

Abstract
Environmentally responsive nanomaterials have been developed for drug delivery applications, in an effort to target and accumulate therapeutic agents at sites of disease. Within a biological system, these nanomaterials will experience diverse conditions which encompass a variety of solute identities and concentrations. In this study, we developed a new quartz crystal microbalance with dissipation (QCM-D) assay, which enabled the quantitative analysis of nanogel swelling, protein adsorption, and biodegradation in a single experiment. As a proof of concept, we employed this assay to characterize non-degradable and biodegradable poly(acrylamide-co-methacrylic acid) nanogels. We compared the QCM-D results to those obtained by dynamic light scattering to highlight the advantages and limitations of each method. We detailed our protocol development and practical recommendations, and hope that this study will serve as a guide for others to design application-specific QCM-D assays within the nanomedicine domain.
Keywords: biodegradable, drug delivery systems, stimuli-sensitive polymers, surfaces and interfaces, swelling
INTRODUCTION
Synthetic nanogels have been previously designed for numerous biosensing and drug delivery applications. However, very little is known about the rheological properties of nanogels. Previous studies by our group1–3 have measured the swelling properties of thin polymer films, and have assumed that those properties correspond to the gels’ behavior at the micro- or nanoscale. Other studies have measured the dynamic swelling of nanogels using dynamic light scattering (DLS).4–6 These measurements have provided an understanding of the intelligence of nanogel systems, and have explained their pH-responsive therapeutic loading and release behavior. However, neither technique can probe the kinetics of nanogel swelling or degradation in response to a stimulus.
Quartz crystal microbalance with dissipation (QCM-D) measurement allows real-time quantification of the viscoelastic properties of a thin layer.7,8 One of the most common applications of QCM-D for biomedical materials has been monitoring layer-by-layer (LbL) self-assembly.9,10 By measuring the frequency and dissipation changes of a quartz substrate during LbL, researchers can confirm the iterative assembly of multilayered structures. Further, by exchanging the running buffer with various salt solutions, researchers have quantified the impact of buffer salts on the films’ rheological properties and dissolution rates.9
QCM-based biosensors are also an active research area, particularly for measuring protein adsorption to surfaces,11,12 or deposition within a supported membrane.13,14 QCM-D is suitable for measuring protein adsorption to thin films and nanomaterials, as it is sufficiently sensitive (ng cm−2) to determine the rate and magnitude of binding events at low analyte concentrations. Further, by infusing a series of protein samples across a range of concentrations, one can obtain an adsorption isotherm and approximate a dissociation constant. Therefore, QCM is a useful tool for materials development applications (e.g., antifouling membranes,15,16 biomaterial coatings,17) and a sensitive detecting element for biosensing applications (e.g., medical diagnostics18–20).
In this work, we developed a QCM-D assay for determining biologically relevant characteristics of nanogels. We developed a protocol for immobilizing nanoscale hydrogels onto planar gold-coated QCM-D sensors. Then, we used the nanogel-coated sensors to measure the nanogels’ swelling, hysteresis, interaction with proteins, and biodegradation. By monitoring both the frequency and dissipation of the quartz sensors, we were able to simultaneously monitor physical phenomena (i.e., swelling, binding, and degradation) and infer mechanistic insight (i.e., surface erosion and bulk degradation). Our purpose was to provide a generalizable protocol, as well as practical guide, for developing custom QCM-D assays that characterize responsive and/or biodegradable nanomaterials. To the best of our knowledge, this is the first report of a method that can quantify each of these relevant biological characteristics of nanogels within a single assay.
A general schematic illustrating our method development is given in Figure 1. We first modified a planar gold sensor chip with cysteamine hydrochloride, to form an amine-terminated monolayer. Carboxylic acid containing hydrogel nanoparticles were then immobilized by zero-length 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) crosslinking. These nanogel-coated sensors were stable in PBS for more than 1 week. We then used the combination of real-time gravimetric and viscoelastic analysis to infer the mechanisms of nanogel swelling (left) or degradation (right) in response to chemical stimuli.
Figure 1.
