

Summary
Intracellular bacterial pathogens inject effector proteins to hijack host cellular processes and promote their survival and proliferation. To systematically map effector-host protein-protein interactions (PPIs) during infection, we generated a library of 32 Salmonella enterica serovar Typhimurium (STm) strains expressing chromosomally encoded affinity-tagged effectors, and quantified PPIs in macrophages and epithelial cells. We identified 446 effector-host PPIs, 25 of which are previously described, and validated 13 by reciprocal co-immunoprecipitations. While effectors converged on the same host cellular processes, most had multiple targets, which often differed between cell types. We demonstrate that SseJ, SseL and SifA modulate cholesterol accumulation at the Salmonella Containing Vacuole (SCV) partially via the cholesterol transporter Niemann-Pick C1 protein, PipB recruits the organelle contact site protein PDZD8 to the SCV, and SteC promotes actin bundling by phosphorylating formin-like proteins. This study provides a method for probing host-pathogen PPIs during infection and a resource for interrogating STm effector mechanisms.
eTOC blurb
Pathogens hijack host cells by injecting effector proteins. In this work, Walch & Selkrig et al., used quantitative proteomics to systematically map the host targets of Salmonella effectors during infection. This effort yielded hundreds of protein-protein interactions, highlighted general effector properties, and uncovered missing links in Salmonella effector biology.
Graphical Abstract
Introduction
To usurp host defenses, intracellular pathogens secrete effector proteins to intercept and modify the host cell, aiming to evade detection by host innate immune receptors and establish a hospitable intracellular niche (Cunha and Zamboni, 2013). In turn, the host has mechanisms to overcome such molecular insults. This evolutionary arms race has driven pathogens to develop remarkably diverse arsenals of effector proteins, as in the case of the bacterium Legionella pneumophila which secretes >300 effectors (Schroeder, 2017). Host-pathogen protein-protein interactions (PPIs) are thereby manifold and key to infection outcome.
Discovering the host targets of an effector is the first step towards deciphering its role in infection. Global yeast two-hybrid studies (Y2H) (Shapira et al., 2009; Uetz et al., 2006) and affinity-tag purification/mass spectrometry (AP/MS) screens (D’Costa et al., 2019; Gordon et al., 2020; Jäger et al., 2011a; Penn et al., 2018; Sontag et al., 2016) have been used to systematically map PPIs at bacterial- and viral-host interfaces. Initial global PPI efforts often resulted in many false-positives and generated skepticism in the community (Rajagopala et al., 2009; Stynen et al., 2012). Yet, as methods and data analysis advanced, large-scale studies helped to resolve the picture of relevant host-pathogen PPIs. This is the case for HIV, where systematic AP-MS revealed the strong and relevant PPIs (Jäger et al., 2011a) out of more than a thousand PPIs reported from just a handful of viral proteins by targeted approaches (Jäger et al., 2011b), thereby fueling new mechanistic insights into HIV biology (Chou et al., 2013; Jäger et al., 2011c). Despite their power, such studies remain limited in their capacity to recapitulate the infection environment. Until now, PPIs have typically been probed within mammalian cells by overexpressing single effectors, in the absence of the pathogen, or by flowing lysates through columns with immobilized effectors. In addition to non-physiological effector levels, such experiments poorly reflect the infection state in vivo due to the absence of infection-relevant rewiring of the host proteome and the absence of additional effectors, which may promote or hinder interactions. Therefore, methods that probe host-pathogen PPIs during infection are still needed.
To identify effector-host PPIs in their native infection context, we developed a proteomics-based methodology to extract Salmonella enterica serovar Typhimurium (STm)-delivered effectors directly from infected cells and quantify their interacting protein partners. We constructed a library of 32 chromosomally-tagged effectors translocated into the host cytoplasm by both Type-3 Secretion Systems, T3SS1 and T3SS2 (Jennings et al., 2017; Ramos-Morales, 2012), and used it to profile effector-host PPIs in both HeLa and RAW264.7 cells. Thereby, we were able to reconstruct an STm-host interactome, spanning 15 effectors and 446 PPIs. Network analysis revealed that diverse effectors target host proteins with related functions, with several effectors converging on the same process, and in some cases even interacting. Despite this, most effectors had multiple targets, often in unrelated host cellular processes. Several PPIs were detected in both cell lines tested, whereas most PPIs were specific to the cellular context. Capitalizing on this resource, we uncovered effector interplay between SseJ, SseL and SifA in cholesterol accumulation at the STm-containing vacuole (SCV), demonstrated that PipB recruits the endoplasmic reticulum (ER)-tethered protein PDZD8 to the SCV, and discovered that the effector kinase SteC promotes actin bundling via interactions with formin-like proteins (FMNL). Overall, we provide a method for probing host-pathogen PPIs in a physiological context, and a rich resource for the discovery of STm effector mechanisms.
Results
Mapping the host targets of Salmonella effectors during infection
We built a library of 32 tagged-effector STm 14028s strains, i.e. nearly all known effectors translocated by T3SS1 and T3SS2 (Table S1). To ensure physiological effector levels, we introduced a C-terminal Strep(2x)-TEV-FLAG(3x) (STF) tag onto the endogenous loci of effector encoding genes (except SifA, see Experimental Procedures). Strains expressing chromosomally tagged effectors were then tested for effector translocation into the cytoplasm of infected epithelial or macrophage cells, two relevant cell types for STm infection (LaRock et al., 2015). As expected, effectors were most abundant at later infection stages (Figure S1A), when intracellular STm loads were high. We detected 20 effectors (2 using T3SS1, 12 using T3SS2 and 6 both) being injected into the cytoplasm of at least one host cell line. These 20 effectors were used in large-scale infections for AP-Quantitative MS analysis (AP-QMS; Figure 1A, Table S1).
Figure 1. AP-QMS pipeline for mapping effector-host PPIs during Salmonella infection.
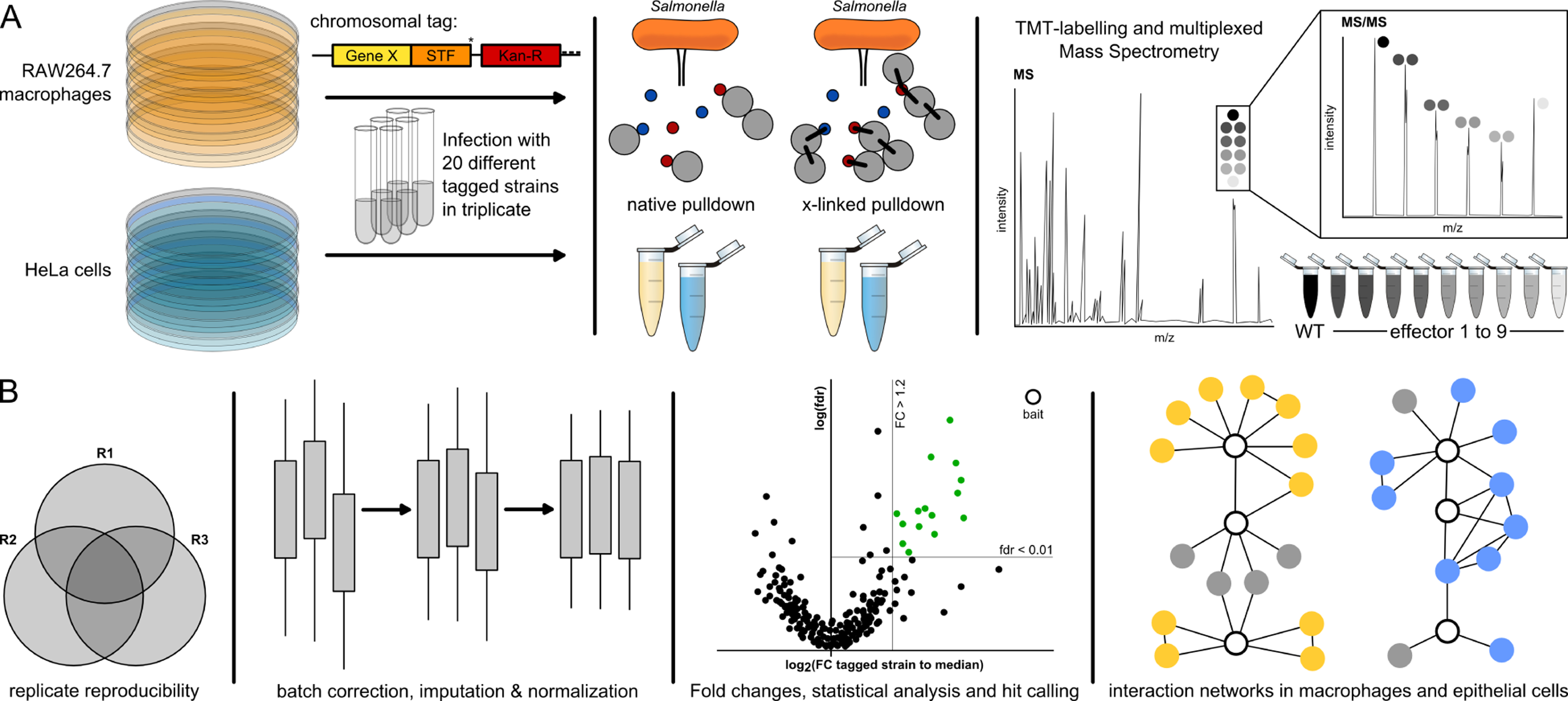
(A) STm strains carrying C-terminally tagged effectors with STF (FLAG(2x)-TEV-STREP(3x)) were used to infect HeLa and RAW264.7 cells at a MOI ~100. Samples were either treated with DSP crosslinker or directly lysed for native anti-FLAG pulldowns. Eluates from the pulldowns were combined in a TMT-10plex labelling run and measured by LC-MS/MS. Elutions from nine different STF-tagged effectors and one untagged wildtype background control were combined.
(B) Only proteins quantified with at least two unique peptides and identified in at least two biological replicates were used for analysis. A protein was a ‘hit’ when the false discovery rate (fdr) was < 1% and exhibited a fold increase of at least 20%. We further refined this list by loosening the fdr requirement to < 5% if a PPI passed the fold change (FC) requirement in both native and crosslinked conditions. Subsequently, only the strongest 20 PPIs per effector with respect to FC or fdr, as well as PPIs detected in both the native and crosslinked pulldowns, were kept for the final hit list. All analyzed data and hits are listed in Table S2. Volcano plots of all pulldowns can be found in Mendeley Data. PPI networks were built from hits and known host functional interactions.
We tested PPIs in two commonly used human epithelial and murine macrophages cell lines for STm infections: HeLa and RAW264.7, to enable comparisons of our dataset with previous global STm-host PPI studies (D’Costa et al., 2019; Sontag et al., 2016) and targeted studies (summarized in (Jennings et al., 2017; LaRock et al., 2015)). We performed FLAG-immunoprecipitation at 20 hours post infection (hpi) under both native (stable interactions) and cross-linking conditions (transient interactions) using the cell permeable cross-linker DSP (Figure 1A). To ensure reproducible quantification of bait and prey proteins relative to background, pulldown eluates were combined in groups of 10 (consisting of 9 distinct effector pulldown eluates and one untagged control; Figure S1B) and analyzed in a single 10-plex Tandem Mass Tag (TMT10 (Werner et al., 2014)) run in biological triplicates (Figure 1A). Only proteins identified with at least two unique peptides and found in at least two biological replicates were used for further analysis (Experimental Procedures; Figure 1B, S1C). Protein enrichments were calculated by comparing the protein abundance (signal sum) in each TMT channel to the median abundance (signal sum) within each TMT10 run for each protein (Figure 1B), which was more robust than comparing to the untagged strain (Figure S1D). Altogether, we detected the bait protein for 11 effectors in both RAW264.7 and HeLa cells, with significant interactions for 12 effectors in RAW264.7 and 9 effectors in HeLa cells (Figure S2A). Volcano plots and input data for all effectors can be found in Mendeley Data. Due to the 20 hpi time point, T3SS2 effectors were more readily detected. The resulting hits for each bait (fold change (FC) ≥ 1.2; False Discovery Rate (fdr) ≤ 0.01, after adjusting stringency for hits in both native and cross-linked conditions and capping the number of hits per effector; see Experimental Procedures) are reported in Table S2 and were used to build PPI networks (Figure 1B).
Across the 2 cell lines and 15 effectors, we detected 462 non-redundant PPIs. Of these, 446 were effector-target PPIs, 15 were the baits themselves and 1 was a clear contaminant (IgG-heavy chain). Of the 446 effector-target PPIs; 421 were effector-host (Table S3) and 25 were effector-bacterial protein interactions. On average, each effector had 19.7 PPIs in RAW264.7 macrophages and 26.4 PPIs in HeLa epithelial cells.
Salmonella effectors target diverse host processes in macrophages and epithelial cells
Using the significant interactions we detected by AP-QMS, and known human, murine or bacterial protein functional interactions (Table S3, (Szklarczyk et al., 2019)), we built two separate PPI networks in RAW264.7 and HeLa cells (Figures 2A and 3A). The networks contained a number of previously characterized PPIs, such as SseJ interacting with the host Rho GTPase proteins RhoA and RhoB (Ohlson et al., 2008), but most interactions reported were previously undescribed. In total, we detected 25 previously reported interactions (Table S3): e.g. PipB2-KLC1/2, PipB2-KIF5B, SseL-OSBP and SseI-Acadm (Auweter et al., 2012; Henry et al., 2006; Sontag et al., 2016) in the two cell lines. We failed to capture some well-described PPIs, such as SifA-SKIP (Diacovich et al., 2009; Jackson et al., 2008; Zhao et al., 2015) and AvrA-MKK7 (Du and Galán, 2009; Jones et al., 2008). False negatives are common in AP-MS protocols and can have multiple causes (Verschueren et al., 2015). In addition, several of the interactions may be indirect and mediated via another host protein (piggybacking is common in AP approaches; (Nesvizhskii, 2012; Teng et al., 2015), which would explain effectors binding to multiple host proteins of the same process.
Figure 2. STm effector-host target physical interactions in RAW264.7 macrophages.
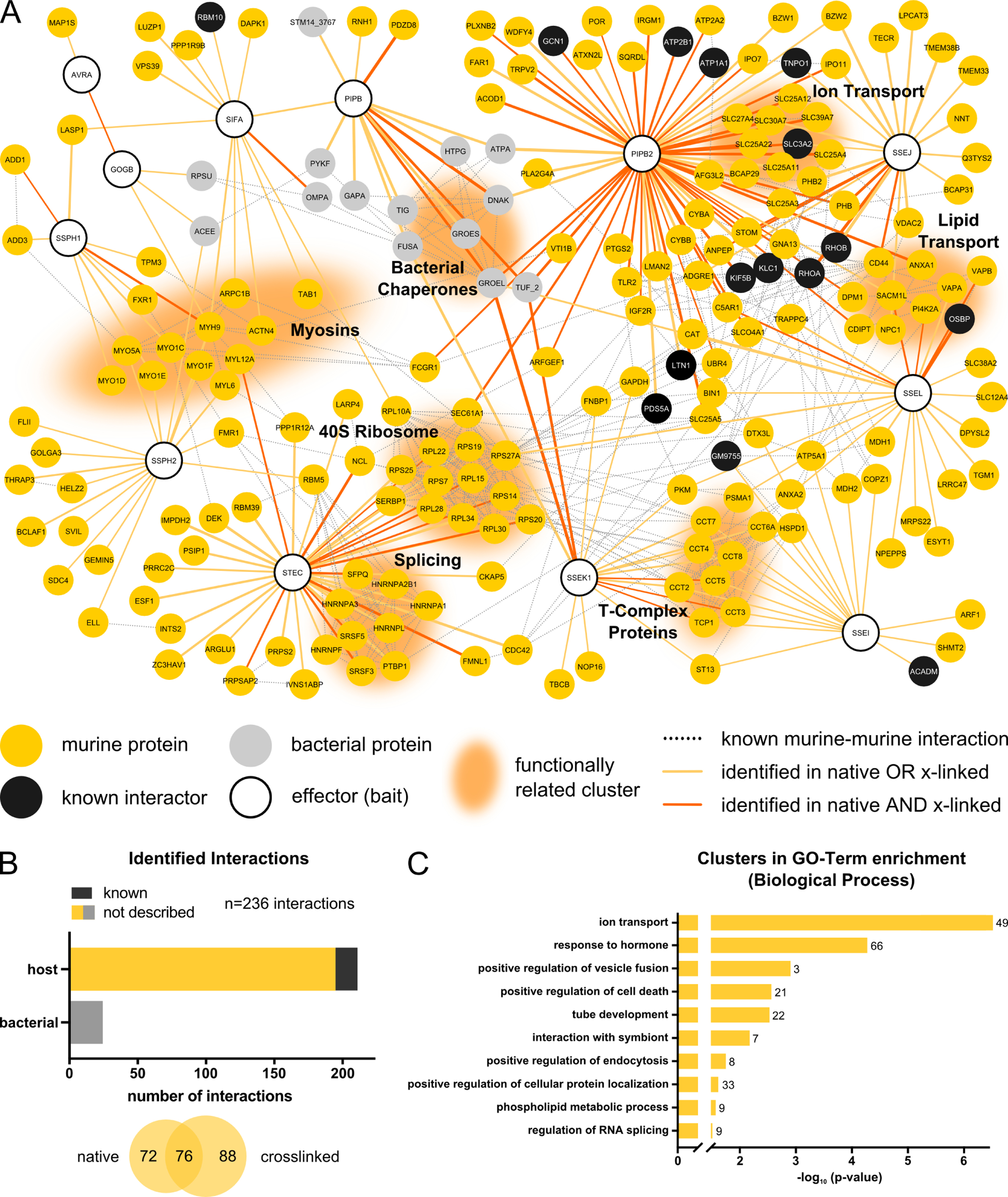
(A) Network of PPIs identified between 12 STm effectors and their target proteins in RAW264.7 cells at 20 hpi. Only effectors identified as bait in AP-QMS and target proteins passing the criteria described in Fig. 1B are shown here. Host proteins from RAW264.7 cells are shown in gold or black (previously identified interactions; Table S3). STm are in grey. The edge color denotes the conditions interaction captured, and the thickness is proportional to the fold change (Log2). Functionally related clusters are grouped and annotated accordingly. Network was generated using Cytoscape version 3.7.2 (Shannon et al., 2003). Murine-murine, as well as bacterial-bacterial functional interactions were extracted from the built-in STRING DB version 11 (Szklarczyk et al., 2019) protein query for Mus musculus and Salmonella with a confidence cutoff of 0.7.
(B) Overview of identified PPIs in RAW264.7 cells at 20 hpi. Hits are grouped according to whether they are of murine or STm origin (upper histogram), or according to whether they were detected in native or cross-linked pulldown samples (lower Venn diagram).
(C) GO-term analysis for enriched processes among all identified PPI partners. GO-term clusters are ordered according to the enrichment significance (negative logarithmic, corrected for multiple testing) (Benjamini and Hochberg, 1995; Bindea et al., 2009) and top 10 GO-clusters are displayed. n signifies the number of proteins present in cluster. Enrichments were normalized to the combined background proteome from AP-QMS experiments. Full enrichments lists can be found in Table S4.
