

Abstract
Interleukin (IL)-15 is a pleiotropic cytokine with multiple roles that improve immune responses to tumor cells. Oncolytic viruses (OV) specifically lyse tumors and activate immune responses. Systemic administration of IL-15 or its complex with the IL-15Rα and chimeric antigen receptor (CAR) natural killer (NK) cells are currently being tested in the clinic. Here we generated a herpes simplex 1-based OV expressing human IL-15/IL-15Rα sushi domain fusion protein (named OV-IL15C), as well as off-the-shelf EGFR-CAR NK cells, and studied their monotherapy and combination efficacy in vitro and in multiple glioblastoma (GBM) mouse models. In vitro, soluble IL-15/IL-15Rα complex was secreted from OV-IL15C-infected GBM cells, which promoted GBM cytotoxicity and improved survival of NK and CD8+ T cells. Frozen, readily available off-the-shelf EGFR-CAR NK cells showed enhanced killing of tumor cells compared to empty vector-transduced NK cells. In vivo, OV-IL15C significantly inhibited tumor growth and prolonged survival of GBM-bearing mice in the presence of CD8+ T cells compared to parental OV. OV-IL15C plus EGFR-CAR NK cells synergistically suppressed tumor growth and significantly improved survival compared to either monotherapy, correlating with increased intracranial infiltration and activation of NK and CD8+ T cells and elevated persistence of CAR NK cells in an immunocompetent model. Collectively, OV-IL15C and off-the-shelf EGFR-CAR NK cells represent promising therapeutic strategies for GBM treatment to improve the clinical management of this devastating disease.
Keywords: oncolytic virus, CAR-NK cells, immunotherapy, GBM, combination therapy
Introduction
Oncolytic viral (OV) therapy has been recognized as a promising approach for cancer treatment. It differs from traditional gene therapy, where a viral vector only serves to deliver a specific gene. The OV itself acts as an immunostimulatory agent that can also selectively replicate in tumor cells resulting in their lysis without harming normal tissues. The concept of OV therapy has existed for decades. Findings over the past two decades support a concept that OV can boost systemic immunity and therefore antitumor immune responses (1). For augmenting OV therapeutic efficacy, various transgenes have been incorporated into the virus, as previously done for T-VEC, the first OV approved by the U.S. FDA for the treatment of melanoma (2).
Interleukin (IL)-15 is a pleiotropic cytokine and plays a key role in the development, homeostasis, activation, and survival of T, natural killer (NK), and NK-T cells (3). IL-15 receptor alpha (IL-15Rα) is one of three receptors required to mediate IL-15 signaling (4). With this understanding, researchers have recently generated IL-15 super cytokine agonists that include IL-15 and complete or partial IL-15Rα to improve in vivo antitumor activities (5,6). Both IL-15 and the IL-15/IL-15Rα complex are actively being tested in phase I and II clinical trials with evidence of immune modulation in patients (7–9). Moreover, engineering viruses to infect tumors and express transgenes, including immunostimulatory molecules such as cytokines, can improve the efficacy of oncolytic virotherapy (10–14).
Another promising approach to control tumor progression is engineering immune cells with a chimeric antigen receptor (CAR), which is an artificially modified fusion protein including an extracellular antigen recognition domain fused to an intracellular signaling domain and designed to enhance the specificity of T cells, NK cells, or other immune cells (15). CAR-modified T cells are currently showing promising efficacy to treat solid tumors, including glioblastoma (GBM) (16,17). Recently, CAR-modified NK cells have also shown early success in lymphoid malignancies (18,19). However, CAR T or CAR NK cells in combination with OV have not yet been widely explored.
Here, we hypothesized that the combination of OV-IL15C with off-the-shelf EGFR-CAR NK cells could improve antitumor efficacy in GBM. The frozen and readily available off-the-shelf EGFR-CAR NK cells were produced from the peripheral blood of individual donors, which are able to target both epidermal growth factor receptor (EGFR) and epidermal growth factor receptor variant III (EGFRvIII). Both OV-IL15C and EGFR-CAR NK cells demonstrated better cytotoxic activity compared to control OV or empty vector (EV)-transduced NK cells in vitro, respectively. In vivo, the combination of OV-IL15C and EGFR-CAR NK cells produced a synergistic antitumor effect and a significant prolongation of survival over either therapy alone, correlating with increased intracranial infiltration and activation of NK and CD8+ T cells.
Materials and Methods
Cell culture
Vero cells (derived from monkey kidney epithelial cells) were used for viral production and plaque assay-based viral titration. Human glioblastoma (GBM) cell lines (U251, LN229, U87vIII) and the mouse GBM cell line CT2A, as well as Vero and human embryonic kidney 293T cells, were cultured in DMEM media (Gibco) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml). GBM30 spheroid cells derived from a GBM patient and modified to express luciferase (named GBM30-luc) were maintained with neurobasal media (DMEM/F12) supplemented with 2% B27 (Gibco), human epidermal growth factor (Stemcell), basic fibroblast growth factor (Stemcell), heparin (Stemcell), and Glutamax (Gibco) in low-attachment cell culture flasks. Primary human cells used for tropism testing were purchased from ScienCell and cultured following the manufacturer’s instructions. U251 cells and GBM30 cells were authenticated by the University of Arizona Genetics Core via STR profiling in January 2015 and March 2018, respectively. Vero cells, 293T cells, CT2A cells, U87vIII cells, and LN229 were not authenticated after receipt. These cell lines were either purchased from ATCC or obtained from Dr. E. Antonio Chiocca’s lab at Harvard University. For all experiments, cells were frozen down in low passage cultures and used within 2–3 passages when thawed. All cell lines were routinely tested for the absence of mycoplasma using the MycoAlert Plus Mycoplasma Detection Kit from Lonza (Walkersville, MD).
Generation of OV-IL15C
Generation of OV-IL15C was performed using the fHsvQuik-1 system as previously described (20). The full-length human IL-15 and IL-15Rα sushi domain DNA sequences were cloned into the pT-oriSIE4/5 plasmid and driven by the herpes simplex virus (HSV) pIE4/5 promoter. For evaluating viral production and infectivity, plaque-forming assays were undertaken with the GBM cell lines U251 and LN229 infected with OV-Q1 or OV-IL15C at an indicated multiplicity of infection (MOI). The plaques in each group were imaged by using Zeiss fluorescent microscopy at different time points post infection.
Detection and quantification of IL-15/IL-15Rα complex produced by OV-IL15-infected cells
An immunoblotting assay was performed to detect the IL-15/IL-15Rα complex. Cell lysates (from virus-infected 293T cells) were prepared with RIPA Lysis and Extraction Buffer (ThermoFisher) containing protease/phosphatase inhibitor cocktail (Halt™). Protein concentration was assessed using a Rapid Gold BCA Protein Assay Kit (Pierce™). Anti-HA-tag antibody (Cell Signaling Technology (Cell Signaling Technology, clone: C29F4) used as a primary antibody was added for overnight incubation at 4°C, followed by incubation with anti-rabbit secondary antibody (Cell Signaling Technology) for one hour at room temperature. β-actin was used as a loading control.
An enzyme-linked immunosorbent assay (ELISA) was performed to quantify the IL-15/IL-15Rα complex secreted into the supernatants from various virus-infected GBM cell lines, using a DuoSet ELISA kit (R&D, Catalog No: DY6924) according to the manufacturer’s instructions.
Assessment of cytotoxicity of NK and CD8+ T cells induced by IL-15/IL-15Rα produced from OV-IL15C-infected GBM cells
Peripheral blood cones were collected from healthy donors after written informed consent in the City of Hope National Medical Center Donor Apheresis Center (DAC) under institutional review board-approved protocols. The studies were conducted in accordance to the Declaration of Helsinki. Primary human NK cells isolated from peripheral blood mononuclear cells (PBMCs) were pre-incubated with 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 cells for 18 hours at 37°C. Primary human CD8+ T cells were isolated and pre-incubated similarly, but the incubation time is 48 instead of 18 hours. A standard 51Cr-release assay was used to measure cytotoxicity levels of the above pre-incubated NK cells and a flow cytometry-based assay using a Fortessa X-20 flow cytometer for the pre-incubated CD8+ T cells. Prior to these assays measuring cytotoxicity levels, the pre-incubated NK cells were cultured with GBM30 or K562 target cells for 4 hours while the pre-incubated CD8+ T cells for 12 hours. The assays were performed in at least three technical replicates with human NK and CD8+ T cells from different donors. The flow cytometry data were analyzed by using Flowjo V10 software (Tree Star, Ashland, OR, USA).
Assessment of in vitro NK and CD8+ T cell survival promoted by IL-15/IL-15Rα that is produced from OV-IL15C-infected GBM cells
Primary human NK and CD8+ T cells were cultured in supernatants from OV-Q1- or OV-IL15C-infected U251 cells in the absence of recombinant human interleukin-2 (rhIL-2) at 37°C. From day 0 to day 4, the number of NK and CD8+ T cells in each group was counted daily by microscopy after staining with Trypan Blue Solution (ThermoFisher). The assays were performed with human NK and CD8+ T cells from different donors in at least three technical replicates for each donor.
Immunoblotting of phospho-STAT5 was used to determine NK and CD8+ T cell activation after 30 min- and 12-hour culture, respectively, in the supernatants from OV-Q1- or OV-IL15C-infected U251 cells. Cell lysates were prepared similarly as for immunoblotting of the IL-15/IL-15Rα complex, as described above. The anti-phospho-STAT5 (Tyr694) (D47E7) XP® Rabbit mAb (Cell Signaling Technology) was used as a primary antibody, and anti-rabbit mAb (Cell Signaling Technology) was used as a secondary antibody.
Expansion of NK cells
Primary human NK cells were isolated from PBMCs, and the purity was assessed by flow cytometry using anti-human CD3 (Miltenyi Biotec) and anti-CD56 mAbs (BD Biosciences). Highly purified NK cells were seeded at 1 × 105 cells/ml and cultured with irradiated (25 Gy) autologous PBMCs (1 × 106 cells/ml) for expansion in culture medium, containing 5% human AB serum, and supplemented with hrIL-2 (NIH, U.S.) (1000 IU/mL) and anti-CD3 (OKT3) (Invitrogen) (10 ng/ml). After five days, the media was completely changed, and cells were resuspended in fresh culture media containing rhIL-2 (1000 U/mL) without anti-CD3 mAb. Thereafter, half of the media was changed and replaced by fresh media with cytokines every 2 days. When the cell density was high, cells were harvested and transferred to larger flasks.