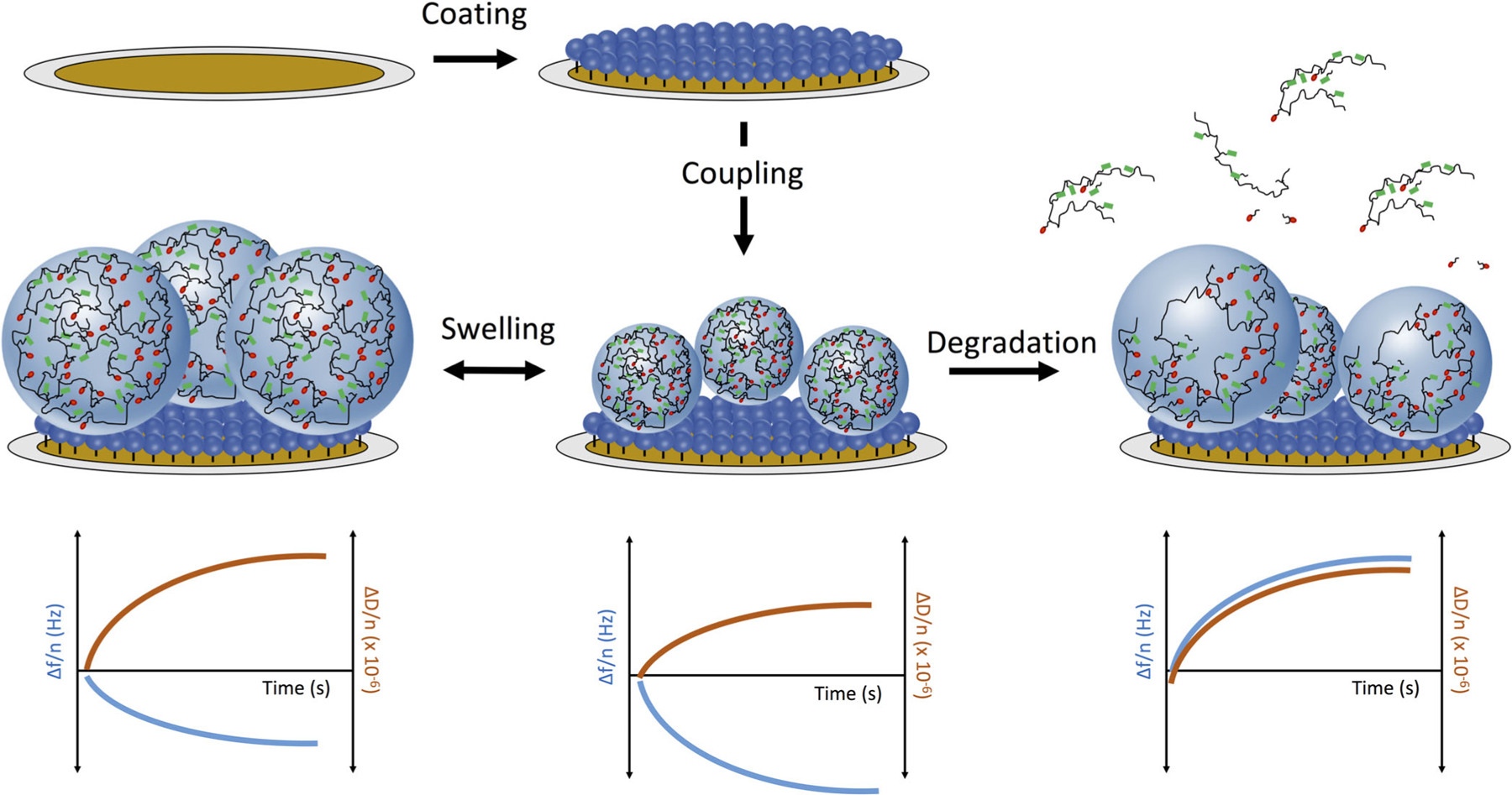
Schematic depiction of experimental approach. A planar gold QCM sensor was first coated with cysteamine hydrochloride (5 mg/mL) to form an amine-terminated monolayer via gold-thiol interactions. Nanogels were covalently immobilized onto the monolayer via EDC-catalyzed zero-length crosslinking. The nanogels’ swelling (left), protein adsorption, or biodegradation (right) processes were characterized through quantification of the dynamic sensor-bound mass (resonance frequency, blue, left axis) or viscoelastic properties (dissipation, red, right axis).
EXPERIMENTAL
Nanogel Synthesis
All chemicals were purchased from Sigma Aldrich or ThermoFisher Scientific and used as received. All buffers were sterile filtered (0.22 μm) prior to use. Poly(acrylamide-co-methacrylic acid) nanogels, crosslinked with methylenebisacrylamide or N,N′-bis(acryloyl)cystamine were synthesized as described by Zhong et al.21 and Clegg et al.,22 respectively.
