

Abstract
Chronic lymphocytic leukemia (CLL) patients who develop Hodgkin lymphoma (HL) have limited survival. No current therapeutic standard of care exists. We conducted a multi-center retrospective study of patients with Hodgkin transformation (HT) of CLL. Clinicobiologic characteristics, treatment type, and survival outcomes were analyzed and compared with historic case series. Ninety-four patients were identified. Median age at HT was 67 years (range, 38-85). Median time from CLL diagnosis to HT was 5.5 years (range, 0-20.2). Prior to HT, patients received a median of two therapies for CLL (range, 0-12). As initial therapy for HT, 61% (n=62) received ABVD-based regimens (adriamycin, bleomycin, vinblastine, and dacarbazine). Seven (7%) patients received hematopoietic cell transplantation (HCT) while in first complete remission (CR1). The median number of treatments for HT per patient was one (range, 0-5) with 59 (61%) patients only receiving one line of therapy. After HT, patients had a median follow-up of 1.6 years (range, 0-15.1). Two-year overall survival (OS) after HT diagnosis was 72% (95% Confidence Interval: 62-83). The patients who received standard ABVD-based therapy had a median OS of 13.2 years. Although limited by small sample size, the patients who underwent HCT for HT in CR1 had a similar 2-year OS (n=7; 67%) compared to patients who did not undergo HCT for HT in CR1 (n=87; 72%; P=0.46). In this multi-center study, HT patients treated with ABVD-based regimens had prolonged survival supporting the use of these regimens as standard of care for these patients.
Introduction
Richter Transformation (RT) is the development of an aggressive lymphoma in the setting of chronic lymphocytic leukemia (CLL), occurring in 5-10% of CLL patients. Clinical outcomes in patients with RT are exceedingly poor with a median overall survival (OS) of 5-8 months from diagnosis.1 In the majority of RT cases, the CLL transforms into diffuse large B-cell lymphoma (DLBCL). Development of Hodgkin lymphoma (HL) in patients with CLL is a rare (<1%), but recognized form of RT.1,2 Based on prior data, the expected OS of CLL patients with HL transformation (HT) appears longer than CLL patients with transformation to DLBCL, with reported survival ranging from 0.8–3.9 years after diagnosis of HT.2-4 In contrast, HT patients have shorter OS (2-year OS of 30-40%) when compared with all patients with de novo HL (2-year OS >90%) after treatment with standard chemotherapy regimens, such as adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD).3-7
There are limited published data about HT patients and most are small series reported from single institutions.3,8-10 To date, there are no clear recommendations for the management of these patients. Extrapolating from data in RT to DLBCL, some groups recommend aggressive therapy with hematopoietic cell transplantation (HCT) after achievement of first complete remission (CR1).3,11 As patients with CLL are typically elderly with comorbidities, undergoing a HCT is infrequently feasible for this patient population. In order to better understand the clinicobiologic features, treatment patterns, and clinical outcomes, we describe the largest reported series of HT patients based upon our inclusive multi-institutional clinical experience.
Methods
After Institutional Review Board approval, CLL patients were retrospectively identified who also developed biopsy-proven, classical HL diagnosed between 2000 and 2018 at 13 United States tertiary cancer centers. The number of patients identified at each center is detailed in the Online Supplementary Table S1. Investigators from each site reviewed pathology records to confirm diagnosis. Clinicobiologic characteristics, treatment type, and survival outcomes for each patient were analyzed (collected variables are listed in the Online Supplementary Table S2). The International Prognostic Score (IPS) for HL and the Richter Scoring System (RSS, scoring validated previously on RT patients with DLBCL) was calculated for each patient at the time of HT diagnosis in patients where data were available (see the Online Supplementary Tables S3 and S4).1,12 Progression-free survival (PFS) was measured from the time of HT diagnosis until relapse, subsequent therapy for HT, or death. OS was measured from the time of HT diagnosis until time of death. PFS and OS estimates were calculated using the Kaplan-Meier method. The log-rank test was used to calculate differences in survival. Cox proportional hazards models and associated Wald tests were used to analyze the relationship between covariates and OS. Logistic regression was used to compare baseline characteristics of patients receiving full dose ABVD versus all other regimens. R statistical computing software version 3.2.1 (The R Foundation for Statistical Computing, Vienna, Austria 2015) was used for statistical analysis.