Retrovirus transduction
To produce retrovirus, GP2–293 cells with a confluency of 70–80% were transfected with a pCIR retrovirus vector expressing an anti-EGFR CAR cassette or with a corresponding empty control plasmid, using Lipofectamine 3000 Reagent (ThermoFisher), as previously described with some modifications (21). The EGFR-CAR cassette sequentially includes a signal peptide, the light chain of an anti-EGFR antibody, a linker, the heavy chain of an anti-EGFR antibody, a hinge, the CD28 costimulatory domain, and CD3ζ. Following the manufacturer’s protocol, RetroNectin (Takara Bio) coating plates were used for 2-hour retroviral infection of the expanded NK cells described in the Expansion of NK cells section with 2,000 × g centrifugation. The infected cells were washed and cultured with rhIL-2 (1,000 IU/ml) for 48 hours prior to the assessment of infection efficiency by flow cytometry. The infected cells were cultured for a certain period before being frozen.
Assessment of EGFR-CAR NK cell activation
For measuring CAR activity, EGFR-CAR NK cells or empty vector-transduced NK cells (EV NK) or untransduced NK cells (UT NK) were incubated with tumor cells at an E/T ratio of 5:1 for 4 hours at 37°C in the presence of anti-CD107a antibody and 1 mg/ml of GolgiPlug™ (BD Biosciences). The cells were harvested, followed by being permeabilized, fixed, and stained with an anti-IFN-γ antibody and an anti-TNF-α antibody before subjected to a flow cytometric analysis. For testing whether the IL-15/IL15-Rα complex secreted by OV-IL15C-infected GBM cells could improve anti-GBM activity of EGFR-CAR NK cells, these CAR NK cells were pre-incubated with 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 cells for 18 hours. The pre-incubated EGFR-CAR NK cells were then washed, counted, and co-cultured with GBM target cells (LN229 or U87vIII) at an E/T ratio of 5:1 for an additional 4 hours in the presence of anti-CD107a antibody and 1 mg/ml of GolgiPlug™. The co-cultured cells were harvested to assess CD107a degranulation and IFN-γ as well as TNF-α production, gated on CD56+ cells.
Animal study
All animal experiments were approved by the City of Hope Institutional Animal Care and Use Committee. Mice were sacrificed when they became moribund with neurologic impairments or obvious weight loss (up to 20%). NOD/SCID/IL-2rg (NSG) and C57BL/6J mice were purchased from The Jackson Laboratory. Details are included in Supplementary Materials.
Assessment of intracranial infiltration and activation of immune cells
6–8-week-old male C57BL/6 mice were implanted with the murine CT2A cell line expressing human EGFR (CT2A-hEGFR). Five days after the CT2A-hEGFR cells were implanted, mice were treated with 2 × 105 pfu OV-IL15C alone, 1 × 106 frozen and unsorted EGFR-CAR NK cells alone, the combination of the two agents, or saline alone. Three days after the treatment, mice were sacrificed to harvest brain tissues to isolate mononuclear cells, as previously described (22,23). The mononuclear cells were subjected to assess NK and T cell infiltration or cultured with PMA (BioLegend) and 1 mg/ml of GolgiPlug™ for 4 hours before assessing the activation capacity of these cytolytic cells by flow cytometry.
Cytokine release array assay
The CT2A-hEGFR mouse model was established and treated as described above. Mice were sacrificed to collect sera for cytokine release array assay using a Quantibody® Mouse Inflammation Array 1 Kit (RayBiotech Life, Inc).
Statistics
Statistical analyses were performed utilizing GraphPad software Prism v.8.0, R.3.3.1, or SAS 9.4. Values are presented as mean ± SEM or SD. Continuous endpoints were compared between two or more groups by a two-sample t test or one-way ANOVA model. The linear mixed model was used to account for the covariance structure due to repeated measures from the same donor. Survival functions were estimated by the Kaplan-Meier method and compared by the log-rank test. P values were adjusted for multiple comparisons by Holm’s procedure. A P value of < 0.05 was considered statistically significant.
Results
Generation of OV-IL15C and quantification of the IL-15/IL-15Rα complex secreted from virally infected GBM cells
We successfully engineered an oncolytic herpes simplex virus (oHSV), referred to as OV-IL15C, which expresses a fusion protein containing human IL-15 and an IL-15Rα sushi domain. The genetic maps of wild-type human HSV-1, parental oHSV control OV-Q1, and OV-IL15C are shown in Fig. 1A. Immunoblotting analysis showed the IL-15/IL-15Rα complex tagged with HA was detected with an expected size of 32 kDa (Fig. 1B). ELISA results indicated that secretion of the IL-15/IL-15Rα complex occurred in a time-dependent manner with approximately 600 pg/ml, 300 pg/ml, 800 pg/ml, 1,000 pg/ml, and 1,200 pg/ml secreted by U251 cells, LN229 cells, GBM30 cells, CT2A cells, and 293T cells, respectively, 96 hours after OV-IL15C infection, while the IL-15/IL-15Rα complex was undetectable in uninfected and OV-Q1-infected cells (Fig. 1C). In order to investigate the capacity of viral production by OV-IL15C, the GBM cell lines were infected with the OVs at different MOIs. The OV-IL15C-infected U251 and LN229 cells produced a similar amount of virus compared to the same cells infected with the control OV-Q1 under either unsaturated or saturated conditions (Fig. 1D), suggesting that OV-IL15C engineering does not affect the capacity for viral production.
Figure 1. Generation of OV-IL15C and quantification of the IL-15/IL-15Rα complex produced by OV-IL15C infection of GBM cells.
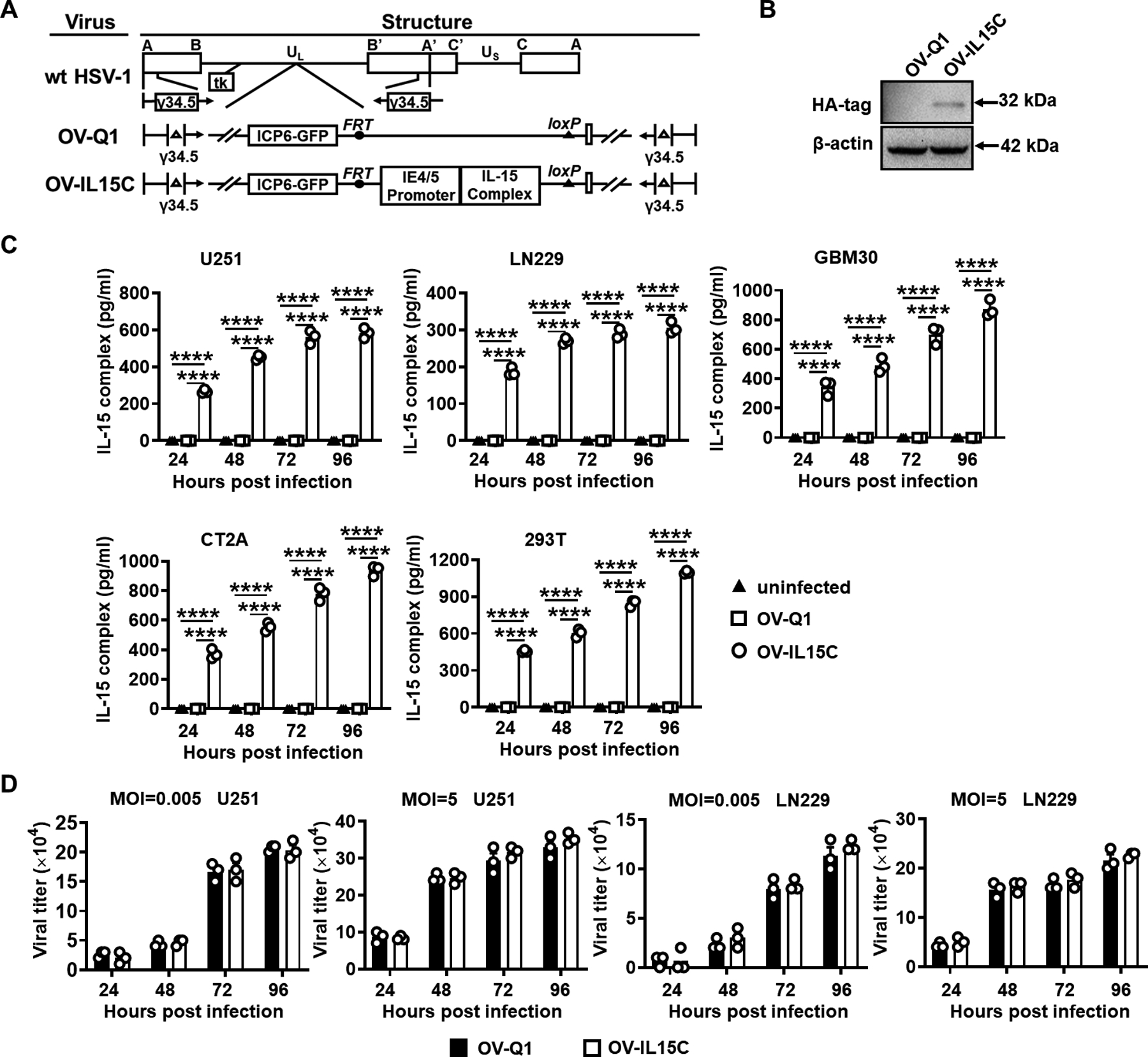
A, Schematic maps of oncolytic viruses used in this study. First: genetic map of wild-type HSV-1 (wt HSV-1). Second: genetic map of the parental oHSV control (OV-Q1) with deletion of γ34.5, dysfunction of ICP6, and insertion of the GFP gene. Third: genetic map of the new oHSV (OV-IL15C) showing the insertion of the human IL-15 and IL-15Rα sushi domain driven by the viral pIE4/5 promoter. B, Immunoblotting to test the expression of the human IL-15/IL-15Rα complex at the protein level. 293T cells were infected by OV-Q1 or OV-IL15C at a MOI of 2. Two days later, cell lysis was collected and used for detection of the HA-tagged IL-15/IL-15Rα complex by immunoblotting with an expected size. β-actin was used as a sample loading control. C, ELISA for the human IL-15/IL-15Rα complex quantification. GBM cell lines (human cell lines U251, LN229, GBM30, murine GBM cell line CT2A) and 293T cells were infected with OV-Q1 or OV-IL15C at a MOI of 0.05. Supernatants from different groups were collected for quantification by a DuoSet ELISA kit. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SEM. ****, P < 0.0001. D, Viral production capacity test. Human GBM cell lines U251 or LN229 were seeded in a 96-well plate and infected by OV-Q1 or OV-IL15C at different MOIs (MOI=0.005 unsaturated or 5 saturated). Twenty-four hours, 48 hours, 72 hours, and 96 hours post infection, GFP-positive plaques were counted with a Zeiss fluorescence microscope.