Figure 3. STm effector-host target physical interactions in HeLa epithelial cells.
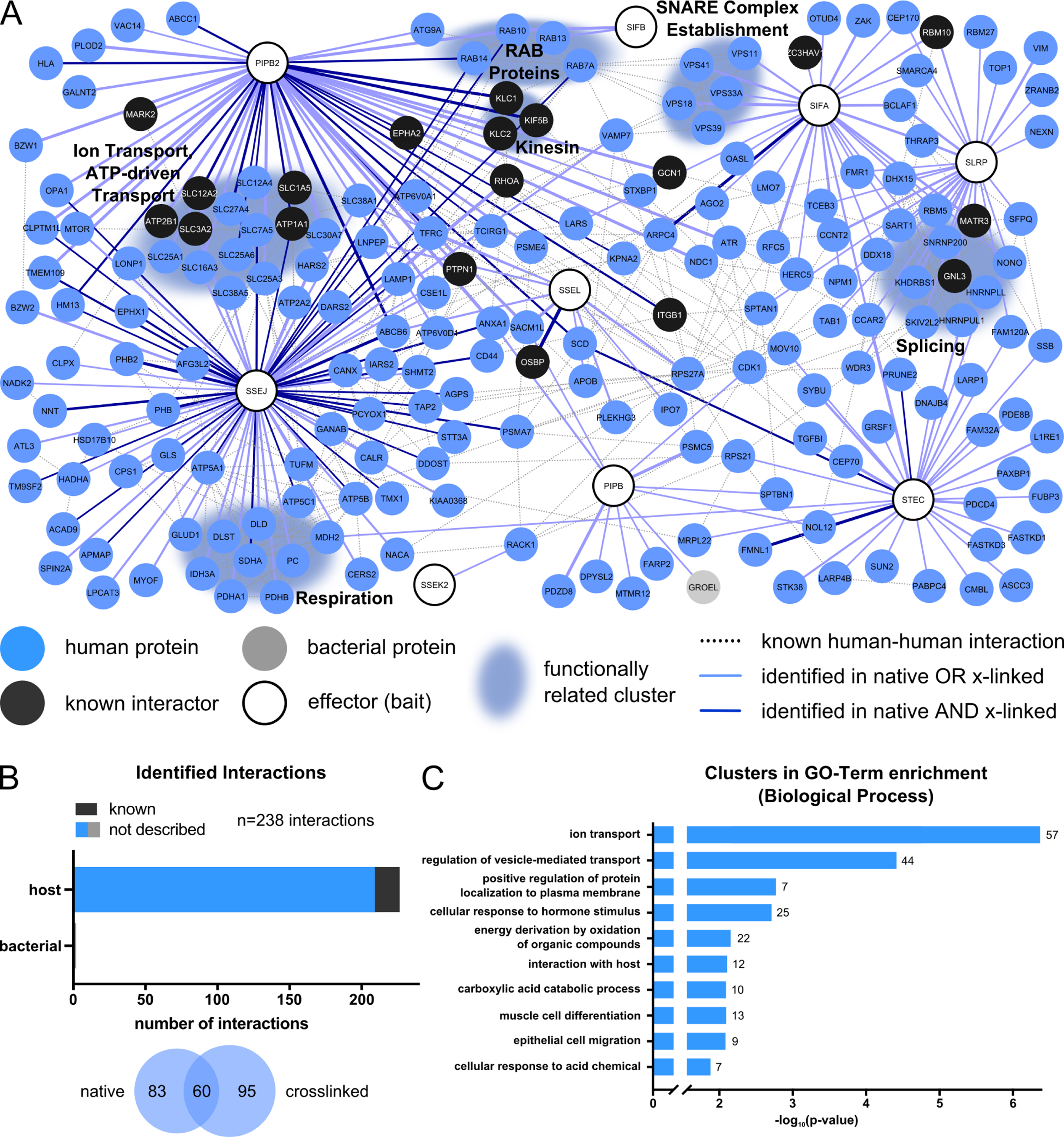
(A) Network of PPIs identified between 9 STm effectors and their target proteins in HeLa cells at 20 hpi. Network is generated and depicted as described in Fig. 2A except HeLa interactors are in blue and human data from STRING was used as input to generate edges.
(B) Overview of identified PPIs in HeLa cells at 20 hpi - as in Fig. 2B.
(C) GO-term analysis for biological processes across the full network as described in Fig. 2C.
Rather than effectors interacting exclusively with a single host protein, we detected several effectors co-purifying with many host targets, such as PipB2, which had 59 in RAW264.7 cells and 48 PPIs in HeLa cells (Figure S2B). This implies that effectors with multiple targets may be common for bacterial pathogens (Hamon et al., 2012; Takahashi-Kanemitsu et al., 2020). These targets frequently belonged to distinct host processes, reinforcing the concept of effector multifunctionality. For example, SteC, a well-known STm effector with kinase activity, was implicated in actin remodeling around the SCV (Poh et al., 2008). Here, SteC in addition to the link to actin (for which we later identify the missing direct interaction partner), displayed several PPIs with host proteins related to mRNA splicing in both cell types, suggesting a potential, additional regulatory role in host-transcription.
To assess the biological processes that effectors target, we overlaid human or murine functional interactions in the networks (Figures 2A and 3A) and performed GO-term enrichment (Figures 2C and 3C, Table S4). Ion transport (PipB2 and SseJ) and vesicle-mediated transport or fusion were among the most enriched targets in both cell lines (Figures 2C, 3C and S2C). Other processes were cell line-specific. Cytoskeleton-dependent intracellular transport (GO-term grouping “ion transport”) was specifically enriched in macrophages, mostly due to SspH1 and SspH2 interacting with myosins and lipid associated processes (Figure 2A and 2C, Table S4). Both processes play important roles in SCV maintenance (Arena et al., 2011; Wasylnka et al., 2008).
A notable feature of both PPI networks was that several effectors converged on the same host protein complexes/processes with myosins, ion transport, lipid transport, 40S ribosome and the T-complex being the most prominent hubs (Figures 2A and 3A). In some cases, multiple effectors targeted the same host protein, such as myosin Myh9, which co-purified with SspH1, SspH2, GogB and SifA in RAW264.7 cells (Figure 2A). This highlights the potential for effector co-operation on the same host cell process (Figure S2D), which may occur simultaneously or in parallel (Ghosh and O’Connor, 2017). Interestingly, we also observed a number of effector-effector interactions (GogB-AvrA, PipB-SifA). Although some may be mediated through common host targets, this reinforces the notion that effectors converge on the same host processes and work cooperatively to hijack them. For example, both AvrA and GogB are known to impose an anti-inflammatory effect on host cells during STm infection. AvrA dampens JNK signaling via MKK7 (Du and Galán, 2009), thereby reducing apoptosis (Jones et al., 2008), whereas GogB acts on NFkB by inhibiting degradation of IFkB (Pilar et al., 2012). Even though no common target for these two effectors is known, the finding that they co-purify suggests they co-regulate inflammation.
An advantage of systematic studies is that common pull-down contaminants are removed during normalization. This allows identification of specific interactions that would normally be disregarded. For example, we detected 25 effector-bacterial PPIs in macrophage cells, e.g. PipB-DnaK, PipB-GroEL, PipB-STM14_3767 (Figure 2B). To exclude that these PPIs are due to partial bacterial lysis during infection or harvesting, we validated the presence of GroEL in the host cytoplasm during infection using a GroEL antibody. Consistent with previous reports showing GroEL is secreted by other bacteria (González-López et al., 2013; McCaig et al., 2013; Pierson et al., 2011; Yang et al., 2011), we detected GroEL in host lysates (Figure S2E). This cannot be explained by bacterial lysis, as another abundant bacterial protein, RecA, was barely detectable in the cell lysate. This suggests that GroEL is secreted into host cells during infection and may play a role in effector maturation in the host cytoplasm. We obtained similar results for STM14_3767, a putative acetyl CoA hydrolase (Figure S2E).
In summary, we recovered both previously described and undescribed PPIs. Most STm effectors have multiple host targets, but in general, effectors converge to target the same host processes, implying that they functionally cooperate during infection.
Strong interactions are validated by reciprocal pull downs on host target proteins
The majority of PPIs we identified were cell line-specific (418 of 446 PPIs, Figure 4A), which prompted us to investigate the underlying reason (Figure S3, Mendeley Data). Approximately one third of the PPIs that were detected specifically in one cell type were due to the lack of detectable expression of that protein in the other cell line (Figure S3G-L). However, most cell-type specific PPIs had similar abundance in both cell lines (Figure S3G & J). The remaining differences can be due to false negatives and/or reflect differences in the STm infection cycle in epithelial cells and macrophages – e.g. STm can escape the SCV and proliferate in the cytoplasm of epithelial cells, but not of macrophages (Castanheira and García-Del Portillo, 2017; Knodler et al., 2010). Hence, effector-host PPIs are largely cell-type specific, and only partially due to differences in protein expression.
Figure 4. Comparison of STm interactomes in RAW264.7 and HeLa cells, and reciprocal PPI validation.
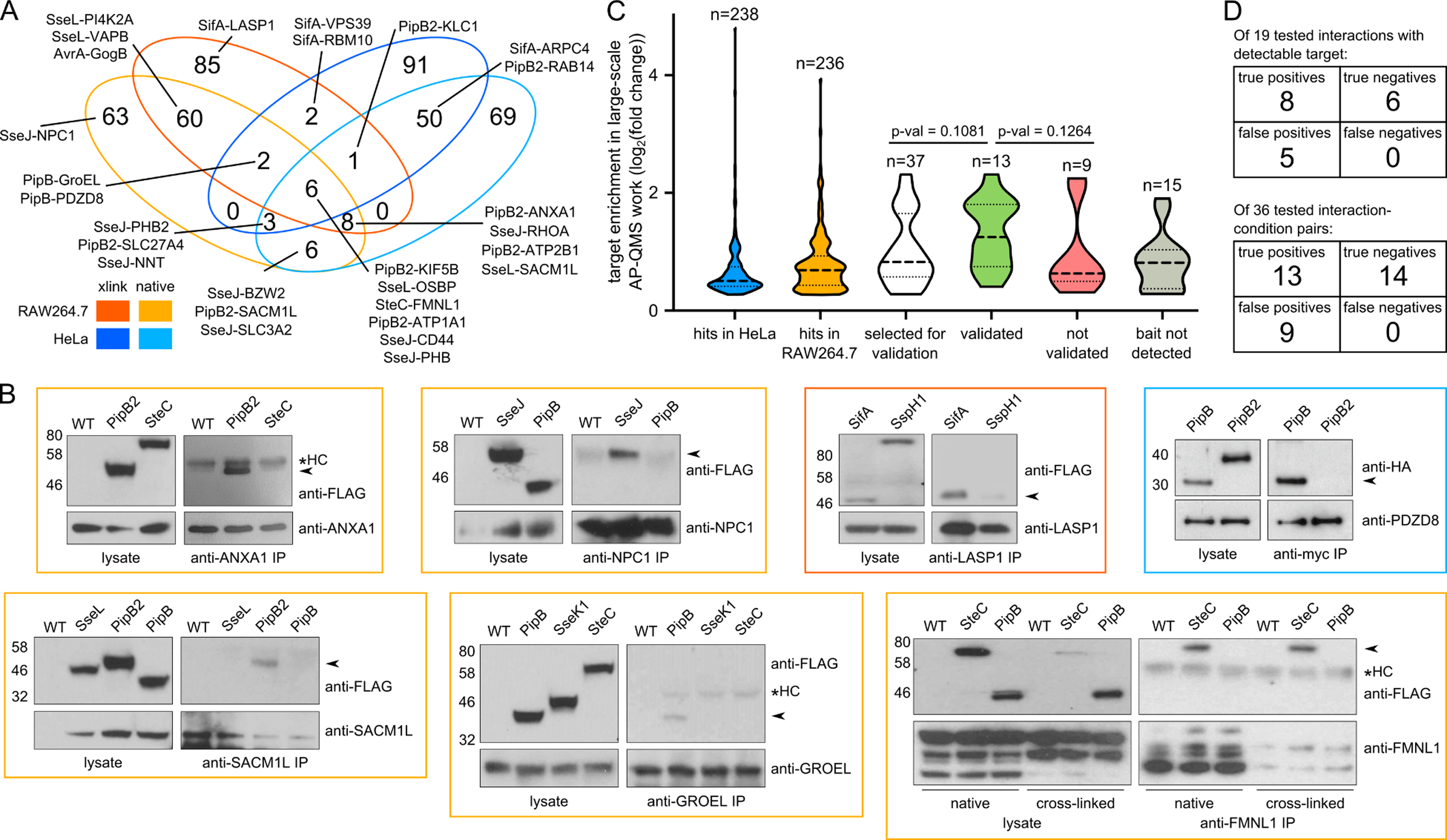
(A) Venn diagram comparison of PPIs across the two cell lines and conditions.
(B) Reciprocal pulldowns using antibodies against host targets were used to validate PPIs detected in the AP-QMS screen at 20 hpi. Effectors with similar expression levels were used in parallel pulldowns as negative controls. The PipB-PDZD8 reciprocal pulldown was performed by infecting PDZD8-myc transfected HeLa cells with STmSL1344 ΔpipB complemented with PipB-2HA in trans; ΔpipB2 expressing PipB2–2HA in trans was used as a negative control. Two independent replicate experiments were performed, except for LASP1 and GroEL pulldowns which were performed once. One exemplary blot per interaction is shown, all blots and raw images are located in the accompanying Mendeley Data. Colored box around the Western Blot image corresponds to the cell background and condition tested (see panel A legend). Validated interactions are indicated by arrows and asterisk denotes antibody heavy chain (HC).
(C) Violin plots of log2 fold enrichments in AP-QMS for all effector-target proteins selected to be tested by reciprocal pulldowns (white), those that could (green) or could not (red) be validated, and those where the bait was not detected in the reciprocal pulldowns (grey). Dotted lines indicate median (bold) and interquartile range (light). Two-tailed Mann-Whitney test was used for significance testing. Enrichments of interactions identified in HeLa cells (blue) and RAW264.7 (orange) are shown for comparison.
(D) Summary of the validation. In the upper table, interactions are considered irrespective of condition or cell line. In this case, 13 PPIs detected in the AP-QMS work, as well as 6 non-cognate controls for a total of 19 interactions was assessed. In the lower table, each cell line and condition is considered as a separate experiment wherein, 22 PPIs from the AP/MS work plus 14 non-cognate controls (i.e. 36 interactions in total) were assessed. All assessed individual reciprocal interactions are listed in Table S5.
Several PPIs were specifically identified in the presence of the crosslinker (Figures 2A-B, 3A-B and 4A). For example, SifA interacted with VPS39 and RBM10 only after crosslinking in both cell types, suggesting that these interactions may be transient. The only partial, though highly significant overlap (p-value < 0.0001, Fisher’s exact test) between native and crosslinked data could have additional reasons: a) loss in the efficiency of bait pulldown after crosslinking; b) increased background/poorer signal-to-noise in crosslinking experiment (Figure S1D, S3C and F); c) differences in sample preparation and increased incubation times in the crosslinking experiment, impacting the recovery of PPIs; and d) false negatives due to stringent thresholds. A number of interactions were conserved across backgrounds and pulldown conditions, indicating strong interactions. Among the conserved interactions, several were previously unrecognized, e.g. SteC-FMNL1, PipB2-ATP1A1, PipB2-ANXA1, SseJ-CD44, SseL-SACM1L or PipB-GroEL.
To assess the uniquely identified interactions in more detail, we selected a subset of 12 host targets, which amounted to 22 distinct effector-host protein interactions – 37 PPIs when counted as separate events if the same interaction occurred in different conditions (native vs. cross-linked, cell line) – and sought to validate their interactions with the respective Salmonella effectors reciprocally (Table S5). The host targets were selected to span a range of interaction enrichment scores. To probe reciprocal interactions, we used immunoprecipitation and antibodies directed against host targets we identified in the original cell line screen, and included non-cognate STm effectors of a similar translocation level to evaluate specificity (Figure 4B). In total, we could immunoprecipitate 7 out of 12 host bait proteins using host-directed antibodies. Among them, we recapitulated the orthogonal pulldown of the STm effector for 8 out of the 13 possible PPI pairs (61.5%, Table S5) in at least one condition, and saw no interaction with any of the 6 tested non-cognate controls for a total of 19 assessed interactions (Figure 4D upper table). 13 of all possible 22 conditions, i.e. 59.1% could be validated via reciprocal immunoprecipitation, with all 14 non-cognate controls displaying no interaction (total number of probed interactions: 36; Table S5; Figure 4D lower table). Note that translocated STm effectors have much lower abundance than host proteins (Selkrig et al., 2020) and these pulldowns were performed in a cell population containing 20–60% infected cells. Consequently, most of the host target protein pool is not bound to the STm effector. Consistent with such an increased difficulty in capturing effector-host PPIs by pulling down host proteins, we observed that stronger interactions in the screen were more readily verifiable (Figure 4C).
We then probed a small subset of STm-host PPIs in primary murine bone marrow derived macrophages (pBMDMs) to explore whether our AP-QMS approach captures interactions that are relevant in primary cells. As STm exhibits low intracellular loads in pBMDMs, we first tested whether endogenously expressed STm effectors could be detected in this setting. Three out of four tested effectors (SseJ-STF, SteC-STF and PipB2-STF, but not SseL-STF) were detected by immunoblot (Figure S4A). To account for the limited source of pBMDMs, we scaled down the input of primary cells by 10-fold and performed AP-QMS for SteC and PipB2. Although we failed to detect the baits, hit proteins assigned to both effectors in RAW264.7 cells (Figure 2A, 3A, Table S2) were also significantly enriched in the pBMDMs pull downs (Figure S4B, C and Mendeley Data). This demonstrates that AP-QMS has the sensitivity to capture interactions in primary cells, despite lower cellular input and STm proliferation, and interactions are similar to those observed in cell lines.
In summary, we recapitulated most of the effector-host PPIs tested using less sensitive reciprocal immunoprecipitations. Furthermore, interactions detected in immortalized cell lines were also enriched in primary cells. Together this suggests that many of the interactions we report here likely also occur in vivo.
SseJ, SseL and SifA regulate cholesterol accumulation on the SCV membrane
We identified several GO-terms related to lipid trafficking in both cell types, and host proteins required for cholesterol trafficking, such as OSBP, NPC1, VAPA/B, SACM1L, to be associated with multiple effectors. These interactions were predominantly mediated by SseJ, SseL and PipB2 (Figure 2A, 3A and S2C). As SseJ esterifies cholesterol (Nawabi et al., 2008), we further probed its connection with cholesterol transport by performing AP-QMS with and without crosslinking in HeLa cells, and analyzed the samples with the corresponding untagged controls in the same TMT run (Figures 5A, S5A, Mendeley Data). Combining all replicates into a single run reduces sample complexity and increases sensitivity for detecting low abundant PPIs. This enabled us to detect a PPI between SseJ and the effector SseL (Figure 5A), which is in line with evidence that these two effectors cooperate to promote SCV stability via direct interactions with OSBP (Auweter et al., 2012; Kolodziejek et al., 2019), a lipid transfer protein that controls cholesterol/PI4P exchange between the ER and Golgi (Mesmin et al., 2017). OSBP co-purified with SseL in all of our AP-QMS setups (Figures 2A, 3A), as well as with SseJ in the targeted TMT run (Figure 5A). Furthermore, other host target proteins involved in lipid transport also co-purified with SseJ (Figures 5A, S5B).
