

Abstract
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease without known cure that primarily affects synovial joints. RA has a prevalence of approximately 1% of the population worldwide. A vicious circle between two critical immune cell types, B cells and neutrophils, develops and promotes disease. Pathogenic anti‐citrullinated protein antibodies (ACPA) directed against a range of citrullinated epitopes are abundant in both plasma and synovial fluid of RA patients. In addition to stimulating numerous cell types, ACPA and other autoantibodies, notably rheumatoid factor, form immune complexes (ICs) that potently activate neutrophils. Attracted to the synovium by abundant chemokines, neutrophils are locally stimulated by ICs. They generate cytokines and release cytotoxic compounds including neutrophil extracellular traps (NETs), strands of decondensed chromatin decorated with citrullinated histones and granule‐derived neutrophil proteins, which are particularly abundant in the synovial fluid. In this way, neutrophils generate citrullinated epitopes and release peptidylarginine deiminase (PAD) enzymes capable of citrullinating extracellular proteins in the rheumatic joint, contributing to renewed ACPA generation. This review article focusses on the central function of citrullination, a post‐translational modification of arginine residues in RA. The discussion includes ACPA and related autoantibodies, somatic hypermutation‐mediated escape from negative selection by autoreactive B cells, promotion of the dominance of citrullinated antigens by genetic and lifestyle susceptibility factors and the vicious circle between ACPA‐producing pathogenic B cells and NET‐producing neutrophils in RA.
Keywords: anti‐citrullinated protein antibodies, B cells, citrullination, dysbiosis, immune complexes, neutrophil extracellular traps, neutrophils, peptidylarginine deiminase, rheumatoid arthritis
This article reviews insights into cross‐talk between B cells and neutrophils in seropositive rheumatoid arthritis and its pathogenesis.
Abbreviations
- ACPA
anti‐citrullinated protein antibodies
- BAFF
B‐cell‐activating factor
- CAIA
collagen antibody‐induced arthritis
- CarP
carbamylated peptide
- CCP
cyclic citrullinated peptide
- CIA
collagen‐induced arthritis
- MPO
myeloperoxidase
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- PAD
peptidylarginine deiminase
- PTPN22
protein tyrosine phosphatase nonreceptor 22
- RA
rheumatoid arthritis
- RF
rheumatoid factor
- SE
shared epitope
- SF
synovial fluid
- SHM
somatic hypermutation
- SNP
single nucleotide polymorphism
RHEUMATOID ARTHRITIS
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease (reviewed in Ref. [1]). This most common form of inflammatory arthritis affects ~1% of the population worldwide and is more prevalent in women than men. RA is a disabling condition characterized by symmetrical inflammation of synovial joints, with small, peripheral joints most commonly affected. The synovial fluid (SF) becomes enriched in leucocytes and cytokines, and the inflamed synovial membrane develops into an inflammatory pannus, an abnormal layer of blood vessel‐containing tissue which invades the space between the bones, covering bones and cartilage. Unless treated, RA erodes the joint cartilage and bone, causing chronic pain, stiffness, progressive loss of function, disability and, once fusion of bones has occurred, lasting deformities. Up to 40% of patients develop extraarticular RA, which ranges from systemic features, such as vasculitis, to affecting individual organs, for example the lung (e.g., interstitial lung disease) or heart (e.g., pericarditis). With the advent of improved and increasingly sophisticated disease‐modifying anti‐rheumatic drugs, RA has become more manageable in recent years. Although it remains incurable, a combination of early intervention, control of inflammation and prevention of joint damage can culminate in reaching a sustained state of remission.
Rheumatoid arthritis is sometimes regarded not as a single disease but a group of related diseases. Although the pathogenesis of RA is complex and remains incompletely understood, it is clear that this is a long, stepwise process which involves the dysregulation of many cell types, all of which make contributions to this disease. Despite their important contributions, cell types, including T cells, osteoclasts, macrophages and fibroblast‐like synoviocytes, are not discussed here. Instead, this review focusses on the interplay of B cells and neutrophils in autoantibody‐driven (seropositive) RA. Following on from the introduction to these two important cell types, and their critical role in the formation of autoantibodies and protein citrullination, which drive RA, we will discuss risk factors and how they link into RA pathogenesis by promoting citrullination and autoantibody formation.
B CELLS
Anti‐citrullinated protein antibodies (ACPA; see below for a detailed discussion) are present in serum of >80% of patients with established RA and in ~50% of those with early RA. These autoantibodies can present as much as a decade prior to the onset of any clinical disease [2, 3, 4 indicative of an early loss of tolerance that initiates disease pathogenesis. During B‐cell development, the antibody repertoire is developed. Tolerance is regulated at the central and peripheral checkpoints, when autoreactive B cells are eliminated, become anergic or undergo B‐cell receptor editing [5]. These processes are, however, less strict than those applying to T cells, and some autoreactive B cells escape. Survival of autoreactive B cells may be aided by their genetic predisposition (see below), and self‐reactive low‐affinity antibodies may be masked on anergic B cells. Moreover, somatic hypermutation (SHM) leading to N‐linked glycosylation of the variable region may permit escape from negative selection [6]. Indeed, glycosylation of the ACPA Fc changes during the transition from pre‐arthritis to arthritis with the appearance of a more pro‐inflammatory glycoform of these autoantibodies [7, 8. In the context of collagen‐induced arthritis (CIA) in the mouse, differential glycosylation of ACPA was shown to affect their pathogenicity [9], with IL‐23 and Th17 cells having key roles in promoting pathogenicity [10].
In addition to pathological autoantibodies, the success of B‐cell depletion therapies targeting CD20 and BAFF [11, 12, 13, 14 identified a critical function of B cells in promoting chronic inflammation in RA. The curious observation that good responsiveness to B‐cell depletion therapy did not correlate with reduced ACPA titres, prompted studies into B cells as drivers of chronic inflammation. Such studies identified continuous antigen‐driven B‐cell activation, proliferating ACPA‐positive memory B cells in both circulation and SF, as well as the production of pro‐inflammatory mediators, notably the neutrophil chemokine IL‐8 by synovial ACPA+ B cells [15], suggestive of crosstalk between pathogenic B cells and neutrophils (Figure 1).
FIGURE 1.
