

Abstract
Previous cell-penetrating peptides (CPPs) generally have low cytosolic delivery efficiencies due to inefficient endosomal escape. In this study, a family of small, amphipathic cyclic peptides was found to be highly efficient CPPs, with cytosolic delivery efficiencies of up to 120% (compared to 2.0% for Tat). These cyclic CPPs bind directly to the plasma membrane phospholipids and enter mammalian cells via endocytosis followed by efficient release from the endosome. Their total cellular uptake efficiency correlates positively with the binding affinity for the plasma membrane, whereas their endosomal escape efficiency increases with the endosomal membrane-binding affinity. The cyclic CPPs induce membrane curvature on giant unilamellar vesicles and budding of small vesicles, which subsequently collapse into amorphous lipid/peptide aggregates. These data suggest that cyclic CPPs exit the endosome by binding to the endosomal membrane and inducing CPP-enriched lipid domains to bud off as small vesicles. Together with their high proteolytic stability, low cytotoxicity, and oral bioavailability, these cyclic CPPs should provide a powerful system for intracellular delivery of therapeutic agents and chemical probes.
Keywords: Cell-penetrating peptide, Cytosolic delivery, Endosomal escape, Cyclic peptide
TOC image
Many hydrophilic compounds such as proteins, peptides, and nucleic acids (e.g., siRNA) possess highly potent and specific biological activities in vitro but cannot be used as therapeutic agents or research tools because they are impermeable to the cell membrane. A major breakthrough was the discovery in the late 1980s that the highly positively charged HIV Tat peptide,47YGRKKRRQRRR57, can translocate across the mammalian cell membrane.1,2 Since then, a large family of short peptides (typically 10–30 residues) capable of penetrating the cell membrane at low μM concentrations without causing significant membrane damage has been identified and collectively called “cell-penetrating peptides” (CPPs).3–5 Moreover, the CPPs have been used to deliver hydrophilic cargos including small-molecule drugs,6,7 peptides,8–10 proteins,11,12 nucleic acids,13–15 and nanoparticles16, 17 into cultured mammalian cells and live organisms. However, further progress of the CPP field has been hampered by two major bottlenecks. First, the mechanism of cell entry by CPPs is incompletely understood and remains a subject of debate. The existing evidence in the literature suggests that at least two different mechanisms are operative for Arg-rich CPPs [e.g., nonaarginine (R9) and Tat]. In the first mechanism (direct translocation), the cationic CPPs interact with membrane phospholipids generating neutral ion pairs, which passively diffuse across the membrane in an energy-independent manner.18–20 In the second mechanism (endocytosis), CPPs bind to cell surface receptors (e.g., heparin sulfate) and/or membrane phospholipids and are internalized by clathrin-mediated endocytosis,21 caveolae/lipid-raft-mediated endocytosis,22,23 and/or receptor-independent macropinocytosis.24,25 It is generally believed that at low CPP concentrations, endocytosis is the primary mechanism for cellular uptake, whereas direct translocation becomes significant at high CPP concentrations (>10 μM),18 but other factors such as the specific sequence of CPP, the cargo size, and the cell type can also affect the mechanism as well as the efficiency of cell entry.26,27
The second and perhaps greater bottleneck is the poor endosomal escape efficiency and lack of mechanistic understanding of the escape process. CPPs that enter cells by endocytosis are initially localized in the early endosomes and in effect still “outside” the cell. To gain access to the cytosol or nucleus, the CPPs must translocate across the endosomal membrane. For most of the previous CPPs, endosomal escape is very inefficient, resulting in entrapment of most of the “internalized” CPPs inside the endosome and low cytosolic delivery efficiency (typically <5%).28,29 Lack of an efficient CPP has in turn hampered the mechanistic study of the escape process and therapeutic applications of CPPs. Although several endosomal escape mechanisms have been proposed,30 their validity awaits further testing.
We31,32 and others33–35 recently discovered that cyclization of Arg-rich CPPs increase their cellular uptake efficiency. In particular, cyclo(FΦRRRRQ) (cFΦR4, where Φ is L-2-naphthylalanine, CPP 1, Table 1) is a highly active CPP, capable of delivering a variety of cargo molecules (e.g., small-molecule dyes, peptides, and proteins) into the cytosol of mammalian cells with efficiencies 4- to 12-fold higher than that of R9, Tat, and penetratin.32 In this work, we synthesized a series of cFΦR4 analogs by modifying the peptide sequence and/or stereochemistry and examined their cell-penetrating activities. These efforts led to several exceptionally active CPPs of greatly improved endosomal escape efficiencies. By using these CPPs as tools, we investigated the mechanism of endosomal escape in cultured mammalian cells and model membranes. The results allowed us to propose a novel mechanistic model for the endosomal escape process.
Table 1.
Sequences and cellular uptake of CPPsa
| CPP | Peptide Sequence | Cellular Uptake (MFIFITC, %) |
|---|---|---|
| 1 | cyclo(FΦRRRRQ) | 100 ± 6 |
| 2 | cyclo(RRΦFRRQ) | 47 ± 10 |
| 3 | cyclo(RRFRΦRQ) | 163 ± 29 |
| 4 | cyclo(FRRRRΦQ) | 59 ± 8 |
| 5 | cyclo(FΦRRRQ) | 97 ± 2 |
| 6 | cyclo(FΦRRRRRQ) | 184 ± 47 |
| 7 | cyclo(FFΦRRRRQ) | 113 ± 6 |
| 8 | cyclo(RFRFRΦRQ) | 98 ± 4 |
| 9 | cyclo(fΦRrRrQ) | 602 ± 77 |
| 10 | cyclo(rRFRΦRQ) | 52 ± 2 |
| 11 | cyclo(fΦRrRrRQ) | 542 ± 33 |
| 12 | cyclo(FfΦRrRrQ) | 681 ± 71 |
| 13 | cyclo(FϕrRrRQ) | 200 ± 38 |
| 14 | cyclo(FWRRRRQ) | 126 ± 8 |
| 15 | cyclo(YΦRRRRQ) | 149 ± 7 |
| 16 | cyclo(HΦRRRRQ) | 53 ± 10 |
| 17 | cyclo(PhgΦRRRRQ) | 128 ± 32 |
| 18 | cyclo(FϕrRrRq) | 206 ± 32 |
| Tat | YGRKKRRQRRR | 32 ± 4 |
| R 9 | RRRRRRRRR | 35 ± 3 |
Φ, L-2-naphthylalanine; ϕ, D-2-naphthylalanine; f, D-phenylalanine; r, D-arginine; k, D-lysine; q, D-glutamine; Phg, L-phenylglycine. See Figure S1A in SI for detailed structures. All values are relative to that of CPP 1 (100%) and represent the mean ± S.D. of three independent experiments.
EXPERIMENTAL SECTION
Materials
Reagents for peptide synthesis and other materials are described in Supporting Information. All of the peptides used in this work were purified by reversed-phase HPLC to at least 95% homogeneity and their authenticity was confirmed by MALDI-TOF mass spectrometry.
Cell Culture
HeLa, NIH 3T3 and A549 cells were maintained in medium consisting of DMEM, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. H1299 cells were grown in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Peptide Synthesis and Labeling
Peptides were synthesized on Rink amide resin LS (0.2 mmol/g) using standard Fmoc chemistry. The typical coupling reaction contained 5 equiv of Fmoc-amino acid, 5 equiv of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) and 10 equiv of diisopropylethylamine (DIPEA) and was allowed to proceed with mixing for 75 min. After the addition of the last (N-terminal) residue, the allyl group on the C-terminal Glu residue was removed by treatment with Pd(PPh3)4, phenylsilane (0.1 and 10 equiv, respectively) in anhydrous DCM (3 × 15 min). The N-terminal Fmoc group was removed by treatment with 20% piperidine in DMF and the peptide was cyclized by treatment with benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP)/HOBt/DIPEA (5, 5, and 10 equiv) in DMF for 3 h. The peptides were deprotected and released from the resin by treatment with 82.5:5:5:5:2.5 (v/v) TFA/thioanisole/water/phenol/ ethanedithiol for 2 h. The peptides were triturated with cold ethyl ether (3x) and purified by reversed-phase HPLC on a C18 column. The purity of product (>98%) was assessed by reversed-phase HPLC equipped with an analytical C18 column. The authenticity of each peptide was confirmed by MALDI-TOF mass spectrometry. To generate fluorescently labelled peptides, an Ne-4-methoxytrityl-L-lysine was added to the C-terminus prior to peptide synthesis. After the solid-phase synthesis was complete but before cleavage, the lysine side chain was selectively deprotected using 1% (v/v) TFA in DCM. The resin was incubated with 5 equiv. of a reactive fluorescent labelling reagent (fluorescein isothiocyanate, Lissamine rhodamine B sulfonyl chloride, or naphthofluorescein succinimidyl ester) and 5 equiv. of DIPEA in DMF overnight. The labeled peptide was deprotected, triturated, purified, and analyzed by MALDI-TOF MS as described above.