Figure 5. NPC1 is recruited to the SCV by SifA, where it physically interacts with SseJ and is functionally antagonized by SseL.
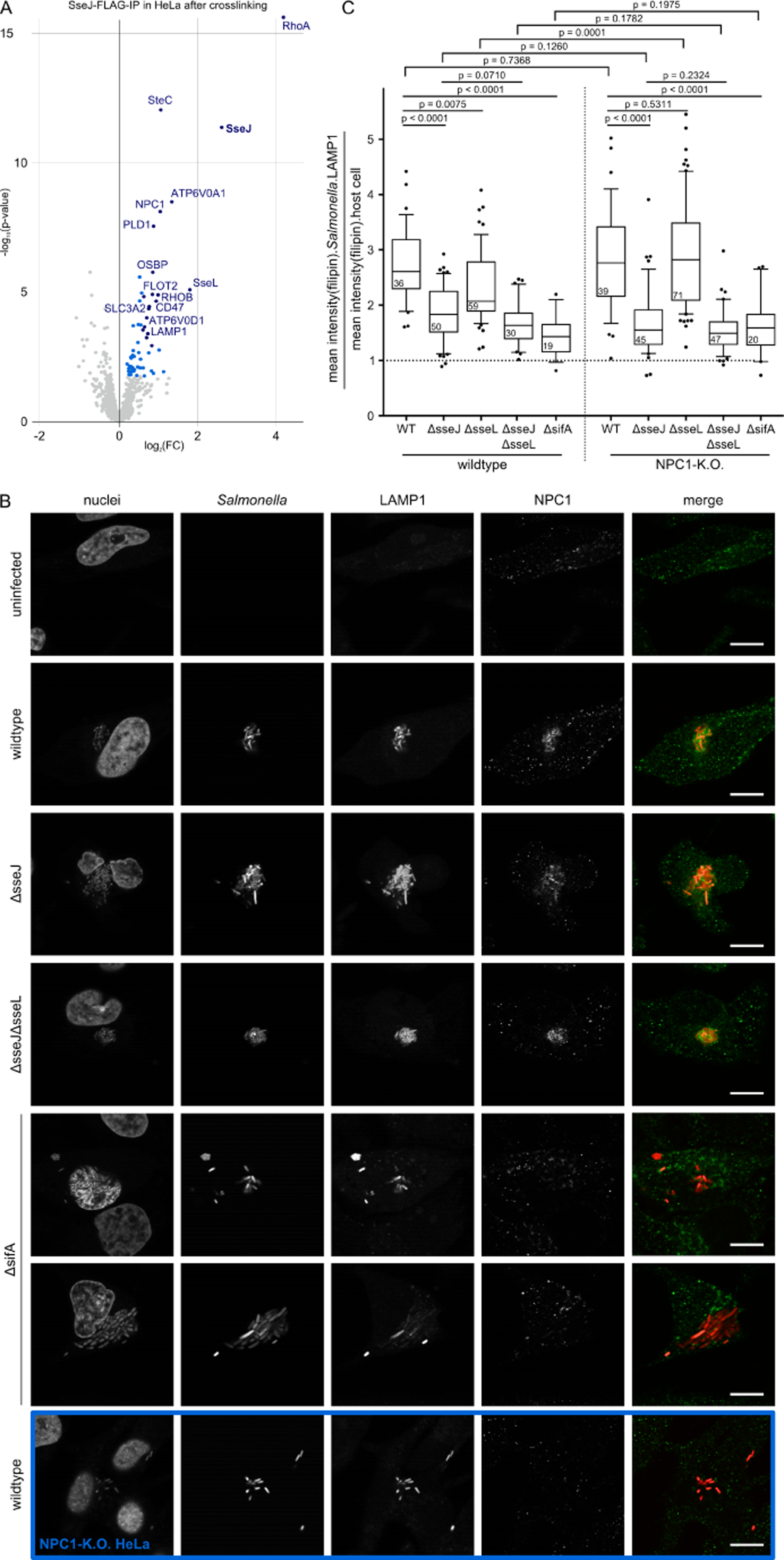
(A) Enrichments after crosslinked pulldown of STF-tagged SseJ in HeLa cells at 20 hpi compared to untagged STm control (native pulldown in Figure S5A). Three replicates for SseJ-STF and wildtype were measured in a single TMT run. Dark blue: FC > 1.5, p-value < 0.001; light blue: FC > 1.2, p-value < 0.01. All hits can be found in the accompanying Mendeley Data.
(B) Confocal microscopy images (60x) of HeLa cells infected with wildtype, ΔsseJ, ΔsseJΔsseL and ΔsifA STm expressing mCherry (pFCcGi) at 12 hpi (MOI = 100). Cells were stained with Hoechst 33342, anti-LAMP1 and anti-NPC1. Merge shows STm in red, NPC1 in green. For ΔsifA two examples are shown – a host cell in which STm grows in the SCV (upper) and one in which the SCV has ruptured and STm is in the cytosol (lower). Identically treated NPC1-K.O. cells were used to assess the anti-NPC1 specificity (blue border). Quantification of NPC1 localization to the SCV is shown in Figure S5C. Scale bar: 10 μm
(C) Accumulation of cholesterol (stained with filipin) at the SCV. A total of 416 manually inspected fields of view (FOVs) across three independent experiments with four technical replicates in each run were analyzed (see Fig. S5D for representative images). The mean filipin intensity in regions of co-localization of intracellular STm with LAMP1 staining (to exclude cytosolic bacteria) was divided by the mean filipin intensity measured within the cell mask. Analysis was performed per FOV (n shown in boxplots). FOVs contained on average 20 infected cells. Boxplots (median and IQR, error bars are according to Tukey) with whiskers spanning Q10 to Q90. Unpaired, two-sided T-test with Welch correction was used to calculate p-values, without correction for multiple testing.
In addition to OSBP, SseJ interacted with the Niemann-Pick disease type C1 protein (NPC1) (Figure 5A, S5A), which is critical for cholesterol egress from the endosomal/lysosomal compartments to the ER (Pfeffer, 2019). To assess whether SseJ or SseL regulate cellular trafficking of NPC1, we examined its localization in HeLa cells infected with wildtype STm and isogenic ΔsseJ, ΔsseJΔsseL mutants by confocal fluorescence microscopy. As we observed previously for ectopically expressed NPC1 (Drecktrah et al., 2008), endogenous NPC1 was also recruited to the SCV at 12 hpi (Figure 5B). This NPC1 recruitment to the SCV was independent of SseJ and SseL (Figure 5B). Since SifA is known to mobilize phagolysosomal membranes to the SCV via PLEKHM1 (McEwan et al., 2015), we tested a ΔsifA mutant, which failed to accumulate NPC1 on the SCV in infected HeLa cells (Figure 5B and S5C). Thus, SifA indirectly recruits NPC1 to the SCV, where it can then physically interact with SseJ.
To dissect further the links of NPC1 and STm in cholesterol trafficking to or from the SCV, we infected HeLa cells with STm and examined cholesterol distribution by filipin staining at 12 hpi (Maxfield and Wüstner, 2012; Wilhelm et al., 2019). Co-localization between cholesterol and the SCV was calculated as the ratio between the mean filipin intensity at the site of the SCV and the overall filipin signal per cell (Figure 5C). Random cholesterol distribution throughout the cell should result in a ratio of 1, filipin enrichment on the SCV in values >1 and exclusion of filipin from the SCV in values <1. Cholesterol was recruited to the SCV upon infection with wildtype STm, but was significantly reduced upon infection with a ΔsseJ mutant, and to a lesser extent upon infection with ΔsseL bacteria (Figure 5C and S5D). The ΔsseJΔsseL double mutant behaved similarly to the ΔsseJ mutant, suggesting that SseJ is the dominant effector influencing cholesterol recruitment and/or retention on the SCV. SCV cholesterol content was lowest in cells infected with STm ΔsifA, suggesting that SifA is vital in this process, possibly through its ability to recruit cholesterol-rich endolysosomal membrane material to the SCV (McEwan et al., 2015).
Cholesterol accumulation at the SCV remained largely unaffected in NPC1 knockout (KO) cells infected with wildtype STm, despite these KO cells having an excessive intracellular accumulation of cholesterol in late endosomes/lysosomes (Figure 5C, S5D) (Tharkeshwar et al., 2017). Interestingly, the ΔsseL mutant no longer conferred a reduction in cholesterol accumulation at the SCV in NPC1 KO cells (Figure 5C). This suggests that SseL promotes SCV cholesterol accumulation via NPC1, with SseL counteracting NPC1’s ability to remove cholesterol from the SCV. Taken together, these findings revealed a complex interplay between the effectors SseJ, SseL and SifA in cholesterol trafficking to the SCV via multiple host target proteins including NPC1 and OSBP.
PipB interacts with PDZD8 and recruits it to the SCV
We detected an interaction between PipB and the PDZ-domain containing protein 8 (PDZD8) in both cell types (Figure 2A, 3A and 4A). We verified the specificity of this interaction in HeLa cells ectopically expressing EGFP-tagged PipB or its effector paralogue EGFP-PipB2. PDZD8 co-immunoprecipitated with EGFP-PipB, but not with EGFP-PipB2 (Figure S6A).
We established that the PipB-PDZD8 interaction is direct by Y2H (Figure 6A-B). To map the PDZD8-PipB interaction more precisely, we tested a series of PipB and PDZD8 truncations by Y2H. Deleting the C-terminal 20 amino acids of PipB (Δ272–291) disrupted PipB-PDZD8 binding, but alone were not sufficient for the interaction with PDZD8, as deletion of the N-terminal 188 amino acids (Δ1–188) also disrupted PDZD8 binding (Figure 6A). A critical segment within the C-terminal 225 amino acids (Δ930–1154) of PDZ8, containing a predicted coiled-coil (CC) domain, was required for the interaction with PipB in Y2H (Figure 6B) and also in HeLa cells expressing an EGFP-PipB(Δ270–291) truncation (Figure 6C). Biochemical subcellular fractionation of HeLa cells revealed that both EGFP-PipB or EGFP-PipB(Δ270–291) co-fractionated with host cell membranes, as did PDZD8 (Figure S6B), and displayed a comparable localization pattern by fluorescence microscopy (Figure 6D).
Figure 6. PipB binds to and recruits PDZD8 to the SCV during infection.
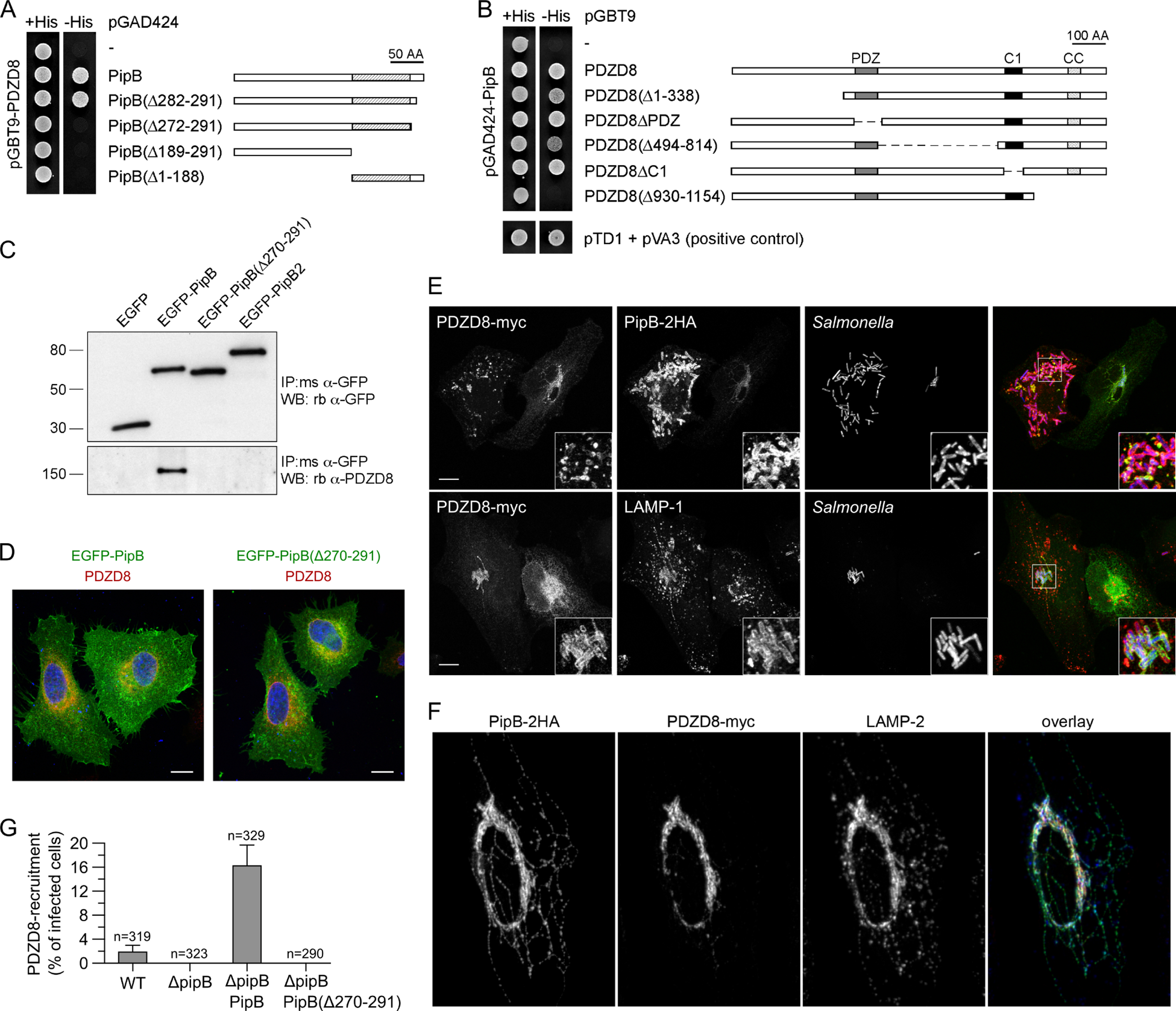
(A) Y2H with truncated versions of PipB. A direct interaction to PDZD8, as indicated by growth in -His conditions, is abolished by deletion of the 20 amino acid C-terminus of PipB.
(B) Y2H with truncated versions of PDZD8. Deletion of the C-terminal 225 amino acids of PDZD8 impairs its interaction with PipB.
(C) Western Blot analysis of immunoprecipitates from HeLa cells transfected with EGFP, EGFP-PipB, EGFP-PipB (Δ270–291) or EGFP-PipB2 fusions. Anti-GFP immunoprecipitates were analyzed by immunoblotting for endogenous PDZD8 using anti-PDZD8 peptide antibodies and anti-GFP antibodies. The PipB-PDZD8 interaction requires the last 22 amino acids of PipB. PipB2 was used as negative control.
(D) HeLa cells were transfected with EGFP-PipB or EGFP-PipB(Δ270–291) and immunostained for endogenous PDZD8 (red). DNA was detected with Hoechst 33342 (blue). Scale bars: 10 μm
(E) Fluorescence microscopy of HeLa cells transfected with a plasmid expressing myc-tagged PDZD8 and infected with STmSL1344 ΔpipB complemented in trans with PipB-2HA (and constitutively expressing mCherry from the chromosome) at 12 hpi. Upper row: Translocated PipB was visualized by immunostaining with anti-HA antibodies and PDZD8 using anti-myc antibodies. Infected cells display a clear redistribution of PDZD8-myc to SCV. Lower row: HeLa cells as per the upper row, immunostained for LAMP-1. Representative image demonstrates a redistribution of PDZD8-myc to the SCV (decorated with LAMP-1), whereas PDZD8-myc remains localized to the ER in uninfected cells. Scale bars: 10 μm.
(F) Fluorescence microscopy images showing that translocated 2xHA-tagged PipB co-localizes with PDZD8. HeLa cells were transfected with a plasmid expressing PDZD8-myc and infected with STmSL1344 ΔpipB complemented in trans with PipB-2HA, and immunostained at 12 hpi. Overlay shows PipB-2HA in green, PDZD8-myc in red and LAMP-2 (decorates SCV and SIFs) in blue. PDZD8 localizes predominantly to the SCV rather than SIFs.
(G) Quantification of PDZD8 recruitment in infected cells. HeLa cells were transfected with a plasmid expressing PDZD8-myc and infected with STmSL1344 wildtype, ΔpipB, ΔpipB complemented with PipB-2HA or PipB-2HA(Δ270–291) for 12 hpi. All strains are constitutively expressing mCherry from the chromosome. PDZD8 localization was scored as the fraction of infected cells displaying recruitment of PDZD8-myc to the SCV. Data from three independent experiments with roughly 100 cells per experiment, n indicates the total cell number. Error bars denote standard deviation. Localization of PDZD8-myc to the SCV depends on full-length PipB expression in trans.
PDZD8 was recently shown to regulate interactions between the ER and late endosomes via ER-mitochondrial contact sites (Elbaz-Alon et al., 2020; Guillén-Samander et al., 2019). Consistently, we observed a partial co-localization between PDZD8 and the ER transmembrane protein, BAP31, the ER luminal disulfide isomerase PDI, and the mitochondrial import receptor TOM20 in resting cells (Figure S6C). We first probed whether endogenous PDZD8 co-localized with EGFP-PipB at the ER, which was partially the case (Figure 6D).
To monitor the PDZD8-PipB interaction in an infection context, we infected HeLa cells ectopically expressing PDZD8-myc with STm ΔpipB complemented with PipB-2HA in trans. Similar to endogenous PDZD8, ectopically expressed PDZD8-myc retained a perinuclear ER-like distribution in uninfected cells (Figure 6E, S6C), indicating overexpression and tagging does not interfere with PDZD8 localization. Strikingly, PDZD8-myc relocalized to PipB-decorated compartments of infected cells (Figure 6E), and to a lesser extent to Salmonella-Induced Filaments (SIFs; LAMP-2) (Figure 6F). Consistent with the C-terminus of PipB mediating binding to PDZD8, ΔpipB PipB (Δ270–291)-2HA bacteria did not recruit PDZD8 to the SCV (Figure 6G). Notably, both PipB-2HA and PipB (Δ270–291)-2HA were effectively translocated into host cells by STm as assessed by immunofluorescence staining, albeit with slightly lower levels for the truncated version (Figure S6D).
Our results demonstrate that PipB recruits PDZD8 to the SCV, with the C-termini of the two proteins being important for their interaction. These results concur with the previous observation that the functional divergence of PipB and PipB2 is linked to their sequence difference in their C-termini (Knodler and Steele-Mortimer, 2005).
