

Abstract
Oncogene-targeted therapy with B-Raf proto-oncogene (BRAF) and mitogen-activated protein kinase kinase (MEK) inhibitors induces a high initial response rate in patients with BRAFV600-mutated melanoma, with a median duration of response of approximately 1 year1–3. Immunotherapy with antibodies to programmed death 1 (PD-1) produces lower response rates but with long response duration. Preclinical models suggest that combining BRAF and MEK inhibitors with PD-1 blockade therapy improves antitumor activity4–6, which may provide additional treatment options for patients unlikely to have long-lasting responses to either mode of therapy alone. We enrolled 15 patients with BRAFV600-mutated metastatic melanoma in a first-in-human clinical trial of dabrafenib, trametinib and pembrolizumab (NCT02130466). Eleven patients (73%) experienced grade 3/4 treatment-related adverse events, the most common being elevation of liver function tests and pyrexia, most of which resolved with drug interruption or discontinuation of either the anti-PD-1 antibody or the targeted therapy combination. Eleven patients (73%; 95% confidence interval = 45–92%) had an objective response, and six (40%; 95% confidence interval = 16–68%) continued with a response at a median follow-up of 27 months (range = 10.3–38.4+ months) for all patients. This study suggests that this triple-combined therapy may benefit a subset of patients with BRAFV600-mutated metastatic melanoma by increasing the frequency of long-lasting antitumor responses.
The oncogene BRAFV600 constitutively activates the mitogen-activated protein kinase (MAPK) pathway in approximately 50% of cutaneous melanomas7. BRAFV600 can be selectively inhibited by BRAF inhibitors, improving progression-free survival (PFS) and overall survival over chemotherapy8,9. The addition of a MEK inhibitor to a BRAF inhibitor improves PFS and overall survival over the use of a BRAF inhibitor alone1–3. The combination of a BRAF and a MEK inhibitor has the additional benefit of reducing toxicities related to BRAF inhibitor-induced paradoxical MAPK activation in BRAF wild-type cells1–3,10. Initial attempts to combine a BRAF inhibitor with immunotherapy resulted in unacceptable toxicity. The combination of the BRAF inhibitor vemurafenib with the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) checkpoint-blocking antibody ipilimumab resulted in hepatic dose-limiting toxicity (DLT), leading to discontinuation of the development of this combination11. The addition of a MEK inhibitor may overcome some of the toxicities of the BRAF inhibitor therapy induced by paradoxical MAPK activation, but there were concerns that it may also have detrimental effects on T-cell function, given the requirement for MAPK signaling after T-cell receptor engagement12.
However, preclinical modeling demonstrated that BRAF and MEK inhibitors could be efficiently combined with immunotherapy agents4–6. In one of these models of a murine BRAFV600E-driven melanoma6, the addition of the MEK inhibitor to combined immunotherapy with the BRAF inhibitor improved antitumor activity by increasing T-cell infiltration, improved in vivo cytotoxicity, decreased tumor-associated macrophages and T regulatory cells, increased melanosomal antigen and major histocompatibility complex (MHC) expression, and increased intratumoral interferon-gamma gene expression. Therefore, rather than negating the benefits of a combination of BRAF inhibitor therapy and immunotherapy, adding a MEK inhibitor had several positive effects. In another model5, short-term treatment combining BRAF and MEK inhibitors enhanced intratumor T-cell infiltration in a BRAFV600E/PTEN−/−-driven melanoma mouse model—a finding that could also be replicated in human biopsies taken within two weeks of the start of therapy—but the intratumoral T-cell infiltration decreased thereafter. When the short-term BRAF and MEK inhibitor therapy was combined with murine anti-programmed death 1 (PD-1) blockade, antitumor activity increased.
Given these data, we sought to determine the activity and safety of a concomitant triple-therapy combination of BRAF and MEK inhibitors with anti-PD-1 in patients with BRAFV600-mutated metastatic melanoma. In this phase 1 trial, a combination of approved doses of the BRAF inhibitor dabrafenib, the MEK inhibitor trametinib and the PD-1-blocking antibody pembrolizumab at 2 mg kg−1 was administered to an initial cohort of three patients in part 1 of this study (Supplementary Table 1). As the first three patients in cohort 1 tolerated the triple therapy over the first 30-d observation period, the study continued to enroll 12 additional patients at the same dosing regimen in part 2, given for a maximum of 24 months (Supplementary Table 1). Patients were recruited from nine sites in four countries (Australia, Canada, New Zealand and the United States). The combined group of 15 patients had a median age of 47 years (range = 24–71 years) (Supplementary Table 2), consistent with previous reports of an association between younger patients and BRAFV600-mutated melanoma13; 13 patients had BRAFV600E-mutated melanoma and two had BRAFV600K-mutated melanoma. There were eight females and seven males. All had stage IV metastatic melanoma, and most had features of poor prognosis, including 11 with M1c disease (non-lung visceral metastases). Of these, four had known levels of lactate dehydrogenase (LDH), all of which were elevated (Supplementary Table 2).
Overall, three patients had DLT that developed within the first month of trial treatment (Supplementary Table 3): one with grade 4 neutropenia that was managed by treatment interruption, followed by restarting the triple therapy with a dose reduction of dabrafenib and trametinib; and two with a grade 4 increase in liver function tests that resolved with treatment interruption. One of these two patients continued dabrafenib and trametinib treatments but discontinued pembrolizumab, and the other patient discontinued the combination of dabrafenib and trametinib but continued pembrolizumab alone off study. The most common toxicity of any grade was pyrexia, occurring in more than 90% of patients, followed by chills, fatigue and diarrhea in 60–67% of patients (Supplementary Table 4). Eleven patients (73%) had grade 3/4 treatment-related adverse events, the most common being pyrexia and elevated liver function test levels (Supplementary Table 4), such as elevation in transaminase levels without concomitant early elevation of bilirubin levels. Treatment-related adverse events led to dose modifications in 14 (93.3%) patients. Of these, 10 (66.7%) patients had grade 3/4 treatment-related adverse events, most commonly increased liver enzyme levels, pyrexia, decreased numbers of white blood cells, and neutropenia. A majority of adverse events resolved within a few weeks, mostly with the temporary interruption of dabrafenib and trametinib (Supplementary Table 5). However, one patient experienced a grade 3 increase in alanine aminotransferase and another had grade 3 pyrexia, neither of which resolved with dose interruption and/or dose reduction (Supplementary Table 5). Thirteen (86.7%) patients received any form of concomitant corticosteroids for the treatment of any adverse event: six patients received a dosage of more than 10 mg d−1 prednisone or its equivalent, representing the only patients with systemic treatment for toxicities with these agents; one patient received prednisone at a supplemental dosage of 10 mg d−1; four patients received topical or inhaled corticosteroids for local toxicities; and two patients received treatment doses of corticosteroids for palliative management of disease progression (Supplementary Table 6). Grade 3 pyrexia and elevation of liver enzymes have also been previously reported in studies involving the dabrafenib and trametinib combination, where pyrexia has been managed through temporary interruption of dosing of dabrafenib, or dabrafenib and trametinib, and by the use of prophylactic glucocorticoids2,3. Administration of the three agents did not alter the expected pharmacokinetics of either agent (Extended Data Fig. 1). Based on the mostly reversible toxicities and favorable pharmacokinetics, the dosing regimen of dabrafenib at 150 mg twice daily, trametinib at 2 mg daily and pembrolizumab at 2 mg kg−1 every 3 weeks was declared the maximum tolerated dose, and taken to part 3 of this study within a phase 2 randomized trial (ClinicalTrials.gov; NCT02130466) (Extended Data Fig. 2).
