

Abstract
The tumor microenvironment (TME) is a highly complex and dynamic ensemble of cells of which a variety of immune cells are a major component. The unparalleled results obtained with immunotherapeutic approaches have underscored the importance of examining the immune landscape of the TME. Recent technological advances have incorporated high-throughput techniques at the single cell level, such as single cell RNA sequencing, mass cytometry, and multi-parametric flow cytometry to the characterization of the TME. Among them, flow cytometry is the most broadly used both in research and clinical settings and multi-color analysis is now routinely performed. The high dimensionality of the data makes the traditional manual gating strategy in 2D scatter plots very difficult. New unbiased visualization techniques provide a solution to this problem. Here we describe the steps to characterize the immune cell compartment in the TME in mouse tumor models by high-parametric flow cytometry, from the experimental setup to the analysis methodology with special emphasis on the use of unsupervised algorithms.
1. Introduction
Immunotherapy has transformed cancer treatment resulting in a paradigm shift: from targeting the tumor to targeting the patient’s immune system. However, despite the unprecedented responses observed, only a subset of patients fully benefits of such approach (Kim & Chen, 2016; Pitt et al., 2016; Sharma, Hu-Lieskovan, Wargo, & Ribas, 2017). A major hurdle for successful anti-cancer therapy is overcoming the immunosuppressive tumor microenvironment (TME) (Hanahan & Coussens, 2012). This underscores the importance of dissecting the immunological landscape of the TME (Binnewies et al., 2018). Recent technological advances applied to the study of the TME have shed light on its complexity and diversity not only among tumor types but also within patients with similar tumor types (Aran et al., 2017; Bindea et al., 2013; Newman et al., 2015).
Both cells from the myeloid and lymphoid lineages can be found in the TME, and studies have shown that defining tumors based on the immune composition of the TME may have predictive value for therapy outcome and disease progression (Herbst et al., 2014; Spranger, 2016). These initial studies have mostly focused on the overall presence of T cells and were based on immuno-histochemistry analysis of tumor sections (Pagès et al., 2018). Due to the highly complex and dynamic nature of the TME, traditional histological and immuno-histochemistry analysis are now being replaced by high-throughput techniques such as multiparametric fluorescence-based flow cytometry (FACS), mass cytometry (CyTOF) and single cell RNA sequencing (scRNAseq). While these high-throughput techniques have increased our capabilities for a deeper understanding of the TME, they have also introduced an overwhelming amount of data, and with it, the need for unbiased high-dimensional data analysis methods.
Given its broad availability in research and diagnostic laboratories all around the world, FACS remains a very popular and versatile phenotyping technique. It allows the study of both surface and intracellular markers at a single cell level. Until not so long ago, flow cytometry data was analyzed in two dimensions by sequential gating performed with pairs of markers and visualized as two-axes plots (scatter, density, pseudocolor plots, etc.) (Bendall, Nolan, Roederer, & Chattopadhyay, 2012). This strategy is very limited when dealing with the rapidly growing number of parameters that can now be measured by FACS. In addition, visualization of pairwise comparisons when working with 20+ parameters is biased by the subjective determination of the gates, and excessively time consuming considering that it leads to 2n number of scatter plots (being n = number of parameters). Most importantly, the segmented observation of the data in a pairwise manner can result in the loss of relevant information, particularly when studying rare populations or subtle changes in fluorescence intensity of certain markers due to a biologically meaningful cause (e.g., cell activation status).
In order to overcome these limitations, several algorithms were developed and adapted for the analysis of high-parametric datasets obtained from CyTOF, scRNAseq and multi-color flow cytometry (Amir et al., 2013; Becht et al., 2019; Chen et al., 2016; Levine et al., 2015; Newell, Sigal, Bendall, Nolan, & Davis, 2012; Van Gassen et al., 2015). These algorithms provide dimensionality reduction visualization methods, which reduce the high-dimensional data (multi-parameter) into a two- or three-dimensions plot, and also unbiased clustering methods that conserve the global information of the dataset (i.e., each parameter and their multivariate relationships). However, several critical steps must be considered for the successful use of these methods to analyze fluorescence-based flow cytometry data, in particular when working with complex cellular sources such as tumor samples.
In this chapter, we provide a detailed description of a method to characterize the immune landscape of the TME of mouse tumor models. This includes a general protocol for tissue processing and important considerations for the optimization of high-parametric flow cytometry panel design, data acquisition, and analysis that can be easily adapted to any tissue of interest. A summary of the steps described are shown in Fig. 1.
Fig. 1.
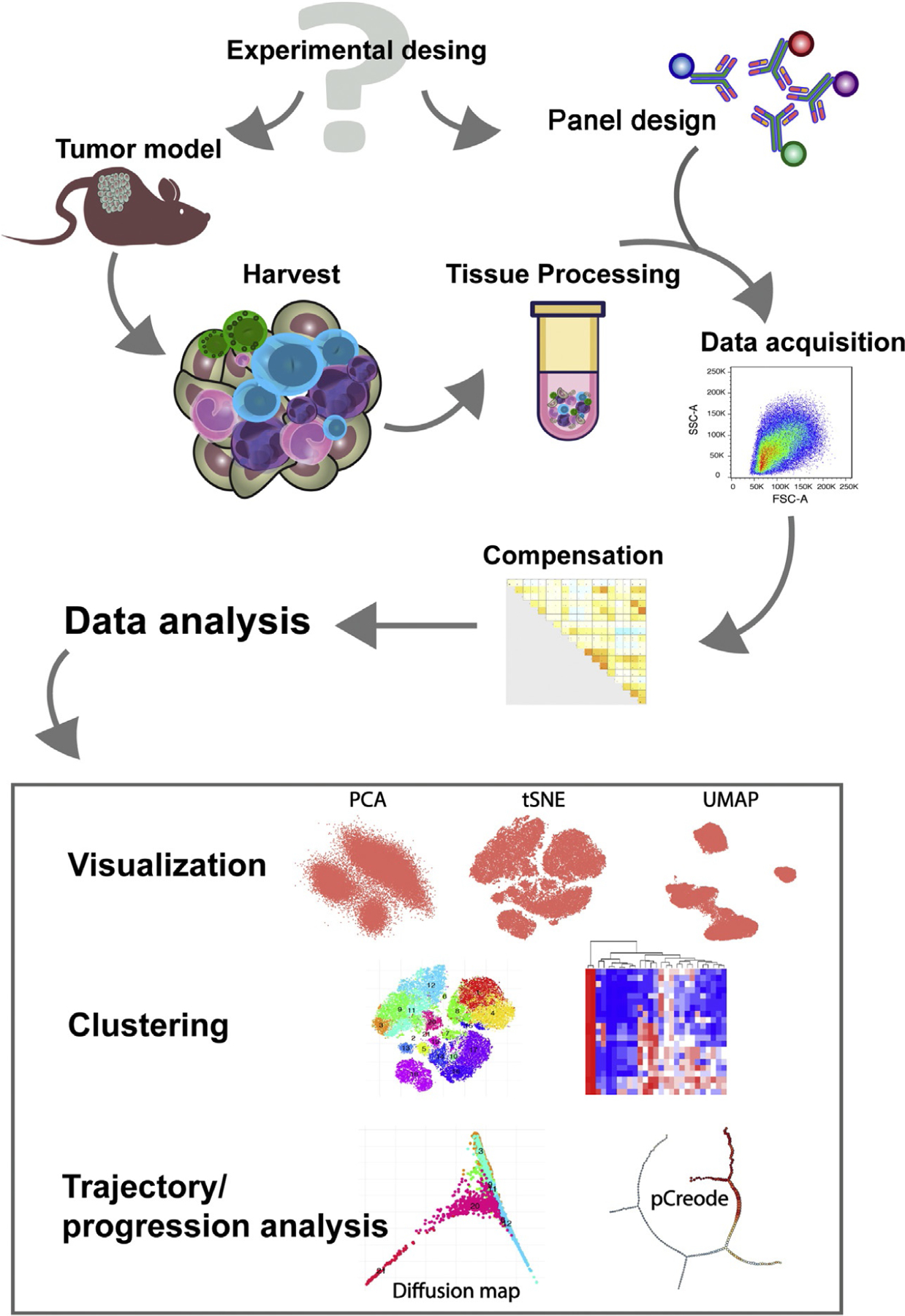
Workflow of experimental design and data analysis of high-parametric flow cytometry by unsupervised algorithms applied to the study of the TME. Schematic representation of the workflow for the characterization of the tumor immune infiltrate by unsupervised high-dimensional flow cytometry.