SteC targets FMNL formins to promote actin polymerization
We identified a PPI between the STm kinase SteC and a formin-like protein, FMNL1, which is highly expressed in macrophages (Yayoshi-Yamamoto et al., 2000). In all conditions and cell lines tested here, FMNL1 co-purified with SteC, and this PPI was also confirmed by reciprocal pulldowns (Figure 4). There are three largely redundant members of the human FMNL family (FMNL1/FMNL2/FMNL3), all sharing the same tripartite domain architecture of an N-terminal regulatory region binding to Rho-family GTPases (e.g. Cdc42), a central polyproline rich stretch for profilin-actin recruitment, and a C-terminal F-actin elongation region (Kühn and Geyer, 2014). Knowing that Cdc42 co-purified with SteC (Figure 2A), we decided to test whether the N-terminal domain of FMNL1 interacted with SteC. We thus purified full-length SteC, a catalytic inactive mutant SteCK256H and the N-terminal domain of FMNL11–385, and tested for complex formation by size-exclusion chromatography. SteC and FMNL1 alone migrated as multimeric species (Figure 7A; blue and orange traces, respectively), but when pre-incubated together, a portion of FMNL11–385 co-migrated with SteC forming a higher molecular weight complex (Figure 7A; green trace). This was also true for SteCK256H. Thus, SteC directly binds to the N-terminus of FMNL1 independent of its kinase activity.
Figure 7. SteC directly targets FMNL proteins to promote actin cytoskeleton rearrangement.
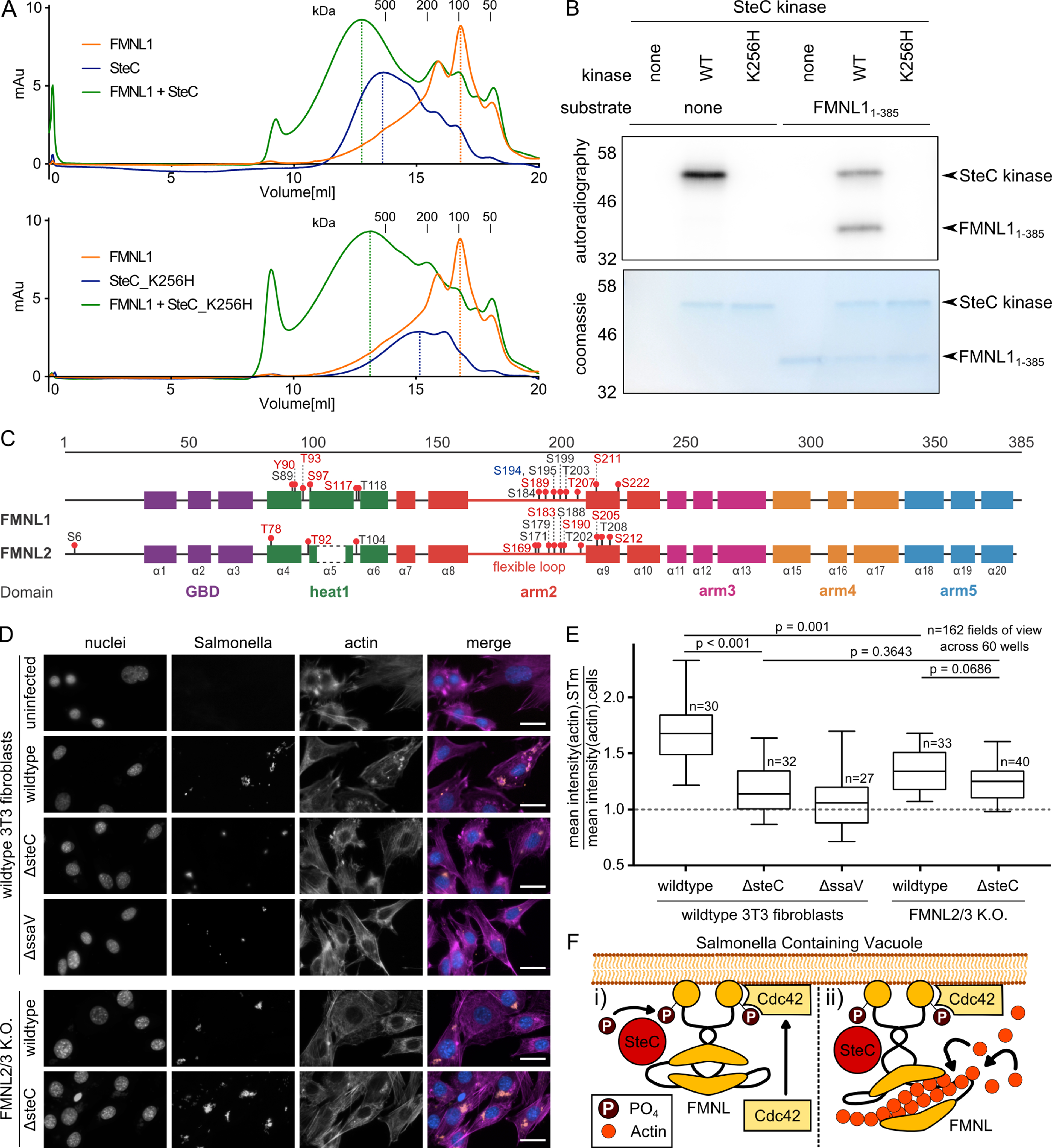
(A) Size exclusion chromatograms obtained from purified recombinant FMNL11–385, SteC (upper panel) or catalytically inactive SteCK256H (lower panel). Pre-incubation of FMNL11–385 with SteC or SteCK256H leads to a leftward shift in elution volume compared to the individual purified proteins, as indicated by the dotted lines.
(B) Autoradiography after in vitro kinase assay. FMNL11–385 was purified and incubated with purified SteC kinase, as well as SteCK256H in the presence of radioactively labelled [32P]-γ-ATP. Protein inputs were visualized by Coomassie blue staining. Only catalytic active SteC is capable of autophosphorylation and of phosphorylating FMNL1.
(C) Protein maps of FMNL1 and FMNL2, including functional regions, secondary structure elements, and phosphosites identified in the in vitro kinase assay, followed by phosphoproteomics. Coloring: Blue: identified in the first replicate (using 50μM ATP), red: identified in the second replicate (5mM ATP) black: identified in both. Phosphorylation by SteC occurs mostly in flexible loops of FMNL1 and FMNL2. Results are found in Table S6.
(D) Representative fluorescence microscopy images after infection of 3T3 fibroblasts (8 hpi) with mCherry-expressing STm strains (MOI 100), and staining with DAPI (blue) and phalloidin (purple). Data from three independent experiments for FMNL2/3 knockout cells (Kage et al., 2017b), and two independent experiments for wildtype 3T3 fibroblasts, spanning 162 FOVs (20 infected cells on average per view). Arrows indicate intracellular STm microcolonies. Scale bar: 30 μm. Quantification is shown in E.
(E) Average actin signal intensity at STm microcolonies divided by overall average actin signal intensity as a measure of actin-STm co-localization. 162 FOVs across 60 wells were analyzed and displayed as boxplots. Boxplots and statistical tests as in Fig. 5C.
(F) Model of SteC-FMNL interaction: (i) SteC directly binds FMNL subfamily formins, and is necessary and sufficient for its phosphorylation. (ii) The interaction between phosphorylated FMNL formins and Cdc42 induces actin polymerization (Kühn et al., 2015).
We then asked whether FMNL formins are direct substrates of SteC by performing an in vitro kinase assay. Consistent with previous reports, SteC was capable of auto-phosphorylation (Poh et al., 2008). In addition, when combined with FMNL11–385, SteC, but not the catalytically inactive SteCK256H, phosphorylated FMNL1 (Figure 7B). To identify the specific FMNL residues that are phosphorylated by SteC, we performed phosphoproteomics after an in vitro kinase assay for both FMNL1 and FMNL2. Thereby we identified the SteC auto-phosphorylation sites and several phosphosites in similar domains of both FMNL proteins (Table S6), many located in the flexible loop of the armadillo repeat region (Figure 7C). Among other sites, S171 (FMNL2) and an equivalent site in FMNL1 (S184) were phosphorylated. A phosphomimetic mutation (S171DD) in FMNL2 has previously been shown to enhance binding to the Rho-family GTPase and FMNL regulator Cdc42 (Kühn et al., 2015).
SteC is required to induce actin bundling around the STm microcolony in 3T3 murine fibroblasts (Imami et al., 2013; Odendall et al., 2012; Poh et al., 2008). We therefore postulated that FMNL proteins may be required for this phenotype, as they are known to promote actin polymerization (Bai et al., 2011; Block et al., 2012; Heimsath and Higgs, 2012). Since FMNL1 had been reported to be absent from 3T3 fibroblasts (Kage et al., 2017a), 3T3 cells disrupted in the more abundant FMNL2 and FMNL3 were used (Kage et al., 2017b). As shown previously (Odendall et al., 2012), actin bundling around the SCV was strictly dependent on SteC in 3T3 fibroblasts (Figure 7D). Interestingly, actin bundling around STm microcolonies was diminished in FMNL2/3 knockout fibroblasts no matter whether infected with STm wildtype or ΔsteC (Figure 7E), suggesting that SteC acts via FMNLs. Despite both STm strains not being able to induce substantial actin bundling in the absence of FMNL2 and FMNL3, there was still some residual bundle formation by SteC. We therefore examined more closely the expression of FMNL subfamily proteins in 3T3 cells using a newly available FMNL1-specific antibody. While FMNL2 and FMNL3 were abundant in control and absent in FMNL2/3 knockout cell lines, as expected, we also detected a high molecular mass variant of FMNL1 in wildtype and knockout cells (Figure S7). We suspect that the residual SteC-dependent actin bundling observed in FMNL2/3 knockout cells is due to this FMNL1 expression. Taken together, these results demonstrate that SteC directly interacts with and phosphorylates FMNL subfamily formins. This could trigger activation of FMNLs by Cdc42, stimulating actin assembly and bundling at sites of STm microcolony formation (Figure 7F).
Discussion
In this work, we describe 446 PPIs, including 25 previously described PPIs, between 15 different STm effectors after infection of two mammalian cell lines. These interactions were identified during infection with physiological effector translocation levels. The majority of detected interactions were cell type-specific (418 PPIs), with only 28 PPIs being conserved across the two cell-types. Although the stringency of our methodology may account for some of the differences, the differential expression levels of host target proteins, and the different infection trajectories in epithelial cells and macrophages are major contributors for this discrepancy. Most effectors co-purified with multiple host targets, several of which were related in function, indicating converging functionalities between effectors. Several of these interactions may be indirect, so further work would be required to define the bona fide physical interactions. Yet the functional relevance of this resource and the ability to detect direct interactions are exemplified by three vignettes of infection biology in cholesterol trafficking, organelle contact site organization and actin rearrangements.
Several prior studies have tried to map STm-host PPIs (Andritschke et al., 2016; Auweter et al., 2011; Patrick et al., 2018; Sontag et al., 2016). All were conducted outside the context of infection and typically after overexpression of a single STm effector inside host cells. For example, in a systematic study, Auweter et al. ectopically expressed a panel of 13 STm effectors in HEK-293T cells and in parallel, expressed and purified 11 STm effectors in E. coli. AP-MS in HEK-293T cells with ectopically expressed effectors or AP-MS on HEK-293T cell proteins bound to immobilized purified effectors in vitro revealed 15 effector-target interactions, two of which (SseJ-RhoA and SseL-OSBP) (Auweter et al., 2011) were also identified in our study.
More recently, BioID was used to study effector-host interactions by tagging a panel of five effectors (PipB2, SseF, SseG, SifA, SopD2) with the biotin ligase BirA and overexpressing fusion proteins in HeLa cells by plasmid-based transfection (D’Costa et al., 2019). In the same study, the authors used AP-MS after ectopic expression of effectors. Although we tagged these same 5 effectors, due to limiting levels of translocated effector protein, we only assessed PipB2 and SifA by AP-QMS. Comparing the two studies, there is some overlap in host-protein targets: 4 proteins for SifA and 16 for PipB2 (Table S3). The overexpressed effectors, the absence of an infection context and the stringent thresholds may account for the differences between the two studies. D’Costa et al. did, however, find similar host processes being targeted by STm effectors as we did (e.g. ion transport, SNARE complex, lipid metabolism, actin-related), underlying the importance of these processes in infection.
Tagging can impede function or localization of some effectors, as previously shown for SifA (Brumell et al., 2002) for which we adjusted the tagging strategy. It will be important to assess whether the C-terminal modification introduced into the rest of the effectors impacts their translocation and/or function. We probed expression and translocation for all effectors, and we could detect 20 effectors in the host cytoplasm (28 were expressed). Although some of the remaining 12 may fail to translocate due to their C-terminal tag, we find it more likely that they are not translocated in sufficient amounts in the cell lines and/or time-points probed here. Introduction of a C-terminal tag may have led, in some cases, to poor stability of otherwise abundant effectors, such as SseF and SseG. Of the 15 translocated effectors we could reproducibly detect by AP-QMS, there were 5 effectors for which we did not detect significantly enriched targets in at least one of the tested cell lines (i.e. GogB, SspH1, SspH2, SseK1 in HeLa cells and SlrP in RAW264.7 cells). In addition to tags compromising function/PPIs, other explanations could include promiscuous or transient interactions (many STm effectors are enzymes) or non-proteinaceous targets (lipid, DNA/RNA, metabolites) (Knodler et al., 2009; McShan et al., 2016; Nawabi et al., 2008). In general, our inability to detect some PPIs which have been described previously, could be due to tagging, infection conditions (20 hpi, stringent pulldowns, exclusion of nuclear fraction), cell lines used, MS-limitations (abundance or detection of prey) or false negatives of the method.
Adding on the previously reported functional cooperation of SseJ and SseL (Kolodziejek et al., 2019), we show that: a) both strongly interact with additional cholesterol trafficking proteins; and b) that SseJ, and to lesser degree SseL, contribute to cholesterol accumulation at the SCV. Furthermore, we show that SifA is required to recruit both cholesterol and NPC1 to the SCV, although we did not detect a direct interaction between SifA and NPC1. Further work is needed to define whether SseJ interacts directly with NPC1, and how SseJ impacts NPC1 function. It seems plausible that both SseJ (sequestration by cholesterol esterification), and SseL (promoting cholesterol influx to the SCV, likely via OSBP-VapA/B), counteract NPC1 to retain cholesterol at the SCV. This is consistent with our observation that SseL-dependent retention at the SCV is fully mitigated in the absence of NPC1.
We also identified a strong interaction between PipB and the host target PDZD8, a protein recently shown to be required for organelle contact site formation. PDZD8 accumulates at contact sites between the ER and late endosomes/lysosomes, together with Rab7 and protrudin (Elbaz-Alon et al., 2020; Guillén-Samander et al., 2019). Interestingly, Rab7 was also enriched in PipB pulldowns, but remained just below our significance thresholds (Mendeley Data). We show that PipB binds to the C-terminal CC domain of PDZD8, which is the same region that mediates the interaction to Rab7 (Elbaz-Alon et al., 2020; Guillén-Samander et al., 2019). It remains to be explored whether Rab7 and PipB compete for PDZD8 binding at the CC domain, or how the PipB-PDZD8 interaction affects the three-way membrane-contact-sites mediated by PDZD8, Rab7 and protrudin (Elbaz-Alon et al., 2020).
One of our strongest interactions was that between SteC and FMNL1. SteC had been linked to actin rearrangements during infection by modulation of MAPK signaling and HSP70 (Imami et al., 2013; Odendall et al., 2012). Yet, the effect attributed to SteC exceeded these interaction partners, indicating a missing piece in the rewiring of host cytoskeletal remodeling by SteC. We identified FMNL subfamily formins as the host targets which bound SteC in vivo and in vitro. We could further show that SteC phosphorylates these formins in vitro at S171 (FMNL2; S184 for FMNL1) and at residues in the same functional region that promote interactions with Cdc42 and thereby actin polymerization (Kühn et al., 2015). Our current model is that SteC directly binds to and phosphorylates FMNL proteins, promoting their interaction with Cdc42 and the recruitment of the complex to the SCV to stimulate actin polymerization (Figure 7F). In agreement with this model, we observed Cdc42 to co-purify with SteC and FMNL1 in murine macrophages (Figure 2A). However, dominant negative versions of Cdc42 were shown in the past to still allow SPI-2-dependent actin assembly (Unsworth et al., 2004). Further work will be required to elucidate whether SteC has any preference for different FMNL subfamily members, the molecular events triggered by binding and phosphorylation of FMNLs by SteC, and whether the SteC-FMNL interaction is linked to the previously reported modulation of MAPK signaling by SteC (Odendall et al., 2012).
In conclusion, we aimed to bridge a technological gap common to host-pathogen PPI studies, which were until now performed exclusively in non-physiological conditions. Our study can nucleate more systematic and unbiased studies of host-pathogen PPIs in a native infection context, providing better insight into the degree and nature of effector cooperation, which is of high relevance in bacterial pathogens with large effector arsenals. Dissecting how pathogens directly modify host pathways via secreted proteins will uncover new aspects of pathogen biology and provide tools and targets to modulate immune responses.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Athanasios Typas (typas@embl.de).
Materials Availability
All bacterial strains generated in this study will be made available on request by the Lead Contact with a completed Materials Transfer Agreement.
Data and Code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD018375. The code and pipelines used for data analysis are available upon request. Raw data files not included in the main manuscript are located at Mendeley Data (DOI: 10.17632/xjb24h7s8j.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Salmonella enterica subsp.
Typhimurium 14028s (STm) and S. Typhimurium SL1344 (STmSL1344) were routinely cultivated in LB Lennox broth with rotation at 37°C overnight. Bacterial antibiotic selection was performed on LB agar containing ampicillin (100 μg/mL) or kanamycin (30 μg/mL) at 37°C. RAW264.7 macrophages (ATCC TIB-71), HeLa epithelial cells (ATCC CCL-2) and NIH 3T3 fibroblasts were cultured at 37°C, 5% CO2 in DMEM containing 4.5g/l (RAW264.7) or 1g/l (HeLa, 3T3 NIH) glucose (Gibco) and 10% FBS. pBMDMs were maintained in RPMI supplemented with m-CSF and 10% FBS as previously described (Selkrig et al., 2020). Cells were passaged at 90% confluency and were not used beyond passage number 15. For cell passaging and seeding, media was removed, cells were washed once in pre-warmed PBS and detached by incubation in 0.05% trypsin-EDTA (for HeLa cells, Thermo Fisher, cat. Nr. 25300054), accutase (for RAW264.7 cells, Thermo Fisher, cat. Nr. A1110501) or 2 mM EDTA, 5% FBS in PBS (for pBMDMs) at 37°C for ~3 min. Complete media was added to the cell suspension and cells were counted using trypan blue staining in a Biorad TC20 automated cell counter. If cells were prepared for infection, the following cell numbers were seeded ~20h prior to the infection: For 96-wells (Zell-Kontakt, cat. Nr. 21315241), 7.5×103 HeLa and 3×104 RAW264.7 cells; 6-wells (Thermo Scientific, cat. Nr. 10119831), 2×105 HeLa and 9×105 RAW264.7 cells; 15cm dishes (Greiner, cat. Nr. 639160), 3.5×106 HeLa and 15.4×106 RAW264.7 cells. For large-scale AP-QMS experiments, five 15cm dishes were seeded per effector per condition, equaling a total cell number of ~75×106 and 17.5×106 for cells for RAW264.7 and HeLa cells, respectively. NIH 3T3 wildtype and derived FMNL2/3 double knockout clones 9.10 and 46.20 were maintained as described previously (Kage et al., 2017b). HeLa cells harboring an NPC1 knockout were maintained as previously described (Tharkeshwar et al., 2017). HeLa and RAW264.7 cells were purchased from ATCC. All cells were subject to routine visual inspection for expected morphological phenotypes. Cells lines were not formally authenticated. RAW264.7 cells are from a male mouse, HeLa cells from a female human and pBMDMs a mixture of male and female.