All but 1 of the 15 patients had a reduction in the sum of the longest diameter of the target lesions compared with baseline (Fig. 1a), with 11 patients (73%; 95% confidence interval (CI) = 45–92%) having a complete or partial response to therapy per investigator review (Supplementary Table 7). One patient (patient 12; Supplementary Table 8) discontinued the triple therapy due to elevation of liver function test levels before the first response assessment. This patient continued off study with only dabrafenib and trametinib, and experienced an objective response, but this patient is not included in Fig. 1b, which shows the longitudinal change in the 14 patients on study with triple therapy who had confirmed responses by repeated computed tomography. Of the 11 patients who had a response to therapy, six (40% overall; 95% CI = 16.34–67.71%) remained in response and five progressed (Fig. 1c and Supplementary Table 8). These five patients progressed at 5, 6, 8, 15 and 22 months from the start of therapy; at data cutoff, three patients were alive on other therapies and two had died. Of the six patients who continued to respond at a median follow-up of 26.9 months (range = 24 to 38+ months), at the time of data cut-off, three were receiving therapy off study, two were receiving triplet therapy and one discontinued pembrolizumab because of toxicities, and continued dabrafenib plus trametinib doublet therapy. The median follow-up of response duration for those patients still in response was 17.1 months (range = 0.03–22.1 months). The Kaplan–Meier estimate of the percentage of responding patients still in response 18 months after the initial response is 58.3%. The median PFS of the whole cohort is 15.4 months (95% CI = 5.4 months to ‘not reached’), and the median overall survival has not been reached (95% CI = 10.3 months to not reached; Extended Data Fig. 3).
Fig. 1 |. Antitumor activity of combined dabrafenib, trametinib and pembrolizumab.
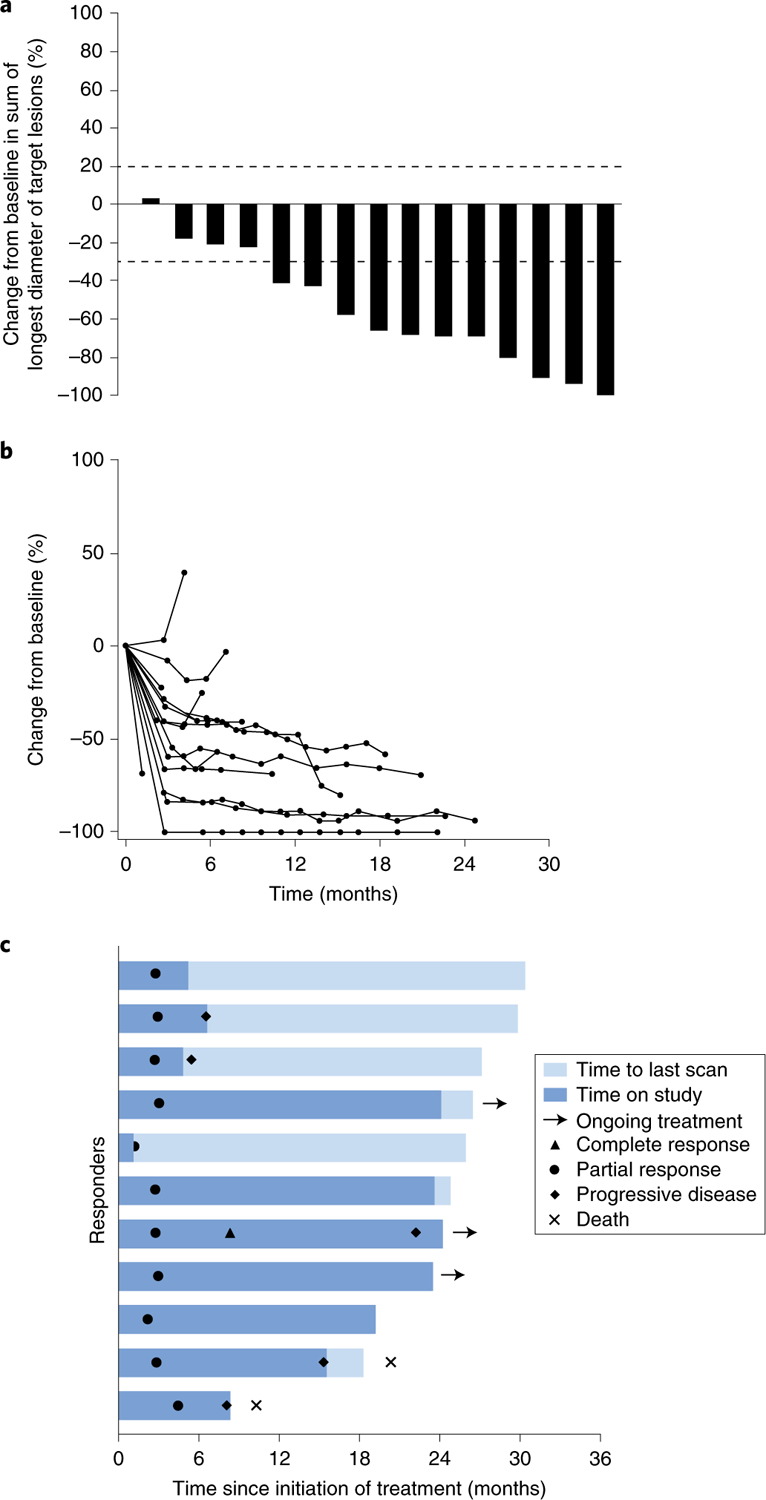
a, Maximum percentage of change from baseline in the sum of the longest diameter of target lesions, as assessed using RECIST version 1.1 by investigator review (n = 15 patients). Dotted lines indicate 20% increase (cutoff for determination of progressive disease) and 30% decrease from baseline (cutoff for determination of partial response per RECIST v1.1 criteria). b, Longitudinal change from baseline in target lesion size (n = 14 patients) in all patients with measurable disease and at least one post-baseline scan. c, Time to response and duration of response in patients with confirmed and unconfirmed response, assessed using RECIST version 1.1 by investigator review (n = 11 patients). Bar length denotes time to last scan. Patients with ongoing treatment continued the triplet or doublet (dabrafenib + trametinib) treatment beyond 2 years, but off study.