Results
Patient characteristics
Ninety-four CLL patients with HT were identified from 13 centers. Median age at initial CLL diagnosis was 60 years (range, 31-84) and 81% of the patients were men. At initial CLL diagnosis, 15% (11 of 81) were Rai 3-4, 67% (25 of 37) had an un-mutated IGHV gene, and 15% (nine of 61) had del(17p). Prior to HT diagnosis, patients had a median of two (range, 0-12) prior therapies for CLL. Seventeen (18%) had no prior CLL treatments. Fortythree (46%) and 25 (27%) patients had received purine analogue- and ibrutinib-based therapy prior to HT, respectively. Median time from CLL diagnosis to HT was 5.5 years (range, 0-20.2). Seven patients had simultaneous diagnosis of CLL and HL.
Table 1.
Baseline Characteristics at Hodgkin transformation of chronic lymphocytic leukemia.
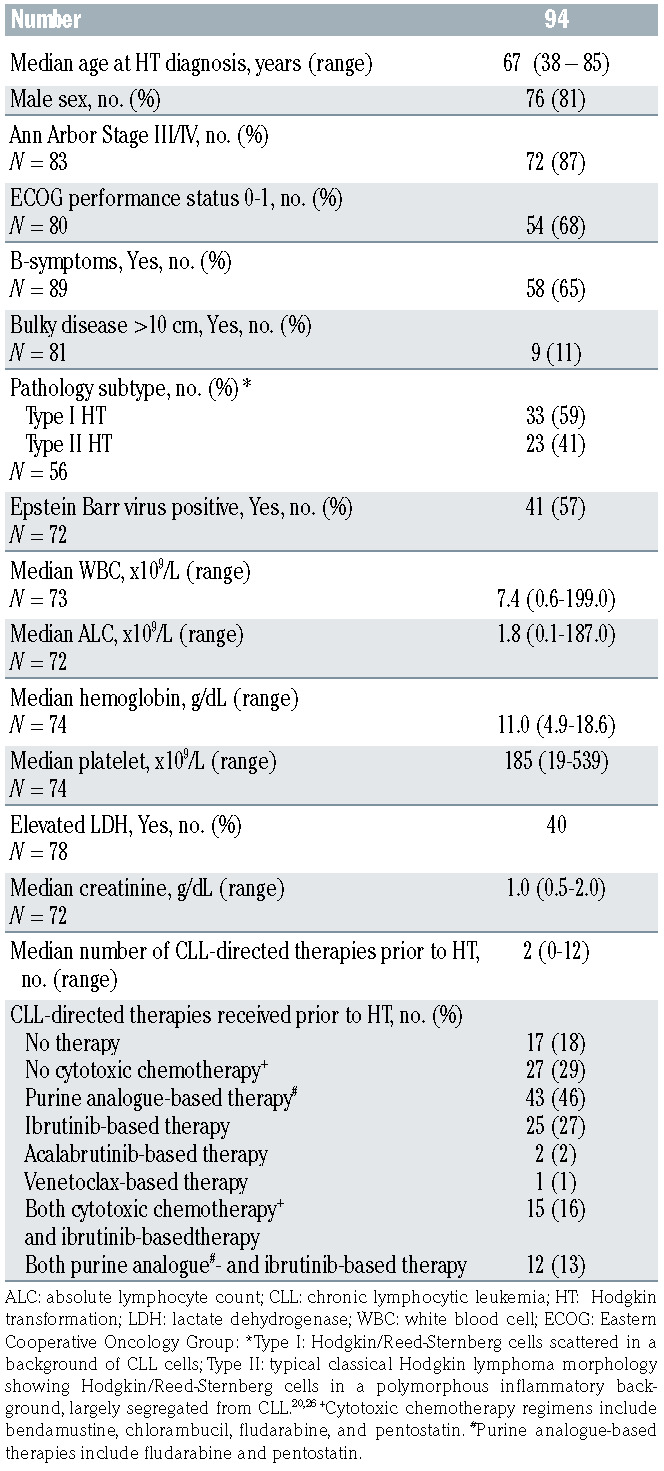
Median age at HT was 67 years (range, 38-85; Table 1). Eighty-seven percent (72 of 83) had Ann Arbor stage III/IV, 15% had Eastern Cooperative Oncology Group (ECOG) performance status of >2, 65% (58 of 89) had Bsymptoms, 11% (nine of 81) had bulky disease (>10 cm), and 62% (36 of 58) had an IPS score of >3 (Table 1). Of 72 evaluable patient samples 41 (57%) tested positive for Epstein Barr virus (EBV). The median number of treatments for HT per patient was one (range, 0-5) with 59 (61%) patients only receiving one line of therapy. Details of regimens received can be found in the Online Supplementary Table S5. As initial therapy for HL, the majority of patients (61%, n=62) received ABVD-based (n=48) or AVD-based regimens (n=14). Of these, anti- CD20 monoclonal antibody was added in seven and BTK inhibitor was added in five (Online Supplementary Table S5). Ten (11%) received a brentuximab vedotin as part of initial therapy. Seven (7%) received an RCHOP-based regimen (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). Six patients (6%) received no therapy for HT due to frailty. Nine (10%) patients received other regimens, which are detailed in the Online Supplementary Table S5. There were 25 patients who received ibrutinib (n=24) or acalabrutinib (n=1) as the CLL treatment immediately prior to the first HT treatment. Of the 25 patients, 17 patients discontinued BTK treatment during the first HT treatment and three of these resumed BTK inhibitor upon completion of HT therapy. Of the nine patients who continued BTK inhibitor therapy concurrently with the first HT treatment, six received AVD, two received rituximab plus AVD, and one received brentuximab vedotin. Twentynine patients received salvage chemotherapy for HT. The majority (n=13) received only one salvage regimen for HT. The most common salvage regimens used were ifosfamide, carboplatin, and etoposide in seven patients and brentuximab vedotin in six patients. Online Supplementary Table S6 describes salvage chemotherapy regimens received by individual patients. Subsequent therapy included autologous HCT and allogeneic HCT in seven (7%) and eleven (12%) patients, respectively. Two (2%) and five (5%) patients received their autologous and allogeneic HCT while in CR1, respectively. Detailed toxicity data are not available for this patient subset.
