes the onset of dermatitis (Gurung et al., 2016). We observed a reduction of neutrophils in the spleens of Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm, despite more severe presentation of chronic proliferative dermatitis. On the other hand, Hoil-1lnzf∗/nzf∗; Sharpincpdm/cpdm displayed an increased frequency of plasma cells in the mesenteric lymph nodes, suggesting that these cells may contribute to the exacerbated skin inflammatory phenotype.
In conclusion, we demonstrate for the first time that the linear ubiquitin binding domain of HOIL-1L positively regulates the TNF-induced NF-κB signaling pathway both in vitro and in vivo (Figure 7). Disrupting the interaction of HOIL-1L-NZF with linear ubiquitin chains by small molecules may constitute a treatment for patients suffering from pathogen-associated septic shock.
Limitations of the study
-
●
HOIL-1L NZF may transiently interact with endogenous linearly ubiquitinated substrates, making endogenous targets difficult to identify.
-
●
Mechanisms of cooperative functions of HOIL-1L NZF and SHARPIN in the regulation of skin inflammation and apoptosis in mice remain open.
STAR★Methods
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myc (9E10) | Covance | MMS-150P |
| Flag (M2) | Sigma | F3165 |
| vinculin | Sigma-Aldrich, | V9131 |
| Tubulin | Abcam | ab15246 |
| ubiquitin P4D1 | Santa Cruz Biotechnology | sc-8017 |
| HOIL-1L | Merck Millipore | MABC576 |
| HOIL-1L | Atlas Antibody | HPA 024185 |
| SHARPIN | Novus | NBP2-04116 |
| Linear ubiquitin (LUB9) | Life Sensors | #AB130), |
| Human HOIP | Sigma | SAB2102031 |
| mouse HOIP homemade | described in Fennell et al., 2020 | |
| IκB-α | Cell Signaling | #4812 |
| pIκB-α | Cell Signaling | #9246 |
| IKK-α/β | Abcam | EPR16628 |
| IKK-α | Bioké | CST2682 |
| NEMO/IKKγ (FL-419) | Santa Cruz | sc-8330 |
| PARP | Cell Signaling | #9542 |
| cleaved caspase-3 | Cell Signaling | #9664 |
| FADD (for immunoprecipitation) | Santa Cruz | sc-271748 |
| FADD (for detection in cell lysates) | Abcam | ab124812 |
| RIPK1 | Cell signaling | #3493 |
| TNF Receptor I | Abcam | Ab 19139 |
| IgG-HRP | Bio-Rad | 170-6516 |
| goat anti-Rabbit IgG-HRP | Dako | P0448 |
| Viability dye eFluor780 | eBioscience | 65-0865-14 |
| CD16/CD32 Fc block | BD Bioscience | 553141 |
| CD23 | eBioscience | 25-0232-82 |
| CD93 | Invitrogen | 62-5892-82 |
| CD19 | BD Biosciences | 563333 |
| CD45 | BioLegend | 103149 |
| CD138 | BD Biosciences | 562935 |
| TCRβ | BD Biosciences | 109229 |
| CD11c | BioLegend | 117335 |
| MHC II | BioLegend | 107608 |
| CD11b | BioLegend | 101257 |
| TCRβ | BioLegend | 109205 |
| CD23 | Biolegend | 101620 |
| DX5 | BD Biosciences | 558295 |
| F4/80 | BioLegend | 123131 |
| B220 | BD Biosciences | 552772 |
| CD8 | BD Biosciences | 553035 |
| NK1.1 | BD Biosciences | 553164 |
| TCRβ | BD Biosciences | 560729 |
| CD23 | BioLegend | 101614 |
| CD21 | BioLegend | 123411 |
| Ly6G | BioLegend | 127618 |
| Ly6C | BD Biosciences | 560595 |
| TCRβ | BD Biosciences | 553170 |
| B220 | BD Biosciences | 562922 |
| CD28 | BioLegend | 122010 |
| CD4 | BioLegend | 100443 |
| CD45 | BD Biosciences | 550994 |
| Bacterial strains | ||
| BL21 (DE3) Escherichia coli | NEB | C2527H |
| DH5-α Escherichia coli | NEB | C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| Protein G Agarose beads | Roche | 1124323301 |
| anti-FLAG (M2) affinity agarose gel | Sigma Aldrich | A2220 |
| Human HOIP recombinant protein | (Fennell et al., 2020) | NA |
| Human HOIL-1L recombinant protein | (Fennell et al., 2020) | NA |
| Human SHARPIN recombinant protein | (Fennell et al., 2020) | NA |
| Critical commercial assays | ||
| ProcartaPlex Immunoassays | (Thermo Fisher Scientific) | Custom made |
| Caspase-Glo 8 Assay Systems | Promega | G8201 |
| Experimental models: cell lines | ||
| HEK293T | ATCC | |
| Experimental models: organisms/strains | ||
| C57BL/KaLawRij Sharpincpdm/cpdm | Seymour et al., 2007 | JAX stock #007599 |
| C57BL/6J Tnfr1-/- (Tnfrsf1atm1Mak/TNFRp55-deficient (B6.129-Tnfrsf1atm1Mak/J)) | Pfeffer et al., 1993 | JAX stock #002818 |
| C57BL/Hoil-1lnzf∗/nzf∗ | Reported in this study | Reported in this study |
| Oligonucleotides | ||
| HOIP C885A forward primer | 5’- GCCCGAGGAGGCGCCATGCACTTTCACTGTACC-3’ | NA |
| HOIP C885A reverse primer | 5’- GGTACAGTGAAAGTGCATGGCGCCTCCTCGGG-3’ | NA |
| HOIL-1L T203A forward primer | 5’-CAGTGCCCCGGGTGCGCCTTCATCAACAAGC-3’ | NA |
| HOIL-1L T203A reverse primer | 5’-GCTTGTTGATGAAGGCGCACCCGGGGCACTG-3’ | NA |