METHOD DETAILS
Media, chemicals and reagents
The following chemicals and reagents used were purchased from Sigma: DMSO (cat. nr. D8418), Triton-X100 (Tx-100), heat inactivated Fetal Bovine Serum (FBS) (F9665–500ML), Phalloidin ATTO-647N (65906), gentamicin (G1914); Gibco: DMEM 4.5 g/L glucose (41965); Roche cOmplete mini EDTA-free protease inhibitors (11873580001); Life Technologies Hoechst 33342 (H3570); Thermo Scientific Pierce™ formaldehyde 16% (w/v) (28908). Antibodies are listed in KRT including the distributor and catalog number.
Bacterial strains and plasmids
All strains used in this study are listed in KRT. Salmonella enterica subsp. Typhimurium 14028s (STm) wildtype was used to generate the tagged effector library as described below. Single gene deletion mutants (ΔsseJ, ΔsseL, ΔsteC, ΔssaV, ΔsifA) were retrieved from a single gene deletion (SGD) collection (Porwollik et al., 2014). Deletion was confirmed by PCR and retransduced into the wildtype STm 14028s background using P22 phage. To generate the ΔsseJΔsseL double mutant, FLP-FRT mediated excision of the antibiotic resistance cassette was performed as previously described (Datsenko and Wanner, 2000), followed by P22 transduction of the second mutated loci. Resulting double mutants were verified by colony PCR. S. Typhimurium SL1344 (STmSL1344) wildtype, ΔpipB and ΔpipB2 strains have been described previously (Hoiseth and Stocker, 1981; Knodler et al., 2002, 2003). For constitutive expression of mCherry from the chromosome, ΔpipB bacteria were transduced with P22 lysate derived from SL1344 wildtype glmS::Ptrc-mCherryST::Cm (Knodler et al., Cell Host Microbe, 2014) and transductants selected on LB agar containing chloramphenicol (30 μg/ml). The Cm resistance cassette was removed by FLP-FRT mediated excision as described above. The complementing plasmids, pPipB-2HA and pPipB2–2HA, are pACYC184 derivatives and have been described previously (Knodler et al., 2002, 2003).
The pPipB(Δ270–291)-2HA plasmid was constructed as follows. The corresponding DNA fragment (including upstream promoter region) was amplified from STmSL1344 genomic DNA with the oligos pipB-Sal (5’ a cgc gtc gac ata ctt tct taa tga gat aaa acg 3’) and pipB269R-Bgl ((5’ gga aga tct acc tgt cag atc ggc tcc tgt 3’). The pipB2 coding sequence was excised from pPipB2–2HA (pACYC184-derivative; (Knodler et al., 2003) with SalI/BglII and replaced with a SalI/BglII-digested PipB(1–269) fragment to create pPipB (Δ270–291)-2HA.
The STm 14028s tagged effector library was generated as follows. To generate the template plasmid (pJPS1), a 2xSTREP-TEV-3xFLAG (STF) tag was cloned into the MCS (EcoRI-HindIII) of pQE30 and designated pMZ2. The pMZ2 plasmid was then used as a PCR template to amplify a 2xSTREP-TEV-3xFLAG tag together with the pKD4 kanamycin resistance cassette using primers JPS26 and JPS27. This amplicon was then T/A cloned into pGEM®-T Easy according to manufacturer’s instructions followed by sequence verification and designation as pJPS1. Purified pJPS1 was used as template plasmid DNA to amplify and introduce a 2xSTREP-TEV-3xFLAG (STF) tag followed by a kanamycin resistance cassette at the C-terminus of chromosomally encoded genes via λ-red recombinase (Datsenko and Wanner, 2000; Uzzau et al., 2001). Clones were selected on LB agar containing kanamycin 30μg/mL and verified PCR and sequencing of the C-terminal region of the targeted gene. The resulting tagged effectors expressed the following C-terminal STF affinity tag sequence; GGAAAGWSHPQFEKGGGSGGGSGGGSWSHPQFEKGENLYFQGADYKDHDGDYKDHDIDYKDDDDK. See Table S1 and KRT for the complete list of effectors targeted and Table S7 for all oligonucleotides used in this study.
To avoid disturbing the C-terminal prenylation motif of the effector sifA (Brumell et al., 2002), the STF tag was chromosomally inserted within the open reading frame between residues D136 and I137 using a two-step selection method related to λ-red recombination was (Kolmsee and Hengge, 2011). Briefly, to generate an STm 14028s strain amenable to pKD45 two-step selection, the endogenous STm ccdAB locus (STM14_5550 and STM14_5550) was deleted via λ-red recombination (Datsenko and Wanner, 2000; Uzzau et al., 2001) using primers JPS38 and JPS39, followed by PCR verification and P22 transduction to a wildtype background and designated STm ΔccdAB::Cm. A fragment of the plasmid pKD45 (Datsenko and Wanner, 2000) encoding a kanamycin-resistance cassette and a ccdB toxin under the control of a rhamnose-inducible promoter was amplified using primer pairs JPS14 and JPS15 containing extensions homologous to the sifA locus (STM14_1400). The resulting amplicon was chromosomally integrated into STm ΔccdAB::Cm using λ-red recombination and selected on LB agar containing 30μg/mL kanamycin (Datsenko and Wanner, 2000). Positive sifA::kan-ccdB transformants were verified by PCR and tested for L-rhamnose sensitivity on M9 minimal agar. Using primers JPS28 and JPS29 and the pJPS1 plasmid as DNA template, an amplicon containing overhangs with sequence homology to sifA and an internal STF sequence was amplified and integrated onto the chromosome using λ-red recombinase (Datsenko and Wanner, 2000). Transformants were counter-selected on M9 minimal agar containing 0.5% L-rhamnose after incubation at 30°C for 2 days and verified by PCR. A list of STF-tagged effectors generated is listed in Table S1, along with summarized test expression behavior in both HeLa and RAW264.7 cells.
For ectopic expression in mammalian cells, the pipB open reading frame was amplified from STmSL1344genomic DNA with the oligonucleotides pipBGFP-N5’ and pipBGFP-N3’2. The amplicon was digested with BglII/SalI and ligated into BglII/SalI-digested pEGFP-C1 (Clontech) to create EGFP-PipB. EGFP-PipB (Δ270–291) was created by amplification with pipB-GFP-N5’ and GFP-PipB-269R, digestion with BglII/SalI and ligation into pEGFP-C1. EGFP-PipB2 has been described previously (Knodler and Steele-Mortimer, 2005). PDZD8 was tagged at the C-terminus with a myc epitope for immunodetection. The coding sequence plus an upstream Kozak sequence were amplified from a PDZD8 cDNA clone, MGC:27107 IMAGE:4837939 (The CCSB Human ORFeome Collection) with the oligonucleotides PDZK8-EcoRI-Kozak and NM_173791-Xho. The amplicon was ligated in EcoRI/XhoI-digested pCMV-Tag 5A (Stratagene) to create pKozak-PDZD8-myc.
For expression in yeast, EcoRI/BglII fragments encoding full length, residues 1–281, residues 1–271, residues 1–188 and residues 189–291 of PipB were amplified from STmSL1344 genomic DNA with the following oligonucleotide pairs and ligated into pGAD424 (Clontech): pGAD-PipB-1F and pGAD-PipB-291R, pGAD-PipB-1F and pGAD-PipB-281R, pGAD-PipB-1F and pGAD-PipB-271R, pGAD-PipB-1F and pGAD-PipB-188R, pGAD-PipB-189F and pGAD-PipB-291R. Full-length and fragments of PDZD8 were PCR amplified as EcoRI/SalI fragments from PDZD8 cDNA (details above). Amplicons were digested and ligated into pGBT9 (Clontech). The following oligonucleotide pairs were used: pGBT9-PDZK8-F and pGBT9-PDZK8-R for pGBT9-PDZD8, pGBT9-PDZK8-F2 and pGBT9-PDZK8-R for pGBT9-PDZD8(Δ1–338), pGBT9-PDZK8-F and pGBT9-PDZK8-R2 for pGBT9-PDZD8(Δ930–1154). Overlap extension PCR (Horton et al., 1989) was used to create pGBT9 constructs that were deleted for residues 368–461 (pGBT9-PDZD8ΔPDZ), residues 494–814 (pGBT9-PDZD8(Δ494–814)) and residues 841–887 (pGBT9-PDZD8ΔC1).
Infection of RAW264.7 macrophages, pBMDMs and HeLa cells
For infection of RAW264.7 or pBMDMs, STm strains were cultured overnight at 37°C in LB broth (Lennox), washed in PBS and added to monolayers at a multiplicity of infection (MOI) of 100. For infections carried out in multiwell plates, the bacteria were centrifuge onto monolayers at 170 g for 5 min to promote bacteria-host cell contact. The infection was performed for 30 min at 37°C, after which the media containing bacteria was removed by aspiration. Cells were washed once in pre-warmed PBS and subsequently cultured at 37°C in DMEM (4.5 g/l glucose) containing 10% FBS and 100μg/ml gentamycin (50μg/ml gentamycin for pBMDMs) to kill remaining extracellular bacteria. After 1 hr, the media was replaced with DMEM containing 10% FBS and 16 μg/ml gentamycin (8 μg/ml gentamycin including m-CSF for pBMDMs) for the remainder of the experiment (this also denotes time point zero). For HeLa and NIH 3T3 fibroblast cell infection, overnight cultures of STm strains were subcultured (300μL overnight culture in 10 ml LB Miller containing relevant antibiotics) and grown for 3.5 hr at 37°C in 100 ml Erlenmeyers at 45 rpm (adapted from (Steele-Mortimer, 2008)). Monolayers were infected with an MOI of 100 or 50 (see figure legend) and the gentamicin protection assays was performed as described above but instead in the presence of DMEM containing 1 g/l glucose (Gibco) and 10% FBS.
Proteomic sample preparation for AP-QMS
For native harvesting, cells were washed twice in PBS at room temperature and lysis buffer (PBS, containing 0.1% Triton-X100 and 1x Protease Inhibitor (cOmplete EDTA free, Roche) was added (300μL for 6-well plates, 5ml for 15cm dish). Cells were put at 4°C for 30 min while shaking gently and subsequently scraped off and resuspended by pipetting. The cell lysate suspension was centrifuged at 4°C for 15 min at 20,000 g to clear the lysate. A small sample of the cleared lysate was saved as “Total” sample, the remaining lysate was directly used for immunoprecipitation. For harvesting after crosslinking, the cells were washed twice in PBS at room temperature and crosslinking buffer (PBS, containing 1mM DSP (Thermo Fisher, cat. nr. 22585)) was added. Crosslinking was performed for 2 hr at 4°C and quenched using 20 mM Tris-HCl at pH 7.5. Cells were washed twice in quenching buffer and subsequently subjected to the lysis protocol described above.
For pulldown, STF-tagged STm effectors were immunoprecipitated using anti-FLAG M2 affinity gel (Sigma, A2220). 50μL of the slurry per sample was washed twice in lysis buffer (centrifugation for 1 min at 4°C and 2655 g). The beads were added to fresh, cleared lysate and incubated for at least 4h (native samples) or overnight (cross-linked samples) at 4°C with rotation. After bead incubation, the suspension was centrifuged at 3220 g for 10 min (4°C) and the supernatant was removed. The beads were washed four times in 1mL of ice cold washing buffer (PBS containing 0.01% Triton-X100) by centrifugation at 2655 g for 1 min (4°C). After the final wash, all remaining buffer was removed and replaced with 40 μL of elution buffer (PBS containing 150 μg/ml 3x FLAG peptide and 0.05% RapiGest). Proteins were eluted by incubating at 4°C for 1h with rotation followed by centrifugation at 7150 g at 4°C and the supernatant containing eluted proteins was removed. The elution step was repeated with an additional 40 μL of elution buffer and pooled with the first elution.
TMT-labeling of AP-QMS samples and Mass Spectrometry
Within each TMT-10plex, untagged control (wildtype), as well as 9 STF-effector strains were assessed in parallel (RAW264.7 run 1: untagged-wildtype, PipB, PipB2, SifA, SseJ, SseL, SspH1, SteC, SlrP, run 2: untagged-wildtype, AvrA, GogB, SipB, SpvC, SseI, SseK1, SspH2, SteA, SteE; HeLa run 1: untagged-wildtype, PipB, PipB2, SifA, SseJ, SseL, SspH1, SspH2, SteC, SlrP, run 2: untagged-wildtype, AvrA, GogB, SifB, SpvC, SseF, SseI, SseK1, SseK2, SteA). For the separate TMT-run of SseJ-STF (Figure 5A, Mendeley Data) and the validation in pBMDMs (Figure S4, Mendeley Data), all three biological replicates were combined in the same TMT-10plex with untagged control (wildtype) in triplicate as control, to which fold changes were calculated as described below.
For each run, all STF-tagged effector strains were seeded and infected at the same time. Prior to LC/MS-MS, 1μL of the elution fractions were used in Western Blot to validate the presence of the effector bait. Total protein concentration was determined using the Pierce Micro BCA kit, according to the manufacturer’s protocol. All samples were adjusted to 10 μg of protein in a 50 μL volume and were subsequently submitted to the EMBL Proteomics Core Facility for sample processing. After reduction of disulfide bridges using 10 mM dithiothreitol at 56°C for 30 min in HEPES buffer (50 mM HEPES, pH 8.5), alkylation was performed using 20 mM 2-chloroacetamide at room temperature in HEPES buffer for 30 min under exclusion of light. Samples were prepared according to the SP3 protocol (Hughes et al., 2019) and trypsinized (sequencing grade, Promega, enzyme to protein ratio 1:50) overnight at 37°C. Subsequently, peptides were recovered in HEPES buffer by collecting supernatant on magnet and combining it with a second elution wash of the magnetic beads with HEPES buffer. Peptides were labelled with TMT10plex (Werner et al., 2014) Isobaric Label Reagent (ThermoFisher) according to the manufacturer’s instructions. In short, 0.8mg reagent was dissolved in 42 μL acetonitrile (100%) and 4 μL of this stock were added to the samples and incubated for 1 h at room temperature. The reaction was then quenched with 5% hydroxylamine for 15 min. Samples were pooled for the TMT-10plex and then further cleaned using OASIS® HLB μElution Plate (Waters). Subsequently, offline high pH reverse phase fractionation was performed on an Agilent 1200 Infinity high-performance liquid chromatography system, using a Gemini C18 column (3 μm, 110 Å, 100 × 1.0 mm, Phenomenex) with 20 mM ammonium formate (pH 10.0) and 100% acetonitrile as mobile phase (Reichel et al., 2016). The first and two last fractions were discarded prior to LC-MS analysis.
AP-QMS Data acquisition
Samples were analyzed on an UltiMate 3000 RSLC nano LC system (Dionex) using a μ-Precolumn C18 PepMap 100 trapping cartridge (5μm, 300 μm i.d. x 5 mm, 100 Å) and a nanoEase™ M/Z HSS T3 column 75 μm x 250 mm C18 as analytical column (1.8 μm, 100 Å, Waters). After trapping with a constant flow of 0.1% formic acid in water at 30 μL/min onto the trapping column for 6 min, elution was carried out via the analytical column at a constant flow of 0.3 μL/min with increasing percentage of solvent (0.1% formic acid in acetonitrile): from 2% to 4% in 4 min, from 4% to 8% in 2 min, then 8% to 28% for a further 96 min, and finally from 28% to 40% in another 10 min. The analytical column was coupled to QExactive plus (Thermo) mass spectrometer and mass-spec was performed according to previously described parameters (Perez-Perri et al., 2018).
AP-QMS Data analysis
IsobarQuant (Franken et al., 2015) and Mascot (v2.2.07) were used to process the acquired data. Peptide search was performed against a Uniprot Homo sapiens proteome database (UP000005640, for HeLa cell samples) or a Uniprot Mus musculus database (UP000000589, for RAW264.7 cell samples), combined with the Salmonella typhimurium (strain 14028s / SGSC 2262) (UP000002695) database containing common contaminants and reversed sequences. The following modifications were included in the search parameters: Carbamidomethyl (C) and TMT10 (K) as fixed modifications, acetyl (protein N-terminus), oxidation (M) and TMT10 (N-terminal) as variable modifications. Mass error tolerance was set as follows: 10ppm for the full scan (MS1) and 0.02Da for MS/MS (MS2) spectra. In addition, a maximum of two missed cleavages were allowed for trypsin, a minimum peptide length of seven amino acids was required and the false discovery rate (fdr) on peptide and protein level was set to 0.01. The output files of IsobarQuant (Franken et al., 2015) were analyzed using the R programming language (ISBN 3–900051-07–0). Only proteins that were quantified with at least two unique peptides and identified in at least two out of three biological replicates were kept for further analysis. The ‘signal_sum’ columns were first annotated to their biological conditions and then a median across all conditions per replicate was computed for each protein. First, potential batch-effects were removed using the ‘removeBatchEffect’ function of the limma package (Ritchie et al., 2015). Second, data were normalized with a variance stabilization normalization (vsn – (Huber et al., 2002)). Finally, missing values were imputed using the impute function (method = “knn”) of the Msnbase package (Gatto and Lilley, 2012). Limma was employed again to test for differential expression. Fold changes with respect to the median of the respective run were calculated for each protein in each pulldown. T-values from the limma output were pasted also into fdrtool (Strimmer, 2008) in order to compute alternative fdrs. In case the standard deviation of the t-values deviated from 1 to a degree that no convergence of statistically significant hits was observed, the q-values from the fdrtool output were used as alternative fdrs. A protein was annotated as a ‘hit’ with an fdr smaller than 1% and a fold increase of at least 20%; this was done for all four datasets (RAW264.7 native, RAW264.7 crosslinked, HeLa native and HeLa crosslinked) independently. This initial hit list was then further refined in multiple steps: 1) PPIs were combined into two datasets, one for each cell line; 2) if a PPI passed the fold change criterion in both conditions (native and crosslinked), the fdr requirement was loosened to fdr ≤ 0.05; 3) the resulting PPIs were ranked according to fdr and according to fold change for each effector and each condition (native and crosslinked); 4) only PPIs that were in the top 20 for either fold change or fdr were called “hit”; 5) in addition, all PPIs that passed the fold change requirement, as well as the loosened fdr requirement in both conditions were called a “hit”. Output from tables from statistical analysis are located in Table S2.
