

Abstract
Background
Skeletal muscle progenitor cells (MPCs) repair damaged muscle postinjury. Pyruvate kinase M2 (PKM2) is a glycolytic enzyme (canonical activity) that can also interact with other proteins (noncanonical activity) to modify diverse cellular processes. Recent evidence links PKM2 to MPC proliferation.
Objectives
This study aimed to understand cellular roles for PKM2 in MPCs and the necessity of PKM2 in MPCs for muscle regeneration postinjury.
Methods
Cultured, proliferating MPCs (C2C12 cells) were treated with a short hairpin RNA targeting PKM2 or small molecules that selectively affect canonical and noncanonical PKM2 activity (shikonin and TEPP-46). Cell number was measured, and RNA-sequencing and metabolic assays were used in follow-up experiments. Immunoprecipitation coupled to proteomics was used to identify binding partners of PKM2. Lastly, an MPC-specific PKM2 knockout mouse was generated and challenged with a muscle injury to determine the impact of PKM2 on regeneration.
Results
When the noncanonical activity of PKM2 was blocked or impaired, there was an increase in reactive oxygen species concentrations (1.6–2.0-fold, P < 0.01). Blocking noncanonical PKM2 activity also increased lactate excretion (1.2–1.6-fold, P < 0.05) and suppressed mitochondrial oxygen consumption (1.3–1.6-fold, P < 0.01). Glutamate dehydrogenase 1 (GLUD1) was identified as a PKM2 binding partner and blocking noncanonical PKM2 activity increased GLUD activity (1.5–1.6-fold, P < 0.05). Mice with an MPC-specific PKM2 deletion did not demonstrate impaired muscle regeneration.
Conclusions
The results suggest that the noncanonical activity of PKM2 is important for MPC proliferation in vitro and demonstrate GLUD1 as a PKM2 binding partner. Because no impairments in muscle regeneration were detected in a mouse model, the endogenous environment may compensate for loss of PKM2.
Keywords: muscle stem cell, muscle progenitor cell, pyruvate kinase M2, PKM2, muscle regeneration, glycolysis, glutamate dehydrogenase, GLUD1
Introduction
Impaired regeneration after skeletal muscle injuries predisposes individuals to functional and metabolic deficiencies that reduce quality of life. Skeletal muscle injuries are repaired by muscle-specific adult stem cells and muscle progenitor cells [MPCs (i.e., Pax7+ cells)], which in response to an injury become activated from quiescence, proliferate, differentiate, and fuse onto damaged muscle fibers. Energy metabolism, including glucose and mitochondrial metabolism, is increasingly recognized as an important contributor to MPC function. In response to an injury, indicators of glycolytic and mitochondrial metabolism increase in MPCs (1, 2). Highlighting the relation between alterations in metabolism and impaired MPC function, MPCs from older rodents and male humans, which have impaired proliferation (3, 4), also have lower oxygen consumption (4, 5). Excitingly, targeting metabolic pathways may have therapeutic value. For example, increasing mitochondrial function can partially restore tissue regeneration in a model of impaired MPC function (6). Accompanying this recognition of the importance of metabolic pathways, numerous metabolic proteins have been explored as putative regulators of MPC function (7, 8).
Pyruvate kinase M2 (PKM2), a metabolic protein, plays a role in MPC function. PKM2 is a product of alternative splicing of the PKM gene, which also encodes PKM1. RNA sequencing revealed that quiescent MPCs dominantly express PKM1, whereas PKM2 is upregulated postactivation (9). Further, immunoblotting analysis showed PKM2 concentrations decrease during MPC differentiation, whereas PKM1 concentrations increase (10). Thus, PKM2 is preferentially expressed during MPC proliferation. Proliferation was impaired in MPCs isolated from mice with a whole-body PKM2 deletion (11). In addition, recombinant PKM2 was reported to increase MPC proliferation (12); however, the function of PKM2 in proliferating MPCs is unknown.
Research in other non-MPC cell types has identified that PKM2 has 2 known major mechanisms of action (Figure 1): 1) enzyme for the conversion of phosphoenolpyruvate to pyruvate in glycolysis (canonical activity); and 2) interacting with other proteins to modulate glucose and mitochondrial metabolism (11, 13–19), inflammation (18, 20–22), and other cellular processes (23) (noncanonical activity). Some researchers speculate that low canonical PKM2 activity causes an accumulation of glycolytic intermediates, which increases flux through anabolic pathways that stem from glycolysis (e.g., serine synthesis pathway, pentose phosphate pathway), increasing proliferation (24). Yet other researchers contend that high noncanonical activity of PKM2 promotes proliferation by interaction with proteins to alter cell metabolism [e.g., c-myc (25)], to coordinate histone modifications (26), or by direct interaction with cell cycle regulators (27, 28). Disentangling the PKM2 activity responsible for increased cell proliferation is difficult because low canonical and high noncanonical activity occur simultaneously, and there may be cell type–specific differences in PKM2 function.
FIGURE 1.
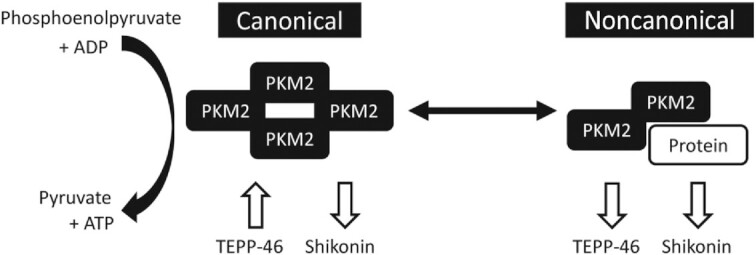
PKM2 has canonical and noncanonical activity and can be manipulated by TEPP-46 or shikonin. PKM2, pyruvate kinase M2.
Building on the expansive knowledge of PKM2 in nonmyogenic cells and the limited knowledge in MPCs, the overarching goal of this research was to define the function of PKM2 in the skeletal muscle regenerative process. The specific goals of this research were to 1) confirm the effect of PKM2 on MPC proliferation; 2) determine whether observed effects of PKM2 are dependent on canonical or noncanonical activity in MPCs; and 3) determine the necessity of PKM2 for recovery from a skeletal muscle injury. To disentangle the contributions of canonical and noncanonical activity of PKM2, we treated MPCs with 2 small molecules that differentially affect activity (Figure 1).
Methods
Cells and treatments
C2C12 cells were purchased from American Type Culture Collection (ATCC). Cells were grown on plates coated with Type 1 Rat Tail Collagen (Corning). Growth medium (GM) was high-glucose DMEM (Gibco), supplemented with GlutaMAX (Gibco) to a final medium concentration of 6 mM, 1% penicillin/streptomycin (Corning), and 10% FBS (VWR). HeLa and HEK293 cells were purchased from ATCC, and were cultured in the same manner and medium as the C2C12 cells. Differentiation medium was high-glucose DMEM (Gibco) supplemented with GlutaMAX to a final concentration of 6 mM, 1% penicillin/streptomycin, and 2% heat-inactivated equine serum (Gibco). Cells were maintained at 37°C in a 5% CO2 atmosphere. When indicated, cells were cultured in glucose-free DMEM (Gibco) with dialyzed FBS. When indicated, cells were given supplemental shikonin (1.4 μM, Cayman Chemicals), TEPP-46 (150 μM, Cayman Chemicals), pyruvate (1 mM, Gibco), aceteoside (100 nM or 2.5 μM, Cayman Chemicals), recombinant PKM2 (0.01, 1, or 50 ng/uL, Abcam), N-acetylcysteine (NAC) (1 μM, VWR), glutathione ethylester (GSHee) (55 μM, Cayman Chemicals), rotenone (500 nM, Cayman Chemicals), oligomycin (2 μM, Cayman Chemicals), trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) (1.5 μM, Cayman Chemicals), or galactose (25 mM, Sigma). Shikonin and TEPP-46 treatments were started ∼18–24 h after seeding, and with the exception of cell number assays, cells were incubated with the compounds or vehicle control (dimethyl sulfoxide) for 8 h.
Knockdown generation
Short hairpin RNA (shRNA) sequences targeting PKM2 were designed by adapting a published sequence from the human to the mouse genome (29). An shRNA plasmid containing the PKM2 targeting sequence (CTACCACTTGCAGCTATTCGA) was purchased from VectorBuilder. The plasmid contained the shRNA sequence downstream of a U6 promoter with ampicillin and G418 resistance. VectorBuilder's scrambled shRNA, which does not recognize sequences in the human or mouse genome, was used as a short hairpin control (shControl). Plasmids were isolated from One Shot TOP10 E. Coli (Invitrogen) grown in Lennox broth containing ampicillin using a Mini Prep Kit (Omega Bio-tek). Plasmids were sequenced to verify sequence fidelity, then delivered to cells using lipofectamine RNAiMAX reagent (Thermo Fisher) following the manufacturer's instructions.
Immunoblot analysis
Immunoblotting was performed as previously described (30). The following proteins were probed using a 1:1000 antibody concentration: PKM1 (7067S, Cell Signaling), PKM2 (4053S, Cell Signaling), α-tubulin (9099S, Cell Signaling), Lamin A/C (2032S, Cell Signaling), Lamin B (12586S, Cell Signaling), glutamate dehydrogenase 1 (GLUD1) (14299-1-AP, ProteinTech), dihydrolipoamide branched-chain transacylase E2 (DBT) (12451-1-AP, ProteinTech), voltage-dependent anion channel (VDAC) (4661S, Cell Signaling), β-tubulin (15115S, Cell Signaling), heme oxygenase 1 (HO-1) (ADI-SPA-896-D, Enzo), or nuclear factor erythroid 2-related factor 2 (NRF2) (12721T, Cell Signaling). Protein expression was normalized to tubulin by dividing band intensity (target/α-tubulin), with the exception of NRF2, which was measured using a nuclear lysate and normalized to Lamin A/C.
RNA extraction and RT-qPCR
RNA was extracted from cultured cells either using the E.Z.N.A.® Total RNA Kit (Omega Biotek) with an on-column DNA digest (Qiagen) or using iScript RT-qPCR sample preparation reagent (BioRad). RNA was reverse transcribed into cDNA using the High Capacity Reverse Transcriptase Kit (Applied Biosystems). qPCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and the following primer sequences: PKM2: CCATCTACCACTTGCAGCTATTC and CACTGCAGCACTTGAAGGA; GLUD1: ATCGGGTGCATCTGAGAAAG and CAGGTCCAATCCCAGGTTATAC; and β-Actin: ACCAGAGGCATACAGGGACA and CTAAGGCCAACCGTGAAAAG (Integrated DNA Technology). Data were collected on a LightCycler 480 (Roche) using a touchdown PCR protocol (95°C for 10 min, 2 cycles of 95°C for 15 s and 62°C for 1 min, 2 cycles of 95°C for 15 s and 60°C for 1 min, 2 cycles of 95°C for 15 s and 58°C for 1 min, 40 cycles of 95°C for 15 s and 56°C for 1 min).
