

Abstract
Lafora body disease (MIM-254780), a glycogen storage disease, characterized by Lafora bodies (deformed glycogen molecules) accumulating in multiple organs, is a rare form of myoclonic epilepsy. It manifests in early adolescent years, initially with seizures and myoclonus, followed by dementia and progressive cognitive decline, ultimately culminating in death within 10 years. In Pakistan so far 5 cases have been reported. Here, we report a new case of Lafora body disease belonging to a consanguineous family from Pakistan. Histopathological analysis confirmed presence of lafora bodies in the patient`s skin. Sanger sequencing revealed novel homozygous 5bp deletion mutation (NM_005670.4; c.359_363delGTGTG) in exon 2 of the EPM2A gene, which was truly segregated in the family. These results will increase our understanding regarding the aetiology of this disorder and will further add to the mutation spectrum of EPM2A gene.
Keywords: Lafora body, EPM2A, consanguineous, deletion mutation, myoclonic epilepsy
INTRODUCTION
Myoclonic epilepsy of Lafora (MIM-254780) is a rare form of progressive myoclonic epilepsy (PME), manifesting in early adolescent years, initially with seizures and myoclonus, followed by dementia and progressive cognitive decline, ultimately culminating in death within 10 years.1 It is a glycogen storage disease, characterized by Lafora bodies (deformed glycogen molecules) accumulating in multiple organs.2 It has a variable prevalence dependent on the geographical area, with higher figures in areas of the world with high rates of consanguinity (Mediterranean area, Asian countries and Middle East), with a worldwide prevalence of about 4/1,000,000 individuals.1,3 In Pakistan so far 5 cases have been reported.4–6 It is an autosomal recessive disease, resulting from mutations in predominantly two genes i.e EPM2A (MIM-607566) and NHLRC1 or EPM2B (MIM-608072), coding for proteins laforin and malin respectively.2 About 250 families have been described, with 42% being attributable to EPM2A gene and remaining to EPM2B gene.1 However, there have been patients in which genetic testing for both these genes was negative, which led to the proposition that either the mutations are in non-coding regulatory regions or the possibility of other genes attributing to this disease.6 This was confirmed later by the identification of a third locus i.e. PRDM8 (MIM-616639), encoding for a protein possibly responsible for translocation of laforin and malin to the nucleus, thereby, ensuring adequate levels in cytoplasm.7 The genetics of Lafora body disease is evolving and identification of different loci would be helpful in understanding the pathogenesis of the disease and devising precision therapies accordingly. Recently, first antibody based drug, VAL-0417 has shown significant potential in degrading the Lafora bodies in-vitro and also reduced the Lafora bodies load in mice model.8 Additionally, a metabolomics approach clearly demonstrated that the drug reverses the physiological effects of Lafora bodies accumulation.8
Here, we report a case of Lafora body disease belonging to a consanguineous family from Pakistan. Histopathological study on axillary skin biopsy from the patient confirmed presence of lafora bodies. Genetic testing by Sanger sequencing showed a novel homozygous 5bp deletion mutation (NM_005670.4; c.359_363delGTGTG) in exon 2 in the EPM2A gene, which was truly segregated in the family. The effect of mutation on protein structure was further analysed through computational tools to see the truncation.
METHODS
A family visiting the Neurology Department of Shifa International Hospital was recruited for this study, after ethical approval from Institutional Review Board/Ethics Committee of Shifa International Hospital, Islamabad, Pakistan (IRB-198-047-2012). The family hailed from Northern Pakistan and comprised of one affected individual. Detailed history and examination was obtained and pedigree generated. Informed written consent was taken from patient’s father for all investigations including skin biopsy and genetic testing. Biopsy was taken from the axillary skin and sent for histopathological testing including period acid-Schiff (PAS) and PAS diastase (PAS-D) staining. Genetic testing was performed by Sanger sequencing. Primer sequences available on request. For computational analysis, protein structure (PDB ID: 4RKK) was obtained from protein data bank (https://www.rcsb.org/). Its structural analysis was performed with PyMol (https://pymol.org/2/).