| HOIL-1L R210A forward primer | 5’-CAACAAGCCCACGGCGCCTGGCTGTGAG-3’ | NA |
| HOIL-1L R210A reverse primer | 5’-ACAGCCAGGCGCCGTGGGCTTGTTGATG -3’ | NA |
| HOIL-1L delta NZF forward primer | 5’ -CGTCGGTCTACGTCACGGACTCGGGGGT-3’ | NA |
| HOIL-1L delta NZF reverse primer | 5’ -TCAAGGGGAGGACGACGACGACGAAGGC-3’ | NA |
| HOIL-1L NZF+RBR forward primer | 5’-CTTGAATTCATGGTGGGCTGGCAG-3’ | NA |
| HOIL-1L NZF+RBR reverse primer | 5’ -CTGCCAGCCCACCATGAATTCAAG -3’ | NA |
| β-actin forward primer | 5’-CGGTTCCGATGCCCTGAGGCTCTT-3’ | NA |
| β-actin reverse primer | 5’-CGT CACACTTCATGATGGAATTGA-3 | NA |
| A20 forward primer | 5’ -AAAGGACTACAGCAGA GCCCAG-3’ | NA |
| A20 reverse primer | 5’-AGAGACATTTCCAGTCCGGTGG-3’ | NA |
| ICAM forward primer | 5’-AAGGAGATCACATTCACGGTG-3’ | NA |
| ICAM reverse primer | 5’-TTTGG GATGGTAGCTGGAAG-3’ | NA |
| IκB-α forward primer | 5’-GCTGAGGCACTTCTGAAAGCTG-3’ | NA |
| IκB-α reverse primer | 5’-TGGACTGGCAGACCTACCATTG-3’ | NA |
| VCAM forward primer | 5’-CTGGGAAGCTGGAACGAAGT-3’ | NA |
| VCAM reverse primer | 5’-GCCAACACTTGACC GTGAC-3’ | NA |
| mouse TNF forward primer | 5’-CATCTTCTCAAAATTCGAGTGACAA-3’ | NA |
| mouse TNF reverse primer | 5’-TGGGAGTAGACAAGGTACAACCC-3’ | NA |
| IL-6 forward primer | 5’-AGCCAG AGTCCTTCAGAGAGA-3’ | NA |
| IL-6 reverse primer | 5’-TGGTCTTGGTCCTTAGCCAC-3’ | NA |
| IL1-β forward primer | 5’- ATGAAAGACGGCACACCCAC-3’ | NA |
| IL1β reverse primer | 5’-CTGCTTGTGAGGTGCTGATG-3’ | NA |
| CCL5 forward primer | 5’-TGC TGCT TTGCCTACCTC TC-3’ | NA |
| CCL5 reverse primer | 5’-CCA C TTCTTCTCTGGGTTGG-3’ | NA |
| gRNA sequence for BbsI overhang (forward primer, gRNA A) | 5’-CACCGCTCACACCCAGGCCGTGT -3’ | NA |
| gRNA sequence for BbsI overhang (forward primer, gRNA B) | 5’ -CACCGTCTCACACCCAGGCCGTG -3’ | NA |
| gRNA sequence for BbsI overhang (reverse primer, gRNA A) | 5’-AAACACACGGCCTGGGTGTGAGC -3’ | NA |
| gRNA sequence for BbsI overhang (reverse primer, gRNA B) | 5’ -AAACCACGGCCTGGGTGTGAGAC -3’ | NA |
| Forward primer for in vitro transcription: | 5’ -TTAATACGACTCACTATAGGG -3’ | NA |
| Reverse primer for in vitro transcription: | 5’ -AAAAGCACCGACTCGGTGCC -3’ | NA |
| Single strand oligonucleotide donor template: | 5’-CCGGGCCCGGCTTTCATCAACAAACC TACTGCGCCTGGGTGTGAGATG -3’ |
NA |
| Forward primer (genotyping Hoil-1lnzf∗/nzf∗ mice) | 5’ -TGGGTTGTCAGCATGTGGTT -3’ | NA |
| Reverse primer (genotyping Hoil-1lnzf∗/nzf∗ mice) | 5’ -GTGGTTCCCTTTCTGGCTCA -3’ | NA |
| Recombinant DNA | ||
| pBABE-puro-Flag-human SHARPIN | (Ikeda et al., 2011) | NA |
| pEGFP-C1-human SHARPIN | (Ikeda et al., 2011) | NA |
| pGEX-6P-1-human HOIP | (Ikeda et al., 2011) | NA |
| pGEX-6P-1-human NEMO | (Fennell et al., 2020) | NA |
| pGEX-6P-1-human HOIL-1L | (Ikeda et al., 2011) | NA |
| pGEX-6P-1-human SHARPIN | (Ikeda et al., 2011) | NA |
| pcDNA3-human-Ubiquitin | (Ikeda et al., 2011) | NA |
| pGEX-4T1-diUbiquitin | (Rahighi et al., 2009) | NA |
| pGEX-4T1-Empty | (Rahighi et al., 2009) | NA |
| pcDNA3-human-HOIL-1L-HA | (Tokunaga et al., 2009) | NA |
| pcDNA3-Myc-human HOIP | (Tokunaga et al., 2009) | NA |
| pGex6P-1-human UbcH7 | (Stieglitz et al., 2012) | NA |
| pET49b-human HOIL-1L (C460A) | (Stieglitz et al., 2012). | NA |
| pcDNA3-Myc-human HOIP (C885A) | (Fennell et al., 2020) | NA |
| pcDNA3-human-HOIL-1L-HA(T203A/R210A) | This study | NA |
| pcDNA3-human-HOIL-1L-HA(C460A) | This study | NA |
| pcDNA3-human-HOIL-1L-HA(ΔNZF) | This study | NA |
| pcDNA3-human-HOIL-1L-HA(NZF+RBR) | This study | NA |
| pNF-κB-Luc (Stratagene) | Promega | E8491 |
| phRL-TK (Promega) | Promega | E6921 |
| Software and algorithms | ||
| FlowJo software c10 | Flowjo | https://www.flowjo.com/solutions/flowjo |
| Prism Graph Pad | Graph Pad | https://www.graphpad.com |
| QuPath v 0.2.3 | (Bankhead et al., 2017) | (Bankhead et al., 2017) |
| Other | ||
| human Flag-TNF | Enzo | ALX-522-008-C050 |
| mouse TNF | Immunotools | 12343017), |
| cycloheximide (CHX) | Sigma Aldrich | C4859 |
| Z-Val-Ala-DL-Asp-fluoromethylketone (z-VAD-OMe-FMK) | Bachem, | N-1560 |
| Lipopolysaccharide (LPS) (from Escherichia coli O11:B4) | Thermo Fisher | L4391 |
| SmaI | Thermo Fisher Scientific | ER0662 |
| Western Blot Luminol Reagent | Santa Cruz | 2048 |
| Ammonium Chloride Potasium (ACK) Lysis buffer | Thermo Fisher Scientific | A104201 |
Resource availability
Lead contact
Further information and requests for resources should be directed to the corresponding lead author, Fumiyo Ikeda (ikeda.fumiyo.375@m.kyushu-u.ac.jp).