Enhancement of NK and CD8+ T cells cytotoxicity and survival by the secreted IL-15/IL-15Rα complex in vitro
Both NK and CD8+ T cells are cytolytic lymphocytes, and their activation by cytokines is critical for their antitumor activities. NK cells against GBM30 cells were found significantly increased in the presence of 10× concentrated supernatants from OV-IL15C-infected GBM cells compared to 10× concentrated supernatants from OV-Q1-infected GBM cells (Fig. 2A). Similarly, the killing ability of CD8+ T cells against GBM30 cells also significantly increased in the presence of enriched supernatants from OV-IL15C-infected GBM cells compared to enriched supernatants from OV-Q1-infected GBM cells (Fig. 2B). Furthermore, the enriched supernatants from OV-IL15C-infected GBM cells significantly prolonged the survival of NK and CD8+ T cells compared to enriched supernatants from OV-Q1-infected GBM cells, correlating to increased levels of phosphorylated STAT5 in both NK and CD8+ T cells (Fig. 2C–F; Supplementary Fig. S1A and S1B).
Figure 2. The IL-15/IL-15Rα complex secreted by OV-IL15C-infected GBM cells enhances cytotoxicity and improves survival of NK and CD8+ T cells in vitro.
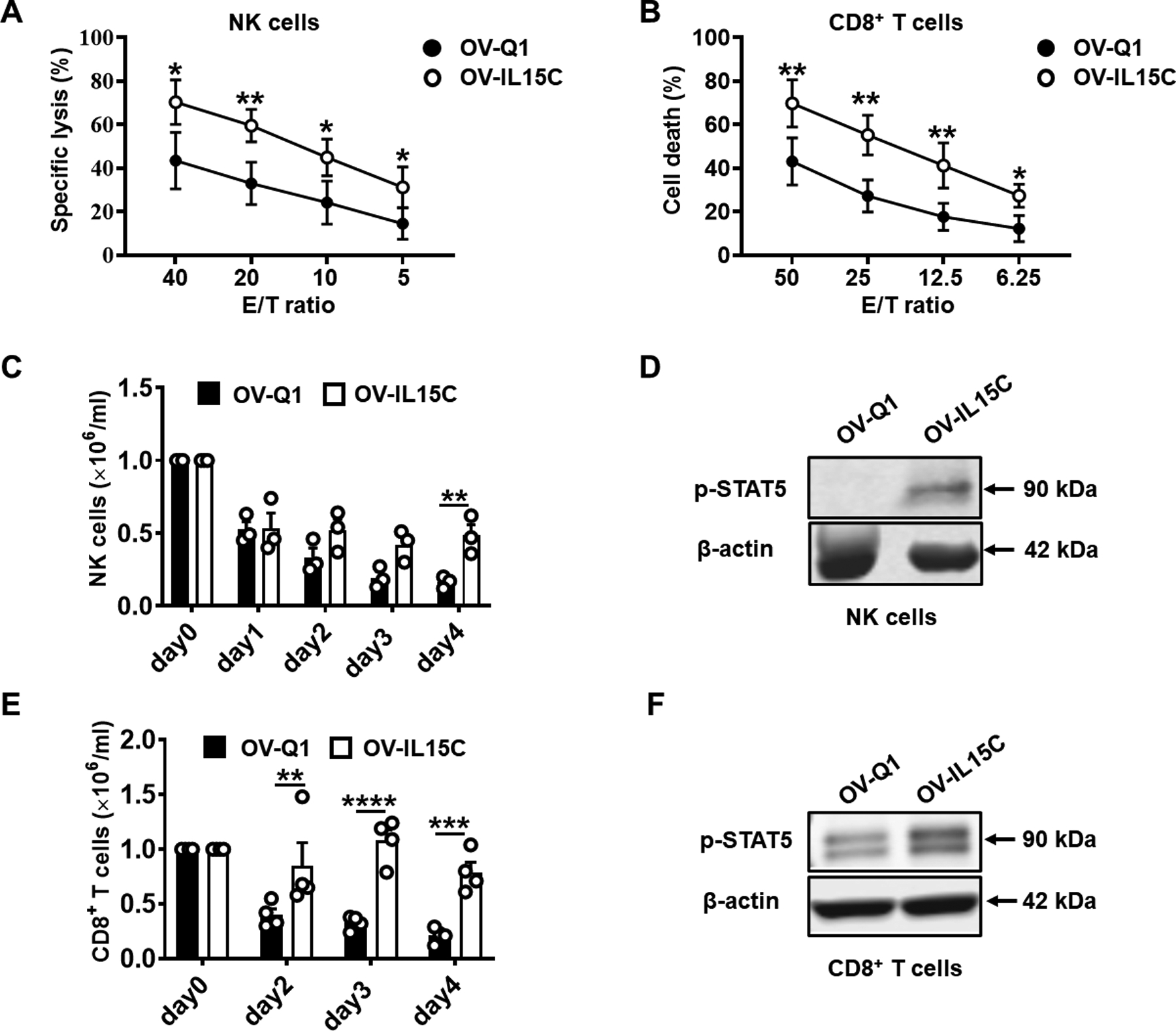
A, 51Cr- release assay. Primary human NK cells were pre-incubated with 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 cells for 18 hours. GBM30 cells were used as target cells and labeled with 51Cr for 1.5 hours, and then co-cultured with the above pre-incubated NK cells at various E/T ratios (40:1, 20:1, 10:1, and 5:1) at 37°C for 4 hours. The results show the average of four different donors. P values are corrected by multiple t tests using the Holm-Sidak method. Values are presented as mean ± SEM. *, P < 0.05; **, P < 0.01. B, Flow cytometry-based cytotoxicity assay. Primary human CD8+ T cells were pre-incubated with 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 cells for 48 hours, followed by co-culturing with the APC-labeled target GBM30 cells at various E/T ratios (50:1, 25:1, 12.5:1, and 6.25:1) at 37°C for 12 hours. The dead cells were stained by SYTOX™ Blue Dead Cell. The results show the average of five different donors. P values are corrected by multiple t tests using the Holm-Sidak method. Values are presented as mean ± SEM. *, P < 0.05; **, P < 0.01. C-F, Primary human NK or CD8+ T cells were cultured in 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 GBM cells in the absence of rhIL-2. On day 0, 1 × 106 NK or CD8+ T cells per well were seeded in a round-bottom 96-well plate. From day 0 to day 4, live cells in each group were counted daily using Trypan Blue exclusion assays. The experiment was repeated with human NK or CD8+ T cells from four different donors. P values are corrected by multiple t tests using the Holm-Sidak method. Values are presented as mean ± SD. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Proteins from NK or CD8+ T cells cultured in 10× concentrated supernatants as above were collected after 0.5 hours or 12 hours of the culture, respectively. 20 μg total proteins for each sample were loaded into the SDS gel. β-actin was used as a sample loading control. Immunoblotting assays for (D) and (F) were repeated with four different donors showing similar data.
OV-IL15C prolongs survival and enhances GBM virotherapy in vivo in a xenograft model
To evaluate the efficacy of OV-IL15C on GBM therapy in vivo, we established an orthotopic xenograft GBM mouse model by intracranially injecting luciferase-expressing GBM30 cells (GBM30-luc) into NOD/SCID/IL-2rg (NSG) mice on day 0. On days 5 and 12, mice received an intratumoral co-injection of OV-Q1 plus human CD8+ T cells or OV-IL15C plus human CD8+ T cells or saline on each day (Fig. 3A). The co-injected human CD8+ T were pre-activated by anti-CD3/anti-CD28 beads. The tumor growth of the mice was monitored by serial in vivo bioluminescence imaging (BLI). Mice treated with OV-IL15C plus human CD8+ T cells showed a significant reduction in tumor burden 8 days (Fig. 3B and C) and 15 days (Fig. 3D and E) post tumor implantation and survived significantly longer (Fig. 3F), compared to mice treated with OV-Q1 plus human CD8+ T cells or saline. To test whether OV-IL15C also enhances GBM therapy in the presence of human NK cells, we re-established the GBM30-luc xenograft GBM model with an intratumoral co-injection of OV-Q1 plus human NK cells or OV-IL15C plus human NK cells, similarly as described above for co-injection of OV and human CD8+ T cells (Supplementary Fig. S2A). However, no significant difference was observed for tumor burden and mouse survival between the OV-Q1 plus human NK cells and the OV-IL15C plus human NK cells group (Supplementary Fig. S2B–D).
Figure 3. Enhancement of GBM virotherapy in vivo by OV-IL15C co-administered with human CD8+ T cells in a xenograft GBM mouse model.