CASE REPORT
A 16-year-old male patient (VI-1) born of consanguineous parents (Figure 1), student of grade 9, was brought to the neurology clinic at Shifa International Hospital with complaints of frequent sudden single jerks of the body and generalized seizures for 1.5 years. He had also lost his ability to walk for 1 year and had progressive cognitive decline over the last 6 months, as well as speech impairment and difficulty in activities of daily living over the past 2 months. The very first symptoms were sudden single jerks of the arms and occasional dropping of objects. This was followed within 6 months by an episode of generalized tonic-clonic convulsion (GTC). When investigated, MRI brain was normal and EEG showed episodes of paroxysmal bursts of high amplitude generalized sharp and slow waves. The patient was seen elsewhere and diagnosed as juvenile myoclonic epilepsy (JME) and started on valproic acid 250 mg orally twice daily and multivitamins.
Figure 1.

Pedigree of a family with a member (VI-1) suffering from Lafora disease.
However, over time, the patient’s myoclonic jerks became more frequent and more intense and involved the whole body, and were particularly marked on voluntary movements. This resulted in falls which eventually led him to need to use a wheel chair. He had a second generalized GTC followed by 4 more episodes in the next 2 months with increasingly short intervals. He was seen by a second neurologist elsewhere and levetiracetam 500 mg twice daily and clonazepam 0.5 mg at night were added.
Over the next 2 months, his family noticed changes in behaviour described as ‘odd childish behaviour’ and ‘irritability over petty issues’, followed within 1.5 months by progressive impairment in his memory and intellectual decline, and he had to be taken out of school. Lacosamide was added and increased to 100 mg twice daily. Seizure frequency increased again. His speech became slower and he would stutter sometimes and developed drooling. Following this, he began to struggle with daily activities and became completely dependent on his mother. At this point in time, he was brought to our neurology outpatient clinic.
Examination revealed a young male of average height and weight, with dysarthric, slow and strained speech. Mini mental state examination (MMSE) score was 12/30. There was action myoclonus and ataxia. Fundi and cranial nerve examination were unremarkable. Limbs had normal muscle bulk, power and reflexes. Based on these findings, he was diagnosed as progressive myoclonic epilepsy (PME) / Lafora body disease.
Sodium valproate 1 g twice daily and levetiracetam 500 mg twice daily were continued. Clonazepam was increased to 2 mg in the morning and 4 mg in the night. Zonisamide 100 mg once daily was added and lacosamide tapered off. He was given written instructions to avoid phenytoin, and in case of acute emergency to report to the local emergency room. An axillary skin biopsy revealed Lafora bodies (Figure 2). Autoimmune encephalitis panel was negative. The patient returned for follow-up at 2 months (i.e 20 months from first symptoms onset), with seizures well controlled. At this point, his latest symptom was swallowing impairment of sufficient severity to recommend gastrostomy.
Figure 2.

(a, b) Axillary skin biopsy of affected patient (VI-1) stained with PAS and PASD respectively. The apocrine sweat glands with cytoplasmic Lafora body (indicated by arrows) are clearly visible in both images.
Genetic testing
Sanger sequencing of the EPM2A and EPM2B coding regions and exon-intron junctions were performed according to procedures described previously.4 We identified a novel homozygous 5 bp deletion mutation (NM_005670.4; c.359_363delGTGTG) in exon 2 in the EPM2A gene (Figure 3a). The variant leads to a frameshift and premature termination codon 29 bp downstream in the same exon (p. Glu120ValfsX10). Parents and one sibling (V-3, V-4, VI-3) were heterozygous for the mutation (Figure 3b). The other sibling (VI-2) did not carry the variant (Figure 3c).
Figure 3.

Sequence chromatograms showing (a) homozygous deletion mutation c.359_363delGTGTG in the EPM2A gene in the affected individual (b) heterozygous carrier and (c) normal wild type sequence.
Structural analysis
For computational structure analysis, the wild type laforin was compared to the mutant protein. Wild type human laforin is composed of N-terminal carbohydrate binding module (CBM) with a C-terminal dual specificity phosphatase (DSP) domain (Figure 4a). The asymmetric unit of the protein has two laforin molecules forming antiparallel dimer mediated by DSP domain (Figure 2a). The tertiary structure maintained by the CBM/DSP orientation is important for its function. Analysis of the novel deletion mutation (c.359_363delGTGTG) through computational tools showed that if any aberrant protein is expressed from the mutated mRNA transcript, it would translate the CBM with C-terminal mutated residue Glu120ValfsX10 (Figure 4b) along with mutated residues.