Materials availability
All materials generated in this study are available to the research community upon request to the lead author, Fumiyo Ikeda (ikeda.fumiyo.375@m.kyushu-u.ac.jp).
Experimental models and subject details
Animals
Mouse lines and husbandry
C57BL/KaLawRij Sharpincpdm/cpdm mice were described elsewhere (Seymour et al., 2007) (JAX stock #007599). C57BL/6J Tnfr1-/- (Tnfrsf1atm1Mak/TNFRp55-deficient (B6.129- Tnfrsf1atm1Mak/J)) were described elsewhere (Pfeffer et al., 1993). Mice were housed under specific pathogen free (SPF) conditions in individually ventilated cages with a HEPA filtered air (TECNIPLAST Green line GM 500) in a 14 hour-light/10 hour-dark cycle. The microbiological status of the mouse colony was monitored by sentinel mice (solid bedding sentinels and contact sentinels). Hoil-1lnzf∗/nzf∗ mice were backcrossed with C57BL/6J mice and subsequently used to generate all the genotypes used in the study and to maintain the line. Hoil-1l+/nzf∗ C57BL/6J female and male mice, or Hoil-1lnzf∗/nzf∗ C57BL/6J female and male mice were crossed with Sharpin+/cpdm male and female mice, which were already backcrossed with C57BL/6J mice. Hoil-1l+/nzf∗; Sharpin+/cpdm male and female mice were crossed to obtain all genotypes used in this study. To maintain this mouse line Hoil-1l+/nzf∗; Sharpin+/cpdm male and female mice obtained from different litters were bred. Hoil-1l+/nzf∗; Sharpin+/cpdm female mice were bred with Tnfr1-/- male mice. Hoil-1l+/nzf∗; Sharpin+/cpdm; Tnfr1+/- male and female mice obtained from these crosses were bred to obtain all genotypes used in this study. To maintain this mouse line, Hoil-1l+/nzf∗; Sharpin+/cpdm; Tnfr1+/- male and female mice and Hoil-1l+ nzf∗ ; Sharpin+/cpdm; Tnfr1-/- male and female mice obtained from different litters were crossed.
All mice were bred and maintained in accordance with ethical animal license protocols complying with the Austrian and European legislation. Animal procedures were covered by the licenses 568809/2013/18, BMWF-V/3b/2020-0.175.109 and GZ 2020-0.648.599.
Generation of Hoil-1lnzf∗/nzf∗ mice
The design of the gRNA was performed using the guide design tool from the Zhang lab (crispr.mit.edu). (Wang et al., 2013). The gRNA template was generated using a forward and reverse oligonucleotide (IDT, HPLC purity) containing BbsI overhangs that were annealed and phosphorylated using T4 Polynucleotide kinase (PNK) (New England Biolabs, M0201S). Next, the px330 plasmid (Addgene plasmid number #42330, a gift from Feng Zhang (Cong et al., 2013) was digested with BbsI and dephosphorylated using calf intestinal alkaline phosphatase (CIP) (NEB, M0290S). The diluted phosphorylated oligonucleotide duplex was ligated into the dephosphorylated px330 plasmid using a T4 ligase (New England Biolabs, M0202M). Next, the ligated product was transformed into Stbl3 E. coli competent cells. The T7 promoter was added to the gRNA template by PCR amplification using primers that contain the T7 promoter sequence and gRNA sequence. The T7-gRNA PCR product was gel purified and used as the template for in vitro transcription (MEGA shortscript T7 kit, Invitrogen AM1345). The in vitro transcribed gRNA was then purified using the Invitrogen MEGAclear kit, AM1908) and eluted in non-DEPC RNAse free water (Ambion, AM9938). Cas9 mRNA was purchased from Sigma (CAS9MRNA-1EA). The single strand donor oligonucleotide contained the double point mutation in HOIL-1L-NZF (T201A/R208A), additionally it also contains a silent mutation of the PAM sequence and a silent mutation that generates a SmaI restriction site that allows genotyping of the mice. A scheme of the targeted strategy can be found in the results section. The primers, the sequence of the repair template and the single strand oligonucleotide donor template used for the generation of the Hoil-1lnzf∗/nzf∗ knockin mouse are listed below: gRNA sequence for BbsI overhang (forward primer): 5’ CACCGCTCACACCCAGGCCGTGT 3’ (gRNA A), 5’ CACCGTCTCACACCCAGGCCGTG 3’ (gRNA B). gRNA sequence for BbsI overhang (reverse primer): 5’ AAACACACGGCCTGGGTGTGAGC 3’ (gRNA A), 5’ AAACCACGGCCTGGGTGTGAGAC 3’ (gRNA B). Forward primer for in vitro transcription: 5’ TTAATACGACTCACTATAGGG 3’. Reverse primer for in vitro transcription: 5’ AAAAGCACCGACTCGGTGCC 3’. Single strand oligonucleotide donor template: 5’CCGGGCCCGGCTTTCATCAACAAACCTACTGCGCCTGGGTGTGAGATG 3’.C57BL/6J female donor mice (3-5-week-old) were treated with 5IU of pregnant mare’s serum gonadotropin (PMSG). 46 hours later the donor mice were injected with 5IU of human chorionic gonadotropin (hCG) (Intervet, GesmbH) to induce super ovulation. Next the females were mated with stud males and checked for plugs. The zygotes were isolated from pregnant females in M2 media and culture in KSOM media. Next a mix consisting of 100 ng/ μl of Cas9 mRNA, 50 ng/μl of gRNA and 200 ng/μl of single stand oligonucleotide diluted in non-DEPC RNase free water (Ambion, AM938) was centrifuged for 1 hour and 30 minutes at 15,000 rpm at 4°C and injected into the cytosol of zygotes (Volume of injection mixture: 50 μl). Following the injection, the zygotes were transferred to pseudo-pregnant females. 3 different founder lines obtained from two different gRNAs were established. The correct sequence for each founder mouse, containing the double point mutations and the silent mutations for genotyping purpose was confirmed by Sanger Sequencing. PCR fragments amplified from genomic DNA extracted from the different founder mice using the Wizard SV genomic DNA extraction kit (Promega, A2360), were cloned into a vector using the TOPO TA cloning kit (Thermo Fisher Scientific).