Cell number and viability assays
Cell number and the percentage of dead cells were determined using an imaging cytometer (Celigo) as previously described (31). Briefly, cells were stained using Hoechst dye (Life Technologies) to determine cell number, whereas the number of dead cells was determined using propidium iodide (Thermo Fisher) staining. Differences in cell number were confirmed after 24 h of treatment with a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
RNA sequencing
RNA was isolated using the column purification method aforementioned. A total of 5 conditions with n = 1 replicate each were analyzed: shControl, short hairpin pyruvate kinase M2 (shPKM2), shikonin, TEPP-46, and a vehicle control. shControl and shPKM2 samples were transfected for 72 h, whereas vehicle control, shikonin, and TEPP-46 were administered for 8 h. RNA quality was determined using an AATI Fragment Analyzer. A NEBNext Poly(A) mRNA magnetic isolation Module (New England Biolabs) was used for polyA + RNA isolation. Next, TruSeq-barcoded libraries were generated using a NEBNext Ultra II RNA Library Prep Kit (New England Biolabs). The library size distribution was determined using a fragment analyzer (Advanced Analytical) and each library was quantified with a Qubit 2.0 (Thermo Fisher). The resulting libraries were pooled and sequenced using a NextSeq500 (Ilumina). At least 20 million reads were generated per library. The resulting reads were processed using TrimGalore version 0.6.0 with cutadapt (32) and fastQC. Reads were mapped to the reference genome (Ensembl GRCm38), then normalized read counts were generated using SARTools (33) and DESEq2 version 1.26.0 (34). Based on the number of replicates in each of the 5 conditions (n = 1), these data were not analyzed statistically. Potentially relevant transcripts were identified as those with a fragments per kilobase million (FPKM) value >1 and a fold change >1.5 occurring in each comparison (shPKM2 compared with shControl; shikonin compared with vehicle control; TEPP-46 compared with vehicle control).
Cellular and metabolic assays
The enzymatic activity of pyruvate kinase was determined using established methods (35). Cellular reactive oxygen species (ROS) were measured as previously described (36). Basal oxygen consumption rate and basal extracellular acidification rate were measured using the Seahorse XFe analyzer (Agilent) as previously described (30). GLUD activity was measured using a commercially available kit (Glutamate Dehydrogenase Activity Assay Kit; Millipore).
Glucose uptake (Glucose Uptake-Glo), lactate excretion (Lactate-Glo), total and oxidized glutathione (GSH/GSSG-Glo), NAD(P), and NAD(P)H (NADP/NADPH-Glo) were measured using commercial kits following the manufacturer's instructions (Promega). All measurements were normalized to cell number, which was measured in a parallel plate as aforementioned.
JC1 staining was performed following manufacturer recommendations (Cayman Chemicals) and reported as a ratio of aggregates to monomers. To measure mitochondrial DNA copy number, DNA was isolated from cultured cells using the High Pure PCR Template Preparation Kit (Roche). Copies of actin and cytochrome c oxidase subunit 1 were measured and the formula 2*2^dCp was used as an indicator of mitochondrial copy number, as previously described (37). The primers used to measure actin were GGAAAAGAGCCTCAGGGCAT and CTGCCTGACGGCCAGG (38). The primers used to measure cytochrome c oxidase subunit 1 were TGCTAGCCGCAGGCATTACT and CGGGATCAAAGAAAGTTGTGTTT (39).
Confocal microscopy
Cells were stained with mitotracker dye (250 nM, Thermo Fisher) for 30 min at 37°C, fixed in 4% paraformaldehyde for 20 min, permeabilized with triton × 100, blocked with 5% goat serum, then incubated with anti-PKM2 antibody (1:250,4053S, Cell Signaling) overnight. The following morning, cells were incubated with Alexafluor 488 goat anti-rabbit antibody (1:500, Thermo Fisher) at 37°C for 60 min and stained with Hoechst (Life Technologies). PKM2 localization was determined using super-resolution structured illumination microscopy (SIM) (Nikon Ti2 eclipse inverted microscope with Hamumatsu Orca Flash 4 Digital Camera), which enables detailed visualization of intracellular structures.
Immunoprecipitation
A volume of 100 μL Dynabeads Protein A (Invitrogen) was added to 5 mg/mL BSA (Fisher) in PBS to reduce nonspecific binding (preblocking). Beads were collected using a magnetic holder and the supernatant was discarded. Beads were washed 2 additional times with 5 mg/mL BSA in PBS. Antibodies (5 μg each) were diluted in 250 uL BSA/PBS and bound to the beads by incubation with head-over-tail rotation at 4°C overnight. The following day, beads were collected on the magnetic holder, and the supernatant was discarded. Beads were washed once in BSA/PBS followed by 2 additional washes in PBS. Antibodies used were anti-PKM2 (4053S, rabbit, Cell Signaling), anti-DBT (12451-1-AP, rabbit, ProteinTech), anti-GLUD1 (14299-1-AP, rabbit, ProteinTech), or rabbit IgG isotype control (31235, Thermo Fisher).
Cells were collected for immunoprecipitation (IP) by rinsing twice with cold PBS, detached using trypsin (VWR), and pelleted by centrifugation at 500 × g for 5 min at 4°C. Cell pellets were resuspended in lysis buffer [10 mM HEPES pH 7.6, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% sodium deoxycholate, phosphatase and protease inhibitors (PhosSTOP and cOmplete, Roche)] and sonicated for 3 s. Cell suspensions were centrifuged at 15,000 × g for 10 min at 4°C, supernatants were collected, and protein concentrations were quantified using a bicinchoninic acid (BCA) assay (Thermo Fisher).
For IP, 1.5 mg protein lysates were diluted to a concentration of 2 μg/μL in lysis buffer [10 mM HEPES pH 7.6, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% sodium deoxycholate, phosphatase and protease inhibitors (PhosSTOP and cOmplete, Roche)] before adding bead slurries prepared as aforementioned. The beads and protein lysates were incubated at 4°C with head-over-tail rotation overnight.
Beads were collected on the magnetic holder and the supernatant removed. Beads were then washed 3 times in wash buffer 1 (50 mM HEPES pH 7.6, 1 mM EDTA, 1% sodium deoxycholate) and twice in wash buffer 2 (50 mM HEPES pH 7.6). Each wash involved removing the tube from the holder and mixing on a vortex.
For samples intended for proteomics analysis, after the final wash, beads were resuspended in 200 mM glycine pH 2.6, incubated for 10 min, and quenched in 90 mM Tris pH 7.5.
For samples intended for immunoblotting, after the final wash, beads were resuspended in 25 μL radioimmunoprecipitation assay (RIPA) buffer with Laemmli buffer, incubated at 95°C for 5 min, cooled briefly on ice, and collected on the magnetic bead holder. The supernatant was collected and analyzed by immunoblotting as aforementioned.
Proteomics analysis
In solution, digestion was performed on each sample separately using S-Trap micro spin columns (ProtiFi) following a Strap protocol as previously described (40, 41) with slight modifications. Protein samples were dried using a speed vac and reconstituted in 25 μL 50 mM triethylammonium bicarbonate (TEAB) pH 8.5, 6 M urea, 2 M thiourea, 1% SDS, then reduced with 10 mM dithiothreitol (DTT) for 1 h at 34°C, alkylated with 60 mM iodoacetamide for 1 h in the dark, and quenched with a final concentration of 33 mM DTT. After quenching, 12% phosphoric acid was added to a final concentration of 1.2%, followed by 1:7 dilution (vol:vol) with 90% methanol, 0.1 M TEAB pH 8.5. The samples were then placed into the spin column and centrifuged at 4000 × g for 30 s at 4°C. Next, columns were washed 3 times with 150 μL 90% methanol, 0.1 M TEAB pH 8.5. Digestion was performed with 25 μL trypsin at 20 ng/μL (1:10 wt:wt) in 50 mM TEAB pH 8.5, which was added to the top of the spin column. Digest solution absorbed into the highly hydrophilic matrix. The spin columns were incubated overnight (16 h) at 37°C. After incubation, the digested peptides were eluted off the S-trap column sequentially with 40 μL each of 50 mM TEAB pH 8.5 followed by 0.2% formic acid (FA) and, finally, 50% acetonitrile (ACN), 0.2% FA. The 3 eluted peptide washes were pooled together and evaporated to dryness by a Speedvac SC110 (Thermo Savant).
The tryptic digests were reconstituted in 22 μL 0.5% FA for nanoLC-ESI (electrospray ionization)-MS/MS analysis, which was carried out using an Orbitrap Fusion™ Tribrid™ (Thermo Fisher) mass spectrometer equipped with a nanospray Flex Ion Source, and coupled with a Dionex UltiMate3000RSLCnano system (Thermo) (40). The tryptic digested peptides (4 μL) were injected onto a PepMap C-18 RP nano trapping column (5 μm, 100 μm i.d. × 20 mm) at a 20-μL/min flow rate for rapid sample loading and then separated on a PepMap C-18 RP nano column (2 μm, 75 μm × 25 cm) at 35°C. The tryptic peptides were eluted in a 90-min gradient of 5% to 38% ACN in 0.1% FA at 300 nL/min, followed by a 7-min ramping to 90% ACN-0.1% FA and an 8-min hold at 90% ACN-0.1% FA. The column was re-equilibrated with 0.1% FA for 25 min before the next run. The Orbitrap Fusion was operated in positive ion mode with spray voltage set at 1.8 kV and source temperature at 275°C. External calibration for Fourier transform (FT), injection time (IT), and quadrupole mass analyzers was performed. In data-dependent acquisition analysis, the instrument was operated using the FT mass analyzer in MS scan to select precursor ions followed by 3-s “Top Speed” data-dependent collision induced dissociation (CID) ion trap MS/MS scans at 1.6 m/z quadrupole isolation for precursor peptides with multiple charged ions above a threshold ion count of 10,000 and normalized collision energy of 30%. MS survey scans were at a resolving power of 120,000 (full width at half maximum at m/z 200) for the mass range of m/z 375–1575. Dynamic exclusion parameters were set at 50 s of exclusion duration with ±10 ppm exclusion mass width. All data were acquired under Xcalibur 3.0 operation software (Thermo Fisher).