We calculated tumor mutational load from whole-exome sequencing (WES) analysis from seven baseline samples, all of which were derived from patients who had a response to therapy (Fig. 2a and Supplementary Tables 9–11). The three patients with high mutational load above the median carried mutations in DNA repair genes: two with missense mutations in TP53 (p.D281G, p.V173G, p.A161V and p.R110C) and one with a nonsense mutation in ATM (p.Q1128*). Secondary MAPK pathway mutations in NRAS, MEK1 or MEK2, which have previously been associated with resistance to BRAF inhibitor therapy14,15, were not observed (Fig. 2a).
Fig. 2 |. Analyses of biopsy specimens from patients treated with dabrafenib, trametinib, and pembrolizumab (patients with available data per Supplementary Table 9).
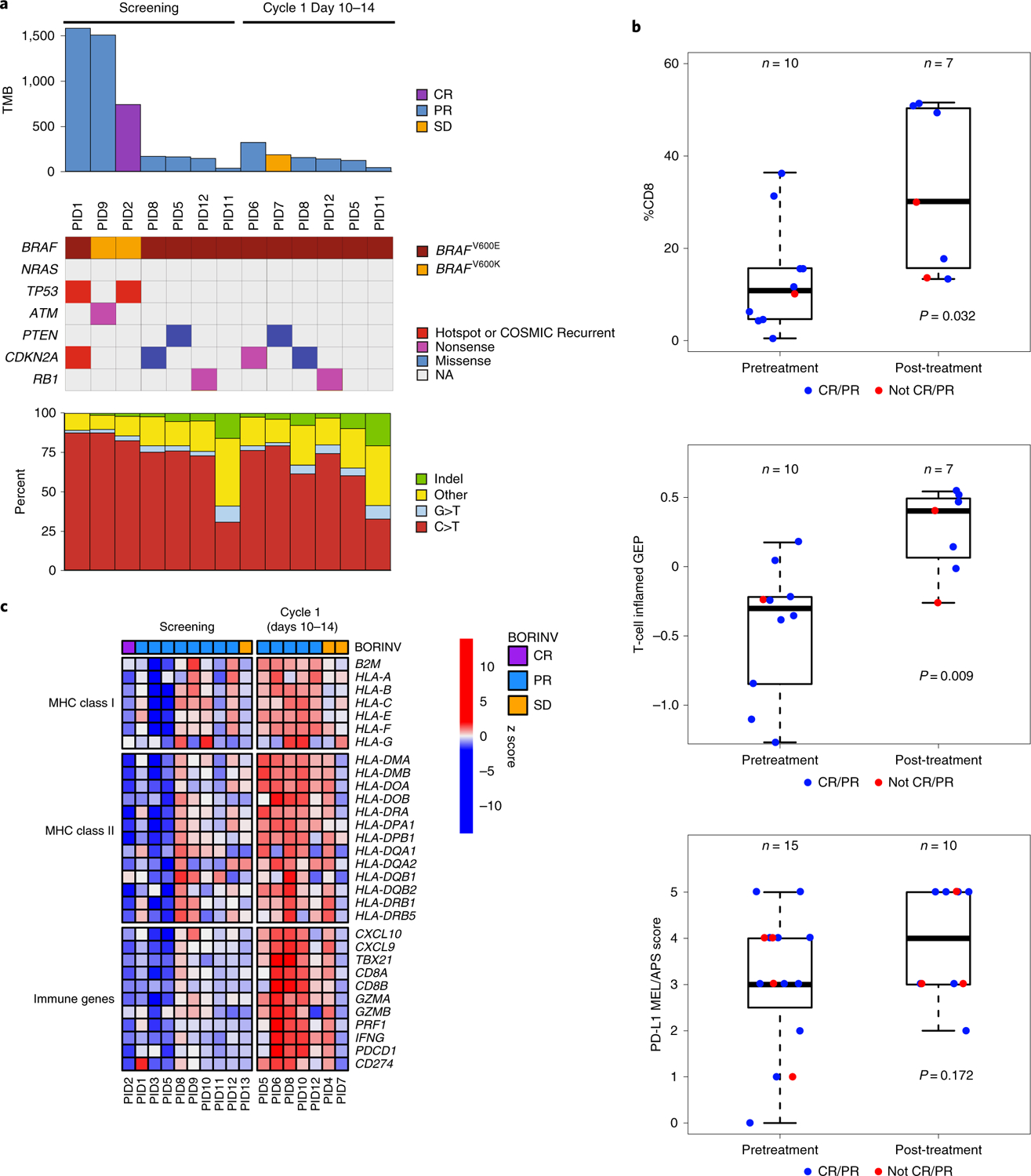
a, Top, bar plot showing the TMB for each sample (n = 9 patients). Middle, color-coded matrix showing the type and presence of BRAF, NRAS and tumor suppressor mutations (n = 9 patients; 7 pretreatment, 6, post-treatment). Recurrent mutations are identified as those occurring in >20 patients in COSMIC. CR, complete response; NA, not applicable; PID, patient ID; PR, partial response; SD, stable disease; TMB, total number of somatic mutations. Bottom, bar plot showing mutation spectra for all samples (n = 9 patients). b, Box plots of CD8 positivity by IHC (top), interferon gene expression profiling using the 18-gene T-cell-inflamed GEP score obtained from RNA-Seq biopsy analyses (middle) and PD-L1 IHC staining using the MEL/Allred proportion score (APS) score (bottom), in pre- and post-treatment biopsy samples (Supplementary Tables 10 and 11). Median, lower and upper quantiles (lower and upper quantiles represented by whiskers) for CD8 positivity in pretreatment samples (n = 10 patients) were 10.8, 5.0 and 15.7, and for post-treatment samples (n = 7 patients) they were 30.1, 15.7 and 50.3, respectively. Median, lower and upper quantiles for GEP score in pretreatment samples (n = 10 patients) were −0.3, −0.73 and −0.23, and for post-treatment samples (n = 7 patients) they were 0.4, 0.06 and 0.49, respectively. Median, lower and upper quantiles for MEL/Allred proportion score for pretreatment samples (n = 15 patients) were 3, 2.5 and 4, and for post-treatment samples (n = 10 patients) they were 4, 3 and 5, respectively. c, Heat map of RNA-Seq analysis showing z scores of individual MHC class I and II, and CD8 T-cell-related immune response genes (n = 10 patients with pretreatment samples; n = 7 patients with post-treatment samples). BORINV, best objective response per investigator review.