2. Cell culture and tumor models
After thawing, mouse tumor cell lines are routinely maintained in standard culture conditions (37°C, 5% CO2) in medium (e.g., Dulbecco’s Modified Eagle Medium (DMEM) or Roswell Park Memorial Institute (RPMI)-1640) supplemented with 10% fetal calf serum (FCS), l-glutamine, and additional supplements (e.g., non-essential amino acids, sodium pyruvate) depending on the cell line (see Note 1).
When cells reach 75–80% confluence, the spent medium is discarded and the cells are gently washed with prewarmed (37°C) Hank’s balanced salt solution (HBSS) or phosphate buffered saline (PBS) to remove remaining FCS. The cell monolayer is then detached with cell dissociation enzymes (e.g., trypsin or TripLE Express) by adding enough volume to cover the monolayer and incubating at 37°C, 5% CO2 for a few minutes (see Notes 2–6). When the cells become loosely attached, they are collected, washed to remove any enzyme remnants, diluted, and reseeded to obtain cells in the exponential growth phase (50–60% confluence) for injection (see Note 7).
For injection, the cells are collected as described above and washed at least twice with HBSS or PBS. Cells are then resuspended in HBSS or PBS at the desired concentration. The number of cells to be injected will depend on the tumor model selected for the study (see Note 8). For subcutaneous injection the volume should not exceed 100–200μL.
For subcutaneous injections, mice are shaved in the lower right flank leaving a square area of around 1in. between the rib cage and the hip clear of hair.
Before tumor injection the skin is cleaned with alcohol (e.g., with an alcohol pad) and time is allowed for the alcohol to evaporate.
The cells should be injected in the center of the shaved and disinfected area using a 27G × 5/8 needle to minimize leakage.
Ideally, harvest will be done when tumors reach 5–10mm. However, the exact size and time point will be tumor model-dependent and should be determined by the investigator during the experimental optimization process.
3. Harvest
Mice are euthanized using CO2 followed by cervical dislocation as per ACUC guidelines (see Note 9).
Mouse fur and skin are cleaned spraying ethanol 70%.
The skin is opened from the center of the abdomen with scissors and then below until the pelvis to uncover the tumor area.
Tumors are collected with a scalpel, making sure of removing the tumor attached to the inner side of the skin.
Tumors are collected into RPMI medium containing 0.5% FCS and stored in wet ice or 4°C until processing (see Note 10).
4. Tumor processing
Keep everything on wet ice or 4°C until digestion. To facilitate the digestion process, transfer the tumor into a petri dish and chop it into small pieces of about 1–2mm. Depending on the tumor characteristics, the appropriate enzymatic digestion process should be setup (see Note 11).
Transfer the digested tumor (single cell suspension) to a new tube passing through a 40μm cell strainer (see Note 12). Quickly wash the cell strainer adding 10mL of cold buffer or medium containing 2–10% of FCS to stop the enzymatic digestion (see Note 13).
Spin the cells down and lyse red blood cells with 1mL of ACK buffer, during 3–5min. Wash with 10mL of FACS buffer (see Note 14).
Resuspend the cells in enough volume of FACS buffer (see Note 15). Separate an aliquot for cell counting if needed and transfer enough cells from the single cell suspension into a 96-well V-bottom plate for staining (see Note 16).
5. Design and validation of high parametric flow cytometry panels
5.1. Panel design
Define your goal: Before designing a flow cytometry panel it is important to define the purpose of the study. If the goal is to have a general overview of the whole immune landscape of the TME, then it will be more appropriate to select markers or combination of markers that allow the unequivocally (as much as possible) definition of different immune cell subsets (e.g., lymphocytes, innate lymphoid cells, monocytes, macrophages, neutrophils, dendritic cells). The more lineage specific markers selected, the more comprehensive the characterization will be. However, the goal may be to evaluate the phenotype and/or activation status of a certain cell type, in which case a “dump” channel can be created to exclude cell populations that are not of interest in the analysis (see Note 17).
Selection of fluorochromes: Once the desired list of markers is defined, the next step is the fluorochrome selection. This is determined based on fluorochrome brightness (staining index), marker expression level and cytometer configuration. The simplest formula is assigning the brightest fluorochromes to the dimmest markers or those expressed at a low density (Mahnke & Roederer, 2007) (see Notes 18 and 19). Staining index values can be obtained from the different antibody supplier companies, and it is highly recommended to obtain the values specific for the instrument that will be used in the study given that fluorescence intensity and resolution may vary among cytometers.
Spillover and spread error: When several markers are needed to define a cell type, the spectral overlap among fluorochromes can be a problem. Certain channels are more prone to suffer from spill over among them. Although compensation normally corrects for this defect, there are cases in which the spectral overlap is so high that the signals result impaired and the resolution is lost (Ashhurst, Smith, & King, 2017). It is recommended to spread the markers that are co-expressed on the same cell along as many lasers as possible, this will minimize the spillover and preserve the characteristics of the signal (see Note 20). In addition to the specific markers, dyes that discriminate live/dead cells and Fc receptor blocking antibodies must be included in the panel.
- Specificity controls: Once the panel is designed, specificity controls are needed in order to set the positive/negative boundaries during analysis. These controls include:
- Isotype control: traditionally considered control for “non-specific” antibody binding, which may result from Fc-mediated binding, cross-reactivity, or non-specific cell adhesion. In order to accurately account for the non-specific binding, the isotype control should be from the same species, same heavy and light chains, conjugated to the same fluorochrome, and have the same fluorophore/antibody ratio (i.e., number of fluorescent molecules bound per antibody). In addition, isotype control antibodies have different activities, which may result in varying levels of non-specific binding. Therefore, finding the ideal isotype control is highly unlikely. Furthermore, isotype controls do not discriminate signal from spectral overlap or cell autofluorescence. For these reasons, and the availability of Fc receptor blocking antibodies, the usefulness of isotype controls is debated and more and more they are being replaced by fluorescence minus one control.
- Fluorescence minus one (FMO): FMO controls consist of all the antibodies present in the mix, except one. The empty channel is the one the FMO controls for. This allows to determine any fluorescence signal that is detected either due to spillover from other channels or cell autofluorescence. This is particularly relevant when working with panels of >6 colors as the major source of background signal is generally due to spectral overlap. FMOs allow to determine the positive/negative boundaries and to maximize the resolution between channels. However, FMOs would not discriminate signal due to non-specific antibody binding.
- Biological control: As it is the case in most experimental setups, proper biological controls should be included. For example, when staining for cytokines or activation markers after stimulation, an unstimulated control should be included. In some cases, these controls are the most appropriate for setting positive/negative boundaries.
5.2. Antibody titration
The optimal antibody concentration to be used in a given panel depends on the expression level of the antigen and the fluorochrome of choice and it needs to be determined experimentally doing a titration. For that purpose, serial dilutions of the antibody are tested and the staining index (SI) is obtained as follows (see Note 21):
where MFIpos: mean fluorescence intensity positive population; MFIneg: mean fluorescence intensity negative population, and SDneg: standard deviation of MFIneg.
5.3. Compensation
Compensation corrects the effects of spillover among different channels and it should be done for each flow cytometry experimental run. Most instruments nowadays have a built-in software to do this automatically, please refer to the manufacturer manual for detailed procedures. Although compensation can be done by the instrument, it is highly recommended to save single-color compensation files to allow for post-acquisition compensation adjustments during analysis. Having adequate single-color compensation controls is key to obtaining a proper compensation matrix that will accurately correct the spillover. Below are basic rules to prepare compensation controls:
Single-color controls need to be as bright or brighter than the experimental stain, and at least 10% of the population should have a positive signal.
Background fluorescence should be similar for positive and negative controls (see Note 22).
The fluorochrome used in the compensation control must match the one used in the experimental sample. This is particularly important when using tandem dyes or dyes detected with the same excitation laser and filters (e.g., FITC vs Alexa Fluor 488).