Figure 4.
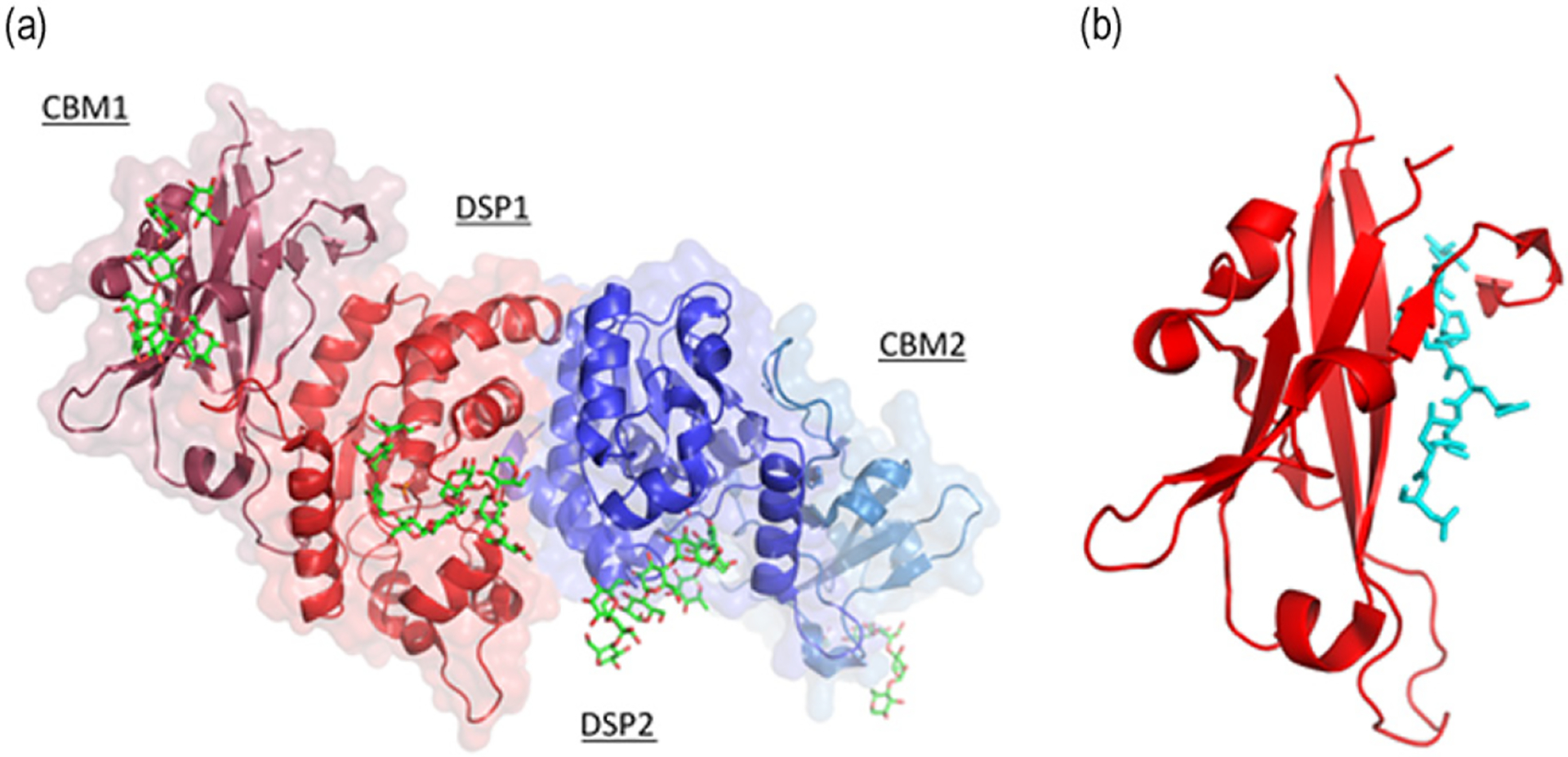
Crystal structure of human laforin protein (PDB ID: 4RKK). (a) Human laforin (dimer) protein showing N-terminal carbohydrate binding module (CBM) and C-terminal dual specificity phosphatase (DSP) domain in complex with maltohexaose (Green sticks) and phosphate in orange. (b) CBM of laforin with C-terminal mutant residues (V120LSPNRTLD128) colored in cyan (Sticks) generated due to mutation Glu120ValfsX10.