Compared with baseline biopsy specimens, one-month on-therapy biopsies had increased CD8 and programmed death ligand 1 (PD-L1) by immunohistochemistry (IHC) analysis, which was confirmed when analysing RNA sequencing (RNA-Seq) data using the previously published T-cell-inflamed 18-gene signature that includes CD8A, CD274 (PD-L1), CXCL9, HLA-E, HLA-DQA1 and HLA-DRB1 (ref. 16) (Fig. 2b and Supplementary Tables 9 and 10)16–20. In further analysis of the RNA-Seq data from baseline with on-treatment biopsy samples regardless of the response status, there was a trend towards an increase in levels of expression of several MHC class I and class II molecules and genes involved in CD8 T-cell function (Fig. 2c and Supplementary Table 9). Similar changes have been previously reported in biopsy specimens of patients treated with BRAF and MEK inhibitor therapy alone5,21,22 and anti-PD-1 monotherapy17,23,24. Increases in the MHC class I expression and immune infiltration induced by the triple therapy support previous work in mouse models showing that this triple combination, including a MEK inhibitor, does not negatively impact the generation of an intratumoral immune response5,6.
In the current study, combined triple therapy of dabrafenib, trametinib and pembrolizumab had a generally manageable toxicity profile consistent with the established profile of the three drugs25–27, with a subset of patients showing long-duration responses1–3,28. A pooled analysis of 563 patients with BRAFV600-mutated advanced melanoma treated with dabrafenib and trametinib in two large randomized trials (COMBI-v and COMBI-d) revealed a median PFS of 11 months (95% CI = 9–13 months)28. Factors indicating poor prognosis were elevated baseline LDH levels and metastases in more than three organs; a subgroup of patients with elevated LDH (≥2 × upper limit of normal) had no complete responses and a median PFS of only 5.5 months28, which defines a patient population that may be suited to receive triple combined or sequential therapy despite the increase in the occurrence of side effects. An additional option for patients with BRAFV600-mutated advanced melanoma is the use of dual immune checkpoint blockade therapy. In a phase 2 randomized trial that reported the response rate and PFS of patients with BRAFV600-mutated advanced melanoma receiving a combination of the anti-PD-1 antibody nivolumab and the anti-CTLA-4 antibody ipilimumab29, the objective response rate was 52% and the median PFS was 8.5 months. There was a 54% rate of drug-related adverse events of grade 3 or 4. A randomized trial that included an arm receiving nivolumab and ipilimumab and another receiving nivolumab alone showed numerically higher rates of PFS with combination therapy compared with nivolumab monotherapy in the majority of patient subgroups, including patients with BRAFV600-mutated melanoma, those with stage M1c disease and those with elevated LDH levels30. Our phase 1 trial had roughly the same rate of patients with poor prognosis factors as in the pivotal trial with pembrolizumab, with the key differences that patients were younger and had BRAFV600 mutations (the median PFS was 5.6 months with pembrolizumab among 556 patients31). Because of the toxicity of concomitant triple therapy, other attempts are testing different sequencing of these same three agents (ClinicalTrials.gov; NCT03149029, NCT02625337 and NCT02858921). Furthermore, concomitant triple-therapy combinations are being pursued in phase 2 and 3 randomized trials32 (ClinicalTrials.gov; NCT02130466, NCT02902042 and NCT02908672). The results of these randomized clinical trials will be used to further assess the benefits of concomitant therapy with BRAF and MEK inhibitors with anti-PD-1 in patients with BRAFV600-mutated advanced melanoma.
In conclusion, combined concomitant triple therapy with a BRAF and a MEK inhibitor and an anti-PD-1 antibody is a feasible treatment approach despite increased toxicity; a subset of patients had a long duration of response without evidence of acquired resistance to the oncogene-targeted therapy at 2 years. This combination may be most suited for the treatment of patients with poor prognostic factors who may not be expected to have long-lasting responses to monotherapy. Additional options for these patients would be different schedules of sequential immunotherapy and targeted therapy, or the use of combined checkpoint inhibitor therapy.
Methods
Clinical study subjects.
Patients included in this analysis were eligible if they were at least 18 years of age with histologically confirmed advanced unresectable stage III or metastatic stage IV melanoma, with at least one measurable lesion as defined by Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1 as observed with computed tomography or magnetic resonance imaging. Additional key eligibility criteria included: the presence of a BRAFV600E or BRAFV600K mutation-positive tumor for treatment with pembrolizumab plus trametinib plus dabrafenib; an Eastern Cooperative Oncology Group performance status of 0 or 1; anticipated life expectancy of at least three months; adequate organ function; the ability to swallow and retain oral medication; and the provision of tissue for biomarker analysis from a newly or recently obtained (within 60 d of the study start) biopsy of an unirradiated tumor. Key exclusion criteria were: current or previous participation within four weeks of the first study treatment with an investigational agent; a BRAF mutation-positive tumor previously treated with systemic therapy for metastatic or advanced melanoma; previous therapy targeting PD-1, PD-L1, BRAF, MEK or the MAPK pathway; a BRAF mutation-positive tumor previously treated with anti-CTLA-4 antibodies; active central nervous system metastases and/or carcinomatous meningitis; a documented history of or active autoimmune disease; a history, or evidence, of interstitial lung disease or active non-infectious pneumonitis; and a known history of human immunodeficiency virus. Data collection started with screening on 3 June 2014 and 29 April 2015, for parts 1 and 2, respectively, and follow-up is ongoing. This study was reviewed and approved by an independent institutional review board at each site, and was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent.
Study design, treatment and end points.