Compensation controls must be treated the same way as the samples, e.g., fixation/permeabilization treatments may alter the fluorochrome.
Antibodies used for single-color controls should be titrated both when using cells or synthetic compensation beads, so that the positive peak is set within the linear range of each detector and the PMT values do not need to be changed (see Note 23).
6. Application Settings
Setting up Application Settings ensures consistency and reproducibility over time across experiments and instruments. Optimal PMT voltages need to be determined for each particular tissue as a function of the tissue’s autofluorescence. Once optimal PMT voltages are determined and saved as the Application Settings, the same setting can be applied to standardize experiments regardless of daily variability of the instrument’s performance (Meinelt et al., 2012) (see Note 24).
- Obtain the robust standard deviation of the electronic noise (rSDEN), and the maximum linear fluorescence (LinMax) for each parameter from the baseline report (see Note 25). The positive and negative signal should be out of these values. The optimal PMT voltages gating in the population of interest should be:
- The PMT values that place the negative unstained population at least 2.5 times above the noise of the cytometer (2.5 × rSDEN).
- The PMT values that keep the positive stained population within the linear range (MFIpos <LinMax).
Once optimal PMT voltages are determined for each parameter in the population of interest, the final settings are saved as Application Setting and used for further experiments (see Notes 26 and 27).
Definition of target values: target values are defined with the established Application Settings using standardized bright beads, like rainbow calibration particles (Thermo Fisher Scientific) or CS&T beads (BD Biosciences) to create a template as shown in Fig. 2. Target values can be used to verify the Application Settings in use and to transfer the Application Setting to a different instrument (provided that the cytometer parameters are similar under the same or different optical configurations) by adjusting the positive gate to that shown in the template.
Fig. 2.
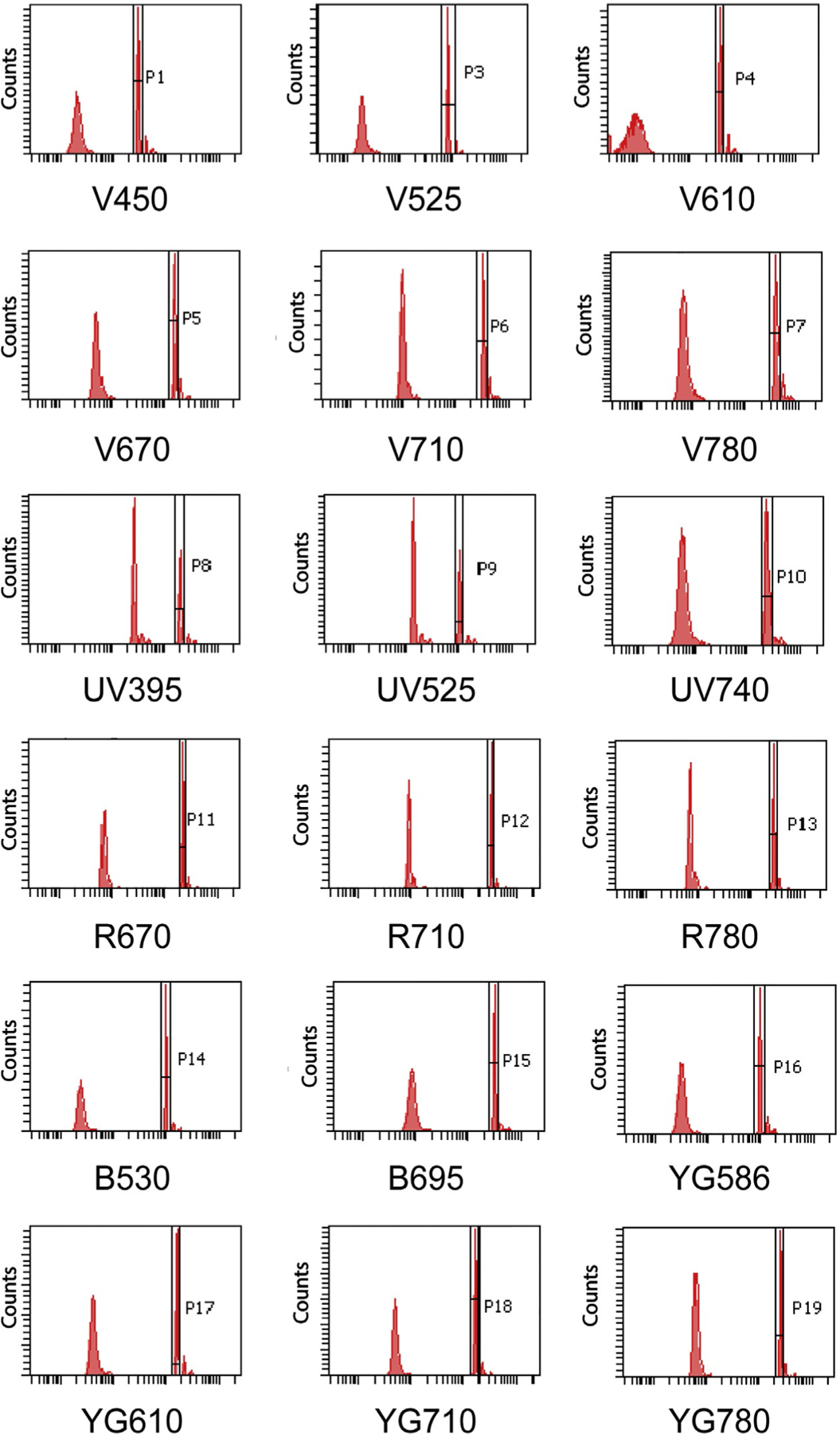
Target value template for a 20-parameter BD LSR Fortessa cytometer. Histograms depict an example of target value for each parameter.
7. Data visualization, clustering and analysis
The traditional flow cytometry data analysis performed by consecutive bivariate gating (i.e., gating strategy) is sufficiently robust when dealing with few parameters. However, this strategy is very limited when analyzing multidimensional data and new dimensionality reduction algorithms can be used for an unbiased data analysis approach. These algorithms can be divided into two main categories: (1) linear dimensionality reduction that conserve the information at the single cell level preserving the global structure of the data but assuming a linear relationship among parameters such as Principal Component Analysis (PCA) (Newell et al., 2012) and (2) non-linear dimensionality reduction in which local distances are prevalent over the global structure of the data such as t-Distributed Stochastic Neighbor Embedding (t-SNE), diffusion maps, and Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP). Each algorithm has different requirements for the size, normalization and/or transformation of the data and should be carefully studied (Amir et al., 2013; Becht et al., 2019). Fig. 3 illustrates a traditional manual gating strategy to define tumor-infiltrating myeloid cells within total CD45+ live cells from a mouse tumor, where only 11 different populations can be defined based on the subjective gating strategy. Below is a brief description of some of the dimensionality reduction, clustering and trajectory methods that can be utilized and how they compare among them and to the traditional gating strategy when applied to the same dataset. See also Table 1 for a list of markers that can be used for this type of analysis and Tables 2–4 for a summary of the methods described below.
Fig. 3.