Table 2.
Baseline chronic lymphocytic leukemia (CLL) characteristics at time of CLL diagnosis: univariate Cox models for overall survival.

Table 3.
Baseline characteristics at time of Hodgkin transformation of chronic lymphocytic leukemia: univariate Cox models for overall survival.

Figure 1.

Overall survival for patients with Hodgkin transformation. (A) Overall survival for patients with Hodgkin transformation (HT) who received prior chronic lymphocytic leukemia (CLL)-directed treatment versus no prior CLL-directed treatment. (B) Overall survival for patients with HT who received prior purine analogue (PA)- based therapy for CLL versus no prior PA-based therapy for CLL.
Survival
After HT diagnosis, patients had a median follow-up of 1.6 years (range, 0.0–15.1). The median time between HT diagnosis and HT treatment was 15 days (interquartile range, 6.5–30.5 days). The median PFS was 21 months (95% Confidence Interval [CI]: 14-58). Estimated 2-year PFS was 48% (95% CI: 38-61; Online Supplementary Figure S1A). There was minimal difference in the estimated 2- year PFS after censoring for subsequent HCT (2-year PFS =48.9%, 95% CI: 38.7– 61.8; Online Supplementary Figure S1B). The median OS was 65 months (95% CI: 34-infinity). Two-year OS after HT diagnosis was 72% (95% CI: 62-83). Five patients (5%) died within 2 months of HT diagnosis.
At the time of initial CLL diagnosis, the baseline characteristics of Rai Stage, IGHV mutational status, or the presence of del(13)(q14), trisomy 12, del(11)(q22.3), or del(17)(p13.1) were not associated with differences in OS after HT (Table 2). Patients who received any CLL-directed therapy (n=80) prior to HT had a significantly lower estimated 2-year OS of 69% (95% CI: 58–82) compared with patients who did not receive any prior CLL-directed therapy (n=17; 93%; 95% CI: 82-100; P=0.02; Figure 1A). Patients who received purine-analogue-based therapy for CLL prior to HT had a significantly lower estimated 2- year OS of 60% (95% CI: 46–79) compared with patients who did not receive purine-analogue-based CLL-directed therapy prior to HT (n=51; 83%; 95% CI: 73–96; P=0.009; Figure 1B). A more detailed depiction of estimated OS by prior CLL-directed therapy is shown in the Online Supplementary Figure S3. The available samples of those patients treated with purine-analogue-based therapy prior to HT (n=34) more frequently demonstrated positivity for EBV (OR 3.24; 95% CI: 1.1-9.9; P=0.02).