KEYNOTE-022 was a three-part phase 1/2 trial in which parts 1 and 2 were open label. This article reports the results from parts 1 and 2, in which the treatment was triple therapy. This multicenter, world-wide study involved pembrolizumab in combination with dabrafenib and/or trametinib in patients with advanced or metastatic melanoma.
Part 1 of this study followed a 3 + 3 design to evaluate safety, tolerability and dosing of pembrolizumab in combination with dabrafenib and trametinib in patients with BRAF-mutant melanoma. A cohort of three to six patients was to be enrolled to receive 2 mg kg−1 pembrolizumab every three weeks on days 1 and 22 of each cycle, plus 150 mg dabrafenib twice daily, plus 2 mg trametinib once daily until study treatment was discontinued (Extended Data Fig. 2). It was planned that these patients would be evaluated for DLT for six weeks from the start of study treatment. If one of the first three patients in this cohort had DLT, the cohort would be expanded to six patients. If two of six patients had DLT, the next cohort of three patients would be enrolled and follow a decreased dosing regimen (Supplementary Table 1, cohort 1). Dose level modifications were allowed based on clinically observed toxicity (Supplementary Table 1). Fifteen patients with BRAF-mutant melanoma were enrolled and treated with pembrolizumab, dabrafenib, and trametinib in parts 1 and 2 of this study.
Patients were treated with pembrolizumab, dabrafenib, and trametinib until documented disease progression, unacceptable toxicity, or investigator or patient decision to discontinue treatment. The maximum allowed duration of treatment with pembrolizumab was 24 months. Patients who had confirmed complete response after at least six months of study treatment and two cycles of study treatment beyond complete response could stop pembrolizumab treatment based on investigator decision, while continuing treatment with trametinib and dabrafenib. Patients who relapsed after complete response or experienced disease progression after stopping pembrolizumab treatment after 24 months were eligible for re-treatment if inclusion criteria for re-treatment were met.
Primary end points for parts 1 and 2 were the determination of safety, tolerability and maximum tolerated dose or maximum administered dose of pembrolizumab in combination with oral dabrafenib and trametinib in patients with advanced BRAF-mutant melanoma (V600E or V600K). Secondary end points for parts 1 and 2 were the pharmacokinetics of pembrolizumab administered intravenously combined with oral dabrafenib and trametinib, and objective response rate by investigator review per RECIST version 1.1. This study is registered at ClinicalTrials.gov (NCT02130466). Exploratory end points included the assessment of biomarkers at baseline and after the administration of pembrolizumab combined with oral dabrafenib and then with trametinib, and the relationship between biomarkers and clinical response.
Assessments.
Response was assessed by radiologic imaging at 12 weeks of study treatment and every 6 weeks thereafter, or whenever clinically indicated, until 18 months. After the 18-month response assessment, response was assessed every 12 weeks or whenever clinically indicated while the patient was on study treatment. Progressive disease was confirmed by repeat imaging at ≥4 weeks. Patients were followed up for survival status by telephone every three months. Adverse events were monitored until 30 d (60 d for serious adverse events) after the last dose of study treatment, and graded per National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
Biomarkers.
PD-L1 expression was assessed in tumor biopsy samples by IHC using the 22C3 PD-L1 IHC assay (Agilent Technologies) and scored on the melanoma (MEL) scale/Allred proportion score (APS) 0–5 scale, as previously described20. MEL/APS scores of 0–1 were defined as PD-L1 negative, and scores of 2–5 were defined as positive. CD8 expression was assessed in tumor biopsy samples by IHC using the mouse monoclonal anti-CD8 antibody (clone C8/144B; number IR623; ready to use (no dilution required))33,34, CD8 Autostainer Link 48 and the Dako EnVision FLEX detection system (Agilent Technologies). Antigen retrieval was performed using the Dako PT Link Pre-Treatment Module with the Dako EnVision FLEX Target Retrieval Solution, High pH. Stained slides were counterstained with hematoxylin. A certified pathologist annotated the tumor area, and image analysis was performed using Definiens image analysis software (Definiens).
For RNA-Seq analyses, RNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tissue using a Roche High Pure FFPET RNA Isolation Kit, and was sequenced on an Illumina HiSeq 4000 at approximately 4 gigabase pairs (Gb) of data per sample. Libraries were prepared with the TruSeq RNA Access Library Prep Kit. The 18-gene T-cell-inflamed gene expression profile (GEP) was analyzed from whole RNA-Seq using the gene set developed as described by Ayers et al.16, and consists of genes related to antigen presentation, chemokine expression, cytolytic activity and adaptive immune resistance, as follows: CCL5, CD27, CD274 (PD-L1), CD276 (B7–H3), CD8A, CMKLR1, CXCL9, CXCR6, HLAD-QA1, HLA-DRB1, HLA-E, IDO1, LAG3, NKG7, PDCD1LG2 (PD-L2), PSMB10, STAT1 and TIGIT. The GEP score was computed by taking a weighted sum of the housekeeping normalized values of the 18 genes on the GEP18 signature. RNA-Seq reads were mapped using HISAT2 version 2.0.4 (ref. 35) and aligned to the hg19 genome using default parameters. Reads were quantified by HTSeq version 0.6.1 (ref. 36) with the intersection-non-empty mode, and counting ambiguous reads if fully overlapping. Raw counts were then normalized to fragments per kilobase of exon per million fragments mapped expression values.
For WES, DNA were extracted from FFPE tissue using an Almac-optimized Qiagen QIAamp DNA FFPE Tissue Kit and sequenced using ACE Cancer Exome Sequencing at Personalis. Tumor samples were sequenced at a minimum of 16 Gb sample−1 and corresponding matched normal blood samples were sequenced at a minimum of 8 Gb sample−1. For analysis of WES, reads were mapped to the hg19 genome using Bowtie 2 using the default parameters. Variant calling was performed using a validated methodology and as published previously37,38. Briefly, duplicate reads from PCR that matched the same genomic interval were removed. Basecalls with Phred quality scores less than Q20 were excluded. Somatic mutations were identified as those with six reads of support, with 10% of the coverage in the tumor, observed on both strands, and that did not occur in the matched normal sample in more than two reads and 2% of the coverage. Mutations were annotated using the Ensembl Variant Effector Predictor (release 92.2). Mutational load was identified as the total number of protein-coding somatic mutations.
Tumor mutation burden (TMB) analysis was performed using the WES analysis. We generated somatic single-nucleotide variant calls; subsequently, single-nucleotide polymorphisms and somatic mutations were removed using the appropriate databases dbSNP version 141 and COSMIC version 68. For each patient, the sum of non-synonymous single-nucleotide variants that passed the aforementioned filters was defined as the TMB.