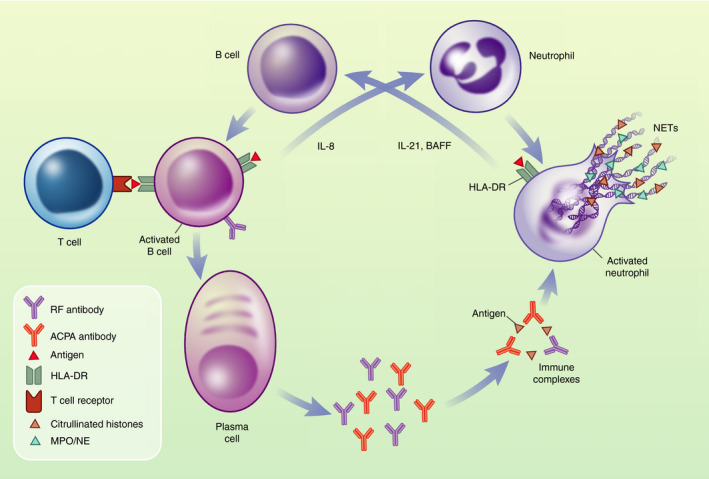
B cells and neutrophils form a vicious circle in RA. Activated B cells release cytokines to crosstalk with other immune cells, with B‐cell‐derived IL‐8 recruiting neutrophils to the synovium [15]. B cells receive T‐cell help with class switching and somatic hypermutation, promoting the development of autoantibodies in a HLA‐DR SE‐dependent fashion. Local plasma cells produce large amounts of autoantibodies including RF and ACPA; these form ICs which activate the complement pathway and promote inflammation, for example by stimulating neutrophils both in the circulation and also in the synovium. Amongst other events, this results in the release of NETs, which are particularly abundant in RA. Myeloperoxidase (MPO), neutrophil elastase (NE) and citrullinated histones are amongst the proteins that decorate NETs [22, 141. Citrullinated histones are thought to act as a continuous source of fresh antigen to B cells, promoting the production of new IgM ACPA. In the synovium, this is promoted by HLA‐DR expressing, activated neutrophils which release cytokines including BAFF and IL‐21, activating B cells [38, 39, 40, 41. In the interest of clarity, RF and ICs are simplified in this cartoon drawing
NEUTROPHILS
Neutrophils, the most abundant circulating leucocyte in humans, play a key role in host defence in killing bacteria and fungi either intracellularly, following phagocytosis, or extracellularly [16, 17. Unstimulated neutrophils circulate for only up to 1 day before homing back to the bone marrow where they undergo apoptosis to be cleared by resident macrophages in an anti‐inflammatory process termed efferocytosis. In contrast, upon activation, the short‐lived neutrophil leaves the blood stream and travels to inflammatory sites, such as the inflamed synovium, following gradients of chemo‐attractants and chemokines [17, 18.
Neutrophils are armed with granules loaded with powerful proteases and highly toxic antimicrobial peptides and possess the ability to generate cytotoxic reactive oxygen species (ROS), which can be released into a phagosome or to the outside of the cell. Neutrophils can moreover release their chromatin as ‘neutrophil extracellular traps’ (NETs; see also below), strands of granule protein‐decorated chromatin that have important functions in host defence and that are highly inflammatory [16, 17. Neutrophilic inflammation can be triggered by microbes and sterile stimuli and can impart serious host tissue injury. Immune complexes (ICs) are powerful pro‐inflammatory stimuli of neutrophils that ligate Fc receptors, and trigger effector functions, including, ROS production, degranulation, NETs, chemokine and cytokine generation [19, 20, 21, 22, all of which are thought to contribute to tissue damage incurred in RA. ICs are key to RA. Depending on their ratio of antibody and antigen, ICs can be soluble or insoluble. Both types are abundant in the SF, with further ICs precipitated onto synovial surfaces.
The SF in RA is characteristically sterile, though containing chemokines and cytokines and infiltrated by a large number of leucocytes (>5000/μl), the majority of which are neutrophils [23, 24. At early stages of (clinical) disease, neutrophils are also recruited into the synovial tissue [25, 26, providing indirect clues about the important role of neutrophils in RA.
Circulating neutrophils from RA patients are characterized by an activated phenotype that is characterized by increased ROS, cytokine, protease and NET production as well as delayed apoptosis [27, 28, 29, 30. Intriguingly, patients with RA, as well as rats in an RA model, were found to harbour circulating low‐density granulocytes, with differential cell surface marker and gene expression signatures that are the subject of ongoing investigation [31, 32, 33. SF neutrophils were reported to be more activated still and having a differential gene expression signature compared to circulating cells from the same patient [34]. SF neutrophils are longer‐lived than circulating neutrophils [35, 36, secrete proteases, and release cytokines and chemokines to activate and recruit further neutrophils [29, 37. SF neutrophils also crosstalk with adaptive immune cells, for example by production of B‐cell activating factor (BAFF), inducing B‐cell proliferation and directly contributing to autoantibody production [38, 39. SF neutrophils moreover acquire the ability to present antigen in an MHC‐II‐dependent fashion and drive CD4+ T‐cell proliferation [40, 41. This is in keeping with observations that the SF containing neutrophil‐derived cytotoxic products, NETs, cytokines and chemokines produced by neutrophils further inflammation [36, 42, 43.
While experiments with laboratory animals and disease models need to be interpreted with caution, mouse models of RA suggest that neutrophils play a key role in this disease. Antibody‐mediated neutrophil depletion abolished development of K/BxN serum transfer arthritis and also of collagen antibody‐induced arthritis (CAIA) [44, 45, and neutrophil depletion after disease onset resulted in steep decline of CIA [46]. In a series of extensive and elegant investigations, the recruitment of neutrophils to the rheumatic joint in the K/BxN serum transfer model was shown to depend on FcγR as well as a cascade of chemokines, cytokines and chemo‐attractants generated by a variety of cells and their respective receptors on neutrophils [47, 48, 49, 50. Meanwhile, IC‐mediated neutrophil gene expression, including that of pro‐inflammatory mediators, is dependent on CARD9‐dependent regulation of the NFκB pathway downstream of FcγR, Src family kinases and Syk [51, 52, 53. Elegant, very recent work with FcγR‐humanized mouse neutrophils revealed that internalization of ICs permits these neutrophils to become antigen‐presenting cells in a FcγR‐dependent fashion [54]. The resulting neutrophils combined dendritic cell (antigen presentation, T‐cell activation, cytokine production) and neutrophil functions (ROS production, phagocytosis) [54].
NETS
Neutrophil extracellular traps are web‐like structures that are released by neutrophils. NETs consist of decondensed chromatin decorated with cytotoxic proteins, citrullinated histones, granule‐derived proteins including neutrophilic proteases, myeloperoxidase (MPO), and antimicrobial peptides. Initially described as a pro‐inflammatory cell death mechanism (NETosis), vital NET release was since also demonstrated [55], with the pathway employed being stimulus‐dependent and varying in its NADPH oxidase dependency as well as cleavage of N‐terminal histone tails [56]. Apart from their crucial function in host immunity, trapping and killing pathogens, NETs have important functions in autoimmune diseases including RA [57, 58. Elevated levels of NETs were identified in RA serum and SF [56, 59, 60.
Peptidyl‐arginine deiminase 4 (PAD4), a leucocyte‐restricted nuclear PAD, mediates histone citrullination, a post‐translational modification of arginine residues during NET formation (Figure 2). NETs represent an important source of citrullinated (and homocitrullinated) epitopes in RA. Indeed, NET‐associated citrullinated histones represent a continuous source of antigen for B cells and promote the localized generation of ACPA (see below for a detailed section on ACPA) that are able to cross‐react with citrullinated histones in ectopic germinal centres in the inflamed synovial joint [59, 61, 62. Plasma cell differentiation and antibody production are promoted in situ by pathogenic, IL‐21‐producing T peripheral helper cells [63]. Not only are ACPA pathogenic by themselves. Potentially aided by RF (see below for a detailed section on RF), which has the capacity to bind several IgG molecules, ACPA form ICs. These ICs fix complement, accumulate in SF and/or deposit on synovial surfaces to amplify inflammation [64, 65, 66. ICs are powerful stimuli of NET release (e.g., [22, 67, 68). In addition, NETs stimulate neutrophils to produce further NETs and to secrete IL‐8 and BAFF [69], promoting the vicious circle between B cells and neutrophils (Figure 1).