Of the 25 patients who developed HT following treatment at any time with ibrutinib, the median time from initiation of ibrutinib to HT was 15.5 months (range, 1.2– 37.7). Ten of these patients had never received a standard chemoimmunotherapy regimen prior to HT. Compared with the patients who did not receive ibrutinib prior to HT, the estimated 2-year OS was slightly lower in the patients who did receive ibrutinib prior to HT (73% vs. 64%) although this finding was not statistically different (P=0.33). The available samples of those patients (n=22) treated with ibrutinib prior to HT tended to be less likely to be positive for EBV than samples from patients not previously treated with ibrutinib (OR 0.52; 95% CI: 0.2-1.6; P=0.21).
At the time of HT diagnosis, the only characteristics associated with OS in univariate analysis were lactate dehydrogenase (LDH) above normal (Hazard Ratio [HR] 4.5; 95% CI: 1.8-11.1; P=0.001), IPS of ≥4 versus <4 (HR 4.8; 95% CI: 1.1-22.0; P=0.043) and RSS of ≥2 versus <2 (HR 5.7 95% CI: 1.8-18.5; P=0.003; Table 3; Figure 2A and B). In the patients who had available data to calculate the RSS (n=57), each increase of the RSS by one point resulted in an increased risk of death (HR 2.3; 95% CI: 1.3-3.9; P=0.004; Figure 2B).
As compared to patients who received ABVD-based regimen (n=48) as first therapy for HT, those who received RCHOP (n=8) had a similar risk of death (HR 1.6, 95% CI: 0.4-5.7; P=0.48; Figure 3A). Conversely, those patients who received AVD-based (n=14; HR 3.0; P=0.04), BV-based (n=9; HR 3.6; P=0.05), or other (n=9; HR 7.67; P<0.001) regimens for first therapy for HT had an increased risk of death compared with patients who received ABVD-based regimen (n=48). This finding still remained significant after adjusting for age of patient at time of first HT therapy (Online Supplementary Table S7). Patients who received ABVD had a median OS of 13.2 years (95% CI: 4.8-infinity). Patients who received ABVD versus all other regimens were more likely to be less than 65 years of age (P=0.05), have a baseline absolute lymphocyte count (ALC) >0.6 (P=0.003), and have an IPS of <4 (P=0.03). There was no difference in OS based on the year that the patient received the first therapy for HT (P=0.81; Online Supplementary Figure S2).
Figure 2.

Overall survival for patients with Hodgkin transformation. Overall survival for patients with Hodgkin transformation categorized by (A) International Prognostic Score (IPS) for Hodgkin lymphoma and (B) Richter Scoring System for Richter transformation.
The patients who underwent HCT for HT in first complete remission had a similar 2-year OS (n=7; 67%; 95% CI: 38-100) to patients who did not undergo HCT for HT in first complete remission (n=87; 72%; 95% CI: 63–84; P=0.46; Figure 3B). Peritransplant mortality did not contribute to shortened survival in this group as the earliest death in this group occurred at 355 days post-transplant.
Discussion
In this retrospective analysis, we describe the largest reported multi-center inclusive series of patients with HT from CLL. Clinical outcomes, including survival, in this series of patients with HT were higher than what has previously been reported for this patient population and strikingly similar to what is historically expected in elderly patients with de novo HL.3,4,6,7 Patients with HT who have received prior CLL-directed therapies (specifically purineanalogue- based treatments), elevated LDH, IPS ≥4, and RSS ≥2 are predicted to have a shorter OS. The OS of patients in this series was higher than what is expected in CLL patients who transform to DLBCL.1,13-15 Unlike patients with RT to DLBCL, the majority of patients only received one line of HL therapy. Only 20% went on to receive HCT and had similar OS to patients who did not receive HCT in CR1. This finding has impact in the standard management of patients with HT as these data challenge the recommendation of HCT in CR1.