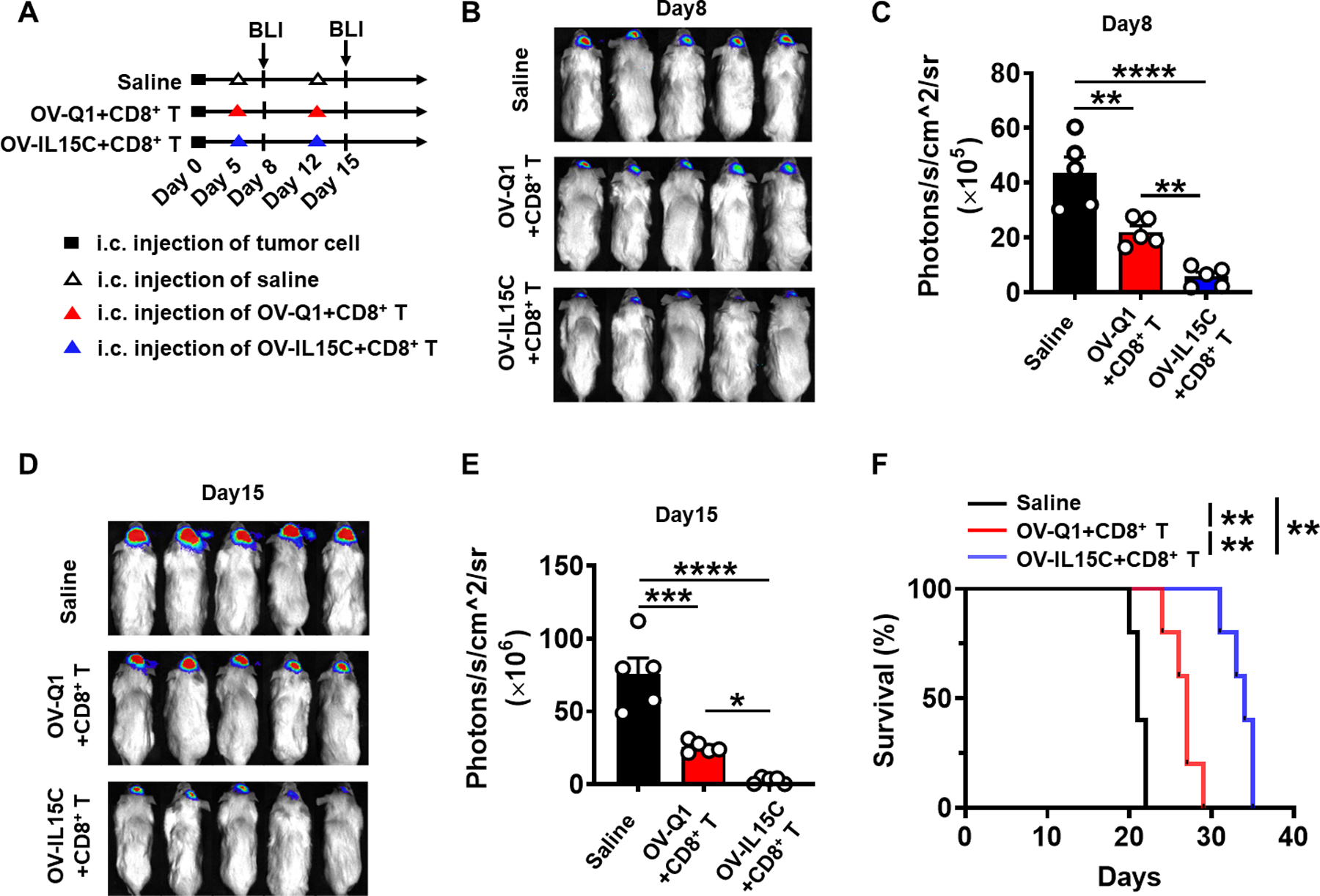
A, Experimental timeline for in vivo study. An orthotopic xenograft GBM mouse model was established by intracranial injection of 1 × 105 luciferase-expressing GBM30 cells (GBM30-luc) into the brain of NOD/SCID/IL-2rg (NSG) mice on day 0. On days 5 and 12, mice were intratumorally injected with 2 × 105 pfu of OV-Q1, OV-IL15C, or saline. The two virus groups were co-administered with 1 × 106 activated CD8+ T cells. n=5 animals for each group. B, On day 8, bioluminescence imaging (BLI) to check brain tumor growth. C, Quantification of BLI in B. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. **, P < 0.01; ****, P < 0.0001. D, BLI to check brain tumor growth on day 15. E, Quantification of BLI in D. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. F, Survival of GBM30-luc bearing mice treated with OV-Q1, OV-IL15C plus CD8+ T cells or saline. Log-rank test was used to compare survival curves. **, P < 0.01.
Off-the-shelf EGFR-CAR NK cells improve anti-GBM activity in vitro and in an in vivo xenograft model
In order to improve NK cell antitumor activity in immunotherapy, we generated CAR NK cells targeting wild-type EGFR and EGFRvIII GBM cells. For this, human peripheral blood NK cells were transduced with an EGFR-CAR retroviral vector (Fig. 4A), followed by further expansion. We were able to generate over 1 × 1011 EGFR-CAR NK cells from the peripheral blood of an individual donor. Cells were frozen and ready to use, so called off-the-shelf, for both in vitro and in vivo experiments after thawing. We assessed cytotoxicity of EGFR-CAR NK cells against the LN229 (EGFR positive cell line) (24) or the EOL-1 (no EGFR or EGFRvIII expression) (25) at different effector/target (E/T) ratios. Across all E/T ratios, EGFR-CAR NK cells exerted superior killing of LN229 cells compared to empty vector (EV)-transduced NK cells (Fig. 4B, left). The EGFR-CAR NK cells were equally efficient as EV-transduced NK cells in killing EOL-1 target cells (Fig. 4B, right). These data indicate that the enhanced killing of LN229 targets by the transduced cells is mediated by the CAR. Furthermore, we measured CD107a degranulation and IFN-γ as well as TNF-α secretion in response to LN229, U87vIII (EGFRvIII positive cell line) (24), and EOL-1 target cell lines, and all these effector functions of EGFR-CAR NK cells were significantly increased against LN229 and U87vIII target cells compared to the EV-transduced NK cells, while the EGFR-CAR NK cells and EV-transduced NK cells showed similar effector function against the EOL-1 cells (Fig. 4C). Next, the luciferase-expressing U87vIII orthotopic GBM model was established as described for the GBM30-luc model, followed by treatment with EGFR-CAR NK cells or EV-transduced NK cells as depicted in Figure 4D. Results showed that a single intracranial infusion of EGFR-CAR NK cells possessed significantly better anti-GBM activity and significantly improved survival of mice bearing GBM tumor, compared to EV-transduced NK or control groups (Figs. 4E–G).
Figure 4. EGFR-CAR NK cells enhance eradication of GBM cells and prolong survival in an in vivo xenograft model.
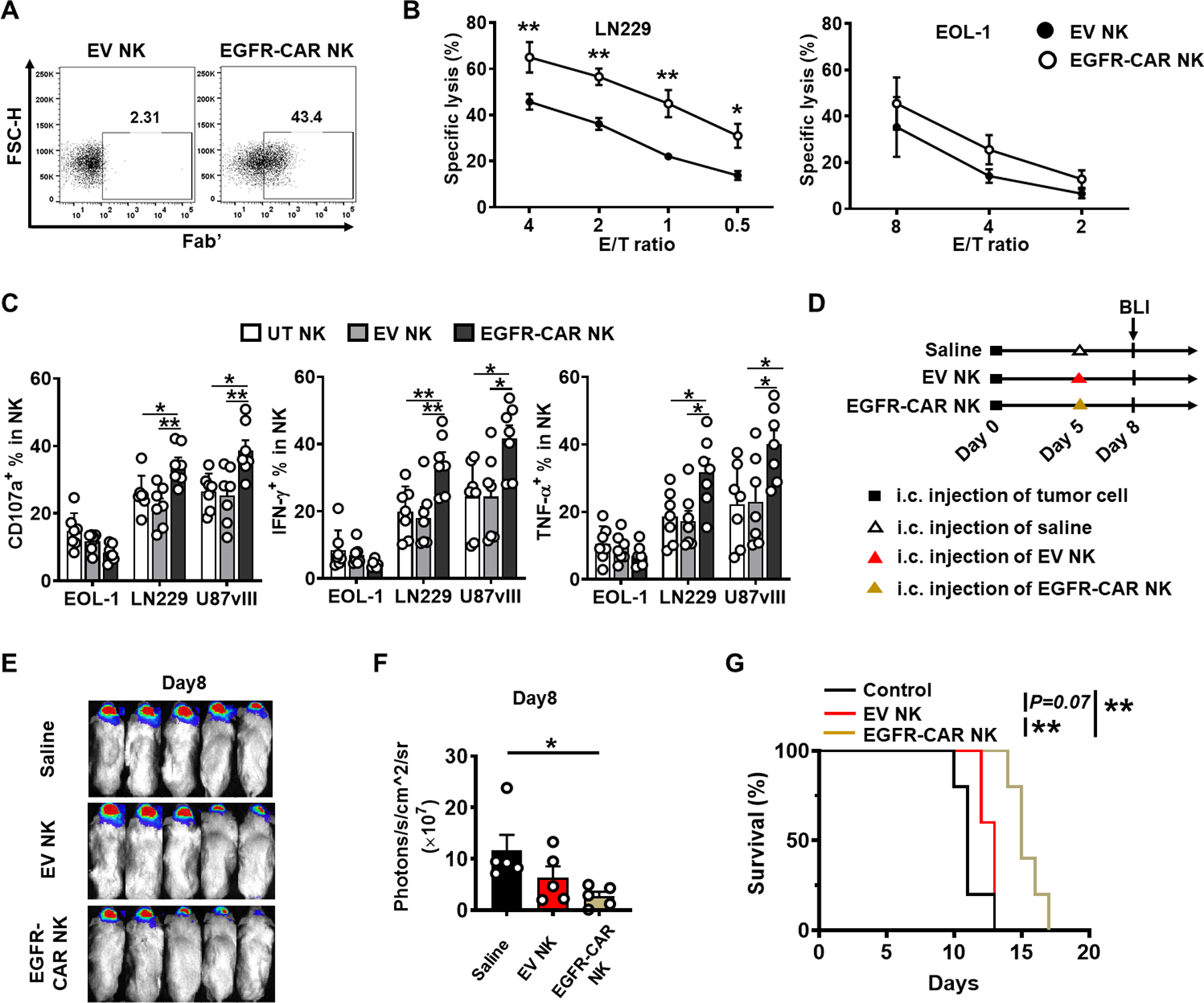
A, Assessment of EGFR-CAR expression 48 hours post retroviral transduction by flow cytometry. Anti-mouse Fab’ antibody was used to stain human anti-EGFR scFv on NK cells. B, Left: unsorted EGFR-CAR NK cells cytotoxicity function against target LN229 (EGFR positive). Right: an eosinophilic leukemia cell line EOL-1 was used as a negative control. P values are corrected by multiple t tests using the Holm-Sidak method. Values are presented as mean ± SEM. *, P < 0.05; **, P < 0.01. The experiment was repeated three times with NK cells isolated from different donors with similar results. C, Degranulation of CD107a as well as secretion of IFN-γ and TNF-α from untransduced (UT), unsorted EV-transduced (EV), or EGFR-CAR transduced NK cells co-cultured with the GBM cell line LN229 (EGFR positive) or U87vIII (EGFRvIII positive). EOL-1 was performed as a negative control for both. The experiment was repeated seven times with NK cells isolated from different donors with similar results. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SEM. *, P < 0.05; **, P < 0.01. D, Experimental timeline for in vivo study. On day 0, a xenograft GBM mouse model was established by intracranial injection of 1 × 105 U87vIII-luc cells into NSG mice. On days 3, mice were intratumorally injected with 1 × 106 EV-transduced NK cells, 1 × 106 EGFR-CAR NK cells, or saline. All transduced cells were unsorted. n=5 animals for each group. E, BLI to check brain tumor growth on day 8. F, Quantification of BLI in E. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05. G, Survival of U87vIII-luc-bearing mice treated with EV-transduced NK cells, EGFR-CAR NK cells, or saline alone. Log-rank test was used to compare animal survival curves. **, P < 0.01.