Network building and GO-term analysis
Networks were built from the hits for both native and crosslinked pulldowns. Known host-host functional interactions (physical and/or functional from genomic context, high-throughput experiments, (conserved) co-expression and previous knowledge), as well as bacterial functional interactions were imported into cytoscape v3.7.2 (Shannon et al., 2003) using STRING protein query (STRING DB version 11 (Szklarczyk et al., 2019)) for the respective organism and a confidence cutoff of 0.7 (see Table S3 for functional interaction network edges of the different organisms). Using a reference list of all the proteins detected in the LC-MS/MS runs for the respective human (HeLa) or rodent (RAW264.7) host, GO-term enrichment for biological processes was performed using ClueGO version 2.5.2 with the cell line specific AP-QMS protein background as reference proteome. GO-term fusion, as well as grouping was enabled using a p-value cutoff of 0.05 after Benjamini Hochberg p-value correction. GO-terms contained in GO level 4 and 5 were searched, requiring at least 3 genes and 15% of genes per term and merging groups if at least 40% of genes and terms overlapped. The leading group term was chosen as the GO-term containing the largest number of genes.
SDS-PAGE and Immunoblotting
For protein separation and detection, the BioRad system, and RunBlue precast gradient gels (expedeon) were used. Prior to loading on the gel, samples were diluted in Laemmli buffer (Laemmli, 1970) containing 100 mM DTT and heated to 98°C for 10 min. Samples were briefly centrifuged and loaded using a Hamilton syringe. SDS-PAGE was performed at a constant voltage of 150V for 50 min. For Western Blot, Immobilon-P PVDF or nitrocellulose membranes were used in a BioRad system (100V for 90 min while keeping the system cool). Subsequently, membranes were blocked in 5% milk in TBST for 1h and incubated in primary antibody diluted 1:1000 (see KRT for manufacturer and origin and antibody dilutions used) overnight. Membranes were washed 3 times for 5 min in TBST and subsequently incubated in secondary antibody conjugated to HRP (see KRT) for 1h in 5% milk in TBST. After washing 3 times for 5 min, exposure using SuperSignalTM West Pico Plus chemiluminescent substrate (Thermo scientific) or Supersignal West Femto Max Sensitivity ECL onto Lucent Blue X-Ray films (advansta) or Kodak film in the dark was used to detect protein bands. Biochemical fractionation of EGFP-PipB and EGFP-PipB (Δ270–291) was performed as previously described (Lau et al., 2019).
Reciprocal pulldown validation
In order to validate PPIs identified from the AP-QMS workflow, we used a panel of 11 host target specific antibodies (see KRT for antibodies used). Per reaction, 50μl slurry of Protein-A beads (Thermo Fisher, cat. nr. 22811) for antibodies produced in rabbit or Protein G beads (Abcam, ab193259) for antibodies produced in mouse or rat, were washed twice in lysis buffer (0.1% Triton-X100 in PBS containing protease inhibitor). For each reaction, 3.5μl of antibody was added to 100μL of washed beads in lysis buffer and incubated at room temperature for 2 hr with constant rotation in order to bind antibodies to the beads. The bead-antibody mixture was applied to the cleared, fresh lysate (obtained as described in the “Proteomic sample preparation section) without removing unbound antibody and incubated at 4°C for 4h. Samples were then centrifuged for 1 min at 2655 g at 4°C and the supernatant was decanted. The antigen-bound beads were washed 3 times in wash buffer (PBS containing 0.01% Triton-X100) by centrifuging at 2655 g at 4°C for 1 min. After the final washing step, supernatants were removed and 100 μL of Laemmli buffer (Laemmli, 1970) containing 100 mM DTT was added to the beads. Samples were heated to 98°C for 10 min followed by centrifugation for 1 min at 20817 g. Eluates were analyzed by immunoblot (see KRT for antibody dilutions).
PipB and PipB2 immunoprecipitations and mass spectrometry
HeLa epithelial cells (ATCC CCL-2) were grown in Eagle’s modified medium (Mediatech) containing 10% heat-inactivated fetal calf serum (Invitrogen) at 37°C with 5% CO2. Cells were seeded in 10 cm tissue-culture treated dishes and transfected with FuGENE 6® reagent (Roche) for 24 hr. Plasmid DNA was prepared using the QIAfilter Plasmid Midi kit (QIAGEN) according to the manufacturer’s instructions. For identification of PipB-specific interacting protein(s) (Figure S6A), eight 10 cm tissue-culture treated dishes of HeLa cells were transfected with pEGFP-C1, pEGFP-PipB or pEGFP-PipB2. Monolayers were washed twice in cold PBS and collected by scraping into PBS. Cells were lysed on ice for 30 min in 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1mM EDTA, 0.1% Nonidet P-40 containing protease inhibitor cocktail set III and phosphatase inhibitor cocktail set II (EMD Biosciences). Samples were centrifuged at 3,000xg for 10 min at 4°C, the post-nuclear supernatant collected and precleared with Protein A agarose for 1 h at 4°C. The supernatant was collected and incubated with mouse anti-GFP clone 3E6 (Molecular Probes) for 1 h at 4°C, followed by the addition of Protein A agarose. Beads were washed four times in lysis buffer and bound proteins eluted with boiling 1.5x SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE on 4–15% gradient gels (BioRad) and visualized with SilverQuest Silver staining kit (Thermo). A 150 kDa band unique to the GFP-PipB immunoprecipitate was excised and sent for LC-MS/MS analysis at the Stanford University Mass Spectrometry (SUMS) Facility. For confirmation of the PipB-PDZD8 interaction under infection conditions (Figure 4B), HeLa cells seeded in 10 cm tissue-culture treated dishes were transfected with PDZD8-myc and infected with the following STmSL1344 strains 12 h later: ΔpipB pPipB-2HA or ΔpipB2 pPipB2–2HA at a MOI of 50 (ten 10 cm dishes per strain). At 12 hpi, monolayers were collected and processed as described above. After 30 min lysis, samples were centrifuged at 6,000xg for 15 min at 4°C (which is sufficient to pellet intact bacteria), the supernatant collected and pre-cleared with Protein A agarose, followed by incubation with mouse anti-myc clone 4A6 agarose conjugate (EMD Millipore). Beads were washed in lysis buffer and bound proteins eluted with boiling 1.5x SDS-PAGE sample buffer. Immunoprecipitates were separated by SDS-PAGE and subject to immunoblotting with affinity purified rabbit polyclonal anti-PDZD8 peptide antibodies (directed against amino acid residues 1101–1118, custom antibody from New England Peptide). and mouse monoclonal anti-HA.11 antibodies (Covance).
Microscopy of F-actin, Filipin and NPC1
Cells were seeded in 96-well glass bottom plates (Greiner CellContact, 30,000 cells per well for RAW264.7, 7,000 cells per well for HeLa or 3T3 fibroblasts). Cells were infected with STm 14028s strains constitutively expressing mCherry from a plasmid (pFCcGi). After infection for the indicated times (see figure legend), cells were washed 3 times in warm PBS and fixed for 45 min at room temperature in 4% (w/v) formaldehyde (Thermo Scientific; 28908) in PBS containing 0.1% Triton-X100. Fixing solution was removed and cells were washed 3 times in cold PBS. Staining with Hoechst (2 μg/ml, Invitrogen, cat. nr. H3570) and Phalloidin ATTO-647N (30.6 μg/ml, Sigma, cat. nr. 65906) were performed in PBS for 1 h at room temperature. After staining, cells were washed 3 times in cold PBS then stored at 4°C in the dark prior to imaging.
For monitoring cholesterol trafficking, HeLa cells that were seeded one day prior at a density of 7,000 cells per well in a 96-well plate were infected as described above. 12 h post infection, cells were washed twice in PBS and fixed in 4% (v/w) formaldehyde in PBS. After two washes in PBS, cells were stained with filipin (10 μg/ml in PBS, Sigma, cat. nr. F4767–1MG) for 30 min, and subsequently with HCS CellMask™ Deep Red Stain (Thermo, cat. nr. H32721) as described by the distributor for another 30 min. Cells were blocked for 1 hr at room temperature in PBS containing 3% Bovine Serum Albumin (Gerbu, cat. nr. 1062,0250 and 1062,9005), and subsequently incubated for 1h at room temperature with Alexa-488-coupled anti-LAMP1 antibody (1:500 in PBS containing 1% BSA, Abcam). Cells were washed twice in PBS and stored at 4°C in the dark.
To stain for NPC1, HeLa cells were infected and fixed as described above. For blocking and permeabilization, cells were incubated in permeabilization buffer (PBS containing 10% goat serum and 0.2% saponin) for 20 minutes at room temperature. Subsequently, cells were incubated in primary antibody (1:500 in permeabilization buffer, Novus biologicals NB400–148) for 45 minutes at room temperature, washed three times in PBS and incubated in secondary antibody (goat anti-rabbit, Alexa 680, 1:1000 in permeabilization buffer, abcam ab175773). LAMP-1 or phalloidin co-staining was performed as described above. Confocal imaging of NPC1 was performed on an Olympus confocal laser scanning microscope (FV3000) using a ×60 oil-immersion objective. Single image planes were captured using the 405 nm laser for excitation of Hoechst 33342, 488 nm for Alexa 488 (LAMP1), 594 nm for mCherry (STm) and 640 nm for Alexa-680 (NPC1).
Widefield imaging was performed on a Nikon Eclipse Ti run with the NIS Elements software (version 4.60) at 10x or 20x magnification. Per well, 9 images (for 10x objective) or 16 images (for 20x objective) were taken at predefined, evenly spaced positions using the following filters: DAPI, FITC, Cy3, Cy5. Images were segmented using the Cell Profiler software (version 3.0.0). For segmentation, a nuclei mask was defined based on the DAPI channel. The identified objects were used to determine cell outlines from phalloidin staining (Cy5 channel). Finally, STm were identified using a fixed threshold from the Cy3- or GFP/FITC-image and filtered with the cell mask. All primary and secondary objects were quantified and further analyzed according to the phenotypic readout (e.g. infection rate, bacterial load) using integrated STm intensity in Cell Profiler (version 3.0.0).
Quantification of co-localization was performed in ImageJ (version 1.51n). A cell mask was created by applying Ostu segmentation to the cell outline image after rolling background subtraction with a radius of 10 pixels (for phalloidin) or Huang segmentation after a rolling background subtraction with a 15-pixel radius (for cytostain). Similarly, a Salmonella mask was created by applying Otsu segmentation after rolling background subtraction with a 10-pixel radius to the Cy3 channel and overlaying it with a LAMP1 mask obtained through Ostu segmentation after a rolling background subtraction with a 10 pixel radius, where applicable. In order to quantify the degree of co-localization, the average intensity of phalloidin, filipin or NPC1-signal within the Salmonella mask was divided by the average intensity within the cell mask by applying the masks to the phalloidin, filipin or NPC1 images, calculating the integrated intensity and normalizing to the size of the cell or Salmonella mask. Random distribution and no co-localization hence yields a mean value of 1, while co-localization of Salmonella and phalloidin or filipin yields a value >1.
Immunofluorescence of PipB and PDZD8
HeLa cells were seeded onto acid-washed coverslips in 24-well plates. Cells were fixed and permeabilized as described previously (Lau et al., 2019). Localization of endogenous PDZD8 was assessed in resting cells seeded on acid-washed coverslips as described above and stained with mouse monoclonal anti-PDI (Stressgen, Cat. No. SPA-891), rabbit anti-PDZD8 (Sigma HPA015716, 1:100 dilution), mouse anti-TOM20 (Santa Cruz clone F-10, 1:100) and rat anti-BAP31 clone CC-1 (Invitrogen Cat. No. MA3–002). Alternatively, HeLa cells were transfected with EGFP-PipB or EGFP-PipB (Δ270–291) for 24 h. Monolayers were incubated with primary antibodies - rabbit polyclonal anti-PDZD8 (HPA015716, Sigma; 1:100 dilution) or mouse anti-PDI (clone RL90, Affinity Bioreagents; 1:200 dilution) – followed by Alexa Fluor-conjugated secondary antibodies. Cells were mounted on glass slides in Mowiol. For PDZD8 detection in infected cells, HeLa cells were transfected with pKozak-PDZD8-myc and infected 18 h later with invasive STmSL1344 ΔpipB pPipB-2HA or ΔpipB pPipB(Δ270–291)-2HA bacteria (in some cases constitutively expressing mCherry from the chromosome) at an MOI of 50 for 10 min. Invasive bacteria were prepared and infection conditions were as described previously (Klein et al., 2017). Monolayers were fixed at 12 hpi, permeabilized and immunostained with the following primary antibodies - rat monoclonal anti-HA (clone 3F10, Roche: 1:250 dilution), mouse monoclonal anti-myc (clone 9B11, Cell Signaling; 1:2000 dilution), rabbit polyclonal anti-LAMP2 (kindly provided by Minoru Fukuda (Fukuda et al., 1988); 1:1000 dilution) or rabbit polyclonal LAMP-1 (Cat. No. L1418, Sigma, 1:200 dilution) – followed by Alexa Fluor-conjugated secondary antibodies. Image acquisition was on a Zeiss LSM510 or LSM710 confocal microscope using sequential acquisition mode through an optical section of 0.25 μm in the z-axis. Images are maximum intensity projections of z-stacks. Quantification of PDZD8 recruitment to the SCV was obtained from three independent experiments, in each of which around 100 cells were analyzed per condition.
Yeast two-hybrid analysis
The AH109 yeast reporter strain was maintained on YPD agar plates. Transformation of AH109 cells with pGAD424- and pGBT9-based constructs by the lithium acetate method was performed following the guidelines in the Matchmaker two-hybrid system (Clontech). Double transformants were isolated on synthetic defined medium lacking leucine and tryptophan. Interaction of fusion proteins was monitored by activation of HIS3 gene transcription following plating on medium lacking histidine, leucine and tryptophan (Mattera et al., 2003).
Protein purification and size exclusion chromatography
Recombinant SteC and a catalytic inactive mutant of SteC (SteC-K256H) were expressed and purified as previously described (Poh et al., 2008). Purified SteC and SteC-K256H were then dialyzed in 20 mM Tris-HCl pH 7.4, 200 mM NaCl overnight at 4°C. Samples were concentrated in a 15 ml Amicon centrifuge column (Ultra 15, 3,000 NMWL cutoff - UCF900324), glycerol was then added to 10% final concentration and samples were snap-frozen and stored at −80°C. An N-terminal recombinant GST fusion of human FMNL1 (1–385) was expressed from pGEX-4T1-tev-FMNL1-A1(1–385) in Rosetta(DE3) plysRare as follows. Briefly, GST fusion proteins were expressed overnight at 250 rpm at 25°C in autoinduction media (Studier, 2005). Cells were harvested and lysed by sonication in lysis buffer (500 mM NaCl, 50 mM Tris/HCl (pH 7.8), 20% glycerol, 100 μg/ml lysozyme and 1x cOmplete mini EDTA-free protease inhibitors). GST-fusions were bound to pre-equilibrated Glutathione Sepharose 4B (GE; 17–0756-01) overnight at 4°C. Beads were then washed thrice with 100 mM NaCl, 50 mM Tris/HCl (pH 7.8), 10% glycerol. GST bound protein was then cleaved using biotinylated thrombin (Merck Millipore; 69672) according to the manufacturer’s instructions overnight at 4°C. Direct interactions between SteC and SteC-K256H with FMNL1 (1–385) were assessed by analytical gel filtration using an Akta FPLC UPC-900 equipped with a Superose 6 Increase 10/300 GL column (Merck). Typically, 500 μg of each protein was loaded onto the column equilibrated with 100 mM NaCl, 50 mM Tris/HCl (pH 7.8), 10% glycerol prior to sample injection. Complex formation was assessed by mixing equimolar amounts of each protein on ice for 5 min at 4°C prior to injection on the column. Optical density was monitored at 280 nm (UV) throughout the experiment. As a reference for molecular mass, a Bio-Rad protein standard (#1511901), covering 1.35 – 670 kDa was used to calculate expected retention times by linear regression. UV traces were combined and visualized in Prism v7.
In vitro kinase assays
Purified recombinant SteC and catalytically inactive SteC-K256H (10μg each) kinases were pre-activated with kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT and 50 μM ATP) for 5 min at 30°C. FMNL1(1–385) and FMNL2 (2–478) were purified as previously described (Kühn et al., 2015). Next, 10 μg of the purified FMNL1 substrate was mixed with Tris-DTT buffer (50 mM Tris-HCl pH 7.5, 2 mM DTT) and added to the pre-activated kinase mix. Radiolabeled [32P]-γ-ATP was added to the mix and incubated for 30 min at 30°C. The reaction was stopped by the addition of 2x Laemmli buffer. Labeled proteins were resolved by SDS-PAGE, transferred to a PVDF membrane and detected by autoradiography. Proteins were visualized by Coomassie staining.
For phosphoproteomics, kinase pre-activation was achieved using 2 μg of SteC and SteC K256H kinases as described above. 8 μg of FMNL1 (1–385) (FMNL1 sample) or 8 μg of FMNL2 (2–478) (FMNL2 sample) or 4 μg of FMNL1 and 4 μg of FMNL2 (FMNL1+FMNL2 sample) were mixed with kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT and 50 μM ATP (first replicate) or 5 mM ATP (second replicate)) and added to the pre-activated kinase. All reactions were incubated at 30°C for 30 min and snap-frozen on dry ice and stored at −80°C. After thawing, HEPES pH 8.5 was added to a final concentration of 100 mM. Reduction/alkylation of cysteine residues was performed by addition of Tris(2-carboxyethyl)phosphine hydrochloride and chloroacetamide (Sigma-Aldrich) at final concentrations of 5 mM and 30 mM, respectively. Trypsin (Sigma-Aldrich) was added at a 1:25 ratio (w/w) and the samples were incubated overnight at room temperature. Samples were then desalted on stage-tips (Rappsilber et al., 2003) prepared in-house and packed with 1 mg of C18 material (ReproSil-Pur 120 C18-AQ 5 μm, Dr Maisch).
LC-MS/MS Phosphoproteomics
Nanoflow LC-MS/MS analysis was performed by coupling an UltiMate 3000 RSLCnano LC system (Thermo Scientific) to a Fusion Orbitrap Lumos mass spectrometer (Thermo Scientific). Dried peptides were resuspended in a loading buffer consisting of 20 mM citric acid (Sigma-Aldrich) and 1% formic acid (Sigma-Aldrich). Peptides were injected, trapped and washed on a precolumn (C18 PepMap 100, 5μm, 300 μm i.d. x 5 mm, 100 Å, Thermo Scientific) for 3 min at a flow rate of 30 μL/min with 100% buffer A (0.1% formic acid in HPLC grade water). Peptides were then transferred into an analytical column (Waters nanoEase HSS C18 T3, 75 μm x 25 cm, 1.8 μm, 100 Å) before separation at a flow rate of 300 nL/min using a 45 min gradient, from 8% to 32% buffer B (0.1% formic acid, 80% acetonitrile, Sigma-Aldrich). Electrospray ionization was performed using a 2.1 kV spray voltage and a transfer capillary temperature of 275°C. The mass spectrometer was operated in data-dependent acquisition mode. Full mass spectra (m/z 300–1500) were acquired in the Orbitrap analyzer at a resolution of 60,000 with an Automated Gain Control (AGC) target value of 4e5 charges and a maximum injection time of 50 ms. The mass spectrometer was operated in Topspeed mode (maximum duty cycle time of 3 s) and precursors were sequentially selected to undergo HCD fragmentation at a normalized collision energy of 30%. The precursor intensity threshold was set to 1e5 and the dynamic exclusion to 8 seconds. MS2 spectra were acquired in the Orbitrap analyzer at a resolution of 30,000 (isolation window of 1.6 Th) with an AGC target value of 1e5 charges and a maximum injection time of 200 ms. Precursors with unassigned charge state as well as charge states of 1+ and ≥ 6+ were excluded from fragmentation.