Patients in our series survived longer than historical series of RT to DLBCL (median OS 65 months vs. 5-8 months).13-17 It is unclear whether this difference is related to underlying disease biology or effectiveness of therapy. Prior molecular studies have revealed that patients with RT to DLBCL that is clonally related to the underlying CLL have much shorter survival than patients with RT to DLBCL that is clonally unrelated to the underlying CLL.18 Therefore, clonally unrelated RT to DLBCL is treated as de novo DLBCL with expected outcomes more consistent with de novo DLBCL.19 Little is known about clonal relation of the HL and CLL cells in patients with HT. One group evaluated tissue samples of 33 HT cases.20 The HL cells were clonally related to the CLL cells in 14 cases and unrelated in 19 cases.20 Interestingly, they found no differences in baseline characteristics or OS with treatment when comparing patients with clonally related versus unrelated HT, which is in contrast to what was previously observed in patients with RT to DLBCL.20,21 Further molecular study of HT patient samples is needed to fully understand the underlying biology of HT, which may help to determine prognosis or guide therapy for these patients in the future.
The dismal survival in patients with RT to DLBCL often prompts clinicians to implement more aggressive therapy for these patients. One group showed that RT patients with DLBCL who underwent allogeneic HCT in CR1 had longer survival than those patients in CR1 who did not receive further therapy or those patients who did not receive HCT until salvage therapy.1 Based on the data for patients with RT to DLBCL, it was extrapolated that patients with HT may require HCT when they achieve CR1.3,11 In our study, the majority of patients (61%) only received one line of HL therapy and only 20% went on to receive HCT (7% while in CR1). Although limited by small numbers, the patients who underwent HCT for HT in CR1 had a similar 2-year OS to patients who did not undergo HCT for HT in CR1. Our data indicate that these patients with HT can have prolonged OS after achieving response to first-line therapy for HT and may not require HCT in CR1. Therefore, our large series does not support the recommendation of HCT in CR1 for this patient population and has major implications in the management of these patients.
In addition to our patient population demonstrating longer survival than previously published series of RT to DLBCL, the OS in our group of patients with HT was higher than what has been previously reported in historic series of patients with HT (2-year OS 72% vs. 30-40%).3,4 Without direct comparison of the baseline characteristics of the patients, it is possible that the historically diagnosed patients had higher risk features such as increased age or higher IPS or RSS compared with the patients in our group. Additionally, it is possible that the increased use of traditional HL treatment such as ABVD in our patients (60%) versus the prior patients (30%) could have contributed to the improved outcomes of our patients.3,4 As only 11% of our patients received targeted HL therapy with BV, it is unclear at this time if novel targeted HL therapy has improved the outcomes of these patients. In fact, in our review patients who received BV-based therapies (n=9) as initial treatment had a higher risk of death than those who received full dose ABVD therapy (n=48). This finding is likely biased by selection of less aggressive induction therapy in less fit patients (older and higher IPS) that were not felt to be able to tolerate full doses of ABVD.
Figure 3.

Overall survival for patients with Hodgkin transformation. Overall survival for patients with Hodgkin transformation (HT) categorized by (A) first treatment for HT and (B) hematopoietic cell transplant (HCT) versus no stem cell transplant (SCT) in first complete remission (CR1). RCHOP: rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; ABVD: adriamycin, bleomycin, vinblastine, and dacarbazine
In contrast to prior reports indicating that patients with HT have a shorter survival than patients with de novo HL, these data from the largest multi-center study actually demonstrate a very similar survival compared to historic controls of patients with de novo HL when matched for age. In our study, the median age at time of HT was 67 years.3,4,6,7 A retrospective study described 95 patients diagnosed with HL over the age of 60 years.6 Similar to our population, the median age of the patients in this analysis was also 67 years and the majority of the patients received ABVD as initial HL treatment (n=67). The 2-year OS reported for this group was 73% and very comparable to the 72% found in our analysis of patients with HT.6 A subset analysis was performed on patients over the age of 60 years treated on the prospective phase III E2496 study, where patients were randomized to ABVD versus Stanford V regimen.7 The median age of patients receiving ABVD on this analysis was 66 years. In this group, the 2-year OS rate was approximately 80%, which is comparable to the 72% found in our current analysis.7 Based on these data, the survival of patients with HT seen in our analysis appear similar to patients with de novo HL of a similar age group.