The combination of OV-IL15C and EGFR-CAR NK cells shows synergistic effects relative to corresponding monotherapies in a xenograft GBM model
The above results showed EGFR-CAR NK cells are superior to EV-transduced NK cells. We next tested whether the IL-15/IL15-Rα complex secreted by OV-IL15C-infected GBM cells can improve EGFR-CAR NK cell activity. For this purpose, after confirming that EGFR-CAR expression and activity were remained after being frozen (Supplementary Fig. S3A and S3B), we thawed frozen EGFR-CAR NK cells and pre-incubated them with 10× concentrated supernatants from OV-Q1- or OV-IL15C-infected U251 cells, followed by co-culture with GBM target cells (LN229 or U87vIII). Results showed that the 10× concentrated supernatants containing the IL-15/IL-15Rα complex secreted from OV-IL15C infected-GBM cells significantly increased or showed a trend of an increase of the secretion of IFN-γ and TNF-α, compared to similarly 10× concentrated supernatants from OV-Q1 infected-GBM cells (Fig. 5A). To test whether the in vitro effects can be translated to in vivo, i.e., whether the combination of EGFR-CAR NK cells with OV-IL15C is superior to its combination with OV-Q1, we utilized the orthotopic GBM30-luc xenograft model (Fig. 5B). Indeed, the combination of OV-IL15C and EGFR-CAR NK cells resulted in a more significant reduction in tumor burden (Fig. 5C and D) and prolonged mouse survival (Fig. 5E), compared to the combination of OV-Q1 and EGFR-CAR NK cells. These in vivo data provide a proof of concept for superior combination immunotherapy of OV-IL15C with CAR NK cells. Next, we investigated whether the combination of OV-IL15C and EGFR-CAR shows a synergistic effect. For this purpose, we re-established the orthotopic GBM30-luc xenograft model and treated it with or without the combination of OV-IL15C and EGFR-CAR NK cells of their monotherapies, as shown in Fig. 5F (4 groups in Fig. 5F–I vs. 3 groups in Fig, 5B–E). Luciferase-based in vivo BLI showed the mice that received either OV-IL15C alone, EGFR-CAR NK cells alone, or the combination of both had significantly inhibited tumor growth, compared to injection with saline control (Fig. 5G, Supplementary Fig. S3C). Importantly, the suppression of tumor growth showed a synergistic effect after treatment with the combination of OV-IL15C and EGFR-CAR NK cells compared to each monotherapy (Fig. 5H). The combination of the two agents also rendered mice to survive significantly longer than those in all other groups (Fig. 5I).
Figure 5. The combination of OV-IL15C and EGFR-CAR NK cells shows better effects than corresponding monotherapies in a xenograft GBM model.
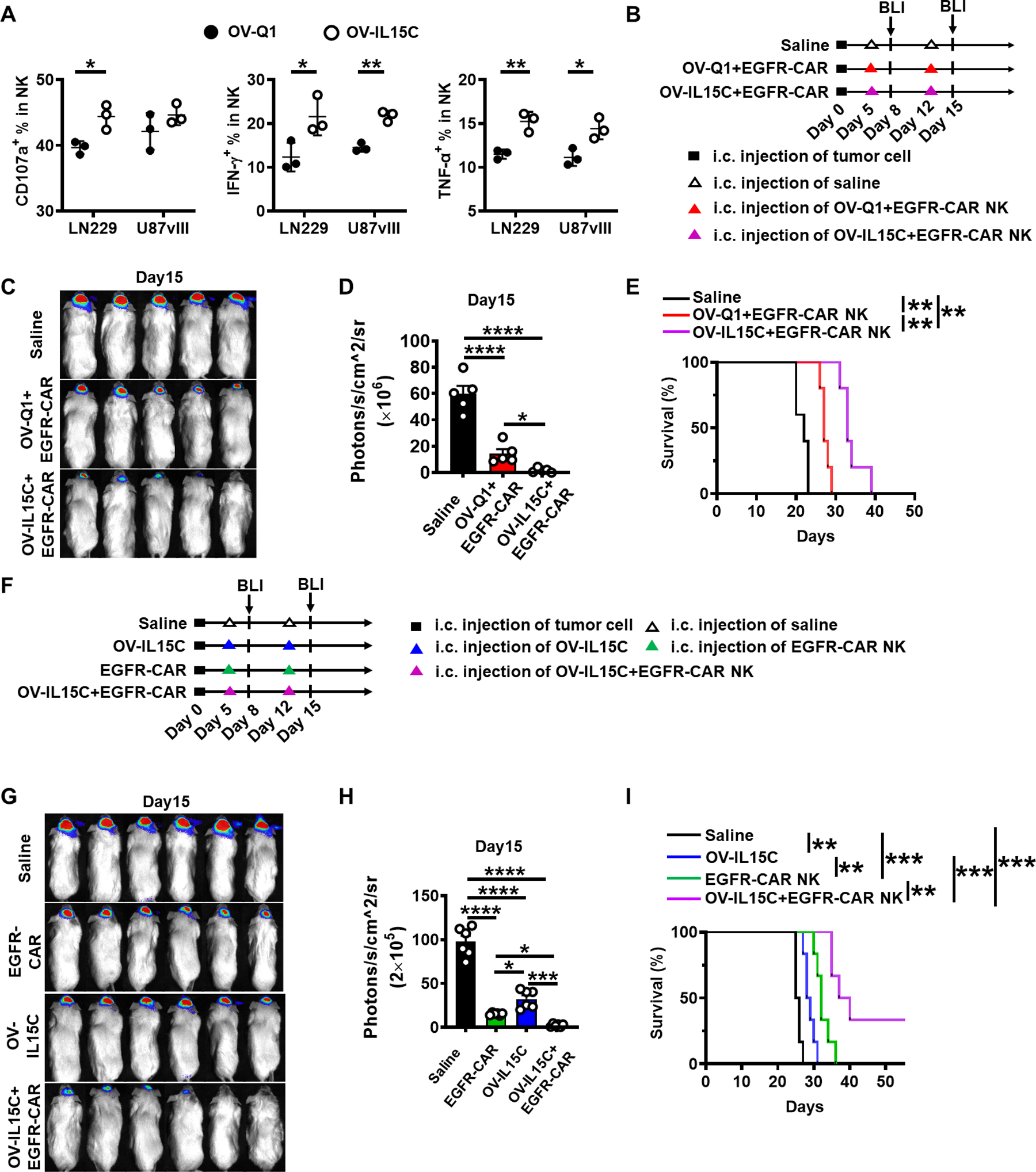
A, Assessment of CD107a degranulation and levels of IFN-γ and TNF-α of frozen EGFR-CAR NK cells. GBM cell lines LN229 and U87vIII were used as target cells. The experiment was repeated three times with different frozen CAR NK cells with similar results. P values were calculated by unpaired t tests. Values are presented as mean ± SEM. *, P < 0.05; **, P < 0.01. B, Experimental timeline for in vivo study. On day 0, a xenograft GBM mouse model was established by intracranial injection of 1 × 105 GBM30-luc cells into NSG mice. On days 5 and 12 after tumor implantation, mice were intratumorally injected with 2 × 105 pfu of OV-Q1 plus 1 × 106 frozen EGFR-CAR NK cells, 2 × 105 pfu of OV-IL15C plus 1 × 106 frozen EGFR-CAR NK cells, or saline alone. All transduced cells were unsorted. n=5 animals for each group. C, BLI of GBM30-luc tumors on day 15. D, Quantification of BLI in C. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05; ****, P < 0.0001. E, Survival of GBM30-luc-bearing mice treated with OV-Q1 or OV-IL15C combined with unsorted frozen EGFR-CAR NK cells or saline. Log-rank test was used to compare animal survival curves. **, P < 0.01. F, The xenograft GBM mouse model was established by intracranial injection of 1 × 105 GBM30-luc cells into NSG mice on day 0. On days 5 and 12, mice were intratumorally injected with 1 × 106 EGFR-CAR NK cells alone, 2 × 105 pfu OV-IL15C alone, 2 × 105 pfu OV-IL15C combined with 1 × 106 EGFR-CAR NK cells or saline as control on each day. n=6 animals for each group. G, BLI of GBM30-luc tumors on day 15. H, Quantification of BLI in G. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. I, Survival of GBM30-luc bearing mice. Log-rank test was used to compare animal survival curves. **, P < 0.01, ***, P < 0.001.