Statistical analysis of biomarkers.
For GEP and TMB, all samples were evaluable. For CD8 IHC, pretreatment and post-treatment samples were available from 12 and 9 patients, respectively; however, only 17 samples were scored and a sample from 1 patient showed <1% CD8+ cells within the tumor. No formal testing of associations was planned comparing the responders versus non-responders for PD-L1 and CD8 IHC or GEP and TMB because of the limited number of available samples and, consequently, the limited power. Descriptive analyses and presentations are provided. To compare biomarkers in terms of pre- versus post-treatment, the biomarker measurements collected pre- and post-treatment were evaluated with a linear mixed-effects model containing a fixed effect for the time point (pre or post) and a random effect for patient. For CD8 and TMB, the measurements were natural log-transformed. The Kenward and Roger method was used to calculate the denominator degrees of freedom for the fixed effects.
Pharmacokinetics.
For pembrolizumab pharmacokinetics, pre- and postdose serum samples were collected on days 1 and 22 of cycle 1 and day 1 of cycle 2. Thereafter, only predose serum samples were collected on day 1 of every alternate cycle. An additional postdose sample was collected between 24 and 96 h after day 1 of cycle 1. All predose serum samples were collected within 24 h before the start of pembrolizumab infusion, and all postdose pharmacokinetics samples were collected within 30 min after end of pembrolizumab infusion.
For trametinib and dabrafenib pharmacokinetics, pre- and postdose serum samples were collected on day 22 of cycle 1 and day 1 of cycle 2. On day 22 of cycle 1, postdose pharmacokinetics samples for trametinib and dabrafenib were drawn at the same time as the postdose pembrolizumab pharmacokinetics sample was drawn (that is, after infusion and approximately 4–6 h after dosing). On day 1 of cycle 2, patients were asked to take their morning dose at home, and a postdose sample was taken during study visit. Thereafter, pharmacokinetics plasma samples were collected on day 1 of every alternate cycle during the study visit. If the visit was in the morning, patients were asked to withhold the morning dose of trametinib and dabrafenib; if the visit was in the afternoon, patients were asked to take the morning dose as usual. DLT is described in Supplementary Protocol 1.
Clinical trial statistical analysis.
PFS, overall survival and duration of response were estimated using the Kaplan–Meier method. Efficacy was analyzed in the intent-to-treat population. Duration of response was analyzed in all confirmed responders. Safety was analyzed in the all-subjects-as-treated population of all patients who received at least one dose of study treatment. Statistical analyses were performed using SAS version 9.4.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Pharmacokinetic concentration-time profiles of pembrolizumab, dabrafenib and trametinib.
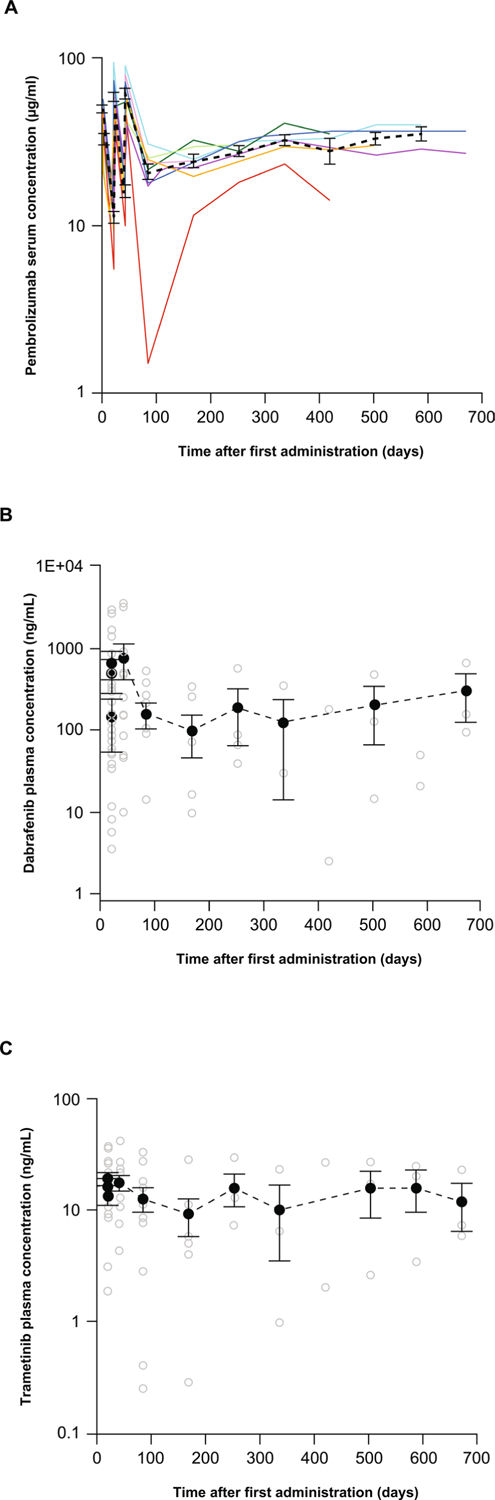
Pharmacokinetic concentration-time profiles of (a) pembrolizumab, (b) dabrafenib and (c) trametinib following administration of 2 mg/kg pembrolizumab intravenously administered together with multiple oral administrations dabrafenib 150 mg twice daily and trametinib 2 mg daily. Individual concentrations/profiles are presented as colored lines for pembrolizumab and open circles for dabrafenib and trametinib. Arithmetic mean concentration-time profiles (±standard error) are presented as dotted black bold lines.
Extended Data Fig. 2 |. Dosing of pembrolizumab in part 2: dose expansion.