QCM-D Materials and Instrumentation
Specialized equipment for QCM-D sensor handing and cleaning is available from Biolin Scientific (Gothenburg, Sweden). The following protocol is optimized for the QCM-D QSense-E4 instrument from Biolin Scientific. All QCM-D reagents should be degassed and filtered prior to use. It is critical not to introduce air into the flow module at any time. Air bubbles will introduce artifacts into your signal. To prevent air bubbles, make sure that the pump is stopped prior to all buffer exchanges. In the event of an air bubble, reverse the pump and remove it before proceeding with the experiment. All components of the QCM-D experiments were conducted at 37 °C.
Sensor and Flow Cell Cleaning
Planar gold sensors were cleaned with a 5:1:1 (volume) mixture of ultrapure water, 25% ammonium hydroxide solution, and 30% hydrogen peroxide solution at 75 °C (5 min). Then, they were rinsed several times with ultrapure water. Finally, the sensors were rinsed with ethanol, dried under a flow of nitrogen, and UV-ozone treated for 10 min (BioForce ProCleaner, BioForce Nanosciences, Salt Lake City, UT). Clean sensors were stored at room temperature for up to a month.
Prior to each experiment, the QCM flow channel and modules were cleaned with warm (42 °C) ultrapure water, followed by sodium dodecyl sulfate (0.5 wt % solution), and ultrapure water again (minimum 5 mL per flow module per wash). These washes were done with a “cleaning sensor” in the flow cell, which was rinsed with water and ethanol, but not subjected to a full cleaning procedure nor used for experimentation.
Sensor Functionalization
It is important to be strategic with the number of unique sensor modifications in a QCM-D experiment. The QSense E4 module (Biolin) can accommodate up to four sensors in parallel. We used, and recommend, three experimental sensors and a blank sensor. The blank sensor was exposed to all sensor coating and treatment steps, but was not coated with nanogels. In quantitative analysis, we subtracted the changes in sensor resonance frequency for the blank sensor from that of the experimental sensors. This corrects for confounding signal due to solute interactions with the sensor itself, rather than attached nanoparticles.
We secured the clean, planar gold sensor in the flow module and flowed 1× PBS at 0.2 mL/min through the flow module. A baseline was established by flowing 1× PBS over the clean sensors for several minutes. Next, an amine-terminated monolayer was formed by flowing cysteamine hydrochloride (10 mg/mL in 1× PBS, 20 min). The cysteamine hydrochloride interacts with the gold-coated quartz sensor via gold-thiol interactions, forming a monolayer. After establishing a new baseline (1× PBS), the nanogels were coupled to the amine-terminated monolayer. The nanogels were suspended in MES buffer, after which the pH was adjusted and the carboxylic acids activated with EDC (2 mg/mL nanogels, 10 mg/mL EDC, in 10 mM MES buffer, pH = 5.5). To ensure the most efficient coupling, it was important to infuse the EDC-activated nanogels immediately after the EDC was added. We infused the entirety of the nanogel stock (5 mL, approximately 25 min) to maximize nanogel coupling, although kinetic analyses revealed that the majority of coupling occurred within the first minute.
Once the nanogels were covalently coupled to the sensor surface, the sensors were ready for characterization experiments. These experiments were typically completed the day following sensor functionalization, although we noted that the nanogel-coated sensors were stable in 1× PBS at room temperature for more than 1 week.
Nanogel Swelling
After a stable baseline reading was established (1× PBS, 37 °C, 0.2 mL/min), we determined the dynamic swelling of the nanogels in response to changes in the buffer pH and ionic strength. A series of buffers with different ionic strength (0.01, 0.1, 0.5, 1, and 5×) or pH values (1× PBS adjusted to pH = 3, 5, 7, 9, and 11 with 1 N NaOH or HCl) were infused at 0.2 mL/min (7 min/buffer). Please note that the pH and ion concentrations of the buffer may lead to intermolecular interactions between salts and the sensor coating (in this case, cysteamine). This is why it was important to include a blank sensor (i.e., a sensor that does not have nanogels). The changes in frequency and dissipation of the blank sensor were subtracted from all sample measurements to determine the contribution of nanogel swelling or hysteresis to the change in sensor resonance frequency.