Flow Cytometry
HeLa cells were cultured in 12-well plates (1.5 × 105 cells per well) overnight. The cells were incubated for 2 h with 5 μM fluorescein isothiocyanate (FITC), rhodamine B (Rho), or naphthofluorescein (NF)-labelled peptide in cellular media. At the end of incubation, the cells were washed with DPBS twice, detached from the plate with 0.25% trypsin, diluted into clear DMEM, pelleted at 250G for 5 min, washed twice with DPBS, resuspended in DPBS, and analyzed on a BD FACS LSR II flow cytometer. For the FITC-labelled peptides, a 488-nm laser was used for excitation and the fluorescence was analyzed in the FITC channel. For the rhodamine-labelled peptides, a 561-nm laser was used for excitation and the fluorescence was analyzed in the PE channel. For NF-labelled peptides, a 633-nm laser was used for excitation and the fluorescence emission was analyzed in the APC channel.
Confocal Microscopy
One mL of HeLa cell suspension (~5 × 104 cells) was seeded in a 35-mm glass-bottomed microwell dish (MatTek) and cultured overnight. For end-stage imaging, cells were gently washed with DPBS twice and treated for 2 h with Rho- or FITC-labelled peptides (5 μM) in phenol-red free, HEPES supplemented DMEM containing 1% FBS in the presence or absence of fluorophore-labeled dextran. After removal of the medium, the cells were gently washed with DPBS twice and imaged on a Nikon A1R live-cell confocal equipped with 100× oil objective or a Visitech Infinity 3 Hawk 2D-array live cell confocal microscope equipped with 60× oil objective. Data were analyzed using NIS-Elemenets AR or MetaMorph Premier.
Fluorescence Polarization (FP) Analysis of CPP Binding to SUVs
A typical FP experiment involved incubating 100 nM fluorescently labeled peptide with varying concentrations of SUV solutions (0–7 mM) in DPBS for 2 h at room temperature. The FP values were measured on a Tecan Infinite M1000 PRO microplate reader, with excitation and emission wavelengths at 470 and 535 nm, respectively, for fluorescein or 530 and 580 nm for rhodamine. EC50 values were determined by plotting the FP values as a function of phospholipid concentrations and fitted to a four-parameter logistic curve with GraphPad PRISM ver.6 software.
Apparent Endosomal Escape Efficiency
The apparent endosomal escape efficiency of each peptide, relative to that of CPP 1, was calculated using equation:
Giant Unilamellar Vesicles (GUVs)
GUVs mimicking the endosomal membrane [50% phosphatidylcholine (PC), 20% phosphatidylethanolamine (PE), 10% phosphatidylinositol (PI), 20% bis(monooleoylglycero)phosphate (BMP), and 0.5% TopFluor-cholesterol (where indicated)] were electroformed. GUVs were harvested, resuspended with 0.5 mg ml−1 Lucifer Yellow (where indicated) at pH 7 or pH 5 and imaged within 4 h. Where indicated, CPP peptides were added and images were obtained continually for 20 min or at 60 min post-addition for end-point imaging. Imaging was performed on a Leica SP2 confocal microscope using a 63× objective.
RESULTS
Structural Variation of cFΦR4 Produces Cyclic CPPs of Improved Efficiency
To gain insights into the structure-activity relationship (SAR) of cyclic CPPs and to potentially obtain CPPs of improved efficiencies, we modified the structure of cFΦR4 (Table 1, peptide 1) by varying its amino acid sequence (Table 1, peptides 2–4 and 14–17), length (peptides 5–8), stereochemistry (peptides 9, 13, and 18), or a combination of the above (peptides 10, 11, and 12). Each peptide also contained an additional lysine on the Gln side chain and was labeled at the lysine side chain with fluorescein isothiocyanate (FITC) (Figure S1A in Supporting Information). HeLa cells were incubated with 5 μM FITC-labeled peptides for 2 h and their cellular uptake efficiencies were assessed by flow cytometry analysis (Table 1). All 17 cFΦR4 analogs are functional CPPs, with activities higher than that of Tat and R9 and 11 of them are more active than cFΦR4. Inspection of the 18 cyclic CPPs and their uptake efficiencies revealed several important trends. First, a combination of two aromatic hydrophobic and three arginine residues is sufficient to generate a functional CPP (e.g., peptide 5), but the uptake efficiency depends critically on its actual sequence (e.g., compare peptides 1–4). Further addition of arginine or hydrophobic residues may either increase (e.g., compare peptides 1 and 5–7) or decrease the CPP activity (e.g., compare peptides 3 and 8), depending on the specific sequence of the resulting CPP. Second, the stereochemical configuration of the arginine and hydrophobic residues greatly impacts the CPP activity, as illustrated by the 6-fold difference in activity between diastereomers cFΦR4 and peptide 9 [cyclo(fΦRrRrQ), where f is D-phenylalanine and r is D-arginine]. Similarly, peptides 11 and 12 are 3- to 6-fold more active than their stereoisomers 6 and 7, respectively (Table 1). Peptides 9 and 18 are a pair of enantiomers and yet have different CPP activities (by 3-fold). These results clearly demonstrate that, like the previous observations with other CPPs,36–38 proper spatial arrangement of the arginine and hydrophobic side chains is critical for obtaining highly efficient cyclic CPPs.
Cyclic CPPs Enter Cells via Endocytosis and Display Low Cytotoxicity
We previously showed that cFΦR4 binds directly to the plasma membrane phospholipids, enters cells via endocytosis, and escapes from the early endosome into the cytosol.32 To determine whether the improved cyclic CPPs also enter cells by endocytosis, we selected two of the most active CPPs, 9 and 12, labeled them with the pH-insensitive dye rhodamine B (Rho), and monitored their entry into HeLa cells by flow cytometry at different temperatures or under ATP-depleted conditions (by treatment with sodium azide and 2-deoxyglucose). Energy depletion decreased the total cellular uptake efficiency of CPPs 1, 9, and 12 by ~80%, while incubation at 4 °C almost completely blocked their cellular entry (≥95% reduction; Figure S2A). We also treated HeLa cells with 3 μM FITC-labeled CPP 9 and monitored the intracellular fluorescence by live-cell confocal microscopy over a 2-h period (Figure 1A and Video S1 in SI). Approximately 20–30 min after the addition of the CPP, mobile fluorescence puncta emerged inside the cells. At ~60 min, diffuse fluorescence became clearly visible, along with the fluorescence puncta. After 90 min, diffuse fluorescence began to dominate the cell interior and finally at 2 h, the cells were almost uniformly labeled with green fluorescence. Remarkably, the CPP 9-treated cells (at 2 h) exhibited stronger fluorescence than the surrounding growth medium, indicating that CPP 9 was actively transported into the cells, causing its concentration inside the cells. To ascertain that the diffuse fluorescence was not a result of photo-induced internalization39 caused by repeated exposure to the laser light during confocal imaging, we treated HeLa cells under the same conditions and imaged only once after the 2-h incubation period. The same diffuse intracellular fluorescence pattern was observed (Figure 1B). In contrast, HeLa cells treated with 3 μM FITC-labeled Tat for 2 h showed only weak, punctate fluorescence inside the cells (Figure 1C). Taken together, the above observations strongly suggest that CPPs 1, 9, and 12 all enter cells through endocytosis followed by endosomal release, although minor contributions by other mechanisms cannot be ruled out.
Figure 1.