DISCUSSION
Lafora body disease is a type of PME, which is a group of diseases characterized by myoclonus, seizures and progressive neurological decline. Lafora body disease is named after the Spanish neurologist Gonzalo Lafora, who in 1911 first observed the inclusions that came to carry his name.1,9 The disease is rare and was historically mainly described in European countries of Spain, Italy and France, but over the past few decades increasing numbers have been identified in Asian countries of Japan, India, Pakistan, as well as the Middle East.3 This is the 6th case being reported from Pakistan and the 4th from our centre.
The clinical manifestation is in a child with no previous developmental problems. Age of onset is usually 14–16 years, but a wide range from 5–20 years has been described.1,3,10 Although our patient falls in this range, the other reported patients from this region tend to have a relatively younger onset, i.e. 10–11 years on average.4–6 Myoclonic jerks are amongst the earliest manifestations, and are more marked on action or by light stimuli. This is the main factor making patients wheelchair dependent in short order.3 The other early symptoms are occipital seizures, presenting as visual impairment/scotomas.1,10 Our patient had myoclonic jerks as the initial symptoms but did not have any history suggestive of occipital seizures.
The other seizure types mostly described are GTCs, atonic, and atypical absence.1,3 These are followed shortly by progressive cognitive deficits (more affected domains are attention, language, memory and executive function), which was demonstrated in the MMSE of our patient. The other features are motor decline (due to motor apraxia and ataxia), leading to difficulty in carrying out tasks of daily living.1,3,10,11 Speech becomes slurred, and even complete mutism eventually occurs.3 The clinical course of disease involves poorly controlled seizures, a vegetative state within 5 years, and death within 10 years due to status epilepticus/ aspiration pneumonia / SUDEP (sudden unexpected death in epilepsy).7,9 Our patient demonstrated the aforementioned sequence, which have been documented in previous data from our region. Neuropsychiatric manifestations in patients from Pakistan have not been reported, at least not emphasized. These were prominent in our patient (Table 1), including behavioural changes, frustration, emotional disturbance, confusion, followed by dementia.3,7 The other noteworthy aspect in our patient is the rapidity of progression of the neurological and functional decline within a period of 1.5 year, which has not been reported in this part of the world. A comparison of our patients with the earlier reported cases from Pakistan is given in Table 1.
Table 1:
Clinical phenotypes of Lafora patients previously reported from Pakistan and their comparison with the present study
| Features | Hashmi et al6 | Aslam et al5 | Ahmad et al4 | Present study | ||
|---|---|---|---|---|---|---|
| Gender | male | male | female | female | male | male |
| Age at onset | N.A | 11 years | 10 years | 11 years | 8 years | 14 years |
| Consanguinity | N.A | + | + | + | ||
| Types of seizures | Myoclonic GTCs Absence seizures | Myoclonic GTCs, Occipital lobe seizures | Myoclonic, GTCs, Atonic, Occipital lobe seizures | Myoclonic GTCs | Myoclonic GTCs | Myoclonic GTCs |
| Signs & Symptoms | Dropping objects, falls, mild cognitive impairment, ataxia | Myoclonic jerks, visual hallucination, GTCs, dysarthria | Flashes of light followed by headache & nausea Later: GTCs and myoclonic jerks | GTC followed by myoclonic jerks and slurring of speech, cognitive decline, dysarthria | GTC Myoclonic jerks | Dropping objects, GTCs Gait difficulty, cognitive decline, behavioural disturbance, dysarthria, ataxia |
| EEG findings | Generalized polyspike & wave discharges | Generalized background slowing and abnormal spikes | Occipital spike wave activity, later generalized polyspike wave discharge | Generalized polyspike & wave discharges with mild background slowing | Generalized spike & wave discharges | Generalized paroxysmal bursts of high amplitude sharp & slow waves |
| Biopsy for Lafora bodies | + | − | + | + | N.A | + |
| Genetic testing | N.A | EPM2A c.262T>G; p.Phe88Val | No mutation identified | EPM2A c.95G>T; p.32Trp>Leu | EPM2A c.95G>T; p.32Trp>Leu | EPM2A c.359_363delGTGTG; p.Glu120ValfsX10 |
| Clinical course | Slow cognitive decline, bed bound after 5 years | Over 3 years developed swallowing difficulty and difficulty in activities of daily living | Had to use wheel-chair after 5 years & bed bound 13 years after diagnosis | Swallowing impairment, developed spasticity in limbs, dependent for daily activities over 3 years | Increased frequency of myoclonic jerks, independent in daily activities but had to leave school | Rapidly progressive cognitive, motor decline. Wheel chair dependent within a year |
N.A: indicates not available; + sign indicates presence while − sign indicates absence of phenotype.