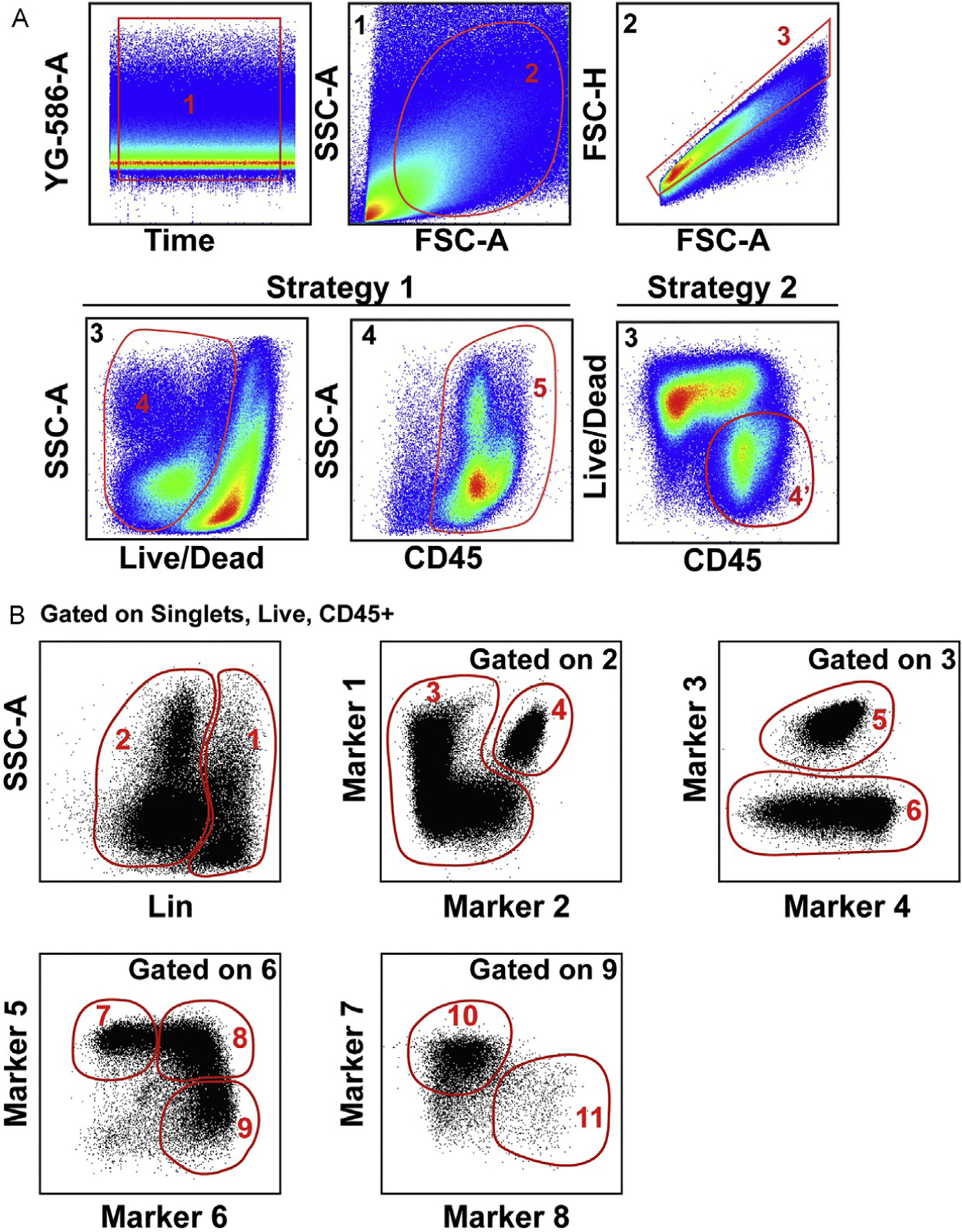
Definition of populations by traditional manual gating strategy applied to the analysis of 20-parameters flow cytometry data of a subcutaneous mouse tumor. (A) Initial gating strategy starting with Gate 1 to select the events acquired with stable flow stream, Gate 2 to exclude cell debris, and Gate 3 to exclude doublets. Strategy 1 and Strategy 2 depict options to select live CD45 positive cells. (B) Example of manual gating strategy to define specific populations starting from Gate 4′ or 5.
Table 1.
Flow cytometry panel (example).
| Marker | Clone | Manufacturer |
|---|---|---|
| CD45.2 | clone 104 | BD Biosciences |
| Ly6G | 1A8 | BD Biosciences |
| CD64 | X54-5/7.1 | BD Biosciences |
| F4/80 | BM8 | BD Biosciences |
| CD24 | M1/69 | BD Biosciences |
| Ly6C | AL-21 | BD Biosciences |
| CX3CR1 | SA011F11 | BioLegend |
| CD103 | 2E7 | BioLegend |
| MHC II | M5/114.15.2 | eBioscience |
| CD68 | FA-11 | BioLegend |
| CD11c | N418 | eBioscience |
| TCRb | H57–597 | eBioscience |
| TCRgd | eBioGL3 | eBioscience |
| CD3 | 145-2C11 | eBioscience |
| NK1.1 | PK136 | eBioscience |
| Ter119 | TER-119 | eBioscience |
| CD19 | eBio1D3 | eBioscience |
| CD135 | A2F10.1 | BD Biosciences |
| CD206 | C068C2 | BioLegend |
| CD11b | M1/70 | eBioscience |
| Siglec F | E50-2440 | BD Biosciences |
| CD209a | MMD3 | Invitrogen |
| LIVE/DEAD Fixable Dead Cell Stain Kit | – | Invitrogen |
| CD16/32 | 2.4G2 | Bioxcell |
| CD103 | 2E7 | BioLegend |
| CD11b | M1/70 | eBioscience |
| CD11c | N418 | eBioscience |
| CD127 | A7R34 | BioLegend |
| CD135 | A2F10.1 | BD Biosciences |
| CD16/32 | 2.4G2 | Bioxcell |
| CD19 | eBio1D3 | eBioscience |
| CD206 | C068C2 | BioLegend |
| CD209a | MMD3 | Invitrogen |
| CD223/Lag3 | C9B7W | BD Biosciences |
| CD24 | M1/69 | BD Biosciences |
| CD25 | PC61 5.3 | eBioscience |
| CD274/PDL1 | MIH5 | BD Biosciences |
| CD279/PD1 | RMP1-30 | BD Biosciences |
| CD3 | 145-2C11 | eBioscience |
| CD4 | GK1.5 | BD Biosciences |
| CD44 | IM7 | BioLegend |
| CD45.2 | clone 104 | BD Biosciences |
| CD62L | MEL-14 | BD Biosciences |
| CD64 | X54-5/7.1 | BD Biosciences |
| CD68 | FA-11 | BioLegend |
| CD8a | 53–6.7 | BioLegend |
| CX3CR1 | SA011F11 | BioLegend |
| F4/80 | BM8 | BD Biosciences |
| Foxp3 | FJK-16s | eBioscience |
| KLRG1 | 2F1 | eBioscience |
| LIVE/DEAD Fixable Dead Cell Stain Kit | – | Invitrogen |
| Ly6C | AL-21 | BD Biosciences |
| Ly6G | 1A8 | BD Biosciences |
| MHC II | M5/114.15.2 | eBioscience |
| NK1.1 | PK136 | eBioscience |
| Siglec F | E50-2440 | BD Biosciences |
| TCRb | H57-597 | eBioscience |
| TCRgd | eBioGL3 | eBioscience |
| Ter119 | TER-119 | eBioscience |
| TIGIT | 1G9 | BD Biosciences |
| Tim3 | RMT3-23 | eBioscience |
Table 2.
Visualization methods.
| Method | Type of method | Environment | Description | Limitations |
|---|---|---|---|---|
| PCA | Visualization, relatedness or trajectory | Many different options, R (included in Cytofkit), Phyton, etc. | It establishes distance and relatedness between populations in the linear space Preserve global distances Fast | Non-linear relatedness not considered Overcrowding of data points |
| tSNE | Visualization | R (included in Cytofkit), Phyton, FlowJo, etc. | Non-linear dimensionality reduction based on the k-neighbor algorithm Not overcrowding of data points Preserve local distances in detriment of global structure | Distance among clusters has no meaning Limited number of events per run Not so fast |
| UMAP | Visualization, some degree of relatedness | R (included in Cytofkit2 but requires Python) Python, and FlowJo Exchange plug-in | Non-linear dimensionality reduction. Preserve local distances, with some global structure Fast | Loss of resolution among populations with little variation Crowding of similar populations |
Table 4.
Differentiation-trajectory methods.
| Method | Environment | Description | Limitations |
|---|---|---|---|
| Diffusion Map | R (included in Cytofkit), and Python package | Preserve global structure Pseudotemporal relatedness Discovery of rare populations Fast | Definition of multi-branch pathways Requires moderate down-sampling |
| pCreode | Phyton package | Preserve global structure Pseudotemporal relatedness Definition of multi-branch pathways | Require extreme down-sampling Loss of rare populations Not so fast |
7.1. Linear dimensionality reduction
PCA is based on the distance and relatedness of different populations and aims to find the directions (components) with the maximum variance in the dataset. One advantage of this method is that the distance in the PCA plot infers relationship among cell populations in the linear space. However, it does not provide information for nonlinear relatedness among the parameters. Fig. 4A right panel depicts the first two components of a PCA analysis of the same dataset shown in Fig. 3.