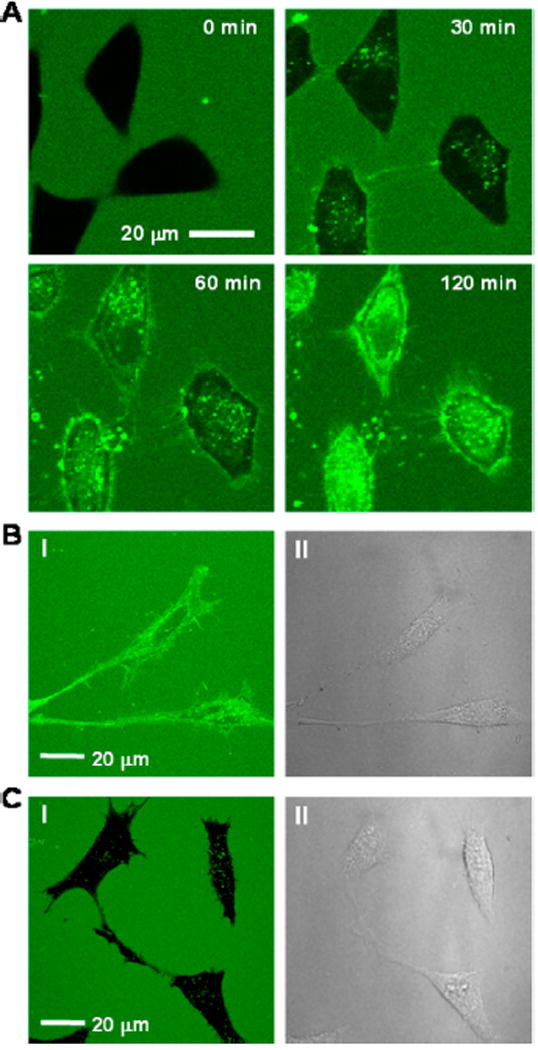
(A) Live-cell confocal microscopic images of HeLa cells after treatment with 3 μM FITC-labeled CPP 9 for indicated periods of time at 37 °C (0–120 min with images taken at 3-min intervals). (B) Same as (A) but imaged only once after 2 h incubation at 37 °C. (C) HeLa cells after treatment with 3 μM FITC-labeled Tat for 2 h at 37 °C. I, FITC fluorescence; II, DIC.
CPPs 1, 9, and 12 were also tested for potential cytotoxicity against four different cell lines (HeLa, NIH 3T3, and lung cancer A549 and H1299 cells) by the MTT assay. No significant cytotoxicity was observed against any of the cells at 50 μM CPP (Figure S2B in SI).
Quantitation of Endosomal Escape and Cytosolic Delivery Efficiencies
The data in Table 1 do not inform what fraction of the “internalized” CPPs reached the cytosol. To quantitate the cytosolic entry of CPPs, we employed a pH-sensitive fluorophore, naphthofluorescein (NF), as the reporter.40 With a pKa of ~7.8, NF is almost completely protonated and non-fluorescent (when excited at ≥590 nm) under acidic conditions, such as inside the endosomes (pH≤6.0). When an NF-labeled CPP escapes from the endosomes into the cytosol (pH~7.4), the large increase in fluorescence intensity is conveniently quantified by flow cytometry analysis. Similarly, the total cellular uptake (in both endosomes and cytosol) can be determined by labeling the CPP with a pH-insensitive dye such as Rho. Comparison of the two fluorescence intensity values allows for a quantitative assessment of the endosomal escape efficiency.
We selected CPPs 1, 2, 4, 7, 9, 11, and 12, which have varying levels of cellular uptake efficiencies (Table 1), for further quantitative assessment. Linear CPPs Tat, R9, and miniature protein 5.3, which was previously reported to have unusually high cytosolic delivery efficiency,41,42 were used as controls. To quantitate the total cellular uptake, each of the 10 CPPs was labeled with Rho through a flexible, hydrophilic linker (miniPEG) to minimize any effect of the dye on the CPP activity (Figure S1B). HeLa cells were treated with 5 μM labeled CPP for 2 h at 37 °C and then analyzed by flow cytometry. The CPPs produced mean fluorescence intensity (MFI) values ranging from 32% to 278% relative to cFΦR4 (which is defined as 100%; Table 2). Among the 10 CPPs tested, CPP 11 was taken up most efficiently by the cells (2.8-fold better than cFΦR4). CPPs 9 and 12 both entered cells 1.5-fold more efficiently than cFΦR4. Notably, the miniature protein 5.3 was taken up by HeLa cells only 1.2-fold better than cFΦR4, whereas Tat and R9 were ~2-fold less efficiently than cFΦR4.
Table 2.
Membrane binding affinities and cellular entry efficiencies of CPPs
| CPP | Total Cellular Uptake (MFIRho, %)a |
Cytosolic Entry (MFINF, %)a |
Apparent Endosomal Escape (γ, %)a |
SUV1 EC50 (μM) |
SUV2 EC50 (μM) |
Cytosolic Delivery Efficiency (%) |
|---|---|---|---|---|---|---|
| 1 | 100 | 100 | 100 | 713 ± 76 | 166 ± 12 | 20 |
| 2 | 38 ± 5 | 37 ± 3 | 98 ± 15 | 2220 ± 510 | 248 ± 18 | 7.5 |
| 4 | 32 ± 6 | 42 ± 4 | 132 ± 26 | 2060 ± 310 | 170 ± 12 | 8.5 |
| 7 | 77 ± 8 | 85 ± 5 | 110 ± 13 | 1300 ± 180 | 139 ± 10 | 17 |
| 9 | 152 ± 13 | 307 ± 15 | 202 ± 20 | 290 ± 60 | 34 ± 4 | 62 |
| 11 | 278 ± 18 | 247 ± 40 | 89 ± 15 | 169 ± 42 | 111 ± 10 | 50 |
| 12 | 149 ± 9 | 598 ± 61 | 402 ± 48 | 231 ± 43 | 16 ± 1 | 121 |
| Tat | 43 ± 3b | 9.9 ± 1.7b | 23 ± 4b | NB | 2580 ± 420 | 2.0c |
| R 9 | 49 ± 3 | 20 ± 2 | 40 ± 5 | >5000 | 880 ± 130 | 4.0 (4.4c) |
| 5.3 | 122 ± 7 | 773 ± 85 | 634 ± 79 | 13.7 ± 3.4 | 7.8 ± 1.0 | 156 (50c) |
Next, to quantitate the cytosolic (and nuclear) delivery efficiencies, the 10 CPPs were similarly labeled with NF, again via a flexible miniPEG linker (Figure S1C). HeLa cells were incubated with the NF-labeled peptides (5 μM) for 2 h at 37 °C and analyzed by flow cytometry. cFΦR4 showed 10- and 5-fold higher cytosolic entry than Tat and R9, respectively (Table 2), in excellent agreement with the previous data determined by other methods.40 Also in agreement with the report of Schepartz and co-workers who directly measured the cytosolic concentrations of several peptides/proteins by fluorescence correlation spectroscopy,42 the miniature protein 5.3 is a remarkably active CPP, having a cytosolic entry efficiency 7.7-fold higher than that of cFΦR4 (or 78-fold better than Tat). Among the cyclic CPPs tested, 9, 11, and 12 have 3.1-, 2.5-, and 6.0-fold higher cytosolic entry efficiencies, respectively, than cFΦR4, whereas CPPs 2, 4, and 7 are less effective. By comparing the total cellular uptake (MFIRho) and cytosolic entry efficiencies (MFINF), we calculated the apparent endosomal escape efficiencies of the 10 CPPs, relative to that of cFΦR4 (which is defined as 100%; Table 2). The results show that while miniature protein 5.3 is highly efficient in endosomal escape (6.3-fold better than cFΦR4), Tat and R9 are not (4.3- and 2.5-fold, respectively, less efficient than cFΦR4). Cyclic CPPs 9 and 12 have substantially improved endosomal escape efficiencies relative to cFΦR4 (2.0- and 4.0-fold, respectively). In contrast, CPP 11 has the most efficient initial uptake (2.8-fold higher than cFΦR4) but exits the endosome less effectively (0.89-fold relative to cFΦR4). Finally, we determined the absolute cytosolic delivery efficiencies (defined as the ratio of cytosolic over extracellular CPP concentration) for the 9 CPPs by using the previously reported value for Tat [2.0%, which was experimentally determined by fluorescence correlation spectroscopy42] as a reference and the relative cytosolic entry efficiencies derived from this work (Table 2). Thus, R9 has a cytosolic delivery efficiency of 4.0%, in agreement with the previously reported value of 4.4%.42 Miniature protein 5.3 has an efficiency of 156%, compared to the reported value of 50% (see below). The 7 cyclic CPPs have cytosolic delivery efficiencies ranging from 7.5% (CPP 2) to 121% (CPP 12) (Table 2).