The combination of OV-IL15C and EGFR-CAR enhances endogenous immune responses and CAR persistence as well as improves GBM therapy in an immunocompetent model
In order to investigate the effects of OV-IL15C on endogenous immune cells, we established an immunocompetent GBM model with an intracranial injection of the murine GBM cell line CT2A. Five days after tumor implantation, mice received an intracranial injection of OV-Q1, OV-IL15C, or saline. Three days after treatment, mice were sacrificed to check the infiltration of the immune cells in both the brain and spleen. Mice treated intracranially with OV-IL15C showed a significant infiltration of endogenous NK and T cells into the brain compared to those treated with OV-Q1 or saline (Fig. 6A); however, no significant difference between OV-IL15C and OV-Q1 was observed in the spleen (Fig. 6B). In both organs, both OVs significantly recruited more or showed a trend of increase of NK and T cells (Fig. 6A and B). Also, OV-IL15C treatment significantly prolonged mouse survival when compared to OV-Q1 or saline treatment (Fig. 6C). To evaluate the benefit of combining OV-IL15C with EGFR-CAR NK cells on immune responses, we modified the immunocompetent GBM model using the murine GBM cell line CT2A expressing human EGFR (CT2A-hEGFR). Five days after tumor implantation, mice were intratumorally treated with OV-IL15C, EGFR-CAR NK cells, the combination of the two agents, or saline. Three days after treatment, mice were sacrificed to check the immune cell infiltration and activation. Blood sera were collected to measure cytokine levels, as detailed below. We found that significantly more endogenous NK and CD8+ T cells infiltrated into the brain when mice were treated with the combination of OV-IL15C and EGFR-CAR NK cells vs. their monotherapies, except for the combination vs. OV-IL15C in T cells, which only showed a trend of an increase (Fig. 6D). The combination also resulted in significantly more TNF-α secretion than EGFR-CAR NK cells rather than OV-IL15C in CD8+ T cells, while similar benefit was not observed in NK cells and both types of cytolytic cells did not show any benefit of the combination regarding IFN-γ production (Fig. 6D). The combination of OV-IL15C and EGFR-CAR NK cells also significantly prolonged mice survival, compared to the other groups (Fig. 6E). To examine whether OV-IL-15C can make EGFR-CAR NK cells persist longer without increasing exhaustion, we re-established the orthotopic CT2A-hEGFR immunocompetent model. Mice were treated with EGFR-CAR NK cells alone or in combination with OV-IL15C five days after tumor implantation. Twenty-four hours, 48 hours, and 72 hours after treatment, we observed that EGFR-CAR NK cells persisted longer (72 hours vs. 48 hours) in the presence of OV-IL15C, compared to EGFR-CAR NK cells alone. On each of three days, more EGFR-CAR NK cells in terms of percentages and/or absolute cell numbers were detected in the brain in the OV-IL15C plus EGFR-CAR group vs. the EGFR-CAR alone group (Fig. 6F). Also, compared to EGFR-CAR NK cells alone, mice treated with the combination of OV-IL15C plus EGFR-CAR NK cells did not show increased cell exhaustion, marked by the expression of PD-1 and Tim-3 (Fig. 6G).
Figure 6. OV-IL15C increases NK and T cells infiltration and activation as well as the persistence of CAR NK cells and improves GBM therapy in the presence of EGFR-CAR NK cells in an immunocompetent model.
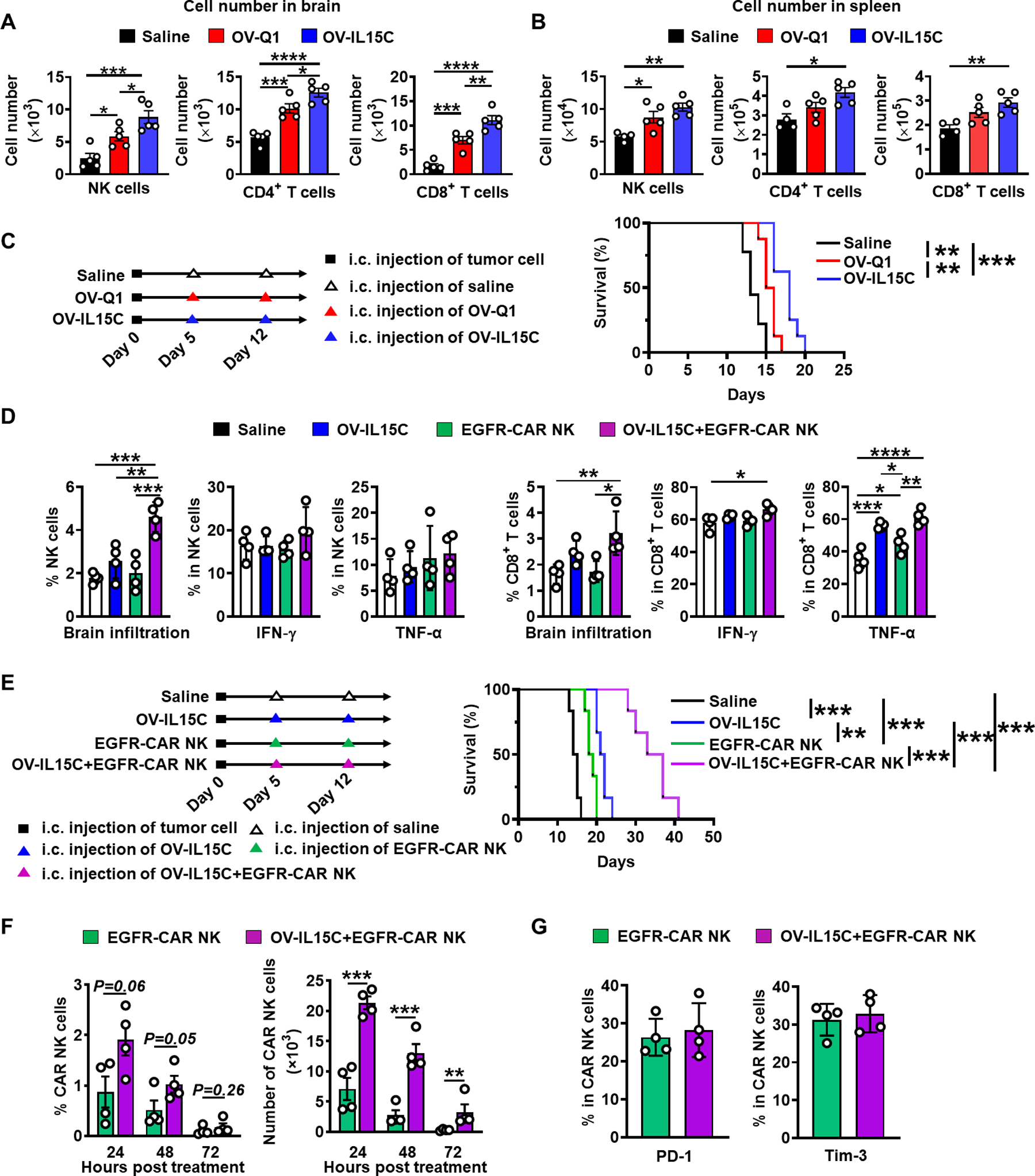
A-B, An immunocompetent GBM mouse model was established by intracranial injecting 1 × 105 CT2A cells into C57BL/6 mice on day 0. n=6 animals for each group. On day 5, mice were intratumorally injected with 2 × 105 pfu of OV-Q1, OV-IL15C, or saline as control. Representative of NK and T cells infiltration in the brain and spleen. P values correct for ordinary one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. C, Left: in vivo experiment schedule. Right: survival of CT2A-bearing mice treated with OV-Q1, OV-IL15C, or saline as control. Log-rank test was used to compare animal survival curves. n=8 animals for each group. **, P < 0.01; ***, P < 0.001. D, The infiltration and activation of NK and CD8+ T cells in the brain. An immunocompetent model implanted with the murine GBM cell line CT2A expressing human EGFR (CT2A-hEGFR) was established on day 0. On day 5, mice were treated with OV-IL15C alone, frozen, and unsorted EGFR-CAR NK cells alone, the combination of the two agents, or saline. Three days later, mice were sacrificed, and brains were collected to assess NK and T cell infiltration as well as activation. P values were calculated after Log10 transformation due to big variations, followed by one-way ANOVA using Holm-Sidak multiple comparisons test. Values are presented as mean ± SD. *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001. E, Left: in vivo experiment schedule. Right: survival of CT2A-hEGFR bearing mice treated with OV-IL15C alone, EGFR-CAR NK cells alone, or combination of the two agents or saline as control. Log-rank test was used to compare animal survival curves. n=6 animals for each group. **, P < 0.01, ***, P < 0.001. F, In vivo persistence of EGFR-CAR NK cells in the presence of OV-IL15C. An immunocompetent model implanted with the murine CT2A-hEGFR GBM cell line was used. Five days after tumor implanted, the mice were treated with frozen EGFR-CAR NK cells alone or in combination with OV-IL15C. Twenty-four, 48 hours or 72 hours after the treatment, mice were sacrificed to check CAR NK cells persistence. P values were calculated after Log10 transformation due to big variations, followed by unpaired t tests. Values are presented as mean ± SD. **, P < 0.01, ***, P < 0.001. G, EGFR-CAR NK cell exhaustion. An immunocompetent model implanted with the murine CT2A-hEGFR GBM cell line was used. Five days later, the mice were treated with frozen EGFR-CAR NK cells alone or in combination with OV-IL15C. Three days after the treatments, mice were sacrificed to determine CAR NK cell exhaustion. Unpaired t tests were used for statistical analysis.
Safety profiling of OV-IL15C and EGFR-CAR NK cells
For evaluating the safety profile of OV-IL15C, an in vitro tropism change assay and an in vivo safety assay were performed. In vitro, no discernible change in tropism was observed following infection with either OV-Q1 or OV-IL15C in primary human oral fibroblasts (HOrF), primary human hepatic sinusoidal endothelial cells (HHSEC), or primary human pulmonary microvascular endothelial cells (HPMEC) (Fig. 7A, top). We also quantified the effects of OV-Q1 and OV-IL15C on infection of these primary human cell lines, and no significant difference was observed. For HPMEC, it is hard to quantify the plaque number because these cells are highly susceptible to oHSV infection, and the formed plaques did not separate very well (Fig. 7A, bottom). To assess in vivo safety of OV-IL15C, BALB/c mice were intracranially injected with wild-type HSV-1 (as a negative control for safety), OV-Q1, or OV-IL15C at a high dose of 1 × 106 pfu. Mice injected with wild-type HSV-1 died rapidly (less than 12 days), while all mice treated with OV-Q1 or OV-IL15C survived for several weeks (Fig. 7B). Since cytokine release syndrome (CRS) is common and major toxicity in CAR T cells, using a Quantibody® Mouse Inflammation array, we measured levels of 37 cytokines in the sera of the mice treated with saline (control), OV-IL-15C, EGFR-CAR NK cells, or the combination of OV-IL15C plus EGFR-CAR NK cells. Results showed that the levels of these cytokines, including IL-6, IL-10, IFN-γ, and GM-CSF, had no significant difference among the 4 treatment groups (Fig. 7C), suggesting that unlikely OV-IL15C, EGFR-CAR NK cells, and their combination will induce CRS in vivo.