Fig. 4.
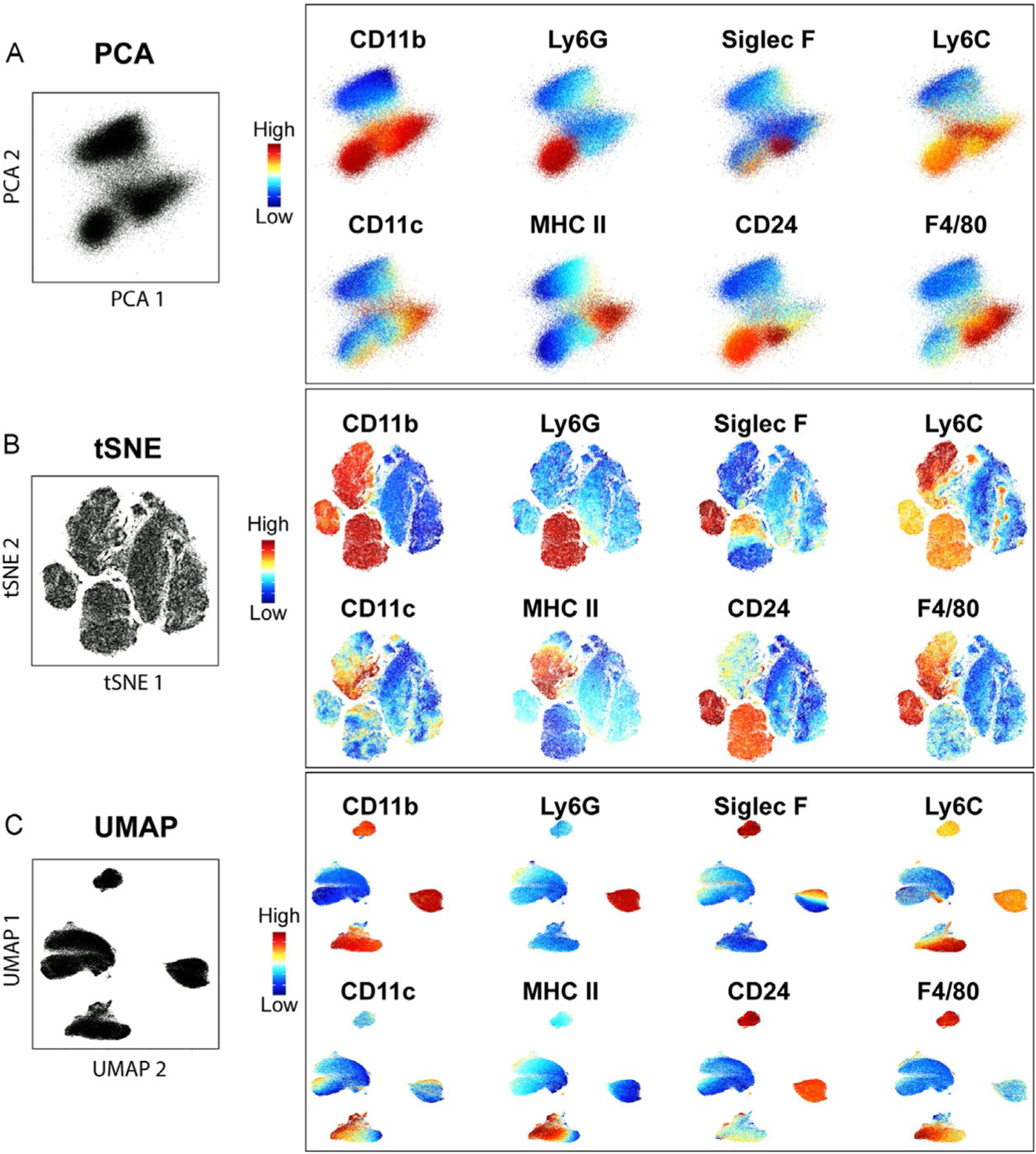
Comparison of different visualization methods. PCA (A), tSNE using perplexity = 50 and iterations = 1000 (B), and UMAP with nearest neighbor = 30 and min_dist = 0.3 (C) showing the expression level of some of the markers used for the analysis overlaid on each plot.
7.2. Non-linear dimensionality reduction
tSNE is based on the k-neighbor algorithm, where the dataset is visualized as a scatter plot and the location of each event (cell) in the plot indicates the similarities with nearby events. While the distance of the events (dots) within a cluster indicates similarity, unlike PCA, the distance between dots from different clusters cannot be used to infer cellular relationship. The same dataset displays more complexity in a tSNE plot than when analyzed by PCA (Fig. 4B). A disadvantage of this method is that only a few hundreds of thousands of events can be run at a given time in most platforms (e.g., Phyton, R, FlowJo) and the required down-sampling of the data may result in the loss of rare populations.
UMAP is similar to tSNE but allows the use of significantly larger datasets and preserves more of the global structure of the data. As shown in Fig. 4C, UMAP increases the distance among cells that are markedly different (distance between clusters), while grouping more those that bear more similarity (fewer clusters). Because it preserves more of the global structure than a tSNE, it allows to some degree to infer cellular relatedness based on cluster distance.
Importantly, the visual clusters generated by these methods are strongly influenced by several parameters that are specific for each (e.g., perplexity, iterations for tSNE and min_dist, nearest neighbors for UMAP), therefore a clear understanding of them is critical for an accurate analysis (Amir et al., 2013; Becht et al., 2019) (see also Table 2).
In all of these analyses, color can be added as an additional dimension. The example shown in Fig. 4 right panels use this additional dimension to show the expression level (color gradient) of some of the markers used for the analysis, which help identify cell types (see below). Alternatively, color can be used to indicate sample(s) of origin for example when several treatment conditions are combined.
7.3. Clustering methods
The methods described above provide a tool for data visualization following dimensionality reduction, however, they do not provide information regarding cell subpopulations. Additional partitioning methods are used to define clusters within the high-dimensional space in an unbiased manner (Weber & Robinson, 2016). Some of the algorithms used for this purpose are briefly described below (see also Table 3).
Table 3.
Clustering methods.
| Method | Environment | Description | Limitations |
|---|---|---|---|
| PhenoGraph | R (included in Cytofkit), Phyton, MATLAB™, FlowJo Exchange plug-in | Based on nearest-neighbor graph, followed by definition of communities as interconnected events | Number of clusters not customizable Not so fast |
| ClusterX | R (defined in Cytofkit) | Based on densities in the tSNE projection map Fast | Limited to tSNE ability to define clusters Number of clusters not customizable |
| FlowSOM | R package (included in Cytofkit), FlowJo Exchange plug-in | Based in Self-Organization Maps (SOM) follow by hierarchical clustering Very fast Number of clusters customizable | For exploratory analysis, hard to determine the number of clusters |
PhenoGraph:
Partitions high-dimensional data into subpopulations based on finding the k-nearest neighbor for each cell (Levine et al., 2015). It uses the parameters (in this case markers expressed on the cell) to define a point in the high-dimensional space and creates a graph/network that represents the phenotypic similarities between cells (neighbors). These neighbors are then grouped into communities which are stratified into clusters. The results provide a quantification (number of clusters) of the general population structure that is based on different cell types and phenotypes within a same cell type. PhenoGraph clusters are depicted as colored and numbered according to their cluster identity and can be displayed on the two-dimensional tSNE or UMAP data visualization scatter plots (Fig. 5A, upper panels). Cluster annotation (i.e., identification of cell types or subtypes) can then be done by obtaining a heatmap representation of the marker expression level (Fig. 5A, bottom panel) or by coloring the scatter plot by marker expression level rather than cluster number (Fig. 4, right panels). Among the advantages of this method are that it can resolve rare populations and that it allows for population discovery. One of the limitations on the other hand is the need to pre-define number of neighbors.
Fig. 5.