To confirm the flow cytometry results, we examined HeLa cells that had been treated with the Rho-labelled CPPs (5 μM for 2 h) by live-cell confocal microscopy (Figure 2). As expected, R9 produced predominantly punctate fluorescence in the cytoplasmic region, consistent with its marginal endosomal escape efficiency (Table 2). In a stark contrast, cells treated with CPPs 9 and 12 showed intense and predominantly diffuse fluorescence throughout the entire cell volume, suggesting that the CPPs efficiently exited the endosomes. In the case of CPP 12, strong accumulation in the nuclear region was observed, likely caused by binding of CPP 12 to nucleic acids in the nucleoli. Cells treated with cFΦR4 or CPP 7 displayed both punctate and diffuse signals in the cytoplasmic region as well as readily visible fluorescence in the nuclear region, as would be expected from their intermediate endosomal escape efficiencies. CPPs 2 and 4 resulted in overall weaker and predominantly punctate fluorescence in the cytoplasmic region. CPP 11 showed intense fluorescence signals (including puncta) in the cytoplasmic region but relatively weak signal inside the nucleus, a pattern consistent with efficient endocytic uptake followed by less efficient endosomal escape. Similar results were obtained when the confocal microscopic experiments were repeated with the FITC-labeled CPPs (Figure S3). We found that while miniature protein 5.3FITC was distributed throughout the cell volume, indicating a facile endosomal escape process,41 strong binding and localization of 5.3FITC to the plasma membrane and intracellular organelles was evident (Figure S3). This may explain the different cytosolic delivery efficiencies determined by the two different methods (156% vs 50%; Table 2). The membrane-bound 5.3 can be detected by flow cytometry but not by fluorescence correlation spectroscopy, which monitors the diffusion of a fluorophore into and out of a small cytoplasmic volume.42
Figure 2.
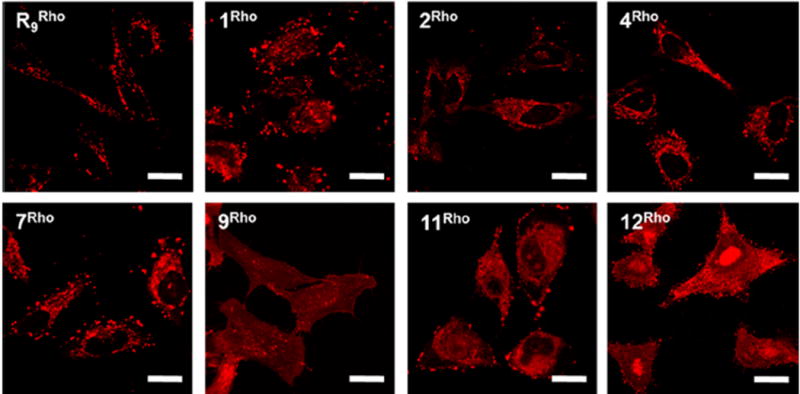
Intracellular distribution of R9 and representative cyclic CPPs. HeLa cells were treated with Rho-labeled CPP (5 μM each) for 2 h at 37 °C, washed, and imaged by live-cell confocal microscopy (without fixation). Scale bars indicate 20 μm.
Total Cellular Uptake Efficiency Correlates with Plasma Membrane Binding
The mechanism of cyclic CPP uptake (i.e., binding to the plasma membrane phospholipids followed by endocytosis) predicts a positive correlation between the total cellular uptake efficiency and the binding affinity to the plasma membrane. To test this hypothesis, we prepared small unilamellar vesicles (SUVs) that mimic the plasma membrane of mammalian cells [45% phosphatidylcholine (PC), 20% phosphatidylethanolamine (PE), 20% sphingomyelin, and 15% cholesterol; SUV1] and measured the binding affinity of the 10 Rho-labeled CPPs in Table 2 for SUV1 by fluorescence polarization (FP) (Figure 3A). The miniature protein 5.3Rho bound most tightly, with an EC50 value (lipid concentration at which half of the CPP is bound) of 13.7 μM (Table 2). The seven cyclic CPPs bound to SUV1 with EC50 values of 169–2060 μM. R9 bound only weakly, with an EC50 value of >5000 μM, while Tat showed no significant binding.32 A plot of the total cellular uptake efficiency against the plasma membrane binding affinity (1/EC50) showed a roughly linear correlation between the two variables for R9 and the cyclic CPPs, but not for Tat or 5.3 (Figure 3B). Note that a number of other factors (e.g. binding to cell-surface proteoglycans or serum proteins) may affect the total cellular uptake efficiency. Tat and R9 have previously been shown to bind with high affinity to the cell surface proteoglycans and are subsequently internalized by endocytosis.43
Figure 3.
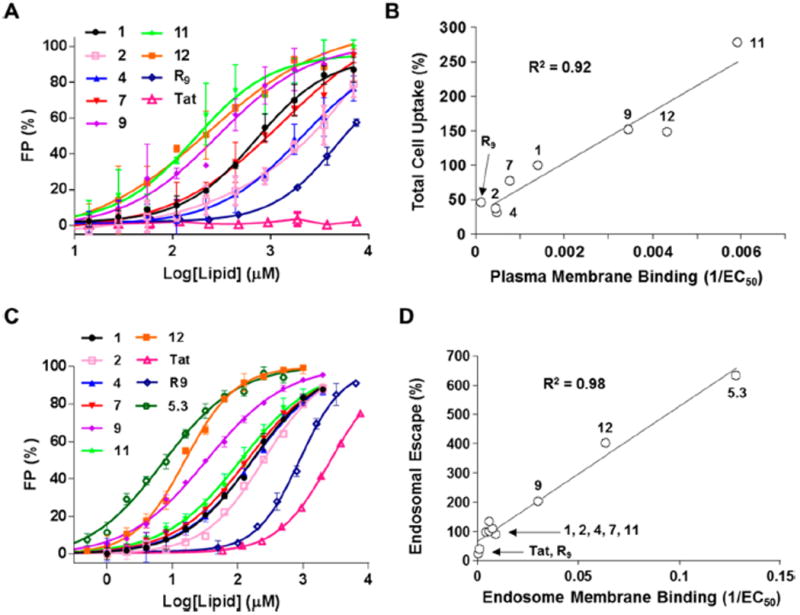
Correlation between membrane binding affinity and total cellular uptake and endosomal escape efficiency. (A) Binding of Rho-labeled CPPs (100 nM each) to SUVs that mimic the plasma outer membrane (SUV1) as monitored by FP assay. (B) Plot of relative total cell uptake efficiency (relative to that of cFΦR4, 100%) against the binding affinity to SUV1 (1/EC50). (C) Binding of Rho-labeled CPPs (100 nM each) to SUVs that mimic the endosomal membrane (SUV2) as monitored by FP assay. (D) Plot of relative apparent endosomal escape efficiency (relative to that of cFΦR4, 100%) against the binding affinity to SUV2 (1/EC50).
Endosomal Escape Efficiency Correlates with Endosomal Membrane Binding
To test whether there is a similar correlation between the endosomal escape efficiencies of CPPs and their binding affinities to the endosomal membrane, we prepared SUVs that mimic the late endosomal membrane of mammalian cells [50% PC, 20% PE, 10% phosphatidylinositol (PI), and 20% bis(monooleoylglycero)phosphate (BMP);44 SUV2] and examined their interaction with the Rho-labeled CPPs (Figure 3C). Compared to SUV1, the cyclic CPPs bound to the negatively charged SUV2 with much higher affinities, having EC50 values of 16–250 μM (Table 2). Again, miniature protein 5.3Rho bound most tightly to SUV2 (EC50 = 7.8 μM), while Tat showed the weakest binding (EC50 = 2580 μM). Remarkably, for all 10 CPPs tested in this study, their endosomal escape efficiency increased linearly with the endosomal membrane-binding affinity (1/EC50) (Figure 3D). To mimic the acidic environment in late endosomes, we also determined the binding affinities of the 10 CPPs for SUV2 at pH 5.5. All of the CPPs bound to SUV2 with increased affinities compared to the neutral pH (Table S1). These results strongly suggest that physical interaction between CPPs and the endosomal membrane phospholipids is a critical step during the endosomal escape process.