Genotyping of Hoil-1lnzf∗/nzf∗ mice
Genomic DNA from Proteinase K-digested mouse toes was extracted using the Wizard SV genomic DNA extraction kit (Promega, A2360) (Fennell et al., 2020). PCR reactions to amplify a fragment containing the target sites were carried out using the forward and reverse primers (Forward primer: 5’ TGGGTTGTCAGCATGTGGTT -3′. Reverse primer: 5′-GTGGTTCCCTTTCTGGCTCA-3′). PCR product was digested with SmaI (Thermo Fisher Scientific, ER0662) for 16 hours. Digested products were analyzed by electrophoresis using 1.5% agarose gels.
Isolation and immortalization of mouse embryonic fibroblasts (MEFs)
Primary MEFs were isolated from E13.5 embryos according to a standard protocol as described previously (Fennell et al., 2020). Briefly, female mice were crossed with a male mouse and checked daily for plugs. When a plug was detected, this was considered developmental stage E0.5. Embryos were isolated at E13.5. The head of the embryos was removed, and the rest of the body was minced with a scalpel and trypsinized for 5 minutes at 37°C. Cells were collected in a falcon tube, centrifuged and plated in a culture dish. To immortalize primary MEFs, pSG5-SV40 largeT antigen plasmid was transfected using GeneJuice Transfection Reagent following the manufacturer's protocol and cells were kept until they became stably proliferative.
Isolation and stimulation of bone marrow derived macrophages (BMDMs)
The bone marrow cells of the tibia and femur of 8 to 12-week-old mice (female and male) were flushed out and differentiated by culturing in DMEM-10% FCS supplemented with mouse colony stimulating factor (M-CSF, 25ng/ml, Peprotech, 315-02) for 5-6 days (Baccarini et al., 1985). For signaling assays, 1x106 BMDM were seeded in 6 well plates, and stimulated with mouse TNF (20ng/ml) after 16 hours of serum starvation or stimulated with LPS (10ng/ml) for the indicated time.
Method details
Tissue culture and transfection
HEK293T (ATCC), primary and immortalized MEF and primary BMDM were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma, D5648) supplemented with fetal calf serum (Thermo Fisher Scientific, 10270106), L-glutamine (Thermo Fisher Scientific, 25030-024), and penicillin–streptomycin (Sigma, P0781) and kept at 37°C and 5% CO2. Transfection of plasmids in MEF and HEK293T was carried out using GeneJuice Transfection Reagent (Merck Millipore, 70967) following the manufacturer's protocol.
NF-κB reporter assay
HEK293T were seeded in a 96-well plate and transfected with pNF-κB-Luc (Stratagene) and phRL-TK (Promega) as well as various HOIP, HOIL-1L and SHARPIN plasmids (as indicated) using GeneJuice. 48 hours post transfection, a Dual-glow luciferase assay (Promega E2940) was performed following the manufacturer's protocol. Synergy H1 hybrid multimode microplate reader (BioTek) was used to measure the luminescence signal. The luciferase signal from each sample was normalized to the corresponding Renilla luciferase signal in each well. All samples were normalized to the control samples transfected with an empty vector (pcDNA3.1-Myc), pNF-κB-Luc and phRK-TK (Ikeda et al., 2011; Fennell et al., 2020).
Cell lysis and immunoprecipitation
Cells were lysed in lysis buffer containing 50 mM HEPES (pH7.4) (Sigma Aldrich, H4034), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 10% Glycerol, 25 mM NAF and 10 μM ZnCl2 which was supplemented with 1 x cOmplete protease inhibitor cocktail (Roche, 11836170001), 1 mM PMSF (Roche, 10837091001) and 10 mM NEM (Sigma- Aldrich, E3876) (Fennell et al., 2020). Immediately after lysis, cells were centrifuged at 15,000 rpm for 15 minutes. Next the supernatant was denatured by adding SDS sample buffer and incubating at 96°C for 3 minutes. Cells lysates were incubated with 1μg of anti-Myc antibody for 2 hours at 4°C, followed by incubation with Protein G beads (Roche, 1124323301) for 2 hours at 4°C. Beads were washed 5-7 times in lysis buffer prior to elution with 30 μl of SDS sample buffer and heated for 3 minutes at 96°C.