Figure 7. Safety profiling of OV-IL15C and EGFR-CAR NK cells.
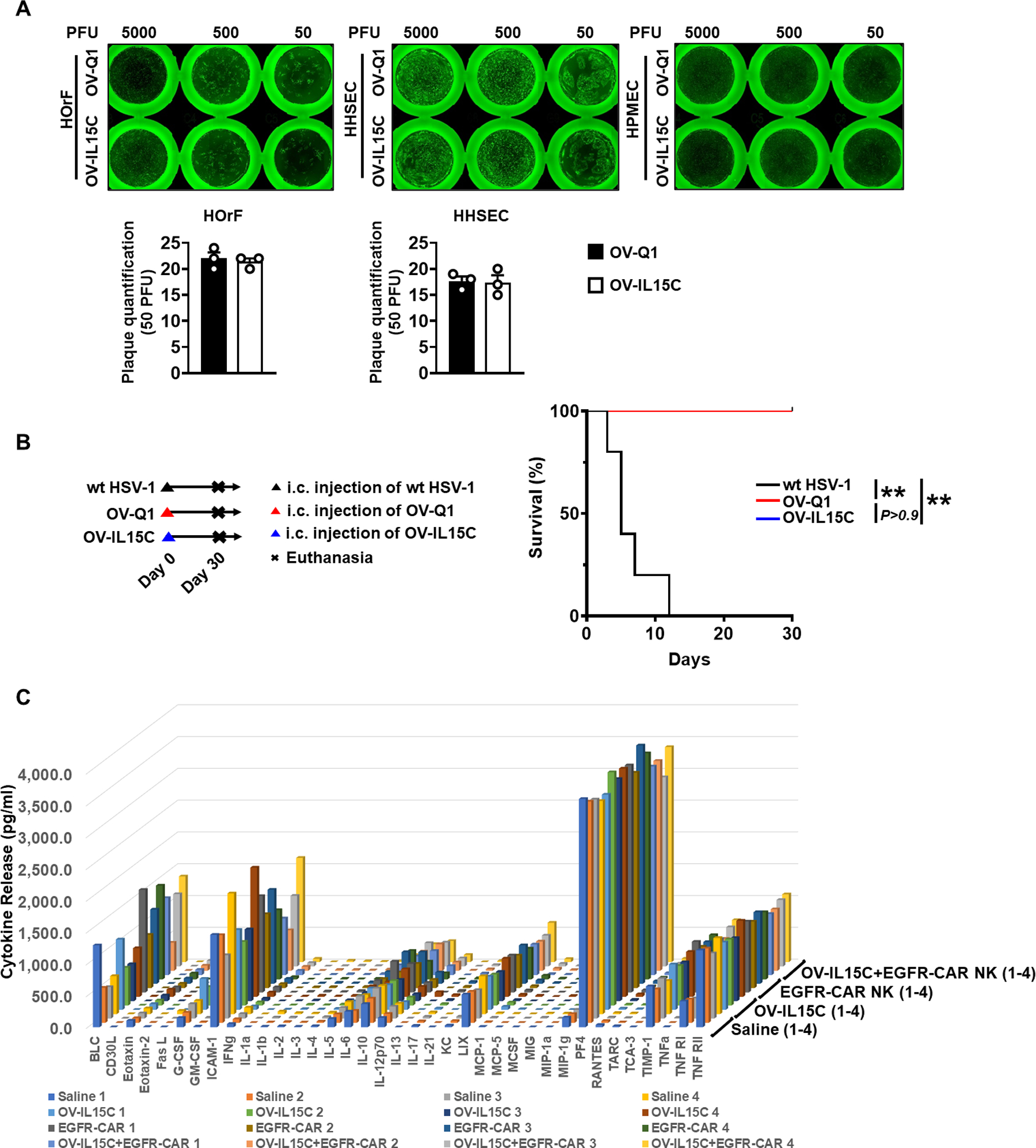
A, Up: plaque forming assays were performed with three different types of primary human cells, including oral fibroblasts (HOrF), hepatic sinusoidal endothelial cells (HHSEC), and pulmonary microvascular endothelial cells (HPMEC). Bottom: quantification of plaque numbers under 50 PFU infection. Forty-eight hours post infection, GFP-positive plaques were counted with a Zeiss fluorescence microscope. Plaque counts of HPMEC were not included because these cells are highly susceptible to oHSV infection, and the formed plaques do not separate very well. The experiment was repeated three times with similar results. B, Survival curves of BALB/c mice treated with wild-type HSV-1 (F strain) and OV-IL15C at the dose of 1 × 106 pfu via intracranial injection. n=5 animals for each group. Log-rank test was used to compare animal survival curves. **, P < 0.01. C, Release assay of various cytokines in the sera from immunocompetent model implanted with the CT2A-hEGFR murine GBM cell line. Mice were treated with OV-IL15C alone, frozen, and unsorted EGFR-CAR NK cells alone, the combination of two agents, or saline alone. Three days later, mice were sacrificed to collect blood sera to measure levels of indicated cytokines by a cytokine release array assay.
Discussion
GBM is the most common and aggressive primary malignant brain tumor in humans without curative therapy at its advanced stages (26). OV is a good approach to treat GBM as its local administration can induce immune infiltration and activation as well as direct lysis of GBM cells (27). OV can also serve as a cargo for local delivery of anti-GBM arsenals. We, therefore, engineered an OV expressing IL-15/IL-15Rα (OV-IL15C) to further improve anti-GBM immune responses. As the IL-15/IL-15Rα complex is released locally due to locoregional administration of OV-IL15C, the virus may not increase too much systemic inflammation but locally produces strong antitumor immunity (28). Among OVs, oHSV has been approved by the U.S. FDA for cancer treatment and is the furthest along in the clinic (29). The OV-IL15C engineered for this study is based on HSV-1, and we demonstrated that it secretes a human IL-15/IL-15Rα complex and is safe in vivo. Accordingly, the complex secreted from OV-IL15C-infected GBM cells could prolong survival and activate both NK and CD8+ T cells in vitro. Consistent with this, the combination therapy of OV-IL15C with CD8+ T cells or EGFR-CAR NK cells significantly improves the therapeutic outcomes in the xenograft and/or immunocompetent GBM mouse models.
IL-15 is an essential cytokine for NK and CD8+ T cell development and function (30). Our group originally discovered that this cytokine is critical in regulating NK cell survival (31). IL-15 complexed with its high-affinity receptor alpha (IL-15Rα) shows promising advantages over IL-15 alone (32). The complex can significantly enhance the half-life and bioavailability of IL-15 (33–35). The IL-15/IL-15Rα complex has a prolonged half-life time in serum (~20 hours) compared to IL-15 (~1 hour) (36–38). This might be more critical in the brain, as historically the brain was recognized as an immune-privileged site (39). In some in vivo mouse models, the IL-15/IL-15Rα complex could cause a rapid and significant regression of glioblastoma and melanoma; however, IL-15 alone could not (40,41). Based on such advantages, engineered cells expressing IL-15/IL-15Rα complex for preclinical and clinical studies have been explored (9). Tumor cells ectopically expressing IL-15/IL-15Rα improves NK cell- and CD8+ T cell-mediated in vivo tumor lysis (42). Intratumoral vaccination of an adenovirus vector expressing IL-15/IL-15Rα inhibits murine breast and prostate cancer (43). Furthermore, CD8+ T cells or dendritic cells (DC) transfected with IL-15/IL-15Rα enhance the antitumor response in vivo (44,45). A previous study showed that engineered an oncolytic vaccinia virus expressing the murine IL-15/IL-15Rα complex elicits potent antitumor immunity (46). Thus, OV-IL15C alone or its combination with another effective agent may maximize the activity of IL-15 and improve oncolytic virotherapy for GBM. However, using an oHSV to express human IL-15/IL-15Rα and the study of how it alone or its combination with another effective therapy modulates the immune response in the tumor microenvironment and the subsequent therapeutic efficacy have not been explored.
CD8+ T cells play an important role in regulating the antitumor immune response. Our results showed that the IL-15/IL-15Rα complex secreted by OV-IL15C-infected GBM cells could significantly enhance cytotoxicity and survival of CD8+ T cells in vitro. We believe that this enhanced cytotoxicity of CD8+ T cells should be antigen non-specific. A previous study demonstrated that IL-15-treated healthy donor T cells kill over 50% of target cells (the P815 cell line), while untreated T cells only kill about 5% of the same target cells (47). Our in vivo xenograft GBM model showed a better therapeutic outcome of OV-IL15C plus CD8+ T cells compared to OV-Q1 plus CD8+ T cells. T cell responses belong to the adaptive immune response, which need time to be ready for fighting tumor cells. In contrast, NK cells belong to the innate immune response and can quickly respond to target cells, including tumor cells, without prior activation. In vitro, we observed that the IL-15/IL-15Rα complex secreted by OV-IL15C-infected GBM cells enhances anti-GBM activity of both CD8+ T cells and NK cells, while in vivo we only observed improved survival by OV-IL15C when combined with CD8+ T cells instead of NK cells. One reason to explain these results may be due to the fact that primary human NK cells have a limited survival period and/or effector function in the brain, especially when they are not armed with a CAR targeting a tumor-associated antigen.
Engineering NK cells with a CAR to effectively treat cancer is necessary, as shown in several preclinical cancer models, including GBM (48–50). However, using NK cells to treat patients with solid tumors remains very limited, in large part due to the inability of NK cells to traffic into tumor tissue as well as the immunosuppressive microenvironment of the tumor. Our previous study demonstrated the efficacy and safety of an intracranial injection of EGFR-CAR-modified NK-92 cells in a GBM orthotopic xenograft model (51). However, NK-92 cells were derived from a lymphoma cell line, it should be irradiated before their infusion into patients, and the irradiation may cause the loss of some antitumor activity (52). In the current study, we used primary human NK cells obtained from peripheral blood mononuclear cells (PBMCs) to engineer EGFR-CAR NK cells. We show EGFR-CAR NK cells derived from peripheral blood and manufactured with expansion by an autologous PBMC condition successfully recognize the EGFR antigen on GBM cells and lead to enhanced anti-GBM activity both in vitro and in vivo. We were able to generate up to 1 × 1011 frozen and ready to use, so called “off-the-shelf” EGFR-CAR products under good laboratory practices-like conditions for all in vitro and in vivo studies, with maintained EGFR-CAR expression as well as good effector function over six months after being frozen.