Cyclic CPPs Do Not Cause Endosomal Membrane Leakage
The proposed mechanisms for CPP release from the endosome often invoke the destruction of or pore formation on the endosomal membrane.30 To test whether cyclic CPPs cause endosomal membrane leakage, HeLa cells were treated with an endocytosis marker, Rho-labeled dextran (dextranRho), in the absence and presence of 5 μM FITC-labeled cyclic CPP 12 and the intracellular distribution of dextranRho was examined by confocal microscopy. While CPP 12 generated strong, diffuse fluorescence throughout the cell, indicating that a significant fraction of CPP 12 had escaped from the endosome into the cytosol, dextranRho remained mostly entrapped inside the endosomes as evidenced by its punctate fluorescence in the cytoplasmic region (Figure S4A). Similar results were obtained with all other cyclic CPPs examined (Figure S3). We also treated HeLa cells with NF-labeled R9 in the absence and presence of unlabeled CPP 12 and quantitated the cytosolic entry of R9NF by flow cytometry. The presence of 5 μM CPP 12 had minimal effect on the cytosolic entry of R9NF (Figure S4B). Finally, we generated LUVs that mimic the late endosomal membrane, loaded them with the small-molecule dye calcein, and tested whether the cyclic CPPs could disrupt the LUV membrane causing the release of calcein from the vesicles. While the addition of 1% Triton X-100 (a positive control) resulted in rapid and complete release of the dye (100%), cyclic CPPs 1, 9, and 12 (each at 20 μM) resulted in 10–20% dye release over a 30-min period (Figure S4C). As a comparison, R9 and penetratin caused 1 and 23% dye release, respectively, under the same conditions. These results strongly argue against the endosomal lysis and pore forming mechanisms, either of which would have caused rapid release of dextranRho and R9NF into the cytosol. The small amount of CPP-induced calcein release from the LUVs is likely caused by budding and collapsing of small vesicles (see below).
Cyclic CPPs Induce Vesicle Budding In Vitro
We chose CPP 12 and giant unilamellar vesicles (GUVs) as a model system to examine the effect of cyclic CPPs on the endosomal membrane structure by confocal microscopy. GUVs that mimic the late endosomal membrane in phospholipid composition were generated by electroformation at pH 7, with 0.5% BODIPY-labeled cholesterol added as a membrane marker (Figure 4A). After the GUVs had been formed, Lucifer yellow was added to the external solution to monitor the GUV membrane integrity. Lowering the external pH to 5.0 had little effect on the GUV structure. However, incubation of the GUVs with 5 μM CPP 12 for 1 h (at pH 7) resulted in a general reduction in the GUV sizes and formation of very small vesicles, apparently as a result of vesicle fission (Figure 4A). Most of the vesicles (~90%) had no visible Lucifer yellow fluorescence inside their lumen, suggesting that CPP 12 did not disrupt the membrane integrity of these vesicles or that of the progenitor GUVs. The remaining vesicles (~10%) contained a spectrum of Lucifer yellow fluorescence intensities inside the lumen, from similar brightness to the external solution to barely detectable (Figure S5). When the experiment was carried out at an external pH of 5.0, the fission and reduction in GUV sizes were more dramatic. In addition, many small, amorphous, and intensely fluorescent aggregates (green) were formed (Figure 4A part III). These aggregates contained a mixture of phospholipids, cholesterol, and CPP 12, since they were intensely fluorescent when any of the three components was fluorescently labeled (data not shown). Finally, increasing the CPP 12 concentration to 20 μM (at pH 5.0) resulted in the total disappearance of the GUVs and the formation of very small vesicles (<1 μm in diameters) as well as numerous lipid/cholesterol/peptide aggregates (Figure 4A part IV). Again, most of the small vesicles contained no detectable Lucifer yellow inside. Similar results were observed with cFΦR4 and CPP 9 (data not shown).
Figure 4.
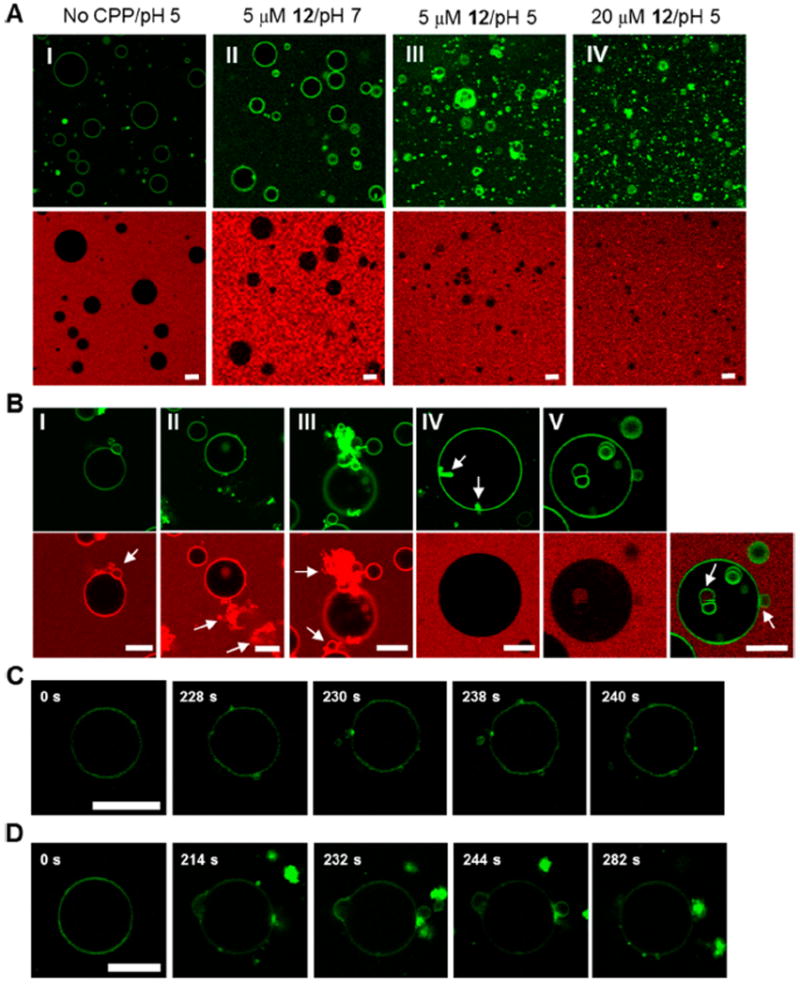
Cyclic CPPs induce membrane budding in vitro. (A) GUVs mimicking the late endosomal membrane composition (with 0.5% of fluorescein-labeled cholesterol) were incubated for 1 h under the indicated conditions in the presence of 0.5 mg ml−1 of Lucifer yellow (shown in red) and imaged by confocal microscopy. Top panel, images from the FITC channel; bottom panel, images from the Lucifer yellow channel. (B) Images of representative GUV structures derived from the experiments in (A). Top panel, images observed from the FITC channel; bottom panel, images observed from the Lucifer yellow channel or the overlap of the above two (part V). Arrows indicate the nascent vesicles and/or lipid-peptide aggregates formed. (C and D) Time lapse images of outward budding/aggregation events upon suspension of GUVs (without fluorescein-labeled cholesterol) in a buffer (pH 5) containing 20 μM FITC-labeled CPP 12. All scale bars indicate 10 μm.