SDS-PAGE and immunoblotting
Briefly, samples were resolved by SDS-PAGE and subsequently transferred to a 0.45 μm nitrocellulose membrane (GE Healthcare, 10600019) (Ikeda et al., 2011; Kumari et al., 2014). The membrane was stained with Ponceau S to assess transfer efficiency. Next, membranes were washed with TBS-Triton X and TBS buffer and incubated in 5% BSA/TBS. Then membranes were incubated with the indicated primary antibodies diluted in 5% BSA/TBS overnight at 4°C. Membranes were incubated with the secondary antibody for 1-2 hours following the manufacturer’s recommendations. The signal was detected using Western Blot Luminol Reagent (Santa Cruz, sc-2048) on high performance chemiluminescence films (GE Healthcare, Amersham Hyperfilm ECL 28-9068-37)
Protein purification
A method is described elsewhere (Ikeda et al., 2011; Asaoka et al., 2016). Briefly, expression plasmids were transformed into BL21 (DE3) Escherichia coli. Bacteria were grown at 37°C in LB media. The expression of GST-tagged fusion proteins was induced by adding IPTG 100 μM (Thermo Fischer Scientific, R0392) at OD600=0.8 in 4 to 6 liters of culture. To induce the expression of HOIP, HOIL-1L and SHARPIN, 100 μM ZnCl2 (Sigma Aldrich, 229997) were added. Bacteria cultures were grown overnight at 18°C. Bacteria were centrifuged, pelleted and resuspended in a buffer containing 100 mM HEPES (Sigma Aldrich, H4034), 600 mM NaCl, 1 mM TCEP-HCl (Thermo Fisher Scientific 20491), pH 7.4, supplemented with recombinant DNASE I (1000 U) (Roche, 0453628001), cOmplete protease EDTA-free inhibitor cocktail (Roche, 11836170001) and 1 mM PMSF (100mM in isopropanol, Roche 10837091001). Subsequently, bacteria were sonicated and 0.5% TritonX-100 was added to the lysate. The lysate was cleared by centrifugation and it was applied to a 5 ml GSTrap FF column (GE Healthcare, 17513101) to purify GST-proteins. PreScission protease (homemade) was used to remove the GST tag. Size exclusion chromatography on gel filtration columns using either the Superdex 200 (16/600) (GE Healthcare, GE28-9893-35) or Superdex 75 (16/600) (GE Healthcare, GE28-9893-33) in a buffer containing 50 mM HEPES (Sigma Aldrich, H4034), 150 mM NaCl, 1 mM TCEP-HCl (Thermo Fisher Scientific, 20491), pH 7.4 was used to resolve the protein eluates. The eluted fractions were subjected to SDS page and stained with InstantBlueTM and the fractions containing the desired proteins were pooled together. The protein concentration was assessed by comparison to BSA standards. The baculovirus used for insect expression of His6 mouse Ube1 in Hi5 cells was purified as previously described (Iwai et al., 1999; Fennell et al., 2020). For GST-empty and GST-Liner di-Ubiquitin, plasmids were expressed in BL21 cells for 16 hours at 25°C and lysed by sonication in the buffer described above. Subsequently, the lysates were incubated 16 hours at 4°C with Glutathione Sepharose 4B (GE Healthcare, GE17-0756-01). Next, beads were washed in 50 mM Tris, 100 mM EDTA, 150 mM NaCl, 0.5% Triton, pH 7.5 and resuspended in a buffer containing 50mM Tris, pH 7.5.
In vitro ubiquitination assay
A method for in vitro ubiquitination assays is described elsewhere (Asaoka et al., 2016; Fennell et al., 2020). Ubiquitin (10 μg) (Sigma-Aldrich, U6253), mouse E1 (Ube1) (150 ng), human Ubch7 (300 ng), human HOIP (5 μg), human SHARPIN (1μg), human HOIL-1L (1 μg), human NEMO (5 μg) and ATP (2 mM) (Roche, 10519979000) were incubated at 37°C for the indicated time in a buffer containing 150 mM NaCl, 20 mM MgCl2 and 50 mM HEPES (Sigma Aldrich, H4034) (pH7.5). Reactions were terminated by adding SDS sample buffer and incubation at 96°C for 1 minute. Samples were subjected to SDS-PAGE followed by immunoblotting.
GST pull-down assay
A method is described elsewhere (Rahighi et al., 2009). Briefly, GST-empty and GST-Linear di ubiquitin were expressed in Escherichia. coli, purified, immobilized on glutathione Sepharose 4B beads (Millipore Sigma, GE 17-075601) and incubated with the recombinant proteins or total cell lysates for 12 hours at 4°C. Next, GST-pulldown samples were washed five times with ice-cold lysis buffer (for total cell lysate samples) or with wash buffer (for recombinant protein samples) (25 mM Tris-HCl buffer (pH 7.2), 20 μM ZnCl2, 1 mM DTT, 100 mM NaCl, and 0.1% Triton X-100) (Sato et al., 2011).
Isolation of TNFR complex I
A method is previously described (Haas et al., 2009; Draber et al., 2015; Fennell et al., 2020). Briefly, 5-20x106 MEFs were seeded in 15 cm dishes. Following 16-hour serum starvation in 0.2% FCS-DMEM, MEFs were treated with 1μg/ml of recombinant Flag-human TNF. Subsequently, cells were washed twice with chilled PBS and lysed in IP-lysis buffer containing (30 mM Tris-HCI (pH 7.4), 120 mM NaCl, 2 mM EDTA, 2 mM KCI, 10% glycerol, 1%Trition X-100, 50 mM NaF, 1 X cOmplete protease inhibitor cocktail (Roche, 11836170001), 1 mM PMSF (Roche, 10837091001), 10 mM NEM (Sigma-Aldrich, E3876), and 5 mM Na3VO4 (Sigma-Aldrich, S6508). Samples were incubated on ice for 30 minutes. Following centrifugation at 15,000rpms for 30 minutes, Flag human TNF (1 μg) was added to the 0-hour control samples. Samples were precleared with Protein G agarose beads for 1 hour at 4°C. anti-FLAG M2 beads (Sigma Aldrich, A2220) were incubated with the pre-cleared samples at 4°C. Samples were washed 6 times with IP-lysis buffer. Samples were eluted by incubation with 2X SDS sample buffer at 96°C for five minutes.