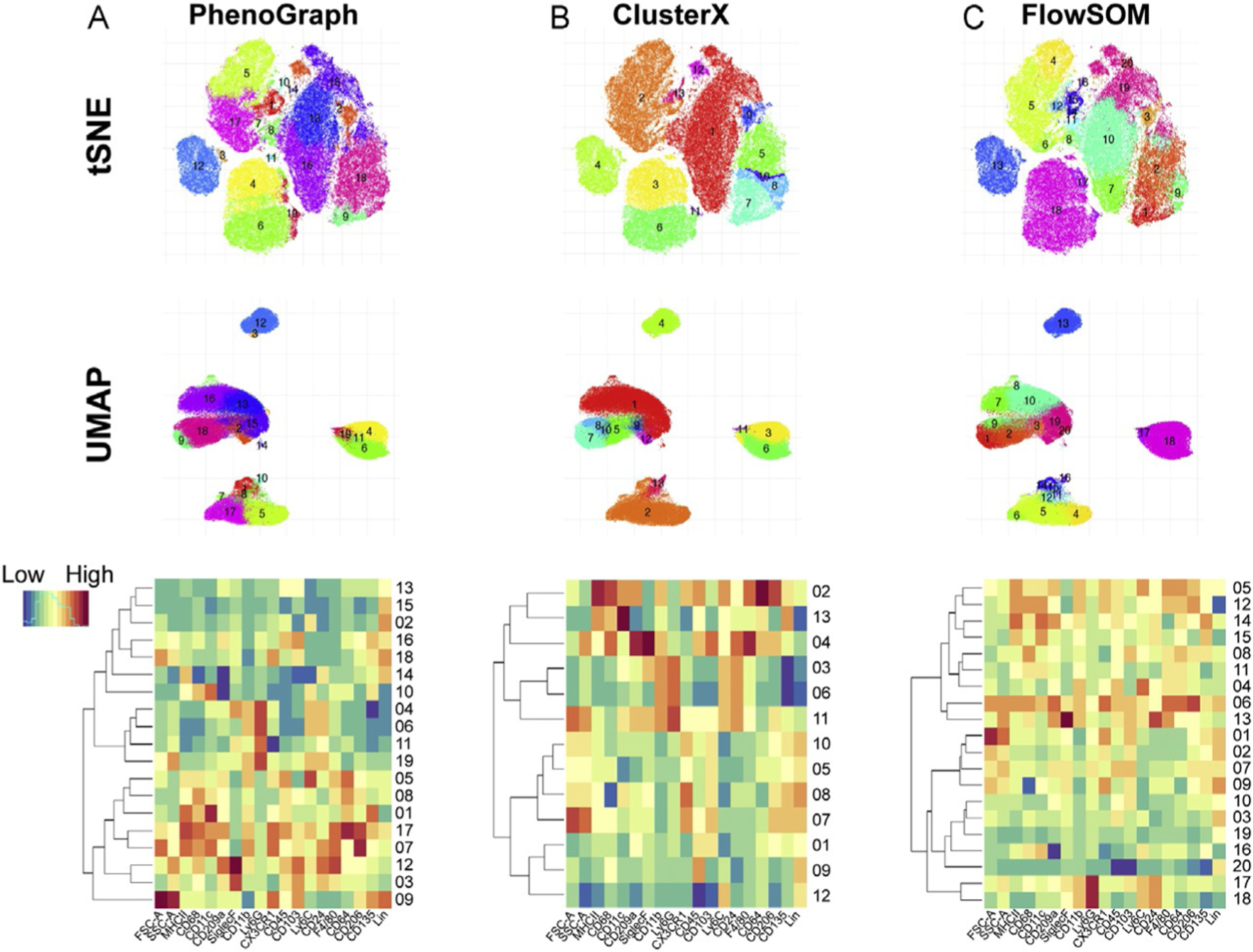
Clustering methods. Visualization of PhenoGraph (k = 30) (A), Cluster X (B), and FlowSOM (k = 20) (C) in tSNE (n = 100,000 events, perplexity = 50, iterations = 1000; top panels), UMAP (n = 100,000 events, nearest neighbor = 30, min_dist = 0.3; middle panels) and heatmap (normalized by rows; bottom panels).
ClusterX:
This method is based on the assumption that cluster centers are characterized by a higher density than the neighbors, and far away from any other points with a higher local density in a pre-defined visualization map (Rodriguez & Laio, 2014). The algorithm depends on relative density rather than the absolute values, and clusters are recognized independently of the dimensionality of the space in which the data points are embedded. This density-based clustering was applied to the t-SNE embedded map in the example and might therefore be limited to the power of the tSNE’s dimensionality reduction in representing the whole variation of the global data into the 2D representation (Fig. 5B, upper panel). Cluster annotation can be performed as indicated for PhenoGraph (Fig. 5B, bottom panel).
FlowSOM:
This is an unsupervised technique for clustering and dimensionality reduction that uses a Self-Organizing Map (SOM) to visualize and interpret the data (Van Gassen et al., 2015). A SOM consists of a grid of nodes, in which a node is a point in the multidimensional input space. The input space is trained so that the nodes closely connected are more similar to each other than to the ones connected through a larger path. During clustering, each point of the dataset is classified with the node that resembles it the best (nearest neighbor). The advantages of this method include the fast runtime, several visualization options, and that it offers one of the best clustering performances allowing users to explore large datasets and the discovery of rare populations. Like with other methods, FlowSOM requires the pre-selection of several parameters and having a clear understanding of them is critical (Van Gassen et al., 2015).
Fig. 5A–C allows for a comparison of the clustering methods mentioned above. The different number of clusters obtained with the different methods may be due to intrinsic limitations of the method or the pre-selected parameters used (e.g., numbers of neighbors and/or clusters pre-defined) (Table 3). All these methods allow for a more granular characterization of the tumor immune microenvironment than the classical bivariate flow cytometry analysis. In addition, one can select specific clusters of interest and run the same or different algorithms for an even more detailed classification.
7.4. Trajectory or differentiation analysis
To infer differentiation paths or cell population relationship, non-linear dimensionality reduction algorithms that preserve the global structure of the dataset are recommended (Table 4). Two options that can be used to analyze cellular relatedness in the tumor microenvironment are:
Diffusion map:
Organize the events in non-linear and complex branches of differentiation still allowing the discovery of rare populations (Haghverdi, Buettner, & Theis, 2015). Diffusion map preserves the global structure and pseudo-temporally order of the cells. It is applicable to datasets with high levels of noise (such as flow cytometry data), samples with heterogeneous densities, and with missing or uncertain values. However, this algorithm falls short in depicting multi-branching transitions. Using the same dataset as in previous examples, Fig. 6A depicts an example of a diffusion map run with equal number of events (see Note 28) from selected clusters defined by UMAP and FlowSOM, containing monocytes, macrophages and dendritic cells. There are at least two branches defined by the transitional expression of markers associated to monocytes, macrophages and dendritic cells projected in the diffusionmap 1 and diffusionmap 2 axes (Fig. 6B). One of them represents the differentiation from Ly6Chi monocytes to macrophages (clusters 4-5-6) and the other resemble the differentiation path from dendritic cell precursor to conventional dendritic cells (cDCs) (clusters 14, 15, and 16). Cluster 12 represents DCs derived from monocytes (moDCs). This analysis also allows to dissect what marker or group of markers drive the differentiation path proposed by the algorithm. As an example, Fig. 6C shows increased or decreased levels of expression of different monocyte and macrophage-associated markers across clusters 4-5-6 in the trajectory defined by diffusionmap 2 axis.
Fig. 6.