To glimpse the intermediate events during the GUV structural changes, we incubated GUVs under milder conditions (e.g., 5 μM CPP 12 and at pH 7) and obtained confocal images at various time points (0 to 60 min). Four main types of intermediate lipid structures/events were observed. A common event involved outward budding of small vesicles from the GUV membrane (Figure 4B part I and V). The newly budded vesicles did not contain Lucifer yellow in their lumen. Another common event was the formation of amorphous aggregates outside the GUVs as described above (Figure 4B part II). In fact, the budding and aggregation events are usually coupled (Figure 4B part III). Although relatively less frequent, formation of lipid aggregates inside the GUV lumen (Figure 4B part IV and S6A) and inward budding of vesicles (Figure 4B part V and S6B) were also observed. Inward budding resulted in the formation of intraluminal vesicles (ILVs) filled with Lucifer yellow. We noticed that some of the ILVs did not contain Lucifer yellow, likely because they were formed during GUV electroformation (i.e., prior to the addition of Lucifer yellow). It appears that the membrane integrity of the GUVs was maintained during all of these intermediate events, as no Lucifer yellow fluorescence was detectable inside the lumen of the GUVs. The varying levels of Lucifer yellow inside the small percentage of vesicles (Figure S5) were likely caused by inward budding and subsequent collapsing of the ILVs and/or the coupled inward budding/aggregation events. The presence of green fluorescence puncta inside some of these vesicles (which likely reflect lipid/cholesterol/peptide aggregates) is consistent with the above scenario. Outward budding and vesicle collapsing would result in the release of a small amount of calcein from the LUVs upon treatment with cyclic CPPs (Figure S4C).
We next mixed GUVs (unlabeled) with 20 μM FITC-labeled CPP 12 at pH 5.0 and imaged the GUVs by confocal microscopy at 2-s intervals for a period of 5 min. Upon mixing, CPP 12 immediately bound to the GUV membrane, which became intensely fluorescent (Figure 4C, t = 0 s and Video S2 in SI). Initially, CPP 12 was uniformly distributed over the entire GUV membrane. However, at 228 s, the membrane-bound CPPs started to cluster together forming multiple highly fluorescent foci on the GUV membrane. Over time, some of these foci turned into small membrane buds, with the CPP concentrated at the regions of increased membrane curvature (Figure 4C, t = 230 s). Some of the membrane buds eventually progressed into budding vesicles, which subsequently pinched off the GUV membrane (Figure 4C, t = 238 and 240 s). Immediately before pinching off, the CPP was concentrated at the narrow neck that connects the budding vesicle and the GUV membrane (Figure 4D and S6C). Compared to budding off of intact vesicles, a more frequent event involved budding vesicles collapsing at the site of budding and the formation of lipid/CPP aggregates at the GUV surface (Figure 4D and Video S3 in SI). Again, immediately before the vesicles collapsed, the CPP was concentrated at the GUV/bud interface. The budded vesicles and the aggregates were greatly enriched in the CPP content relative to the GUV surface, which gradually became depleted in CPP content and barely visible under the imaging conditions. Concomitantly, the GUVs experienced profound structural changes, as their membranes became much more fluidic and flexible.
Cyclic CPPs Are Orally Bioavailable
The excellent proteolytic stability of small cyclic peptides and the high membrane permeability of the cyclic CPPs suggest that they may be orally bioavailable. We thus performed preliminary studies on the pharmacokinetic (PK) properties of cFΦR4 and CPP 9. Figure 5 shows the plasma concentration versus time curves of cFΦR4 in mice following intravenous injection (IV) and oral gavage (PO) at 1.5 and 40 mg kg−1 doses, respectively. Non-compartmental analysis of the mouse PK data demonstrated a plasma elimination half-life of ~1 h, a total plasma clearance of 0.08 ml min−1, and a terminal volume of distribution of 7.51 ml (Table S2). These data suggest that cFΦR4’s distribution is limited to approximately total body water but greater than the extracellular fluid.45 CPP plasma clearance values are much lower than mouse hepatic and renal blood flow (1.8 and 1.3 ml min−1, respectively) but approach glomerular filtration rates (0.25 ml/min). These results suggest that cFΦR4 can penetrate the intracellular space in vivo, consistent with its in vitro properties. Remarkably, when given orally at a dosage of 40 mg kg−1, a maximal plasma concentration (Cmax) of 3.2 μM was achieved within the first hour after administration and resulted in 4% oral bioavailability for cFΦR4. CPP 9 demonstrated similar oral bioavailability (data not shown).
Figure 5.
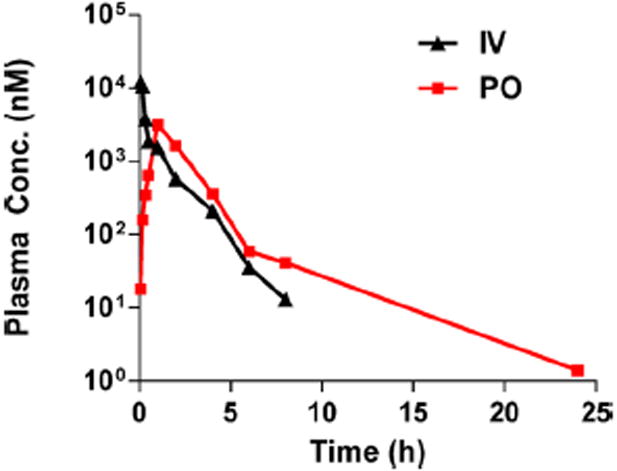
Pharmacokinetic properties of cFΦR4. Plasma concentrations versus time for cFΦR4 after oral administration of 40 mg kg−1 in PBS versus 1.5 mg kg−1 IV route to mice.
DISCUSSION
In this study, structural variation of cFΦR4 resulted in a family of cyclic CPPs that are up to 60-fold more active than Tat and R9, two of the most active and widely used CPPs. With cytosolic delivery efficiencies of 62–120%, CPP 9 and 12 represent, to our knowledge, the most active CPPs reported to date. The only other system having a similar level of efficiency is the 36-residue miniature protein 5.3 (156%) reported by Schepartz and co-workers.41,42 For both cyclic CPPs and the miniature protein, the high cytosolic delivery efficiency is primarily the result of a vastly improved endosomal escape process. Compared to conventional CPPs and miniature protein 5.3, which are linear peptides comprising of proteinogenic amino acids, the cyclic CPPs are much more stable against proteolysis due to conformational rigidity. Indeed, CPPs 1 and 9 demonstrated oral bioavailability, making them the first validated examples of orally bioavailable CPPs. The 4% bioavailability (for cFΦR4) is much higher than that of most orally administered peptidyl drugs currently on the market (generally <1–2%).46 Further studies are required to characterize the precise means of CPP elimination and evaluate potential relationships between in vitro properties and in vivo oral absorption. These features should make the cyclic CPPs an excellent scaffold for therapeutic as well as research applications.
We have previously established that cFΦR4 binds directly to the plasma membrane phospholipids and enters mammalian cells by endocytosis.32 The greatly reduced cellular uptake of other cyclic CPPs at 4 °C or under energy-depletion conditions suggests that they also enter cells by the same endocytic mechanisms. This, of course, raises the question of how they exit the endosome, a major unsolved issue in the CPP field. Previously proposed mechanisms generally involved some form of endosomal membrane disruption, ranging from total destruction of the endosome [e.g., proton sponge effect] to transient pore formation.29,30 However, these mechanisms cannot explain all of the experimental observations. For example, complete destruction of the endosomal compartment would result in significant toxicity, but most of the Arg-rich CPPs (including cyclic CPPs) have been found to be relatively nontoxic to cultured cells. Disruption of the endosomal membrane would also increase the release of endocytosis markers such as dextranRho or R9NF, which was not observed with cyclic CPPs (Figure S3 and S4). While formation of transient pores or inverted micelles may allow the release of CPPs alone or CPPs conjugated to small-molecule cargos, efficient release of CPPs conjugated to macromolecular cargos such as proteins would be more challenging.