qRT-PCR
This method is described elsewhere (Asaoka et al., 2016; Fennell et al., 2020). 1x106 primary BMDMs or 1x106 MEFs were serum starved in 0.2% FBS-DMEM for 15-16 hours and then treated with mouse TNF (20 ng/ml) for the indicated times. Samples were washed with chilled PBS and RNA was extracted using TRIzol (Life Technologies, 15596018) and treated with TURBO DNA-free kit (Invitrogen, AM1907). cDNA was generated using oligo (dT) 18 primer (New England Biolabs, 513165) and SuperScript II Reverse Transcriptase (Invitrogen, 18064-014) following the manufacturer’s protocol. Real time quantitative PCR was performed in a CFX 96 BioRad CFX 96 Real-Time PCR detection instrument with GoTaq qRT-PCR master mix (Promega, A6002). The sequence of the primers used in this study are: β-actin forward primer 5’-CGGTTCCGATGCCCTGAGGCTCTT-3’, β-actin reverse primer 5’ CGT CACACTTCATGATGGAATTGA-3. A20: forward primer 5’ AAAGGACTACAGCAGA GCCCAG-3’, A20 reverse primer 5’-AGAGACATTTCCAGTCCGGTGG-3’. ICAM forward primer 5’-AAGGAGATCACATTCACGGTG-3’, ICAM reverse primer 5’-TTTGG GATGGTAGCTGGAAG-3’, IκB-α forward primer 5’-GCTGAGGCACTTCTGAAAGCTG-3’, IκB-α reverse primer 5’-TGGACTGGCAGACCTACCATTG-3’. VCAM forward primer 5’-CTGGGAAGCTGGAACGAAGT-3’, VCAM reverse primer 5’-GCCAACACTTGACC GTGAC-3’. mouse TNF forward primer 5’ CATCTTCTCAAAATTCGAGTGACAA, mouse TNF reverse primer 5’ TGGGAGTAGACAAGGTACAACCC. IL-6 forward primer 5’-AGCCAG AGTCCTTCAGAGAGA-3’, IL-6 reverse primer 5’-TGGTCTTGGTCCTTAGCCAC-3’. IL1-β forward primer 5’- ATGAAAGACGGCACACCCAC-3’, IL1β reverse primer 5’-CTGCTTGTGAGGTGCTGATG-3’CCL5 forward primer 5-TGC TGCT TTGCCTACCTC TC-3’, CCL5 reverse primer 5’-CCA C TTCTTCTCTGGGTTGG-3’.
Cell death analysis by viability marker staining
MEFs were seeded at 80% confluency 12-16 hours prior to stimulation. MEFs were stimulated with mTNF (100ng/ml) for the indicated timepoints. Subsequently, supernatants and adherent cells were collected by centrifugation after trypsinization and resuspended in PBS containing 5% FCS and 2mM EDTA. Next, these harvested MEFs were stained with the viability dye eFluor 780 (eBioscience, 65-0865-14) (1/1000). eFluor780 positive cells were enumerated by flow cytometry (LSR Fortessa flow cytometer (BD). The data was analyzed using the FlowJo software.
Caspase-8 activity assay
A method is described elsewhere (Kumari et al., 2014; Fennell et al., 2020). Briefly, MEFs were seeded at a density of 5x104 cells/well in a 96-well white plate (Thermo Fisher Scientific, 136101) and treated with mouse TNF (100 ng/ml) and cycloheximide (1 μg/ml) for the indicated timepoints. Caspase Glow 8 assay system (Promega, G8202) was used to measure caspase-8 activity following the manufacturer’s instructions. Synergy H1 microplate reader (Fisher Scientific) was used to measure the signal.
Peritoneal lavage
Mice were deeply anesthetized with 150 μg/g ketamine (Ketamidor, Richter pharma) and 15 μg/g xylazine (Sedaxylan, Dechra). Peritoneal cavity was cut open and the peritoneum was flushed with 7-8 ml of harvest solution (PBS, 0,02% EDTA). The peritoneal lavage was retrieved and kept on ice until further processing.
TNF-induced shock
8 to10-week-old sex-matched mice were injected intravenously (i.v.) via the tail vein with 450 μg/kg of endotoxin-free recombinant mouse TNF (ImmunoTools) in 200 μl of PBS (pH 6.8) (Tortola et al., 2016). Mouse body temperature was recorded with an infrared thermometer (BIO-IRB153, Bioseb) by placing it 2-3 cm from the rectum (Mei et al., 2018). The weight, of the mice was monitored for 12 hours.
LPS-induced septic shock
8 to 10-week old sex-matched mice were injected intraperitoneally (i.p.) with 30 mg/kg of E. coli 0111: B4 LPS (L4391, Sigma) dissolved in 0.9% NaCl (Lamkanfi et al., 2009). Mice body temperature was recorded with an infrared thermometer (BIO-IRB153, Bioseb) by placing it 2-3 cm from the rectum (Mei et al., 2018). The weight and survival of the mice were monitored after the LPS-injection.
Flow cytometry
The spleen and mesenteric lymph nodes were harvested from 4-week-old mice. The peritoneal exudate was harvested as descried below. Spleen homogenates were lysed with Ammonium Chloride Potassium (ACK) lysis buffer to lyse red blood cells. Subsequently, cells were stained with the viability dye eFluor780 (eBioscience, 65-0865-14) at 4°C for 15 minutes. Cells were washed and blocked with CD16/CD32 Fc block (BD Bioscience, 553141) at 4°C for 10 minutes. The following antibodies were used for staining: CD23 (eBioscience, 25-0232-82), CD93 (Invitrogen, 62-5892-82), CD19 (BD Biosciences, 563333), CD45 (BioLegend, 103149), CD138 (BD Biosciences, 562935), TCRβ (BD Biosciences,109229), CD11c (BioLegend, 117335), MHC II (BioLegend, 107608), CD11b (BioLegend, 101257), TCRβ (BioLegend, 109205), CD23 (Biolegend, 101620), DX5 (BD Biosciences, 558295), F4/80 (BioLegend, 123131), B220 (BD Biosciences, 552772), CD8 (BD Biosciences, 553035), NK1.1 (BD Biosciences, 553164), TCRβ (BD Biosciences, 560729), CD23 (BioLegend, 101614), CD21 (BioLegend, 123411), Ly6G (BioLegend, 127618), Ly6C (BD Biosciences, 560595), TCRβ (BD Biosciences, 553170), B220 (BD Biosciences, 562922), CD28 (BioLegend, 122010), CD4 (BioLegend, 100443), CD45 (BD Biosciences, 550994). The samples were acquired using an LSR Fortessa flow cytometer (BD). The data was analyzed using the FlowJo software.