Resulting spectra were searched against the Swiss-Prot mouse database (UP000000589, November 2019) with the Lehtiö Lab Data-Dependent Acquisition Mass Spectrometry Proteomics Pipeline version 1.4 (42) applying MSGF+ and Percolator. The search was performed specifying fixed carbamidomethyl modifications of all cysteine residues, variable oxidation of all methionine residues, and a precursor tolerance of 10 ppm. Only fully tryptic peptides were matched with no limit on the number of missed cleavages. A target-decoy strategy was applied to achieve peptide spectral match (PSM) and peptide false discovery rates (FDRs) of 1% (43, 44) and the picked FDR method (45) was applied to achieve a protein FDR of 1%. No spectra were omitted based on predesignated contaminants.
siRNA transfection
To knock down GLUD1 concentrations, C2C12s were reverse transfected with 25 nM small interfering RNA (siRNA) targeting GLUD1 [siGLUD1, On-TARGETplus Mouse Glud1 (14661) siRNA—SMARTpool] or a nontargeting control (siControl, ON-TARGETplus Non-targeting pool) with Dharmafect 1 (T-2001-01 Horizon Bioscience). The following day, vehicle control, shikonin, or TEPP-46 treatments were started. Knockdown effectiveness was measured at 48 h of siRNA transfection and cell number was measured at 60 h of shikonin/TEPP-46 treatment.
Subcellular fractionation
Subcellular fractionation of the nucleus away from the remainder of the cell was performed following the REAP method (46). Lysates were analyzed by immunoblotting as aforementioned.
To isolate mitochondria and cytoplasmic fractions, cultured cells were scraped and collected in PBS. Cells were pelleted by centrifugation (500 × g for 5 min at 4°C), washed in PBS, and pelleted again. After aspirating the supernatant, the pellet was flash frozen in liquid nitrogen, and thawed on ice. Next, 3 mL hypotonic solution [83 mM sucrose (Sigma), 10 mM MOPS (Mallinckrodt) pH 7.2] was added to each sample. After a 10-min incubation on ice, samples were passed through a tight pestle clearance dounce homogenizer 25 times. Next, 3 mL hypertonic solution was added (250 mM sucrose, 30 mM MOPS pH 7.2) and samples were centrifuged at 1000 × g for 5 min at 4°C to pellet nuclei and unlysed cells. The pellet was discarded and supernatant was collected in a new tube. The supernatant was recentrifuged to remove any remaining nuclei and unlysed cells. To separate the mitochondria and cytosol, the supernatant was centrifuged at 9000 × g for 12 min at 4°C. After the centrifugation, the supernatant was collected as a cytosolic fraction. The mitochondrial pellet was resuspended in wash buffer [320 mM sucrose, 1 mM EDTA (Fisher), 10 mM Tris-HCl (Amresco), pH 7.2]. The mitochondrial pellet was washed 5 times. Each wash step involved resuspending the pellet in wash buffer and centrifugation at 9000 × g for 12 min at 4°C. After the last wash step, the pellet was resuspended in RIPA buffer. Immunoblotting was performed as aforementioned.
Mouse model generation
All studies involving mice were approved by the Cornell University Institutional Animal Care and Use Committee. PKM2fl/fl mice [Jackson Laboratory, stock number 024048 (47)] were bred with Pax7creER mice [Jackson Laboratory, stock number 017763 (48)] and Rosa26-tdTomato [red fluorescent protein (RFP); Jackson Laboratory, stock number 007914 (49)]. Resulting mice were Pax7-PKM2fl/fl; RFP+ [PKM2 knockout (KO) mice] and Pax7-PKM2wt/wt; RFP+ (control mice). Mice were bred in-house and maintained on a 14-h light, 10-h dark schedule. For models of muscle development, 10-d-old pups were injected once with tamoxifen (Cayman Chemicals, 1 mg/kg IP) suspended in sunflower oil (Sigma). Mice (4–7/genotype) were killed at 30 d old. Tibialis anterior (TA) wet weight was measured using a standard laboratory scale and normalized to tibia bone length, which was measured with a ruler. For models of regeneration after an injury, 8-wk-old mice were injected on 2 consecutive days with tamoxifen (Cayman Chemicals, 50 mg/kg IP). The following day, the TA muscle, a common muscle examined in injury studies (1, 36), was injected with 50 μL 1.2% barium chloride (VWR) under anesthesia with isoflurane. Mice were killed at 10 or 21 d postinjury (4–6 mice per sex per genotype). Both male and female mice were used for all experiments.
Sectioning and staining
The TA was mounted to cork using optimal cutting temperature compound (Tissue-Tek) mixed with tragacanth powder (Alfa Aesar) and frozen in isopentane over liquid nitrogen. Samples were sectioned on a cryostat (Leica) into 7-μM sections. Samples were stained with hematoxylin and eosin (H&E), or antibodies targeting proteins of interest, specifically laminin and Pax7. In uninjured 30-d-old mice, myofiber cross-sectional area (CSA) was measured using H&E-stained images. In injured mice, myofiber CSA was measured using laminin-stained images, to better capture disruption of myofiber size/integrity in response to injury. Laminin was stained using anti-Laminin primary antibody (Sigma) and Alexafluor 488 secondary antibody (Thermo Fisher). Pax7 staining was performed using an Alexafluor 594 tyramide kit (Invitrogen) as previously described (36). For each mouse and outcome, 4 representative images were quantified. To determine CSA, a minimum of 1200 fibers were measured for each mouse and for Pax7 cell number, all Pax7+ cells in the section were counted. Images were collected on a Zeiss 710 confocal or Olympus upright BX-50 microscope with Motic Image Plus 2.0 software.
Grip strength testing and NMR procedure
Grip strength was measured using a grip strength apparatus (BIOSEB) at postnatal day 30. Mice were lifted from their cage by the tail and placed in proximity to a metal grid, onto which the mice gripped with their forelimbs. Next, the researcher pulled the mouse gently away from the grid and the maximum force generated by the mouse before it was pulled from the grid was measured by the instrument. Each mouse was tested 5 times and the maximum strength across the trials was recorded. Body composition of mice was measured using NMR (Bruker Minispec LF65).
MPC isolation from control and PKM2 KO mice
Mice were killed with carbon dioxide followed by cervical dislocation. All hindlimb muscles were isolated from the mouse and temporarily incubated in low-glucose DMEM (Gibco). After all mice had been killed and muscle collected, samples were moved into a biological safety cabinet and minced using a new razor blade. Samples were then washed in PBS. Next, collagenase (Roche) enzyme, resuspended in low-glucose DMEM, was added to the tissue and samples were incubated in a 37°C water bath for 30 min. Eight units of dispase enzyme (Sigma) were added to the sample, which was incubated for an additional hour with mixing on a vortex every 10 min to enhance digestion. Next, samples were passed through a 70-μm filter, digestion enzymes were quenched by the addition of mouse GM [41.5% low-glucose DMEM, 41.5% Ham's F10 (Gibco), 15% FBS, 1% GlutaMAX, 1% penicillin/streptomycin (Corning)], and cells were pelleted by centrifugation at 500 × g for 5 min at room temperature. The resulting cell pellet was incubated in RBC lysis buffer (BioLegend) on ice for 5 min, quenched with medium, and centrifuged to pellet the cells again. The cell pellet was next resuspended in a negative selection antibody cocktail [anti-CD45 (BioLegend), anti-CD31 (BioLegend), anti-CD11b (BioLegend), anti-SCA1 (BioLegend)] in PBS containing 10% goat serum. Samples were incubated with the antibody cocktail on ice for 20 min, diluted in PBS with 10% goat serum, and pelleted. Samples were then incubated with MagniSort SAV Negative Selection Beads (Invitrogen) on a head-over-tail rotator for 10 min and placed on a magnetic stand for 5 min to pellet the beads. The resulting solution was added to GM and centrifuged at 500 × g for 5 min at room temperature to pellet the cells. The resulting cell pellet was resuspended in GM and plated onto non-collagen-coated plates to encourage plating of fibroblasts in conditions that are not optimal for MPC attachment. After 1 h the supernatant was collected from the plate and centrifuged at 500 × g for 5 min at room temperature, then resuspended and seeded in mouse GM with basic fibroblast growth factor (bFGF, 2.5 ng/mL; Promega). When indicated, mouse MPCs were cultured in an alternative medium modeled after a previous publication [89% Hams F10, 1% penicillin/streptomycin, 10% fetal calf serum (Hyclone), 2.5 ng/mL bFGF] (11). Cell number was determined using the Celigo analyzer by counting red cells, because the mouse MPCs express RFP. To address mouse-to-mouse variation in initial cell number, a ratio of cell number at 96 h:48 h was used as an indicator of cell proliferation rate.
Statistics
Statistical analysis was performed in GraphPad Prism 9.0.0. Protein concentrations of PKM1 and PKM2 across time were compared using a 1-factor ANOVA, with Tukey's multiple-comparisons post hoc test. The impact of shPKM2 compared with shControl on MPC number over time was compared using a 2-factor ANOVA with main effects of time and shRNA. Sidak's multiple-comparisons test was used to compare the impact of shRNA within each time point. The impact of shPKM2/shControl or vehicle control/shikonin and supplemental pyruvate on MPC number was determined using a 2-factor ANOVA with main effects of treatment and pyruvate. The impact of shikonin or TEPP-46 on cell number, or the percentage of dead cells, was determined using a 2-factor ANOVA with main effects of time and treatment. Sidak's multiple-comparisons test was used to compare the impact of treatment within each time point.
Pyruvate kinase enzyme activity, MTT absorbance, ROS concentrations, HO-1 protein concentrations, NRF2 protein concentrations, glucose uptake, lactate excretion, basal extracellular acidification rate, basal oxygen consumption rate, JC1 aggregate:monomer ratio, mitochondrial DNA copy number, and GLUD activity were compared between vehicle control and small-molecule (shikonin or TEPP-46) treated cultures with an unpaired t test (1-sided test for ROS, HO-1, and NRF2 concentrations). The impact of NAC and GSHee on cell number, or siGLUD1 and siControl, was determined using a 2-factor ANOVA with main effects of PKM2 inhibitor treatment and NAC/GSHee or vehicle control, or siGLUD1 and siControl. If a significant interaction was observed, Sidak's multiple-comparisons test was used to compare the effect of treatment within each PKM2 inhibitor/vehicle treatment group.
The impact of glucose availability (glucose containing, galactose containing, glucose free) and mitochondrial inhibitors (control compared with small-molecule inhibitor) was compared using a 2-factor ANOVA with main effects of time and medium/treatment. Sidak's multiple-comparisons test was used to determine the impact of treatment within each time point.
Cell number ratios in mouse MPCs were compared using a 1-factor ANOVA. Normalized TA wet weights, grip strength, percentage lean mass, percentage body fat, mean fiber CSA, and number of Pax7 positive cells were compared between wild-type (WT) and PKM2 KO mice using an unpaired t test. For all results, significance was determined as P < 0.05 and P < 0.1 was considered a trend.