FIGURE 2.
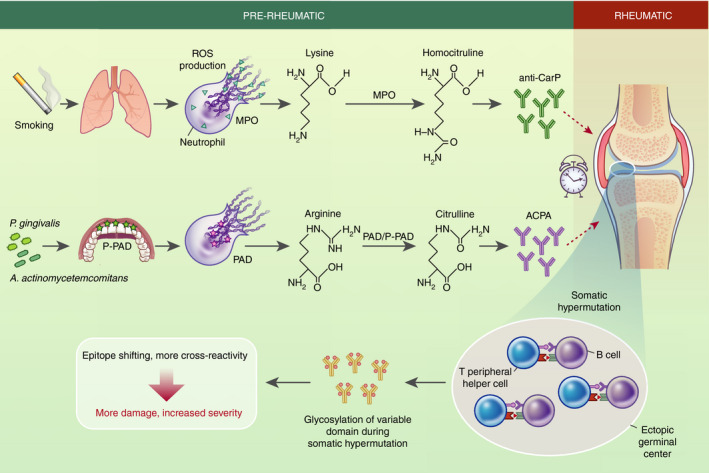
Citrullination and homocitrullination underpin the pathogenesis of RA. Pathogenic autoantibodies including ACPA and anti‐CarP in pre‐rheumatic patients precede the onset of clinical symptoms in RA by up to a decade. In addition to a genetic disposition, triggers at mucosal sites are thought to play a key role in these early events. Smoking‐induced lung inflammation can promote neutrophil‐derived NETs in the lung form and exteriorization of myeloperoxidase (MPO). Together with smoke‐derived cyanate, homocitrullination (also known as carbamylation) of lysine causes the generation of homocitrulline, indirectly promoting the generation of anti‐carP antibodies [86]. Alternative scenarios involve microbial dysbiosis at mucosal surfaces, such as the gingival tissues. Two causative pathogens in gingivitis, P. gingivalis and A. actinomycetemcomitans can promote citrullination of antigens by employing bacterial P‐PAD and neutrophil PAD4, respectively [95, 100. Citrullination is a process by which arginine is enzymatically converted to citrulline, generating a highly immunogenic antigen [62]. The events leading to the onset of symptomatic disease years later remain obscure, with possibilities including infection and/or local trauma. In the rheumatic phase, autoantibody maturation occurs locally in the inflamed synovial tissue. B‐cell SHM involving crosstalk of pathogenic B cells and pathogenic peripheral T helper cells occurs in ectopic germinal centres [63]. Strikingly, SHM results in characteristic glycosylation of the antibodies variable domain. This unusual form of SHM does not result in affinity maturation but instead drives epitope shifting and cross‐reactivity of ACPA [110, 115, ultimately resulting in more tissue damage and increased disease severity
Having introduced neutrophils and B cells, and how they activate one another in established disease, the next section of this review article will discuss genetic and lifestyle factors that predispose to RA. The focus lies on how these factors promote post‐translational modification such as citrullination and/or the NET generation by neutrophils, in turn promoting B‐cell generation of autoantibodies, in particular ACPA, promoting RA pathogenesis.
GENETIC FACTORS
While it is still unclear why some people ultimately develop RA, there are well‐documented genetic and environmental risk factors. The most significant genetic risk factor identified to date is the class II major histocompatibility locus, with so‐called shared epitope (SE) containing alleles increasing the risk of developing seropositive RA according to epidemiological studies [70, 71. SE sequences 70QKRAA74, 70QRRAA74 or 70RRRAA74 within HLA‐DRB1 are involved in shaping the peptide‐binding pocket of the HLA molecule. The presence of two positively charged residue (lysine, arginine) in residues 71–73 increases the binding capacity of citrullinated peptides over native peptides [72, 73. In addition, citrullinated peptides were also shown to display enhanced binding to HLA‐DQ [74]. Altogether, these mechanisms achieve that citrullinated peptides are preferentially presented, and activate CD4+ T‐cell responses, which in turn promote the immune response by helping ACPA‐producing B‐cell antibody maturation (class switching and somatic hypermutation).
Additional genetic risk factors have been attributed to single nucleotide polymorphisms (SNPs) in a range of genes. The most prominent of these is a SNP, C1858T, in the leucocyte‐restricted protein tyrosine phosphatase nonreceptor 22 (PTPN22) which encodes the R620W variant [75]. This allele and especially C1858T homozygosity cause an elevated risk of developing RA, earlier disease onset and more aggressive disease, with RF positivity conferring increased odds. R620W PTPN22 was reported to drive blunted BCR signalling and reduced B‐cell apoptosis, resulting in increased escape of poly‐ and autoreactive B cells from central and peripheral tolerance in humans and mouse models [76, 77, 78. PTPN22 is most highly expressed in neutrophils, with R620W reported to promote transendothelial migration and ROS production in human neutrophils [79], while in a mouse model Ptpn22 deficiency caused decreased pro‐inflammatory responses to IC stimulation without affecting neutrophil recruitment to inflammatory sites [80]. PTPN22 was moreover shown to physically interact with and inhibit PAD4 in a phosphatase‐independent fashion. Indeed, R620W PTPN22 promoted enhanced citrullination which resulted in increased NET production by neutrophils and in defective Th2 and Th17 cytokine production by peripheral blood‐derived mononuclear cells [81, 82.
LIFESTYLE: SMOKING
The major environmental factor associated with developing RA is smoking, which in combination with the HLA‐DR SE confers a significantly increased susceptibility to ACPA‐positive RA according to epidemiological studies (e.g., [83, 84). This suggests that smoking may provide an external trigger for those already carrying genetic risk factors to develop RA, and identifies the lung as an important mucosal site in RA development (Figure 2). Indeed smokers' lung biopsies were characterized by upregulated PAD2/4 expression and increased protein citrullination [85, 86. Recently, enhanced carbamylation and anti‐CarP (see below) were also found in smokers and smoke‐exposed laboratory mice [87, 88, directly implying neutrophils. Interestingly, neutrophils isolated from smokers were moreover shown to be more prone to NET production in a nicotine‐dependent fashion. In experimental mice, too, nicotine promoted NET production and caused more severe disease scores in CIA [89, 90.