AST/ALT measurements
12 hours post i.p. LPS injection (30 mg/kg), mice were deeply anesthetized with 150 μg/g ketamine (Ketamidor, Richter pharma) and 15 μg/g xylazine (Sedaxylan, Dechra). Blood was withdrawn from the vena cava with a 25-gouge heparinized needle connected to a syringe containing 50 μl of 100 U/mL heparin (Sigma-Aldrich) to prevent coagulation and the total final volume was noted. Subsequently, blood was centrifuged at 12000xg for 5 minutes and the serum was collected. The concentration of liver damage marker AST and ALT were determined by the veterinary in vitro diagnostics laboratory InVitro GmbH and normalized to the volume of withdrawn blood.
Cytokine analysis
To measure cytokine levels in the serum of mice, ProcartaPlex Immunoassays (Thermo Fisher Scientific) were used. For mice subjected to LPS-induced septic shock, blood was withdrawn as described in the AST/ALT measurements. For mice subjected to TNF-induced shock, blood was withdrawn from the submandibular vein using a lancet and collected in a BD Microtainer blood collection tube to avoid coagulation. Subsequently, blood was centrifuged at 6000xg for 3 minutes and plasma was collected. Samples were measured in duplicates following the manufacturer’s instructions. The fluorescence intensity of each sample was read in a Luminex analyzer.
Histopathological analysis
A method is described elsewhere (Kumari et al., 2014). Briefly, mouse tissues were fixed in 10% neutral buffered formalin (Sigma, HT501128) for 12-16 hours, embedded in paraffin and sectioned for hematoxylin and eosin (H&E) staining using an automated stainer (Microm HMS 740). An automated stainer (Bond III, Leica) was used for immunohistochemistry. Primary antibodies for immunohistochemistry are keratin 14 (KRT14) (Sigma Aldrich, SAB4501657, 1:200) cleaved caspase-3 (Cell Signaling, 9661, 1:100), Ly6G (Abcam, ab2557, 1:500), CD3 (Abcam ab49943, 1:200) and B220/CD45R (BD Biosciences 550286, 1:100). The secondary antibodies used are goat anti-rabbit IgG (Dako, E0432 1:500), anti-rat IgG (Abcam, ab6733, 1:500). Signal was detected using the Leica Bond Intense R Detection system. A board-certified veterinary comparative pathologist evaluated the slides using a Zeiss Axioscope 2 MOT microscope and images were acquired with a SPOT Insight color camera (SPOT Imaging, Diagnostic Instrument, Inc.).
For skin thickness measurements, whole slide images of KRT14-stained sections were evaluated using the Panoramic Viewer software. From each digital scene spanning 4 mm, 10 non-contiguous, representative foci were selected, and the thickness of the entire epidermis was measured in each focus. For quantification of CD3 positive cells in the skin, QuPath v 0.2.3, an open source image analysis tool was used (Bankhead et al., 2017). Annotations of skin sections were generated with a pixel classifier after detecting the tissue with simple thresholder for the hematoxylin channel. The annotations were manually verified and refined by a pathologist. Positive cell detection was performed by setting the optical density sum for the Cell DAB OD mean score compartment. The threshold was applied to all images within the workspace with a command history script.
Quantification and statistical analysis
Statistical analysis was performed using Prism 8 software (GraphPad). The mean values with standard deviation are shown. Parametric Ttest was used for normally distributed datasets. ANOVA was used to analyze repeated measurements over time. The significance and confidence level were set at 0.05, and p values are indicated in each figure legends as well as the number of replicates used to calculate statistics.
Acknowledgments
We thank all the members of the Ikeda lab, especially Alan Rodríguez Carvajal, and the Penninger lab for their support and constructive discussions on the project. We thank molecular biology services, bio-optics, bioinformatics, transgenic services (the IMP-IMBA core facilities), the Histopathology facility and the Protein Technologies facility (part of the Vienna Biocenter Core Facilities) for their technical support. We thank Margit Jaschke and Melanie Gierer (Thermo Fisher Scientific) for their support with the ELISA measurement and René Rauschmeier (IMP) for i.v. injections. J.M.P. is supported by the Austrian Federal Ministry of Education, Science and Research, the Austrian Academy of Sciences, and the City of Vienna and grants from the Austrian Science Fund (FWF) Wittgenstein award (Z 271-B19), the T. von Zastrow foundation, a Canada 150 Research Chairs Program (F18-01336), and CIHR grant. P.K. is supported by the Austrian Science Fund (FWF) grants P31848-B and P33000-B. Research in the Ikeda Lab is supported by JSPS KAKENHI Grant Number JP18K19959, JP21H04777, JP21H00288 and the Austrian Academy of Sciences. We also thank Angela Andersen from the Life Science Editors for editing the manuscript.
Author contributions
C.G.-D. and F.I. designed the experiments, prepared the figures, and wrote the manuscript. C.G.-D., G.J., K.S., L.D., A.B., K.E., J.A., M.S., and L.M.F. performed experiments and analyzed data. A.K. performed histopathology analysis. A.B., K.E., P.K., and A.H. contributed to in vivo experiments and provided essential technical advice. A.B. designed the graphical abstract. J.M.P. and F.I. supervised author students and secured funding. F.I. conducted the overall project.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103241.
Supplemental information
Data and code availability
The data generated in this study are available upon request to the Lead Contact Fumiyo Ikeda (fumiyo.ikeda@bioreg.kyushu-u.ac.jp). This study did not generate any code.