For older patients with de novo HL, clinical outcomes have improved over time.22 This finding was attributed to the adoption and development in expertise of ABVD administration and willingness of clinicians to offer combination chemotherapy to older patients. In much the same way, recent development of novel therapies, such as ibrutinib, have significantly improved survival for patients with CLL, a disease predominately of older patients.23,24 A single-center review found that RT to DLBCL was most likely to occur during the first 2 years of therapy (7% at 2 years).25 This is similar to our finding that of the 25 patients who developed HT following treatment with ibrutinib, the median time from initiation of ibrutinib to HT was 15.5 months. There was no statistically significant difference in 2-year OS between the patients who did and did not receive ibrutinib prior to HT. In five of our cases, clinicians opted to continue ibrutinib therapy to control the patient’s CLL while adding chemotherapy to treat the HL. At this time, the full effect of ibrutinib and other novel agents on risk for HT is unclear.
Although prior ibrutinib therapy did not contribute to reduced survival, we found that prior purine-analoguebased therapies for CLL prior to HT led to reduced survival. This finding has been reported by other groups and is possibly attributed to immunosuppression resulting from purine-analogue therapy.8 In our limited data set, this attribution is supported by the fact that patients who received prior fludarabine-based therapy were more likely to have EBV-positive HT. Interestingly, other traditional baseline characteristics of CLL that have been associated with survival in CLL, such as Rai Stage, unmutated IGHV and karyotype, did not correlate with survival after HT. In addition to prior purine-analogue therapy, the major factors that were identified to predict poor survival for patients with HT were elevated LDH, IPS, and RSS. These factors may be useful in counseling patients with this diagnosis. Interpretation of our data is limited by the intrinsic nature of retrospective analyses. Although individual centers locally confirmed the diagnosis of HT, central review of samples was not feasible secondary to limited quantity of sample and a small number of patients from many centers. We did not search all patients with consecutive HL diagnoses at each center, but searched for CLL patients who subsequently developed HL. As such, it is possible this may have introduced selection bias to our results that may lead to overestimation of patient outcomes. Subset analyses of this patient population were limited secondary to small numbers of patients. Our study examined patients treated at tertiary referral cancer centers and results may not be generalizable to patients treated in the community setting. Despite these limitations, our series is notable as the largest reported cohort of this rare group of patients with HT.
In summary, this series of patients with HT from CLL reveals similar survival to what is expected in patients with de novo HL when compared with age- and treatmentmatched HL patients in previously published studies. These data support that HT patients have longer survival compared with historic controls of patients with RT from DLBCL and do not require allogeneic HCT in CR1. Based on this series, HT patients should be treated with regimens used to treat de novo HL. The effects of novel targeted HL and CLL therapies on the outcomes of patients with HT is unclear at this time, and further study of this rare population, ideally in prospective clinical trials, is required to fully define the optimum management of these patients.
Supplementary Material
Funding Statement
Funding: National Cancer Institute (R35 CA198183; K23 CA212271), Four Winds Foundation, Connie Brown CLL Fund, Kevin Sullivan Foundation, and the D. Warren Brown Foundation.
References
- 1.Tsimberidou AM, O'Brien S, Khouri I, et al. Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol. 2006;24(15):2343-2351. [DOI] [PubMed] [Google Scholar]
- 2.Parikh SA, Habermann TM, Chaffee KG, et al. Hodgkin transformation of chronic lymphocytic leukemia: incidence, outcomes, and comparison to de novo Hodgkin lymphoma. Am J Hematol. 2015;90(4):334-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsimberidou AM, O'Brien S, Kantarjian HM, et al. Hodgkin transformation of chronic lymphocytic leukemia: the M. D. Anderson Cancer Center experience. Cancer. 2006;107(6):1294-1302. [DOI] [PubMed] [Google Scholar]
- 4.Bockorny B, Codreanu I, Dasanu CA. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: a retrospective analysis of world literature. Br J Haematol. 2012;156(1):50-66. [DOI] [PubMed] [Google Scholar]