Results
Noncanonical PKM2 activity supported MPC proliferation
Building on previous reports (10, 50), we found that PKM2 expression declined during later stages of differentiation (3–6 d) compared with concentrations in the growth stage or at 1 d of differentiation (Supplemental Figure 1A). Conversely, PKM1 increased from proliferation through differentiation (Supplemental Figure 1B).
Knockdown of PKM2 (shPKM2, 72% reduction in PKM2 mRNA concentration) reduced cell number compared with shControl (Figure 2A). This genetic approach reduced both canonical and noncanonical PKM2 activity. We reasoned that if canonical activity was required for MPC proliferation, lower production of pyruvate, as a consequence of lower PKM2 concentrations, may limit cell proliferation. Although providing supplemental pyruvate showed a trend toward increased MPC proliferation (P < 0.1) in both shPKM2 and shControl cultures (Figure 2B), supplemental pyruvate did not rescue the lower cell number in cultures treated with shPKM2.
FIGURE 2.

The noncanonical activity of PKM2 promotes MPC proliferation. (A) Cell number in cultures transfected with shControl or shPKM2 (n = 3). (B) Cell number in cultures transfected with shControl or shPKM2 and supplemental pyruvate (n = 4). (C) PK enzyme activity (n = 4), (D) cell number (n = 3), (E) MTT absorbance (n = 4), and (F) percentage of dead cells (n = 4) in vehicle control or shikonin-treated cultures. (G) Cell numbers in cultures treated with vehicle control or shikonin and pyruvate (n = 3). (H) PK enzyme activity (n = 4), (I) cell number (n = 3), (J) MTT absorbance (n = 4), and (K) percentage of dead cells (n = 5) in vehicle control or TEPP-46 treated cultures. Values are mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (asterisks indicate significant effects). AU, arbitrary units; GM, growth medium; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; PK, pyruvate kinase; shControl, short hairpin control; shPKM2, short hairpin pyruvate kinase M2.
To more specifically probe the contributions of noncanonical and canonical PKM2, we treated MPCs with 2 small molecules. The first, shikonin, blocks canonical (Figure 2C) and noncanonical PKM2 activity (15). Treating with shikonin reduced cell number (Figure 2D) and MTT absorbance, an indirect indicator of cell number (Figure 2E), compared with the vehicle control. Cell death did not vary substantially between shikonin-treated and control cells (Figure 2F). As observed in the shPKM2 model, supplemental pyruvate increased cell number in both the vehicle control and shikonin-treated cultures, but supplemental pyruvate did not rescue the impact of shikonin (Figure 2G). The second small molecule, TEPP-46, increases canonical activity, as verified by the increase in pyruvate kinase enzymatic activity (Figure 2H), at the expense of noncanonical activity (16). TEPP-46 treatment reduced cell number (Figure 2I) and MTT absorbance (Figure 2J), although it did not alter the percentage of dead cells (Figure 2K). Based on these results, we posit that the noncanonical activity of PKM2 supports MPC proliferation.
Interestingly, although a previous report demonstrated that the small molecule acteoside can increase PKM2 concentrations and MPC proliferation and providing supplemental PKM2 can increase MPC proliferation (12), we were unable to reproduce either of these effects (Supplemental Figure 1C, D).
RNA sequencing revealed oxidative stress regulation by PKM2
To give insight into pathways altered by PKM2 in proliferating MPCs, RNA-sequencing data were compared between shPKM2 and shControl cells, as well as vehicle control and shikonin or TEPP-46. Many of the genes that were upregulated >1.5-fold across all 3 models encoded protein products involved in oxidative stress regulation including glutamate-cysteine ligase catalytic subunit (Gclc), glutathione S-transferase α (Gsta)1, Gsta4, glutathione S-transferase (Gm3776), heme oxygenase 1 (Hmox1), and solute carrier family 7 member 11 (slc7a11) (Figure 3A). This finding motivated us to examine oxidative stress when noncanonical PKM2 activity was impaired. ROS, a key contributor to oxidative stress, were increased after shikonin or TEPP-46 treatment (Figure 3B). In addition, HO-1, a mediator of cellular stress responses which is the protein product of Hmox1, was increased after either shikonin or TEPP-46 treatment (Figure 3C). Many of the transcripts upregulated in the RNA-sequencing data set were targets of NRF2 [alcohol dehydrogenase 7, Gsta1, Gsta4, Gclc, Gm3776, Hmox1, HIV-1 Tat interactive protein 2, slc7a11 (51–53)], a master regulator of cellular antioxidant capacity; therefore, we next measured NRF2 concentrations. NRF2 was increased after either shikonin or TEPP-46 treatment (Figure 3D). In addition, targets identified encode proteins involved in glutathione synthesis and recycling (Gclc, Gsta1, Gsta4, Slc7a11). Interestingly, whereas shikonin increased total glutathione and oxidized glutathione (GSSG) concentrations (Supplemental Figure 2A, B), no change was observed with TEPP-46. In addition, NAD(P)H, the cofactor involved in glutathione recycling (from GSSG to reduced glutathione), was increased with shikonin treatment, but not TEPP-46 treatment (Supplemental Figure 2C, D). Providing cell-permeable glutathione (GSHee) or NAC, a precursor to glutathione, did not rescue cell number in shikonin- or TEPP-46-treated cultures (Supplemental Figure 2E) and, in fact, NAC was toxic at even low doses.
FIGURE 3.
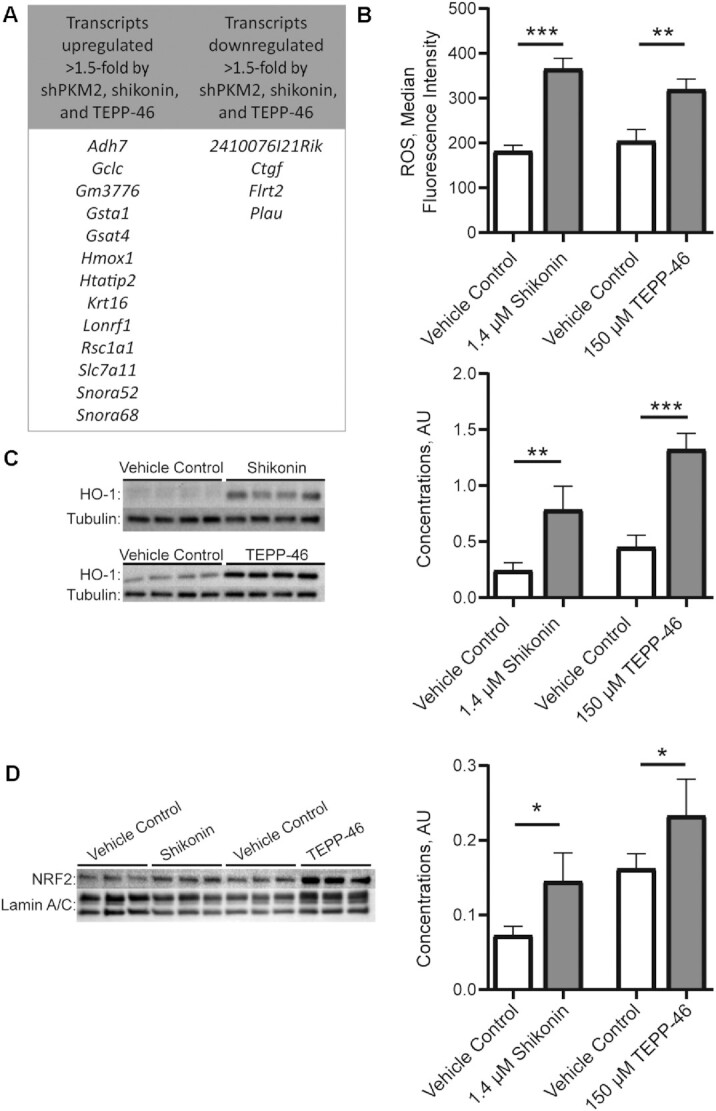
RNA sequencing and follow-up measurements of oxidative stress in models of inhibited pyruvate kinase M2 activity in MPCs. (A) Transcripts up- or downregulated >1.5-fold in shPKM2 compared with short hairpin control, and shikonin or TEPP-46 compared with a vehicle control. (B) ROS (n = 3), (C) HO-1 protein concentrations (n = 4), and (D) NRF2 protein concentrations (n = 3) in vehicle control, shikonin, and TEPP-46 treated cultures. Values are mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (asterisks indicate significant effects). Adh7, alcohol dehydrogenase 7; AU, arbitrary units; Ctgf, connective tissue growth factor; Flrt2, fibronectin leucine rich transmembrane protein 2; Gclc, glutamate-cysteine ligase catalytic subunit; Gm3776, glutathione S-transferase; Gsta, glutathione S-transferase α; hmox1, heme oxygenase 1; HO-1, heme oxygenase 1; Htatip2, HIV-1 Tat interactive protein 2; Krt16, keratin 16; Lonrf1, LON peptidase N-terminal domain and ring finger 1; NRF2, nuclear factor erythroid 2-related factor 2; Plau, plasminogen activator urokinase; ROS, reactive oxygen species; Rsc1a1, regulator of solute carriers 1; slc7a11, solute carrier family 7 member 11; shPKM2, short hairpin pyruvate kinase M2; Snora, small nucleolar RNA H/ACA box.
PKM2 altered glucose uptake and mitochondrial respiration, fundamental processes for MPC proliferation
Across many cell types, PKM2 has been shown to regulate glucose and mitochondrial metabolism. Although, whether PKM2 is stimulatory or inhibitory varies among different cell types. Culturing MPCs in glucose-free or galactose-containing media prevents MPC proliferation (Figure 4A). Similarly, blocking complex 1 (rotenone), the ATP synthase (oligomycin), or inducing mitochondrial uncoupling (FCCP) reduced MPC proliferation (Figure 4B). Thus, both glucose metabolism and mitochondrial respiration are essential for MPC proliferation and altering these requirements could underlie the role of PKM2 in MPC proliferation. Treating with shikonin or TEPP-46 reduced glucose uptake (Figure 4C). Interestingly, extracellular acidification rate (Figure 4D) and lactate excretion (Figure 4E) were increased after either shikonin or TEPP-46 treatment. This suggests that despite lower glucose uptake, glucose or pyruvate from other substrates may be preferentially shunted to lactate in the absence of noncanonical PKM2 activity. Shikonin and TEPP-46 treatment also reduced basal oxygen consumption rate (Figure 4F). This decrease was accompanied by a reduction in mitochondrial membrane potential (Figure 4G), but no change was observed in mitochondrial copy number (Figure 4H). Thus, the noncanonical activity of PKM2 is important for maintaining mitochondrial respiration.
FIGURE 4.