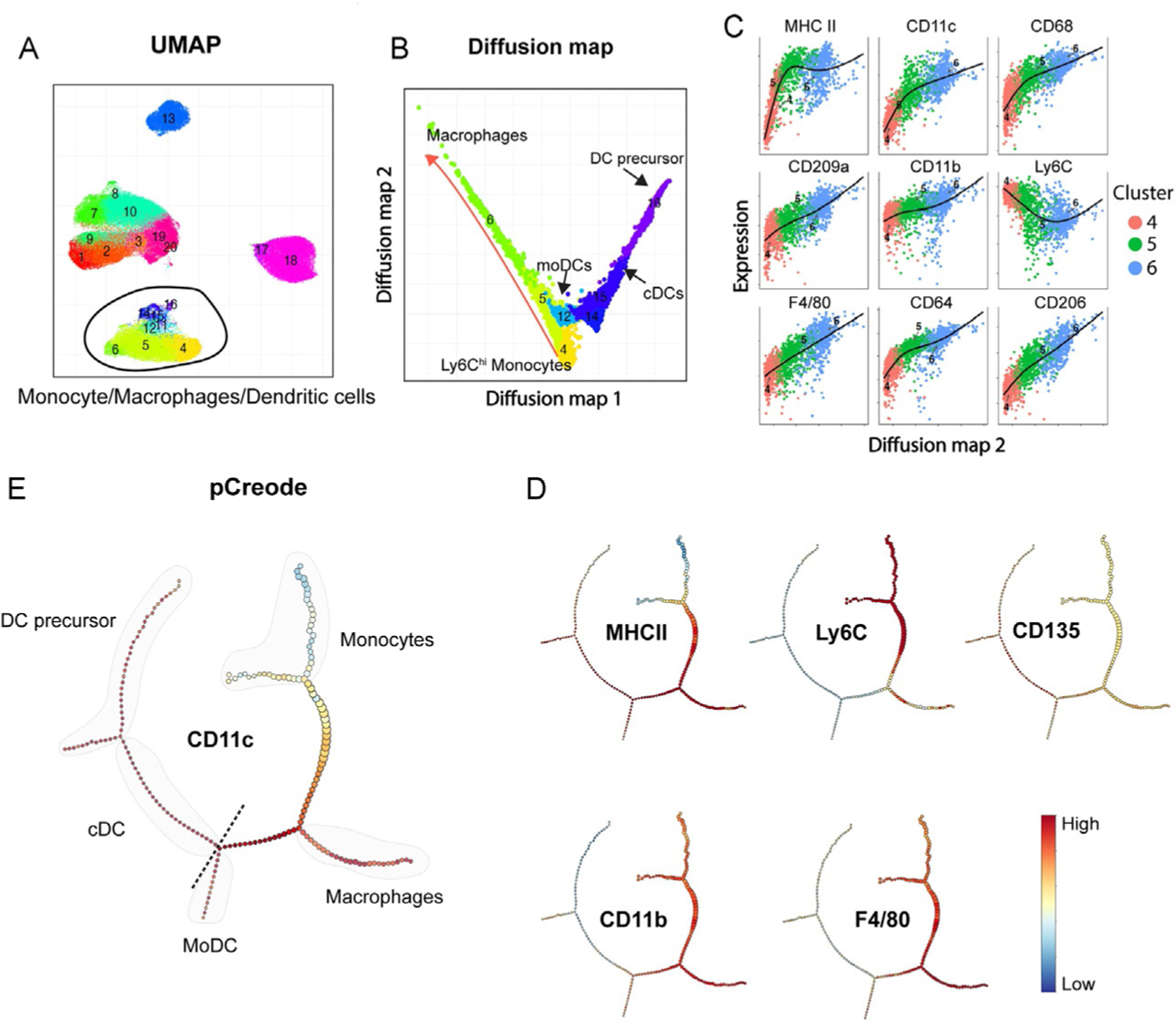
Trajectory analysis of monocytes, macrophages, and dendritic cells in the TME. (A) UMAP visualization from Fig. 5C showing clusters selected for diffusion map analysis. (B) 2D representation of the diffusion map analysis with an overlay of FlowSOM clusters. (C) Expression level of markers across clusters 4-5-6. (D) pCreode analysis from the same clusters from (B) (plot ranked 0 from n = 10 runs). (E) Overlay of markers in pCreode from (D).
pCreode:
Is an unsupervised algorithm that compares graphs with different topologies and infers a statistically significant hierarchy of cell dynamics that define a developmental trajectory based on the parameters used in the analysis (Herring et al., 2018). An advantage of pCreode is that it allows for the visualization of multi-branching transitions (Fig. 6D) as well as the overlay of different markers to facilitate the interpretation (Fig. 6E). However, it requires a much stronger down-sampling, which results in the loss of information (rare populations). Another advantage of pCreode over diffusion map is that the node size indicates the abundance of cells within each differentiation transitional state.
Regardless of the robustness or limitations of the method of choice to define differentiation trajectories, the biological meaning of the data needs to be confirmed experimentally.
8. Basic steps for high dimensional flow cytometry data analysis
Export the files in FCS format and open them in any suitable software (e.g., FlowJo, Cytobank).
For each individual sample, plot the parameter in the last position of the laser delay settings (e.g., YG-586-A) against Time. Select the segment of Time in which the data looks constant (as shown in Fig. 3A—gate 1) (see Notes 29 and 30).
From the Time gate, select by forward (FSC) and side scatter (SSC) the gate that defines the populations of interest (Fig. 3A—gate 2).
Exclude doublets via any of the methods available (FSC-H vs FSC-A or SSC-W vs SSC-A and FSC-W vs FCS-A) (Fig. 3A—gate 3) (see Note 31).
Transform all fluorescent markers to biexponential normalization given that this transformation will avoid the artifacts due to logarithmic normalization around zero.
Select the live cells with the L/D marker and CD45+ cells (tumor infiltrating-immune cells, Fig. 3A—Gates 4 and 5 in Strategy 1 or Gate 4′ in Strategy 2).
Perform quality control of the compensation matrix automatically generated during data acquisition by comparing single pairwise markers (N × N plot in a FlowJo Layout). Fine tune any obviously under or over-compensated markers as needed (see Note 32).
Export the population of interest (e.g., CD45+ Live cells) or continue with the gating strategy using the dump channels to exclude any populations that are not of interest (see Note 33).
Prior to running any algorithms, verify that staining, processing and acquisition have no artifacts or defects. The use of specificity controls is crucial for this step (see Section 5).
Depending on the algorithm of choice, down-sample the properly compensated data to a fix value that will be equal in every sample/group to be analyzed.
Concatenate the data including the same number of events from each sample from every group to be analyzed (see Note 34). For example, for a total of 100,000 events to be analyzed from two treatment conditions (e.g., untreated vs therapy) with 5 samples per condition: 50,000 events from untreated group = 10,000 events per untreated tumor × 5 samples; 50,000 events from therapy group = 10,000 events per treated tumor × 5 samples (see Notes 35 and 36).
Further steps will depend on the dimensionality reduction, clustering and/or trajectory analysis method of choice.
9. Concluding remarks
One of the biggest challenges in the analysis of high-parametric flow cytometry data is the correct definition of cell populations. While under homeostatic conditions and with few parameters this may seem accurate and indisputable, under pathological situations such as in the TME and with high-parametric data, it can be an arduous job. In this situation, traditional manual gating strategies become time consuming and biased and valuable information can be missed. Thus, an unsupervised approach based on the whole dimension of the data is needed for a comprehensive and objective characterization of the TME.
When applying these algorithms, user-defined settings are important variables that affect the data output. Therefore, a good understanding of each method and what each pre-defined parameter means is critical for the success of the analysis. Additionally, the quality of the compensated data, proper down-sampling, and the markers selected for the dimensionality reduction methods will also influence the outcome. It is likely that several trial runs will be needed to find the optimal conditions for different experimental datasets for the best representations that are biologically relevant. Many of these algorithms are also sensitive to sample-to-sample variations. Therefore, standardized methods should be implemented throughout the whole process: from the tissue harvest and processing through the flow data acquisition. A common practice used in CyTOF analysis that can be applied to flow cytometry, is the barcoding of samples that are then acquired in a single tube minimizing batch effects. Although this is still somehow limited by the less abundant number of parameters available for barcoding in flow cytometry, newer instruments and novel improved fluorochromes will soon allow the detection of up to 50 parameters and reducing compensation difficulties.
An advantage of the unbiased analysis approach for high-dimensional data is the possibility to identify novel cell populations. It is important, however, to keep in mind that such populations may represent a different activation/maturation state rather than a new cell type. Further experimental approaches are needed to formally determine their origin/identity, particularly given our still limited knowledge of the TME. Another useful feature of these analyses is that selecting a cluster from a tSNE and back-gating using the pairwise comparison approach can help select the best 4–6 markers that define a given population to be used for purification (e.g., FACS sorting) and further characterization (e.g., functional and transcriptional analysis).
Because all methods have their advantages and disadvantages, and they may provide different type of information, a combination of different approaches should be used for a more thorough analysis. Here we just exemplified a few of them, however, other algorithms are currently available and new ones are constantly being optimized.
10. Notes
Optimal culture conditions should be determined for the specific tumor cell line of interest. Recommendations from the American Type Culture Collection (ATCC, Manassas, VA, USA) can be used as a starting point.