- 5.Ramchandren R, Advani RH, Ansell SM, et al. Brentuximab vedotin plus chemotherapy in North American patients with newly diagnosed stage III or IV Hodgkin lymphoma. Clin Cancer Res. 2019;25(6):1718-1726. [DOI] [PubMed] [Google Scholar]
- 6.Evens AM, Helenowski I, Ramsdale E, et al. A retrospective multicenter analysis of elderly Hodgkin lymphoma: outcomes and prognostic factors in the modern era. Blood. 2012;119(3):692-695. [DOI] [PubMed] [Google Scholar]
- 7.Evens AM, Hong F, Gordon LI, et al. The efficacy and tolerability of adriamycin, bleomycin, vinblastine, dacarbazine and Stanford V in older Hodgkin lymphoma patients: a comprehensive analysis from the North American intergroup trial E2496. Br J Haematol. 2013;161(1):76-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parikh SA, Rabe KG, Ding W, et al. Hodgkin Transformation of chronic lymphocytic leukemia (CLL): Mayo Clinic experience. Blood. 2013;122(21):1642-1642. [Google Scholar]
- 9.Fayad L, Robertson LE, O'Brien S, et al. Hodgkin's disease variant of Richter's syndrome: experience at a single institution. Leuk Lymphoma. 1996;23(3-4):333-337. [DOI] [PubMed] [Google Scholar]
- 10.Kazmierczak M, Kroll-Balcerzak R, Balcerzak A, et al. Hodgkin lymphoma transformation of chronic lymphocytic leukemia: cases report and discussion. Med Oncol. 2014;31(1):800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kharfan-Dabaja MA, Kumar A, Hamadani M, et al. Clinical practice recommendations for use of allogeneic hematopoietic cell transplantation in chronic lymphocytic leukemia on behalf of the Guidelines Committee of the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2016; 22(12): 2117-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin's disease. International Prognostic Factors Project on Advanced Hodgkin's Disease. N Engl J Med. 1998;339(21):1506-1514. [DOI] [PubMed] [Google Scholar]
- 13.Tsimberidou AM, O'Brien SM, Cortes JE, et al. Phase II study of fludarabine, cytarabine (Ara-C), cyclophosphamide, cisplatin and GM-CSF (FACPGM) in patients with Richter's syndrome or refractory lymphoproliferative disorders. Leuk Lymphoma. 2002;43(4):767-772. [DOI] [PubMed] [Google Scholar]
- 14.Robertson LE, Pugh W, O'Brien S, et al. Richter's syndrome: a report on 39 patients. J Clin Oncol. 1993;11(10):1985-1989. [DOI] [PubMed] [Google Scholar]
- 15.Dabaja BS, O'Brien SM, Kantarjian HM, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin (daunoXome), and dexamethasone (hyperCVXD) regimen in Richter's syndrome. Leuk Lymphoma. 2001;42(3):329-337. [DOI] [PubMed] [Google Scholar]
- 16.Tsimberidou AM, Kantarjian HM, Cortes J, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone plus rituximab and granulocyte- macrophage-colony stimulating factor (GM-CSF) alternating with methotrexate and cytarabine plus rituximab and GM-CSF in patients with Richter syndrome or fludarabine- refractory chronic lymphocytic leukemia. Cancer. 2003;97(7):1711-1720. [DOI] [PubMed] [Google Scholar]
- 17.Harousseau JL, Flandrin G, Tricot G, Brouet JC, Seligmann M, Bernard J. Malignant lymphoma supervening in chronic lymphocytic leukemia and related disorders. Richter's syndrome: a study of 25 cases. Cancer. 1981;48(6):1302-1308. [DOI] [PubMed] [Google Scholar]
- 18.Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142(2):202-215. [DOI] [PubMed] [Google Scholar]
- 19.Wierda WG, Byrd JC, Abramson JS, et al. NCCN Guidelines insights: chronic lymphocytic leukemia/small lymphocytic lymphoma, version 2.2019. J Natl Compr Canc Netw. 2019;17(1):12-20. [DOI] [PubMed] [Google Scholar]
- 20.Xiao W, Chen WW, Sorbara L, et al. Hodgkin lymphoma variant of Richter transformation: morphology, Epstein-Barr virus status, clonality, and survival analysiswith comparison to Hodgkin-like lesion. Hum Pathol. 2016;55:108-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011; 117(12): 3391-3401. [DOI] [PubMed] [Google Scholar]
- 22.Brenner H, Gondos A, Pulte D. Ongoing improvement in long-term survival of patients with Hodgkin disease at all ages and recent catch-up of older patients. Blood. 2008;111(6):2977-2983. [DOI] [PubMed] [Google Scholar]
- 23.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woyach JA, Ruppert AS, Guinn D, et al. BTK. J Clin Oncol. 2017;35(13):1437-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohno T, Smir BN, Weisenburger DD, Gascoyne RD, Hinrichs SD, Chan WC. Origin of the Hodgkin/Reed-Sternberg cells in chronic lymphocytic leukemia with "Hodgkin's transformation". Blood. 1998; 91(5):1757-1761. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.