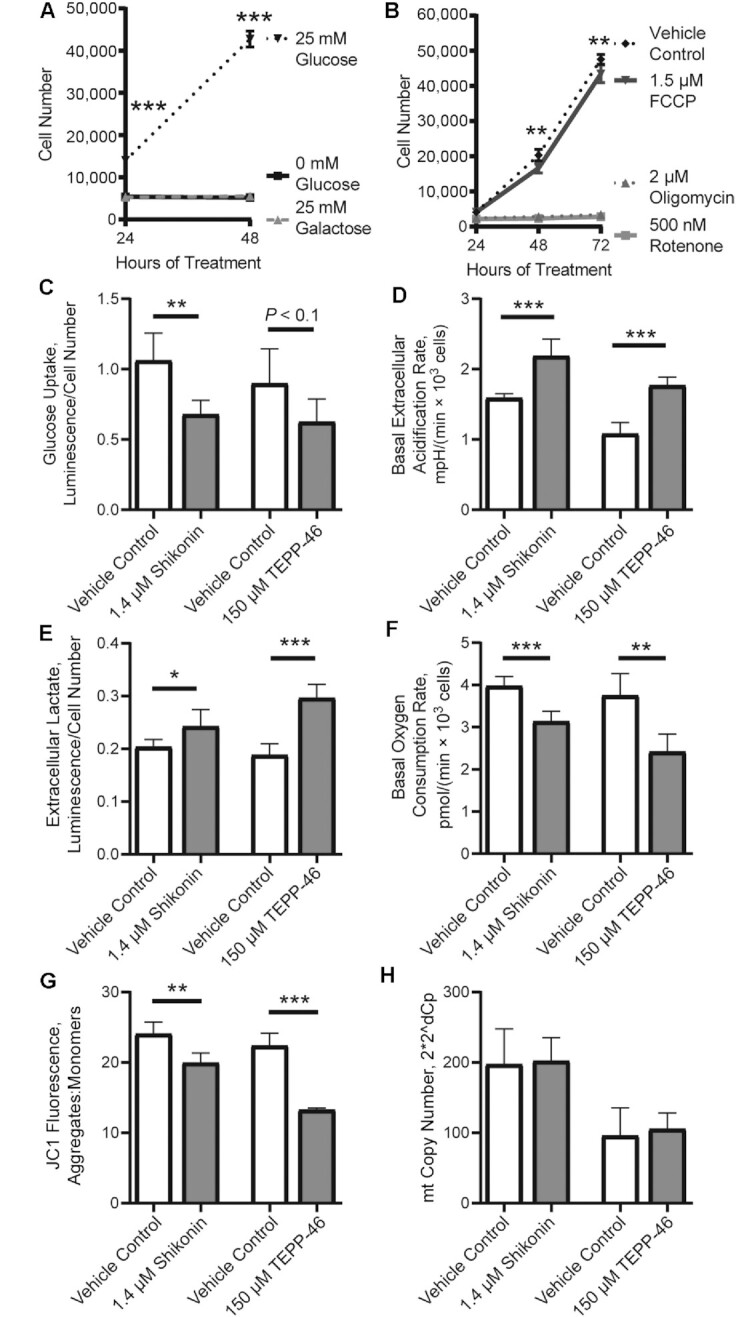
Metabolic requirements for MPC proliferation and alterations in metabolism by altering noncanonical pyruvate kinase M2 activity. (A) Cell number in cultures given glucose-free medium, glucose-free medium with glucose added back, or glucose-free medium with added galactose (n = 6). (B) Cell number in cultures given a vehicle control, FCCP, oligomycin, or rotenone (n = 3). (C) Glucose uptake (n = 5), (D) extracellular acidification (n = 5), (E) extracellular lactate (n = 6), (F) basal oxygen consumption rate (n = 5), (G) JC1 aggregate:monomer ratio (n = 5), and (H) mtDNA copy number (n = 5) in cultures treated with vehicle control, shikonin, or TEPP-46. Values are mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (asterisks indicate significant effects). FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone; mt, mitochondrial.
PKM2 interacted with GLUD1 in proliferating MPCs
Previous research identified interactions of PKM2 with transcription factors, and the nuclear localization of PKM2 is thought to be important for PKM2-driven alterations in proliferation and metabolism (28). Interestingly, unlike HEK293 and Hela cells, which were measured as assay controls, MPCs did not have a measurable fraction of nuclear PKM2 detectable by immunoblotting (Figure 5A, Supplemental Figure 3); however, cellular localization of PKM2 with SIM revealed a small fraction of PKM2 in the nucleus (Figure 5B).
FIGURE 5.
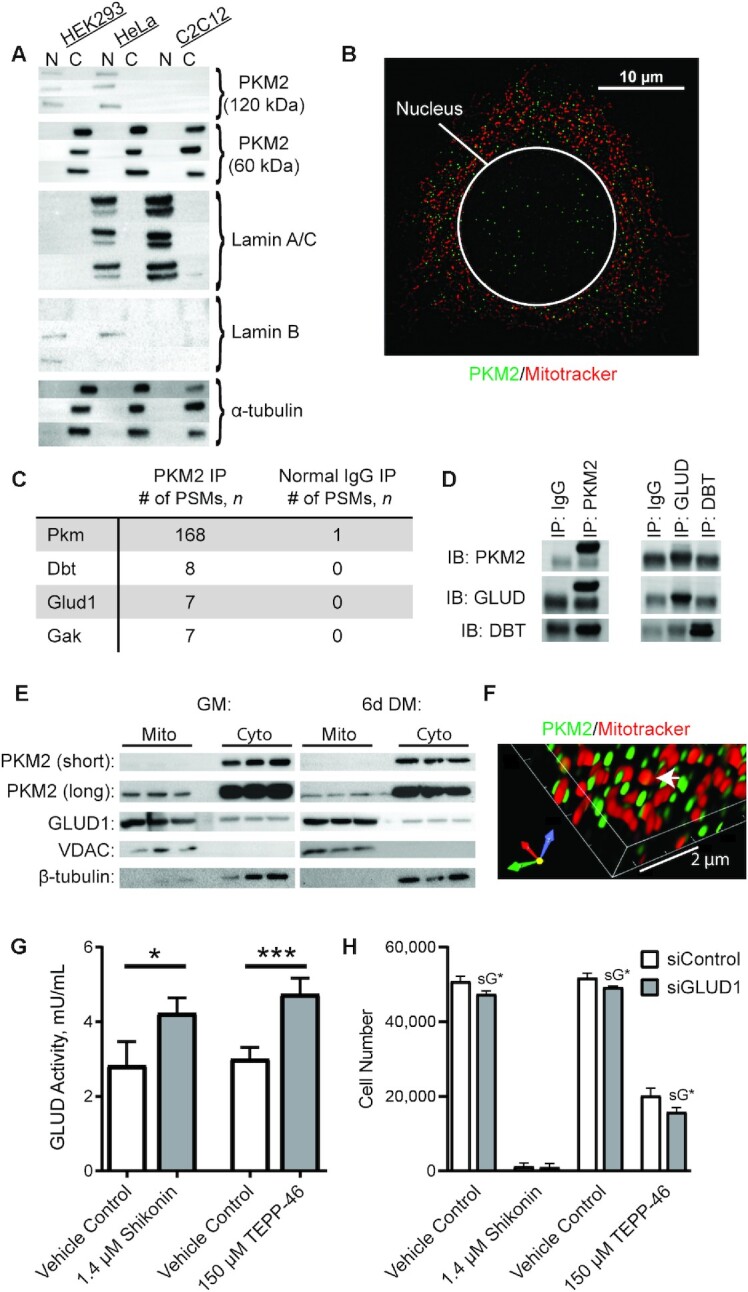
PKM2 localization and binding partners in MPCs. (A) Immunoblotting of PKM2 and cytosolic (tubulin) and nuclear (Lamin A/C and lamin B) markers in MPCs, HeLa cells, and HEK293 cells. N indicates nuclear fraction and C indicates rest of cell (n = 3). (B) Confocal microscopy of Hoechst, mitotracker, and PKM2 in MPCs. (C) Top 3 candidate PKM2 interacting proteins identified using nontargeted MS-based proteomic analysis. (D) Validation of PKM2 binding partners by IP coupled to immunoblotting. (E) Subcellular fractionation of cytosolic and mitochondrial fractions (n = 3 replicates). (F) Co-staining of PKM2 and Mitotracker imaged using structured illumination microscopy. (G) GLUD activity in vehicle control, shikonin, or TEPP-46 treated cultures (n = 4). (H) Cell number in cultures treated with vehicle control, shikonin, or TEPP-46 and siControl or siGLUD1 (n = 4). Values are mean ± SD. *P < 0.05, ***P < 0.001 (asterisks indicate significant effects). sG*, effect of siGLUD1. Dbt, dihydrolipoamide branched-chain transacylase E2; DM, differentiation medium; Gak, cyclin G associated kinase; Glud1, glutamate dehydrogenase 1; GM, growth medium; IB, immunoblot; IP, immunoprecipitation; MPC, muscle progenitor cell; Pkm, pyruvate kinase; PKM2, pyruvate kinase M2; PSM, peptide spectral match; siControl, small interfering RNA non-targetting control; siGLUD1, small interfering RNA targetting GLUD1; VDAC, voltage-dependent anion channel.
Co-IP was used to enrich for PKM2 and its interacting partners, and the sample was analyzed using MS-based proteomics to infer the identity of PKM2 interactors (Supplemental Tables 1–3). As expected, PKM was the most abundant species identified in the sample (168 PSMs), followed by DBT (8 PSMs), a protein involved in branched-chain amino acid catabolism; GLUD1 (7 PSMs), a protein that catalyzes the oxidative deamination of glutamate to α-ketoglutarate; and cyclin G associated kinase (GAK, 7 PSMs), a protein that regulates cyclin G (Figure 5C). To follow up, we immunoprecipitated PKM2 and immunoblotted for GLUD1 and DBT, or immunoprecipitated GLUD1 and DBT followed by immunoblotting for PKM2. These results revealed an interaction of PKM2 and GLUD1, but did not provide confirmation of a PKM2-by-DBT interaction (Figure 5D). Therefore, additional studies focused on the interaction of PKM2 with GLUD1.
To determine the subcellular location where PKM2 and GLUD1 may interact, an additional subcellular fractionation was performed that yielded cytosolic and mitochondrial fractions. The assay was performed in GM or after 6 d in differentiation medium, a time point when PKM2 concentrations are declining. PKM2 was dominantly localized to the cytosol, although with a longer exposure time, a mitochondrial fraction was visualized (Figure 5E). In addition, SIM confirmed some localization of PKM2 within mitochondria (Figure 5F). This is consistent with glioblastoma and HCT116 cells, where mitochondrial PKM2 has also been observed, under various physiological conditions (e.g., oxidative stress, glucose starvation) (54, 55). In addition, GLUD1, in addition to the anticipated mitochondrial fraction, showed a notable cytoplasmic fraction. Interestingly, blocking noncanonical PKM2 activity with shikonin or TEPP-46 increased GLUD1 enzymatic activity (Figure 5G).