MaxQuant software (version 1.6.2.3 (Cox and Mann, 2008)) was used to process the raw data files, which were searched against a database consisting of FMNL1, FMNL2 and SteC proteins as well as commonly observed contaminants. The following parameters were used for the database search: trypsin digestion with a maximum of 3 missed cleavages, fixed carbamidomethylation of cysteine residues, variable oxidation of methionine residues as well as variable phosphorylation of serine/threonine/tyrosine residues and variable N-terminal acetylation. Mass tolerance was set to 4.5 ppm at the MS1 level and 20 ppm at the MS2 level. False discovery rate was set to 1%, the minimum peptide length to 7 residues, a score cut-off of 40 was used for modified peptides, and the match between runs option was used with a retention match time window of 2 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical testing was applied throughout this study to compare experimental conditions and determine significant deviations. In each instance, the statistical test that was used, as well as the program or package that was applied are indicated. In brief, for assessing the significance of enrichments in AP/QMS, the limma (Ritchie et al., 2015) and fdrtool (Strimmer, 2008) packages were implemented using the R programming language, as described in further details in the corresponding STAR method section. All other statistical tests to determine significance were selected according to the appropriate assumptions (normality of the distributions, similarity of the variance, type of question and null-hypothesis) and were conducted in GraphPad Prism v7. The specification of the test used, as well as the metrics and ranges displayed in the plots are indicated in the figure legend or within the text.
Supplementary Material
Table S1. STm tagged effector library; list of effectors and performance. Related to STAR Methods and Figure 1.
Table S3. Summary of previously published PPIs during STm infection recovered by AP-QMS and STRING DB interactions used for network construction. Related to Figures 2 and 3.
Table S4. GO-terms and GO clusters identified in RAW264.7 and HeLa cells. Related to Figures 2, 3 and S2.
Table S5. PPIs chosen for validation with reciprocal pulldown. Related to Figure 4.
Table S6. Identified phosphosites in in vitro kinase assay of SteC with FMNL1 and FMNL2. Related to Figure 7C.
Table S7. Complete list of oligonucleotides used for this study. Related to STAR methods and Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-FMNL1 (produced in rabbit) | LS Bio | Cat. Nr. LS-C401690; RRID:AB_11210956 |
| anti-ANXA1 (produced in rabbit) | proteintech | Cat. Nr. 21990–1-AP; RRID:AB_11182596 |
| anti-MAP1S (produced in mouse) | Precision antibodies | Cat. Nr. AG10006 |
| anti-TCP1 (produced in rat) | Thermo Fisher | Cat. Nr. MA3–026; RRID:AB_2303275 |
| anti-LASP1 (produced in rabbit) | proteintech | Cat. Nr. 10515–1-AP; RRID:AB_2280966 |
| anti-PHB1 (produced in rabbit) | Cell Signalling | Cat. Nr. #2426; RRID:AB_823689 |
| anti-SACM1L (produced in rabbit) | proteintech | Cat. Nr. 13033–1-AP; RRID:AB_2301284 |
| anti-BCLAF1 (produced in rabbit) | Millipore | Cat. Nr. AB10546; RRID:AB_10806089 |
| anti-VPS39 (produced in rabbit) | proteintech | Cat. Nr. 16219–1-AP; RRID:AB_11043179 |
| anti-GroEL (produced in rabbit) | kind donation from Bukau lab | N/A |
| anti-NPC1 (produced in rabbit) | Thermo Fisher | Cat. Nr. PA1–16817; RRID:AB_2298492 |
| anti-FMNL2 (produced in mouse) | abcam | Cat. Nr. ab57963; RRID:AB_941625 |
| anti-Histone H3 (D1H2) (produced in rabbit) | Cell Signalling | Cat. Nr. #4499S; RRID:AB_10544537 |
| anti-RecA (produced in rabbit) | abcam | Cat. Nr. ab63797; RRID:AB_1142554 |
| anti-GAPDH (D16H11) (HRP-conjugate, produced in rabbit) | Cell Signalling | Cat. Nr. #8884; RRID:AB_11129865 |
| anti-FLAG M2 (produced in mouse) | Sigma | Cat. Nr. F1804; RRID:AB_262044 |
| anti-RNA polymerase Sigma 70 (RpoD) [2G10] | Acris antibodies | Cat. Nr. GTX12088; RRID:AB_384272 |
| anti-LAMP1 (produced in rat, Alexa-488 conjugate) | abcam | Cat. Nr. ab24871; RRID:AB_2134490 |
| anti-LAMP1 (polyclonal, produced in rabbit) | Sigma | Cat. Nr. L1418; RRID:AB_477157 |
| anti-LAMP2 (polyclonal, produced in rabbit) | courtesy of M. Fukuda | N/A |
| anti-NPC1 (polyclonal, produced in rabbit) | Novus biologicals | Cat. Nr. NB400–148; RRID:AB_10001101 |
| anti-PDZD8 (amino acid residues 1101–1118), rabbit affinity purified | New England Peptide (custom) | N/A |
| anti-PDZD8 (polyclonal, produced in rabbit) | Sigma | Cat. Nr. HPA015716; RRID:AB_1855162 |
| anti-PDZD8 (polyclonal, produced in rabbit) | Sigma | Cat. Nr. HPA051610; RRID:AB_2681549 |
| anti-BAP31 (monoclonal, produced in mouse) | Invitrogen | Cat. Nr. MA3–002; RRID:AB_325095 |
| anti-PDI (produced in mouse) | Affinity Bioreagents | Cat. Nr. clone RL90 |
| anti-PDI (monoclonal, produced in mouse) | Stressgen | Cat. Nr. SPA-891; RRID:AB_2039450 |
| anti-TOM20 (monoclonal, clone F-10, produced in mouse) | Santa Cruz | Cat. Nr. sc-17764; RRID:AB_628381 |
| anti-Hsp27 (monoclonal, clone G31, produced in mouse) | Cell Signaling | Cat. Nr. #2402; RRID:AB_331761 |
| anti-calnexin (polyclonal, produced in rabbit) | Enzo | Cat. Nr. ADI-SPA-860; RRID:AB_10616095 |
| anti-lamin A (monoclonal, produced in mouse) | Cell Signaling | Cat. Nr. 133A2; RRID:AB_2800093 |
| secondary anti-rabbit (Alexa-680 coupled, produced in goat) | abcam | Cat. Nr. ab175773 |
| secondary anti-rat (HRP-conjugate, produced in rabbit) | abcam | Cat. Nr. ab6734; RRID:AB_955450 |
| secondary anti-mouse (HRP-conjugate, produced in goat) | Sigma | Cat. Nr. HVZ-A4416–1ML |
| secondary anti-rabbit (HRP-conjugate, produced in donkey) | Sigma/GE | Cat. Nr. NA934–1ML; RRID:AB_772206 |
| anti-rabbit IgG horseradish peroxidase | Cell Signalling | Cat. Nr. #7071; RRID:AB_2099234 |
| anti-GFP (monoclonal, clone 3E6, produced in mouse) | Molecular Probes | Cat. Nr. mAb 3E6; RRID:AB_2313858 |
| anti-GFP (polyclonal, produced in rabbit) | Molecular Probes | Cat. Nr. A-11122; RRID:AB_221569 |
| rat monoclonal anti-HA high affinity, clone 3F10 | Roche | Cat. Nr. #3F10; RRID:AB_2314622 |
| mouse monoclonal anti--HA.11 | BioLegend | N/A |
| mouse monoclonal anti-myc, clone 9B11 | Cell Signaling | Cat. Nr. #2040; RRID:AB_2148465 |
| Bacterial and virus strains | ||
| TOP10 F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(StrR) endA1 λ | Invitrogen | N/A |
| Rosetta(DE3) plysRare | Novagen | N/A |
| wild type Salmonella enterica serovar Typhimurium 14028s | Porwollik et al., 2014 | N/A |
| STm 14028S gogB:STF-kan | this study | N/A |
| STm 14028S sseK1:STF-kan | this study | N/A |
| STm 14028S sseJ:STF-kan | this study | N/A |
| STm 14028S ΔccdAB::Cm, sifA:STF | this study | N/A |
| STm 14028S pipB:STF-kan | this study | N/A |
| STm 14028S pipB2:STF-kan | this study | N/A |
| STm 14028S sifB:STF-kan | this study | N/A |
| STm 14028S slrP:STF-kan | this study | N/A |
| STm 14028S sopD2:STF-kan | this study | N/A |
| STm 14028S spvB:STF-kan | this study | N/A |
| STm 14028S spvC:STF-kan | this study | N/A |
| STm 14028S srgE:STF-kan | this study | N/A |
| STm 14028S sseF:STF-kan | this study | N/A |
| STm 14028S sseG:STF-kan | this study | N/A |
| STm 14028S sseI:STF-kan | this study | N/A |
| STm 14028S sipA:STF-kan | this study | N/A |
| STm 14028S sopB:STF-kan | this study | N/A |
| STm 14028S sseK2:STF-kan | this study | N/A |
| STm 14028S sseL:STF-kan | this study | N/A |
| STm 14028S sspH1:STF-kan | this study | N/A |
| STm 14028S sspH2:STF-kan | this study | N/A |
| STm 14028S steA:STF-kan | this study | N/A |
| STm 14028S steB:STF-kan | this study | N/A |
| STm 14028S steC:STF-kan | this study | N/A |
| STm 14028S sptP:STF-kan | this study | N/A |
| STm 14028S steE:STF-kan | this study | N/A |
| STm 14028S cigR:STF-kan | this study | N/A |
| STm 14028S avrA:STF-kan | this study | N/A |
| STm 14028S sipB:STF-kan | this study | N/A |
| STm 14028S sipC:STF-kan | this study | N/A |
| STm 14028S sopA:STF-kan | this study | N/A |
| STm 14028S steD:STF-kan | this study | N/A |
| STm 14028S ΔsseJ:: | this study | N/A |
| STm 14028S ΔsseL:: | this study | N/A |
| STm 14028S ΔsteC:: | this study | N/A |
| STm 14028S ΔsseJ::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S ΔsseL::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S ΔsteC::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S ΔsseJ::ΔsseL:: | this study | N/A |
| STm 14028S ΔssaV::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S ΔprgK::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S ΔsifA::kan, retransduced into wildtype using P22 | Porwollik et al., 2014 | N/A |
| STm 14028S, pFCcGi | this study | N/A |
| STm 14028S ΔsseJ::, pFCcGi | this study | N/A |
| STm 14028S ΔsseL::, pFCcGi | this study | N/A |
| STm 14028S ΔsteC::, pFCcGi | this study | N/A |
| STm 14028S ΔsseJ::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsseJ::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsseL::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsseL::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsteC::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsteC::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsseJ::ΔsseL::, pFCcGi | this study | N/A |
| STm 14028S ΔssaV::kan, pFCcGi | this study | N/A |
| STm 14028S ΔprgK::kan, pFCcGi | this study | N/A |
| STm 14028S ΔsifA::kan, pFCcGi | this study | N/A |
| SL1344 wild type | Hoiseth and Stocker 1981 | N/A |
| STm SL1344 ΔpipB | Knodler et al., 2002 | N/A |
| STm SL1344 ΔpipB pPipB-2HA | Knodler et al., 2002 | N/A |
| STm SL1344 ΔpipB glmS::Ptrc-mCherryST::FRT | this study | N/A |
| STm SL1344 ΔpipB glmS::Ptrc-mCherryST::FRT pPipB-2HA | this study | N/A |
| STm SL1344 ΔpipB glmS::Ptrc-mCherryST::FRT pPipB(Δ270–291)-2HA | this study | N/A |
| STm SL1344 ΔpipB2 | Knodler et al., 2003 | N/A |
| STm SL1344 ΔpipB2 pPipB2–2HA | Knodler et al., 2003 | N/A |
| Deposited data | ||
| Additional and original data for Western Blots, limma analyses, all plots and other MS runs | this study | Mendeley Data DOI: 10.17632/xjb24h7s8j.1 |
| All original proteomic data | this study | PRIDE repository: PXD018375 |
| STRING DB v11 | Szklarczyk et al., 2019 | https://string-db.org/ |
| UniProt (UP000005640, UP000000589, UP000002695) | The UniProt Consortium, 2021 | https://www.uniprot.org/ |
| Experimental models: cell lines | ||
| HeLa (Wildtype) | ATCC | ATCC CCL-2; RRID:CVCL_0030 |
| HeLa (NPC1-K.O.) | provided by Dr. Willem Annaert (VIB, KU-Leuven) | N/A |
| RAW264.7 (Wildtype) | ATCC | ATCC TIB-71; RRID:CVCL_0493 |
| 3T3 (Wildtype) | provided by Prof. Dr. Klemens Rottner | N/A |
| 3T3 (FMNL2/3-K.O.) | provided by Prof. Dr. Klemens Rottner | N/A |
| pBMDM (Wildtype mice) | bone marrow provided by Prof. Dr. Wolf-Dietrich Hardt (ETH) | N/A |
| Oligonucleotides | ||
| See Table S7 for a full list of oligonucleotides used in this study. Further information on all oligos is provided on Mendeley Data (DOI: 10.17632/xjb24h7s8j.1) | ||
| Recombinant DNA | ||
| pFCcGi: Constitutive expression of mCherry(FC), AmpR | Figueira et al., 2013 | RRID:Addgene_59324 |
| pGEM-T Easy: AmpR, T/A cloning vector | Invitrogen | Cat. Nr. PR-A1360 |
| pQE30: AmpR | Qiagen | N/A |
| pMZ2: plasmid containing 2x Myc-TEV-FLAG tag and KanR gene from pKD4 cloned into the EcoRI/HindIII site of pQE-30, AmpR | this work | N/A |
| pJSP1: pGEM-Teasy backbone containing 2x STREP-TEV-3xFLAG tag and KanR gene from pKD4, AmpR | this work | N/A |
| pKD45: plasmid carrying neo-ccdB cassette, R6K ori | Datsenko & Wanner 2000 | N/A |
| pKD3: plasmid carrying FRT-cat-FRT cassette, CmR, AmpR | Datsenko & Wanner 2000 | RRID:Addgene_45604 |
| pKD4: plasmid carrying FRT-neo-FRT cassette, KanR, AmpR | Datsenko & Wanner 2000 | RRID:Addgene_45605 |
| pKD46: λ Red recombinase expression vector, AmpR | Datsenko & Wanner 2000 | N/A |
| pCP20: Plasmid expressing FLP recombinase, AmpRs, ts rep | Datsenko & Wanner 2000 | N/A |
| pACYC184: CmR, TetR | New England Biolabs | GenBank Accession #: X06403 |
| pPipB-2HA: plasmid expressing c-terminal HA tagged PipB | Knodler et al. 2003 | N/A |
| pPipB2–2HA: plasmid expressing c-terminal HA tagged PipB2 | Knodler et al. 2003 | N/A |
| EGFP-PipB: expression of GFP tagged PipB | this work | N/A |
| EGFP-PipB(Δ270–291): expression of GFP tagged PipB(Δ270–291) | this work | N/A |
| EGFP-PipB2: expression of GFP tagged PipB2 | Knodler and Steele-Mortimer, 2005 | N/A |
| pKozak-PDZD8-myc: ectopic expression of PDZD8 expressing a c-terminal myc tag | this work | N/A |
| pCMV-Tag 5A: mammalina expression vector, KanR | Stratagene | N/A |
| pGAD424: generation of gene fusions for Y2H | Clontech | N/A |
| pGAD424-PipB: plasmid expressing PipB for Y2H | this work | N/A |
| pGAD424-PipB(1–281): plasmid expressing PipB(1–281) for Y2H | this work | N/A |
| pGAD424-PipB(1–271): plasmid expressing PipB(1–271) for Y2H | this work | N/A |
| pGAD424-PipB(1–188): plasmid expressing PipB(1–188) for Y2H | this work | N/A |
| pGAD424-PipB(189–291): plasmid expressing PipB(189–291) for Y2H | this work | N/A |
| pGBT9: generation of gene fusions for Y2H | Clontech | N/A |
| pGBT9-PDZD8: plasmid expressing PDZD8 for Y2H | this work | N/A |
| pGBT9-PDZD8(Δ930–1154): plasmid expressing PDZD8(Δ930–1154) for Y2H | this work | N/A |
| pGBT9-PDZD8(Δ1–338): plasmid expressing PDZD8(Δ1–338) for Y2H | this work | N/A |
| pGBT9-PDZD8ΔPDZ: plasmid expressing PDZD8ΔPDZ for Y2H | this work | N/A |
| pGBT9-PDZD8(Δ494–814): plasmid expressing PDZD8(Δ494–814) for Y2H | this work | N/A |
| pGBT9-PDZD8ΔC1: plasmid expressing PDZD8ΔC1 for Y2H | this work | N/A |
| Software and algorithms | ||
| Inkscape v1.0.2 | Free Software Foundation, Inc. | https://inkscape.org |
| GraphPad Prism v7 | GraphPad Software, San Diego, California USA | https://www.graphpad.com/scientific-software/prism/ |
| Cell Profiler v3.0.0 | McQuin et al. 2018 | https://cellprofiler.org/ |
| cytoscape v3.7.2 | Shannon et al. 2003 | https://cytoscape.org/ |
| clueGO plug-in in cytoscape (v2.5.2) | Bindea et al. 2009 | httphttp://apps.cytoscape.org/apps/cluego |
| ImageJ v1.51n | Schneider et al. 2012 | https://imagej.net/Welcome |
| MaxQuant v1.6.2.3 | Cox and Mann, 2008 | https://www.maxquant.org/ |
| IsobarQuant | Franken et al., 2015 | https://github.com/protcode/isob |
| Mascot v2.2.07 | Perkins et al. 1999 | https://www.matrixscience.com/ |
| R programming language (ISBN 3–900051-07–0) | R Core team, 2017 | https://www.r-project.org/ |
Highlights.