DYSBIOSIS: EXAMPLE PERIODONTITIS
Links between the disruption of the beneficial relationship between commensal bacteria and the host at mucosal surfaces were documented in inflammation, with dysbiosis of the oral microbiota and periodontitis most clearly associated with RA pathogenesis [91] (Figure 2). A statistically significant association between RA and periodontitis was shown in a number of clinical studies, with RA patients with severe periodontitis suffering from more severe RA [92]. Associations between ACPA and severity of periodontitis in RA and pre‐RA patients were also described [93, 94. A possible explanation for this observation rests with two key periodontal pathogens. Porphyromonas gingivalis expresses a bacterial deiminase, PPAD, which citrullinates C‐terminal arginines of bacterial and host origin in a calcium‐independent fashion, potentially contributing to the breaking of tolerance [95]. In the context of CIA in the mouse, P. gingivalis PPAD activity could increase inflammatory arthritis [96, 97. Notably, P. gingivalis also affects the cytokine response of gingival epithelial cells, driving recruitment of Th17 cells and neutrophils via CCL20 and CXCL8. P. gingivalis was further shown to trigger release of non‐bactericidal NET production by neutrophils, encouraging microbial growth and increasing citrullinated antigens in the periodontal space [98, 99. In a separate mechanism, Aggregatibacter actinomycetemcomitans was shown to use its pore‐forming leucotoxins to induce hypercitrullination and NET formation by neutrophils, which in turn increased ACPA in the periodontal space [100].
AUTOANTIBODIES
As laid out above, the earliest and perhaps most conspicuous feature of RA are autoantibodies, which can be present years or even decades prior to onset of clinical symptoms (pre‐RA), with epitope spreading and expansion of autoantibodies occurring prior to the onset of clinical disease.
Rheumatoid factor
Rheumatoid factor refers to antibodies directed against the Fc region of IgG and was described in the 1940s. Despite its high prevalence in RA (up to 80% of patients) and positive association with more severe disease progression, RF is not restricted to RA, making it an unreliable diagnostic marker (reviewed in Ref. [1]). RF can undergo class switching, with IgM and IgA RF most commonly observed in RA. RF moreover undergoes somatic hypermutation and affinity maturation in RA. By recognizing IgG, RF is perfectly suited to forming large ICs, and to promoting deposition of complement to improve clearance of excess antibodies, the likely function of natural RF [101]. However, as laid out above, ICs also play a key role in promoting persistent inflammation including via neutrophils.
Anti‐citrullinated protein antibodies
Anti‐citrullinated protein antibodies represent a second class of highly prevalent autoantibodies found in RA. ACPAs are present in the serum of 80–90% of patients with established RA and in up to 20% of their first‐degree relatives [102]. For diagnostic purposes, presence of ACPA in the serum is detected by using cyclic citrullinated peptide (CCP2/CCP3) assays, where synthetic CCPs are used that were optimized for optimal ACPA capture. Serum ACPA react with peptides derived from a range of citrullinated protein antigens that are found in the rheumatic joints, including fibrin vimentin, α‐enolase and histones [62, 103, 104, 105, 106, with a high degree of cross‐reactivity between substrates observed for individual monoclonal ACPAs [107]. Interestingly, it was recently suggested that improved screening might detect autoantibodies in seronegative RA patients that do not cross‐react well with the CCPs used in current clinical testing [108]. Although the presence of ACPA in a person without clinical symptoms does not predict that they will be developing RA, contrasting with RF, the presence of ACPA is highly specific to RA, making them a useful diagnostic tool. ACPA is moreover indicative of more severe disease progression [109]. In RA, ACPA associate with RF and the HLA SE, as laid out above.
Anti‐citrullinated protein antibodies undergo class switching, extensive somatic hypermutation as well as conspicuous variable region glycosylation and epitope spreading, leading to cross‐reactive antibodies (Figure 2). However, affinity maturation is limited and even serum IgG ACPA is characterized by low binding affinity for citrullinated protein [110, 111, 112, 113, 114, 115. Interestingly, despite the short half‐life of 5.9 days of IgM in RA [116], and the fact that long‐lived IgM‐secreting plasma cells are not described in humans, IgM ACPA continues to be present in the serum of RA patients, or can occur at later stages in previously IgM ACPA‐negative patients [117, 118. This suggests that new citrullinated antigen‐specific B cells continuously generate new ACPA, implying the continued generation of fresh citrullinated antigens, for example due to neutrophil‐mediated production of NETs [57, 59, 60.
Anti‐CarP
Processes related to citrullination also lead to secondary modification of protein epitopes that are similarly antigenic. Carbamylation, also known as homocitrullination, is the post‐translational modification of lysine to homocitrulline (Figure 2). Unlike citrullination, carbamylation is a chemical modification that does not rely on an enzyme, but occurs on the presence of cyanate as a myeloperoxidase‐dependent oxidation of thiocyanate which is abundant in smokers [86]. Carbamylated peptides were predicted to bind SE alleles [119]. Anti‐CarP (carbamylated peptide) antibodies are directed against carbamylated proteins and frequently cross‐react with citrullinated proteins [119]. Anti‐CarP are present in 45% of RA patients including some of those who are ACPA‐negative. Like ACPA, anti‐CarP antibodies are very specific to RA, present in the serum up to 10 years before the onset of clinical symptoms, and are associated with NETs [58, 120.
Anti‐PAD antibodies
Neutrophils express three PAD enzymes, PAD2/3/4. Extracellular, active PAD was observed in cell‐free SF of RA patients [121]. Neutrophil NETosis was found to result in release of free PAD2/4 in vitro, raising the possibility that dying neutrophils may be the source of free PAD2/4 [122]. However, neutrophils were also found to spontaneously secrete or expose PAD2/4 [123], providing an alternative explanation for the citrullination of extracellular targets in the rheumatic joint. Interestingly, PADs themselves also serve as RA autoantigens. Indeed, ~30% of RA patient, but not control sera, were found to be anti‐PAD4 positive, with anti‐PAD4 positivity occurring prior to (clinical) disease onset and being a marker of severe disease [124, 125, 126. PAD4 requires >100 μM Ca2+ to display any catalytic activity in vitro and mM Ca2+ concentrations for optimal activity. This vastly exceeds the Ca2+ concentration of the resting cell [127], but can be achieved following activation and opening of Ca2+ channels [128]. The fact that histone citrullination occurs physiologically to regulate gene expression [129] moreover suggests the existence of additional mechanisms that allow PAD enzymes to be active at lower Ca2+. Fascinatingly, a subset of cross‐reactive anti‐PAD3/4 antibodies were shown to inducing a conformational change into PAD4 when binding to it, rendering it hyperactive by which vastly reduced its Ca2+ requirement to a physiological level [130, 131. Presence of these truly pathogenic anti‐PAD3/4 antibodies correlates with particularly aggressive disease.