In mammary epithelial cells, GLUD1 is more highly expressed in quiescent than in proliferating cells, and overexpressing GLUD1 reduces cell proliferation (56). To determine whether PKM2 promotes MPC proliferation through inhibiting GLUD1 activity, MPCs were treated with an siRNA targeting GLUD1 (siGLUD1) or control (siControl) and with shikonin and TEPP-46. The siGLUD1 treatment resulted in a 93% knockdown of GLUD1. Cell number was slightly suppressed by siGLUD1, a finding that may be expected owing to similar findings in lung and breast cancer cells (57). siGLUD1 cells were not protected from reductions in cell number by shikonin or TEPP-46 (Figure 5H). The specific effects of GLUD1 on MPC function should be addressed in future research.
Skeletal muscle stem cell–specific PKM2 was dispensable for muscle development and regeneration in vivo
To assess the role of skeletal muscle stem cell–specific PKM2 in vivo, Pax7-CreER mice were bred to Rosa26-tdTomato mice (RFP) and PKM2fl/fl mice generating Pax7-PKM2wt/wt; RFP+ (WT) and Pax7-PKM2fl/fl; RFP+ (KO) mice (Figure 6A, Supplemental Figure 4). After isolation and culture of MPCs from these mice we confirmed PKM2 deletion in KO mice and expression of RFP in both WT and KO mice (Figure 6B, C). However, no difference in cell number was evident between the WT and KO MPCs, suggesting that MPCs from KO mouse adapted to the loss of PKM2. This finding is contrary to inhibition of PKM2 in vitro and previous results in MPCs from whole-body PKM2 KO mice (11). To verify that observed differences in cell proliferation were not due to differences in culture medium, we compared the proliferation of KO and WT cells in different media. When cultured in either the medium our laboratory commonly uses for mouse MPCs (Ham's F10/low-glucose DMEM/15% FBS) or a medium previously used, in which differences in MPC proliferation were identified between WT and whole-body PKM2 KO mice [Ham's F10, 10% fetal calf serum (11)], no differences were observed in MPC proliferation (Supplemental Figure 5).
FIGURE 6.

Generation of an MPC-specific PKM2 KO mouse and measurements of muscle development. (A) Pax7-PKM2wt/wt; RFP+ (WT) and Pax7-PKM2fl/fl; RFP+ (KO) mice were generated. (B, C) Isolation of MPCs after in vivo tamoxifen injection confirmed successful PKM2 deletion (n = 2/genotype). (D) Ten-day-old pups were injected with tamoxifen and aged to 30 d of age for measurements (n = 7 WT and n = 4 KO). (E) TA wet weight normalized to tibia bone length, (F) maximum grip strength, (G) percentage lean mass, (H) percentage fat mass, (I) fiber CSA distribution, and (J) Pax7+ cell number were compared between WT (n = 7) and KO (n = 4) mice. Yellow arrows show Pax7+ cells. Values are mean ± SD. AU, arbitrary units; CSA, cross-sectional area; KO, knockout; MPC, muscle progenitor cell; Pax7, paired box 7; PKM2, pyruvate kinase M2; RFP, red fluorescent protein; TA, tibialis anterior; WT, wild-type.
To assess the role of PKM2 in muscle development, mice were injected with tamoxifen at 10 d of age and assessed at 30 d of age, capturing a period of time when Pax7-positive cells fuse onto muscle fibers contributing to postnatal muscle growth (58) (Figure 6D). WT and KO mice did not differ in muscle size (Figure 6E), maximum grip strength (Figure 6F), percentage lean mass (Figure 6G), or percentage body fat (Figure 6H). Similarly, mean fiber CSA was not different between WT and KO mice (Figure 6I, Supplemental Figure 6). The number of muscle stem cells per unit of muscle area also did not differ between WT and KO mice (Figure 6J).
To assess the role of PKM2 in muscle regeneration, WT and PKM2 KO mice were aged to 8 wk, injected with tamoxifen, and muscle injury was induced by intramuscular injection with barium chloride (Figure 7A). At 10 d postinjury, RFP labeling in the muscle fibers confirmed fusion of Pax7+ cells, indicating tissue regeneration. At 10 d postinjury, WT and KO mice did not differ in fiber CSA (Figure 7B, C). Similarly, the fiber CSAs did not differ between WT and KO mice at 21 d postinjury (Figure 7D, E). After a muscle injury, a subset of MPCs commit to the muscle lineage, whereas others self-renew to maintain the stem cell pool. To determine self-renewal potential, Pax7+ cell numbers were compared between WT and KO mice at 21 d postinjury. The number of Pax7+ cells did not differ between WT and PKM2 KO mice (Figure 7F, G), thus MPC self-renewal is not negatively affected by loss of PKM2.
FIGURE 7.
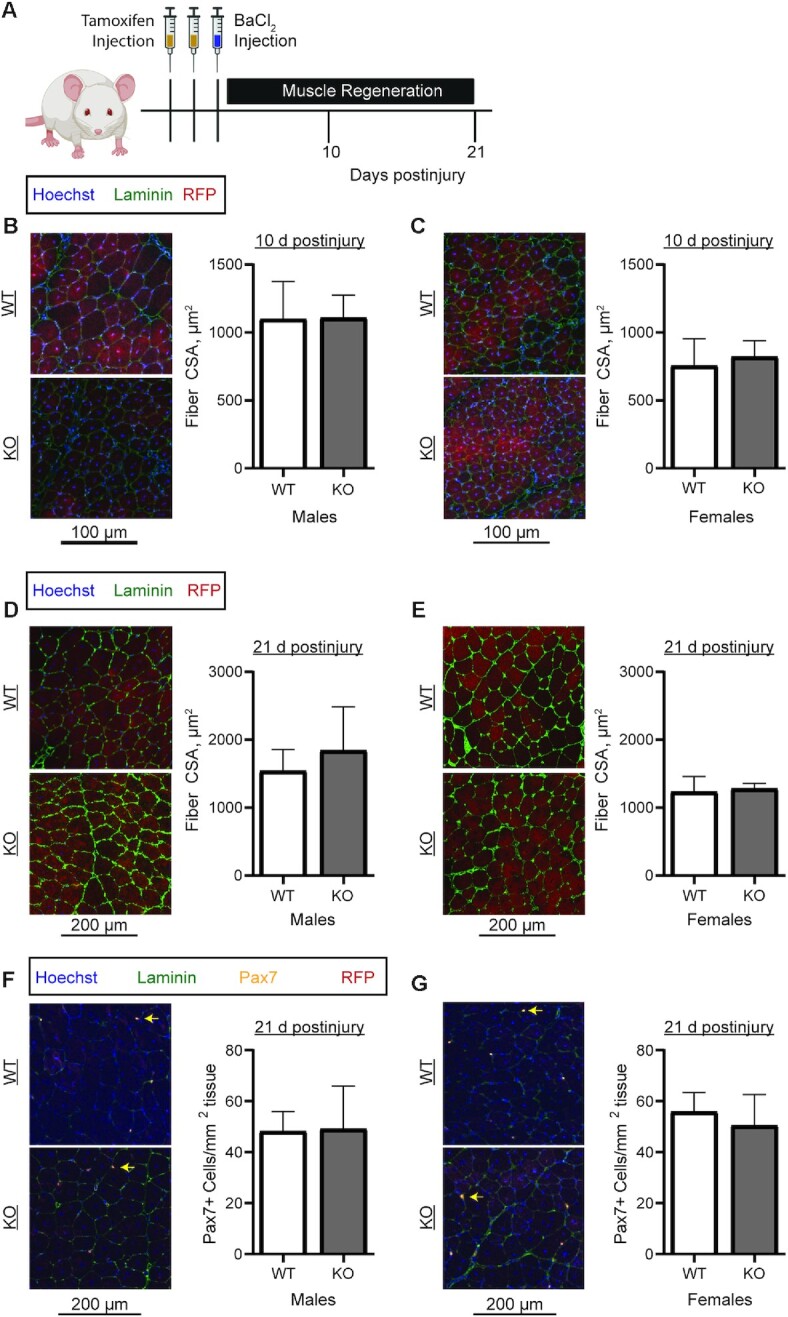
Assessment of regenerative capacity in WT and PKM2 KO mice. (A) Eight-week-old mice were injected twice with tamoxifen on 2 consecutive days and with barium chloride on the third day. Mice were killed at 10 and 21 d postinjury. (B, C) Mean myofiber CSA at 10 d postinjury in (B) male (n = 5/genotype) and (C) female (WT, n = 6; KO, n = 4) WT and KO mice. (D, E) Mean myofiber CSA at 21 d postinjury in (D) male (n = 5/genotype) and (E) female (WT, n = 6; KO, n = 4) WT and KO mice. (F, G) Pax7+ cell number at 21 d postinjury in (F) male (n = 5/genotype) and (G) female (WT, n = 6; KO, n = 4) WT and KO mice. Yellow arrows show Pax7+ cells. Values are mean ± SD. CSA, cross-sectional area; KO, knockout; Pax7, paired box 7; RFP, red fluorescent protein; WT, wild-type.
Discussion
In the present study, by combining multiple approaches that differentially affect canonical and noncanonical PKM2 activity, we were able to ascribe the effect of PKM2 on MPC proliferation to the noncanonical activity of PKM2. Further, we identified GLUD1 as a novel PKM2 binding partner in MPCs. A relation between PKM2 and cell proliferation has previously been explored across many cell types. Whereas in most cells, reducing PKM2 blunts cell proliferation (13, 21, 22, 59, 60), in a subset of cell types inhibiting PKM2 has no effect or a positive effect on proliferation (29, 61). Our findings confirmed previous findings that inhibiting PKM2 retards MPC proliferation (11). However, unlike a previous study (12), we did not observe an increase in MPC proliferation after supplementation with recombinant PKM2. Differential responses between the studies may be due to the modest magnitude of proliferation with recombinant PKM2 in the original study.