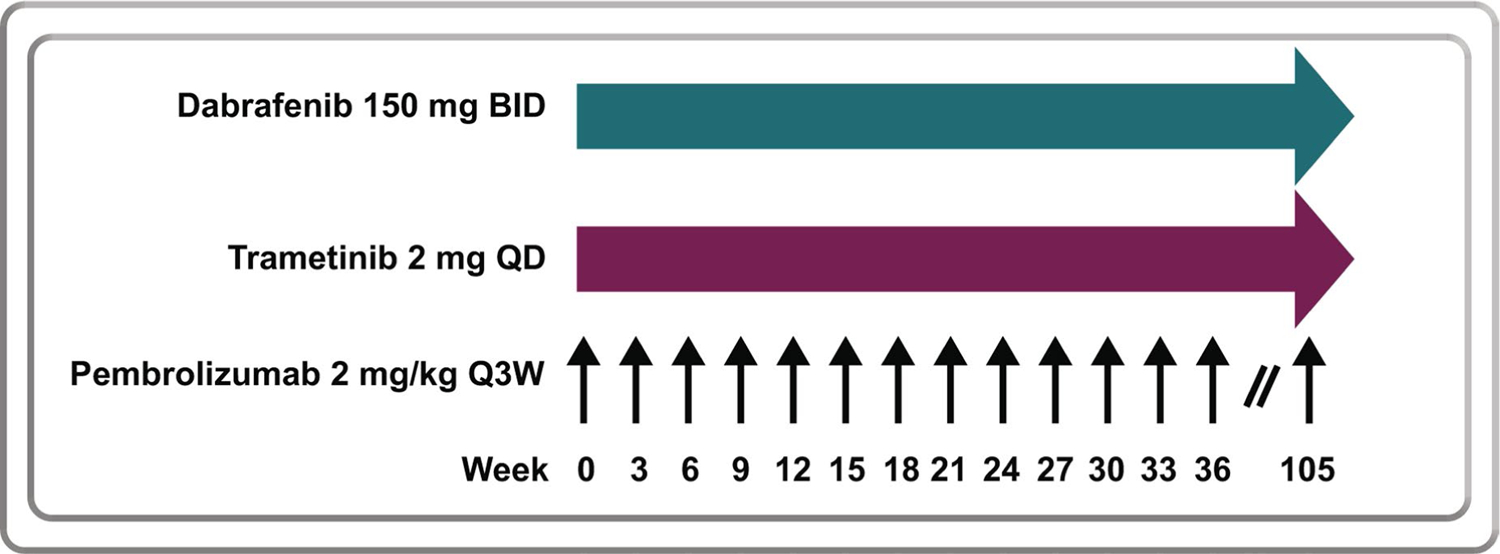
Pembrolizumab 2 mg/kg Q3W, trametinib 2 mg QD, and dabrafenib 150 mg BID in the dose expansion phase. BID, twice daily; Q3W, every 3 weeks; QD, once daily.
Extended Data Fig. 3 |. Kaplan-Meier estimates of progression-free survival and overall survival.
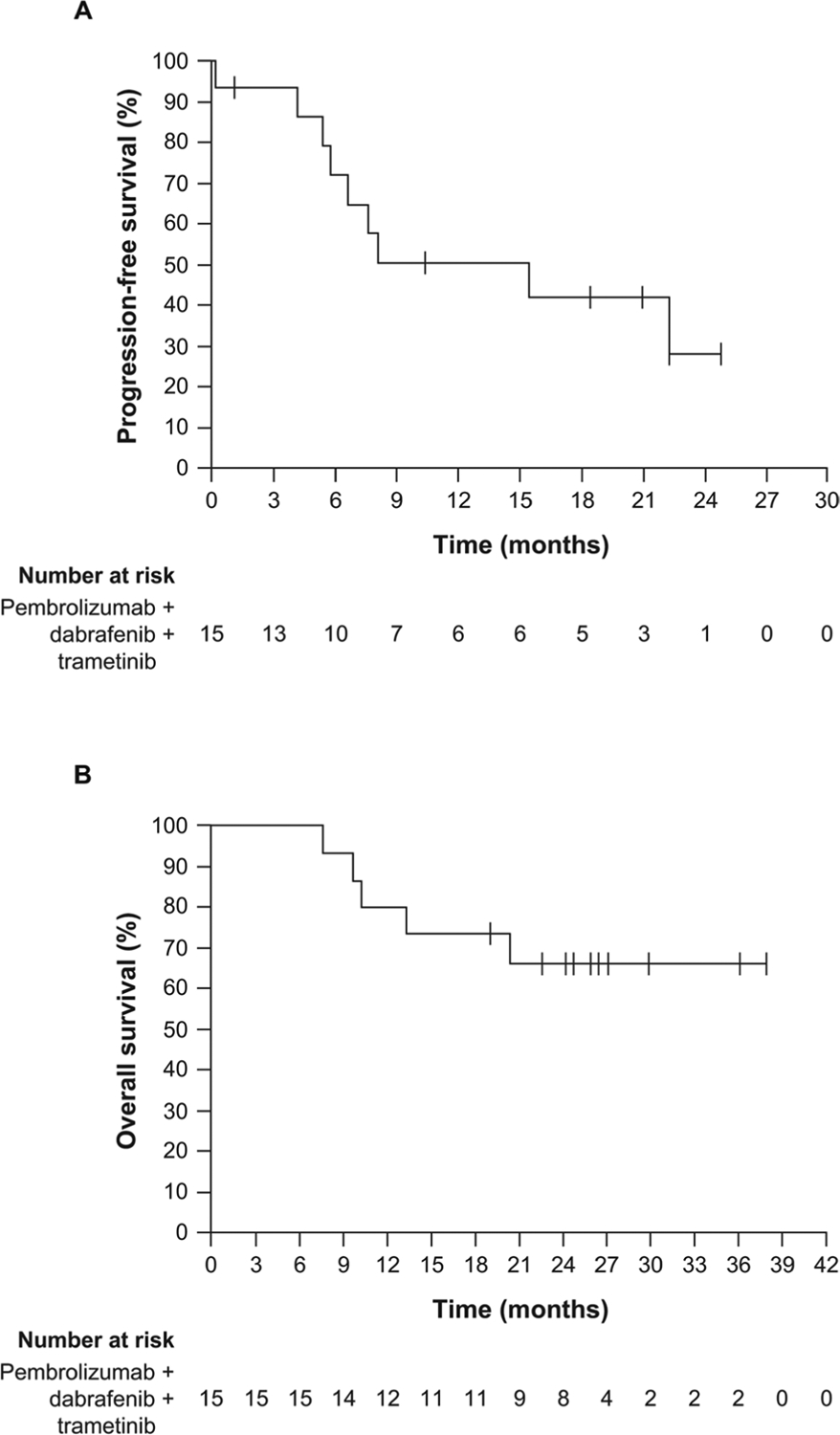
(a) Progression-free survival and (b) overall survival.