A method to map Salmonella effector-host protein-protein interactions during infection
Salmonella effectors are multifunctional, most targeting more than one host process
Effectors can converge on same host process and act in cell-type specific manner
SteC promotes actin bundling by phosphorylating formin-like proteins
Acknowledgements
We thank W. Annaert (VIB, KU-Leuven) for the NPC1 knockout, B. Bukau (ZMBH, Heidelberg) for the GroEL antibody, W. D. Hardt (ETH, Zürich) for rodent bone marrow, F. Kage for advice on FMNL2/3 knockout cells, and S. Kaspar (EMBL) for statistics consultation. JS and CP were partially supported by fellowships from the EMBL Interdisciplinary Postdoc (EIPOD) program; Marie Skłodowska-Curie Actions COFUND (grant number 291772); MG by a grant from the DFG (GE 976/10–1); OSM by the Intramural Research Program of the NIAID (ZIA AI000909); KR by intramural funds from the Helmholtz Society. This work was funded by EMBL core funding.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andritschke D, Dilling S, Emmenlauer M, Welz T, Schmich F, Misselwitz B, Rämö P, Rottner K, Kerkhoff E, Wada T, et al. (2016). A Genome-Wide siRNA Screen Implicates Spire1/2 in SipA-Driven Salmonella Typhimurium Host Cell Invasion. PLoS One 11, e0161965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena ET, Auweter SD, Antunes LCM, Vogl AW, Han J, Guttman JA, Croxen MA, Menendez A, Covey SD, Borchers CH, et al. (2011). The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect. Immun 79, 4392–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Bhavsar AP, de Hoog CL, Li Y, Chan YA, van der Heijden J, Lowden MJ, Coombes BK, Rogers LD, Stoynov N, et al. (2011). Quantitative mass spectrometry catalogues Salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J. Biol. Chem 286, 24023–24035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Yu HB, Arena ET, Guttman JA, and Finlay BB (2012). Oxysterol-binding protein (OSBP) enhances replication of intracellular Salmonella and binds the Salmonella SPI-2 effector SseL via its N-terminus. Microbes Infect. 14, 148–154. [DOI] [PubMed] [Google Scholar]
- Bai SW, Herrera-Abreu MT, Rohn JL, Racine V, Tajadura V, Suryavanshi N, Bechtel S, Wiemann S, Baum B, and Ridley AJ (2011). Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, and Galon J. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasche S, Arens S, Ceol A, Siszler G, Schmidt MA, Häuser R, Schwarz F, Wuchty S, Aloy P, Uetz P, et al. (2014). The EHEC-host interactome reveals novel targets for the translocated intimin receptor. Sci. Rep 4, 7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block J, Breitsprecher D, Kühn S, Winterhoff M, Kage F, Geffers R, Duwe P, Rohn JL, Baum B, Brakebusch C, et al. (2012). FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr. Biol 22, 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell JH, Goosney DL, and Finlay BB (2002). SifA, a type III secreted effector of Salmonella typhimurium, directs Salmonella-induced filament (Sif) formation along microtubules. Traffic 3, 407–415. [DOI] [PubMed] [Google Scholar]
- Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, et al. (2007). Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. U. S. A 104, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanheira S, and García-Del Portillo F. (2017). Salmonella Populations inside Host Cells. Front. Cell. Infect. Microbiol 7, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Upton H, Bao K, Schulze-Gahmen U, Samelson AJ, He N, Nowak A, Lu H, Krogan NJ, Zhou Q, et al. (2013). HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc. Natl. Acad. Sci. U. S. A 110, E123–E131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cunha LD, and Zamboni DS (2013). Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front. Cell. Infect. Microbiol 3, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa VM, Coyaud E, Boddy KC, Laurent EMN, St-Germain J, Li T, Grinstein S, Raught B, and Brumell JH (2019). BioID screen of Salmonella type 3 secreted effectors reveals host factors involved in vacuole positioning and stability during infection. Nat Microbiol. [DOI] [PubMed] [Google Scholar]
- Diacovich L, Dumont A, Lafitte D, Soprano E, Guilhon A-A, Bignon C, Gorvel J-P, Bourne Y, and Méresse S. (2009). Interaction between the SifA virulence factor and its host target SKIP is essential for Salmonella pathogenesis. J. Biol. Chem 284, 33151–33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drecktrah D, Levine-Wilkinson S, Dam T, Winfree S, Knodler LA, Schroer TA, and Steele-Mortimer O. (2008). Dynamic behavior of Salmonella-induced membrane tubules in epithelial cells. Traffic 9, 2117–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, and Galán JE (2009). Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog. 5, e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Guo Y, Segev N, Harel M, Quinnell DE, Geiger T, Avinoam O, Li D, and Nunnari J. (2020). PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat. Commun 11, 3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken H, Mathieson T, Childs D, Sweetman GMA, Werner T, Tögel I, Doce C, Gade S, Bantscheff M, Drewes G, et al. (2015). Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc 10, 1567–1593. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Viitala J, Matteson J, and Carlsson SR (1988). Cloning of cDNAs encoding human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Comparison of their deduced amino acid sequences. J. Biol. Chem 263, 18920–18928. [PubMed] [Google Scholar]
- Gatto L, and Lilley KS (2012). MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28, 288–289. [DOI] [PubMed] [Google Scholar]
- Ghosh S, and O’Connor TJ (2017). Beyond Paralogs: The Multiple Layers of Redundancy in Bacterial Pathogenesis. Front. Cell. Infect. Microbiol 7, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-López MA, Velázquez-Guadarrama N, Romero-Espejel ME, and Olivares-Trejo J. de J. (2013). Helicobacter pylori secretes the chaperonin GroEL (HSP60), which binds iron. FEBS Lett. 587, 1823–1828. [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén-Samander A, Bian X, and De Camilli P. (2019). PDZD8 mediates a Rab7-dependent interaction of the ER with late endosomes and lysosomes. Proc. Natl. Acad. Sci. U. S. A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon MA, Ribet D, Stavru F, and Cossart P. (2012). Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 20, 360–368. [DOI] [PubMed] [Google Scholar]
- Heimsath EG Jr, and Higgs HN (2012). The C terminus of formin FMNL3 accelerates actin polymerization and contains a WH2 domain-like sequence that binds both monomers and filament barbed ends. J. Biol. Chem 287, 3087–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Couillault C, Rockenfeller P, Boucrot E, Dumont A, Schroeder N, Hermant A, Knodler LA, Lecine P, Steele-Mortimer O, et al. (2006). The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. U. S. A 103, 13497–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiseth SK, and Stocker BA (1981). Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, and Pease LR (1989). Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Huber W, von Heydebreck A, Sültmann H, Poustka A, and Vingron M. (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96–S104. [DOI] [PubMed] [Google Scholar]
- Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, and Krijgsveld J. (2019). Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 14, 68–85. [DOI] [PubMed] [Google Scholar]
- Imami K, Bhavsar AP, Yu H, Brown NF, Rogers LD, Finlay BB, and Foster LJ (2013). Global impact of Salmonella pathogenicity island 2-secreted effectors on the host phosphoproteome. Mol. Cell. Proteomics 12, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LK, Nawabi P, Hentea C, Roark EA, and Haldar K. (2008). The Salmonella virulence protein SifA is a G protein antagonist. Proc. Natl. Acad. Sci. U. S. A 105, 14141–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. (2011a). Global landscape of HIV-human protein complexes. Nature 481, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Gulbahce N, Cimermancic P, Kane J, He N, Chou S, D’Orso I, Fernandes J, Jang G, Frankel AD, et al. (2011b). Purification and characterization of HIV-human protein complexes. Methods 53, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al. (2011c). Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings E, Thurston TLM, and Holden DW (2017). Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms And Physiological Consequences. Cell Host Microbe 22, 217–231. [DOI] [PubMed] [Google Scholar]
- Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, and Neish AS (2008). Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 3, 233–244. [DOI] [PubMed] [Google Scholar]
- Kage F, Winterhoff M, Dimchev V, Mueller J, Thalheim T, Freise A, Brühmann S, Kollasser J, Block J, Dimchev G, et al. (2017a). FMNL formins boost lamellipodial force generation. Nat. Commun 8, 14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage F, Steffen A, Ellinger A, Ranftler C, Gehre C, Brakebusch C, Pavelka M, Stradal T, and Rottner K. (2017b). FMNL2 and −3 regulate Golgi architecture and anterograde transport downstream of Cdc42. Sci. Rep 7, 9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Powers TR, and Knodler LA (2017). Measurement of Salmonella enterica Internalization and Vacuole Lysis in Epithelial Cells. Methods Mol. Biol 1519, 285–296. [DOI] [PubMed] [Google Scholar]
- Knodler LA, and Steele-Mortimer O. (2005). The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell 16, 4108–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler LA, Celli J, Hardt W-D, Vallance BA, Yip C, and Finlay BB (2002). Salmonella effectors within a single pathogenicity island are differentially expressed and translocated by separate type III secretion systems. Mol. Microbiol 43, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Vallance BA, Hensel M, Jäckel D, Finlay BB, and Steele-Mortimer O. (2003). Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol. Microbiol 49, 685–704. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Winfree S, Drecktrah D, Ireland R, and Steele-Mortimer O. (2009). Ubiquitination of the bacterial inositol phosphatase, SopB, regulates its biological activity at the plasma membrane. Cell. Microbiol 11, 1652–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, and Steele-Mortimer O. (2010). Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. U. S. A 107, 17733–17738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmsee T, and Hengge R. (2011). Rare codons play a positive role in the expression of the stationary phase sigma factor RpoS (σ(S)) in Escherichia coli. RNA Biol. 8, 913–921. [DOI] [PubMed] [Google Scholar]
- Kolodziejek AM, Altura MA, Fan J, Petersen EM, Cook M, Brzovic PS, and Miller SI (2019). Salmonella Translocated Effectors Recruit OSBP1 to the Phagosome to Promote Vacuolar Membrane Integrity. Cell Rep. 27, 2147–2156.e5. [DOI] [PubMed] [Google Scholar]
- Kühn S, and Geyer M. (2014). Formins as effector proteins of Rho GTPases. Small GTPases 5, e29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn S, Erdmann C, Kage F, Block J, Schwenkmezger L, Steffen A, Rottner K, and Geyer M. (2015). The structure of FMNL2–Cdc42 yields insights into the mechanism of lamellipodia and filopodia formation. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- LaRock DL, Chaudhary A, and Miller SI (2015). Salmonellae interactions with host processes. Nat. Rev. Microbiol 13, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau N, Haeberle AL, O’Keeffe BJ, Latomanski EA, Celli J, Newton HJ, and Knodler LA (2019). SopF, a phosphoinositide binding effector, promotes the stability of the nascent Salmonella-containing vacuole. PLoS Pathog. 15, e1007959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Arighi CN, Lodge R, Zerial M, and Bonifacino JS (2003). Divalent interaction of the GGAs with the Rabaptin-5-Rabex-5 complex. EMBO J. 22, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, and Wüstner D. (2012). Analysis of cholesterol trafficking with fluorescent probes. Methods Cell Biol. 108, 367–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaig WD, Koller A, and Thanassi DG (2013). Production of outer membrane vesicles and outer membrane tubes by Francisella novicida. J. Bacteriol 195, 1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Richter B, Claudi B, Wigge C, Wild P, Farhan H, McGourty K, Coxon FP, Franz-Wachtel M, Perdu B, et al. (2015). PLEKHM1 regulates Salmonella-containing vacuole biogenesis and infection. Cell Host Microbe 17, 58–71. [DOI] [PubMed] [Google Scholar]
- McShan AC, Anbanandam A, Patnaik S, and De Guzman RN (2016). Characterization of the Binding of Hydroxyindole, Indoleacetic acid, and Morpholinoaniline to the Salmonella Type III Secretion System Proteins SipD and SipB. ChemMedChem 11, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas-Gervais S, and Antonny B. (2017). Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J. 36, 3156–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawabi P, Catron DM, and Haldar K. (2008). Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol 68, 173–185. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI (2012). Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 12, 1639–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendall C, Rolhion N, Förster A, Poh J, Lamont DJ, Liu M, Freemont PS, Catling AD, and Holden DW (2012). The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson MB, Huang Z, Alto NM, Blanc M-P, Dixon JE, Chai J, and Miller SI (2008). Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 4, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick KL, Wojcechowskyj JA, Bell SL, Riba MN, Jing T, Talmage S, Xu P, Cabello AL, Xu J, Shales M, et al. (2018). Quantitative Yeast Genetic Interaction Profiling of Bacterial Effector Proteins Uncovers a Role for the Human Retromer in Salmonella Infection. Cell Syst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn BH, Netter Z, Johnson JR, Von Dollen J, Jang GM, Johnson T, Ohol YM, Maher C, Bell SL, Geiger K, et al. (2018). An Mtb-Human Protein-Protein Interaction Map Identifies a Switch between Host Antiviral and Antibacterial Responses. Mol. Cell 71, 637–648.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Perri JI, Rogell B, Schwarzl T, Stein F, Zhou Y, Rettel M, Brosig A, and Hentze MW (2018). Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun 9, 4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR (2019). NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem 294, 1706–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T, Matrakas D, Taylor YU, Manyam G, Morozov VN, Zhou W, and van Hoek ML (2011). Proteomic characterization and functional analysis of outer membrane vesicles of Francisella novicida suggests possible role in virulence and use as a vaccine. J. Proteome Res 10, 954–967. [DOI] [PubMed] [Google Scholar]
- Pilar AVC, Reid-Yu SA, Cooper CA, Mulder DT, and Coombes BK (2012). GogB is an anti-inflammatory effector that limits tissue damage during Salmonella infection through interaction with human FBXO22 and Skp1. PLoS Pathog. 8, e1002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, and Holden DW (2008). SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell. Microbiol 10, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porwollik S, Santiviago CA, Cheng P, Long F, Desai P, Fredlund J, Srikumar S, Silva CA, Chu W, Chen X, et al. (2014). Defined single-gene and multi-gene deletion mutant collections in Salmonella enterica sv Typhimurium. PLoS One 9, e99820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala SV, Hughes KT, and Uetz P. (2009). Benchmarking yeast two-hybrid systems using the interactions of bacterial motility proteins. Proteomics 9, 5296–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Morales F. (2012). Impact of Salmonella enterica Type III Secretion System Effectors on the Eukaryotic Host Cell. International Scholarly Research Notices 2012. [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M. (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Reichel M, Liao Y, Rettel M, Ragan C, Evers M, Alleaume A-M, Horos R, Hentze MW, Preiss T, and Millar AA (2016). In Planta Determination of the mRNA-Binding Proteome of Arabidopsis Etiolated Seedlings. Plant Cell 28, 2435–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder GN (2017). The Toolbox for Uncovering the Functions of Legionella Dot/Icm Type IVb Secretion System Effectors: Current State and Future Directions. Front. Cell. Infect. Microbiol 7, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkrig J, Li N, Hausmann A, Mangan MSJ, Zietek M, Mateus A, Bobonis J, Sueki A, Imamura H, El Debs B, et al. (2020). Spatiotemporal proteomics uncovers cathepsin-dependent macrophage cell death during Salmonella infection. Nat Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. (2009). A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139, 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag RL, Nakayasu ES, Brown RN, Niemann GS, Sydor MA, Sanchez O, Ansong C, Lu S-Y, Choi H, Valleau D, et al. (2016). Identification of Novel Host Interactors of Effectors Secreted bySalmonellaandCitrobacter. mSystems 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Mortimer O. (2008). Infection of epithelial cells with Salmonella enterica. Methods Mol. Biol 431, 201–211. [DOI] [PubMed] [Google Scholar]
- Strimmer K. (2008). fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462. [DOI] [PubMed] [Google Scholar]
- Studier FW. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Stynen B, Tournu H, Tavernier J, and Van Dijck P. (2012). Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol. Mol. Biol. Rev 76, 331–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Kanemitsu A, Knight CT, and Hatakeyama M. (2020). Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol 17, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng B, Zhao C, Liu X, and He Z. (2015). Network inference from AP-MS data: computational challenges and solutions. Brief. Bioinform 16, 658–674. [DOI] [PubMed] [Google Scholar]
- Tharkeshwar AK, Trekker J, Vermeire W, Pauwels J, Sannerud R, Priestman DA, Te Vruchte D, Vints K, Baatsen P, Decuypere J-P, et al. (2017). A novel approach to analyze lysosomal dysfunctions through subcellular proteomics and lipidomics: the case of NPC1 deficiency. Sci. Rep 7, 41408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Dong Y-A, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, et al. (2006). Herpesviral protein networks and their interaction with the human proteome. Science 311, 239–242. [DOI] [PubMed] [Google Scholar]
- Unsworth KE, Way M, McNiven M, Machesky L, and Holden DW. (2004). Analysis of the mechanisms of Salmonella-induced actin assembly during invasion of host cells and intracellular replication. Cell. Microbiol 6, 1041–1055. [DOI] [PubMed] [Google Scholar]
- Uzzau S, Figueroa-Bossi N, Rubino S, and Bossi L. (2001). Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U. S. A 98, 15264–15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren E, Von Dollen J, Cimermancic P, Gulbahce N, Sali A, and Krogan NJ. (2015). Scoring Large-Scale Affinity Purification Mass Spectrometry Datasets with MiST. Curr. Protoc. Bioinformatics 49, 8.19.1–8.19.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylnka JA, Bakowski MA, Szeto J, Ohlson MB, Trimble WS, Miller SI, and Brumell JH. (2008). Role for myosin II in regulating positioning of Salmonella-containing vacuoles and intracellular replication. Infect. Immun 76, 2722–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M, and Savitski MM. (2014). Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem 86, 3594–3601. [DOI] [PubMed] [Google Scholar]
- Wilhelm LP, Voilquin L, Kobayashi T, Tomasetto C, and Alpy F. (2019). Intracellular and Plasma Membrane Cholesterol Labeling and Quantification Using Filipin and GFP-D4. In Intracellular Lipid Transport: Methods and Protocols, Drin G, ed. (New York, NY: Springer New York; ), pp. 137–152. [DOI] [PubMed] [Google Scholar]
- Yang C-K Ewis HE, Zhang X, Lu C-D, Hu H-J, Pan Y, Abdelal AT, and Tai PC. (2011). Nonclassical protein secretion by Bacillus subtilis in the stationary phase is not due to cell lysis. J. Bacteriol 193, 5607–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayoshi-Yamamoto S, Taniuchi I, and Watanabe T. (2000). FRL, a novel formin-related protein, binds to Rac and regulates cell motility and survival of macrophages. Mol. Cell. Biol 20, 6872–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Moest T, Zhao Y, Guilhon A-A, Buffat C, Gorvel J-P, and Méresse S. (2015). The Salmonella effector protein SifA plays a dual role in virulence. Sci. Rep 5, 12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. STm tagged effector library; list of effectors and performance. Related to STAR Methods and Figure 1.
Table S3. Summary of previously published PPIs during STm infection recovered by AP-QMS and STRING DB interactions used for network construction. Related to Figures 2 and 3.
Table S4. GO-terms and GO clusters identified in RAW264.7 and HeLa cells. Related to Figures 2, 3 and S2.
Table S5. PPIs chosen for validation with reciprocal pulldown. Related to Figure 4.
Table S6. Identified phosphosites in in vitro kinase assay of SteC with FMNL1 and FMNL2. Related to Figure 7C.
Table S7. Complete list of oligonucleotides used for this study. Related to STAR methods and Key Resource Table.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD018375. The code and pipelines used for data analysis are available upon request. Raw data files not included in the main manuscript are located at Mendeley Data (DOI: 10.17632/xjb24h7s8j.1).