Excitingly, this research uncovered GLUD1 as a novel PKM2 binding partner. GLUD1 supports oxidative deamination of glutamate to α-ketoglutarate (62). In addition to GLUD1, glutamate can be converted to α-ketoglutarate through the activity of transaminase enzymes that shuttle the glutamate nitrogen into biosynthetic intermediates, a process that is favored in proliferating cells. In mammary epithelial cells, the transition from a highly proliferative to quiescent state is associated with an increase in GLUD1 activity and decrease in transaminase activity (56). Further, overexpression of GLUD1 in these epithelial cells reduced cellular synthesis of nonessential amino acids, suggesting decreased transaminase enzyme activity (56). Our data showed that blocking PKM2 increases GLUD1 activity, which suggests that PKM2 inhibits GLUD1 activity during MPC proliferation, potentially allowing more glutamate to be readily available for transaminase enzymes. In support of this hypothesis, during the transition from MPC quiescence to activation, when PKM2 concentrations were previously shown to increase (4.9-fold mRNA concentration), there was a corresponding increase in transaminase enzyme concentrations [glutamic-oxaloacetic transaminase 1 and 2 (3.2-fold and 2.0-fold, respectively), phosphoserine aminotransferase 1 (8.2-fold), and branched-chain aminotransferase 1 (5.4-fold) mRNA concentration] (9). GLUD1 was not reported in this data set, thus whether GLUD1 concentrations also change is unknown. PKM2 is well recognized for coordinating glucose carbon flux into biosynthetic outputs (63). By interacting with GLUD1, PKM2 may also coordinate the shuttling of glutamine nitrogen into biosynthetic intermediates, especially for amino acid synthesis (Figure 8). In addition, GLUD1 and its byproduct ammonia have previously been implicated as upstream regulators of phenotypes we identified in cells with disrupted PKM2 activity including alterations to oxidative stress, glycolysis, and mitochondrial metabolism (64–66). Future research in MPCs is needed to identify causal links between binding partners and cell phenotypes.
FIGURE 8.

Summary of findings and potential implications for other cellular pathways. GLUD1, glutamate dehydrogenase 1; PKM2, pyruvate kinase M2; TCA, tricarboxylic acid.
Our data suggest that disruption of PKM2 activity in proliferating MPCs induces oxidative stress. Similar to our findings, in cultured liver cancer cells (HepG2 and SMMC-7721), blocking PKM2 increased ROS (67). ROS can be managed by the antioxidant glutathione, and in our RNA-sequencing data set, several genes encoding proteins for glutathione metabolism were upregulated. In livers from PKM2-null mice, glutathione biosynthesis is the top upregulated pathway, supporting a relation between PKM2 and glutathione (68). Glutathione converts from the oxidized state back to the reduced state through the action of NAD(P)H, a cofactor that is regenerated from NAD(P) through the pentose phosphate pathway. In nonmyogenic cells, PKM2 was positively associated with NAD(P)H (69), the first enzyme of the pentose phosphate pathway (glucose-6-phosphate dehydrogenase) (70–72), and other metabolites generated through the pentose phosphate pathway (11, 73). Interestingly, we observed increased total glutathione and GSSG, as well as higher NAD(P)H concentrations, in shikonin- but not TEPP-46-treated cultures. Because shikonin blocks canonical PKM2 and TEPP-46 forces canonical PKM2, this difference likely reflects the differential effects of the activity of TEPP-46 and shikonin. One possibility is that by blocking canonical PKM2 activity, shikonin may increase availability of glycolytic intermediates and thus flux through the pentose phosphate pathway to regenerate NAD(P)H. Alternatively, NAD(P)H could be generated through malic enzyme (74), the enzyme which converts malate to pyruvate, which could be an important source of pyruvate and further lactate in shikonin-treated cells. Thus, inhibiting noncanonical PKM2 activity causes cellular oxidative stress, and the response to this stress may partly depend on whether the canonical activity is also inhibited.
The relation between PKM2 and glucose/mitochondrial metabolism is heterogeneous across cell types. For example, although PKM2 is associated with changes in glucose uptake and lactate production, the directionality of this relation is variable among different cell types (11, 13–15, 17, 23, 61, 75). Similarly, cell-type variability exists in the relation between PKM2 and mitochondrial phenotypes (18, 23, 76). To our knowledge, a detailed investigation into cell-type-specific differences that underlie this variability is lacking. Given the relations between PKM2 and metabolism and between MPC proliferation and metabolism, we measured indicators of glucose and mitochondrial metabolism in MPCs. We identified that blocking the noncanonical activity of PKM2 increased lactate production, suggesting glucose may be converted to lactate instead of being shunted into biosynthetic pathways that stem from glycolysis, such as nucleotide biosynthesis. In addition, increased conversion of pyruvate into lactate may reduce pyruvate availability for the tricarboxylic acid cycle, resulting in the observed reduction in mitochondrial membrane potential and oxygen consumption rate.
Surprisingly, mice with an MPC-specific PKM2 deletion did not exhibit impaired muscle development and regeneration in vivo, an important and previously unidentified finding in skeletal muscle. The results are consistent with findings in cultured colon cancer cells and a colon tumor xenograft (both using HCT116 cells). In the cultured colon cancer cells, an shRNA vector targeting both PKM1 and PKM2 reduced cell proliferation. However, in vivo the shRNA vector had no impact on xenograft tumor growth (29). One possible and likely explanation is a difference in nutrient availability in cell culture models compared with the in vivo environment, where the circulation can supply missing metabolites. An interesting and unresolved question is whether a whole-body PKM2 KO mouse would have impaired regenerative capacity, recognizing that MPCs from a whole-body PKM2 KO mouse had severely impaired proliferation in culture (11). GLUD1, the PKM2 binding partner identified in this study, is involved in maintaining intramuscular glutamine concentrations through a role in macrophages, and interestingly GLUD1 deletion in macrophages accelerates recovery from a muscle injury (77). Others have demonstrated that PKM2 concentrations increase in activated macrophages (18), and inhibiting noncanonical PKM2 activity, with shikonin (78) or TEPP-46 (18, 79), decreases proinflammatory cytokine production. It is unknown whether, similar to our results in MPCs, GLUD1 and PKM2 are binding partners in macrophages. But it is conceivable that PKM2 may inhibit GLUD1 in macrophages and increase glutamine availability to MPCs, which would support MPC function and muscle regeneration. It is also possible that a minimum amount of proliferation is necessary for regeneration, and this amount is met in PKM2 KO animals. In addition, other muscle groups, with a different fiber type composition, may have displayed a different result.
In conclusion, our research highlights the noncanonical activity of PKM2 as important for proliferation in vitro and identifies GLUD1 as a novel PKM2 binding partner. Although we identified a role for PKM2 in promoting MPC proliferation in a cell culture model, this phenotype did not translate into muscle development or postinjury tissue regeneration in vivo, suggesting that the disturbance that disrupts MPC proliferation in vitro is satisfied by the endogenous environment in vivo, at least in young healthy animals. Although our work does not support PKM2 as an essential protein for MPC function in vivo, it emphasizes the need to simultaneously consider extracellular nutrient availability with intracellular protein function when exploring metabolic enzymes. Overall, our data identify the function of PKM2 in MPC proliferation, but future research is required to delineate the adaptive mechanism used by MPCs when PKM2 is absent in vivo.
Supplementary Material
Acknowledgments
We acknowledge the Cornell BRC Imaging Facility, which received funding through NIH S10RR025502 for equipment used in this study, the Cornell Flow Cytometry Facility, and the Cornell Transcriptional Regulation and Expression Facility. We thank the Proteomics Facility of Cornell University for providing the MS data and NIH SIG grant 1S10 OD017992-01 grant support for the Orbitrap Fusion mass spectrometer. Further, research reported in this publication was supported by the UAB High Resolution Imaging Facility. Figure diagrams containing mice were created using BioRender. Rebekah Epstein helped with image quantification. The authors’ responsibilities were as follows—JEB and AT-M: designed the research and wrote the paper; JEB, BJG, AB, and AT-M: conducted the research; DB, MSF, and NMV: provided essential materials; JEB, EP, NMV, and AT-M: analyzed the data or performed statistical analysis; AT-M: had primary responsibility for the final content; and all authors: read and approved the final manuscript.
Notes
Supported by National Science Foundation Graduate Research Fellowship Program grant DGE-1650441 (to JEB). BJG was funded by a Canadian Institutes for Health Research Doctoral Foreign Study Award. AT-M received funding through NIH U01AR071133I. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Author disclosures: the authors report no conflicts of interest.
Supplemental Figures 1–6 and Supplemental Tables 1–3 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn.
Abbreviations used: ACN, acetonitrile; ATCC, American Type Culture Collection; bFGF, basic fibroblast growth factor; CSA, cross-sectional area; DBT, dihydrolipoamide branched-chain transacylase E2; DTT, dithiothreitol; FA, formic acid; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone; FDR, false discovery rate; FT, Fourier transform; Gclc, glutamate-cysteine ligase catalytic subunit; GLUD1, glutamate dehydrogenase 1; GM, growth medium; Gm3776, glutathione S-transferase; GSHee, glutathione ethylester; GSSG, oxidized glutathione; Gsta, glutathione S-transferase α; H&E, hematoxylin and eosin; hmox1, heme oxygenase 1; HO-1, heme oxygenase 1; IP, immunoprecipitation; KO, knockout; MPC, muscle progenitor cell; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; NAC, N-acetylcysteine; NRF2, nuclear factor erythroid 2-related factor 2; PKM2, pyruvate kinase M2; PSM, peptide spectral match; RFP, red fluorescent protein; RIPA, radioimmunoprecipitation assay; ROS, reactive oxygen species; shControl, short hairpin control; shPKM2, short hairpin pyruvate kinase M2; shRNA, short hairpin RNA; siControl, small interfering RNA non-targetting control; siGLUD1, small interfering RNA targetting GLUD1; SIM, structured illumination microscopy; siRNA, small interfering RNA; slc7a11, solute carrier family 7 member 11; TA, tibialis anterior; TEAB, triethylammonium bicarbonate; WT, wild-type.
Contributor Information
Jamie E Blum, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Brandon J Gheller, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Abby Benvie, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Martha S Field, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Elena Panizza, Department of Molecular Medicine, Cornell University, Ithaca, NY, USA.
Nathaniel M Vacanti, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Daniel Berry, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA.