CONCLUDING REMARKS
Despite decades of research into RA, the trigger that precipitates the original break of tolerance and the nature of additional events that initiate clinical disease remain obscure. It is clear, however, that a combination of genetic predisposition, lifestyle choices and dysbiosis can all contribute to culminate in clinical disease. Citrullination, much of it likely to be neutrophil‐derived, and ACPA lie at the heart of the initial break of tolerance, and subsequently, a vicious circle between neutrophils and B‐cell‐derived autoantibodies/ICs plays out. Neutrophils are often portrayed as uniquely pro‐inflammatory; however, this is likely not the whole truth. Not only do neutrophils clear insoluble ICs from biological fluids, thereby reducing these highly pro‐inflammatory stimuli, but the same ICs also induce neutrophil apoptosis [132, 133, 134, a cell death that promotes the resolution of inflammation [135]. A separate intriguing notion is moreover that physiologically citrullination may occur in order to limit excessive inflammation. Free histones are extremely cytotoxic [136]; however, their citrullination in NETs renders them more susceptible to proteolytic degradation. It also reduces their ability to stimulate further NET production [137, 138. In a similar way, citrullination may confer the host with a degree of protection from the highly toxic antibacterial peptide LL37 [139, 140.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTION
UK and SV wrote the manuscript and designed the figures.
ACKNOWLEDGEMENTS
We thank Alison Schroeer for redrawing our figures and apologize to the authors of the numerous primary research papers that could not be cited here due to space limitations.
Karmakar U, Vermeren S. Crosstalk between B cells and neutrophils in rheumatoid arthritis. Immunology. 2021;164:689–700. 10.1111/imm.13412
Funding information
UK holds a Versus Arthritis PhD Scholarship (21577); SV is funded by the MRC (MR/S008020/1).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
We need to talk about neutrophils. Immunology 2021, 164: 655‐656.
Neutrophil‐T cell crosstalk in inflammatory bowel disease. Immunology 2021, 164: 657‐664.
Neutrophils in pregnancy: New insights into innate and adaptive immune regulation. Immunology 2021, 164: 665‐676.
Neutrophils in secondary lymphoid organs. Immunology 2021, 164: 677‐688.
Our evolving view of neutrophils in defining the pathology of chronic lung disease. Immunology 2021, 164: 701‐721.
REFERENCES
- 1. Chang MH, Nigrovic PA. Antibody‐dependent and ‐independent mechanisms of inflammatory arthritis. JCI Insight. 2019;4:e125278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brink M, Hansson M, Mathsson‐Alm L, Wijayatunga P, Verheul MK, Trouw LA, et al. Rheumatoid factor isotypes in relation to antibodies against citrullinated peptides and carbamylated proteins before the onset of rheumatoid arthritis. Arthritis Res Ther. 2016;18:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst‐Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–6. [DOI] [PubMed] [Google Scholar]
- 4. Rantapaa‐Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–9. [DOI] [PubMed] [Google Scholar]
- 5. Melchers F. Checkpoints that control B cell development. J Clin Invest. 2015;125:2203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, et al. Redemption of autoantibodies on anergic B cells by variable‐region glycosylation and mutation away from self‐reactivity. Proc Natl Acad Sci USA. 2014;111:E2567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ercan A, Cui J, Chatterton DE, Deane KD, Hazen MM, Brintnell W, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010;62:2239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rombouts Y, Ewing E, van de Stadt LA, Selman MH, Trouw LA, Deelder AM, et al. Anti‐citrullinated protein antibodies acquire a pro‐inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. 2015;74:234–41. [DOI] [PubMed] [Google Scholar]
- 9. Ohmi Y, Ise W, Harazono A, Takakura D, Fukuyama H, Baba Y, et al. Sialylation converts arthritogenic IgG into inhibitors of collagen‐induced arthritis. Nat Commun. 2016;7:11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, et al. Regulation of autoantibody activity by the IL‐23‐TH17 axis determines the onset of autoimmune disease. Nat Immunol. 2017;18:104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edwards JC, Szczepanski L, Szechinski J, Filipowicz‐Sosnowska A, Emery P, Close DR, et al. Efficacy of B‐cell‐targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–81. [DOI] [PubMed] [Google Scholar]
- 12. Chatzidionysiou K, Lie E, Nasonov E, Lukina G, Hetland ML, Tarp U, et al. Highest clinical effectiveness of rituximab in autoantibody‐positive patients with rheumatoid arthritis and in those for whom no more than one previous TNF antagonist has failed: pooled data from 10 European registries. Ann Rheum Dis. 2011;70:1575–80. [DOI] [PubMed] [Google Scholar]
- 13. Porter D, van Melckebeke J, Dale J, Messow CM, McConnachie A, Walker A, et al. Tumour necrosis factor inhibition versus rituximab for patients with rheumatoid arthritis who require biological treatment (ORBIT): an open‐label, randomised controlled, non‐inferiority, trial. Lancet. 2016;388:239–47. [DOI] [PubMed] [Google Scholar]
- 14. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. 2021;20:179–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kristyanto H, Blomberg NJ, Slot LM, van der Voort EIH, Kerkman PF, Bakker A, et al. Persistently activated, proliferative memory autoreactive B cells promote inflammation in rheumatoid arthritis. Sci Transl Med. 2020;12:eaaz5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. 2014;15:602–11. [DOI] [PubMed] [Google Scholar]
- 17. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: new insights and open questions. Sci Immunol. 2018;3:eaat4579. [DOI] [PubMed] [Google Scholar]
- 18. Michael M, Vermeren S. A neutrophil‐centric view of chemotaxis. Essays Biochem. 2019;63:607–18. [DOI] [PubMed] [Google Scholar]
- 19. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev. 2015;268:25–51. [DOI] [PubMed] [Google Scholar]
- 20. Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex‐mediated neutrophil recruitment and tissue injury. Circulation. 2009;120:2012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fossati G, Bucknall RC, Edwards SW. Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: changes in functional responses induced by priming with cytokines. Ann Rheum Dis. 2002;61:13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcalphaRI. J Immunol. 2016;197:4552–9. [DOI] [PubMed] [Google Scholar]
- 23. Fawthrop F, Hornby J, Swan A, Hutton C, Doherty M, Dieppe P. A comparison of normal and pathological synovial fluid. Br J Rheumatol. 1985;24:61–9. [DOI] [PubMed] [Google Scholar]
- 24. Freemont AJ, Denton J. Disease distribution of synovial fluid mast cells and cytophagocytic mononuclear cells in inflammatory arthritis. Ann Rheum Dis. 1985;44:312–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palmer DG, Hogg N, Revell PA. Lymphocytes, polymorphonuclear leukocytes, macrophages and platelets in synovium involved by rheumatoid arthritis. A study with monoclonal antibodies. Pathology. 1986;18:431–7. [DOI] [PubMed] [Google Scholar]
- 26. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. 1997;40:217–25. [DOI] [PubMed] [Google Scholar]