A recent study suggested that IL-15 preserves the CAR T cell Tscm (T-stem cell memory phenotype) and improves their metabolic fitness (53). However, there are at least two advantages for combining OV-IL15C with CAR NK cells instead of CAR T cells: (1) CAR NK cells can be allogeneic, off-the-shelf, and thus the cost for manufacturing will be lower compared to autologous CAR T cells; (2) one of the major concerns with CAR T cell therapy is near-lethal or lethal toxicity, such as cytokine release syndrome (CRS) and inflammatory encephalomyelitis. Without data from the clinic, we are unsure whether the IL-15/IL-15Rα complex produced by our oncolytic virus will worsen CRS of CAR T cells, considering that oncolytic virus itself may potentially further activate immune cells above the level of CRS. The addition of IL-15 may further exacerbate inflammatory or toxic conditions for patients. In contrast, NK cells do not have “memory” in the traditional sense of T cells. Therefore, the likelihood of massive clonal expansion upon exposure or re-exposure to tumor-associated antigens is extremely small, likely making CAR NK cell therapy much less toxic and better tolerated than CAR T cell therapy. The recent clinical trial showed that the infusion of anti-CD19 CAR NK cells expressing IL-15 did not induce an increase of the levels of IL-6 or CRS in 11 patients with lymphoid cancer (18). Our in vivo safety data demonstrated that there was no significant difference in levels of 37 cytokines, including IL-6, among combination therapy of OV-IL15C with EGFR-CAR NK cells or their monotherapies.
GBM is a very heterogeneous cancer. Even in the same individual patient, some tumor cells express wild-type EGFR, some express EGFRvIII, some express both genes, and some do not express either (54). EGFR-CAR NK cells directly target EGFR and EGFRvIII positive tumor cells, while OV has selectivity for tumor cells vs. normal cells and can therefore kill tumor cells lacking EGFR and EGFRvIII expression. EGFR-CAR NK cells and OV both can launch endogenous immune responses to tumor cells (20,21,55–57). OV may also enhance intracranial infiltration of EGFR-CAR NK cells that are systemically intravenous administered, as we previously showed for endogenous NK cells in our animal models (58). Furthermore, EGFR-CAR NK cells persist longer in the presence of OV-IL15C compared to EGFR-CAR NK cells alone without showing exhaustion in mice, resulting in an enhanced anti-GBM activity. All of these effects converge to transform a “cold” TME with few immune effector cells into a “hot” TME with increased immune cells and provide a strong rationale to combine OV, or OV-IL15C, with EGFR-CAR NK cells to target heterogeneous GBM.
In summary, we developed an innovative and promising method by intracranial co-injection of OV-IL15C with off-the-shelf EGFR-CAR-modified primary human NK cells to target GBM. Our current results provide strong experimental proof applicable for future clinical application.
Supplementary Material
Statement of Significance.
The combination of an oncolytic virus expressing the IL-15/IL-15Rα complex and frozen, ready-to-use anti-EGFR-CAR NK cells elicits strong anti-tumor responses in glioblastoma.
Acknowledgments
This work was supported by grants from the NIH (NS106170, AI129582, CA247550, and CA223400 to J. Yu; CA210087, CA068458, and CA163205 to M.A. Caligiuri), the Leukemia & Lymphoma Society (1364-19 to J. Yu), and The California Institute for Regenerative Medicine (DISC2COVID19-11947 to J. Yu).
Footnotes
Disclosure of Potential Conflicts of Interest: M.A. Caligiuri and J. Yu are co-founders of CytoImmune, Inc.
References
- 1.Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB, et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther 1999;10:2741–55 [DOI] [PubMed] [Google Scholar]
- 2.Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. Journal for ImmunoTherapy of Cancer 2016;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol 1999;17:19–49 [DOI] [PubMed] [Google Scholar]
- 4.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity 2002;17:537–47 [DOI] [PubMed] [Google Scholar]
- 5.Van den Bergh JMJ, Lion E, Van Tendeloo VFI, Smits ELJM. IL-15 receptor alpha as the magic wand to boost the success of IL-15 antitumor therapies: The upswing of IL-15 transpresentation. Pharmacology & Therapeutics 2017;170:73–9 [DOI] [PubMed] [Google Scholar]
- 6.Wu J IL-15 Agonists: The Cancer Cure Cytokine. J Mol Genet Med 2013;7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi H, Dubois S, Sato N, Sabzevari H, Sakai Y, Waldmann TA, et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood 2005;105:721–7 [DOI] [PubMed] [Google Scholar]
- 8.Morris JC, Ramlogan-Steel CA, Yu P, Black BA, Mannan P, Allison JP, et al. Vaccination with tumor cells expressing IL-15 and IL-15Ralpha inhibits murine breast and prostate cancer. Gene Ther 2014;21:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Y, Luan L, Patil NK, Sherwood ER. Immunobiology of the IL-15/IL-15Ralpha complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev 2017;38:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med 2019;216:1986–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhan Y, Lew AM, Chopin M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front Immunol 2019;10:2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P, et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J Immunol 2017;199:1886–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng H, Bu HF, Liu F, Wu L, Pfeifer K, Chou PM, et al. In Inflamed Intestinal Tissues and Epithelial Cells, Interleukin 22 Signaling Increases Expression of H19 Long Noncoding RNA, Which Promotes Mucosal Regeneration. Gastroenterology 2018;155:144–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D’Costa K, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med 2012;209:2049–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discovery 2013;3:388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 2020;382:545–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol 2020;13:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu B, Ma R, Russell L, Yoo JY, Han J, Cui H, et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016;7:27764–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Chen X, Chu J, Xu B, Meisen WH, Chen L, et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res 2015;75:5273–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu B, Ma R, Russell L, Yoo JY, Han J, Cui H, et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol 2018: 10.1038/nbt.4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Water JAJM, Bagci-Onder T, Agarwal AS, Wakimoto H, Roovers RC, Zhu Y, et al. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proceedings of the National Academy of Sciences 2012;109:16642–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahmud H, Kornblau SM, Ter Elst A, Scherpen FJG, Qiu YH, Coombes KR, et al. Epidermal growth factor receptor is expressed and active in a subset of acute myeloid leukemia. J Hematol Oncol 2016;9:64– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803–20 [DOI] [PubMed] [Google Scholar]
- 27.Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J 2012;18:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Molecular Therapy - Oncolytics 2019;13:14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am J Clin Dermatol 2017;18:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 transpresentation promotes human NK cell development and differentiation in vivo. J Exp Med 2009;206:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai CF, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest 1997;99:937–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortier E, Quéméner A, Vusio P, Lorenzen I, Boublik Y, Grötzinger J, et al. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem 2006;281:1612–9 [DOI] [PubMed] [Google Scholar]
- 33.Van den Bergh JM, Lion E, Van Tendeloo VF, Smits EL. IL-15 receptor alpha as the magic wand to boost the success of IL-15 antitumor therapies: The upswing of IL-15 transpresentation. Pharmacol Ther 2017;170:73–9 [DOI] [PubMed] [Google Scholar]
- 34.Stoklasek TA, Schluns KS, Lefrancois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol 2006;177:6072–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Epardaud M, Elpek KG, Rubinstein MP, Yonekura A-r, Bellemare-Pelletier A, Bronson R, et al. Interleukin-15/Interleukin-15Rα Complexes Promote Destruction of Established Tumors by Reviving Tumor-Resident CD8+ T Cells. Cancer Research 2008;68:2972–83 [DOI] [PubMed] [Google Scholar]
- 36.Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, et al. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci U S A 2006;103:9166–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoklasek TA, Schluns KS, Lefrançois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol 2006;177:6072–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kokaji AI, Hockley DL, Kane KP. IL-15 transpresentation augments CD8+ T cell activation and is required for optimal recall responses by central memory CD8+ T cells. J Immunol 2008;180:4391–401 [DOI] [PubMed] [Google Scholar]
- 39.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev 2006;213:48–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaston DC, Odom CI, Li L, Markert JM, Roth JC, Cassady KA, et al. Production of bioactive soluble interleukin-15 in complex with interleukin-15 receptor alpha from a conditionally-replicating oncolytic HSV-1. PLoS One 2013;8:e81768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res 2008;68:2972–83 [DOI] [PubMed] [Google Scholar]
- 42.Rowley J, Monie A, Hung CF, Wu TC. Inhibition of tumor growth by NK1.1+ cells and CD8+ T cells activated by IL-15 through receptor beta/common gamma signaling in trans. J Immunol 2008;181:8237–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris JC, Ramlogan-Steel CA, Yu P, Black BA, Mannan P, Allison JP, et al. Vaccination with tumor cells expressing IL-15 and IL-15Rα inhibits murine breast and prostate cancer. Gene Ther 2014;21:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowley J, Monie A, Hung CF, Wu TC. Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis. Eur J Immunol 2009;39:491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steel JC, Ramlogan CA, Yu P, Sakai Y, Forni G, Waldmann TA, et al. Interleukin-15 and its receptor augment dendritic cell vaccination against the neu oncogene through the induction of antibodies partially independent of CD4 help. Cancer Res 2010;70:1072–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol Ther 2018;26:2476–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.White L, Krishnan S, Strbo N, Liu H, Kolber MA, Lichtenheld MG, et al. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV). Blood 2006;109:3873–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 2016;375:2561–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res 2014;2:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33:2780–8 [DOI] [PubMed] [Google Scholar]
- 51.Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci Rep 2015;5:11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res 2018;8:1083–9 [PMC free article] [PubMed] [Google Scholar]
- 53.Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res 2019;7:759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013;24:438–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Frontiers in immunology 2017;8:533– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Frontiers in immunology 2019;10:2683– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Vloten JP, Workenhe ST, Wootton SK, Mossman KL, Bridle BW. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. The Journal of Immunology 2018;200:450–8 [DOI] [PubMed] [Google Scholar]
- 58.Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med 2012;18:1827–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.