References
- Asaoka T. Linear ubiquitination by LUBEL has a role in Drosophila heat stress response. EMBO Rep. 2016;17:1624–1640. doi: 10.15252/embr.201642378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaoka T., Ikeda F. New insights into the role of ubiquitin networks in the regulation of antiapoptosis pathways. Int. Rev. Cell Mol. Biol. 2015;318:121–158. doi: 10.1016/bs.ircmb.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Baccarini M., Bistoni F., Lohmann-Matthes M.L. In vitro natural cell-mediated cytotoxicity against Candida albicans: macrophage precursors as effector cells. J. Immunol. 1985;134:2658–2665. [PubMed] [Google Scholar]
- Bankhead P. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012 doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015 doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P., Dixit V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016 doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Carvajal A.R. The linear ubiquitin chain assembly complex LUBAC generates heterotypic ubiquitin chains. eLife. 2021;10 doi: 10.7554/eLife.60660. e60660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas T. The inflammatory caspases-1 and -11 mediate the pathogenesis of dermatitis in sharpin-deficient mice. J. Immunol. 2015;195:2365–2373. doi: 10.4049/jimmunol.1500542. [DOI] [PubMed] [Google Scholar]
- Draber P. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 2015;13:2258–2272. doi: 10.1016/j.celrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell L.M. Site-specific ubiquitination of the E3 ligase HOIP regulates apoptosis and immune signaling. EMBO J. 2020;39 doi: 10.15252/embj.2019103303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell L.M., Rahighi S., Ikeda F. Linear ubiquitin chain-binding domains. FEBS J. 2018;285:2746–2761. doi: 10.1111/febs.14478. [DOI] [PubMed] [Google Scholar]
- Fuseya Y. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 2020;22:663–673. doi: 10.1038/s41556-020-0517-9. [DOI] [PubMed] [Google Scholar]
- Gerlach B. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- Gómez-Díaz C., Ikeda F. Roles of ubiquitin in autophagy and cell death. Semin. Cell Dev. Biol. 2019;93:125–135. doi: 10.1016/j.semcdb.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P., Sharma B.R., Kanneganti T.D. Distinct role of IL-1β in instigating disease in Sharpin cpdm mice. Sci. Rep. 2016;6 doi: 10.1038/srep36634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas T.L. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Ikeda F. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature. 2011;471:637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K. Identification of the von Hippel-Lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. 1999;96:12436–12441. doi: 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justus S.J., Ting A.T. Cloaked in ubiquitin, A killer hides in plain sight: the molecular regulation of RIPK1. Immunol. Rev. 2015;266:145–160. doi: 10.1111/imr.12304. [DOI] [PubMed] [Google Scholar]
- Karki R. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021 doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kelsall I.R. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. U S A. 2019;116:13293–13298. doi: 10.1073/pnas.1905873116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife. 2014 doi: 10.7554/eLife.03422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M. Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival. Blood. 2009;113:2742–2745. doi: 10.1182/blood-2008-09-178038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity. 2018;49:42–55.e6. doi: 10.1016/j.immuni.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M.W. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J. Body temperature measurement in mice during acute illness: implantable temperature transponder versus surface infrared thermometry. Sci. Rep. 2018;8:3526. doi: 10.1038/s41598-018-22020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastase M.-V. An essential role for SHARPIN in the regulation of caspase 1 activity in sepsis. Am. J. Pathol. 2016;186:1206–1220. doi: 10.1016/j.ajpath.2015.12.026. [DOI] [PubMed] [Google Scholar]
- Pahl H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Peltzer N. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014 doi: 10.1016/j.celrep.2014.08.066. [DOI] [PubMed] [Google Scholar]
- Peltzer N. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature. 2018;557:112–117. doi: 10.1038/s41586-018-0064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer N., Walczak H. Cell death and inflammation – a vital but Dangerous Liaison. Trends Immunol. 2019 doi: 10.1016/j.it.2019.03.006. [DOI] [PubMed] [Google Scholar]
- Pfeffer K. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-C. [DOI] [PubMed] [Google Scholar]
- Rahighi S. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Rickard J.A. TNFR1-dependent cell death drives infammation in Sharpin-defcient mice. Elife. 2014;2014 doi: 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers M.A. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014;211:1333–1347. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K. Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K., Iwai K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol.l Rev. 2015;266:175–189. doi: 10.1111/imr.12308. [DOI] [PubMed] [Google Scholar]
- Sato Y. Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. U S A. 2011;108:20520–20525. doi: 10.1073/pnas.1109088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour R.E. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 2007;8:416–421. doi: 10.1038/sj.gene.6364403. [DOI] [PubMed] [Google Scholar]
- Sharma B.R. Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ. 2019;26:741–750. doi: 10.1038/s41418-018-0159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan K., Ruddle N., Schreiber R. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J. Immunol. 1989;142:11 3884–3893. [PubMed] [Google Scholar]
- Shimizu S. Differential involvement of the Npl4 zinc finger domains of SHARPIN and HOIL-1L in linear ubiquitin chain assembly complex-mediated cell death protection. Mol. Cell Biol. 2016;36:1569–1583. doi: 10.1128/MCB.01049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz B. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012;13:840–846. doi: 10.1038/embor.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- Tokunaga F. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- Tortola L. The tumor suppressor Hace1 is a critical regulator of TNFR1-mediated cell fate. Cell Rep. 2016;15:1481–1492. doi: 10.1016/j.celrep.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle J. A study of cecal ligation and Puncture-induced sepsis in tissue-specific tumor necrosis factor receptor 1-deficient mice. Front. Immunol. 2019;10:2574. doi: 10.3389/fimmu.2019.02574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster J.D., Vucic D. The balance of TNF mediated pathways regulates inflammatory cell death signaling in Healthy and Diseased tissues. Front. Cell Dev. Biol. 2020 doi: 10.3389/fcell.2020.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt A., Vucic D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017 doi: 10.1038/cdd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request to the Lead Contact Fumiyo Ikeda (fumiyo.ikeda@bioreg.kyushu-u.ac.jp). This study did not generate any code.