Anna Thalacker-Mercer, Division of Nutritional Sciences, Cornell University, Ithaca, NY, USA; Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
References
- 1. Pala F, Di Girolamo D, Mella S, Yennek S, Chatre L, Ricchetti M, Tajbakhsh S. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J Cell Sci. 2018;131;jcs212977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai C-Ret al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature. 2014;510:393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chakravarthy MV, Davis BS, Booth FW. IGF-I restores satellite cell proliferative potential in immobilized old skeletal muscle. J Appl Physiol. 2000;89:1365–79. [DOI] [PubMed] [Google Scholar]
- 4. Riddle ES, Bender EL, Thalacker-Mercer AE. Expansion capacity of human muscle progenitor cells differs by age, sex, and metabolic fuel preference. Am J Physiol Cell Physiol. 2018;315:C643–C52. [DOI] [PubMed] [Google Scholar]
- 5. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold Ret al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–43. [DOI] [PubMed] [Google Scholar]
- 6. Sahu A, Mamiya H, Shinde SN, Cheikhi A, Winter LL, Vo NV, Stolz D, Roginskaya V, Tang WY, St Croix Cet al. Age-related declines in α-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat Commun. 2018;9:4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hori S, Hiramuki Y, Nishimura D, Sato F, Sehara-Fujisawa A. PDH-mediated metabolic flow is critical for skeletal muscle stem cell differentiation and myotube formation during regeneration in mice. FASEB J. 2019;33:8094–109. [DOI] [PubMed] [Google Scholar]
- 8. Das S, Morvan F, Morozzi G, Jourde B, Minetti GC, Kahle P, Rivet H, Brebbia P, Toussaint G, Glass DJet al. ATP citrate lyase regulates myofiber differentiation and increases regeneration by altering histone acetylation. Cell Rep. 2017;21:3003–11. [DOI] [PubMed] [Google Scholar]
- 9. Ryall JG, Dell'Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco Met al. The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lunt SY, Muralidhar V, Hosios AM, Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo PNet al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell. 2015;57:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kodani A, Kikuchi T, Tohda C. Acteoside improves muscle atrophy and motor function by inducing new myokine secretion in chronic spinal cord injury. J Neurotrauma. 2019;36:1935–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prakasam G, Singh RK, Iqbal MA, Saini SK, Tiku AB, Bamezai RNK. Pyruvate kinase M knockdown–induced signaling via AMP-activated protein kinase promotes mitochondrial biogenesis, autophagy, and cancer cell survival. J Biol Chem. 2017;292:15561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–306. [DOI] [PubMed] [Google Scholar]
- 16. Anastasiou D, Yu Y, Israelsen WJ, Jiang J-K, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha Aet al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iansante V, Choy PM, Fung SW, Liu Y, Chai J-G, Dyson J, Del Rio A, D'Santos C, Williams R, Chokshi Set al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun. 2015;6:7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo-Fernandez R, Johnston DGWet al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:347. [DOI] [PubMed] [Google Scholar]
- 19. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, Yorek MA, Wu I-H, Lockhart S, Coppey LJet al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23:753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alves-Filho JC, Palsson-McDermott EM. Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol. 2016;7:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwon O-H, Kang T-W, Kim J-H, Kim M, Noh S-M, Song K-S, Yoo H-S, Kim W-H, Xie Z, Pocalyko Det al. Pyruvate kinase M2 promotes the growth of gastric cancer cells via regulation of Bcl-xL expression at transcriptional level. Biochem Biophys Res Commun. 2012;423:38–44. [DOI] [PubMed] [Google Scholar]
- 22. Yang P, Li Z, Li H, Lu Y, Wu H, Li Z. Pyruvate kinase M2 accelerates pro-inflammatory cytokine secretion and cell proliferation induced by lipopolysaccharide in colorectal cancer. Cell Signal. 2015;27:1525–32. [DOI] [PubMed] [Google Scholar]
- 23. Salama SA, Mohammad MA, Diaz-Arrastia CR, Kamel MW, Kilic GS, Ndofor BT, Abdel-Baki MS, Theiler SK. Estradiol-17β upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-α and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J Clin Endocrinol Metab. 2014;99:3790–9. [DOI] [PubMed] [Google Scholar]
- 24. Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A. 2012;109:6904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gupta A, Ajith A, Singh S, Panday RK, Samaiya A, Shukla S. PAK2–c-Myc–PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis. 2018;9:825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, Wei C, Guo F, Chen Yet al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014;53:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang W, Lu Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle. 2013;12:3343–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WRet al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A. 2013;110:489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blum JE, Gheller BJ, Hwang S, Bender E, Gheller M, Thalacker-Mercer AE. Consumption of a blueberry-enriched diet by women for 6 weeks alters determinants of human muscle progenitor cell function. J Nutr. 2020;150:2412–8. [DOI] [PubMed] [Google Scholar]
- 31. Gheller BJ, Blum J, Soueid-Baumgarten S, Bender E, Cosgrove BD, Thalacker-Mercer A. Isolation, culture, characterization, and differentiation of human muscle progenitor cells from the skeletal muscle biopsy procedure. J Vis Exp. 2019;(150):e59580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.j. 2011;17(1):10–12. [Google Scholar]
- 33. Varet H, Brillet-Gueguen L, Coppee JY, Dillies MA. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One. 2016;11:e0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. 2014;542:91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gheller BJ, Blum JE, Lim EW, Handzlik MK, Hannah Fong EH, Ko A, Khanna S, Gheller ME, Bender EL, Alexander MSet al. Extracellular serine and glycine are required for mouse and human skeletal muscle stem and progenitor cell function. Mol Metab. 2020;43:101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, Meyer JN. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015;1241:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aiken CE, Cindrova-Davies T, Johnson MH. Variations in mouse mitochondrial DNA copy number from fertilization to birth are associated with oxidative stress. Reprod Biomed Online. 2008;17:806–13. [DOI] [PubMed] [Google Scholar]
- 39. Frangini M, Franzolin E, Chemello F, Laveder P, Romualdi C, Bianchi V, Rampazzo C. Synthesis of mitochondrial DNA precursors during myogenesis, an analysis in purified C2C12 myotubes. J Biol Chem. 2013;288:5624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang Y, Anderson E, Zhang S. Evaluation of six sample preparation procedures for qualitative and quantitative proteomics analysis of milk fat globule membrane. Electrophoresis. 2018;39:2332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zougman A, Selby PJ, Banks RE. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 2014;14:1006–10. [DOI] [PubMed] [Google Scholar]
- 42. Boekel J, Peltzer A. lehtiolab/ddamsproteomics: Release 1.3. (2019). doi: 10.5281/zenodo.3557399. [DOI]
- 43. Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. [DOI] [PubMed] [Google Scholar]
- 44. Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Savitski MM, Wilhelm M, Hahne H, Kuster B, Bantscheff M. A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol Cell Proteomics. 2015;14:2394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K. REAP: a two minute cell fractionation method. BMC Res Notes. 2010;3:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JWet al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones ARet al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harada Y, Nakamura M, Asano A. Temporally distinctive changes of alternative splicing patterns during myogenic differentiation of C2C12 cells. J Biochem. 1995;118:780–90. [DOI] [PubMed] [Google Scholar]
- 51. Panieri E, Telkoparan-Akillilar P, Suzen S, Saso L. The NRF2/KEAP1 axis in the regulation of tumor metabolism: mechanisms and therapeutic perspectives. Biomolecules. 2020;10:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gomez JC, Dang H, Martin JR, Doerschuk CM. Nrf2 modulates host defense during Streptococcus pneumoniae pneumonia in mice. J Immunol. 2016;197:2864–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, Yin Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019;10:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H, Li C, Yang F, Zeng R, Wei Pet al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017;27:329–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, Gygi SP, Selfors LM, Brugge JS. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. 2016;23:867–80. [DOI] [PubMed] [Google Scholar]
- 57. Jin L, Li D, Alesi GN, Fan J, Kang H-B, Lu Z, Boggon TJ, Jin P, Yi H, Wright ERet al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27:257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gattazzo F, Laurent B, Relaix F, Rouard H, Didier N. Distinct phases of postnatal skeletal muscle growth govern the progressive establishment of muscle stem cell quiescence. Stem Cell Reports. 2020;15:597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, Simmet T, Seufferlein T. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol Cancer. 2016;15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang C, Jiang J, Ji J, Cai Q, Chen X, Yu Y, Zhu Z, Zhang J. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci Rep. 2017;7:2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tech K, Tikunov AP, Farooq H, Morrissy AS, Meidinger J, Fish T, Green SC, Liu H, Li Y, Mungall AJet al. Pyruvate kinase inhibits proliferation during postnatal cerebellar neurogenesis and suppresses medulloblastoma formation. Cancer Res. 2017;77:3217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology (Basel). 2017;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell. 2011;42:713–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kala G, Hertz L. Ammonia effects on pyruvate/lactate production in astrocytes—interaction with glutamate. Neurochem Int. 2005;47:4–12. [DOI] [PubMed] [Google Scholar]
- 65. Wang F, Chen S, Jiang Y, Zhao Y, Sun L, Zheng B, Chen L, Liu Z, Zheng X, Yi Ket al. Effects of ammonia on apoptosis and oxidative stress in bovine mammary epithelial cells. Mutagenesis. 2018;33:291–9. [DOI] [PubMed] [Google Scholar]
- 66. Lorin S, Tol MJ, Bauvy C, Strijland A, Pous C, Verhoeven AJ, Codogno P, Meijer AJ. Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy. 2013;9:850–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wong CC-L, Au SL-K, Tse AP-W, Xu IM-J, Lai RK-H, Chiu DK-C, Wei LL, Fan DN-Y, Tsang FH-C, Lo RC-Let al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS One. 2014;9:e115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks Tet al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016;30:1020–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kumar B, Bamezai RN. Moderate DNA damage promotes metabolic flux into PPP via PKM2 Y-105 phosphorylation: a feature that favours cancer cells. Mol Biol Rep. 2015;42:1317–21. [DOI] [PubMed] [Google Scholar]
- 70. Pu H, Zhang Q, Zhao C, Shi L, Wang Y, Wang J, Zhang M. Overexpression of G6PD is associated with high risks of recurrent metastasis and poor progression-free survival in primary breast carcinoma. World J Surg Oncol. 2015;13:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dong T, Kang X, Liu Z, Zhao S, Ma W, Xuan Q, Liu H, Wang Z, Zhang Q. Altered glycometabolism affects both clinical features and prognosis of triple-negative and neoadjuvant chemotherapy-treated breast cancer. Tumour Biol. 2016;37:8159–68. [DOI] [PubMed] [Google Scholar]
- 72. Yang L, Hou Y, Yuan J, Tang S, Zhang H, Zhu Q, Du Y-e, Zhou M, Wen S, Xu Let al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget. 2015;6:25755–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DSet al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maddocks OD, Labuschagne CF, Vousden KH. Localization of NADPH production: a wheel within a wheel. Mol Cell. 2014;55:158–60. [DOI] [PubMed] [Google Scholar]
- 75. Xie J, Dai C, Hu X. Evidence that does not support pyruvate kinase M2 (PKM2)-catalyzed reaction as a rate-limiting step in cancer cell glycolysis. J Biol Chem. 2016;291:8987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. 2019;10:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shang M, Cappellesso F, Amorim R, Serneels J, Virga F, Eelen G, Carobbio S, Rincon MY, Maechler P, De Bock Ket al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. 2020;587(7835):626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang Het al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TLet al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.