Initial steps in diagnosis are interpretation from clinical findings and EEG features of background slowing, paroxysmal bursts of irregular generalized epileptiform discharges, a normal MRI brain and skin biopsy revealing Lafora bodies, with ultimate confirmation by genetic testing.10,12 It is important to diagnose the disease early, as ample knowledge about the disease is one of the biggest contributing factors, in compliance with anti-epileptic drugs.13 Most commonly PME is misdiagnosed as JME and subsequently mismanaged. This happened in our patient and was also shown in a recent study from Germany, which concluded that in refractory epilepsies, genetic testing should be considered early.14
It is vital that both the physician taking the skin biopsy specimen and the one interpreting the histopathology have a high level of expertise.10 There is frequent false-positivity in this disease for two reasons. Firstly, because it is rare, most pathologists see too few cases to be guided by experience. The axillary glands contain normal secretory material that also stains with PAS, and it takes some experience of careful review of the literature to distinguish the Lafora bodies from normal contents of apocrine glands.15
Genetic testing is the gold standard for diagnosis. Two genes are mainly responsible for the pathology of Lafora Body disease, EPM2A (MIM-607566) and EPM2B or NHLRC1 (MIM-608072).1 EPM2A codes for the laforin phosphatase and NHLRC1 for the malin ubiquitin E3 ligase.2 Both proteins are involved in glycogen metabolism and the dysfunction of either results in aggregation of malstructured and insoluble glycogen that accumulate into the disease’s pathogenic and pathognomonic inclusions.3 Over 100 different mutations of these 2 genes have been identified so far.15 Some are missense, some non-sense, some exon and some entire gene deletion.15,16 A third gene, PRMD8 (MIM-616639) has been reported to be involved in an earlier onset from of the disease in one English family of Pakistani origin.7
In our patient, a novel homozygous 5 bp deletion mutation (NM_005670.4; c.359_363delGTGTG) was identified in exon 2 in EPM2A gene by Sanger sequencing. The novel mutation (p. Glu120ValfsX10) identified in the present study is located in the in-between region of the protein’s two main domains, the N-terminal carbohydrate binding module (CBM; residues 1–124) and the C-terminal dual-specificity phosphatase domain (DSP; residues 157–326) (Figure 2a). It results in the production of truncated protein containing N-terminal 130 amino acids only, assuming the mRNA is not eliminated by nonsense-mediated decay (NMD).17,18
A truncated protein would lack the important phosphatase domain.19,20 Interestingly it was recently clearly demonstrated that this phosphatase activity is not essential in mitigating the disease, and that laforin’s critical function is conveyed through its interaction with malin. If a truncated protein is present, it would be expected that the interaction with malin is lost, and the latter’s possible function in regulating its substrates glycogen synthase, glycogen debranching enzyme and the glycogen synthase regulator protein R5 (PTG).21–23
In conclusion, we report a novel homozygous 5 bp deletion mutation (NM_005670.4; c.359_363delGTGTG) in the EPM2A gene in a consanguineous family from Pakistan. Lafora body disease, although rare, has been increasingly recognized in Asian countries. Recently antibody based drug, VAL-0417 has shown significant potential by degrading the Lafora bodies both in-vitro and in-vivo in Epm2a_/_mice. Early diagnosis coupled with such precision therapies will provide significant clinical benefit to Lafora body disease patients in near future.