Supplementary Material
Acknowledgements
Funding for this research was provided by Merck Sharpe & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. The authors thank the patients and their families, all primary investigators and site personnel for participation in the study. The authors thank S. Ebbinghaus and S. Diede for study design and oversight, M. Bucci, J. Siegel and N. Cote for data acquisition, A. L. Webber for biomarker analysis, M. Nebozhyn for GEP analysis, R. Cristescu for WES analysis; Y. Cui for CD8 analysis, and R. Mogg for biomarker analysis. Editorial assistance was provided by D. Mitra of the ApotheCom pembrolizumab team. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. A.R., J.T. and C.S.G. were funded by NIH grants R35 CA197633 and P01 CA168585, and by the Parker Institute for Cancer Immunotherapy.
Competing interests
A.R. received personal fees for consulting from Amgen, Bristol-Myers Squibb, Chugai, Genentech–Roche, Merck–MSD and Novartis, and is on the scientific advisory board of, and holds stock in, Advaxis, Apricity, Arcus, Bioncotech, Compugen, CytomX, Five Prime, FLX Bio, ImaginAb, Isoplexis, Kite-Gilead, Merus, Rgenix, Lutris, PACT Pharma and Tango Therapeutics. V.A. received personal fees and non-financial support from Bristol-Myers Squibb and Merck Sharpe & Dohme, and personal fees only from Merck Serono, Novartis and Pierre Fabre. W.H.M. received personal fees from Bristol-Myers Squibb, Novartis, GlaxoSmithKline, Amgen and Roche. M.S.C. received personal fees from Merck Sharpe & Dohme, Novartis, Bristol-Myers Squibb and Pierre Fabre. R.F. received personal fees from Merck Sharpe & Dohme. G.V.L. received personal fees from Aduro, Amgen, Array, Bristol-Myers Squibb, Merck Sharpe & Dohme, Novartis, Oncosec and Pierre Fabre. F.S.H. reports receiving personal fees from Merck, Bristol-Myers Squibb, EMD Serono, Celldex, Sanofi, and Novartis; other from Merck to his institution and he has a patent on MICA related disorders for which he receives royalties. B.M. reports employment and stock options at Novartis and stock options at GlaxoSmithKline. B.H.M., Q.Z., R.G. and N.I. report employment at Merck Sharp & Dohme. N.I. also holds stocks in Merck & Co. and GlaxoSmithKline. O.H. reports personal fees from Merck, during the conduct of the study; personal fees from Amgen, Novartis, Roche, Bristol-Myers Squibb, Genenetch, and contracted institutional support from AstraZeneca, Bristol-Myers Squibb, Celldex, Genentech, Immunocore, Incyte, Merck, MerckSerono, MedImmune, Novartis, Pfizer, Rinat and Roche. All other authors have no competing interests.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41591-019-0476-5.
Extended data is available for this paper at https://doi.org/10.1038/s41591-019-0476-5.
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-019-0476-5.
Data availability
To protect the privacy and confidentiality of the 22 patients in this small study, RNA-Seq and WES data supporting the GEP and TMB analyses have not been made publicly available in a repository and are instead provided in the Supplementary Material linked to this article. Patient BRAF mutation status, GEP and TMB scores, and available RNA-Seq and WES data are listed in Supplementary Table 10, and anonymized patient-level somatic mutation data are provided in Supplementary Table 11. Requests for access to patient-level clinical data from the KEYNOTE trial in this study can be submitted through the EngageZone site (http://engagezone.msd.com/ds_documentation.php) or via email (dataaccess@merck.com) per Merck’s data-sharing policy. Research proposals involving predictive biomarker data should include statistical analysis plans that describe a prespecified hypothesis and accompanying statistical power calculation. Studies of de novo biomarker discovery must additionally include descriptions of independent training and validation sets.
References
- 1.Larkin J et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med 371, 1867–1876 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Long GV et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med 371, 1877–1888 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Robert C et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med 372, 30–39 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Homet Moreno B, Mok S, Comin-Anduix B, Hu-Lieskovan S & Ribas A Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 5, e1052212 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deken MA et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 5, e1238557 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu-Lieskovan S et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med 7, 279ra41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akbani R et al. Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman PB et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med 364, 2507–2516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauschild A et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Su F et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med 366, 207–215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribas A, Hodi FS, Callahan M, Konto C & Wolchok J Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med 368, 1365–1366 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Vella LJ et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res 2, 351–360 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Long GV et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol 29, 1239–1246 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Shi H et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Allen EM et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 4, 94–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayers M et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizvi NA et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hugo W et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daud AI et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J. Clin. Oncol 34, 4102–4109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilmott JS et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res 18, 1386–1394 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Frederick DT et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res 19, 1225–1231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen PL et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilain RE et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin. Cancer Res 23, 5024–5033 (2017). [DOI] [PubMed] [Google Scholar]
- 25.KEYTRUDA® (Pembrolizumab) for Injection, for Intravenous Use (Merck Sharp & Dohme Corp., 2019). [Google Scholar]
- 26.Tafinlar 50 mg Hard Capsules (Novartis Europharm, 2015). [Google Scholar]
- 27.MEKINIST (Trametinib) Tablets, for Oral Use (GlaxoSmithKline, 2018). [Google Scholar]
- 28.Schadendorf D et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 82, 45–55 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Postow MA et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med 372, 2006–2017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolchok JD et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 377, 1345–1356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schachter J et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390, 1853–1862 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Ribas A et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol 33, 3003 (2015). [Google Scholar]
- 33.Lyle S et al. Human hair follicle bulge cells are biochemically distinct and possess an epithelial stem cell phenotype. J. Investig. Dermatol. Symp. Proc 4, 296–301 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Nuckols JD, Shea CR, Horenstein MG, Burchette JL & Prieto VG Quantitation of intraepidermal T-cell subsets in formalin-fixed, paraffin-embedded tissue helps in the diagnosis of mycosis fungoides. J. Cutan. Pathol 26, 169–175 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grasso CS et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 8, 730–749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasso CS et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To protect the privacy and confidentiality of the 22 patients in this small study, RNA-Seq and WES data supporting the GEP and TMB analyses have not been made publicly available in a repository and are instead provided in the Supplementary Material linked to this article. Patient BRAF mutation status, GEP and TMB scores, and available RNA-Seq and WES data are listed in Supplementary Table 10, and anonymized patient-level somatic mutation data are provided in Supplementary Table 11. Requests for access to patient-level clinical data from the KEYNOTE trial in this study can be submitted through the EngageZone site (http://engagezone.msd.com/ds_documentation.php) or via email (dataaccess@merck.com) per Merck’s data-sharing policy. Research proposals involving predictive biomarker data should include statistical analysis plans that describe a prespecified hypothesis and accompanying statistical power calculation. Studies of de novo biomarker discovery must additionally include descriptions of independent training and validation sets.