- 27. Eggleton P, Wang L, Penhallow J, Crawford N, Brown KA. Differences in oxidative response of subpopulations of neutrophils from healthy subjects and patients with rheumatoid arthritis. Ann Rheum Dis. 1995;54:916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto T, Kaneko T, Seto M, Wada H, Kobayashi T, Nakatani K, et al. The membrane proteinase 3 expression on neutrophils was downregulated after treatment with infliximab in patients with rheumatoid arthritis. Clin Appl Thromb Hemost. 2008;14:186–92. [DOI] [PubMed] [Google Scholar]
- 29. Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in expression of membrane TNF, NF‐{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis. 2011;70:537–43. [DOI] [PubMed] [Google Scholar]
- 30. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014;16:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoffmann MH, Bruns H, Backdahl L, Neregard P, Niederreiter B, Herrmann M, et al. The cathelicidins LL‐37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Ann Rheum Dis. 2013;72:1239–48. [DOI] [PubMed] [Google Scholar]
- 32. Wright HL, Makki FA, Moots RJ, Edwards SW. Low‐density granulocytes: functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J Leukoc Biol. 2017;101:599–611. [DOI] [PubMed] [Google Scholar]
- 33. Hacbarth E, Kajdacsy‐Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986;29:1334–42. [DOI] [PubMed] [Google Scholar]
- 34. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol. 2020;11:584116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raza K, Scheel‐Toellner D, Lee CY, Pilling D, Curnow SJ, Falciani F, et al. Synovial fluid leukocyte apoptosis is inhibited in patients with very early rheumatoid arthritis. Arthritis Res Ther. 2006;8:R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cascao R, Rosario HS, Souto‐Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmun Rev. 2010;9:531–5. [DOI] [PubMed] [Google Scholar]
- 37. Talbot J, Bianchini FJ, Nascimento DC, Oliveira RD, Souto FO, Pinto LG, et al. CCR2 expression in neutrophils plays a critical role in their migration into the joints in rheumatoid arthritis. Arthritis Rheumatol. 2015;67:1751–9. [DOI] [PubMed] [Google Scholar]
- 38. Assi LK, Wong SH, Ludwig A, Raza K, Gordon C, Salmon M, et al. Tumor necrosis factor alpha activates release of B lymphocyte stimulator by neutrophils infiltrating the rheumatoid joint. Arthritis Rheum. 2007;56:1776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, et al. Proinflammatory mediators elicit secretion of the intracellular B‐lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood. 2005;105:830–7. [DOI] [PubMed] [Google Scholar]
- 40. Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. 2003;48:2796–806. [DOI] [PubMed] [Google Scholar]
- 41. Vono M, Lin A, Norrby‐Teglund A, Koup RA, Liang F, Lore K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood. 2017;129:1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10:593–601. [DOI] [PubMed] [Google Scholar]
- 43. O'Neil LJ, Kaplan MJ. Neutrophils in rheumatoid arthritis: breaking immune tolerance and fueling disease. Trends Mol Med. 2019;25:215–27. [DOI] [PubMed] [Google Scholar]
- 44. Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–8. [DOI] [PubMed] [Google Scholar]
- 45. Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in anti‐type II collagen antibody and lipopolysaccharide‐induced arthritis. Immunology. 2006;119:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, et al. A key role for G‐CSF‐induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112:5193–201. [DOI] [PubMed] [Google Scholar]
- 47. Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, et al. Lipid‐cytokine‐chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sadik CD, Kim ND, Iwakura Y, Luster AD. Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcgammaR signaling. Proc Natl Acad Sci USA. 2012;109:E3177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyabe Y, Miyabe C, Murooka TT, Kim EY, Newton GA, Kim ND, et al. Complement C5a receptor is the key initiator of neutrophil adhesion igniting immune complex‐induced arthritis. Sci Immunol. 2017;2:eaaj2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miyabe Y, Miyabe C, Mani V, Mempel TR, Luster AD. Atypical complement receptor C5aR2 transports C5a to initiate neutrophil adhesion and inflammation. Sci Immunol. 2019;4:eaav5951. [DOI] [PubMed] [Google Scholar]
- 51. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. 2011;187:4319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kovacs M, Nemeth T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med. 2014;211:1993–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nemeth T, Futosi K, Sitaru C, Ruland J, Mocsai A. Neutrophil‐specific deletion of the CARD9 gene expression regulator suppresses autoantibody‐induced inflammation in vivo. Nat Commun. 2016;7:11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mysore V, Cullere X, Mears J, Rosetti F, Okubo K, Liew PX, et al. FcgammaR engagement reprograms neutrophils into antigen cross‐presenting cells that elicit acquired anti‐tumor immunity. Nat Commun. 2021;12:4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133:2178–85. [DOI] [PubMed] [Google Scholar]
- 56. Pieterse E, Rother N, Yanginlar C, Gerretsen J, Boeltz S, Munoz LE, et al. Cleaved N‐terminal histone tails distinguish between NADPH oxidase (NOX)‐dependent and NOX‐independent pathways of neutrophil extracellular trap formation. Ann Rheum Dis. 2018;77:1790–8. [DOI] [PubMed] [Google Scholar]
- 57. Carmona‐Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. 2020;5:e139388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. O'Neil LJ, Barrera‐Vargas A, Sandoval‐Heglund D, Merayo‐Chalico J, Aguirre‐Aguilar E, Aponte AM, et al. Neutrophil‐mediated carbamylation promotes articular damage in rheumatoid arthritis. Sci Adv. 2020;6:eabd2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Bont CM, Stokman MEM, Faas P, Thurlings RM, Boelens WC, Wright HL, et al. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun. 2020;113:102484. [DOI] [PubMed] [Google Scholar]
- 61. Dwivedi N, Neeli I, Schall N, Wan H, Desiderio DM, Csernok E, et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 2014;28:2840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. 2016;75:1866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sokolove J, Johnson DS, Lahey LJ, Wagner CA, Cheng D, Thiele GM, et al. Rheumatoid factor as a potentiator of anti‐citrullinated protein antibody‐mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hecht C, Englbrecht M, Rech J, Schmidt S, Araujo E, Engelke K, et al. Additive effect of anti‐citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann Rheum Dis. 2015;74:2151–6. [DOI] [PubMed] [Google Scholar]
- 66. Laurent L, Anquetil F, Clavel C, Ndongo‐Thiam N, Offer G, Miossec P, et al. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis‐specific immune complexes containing anticitrullinated protein antibodies. Ann Rheum Dis. 2015;74:1425–31. [DOI] [PubMed] [Google Scholar]
- 67. Behnen M, Leschczyk C, Moller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac‐1. J Immunol. 2014;193:1954–65. [DOI] [PubMed] [Google Scholar]
- 68. Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, et al. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood. 2012;120:4421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Domer D, Walther T, Moller S, Behnen M, Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol. 2021;12:636954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Okada Y, Kim K, Han B, Pillai NE, Ong RT, Saw WY, et al. Risk for ACPA‐positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum Mol Genet. 2014;23:6916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van der Woude D, Lie BA, Lundstrom E, Balsa A, Feitsma AL, Houwing‐Duistermaat JJ, et al. Protection against anti‐citrullinated protein antibody‐positive rheumatoid arthritis is predominantly associated with HLA‐DRB1*1301: a meta‐analysis of HLA‐DRB1 associations with anti‐citrullinated protein antibody‐positive and anti‐citrullinated protein antibody‐negative rheumatoid arthritis in four European populations. Arthritis Rheum. 2010;62:1236–45. [DOI] [PubMed] [Google Scholar]