ACKNOWLEDGEMENTS
We are very thankful to the family members for their valuable participation.
DISCLOSURE
Financial support:
This work was funded in part by the National Institutes of Health under award number P01NS097197. B.A.M. holds the University of Texas Southwestern Jimmy Elizabeth Westcott Chair in Pediatric Neurology.
Footnotes
Conflict of interest: None
REFERENCES
- 1.Desdentado L, Espert R, Sanz P, Tirapu-Ustarroz J. Enfermedad de Lafora: revision de la bibliografia [Lafora disease: a review of the literature]. Rev Neurol 2019;68(2): 66–74. [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez-Martin P, Lahuerta M, Viana R, Knecht E, Sanz P. Regulation of the autophagic PI3KC3 complex by laforin/malin E3-ubiquitin ligase, two proteins involved in Lafora disease. Biochim Biophys Acta Mol Cell Res 2020;1867(2):118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen AC, Andermann E. Progressive myoclonus epilepsy, Lafora Type. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds: GeneReviews®. Seattle (WA): University of Washington, Seattle; December 28, 2007. [PubMed] [Google Scholar]
- 4.Ahmad A, Dad R, Ullah MI, et al. Clinical and genetic studies in patients with Lafora disease from Pakistan [published correction appears in J Neurol Sci 2017;375:281]. J Neurol Sci. 2017; 373: 263–7. [DOI] [PubMed] [Google Scholar]
- 5.Aslam Z, Lee E, Badshah M, Naeem M, Kang C. Whole exome sequencing identified a novel missense mutation in EPM2A underlying Lafora disease in a Pakistani family. Seizure 2017;51:200–203. [DOI] [PubMed] [Google Scholar]
- 6.Hashmi M, Saleem F, Mustafa MS, Sheerani M, Ehtesham Z, Siddiqui K. Role of levetiracetam in refractory seizures due to a rare progressive myoclonic epilepsy: Lafora body disease. BMJ Case Rep 2010; 2010:bcr0120102653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbull J, Girard JM, Lohi H, et al. Early-onset Lafora body disease. Brain 2012;135(Pt 9):2684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brewer MK, Uittenbogaard A, Austin GL, et al. Targeting pathogenic Lafora bodies in Lafora disease using an antibody-enzyme fusion. Cell Metab 2019;30(4):689–705.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gentry MS, Afawi Z, Armstrong DD, et al. The 5th International Lafora Epilepsy Workshop: Basic science elucidating therapeutic options and preparing for therapies in the clinic. Epilepsy Behav 2020;103(Pt A):106839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull J, Tiberia E, Striano P, et al. Lafora disease. Epileptic Disord 2016;18(S2):38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baykan B, Striano P, Gianotti S, et al. Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation. Epilepsia. 2005;46(10):1695–7. [DOI] [PubMed] [Google Scholar]
- 12.Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol 2018;14(10):606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chinnaiyan S, Narayana S, Nanjappa VP. Adherence to antiepileptic therapy in adults. J Neurosci Rural Pract 2017;8(3):417–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin S, Strzelczyk A, Lindlar S, et al. Drug-resistant juvenile myoclonic epilepsy: Misdiagnosis of progressive myoclonus epilepsy. Front Neurol 2019;10:946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kecmanović M, Keckarević-Marković M, Keckarević D, Stevanović G, Jović N, Romac S. Genetics of Lafora progressive myoclonic epilepsy: current perspectives. Appl Clin Genet 2016;9:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat 2009;30(5):715–23. [DOI] [PubMed] [Google Scholar]
- 17.Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet. 2006; 7:103–24. [DOI] [PubMed] [Google Scholar]
- 18.Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 2014; 24(9):506–14. [DOI] [PubMed] [Google Scholar]
- 19.Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem 2006;281(41):30412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tagliabracci VS, Turnbull J, Wang W, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A 2007;104(49):19262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vilchez D, Ros S, Cifuentes D, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 2007; 10: 1407–13. [DOI] [PubMed] [Google Scholar]
- 22.Solaz-Fuster MC, Gimeno-Alcañiz JV, Ros S, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet 2008;17(5):667–78. [DOI] [PubMed] [Google Scholar]
- 23.Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem 2008;283(7):4069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]