Cells should not be used at low (<40%) or high (>80%) confluence as this may lead to the selection of subclones or changes in the metabolic status of the cells, respectively.
Porcine Trypsin/EDTA is commonly used to detach adherent cells. TripLE reagents (Gibco-Thermo Fisher Scientific) are animal origin-free recombinant dissociation enzymes that better preserve the integrity of cell surface molecules.
The incubation time has to be optimized for each cell line and it should be just enough to have the cells loosely attached.
If Trypsin is used, FCS containing medium should be used immediately to inhibit its activity. TripLE reagents can be inhibited by dilution.
Non-adherent cells can be collected by gently pipetting and transferring to a sterile tube, centrifuged down and resuspended in fresh medium at the appropriate concentration to be reseeded.
The dilution factor will depend on the cell type and size of the tissue culture flask of choice.
Careful setup of the tumor model of interest is strongly recommended before any further analysis. This includes determining the cell concentration, volume of injection, timing, etc.
Euthanasia should be performed following appropriate institutional guidelines.
If extended storage of the tumors before processing is needed (i.e., tissue processing cannot be performed immediately after harvest), it is recommended to collect and keep the tumors in PBS containing higher concentration of FCS (2–10%) to avoid pH fluctuations. Tissues should then be transferred to the digestion medium at the time of processing being careful not to increase the FCS content of the digestion medium as it will block the enzymatic activity. It is important to consider that the longer storage may impact on immune cell populations differently, thus affecting the overall composition of the infiltrate and biasing the results. Therefore, long storage periods should be avoided as much as possible.
An example of a digestion cocktail used in our lab is as follows: 200U/mL of Collagenase IV (Gibco) and 0.1mg/mL of DNase I (Roche). Tumors are incubated 10min in a water bath at 37°C and then transfer to a rotator inside an incubator (37°C, 5% CO2) for extra 50min of digestion.
To avoid clogs during flow cytometry, cells should be filtered again passing through a 40–50μm cell strainer or mesh after staining and prior to running the samples in the flow cytometer.
Stopping the enzymatic reaction is an important step to reduce cell death.
An example of FACS buffer: PBS/2mM EDTA/2% FCS.
The volume should be determined based on the tumor size/number of cells obtained for the tumor model of interest.
Using amount of tissue (e.g., 50mg of tumor) as a reference rather than cell number helps account for dead cells in the mix and avoid artifacts in the staining.
A fluorophore with high spectral overlap into other channels can be used for the dump because that channel will be excluded early on in the analysis.
Certain cell types are highly autofluorescent (e.g., myeloid cells). This should be considered when selecting the appropriate fluorochrome for a given marker as a true positive signal may be masked by the autofluorescence if using a weak fluorochrome.
Brilliant Violet dyes interact with each other quenching their signal. Using buffers that block the interaction of these dyes is strongly recommended.
Information about the spread characteristics of each fluorochrome can be obtained from the supplier’s specifications.
Other metric to assess reagent brightness is the separation index (Ashhurst et al., 2017).
It is recommended to include both positive and negative populations within the same tube. This can be done either with cells, in which case only a fraction of them will express the marker used for the single-color control, or with synthetic compensation beads by mixing positive and negative beads (some companies offer a premade cocktail containing reactive and non-reactive compensation beads).
The linear range of each detector can be obtained from the CS&T baseline.
If the cytometer baseline changes, a new application setting needs to be determined.
The rSDEN is the background noise level of each parameter due to the electronic system.
It is recommended to run a full compensation and one fully stained tumor sample to verify there are no excessive spillovers and all parameters are within the linear range. If PMT voltages need to be modified, a new application setting with the updated values should be saved and a new compensation acquired.
If needed, PMT voltages can be increased as long as their values stay within the linear range before saving the Application Settings. However, it is not recommended to decrease the values given that they were set to the minimum possible.
It is important to keep in mind that by using the same number of events from each cluster, information related to frequency/abundance of each population is lost.
Avoid including the beginning and end of the run when the sheath flow is less stable. Also, exclude any defective region, for instance, because of a clog during acquisition. This should be applied when doing manual gating as well.
Do not run samples at high speed when they contain too much debris and use a higher FSC threshold during acquisition to record mainly the events of interest.
After this step, if the tumor cells are labeled (e.g., GFP, YFP), that channel can be used against FSC or SSC to exclude tumor cells from the analysis.
Even when the manual compensation is not recommended, obvious over and under compensations could lead to data misinterpretation and need to be corrected before proceeding with further analysis.
It is recommended to run the first analysis with total CD45+ live cells to obtain a big picture of the staining and subsets segregation before further analysis.
When comparing more than one treatment condition, the analysis should be run in concatenated groups to avoid batch effects.
The number of events per sample will depend on the algorithm of choice.
Always keep a computational barcoding of the samples (e.g., in FlowJo adding an extra keyword, as “SampleID”) which will be useful to determine any batch effect or sample abnormality, and group identification.
Acknowledgment
This work was supported by the Intramural Research Program of the U.S. National Institutes of Health National Cancer Institute.
References
- Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature Biotechnology, 31(6), 545–552. 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, et al. (2017). Comprehensive analysis of normal adjacent to tumor transcriptomes. Nature Communications, 8(1). 1077. 10.1038/s41467-017-01027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashhurst TM, Smith AL, & King NJC (2017). High-dimensional fluorescence cytometry. Current Protocols in Immunology, 119, 5.8.1–5.8.38. 10.1002/cpim.37. [DOI] [PubMed] [Google Scholar]
- Becht E, Mcinnes L, Healy J, Dutertre C, Kwok IWH, Ng LG, et al. (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology, 37(1), 38–44. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, & Chattopadhyay PK (2012). A deep profiler’s guide to cytometry. Trends in Immunology, 33(7), 323–332. 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity, 39(4), 782–795. 10.1016/J.IMMUNI.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine, 24(5), 541–550. 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, & Chen J (2016). Cytofkit: A bioconductor package for an integrated mass cytometry data analysis pipeline. PLoS Computational Biology, 12(9), 1–17. 10.1371/journal.pcbi.1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Buettner F, & Theis FJ (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics, 31(18), 2989–2998. 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
- Hanahan D, & Coussens LM (2012). Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell, 21(3), 309–322. 10.1016/J.CCR.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature, 515(7528), 563–567. 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CA, Banerjee A, Mckinley ET, Gerdes MJ, Coffey RJ, Lau KS, et al. (2018). Unsupervised trajectory analysis of single-cell RNA-seq and imaging data reveals alternative tuft cell origins in the gut. Cell Systems, 6(1), 37–51.e9. 10.1016/j.cels.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, & Chen DS (2016). Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Annals of Oncology, 27(8), 1492–1504. 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Downing JR, Pe D, Nolan GP, et al. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell, 162(1), 184–197. 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke YD, & Roederer M (2007). Optimizing a multicolor immunophenotyping assay. Clinics in Laboratory Medicine, 27(3), 469–485. 10.1016/J.CLL.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinelt E, Reunanen M, Edinger M, Jaimes M, Stall A, Sasaki D, et al. (2012). Technical bulletin: Standardizing application setup across multiple flow cytometers using BD FACSDiva™ version 6 software. BD Biosciences. [Google Scholar]
- Newell EW, Sigal N, Bendall SC, Nolan GP, & Davis MM (2012). Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity, 36(1), 142–152. 10.1016/J.IMMUNI.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. (2015). Robust enumeration of cell subsets from tissue expression profiles. Nature Methods, 12(5), 453–457. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, et al. Galon J, (2018). International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet (London, England), 391(10135), 2128–2139. 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. (2016). Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity, 44(6), 1255–1269. 10.1016/J.IMMUNI.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, & Laio A (2014). Machine learning. Clustering by fast search and find of density peaks. Science (New York, NY), 344(6191), 1492–1496. 10.1126/science.1242072. [DOI] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, & Ribas A (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell, 168(4), 707–723. 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S (2016). Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. International Immunology, 28(8), 383–391. 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A, 87(7), 636–645. 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- Weber LM, & Robinson MD (2016). Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry, 89, 1084–1096. 10.1002/cyto.a.23030. [DOI] [PubMed] [Google Scholar]