- 72. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high‐affinity peptide interaction with the rheumatoid arthritis‐associated HLA‐DRB1*0401 MHC class II molecule. J Immunol. 2003;171:538–41. [DOI] [PubMed] [Google Scholar]
- 73. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA‐DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210:2569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kampstra AS, van Heemst J, Moustakas AK, Papadopoulos GK, Huizinga TW, Toes RE. The increased ability to present citrullinated peptides is not unique to HLA‐SE molecules: arginine‐to‐citrulline conversion also enhances peptide affinity for HLA‐DQ molecules. Arthritis Res Ther. 2016;18:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mustelin T, Bottini N, Stanford SM. The contribution of PTPN22 to rheumatic disease. Arthritis Rheumatol. 2019;71:486–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schickel JN, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, et al. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol. 2016;1:aaf7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Metzler G, Dai X, Thouvenel CD, Khim S, Habib T, Buckner JH, et al. The autoimmune risk variant PTPN22 C1858T alters B cell tolerance at discrete checkpoints and differentially shapes the naive repertoire. J Immunol. 2017;199:2249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, et al. The autoimmune‐associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. 2015;74:1588–95. [DOI] [PubMed] [Google Scholar]
- 80. Vermeren S, Miles K, Chu JY, Salter D, Zamoyska R, Gray M. PTPN22 is a critical regulator of Fcgamma receptor‐mediated neutrophil activation. J Immunol. 2016;197:4771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chang HH, Dwivedi N, Nicholas AP, Ho IC. The W620 polymorphism in PTPN22 disrupts its interaction with peptidylarginine deiminase type 4 and enhances citrullination and NETosis. Arthritis Rheumatol. 2015;67:2323–34. [DOI] [PubMed] [Google Scholar]
- 82. Chang HH, Liu GY, Dwivedi N, Sun B, Okamoto Y, Kinslow JD, et al. A molecular signature of preclinical rheumatoid arthritis triggered by dysregulated PTPN22. JCI Insight. 2016;1:e90045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA‐DR (shared epitope)‐restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. [DOI] [PubMed] [Google Scholar]
- 84. Sugiyama D, Nishimura K, Tamaki K, Tsuji G, Nakazawa T, Morinobu A, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta‐analysis of observational studies. Ann Rheum Dis. 2010;69:70–81. [DOI] [PubMed] [Google Scholar]
- 85. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. 2008;67:1488–92. [DOI] [PubMed] [Google Scholar]
- 86. Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–84. [DOI] [PubMed] [Google Scholar]
- 87. Lugli EB, Correia RE, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non‐smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther. 2015;17:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ospelt C, Bang H, Feist E, Camici G, Keller S, Detert J, et al. Carbamylation of vimentin is inducible by smoking and represents an independent autoantigen in rheumatoid arthritis. Ann Rheum Dis. 2017;76:1176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hosseinzadeh A, Thompson PR, Segal BH, Urban CF. Nicotine induces neutrophil extracellular traps. J Leukoc Biol. 2016;100:1105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen‐induced arthritis. Rheumatology. 2017;56:644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cheng Z, Do T, Mankia K, Meade J, Hunt L, Clerehugh V, et al. Dysbiosis in the oral microbiomes of anti‐CCP positive individuals at risk of developing rheumatoid arthritis. Ann Rheum Dis. 2021;80:162–8. [DOI] [PubMed] [Google Scholar]
- 92. Potempa J, Mydel P, Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol. 2017;13:606–20. [DOI] [PubMed] [Google Scholar]
- 93. Gonzalez‐Febles J, Rodriguez‐Lozano B, Sanchez‐Piedra C, Garnier‐Rodriguez J, Bustabad S, Hernandez‐Gonzalez M, et al. Association between periodontitis and anti‐citrullinated protein antibodies in rheumatoid arthritis patients: a cross‐sectional study. Arthritis Res Ther. 2020;22:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mankia K, Cheng Z, Do T, Hunt L, Meade J, Kang J, et al. Prevalence of periodontal disease and periodontopathic bacteria in anti‐cyclic citrullinated protein antibody‐positive at‐risk adults without arthritis. JAMA Netw Open. 2019;2:e195394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha‐enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010;62:2662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 2013;9:e1003627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jung H, Jung SM, Rim YA, Park N, Nam Y, Lee J, et al. Arthritic role of Porphyromonas gingivalis in collagen‐induced arthritis mice. PLoS One. 2017;12:e0188698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Aliko A, Kaminska M, Bergum B, Gawron K, Benedyk M, Lamont RJ, et al. Impact of porphyromonas gingivalis peptidylarginine deiminase on bacterial biofilm formation, epithelial cell invasion, and epithelial cell transcriptional landscape. Sci Rep. 2018;8:14144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bryzek D, Ciaston I, Dobosz E, Gasiorek A, Makarska A, Sarna M, et al. Triggering NETosis via protease‐activated receptor (PAR)‐2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PLoS Pathog. 2019;15:e1007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans‐induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. 2016;8:369ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Posnett DN, Edinger J. When do microbes stimulate rheumatoid factor? J Exp Med. 1997;185:1721–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. El‐Gabalawy HS, Robinson DB, Hart D, Elias B, Markland J, Peschken CA, et al. Immunogenetic risks of anti‐cyclical citrullinated peptide antibodies in a North American Native population with rheumatoid arthritis and their first‐degree relatives. J Rheumatol. 2009;36:1130–5. [DOI] [PubMed] [Google Scholar]
- 103. Masson‐Bessiere C, Sebbag M, Girbal‐Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial targets of the rheumatoid arthritis‐specific antifilaggrin autoantibodies are deiminated forms of the alpha‐ and beta‐chains of fibrin. J Immunol. 2001;166:4177–84. [DOI] [PubMed] [Google Scholar]
- 104. Mathsson L, Mullazehi M, Wick MC, Sjoberg O, van Vollenhoven R, Klareskog L, et al. Antibodies against citrullinated vimentin in rheumatoid arthritis: higher sensitivity and extended prognostic value concerning future radiographic progression as compared with antibodies against cyclic citrullinated peptides. Arthritis Rheum. 2008;58:36–45. [DOI] [PubMed] [Google Scholar]
- 105. Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, et al. Identification of citrullinated alpha‐enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis. 2014;73:1414–22. [DOI] [PubMed] [Google Scholar]