The availability of CPPs with high as well as varying levels of endosomal escape efficiencies provided us a unique opportunity to investigate the mechanism of endosomal release. The results of these studies allowed us to propose an alternative endosomal release mechanism, in which the CPPs bind directly to the luminal side of the endosomal membrane, inducing membrane curvature and budding of the CPP-enriched lipids as small vesicles (Figure 6). During a budding process, the budding neck involves severe membrane distortion (i.e., formation of acute negative Gaussian curvatures) and represents the highest-energy state (the “transition state”).47 By selectively binding to the budding neck, the CPPs reduce the energy barrier and accelerate the rate of the budding event. Endosomal acidification further facilitates this budding event by increasing the binding affinity of arginine-rich CPPs for the membrane (Table S1). After budding off the endosomal membrane, the small vesicles presumably become less stable as the pH gradient across their membrane dissipates (we assume that the proton pumps are not sorted to the small vesicles) and collapse to release the luminal contents into the cytosol. pH gradients generated across artificial membranes last for minutes to hours depending on the lipid composition,48 indicating that protons move across lipid bilayers rather easily. Late endosome membrane is rich in negatively charged lipid BMP which, together with PI, should further enhance the binding of cationic CPPs to the endosomal membrane (Table 2, SUV2), thereby facilitating vesicle budding as well as concentrating the CPPs to the budding vesicles (Figure 4). Note that budding into the endosome (i.e., formation of multivesicular bodies) as well as outward budding from the endosome are well-established phenomena.49–51 By synchrotron small angle X-ray scattering analysis, Zhao et al. have previously shown that our cyclic CPPs are effective in generating negative Gaussian curvatures on artificial membranes.52 The cyclic CPPs differ from classical CPPs (e.g., Tat and R9) by containing both arginine and hydrophobic residues, both of which are critical for the CPP activity and formation of negative Gaussian curvatures.31,51 It was hypothesized that insertion of the hydrophobic side chains in between phospholipid molecules generates positive curvatures, whereas the bidentate hydrogen-bond interactions between the guanidinium groups and the lipid phosphates induce negative curvatures.53 It should be noted that our GUV studies involved adding CPPs to the external solution, whereas during endosomal escape, the CPP is inside the endosome (at least at the beginning). Our attempts to generate GUVs loaded with cyclic CPPs were unsuccessful. We note that the negative Gaussian curvature at the budding neck is symmetrical and predict that addition of CPPs to the luminal solution would similarly stabilize the budding neck structure and induce both outward and inward budding events. In this regard, an amphipathic peptide derived from the influenza virus M2 protein has previously been shown to generate negative Gaussian curvature on artificial membranes54 and induce both inward and outward budding of small vesicles from GUVs.47 Penetratin was also reported to cause the “fragmentation” of GUVs into smaller vesicles and the formation of lipid/peptide aggregates on both the outside and inside surfaces of GUVs.55–58 In vitro generation of a pH gradient across an LUV membrane (acidic inside) was shown to be sufficient to drive certain CPPs across phospholipid bilayers, indicating that endosomal escape can occur in the absence of other cellular machinery such as proteins.48,59
Figure 6.
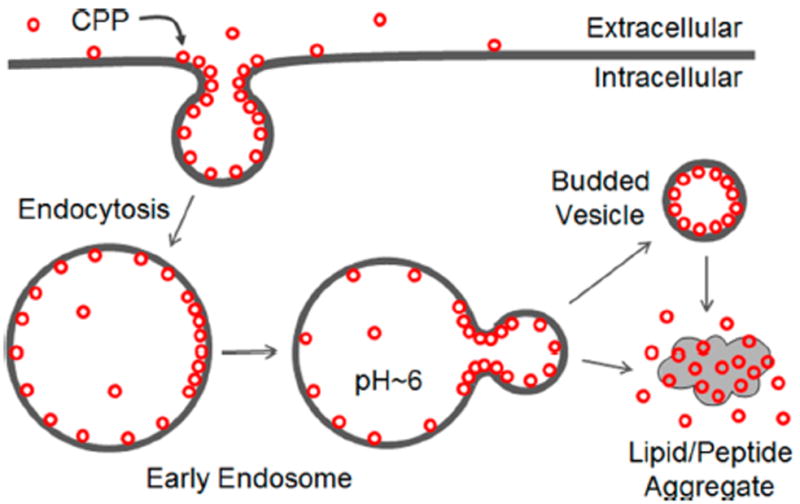
Proposed mechanism of CPP uptake and endosomal escape.
Our model is consistent with all of the experimental observations to date. It readily explains how large protein cargos attached to CPPs can exit the endosome in their native states.12,32 It also explains why most CPPs have minimal toxicity to cultured mammalian cells, since their release via vesicle budding does not compromise the integrity of the endosomal membrane (Figure 4). Our model predicts that peptides with higher binding affinity for the endosomal membrane would have greater endosomal release efficiency, which is exactly what we observed experimentally (Figure 3C, D). Our model explains why cyclic peptides are particularly effective CPPs whereas the corresponding linear peptides are not,31 because the entropic advantage associated with the more rigid structures of cyclic peptides allows them to bind to the fluidic membrane phospholipids more tightly than their linear counterparts. We previously observed that for endocyclic delivery of peptidyl cargos by cFΦR4, the delivery efficiency decreased dramatically as the cargo size (and thus the ring size) increased.31 Presumably, the larger rings are more conformationally flexible and bind to both plasma and endosomal membranes with lower affinities. It provides a simple explanation for the SAR of cyclic CPPs. The cyclic CPPs in Table 1 differ from each other by having different number of arginine/hydrophobic residues and/or different spatial orientation of the residues. Consequently, they bind to the plasma and endosomal membranes with different affinities and have different initial uptake and endosomal escape efficiencies. Since the correlation between endosomal escape efficiency and endosomal membrane-binding affinity applies to Tat, R9, cyclic CPPs, and the miniature protein 5.3, we hypothesize that the vesicle budding mechanism is operative for most (if not all) CPPs that enter cells by endocytosis. In fact, our model provides a plausible explanation for the different time points at which CPPs exit the endosome. cFΦR4 and 5.3 can exit from the early endosome because they bind to the endosomal membrane with sufficient affinity under neutral pH and do not require the acidic pH of the late endosome for membrane binding and vesicle budding.32,41,42 In contrast, Tat does not bind the early endosomal membrane tightly enough at neutral pH and therefore cannot exit from the early endosome. When the pH reaches 4.5–5.5 in late endosomes, Tat gains sufficient membrane binding and induces vesicle budding. R9 binds to the endosomal membrane with an intermediate affinity between those of cFΦR4/5.3 and Tat and thus exits the endosomal pathway later than cFΦR4/5.3 do but earlier than Tat does.32,41,42 Finally, it has not escaped our attention that, at sufficiently high concentration and under proper conditions, CPPs may operate on the plasma membrane by the same mechanism, resulting in their immediate availability in the cytosol (i.e. direct translocation18–20, 60–64). Thus, vesicle budding and/or membrane aggregation may represent a unifying mechanism for all CPPs.
In conclusion, we have discovered a family of exceptionally active and metabolically stable cyclic CPPs and elucidated their mechanism of cellular entry. Coupled with their low cytotoxicity and ease of preparation, these cyclic CPPs should provide a very useful class of general membrane transporters for cytosolic delivery of chemical probes and therapeutic agents.
Supplementary Material
Acknowledgments
We thank Drs. P. Upadhyaya and S. Cole for technical assistance.
Funding
This work was supported by the National Institutes of Health (GM110208 and GM062820), the Medical Research Council [JSR: MR/L00870X/1 and JSR: MR/L018578/1] and the European Union Seventh Framework Program [JSR: FP7-PEOPLE-2012-CIG: 333955].
Footnotes
Supporting Information
Experimental details and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
D.P. and Z.Q. are founders of CycloPorters, LLC.