- 107. Steen J, Forsström B, Sahlström P, Odowd V, Israelsson L, Krishnamurthy A, et al. Recognition of amino acid motifs, rather than specific proteins, by human plasma cell‐derived monoclonal antibodies to posttranslationally modified proteins in rheumatoid arthritis. Arthritis Rheumatol. 2019;71:196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Reed E, Hedstrom AK, Hansson M, Mathsson‐Alm L, Brynedal B, Saevarsdottir S, et al. Presence of autoantibodies in “seronegative” rheumatoid arthritis associates with classical risk factors and high disease activity. Arthritis Res Ther. 2020;22:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol. 2012;8:144–52. [DOI] [PubMed] [Google Scholar]
- 110. van der Woude D, Rantapaa‐Dahlqvist S, Ioan‐Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, et al. Epitope spreading of the anti‐citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis. 2010;69:1554–61. [DOI] [PubMed] [Google Scholar]
- 111. Suwannalai P, van de Stadt LA, Radner H, Steiner G, El‐Gabalawy HS, Zijde CM, et al. Avidity maturation of anti‐citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012;64:1323–8. [DOI] [PubMed] [Google Scholar]
- 112. Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity maturation drives epitope spreading and generation of proinflammatory anti‐citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2018;70:1946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lu DR, McDavid AN, Kongpachith S, Lingampalli N, Glanville J, Ju CH, et al. T cell‐dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol. 2018;70:1732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lloyd KA, Steen J, Amara K, Titcombe PJ, Israelsson L, Lundstrom SL, et al. Variable domain N‐linked glycosylation and negative surface charge are key features of monoclonal ACPA: implications for B‐cell selection. Eur J Immunol. 2018;48:1030–45. [DOI] [PubMed] [Google Scholar]
- 115. Vergroesen RD, Slot LM, Hafkenscheid L, Koning MT, van der Voort EIH, Grooff CA, et al. B‐cell receptor sequencing of anti‐citrullinated protein antibody (ACPA) IgG‐expressing B cells indicates a selective advantage for the introduction of N‐glycosylation sites during somatic hypermutation. Ann Rheum Dis. 2018;77:956–8. [DOI] [PubMed] [Google Scholar]
- 116. Levy J, Barnett EV, MacDonald NS, Klinenberg JR. Altered immunoglobulin metabolism in systemic lupus erythematosus and heumatoid arthritis. J Clin Invest. 1970;49:708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Verpoort KN, Jol‐van der Zijde CM, Papendrecht‐van der Voort EA, Ioan‐Facsinay A, Drijfhout JW, van Tol MJ, et al. Isotype distribution of anti‐cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum. 2006;54:3799–808. [DOI] [PubMed] [Google Scholar]
- 118. Lakos G, Soos L, Fekete A, Szabo Z, Zeher M, Horvath IF, et al. Anti‐cyclic citrullinated peptide antibody isotypes in rheumatoid arthritis: association with disease duration, rheumatoid factor production and the presence of shared epitope. Clin Exp Rheumatol. 2008;26:253–60. [PubMed] [Google Scholar]
- 119. Scinocca M, Bell DA, Racape M, Joseph R, Shaw G, McCormick JK, et al. Antihomocitrullinated fibrinogen antibodies are specific to rheumatoid arthritis and frequently bind citrullinated proteins/peptides. J Rheumatol. 2014;41:270–9. [DOI] [PubMed] [Google Scholar]
- 120. Shi J, van de Stadt LA, Levarht EW, Huizinga TW, Hamann D, van Schaardenburg D, et al. Anti‐carbamylated protein (anti‐CarP) antibodies precede the onset of rheumatoid arthritis. Ann Rheum Dis. 2014;73:780–3. [DOI] [PubMed] [Google Scholar]
- 121. Kinloch A, Lundberg K, Wait R, Wegner N, Lim NH, Zendman AJ, et al. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 2008;58:2287–95. [DOI] [PubMed] [Google Scholar]
- 122. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 2015;67:3135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhou Y, Chen B, Mittereder N, Chaerkady R, Strain M, An LL, et al. Spontaneous secretion of the citrullination enzyme PAD2 and cell surface exposure of PAD4 by neutrophils. Front Immunol. 2017;8:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Umeda N, Matsumoto I, Kawaguchi H, Kurashima Y, Kondo Y, Tsuboi H, et al. Prevalence of soluble peptidylarginine deiminase 4 (PAD4) and anti‐PAD4 antibodies in autoimmune diseases. Clin Rheumatol. 2016;35:1181–8. [DOI] [PubMed] [Google Scholar]
- 125. Harris ML, Darrah E, Lam GK, Bartlett SJ, Giles JT, Grant AV, et al. Association of autoimmunity to peptidyl arginine deiminase type 4 with genotype and disease severity in rheumatoid arthritis. Arthritis Rheum. 2008;58:1958–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kolfenbach JR, Deane KD, Derber LA, O'Donnell CI, Gilliland WR, Edison JD, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:2633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Damgaard D, Senolt L, Nielsen MF, Pruijn GJ, Nielsen CH. Demonstration of extracellular peptidylarginine deiminase (PAD) activity in synovial fluid of patients with rheumatoid arthritis using a novel assay for citrullination of fibrinogen. Arthritis Res Ther. 2014;16:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Arandjelovic S, McKenney KR, Leming SS, Mowen KA. ATP induces protein arginine deiminase 2‐dependent citrullination in mast cells through the P2X7 purinergic receptor. J Immunol. 2012;189:4112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Fuhrmann J, Thompson PR. Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem Biol. 2016;11:654–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Darrah E, Giles JT, Ols ML, Bull HG, Andrade F, Rosen A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci Transl Med. 2013;5:186ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Demoruelle MK, Wang H, Davis RL, Visser A, Hoang J, Norris JM, et al. Anti‐peptidylarginine deiminase‐4 antibodies at mucosal sites can activate peptidylarginine deiminase‐4 enzyme activity in rheumatoid arthritis. Arthritis Res Ther. 2021;23:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Karmakar U, Chu JY, Sundaram K, Astier AL, Garside H, Hansen CG, et al. Immune complex‐induced apoptosis and concurrent immune complex clearance are anti‐inflammatory neutrophil functions. Cell Death Dis. 2021;12:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chu JY, Dransfield I, Rossi AG, Vermeren S. Non‐canonical PI3K‐Cdc42‐Pak‐Mek‐Erk signaling promotes immune‐complex‐induced apoptosis in human neutrophils. Cell Rep. 2016;17:374–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gamberale R, Giordano M, Trevani AS, Andonegui G, Geffner JR. Modulation of human neutrophil apoptosis by immune complexes. J Immunol. 1998;161:3666–74. [PubMed] [Google Scholar]
- 135. Arienti S, Barth ND, Dorward DA, Rossi AG, Dransfield I. Regulation of apoptotic cell clearance during resolution of inflammation. Front Pharmacol. 2019;10:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep. 2020;31:107602. [DOI] [PubMed] [Google Scholar]
- 137. Shi L, Aymonnier K, Wagner DD. Neutrophil stimulation with citrullinated histone H4 slows down calcium influx and reduces NET formation compared with native histone H4. PLoS One. 2021;16:e0251726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Knopf J, Leppkes M, Schett G, Herrmann M, Munoz LE. Aggregated NETs sequester and detoxify extracellular histones. Front Immunol. 2019;10:2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Koziel J, Bryzek D, Sroka A, Maresz K, Glowczyk I, Bielecka E, et al. Citrullination alters immunomodulatory function of LL‐37 essential for prevention of endotoxin‐induced sepsis. J Immunol. 2014;192:5363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wong A, Bryzek D, Dobosz E, Scavenius C, Svoboda P, Rapala‐Kozik M, et al. A novel biological role for peptidyl‐arginine deiminases: citrullination of cathelicidin LL‐37 controls the immunostimulatory potential of cell‐free DNA. J Immunol. 2018;200:2327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]