References
- 1.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 2.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 3.Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 4.Stanzl EG, Trantow BM, Vargas JR, Wender PA. Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc Chem Res. 2013;46:2944–2954. doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Futaki S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv Drug Deliv Rev. 2005;57:547–558. doi: 10.1016/j.addr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Nori A, Jensen KD, Tijerina M, Kopecková P, Kopecek J. Tat-conjugated synthetic macromolecules facilitate cytoplasmic drug delivery to human ovarian carcinoma cells. Bioconjug Chem. 2003;14:44–50. doi: 10.1021/bc0255900. [DOI] [PubMed] [Google Scholar]
- 7.Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat Med. 2000;6:1253–1257. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]
- 8.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med. 2002;8:808–815. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 9.Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med. 2004;10:305–309. doi: 10.1038/nm994. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular Transporters for Peptides: Delivery of a Cardioprotective Epsilon PKC Agonist Peptide into Cells and Intact Ischemic Heart Using a Transport System, R-7. Chem Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 11.Pooga M, Kut C, Kihlmark M, Hällbrink M, Fernaeus S, Raid R, Land T, Hallberg E, Bartfai T, Langel U. Cellular translocation of proteins by transportan. FASEB J. 2001;15:1451–1453. doi: 10.1096/fj.00-0780fje. [DOI] [PubMed] [Google Scholar]
- 12.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi A, Akuta T, Okuyama H, Senda T, Yokoi H, Inokuchi H, Fujita S, Hayakawa T, Takeda K, Hasegawa M, Nakanishi M. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J Biol Chem. 2001;276:26204–26210. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- 14.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GGM. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome–DNA complexes. Proc Natl Acad Sci USA. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Wu H, McBride JL, Jung K-E, Hee Kim M, Davidson BL, Kyung Lee S, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 16.Josephson L, Tung CH, Moore A, Weissleder R. High-Efficiency Intracellular Magnetic Labeling with Novel Superparamagnetic-Tat Peptide Conjugates. Bioconjug Chem. 1999;10:186–191. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Zhang Q, Remsen EE, Wooley KL. Nanostructured materials designed for cell binding and transduction. Biomacromolecules. 2001;2:362–368. doi: 10.1021/bm015515c. [DOI] [PubMed] [Google Scholar]
- 18.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 19.Hirose H, Takeuchi T, Osakada H, Pujals S, Katayama S, Nakase I, Kobayashi S, Haraguchi T, Futaki S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol Ther. 2012;20:984–993. doi: 10.1038/mt.2011.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ter-Avetisyan G, Tünnemann G, Nowak D, Nitschke M, Herrmann A, Drab M, Cardoso MC. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–3378. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 22.Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 23.Fittipaldi A, Ferrari A, Zoppé M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K, Negishi M, Nomizu M, Sugiura Y, Futaki S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2007;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- 26.Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–172. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Mueller J, Kretzschmar I, Volkmer R, Boisguerin P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug Chem. 2008;19:2363–2374. doi: 10.1021/bc800194e. [DOI] [PubMed] [Google Scholar]
- 28.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals. 2012;5:1177–1209. doi: 10.3390/ph5111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Qian Z, Liu T, Liu YY, Briesewitz R, Barrios AM, Jhiang SM, Pei D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem Biol. 2013;8:423–431. doi: 10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner NG, Luechapanichkul R, Barrios AM, Pei D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry. 2014;53:4034–4046. doi: 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lättig-Tünnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat Commun. 2011;2:453–459. doi: 10.1038/ncomms1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal D, Nasrolahi Shirazi A, Parang K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew Chem Int Ed. 2011;50:9633–9637. doi: 10.1002/anie.201102572. [DOI] [PubMed] [Google Scholar]
- 35.Traboulsi H, Larkin H, Bonin MA, Volkov L, Lavoie CL, Marsault É. Macrocyclic cell penetrating peptides: a study of structure-penetration properties. Bioconjug Chem. 2015;26:405–411. doi: 10.1021/acs.bioconjchem.5b00023. [DOI] [PubMed] [Google Scholar]
- 36.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc Natl Acad Sci USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marelli UK, Bezençon J, Puig E, Ernst B, Kessler H. Enantiomeric Cyclic Peptides with Different Caco‐2 Permeability Suggest Carrier‐Mediated Transport. Chem Eur J. 2015;21:8023–8027. doi: 10.1002/chem.201501270. [DOI] [PubMed] [Google Scholar]
- 38.Purkayastha N, Eyer K, Robinson T, Dittrich PS, Beck AK, Seebach D, Kolesinska B, Cadalbert R. Enantiomeric and diastereoisomeric (mixed) L/ D-octaarginine derivatives - a simple way of modulating the properties of cell-penetrating peptides. Chem Biodivers. 2013;10:1165–1184. doi: 10.1002/cbdv.201300180. [DOI] [PubMed] [Google Scholar]
- 39.Ohtsuki T, Miki S, Kobayashi S, Haraguchi T, Nakata E, Hirakawa K, Sumita K, Watanabe K, Okazaki S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci Rep. 2015;5:18577. doi: 10.1038/srep18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian Z, Dougherty PG, Pei D. Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluorophore. Chem Commun. 2015;51:2162–2165. doi: 10.1039/c4cc09441g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Appelbaum JS, LaRochelle JR, Smith BA, Balkin DM, Holub JM, Schepartz A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem Biol. 2012;19:819–830. doi: 10.1016/j.chembiol.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LaRochelle JR, Cobb GB, Steinauer A, Rhoades E, Schepartz A. Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-arg proteins and stapled peptides. J Am Chem Soc. 2015;137:2536–2541. doi: 10.1021/ja510391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–597. doi: 10.1016/j.addr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Yang ST, Zaitseva E, Chernomordik LV, Melikov K. Cell-penetrating peptide induces leaky fusion of liposomes containing late endosome-specific anionic lipid. Biophys J. 2010;99:2525–2533. doi: 10.1016/j.bpj.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 46.Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madani F, Abdo R, Lindberg S, Hirose H, Futaki S, Langel U, Gräslund A. Modeling the endosomal escape of cell-penetrating peptides using a transmembrane pH gradient. Biochim Biophys Acta. 2013;1828:1198–1204. doi: 10.1016/j.bbamem.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurley JH, Boura E, Carlson LA, Różycki B. Membrane budding. Cell. 2010;143:875–887. doi: 10.1016/j.cell.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 52.Zhao K, Choe UJ, Kamei DT, Wong GCL. Enhanced activity of cyclic transporter sequences driven by phase behavior of peptide–lipid complexes. Soft Matter. 2012;8:6430–6433. doi: 10.1039/C2SM25405K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mishra A, Lai GH, Schmidt NW, Sun VZ, Rodriguez AR, Tong R, Tang L, Cheng J, Deming TJ, Kamei DT, et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc Natl Acad Sci USA. 2011;108:16883–16888. doi: 10.1073/pnas.1108795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt NW, Mishra A, Wang J, DeGrado WF, Wong GC. Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission. J Am Chem Soc. 2013;135:13710–13719. doi: 10.1021/ja400146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amand HL, Bostrom CL, Lincoln P, Norden B, Esbjorner EK. Binding of cell-penetrating penetratin peptides to plasma membrane vesicles correlates directly with cellular uptake. Biochim Biophys Acta. 2011;1808:1860–1867. doi: 10.1016/j.bbamem.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 56.Lamazière A, Wolf C, Lambert O, Chassaing G, Trugnan G, Ayala-Sanmartin J. The homeodomain derived peptide penetratin induces curvature of fluid membrane domains. PLoS ONE. 2008;3:e1938. doi: 10.1371/journal.pone.0001938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Säälik P, Niinep A, Pae J, Hansen M, Lubenets D, Langel Ü, Pooga M. Penetration without cells: membrane translocation of cell-penetrating peptides in the model giant plasma membrane vesicles. J Control Release. 2011;153:117–125. doi: 10.1016/j.jconrel.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 58.Thorén PEG, Persson D, Esbjörner EK, Goksör M, Lincoln P, Nordén B. Membrane Binding and Translocation of Cell-Penetrating Peptides. Biochemistry. 2004;43:3471–3489. doi: 10.1021/bi0360049. [DOI] [PubMed] [Google Scholar]
- 59.Magzoub M, Pramanik A, Gräslund A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry. 2005;44:14890–14897. doi: 10.1021/bi051356w. [DOI] [PubMed] [Google Scholar]
- 60.Wheaten SA, Ablan FD, Spaller BL, Trieu JM, Almeida PF. Translocation of cationic amphipathic peptides across the membranes of pure phospholipid giant vesicles. J Am Chem Soc. 2013;135:16517–16525. doi: 10.1021/ja407451c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Islam MZ, Ariyama H, Alam JM, Yamazaki M. Entry of cell-penetrating peptide transportan 10 into a single vesicle by translocating across lipid membrane and its induced pores. Biochemistry. 2014;2014(53):386–396. doi: 10.1021/bi401406p. [DOI] [PubMed] [Google Scholar]
- 62.Henriques ST, Huang YH, Chaousis S, Sani MA, Poth AG, Separovic F, Craik DJ. The Prototypic Cyclotide Kalata B1 Has a Unique Mechanism of Entering Cells. Chem Biol. 2015;22:1087–1097. doi: 10.1016/j.chembiol.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 63.Nischan N, Herce HD, Natale F, Bohlke N, Budisa N, Cardoso MC, Hackenberger CP. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew Chem Int Ed. 2015;54:1950–1953. doi: 10.1002/anie.201410006. [DOI] [PubMed] [Google Scholar]
- 64.Ben-Dov N, Korenstein R. Enhancement of Cell Membrane Invaginations, Vesiculation and Uptake of Macromolecules by Protonation of the Cell Surface. PLoS ONE. 2012;7:e35204. doi: 10.1371/journal.pone.0035204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.