Model Protein Adsorption
Protein adsorption experiments quantified the extent to which protein bound to the nanogels. By quantifying protein adsorption at several equilibrium concentrations, we obtained a protein adsorption isotherm and estimated the protein-polymer dissociation constant. First, we established a baseline by flowing 1× PBS (pH = 7.4, 37 °C) at 0.2 mL/min. Simultaneously, we prepared a fresh dilution series of lysozyme (hen egg white, Sigma Aldrich) in 1× PBS. The lysozyme stocks were infused, from low to high concentration, over both the nanogel decorated and unmodified sensors. In between each protein infusion, we flowed 1× PBS buffer without lysozyme to quantify dissociation. Each infusion was approximately 7 min in duration.
Biodegradation
Finally, we quantified the biodegradation kinetics of disulfide-crosslinked nanogels in response to the presence of a reducing agent. Non-degradable nanogels, which were crosslinked with methylenebisacrylamide, were used as a control. First, a baseline was established (1× PBS, 37 °C, 0.2 mL/min). Simultaneously, we prepared a fresh stock of reducing agent [10 mM dithiothreitol (DTT) in 1× PBS, adjusted to pH = 7.4]. We infused the reducing agent at 0.2 mL/min until the frequency and dissipation of the degradable nanogel sensors stabilized. For our systems, this took approximately 20 min. Then, we established a new baseline reading (1× PBS, 37 °C, 0.2 mL/min) to determine the total mass loss. As a secondary confirmation of biodegradation, we then attempted to replicate a portion of the swelling experiment. We concluded that degradation was complete if there was no detectable swelling response to solution ionic strength.
Calculations and Assumptions
For each experiment (i.e., swelling, protein adsorption, and degradation), the initial resonance frequency and dissipation values were subtracted from all measurements, setting the baseline to zero. The frequency and dissipation shifts for the blank sensor were also subtracted from each sample sensors. The resonance frequency measurements were converted to adsorbed mass using the Sauerbrey equation:
where Δm is the change in mass bound to the sensor (in ng/cm2), C is a constant determined by the intrinsic properties of quartz (C = 17.7 ng cm−2 Hz−1 for a 5 MHz crystal), and n is the overtone number (i.e., 1, 3, 5, 7, 9, 11, or 13).
The Sauerbrey approximation was developed for quantifying the deposition of rigid species onto a quartz sensor in a vacuum, and assumes that the sensor-bound mass is similar in density and stiffness to the underlying quartz. In the present study, soft nanoparticles (i.e., acrylamide and methacrylic acid, crosslinked into nanoscale networks) are bound to the sensor in aqueous media. As described by Denison23 and Behrndt,24 the Sauerbrey equation can be applied in our application because the bound nanogel film is sufficiently thin and the changes in resonance frequency are much less than 2% of the resonance frequency of the blank sensor.
The dissipation measurement is a ratio of the lost (i.e., viscous) to stored (i.e., elastic) energy during the oscillation of the crystal. It is calculated using the following relationship:
where D is the dissipation measurement, El is the loss energy, Es is the stored energy, ƒ is the frequency measurement, and τ is the time constant for the amplitude decay of the quartz crystal when the driving voltage is turned off.25
As the bound layer undergoes a physical transition that increases its viscous behavior (e.g., swelling, degradation of crosslinks) the dissipation increases. Conversely, as the sensor-bound layer increases in elasticity (e.g., hysteresis resulting in a loss of bound water), the dissipation decreases. In this study, we used dissipation measurements to perform qualitative analyses on the physical transitions of the bound nanogel layer.
All swelling and biodegradation experiments were performed in triplicate. For plots, representative data are shown. The data were collected using the QSoft software (Biolin), exported using the QTools software (Biolin), and plotted and analyzed in PRISM (GraphPad).
RESULTS AND DISCUSSION
Nanogel Synthesis and Characterization
Poly(acrylamide-co-methacrylic acid) nanogels were synthesized with both non-degradable (methylenebisacrylamide) and degradable (N,N′-bis(acryloyl)cystamine) crosslinking agents. Assuming that monomer incorporation was proportional to the monomer feed, the final nanogels contained 22.5 mol% methacrylic acid, 75 mol% acrylamide, and 2.5 mol% crosslinker. These formulations have been characterized extensively in previous reports.21,22
Formulation characterization data are given in Figure S1 in Supporting Information. The non-degradable nanogels were smaller in hydrodynanic diameter than the degradable nanogels (76.1 ± 8.2 and 83.1 ± 9.8 nm, respectively, p < 0.05) [Figure S1(a); Supporting Information]. While these diameter values are similar, the small difference can be attributed to a more efficient incorporation of methylenebisacrylamide than N,N′-bis(acryloyl)cystamine. This result is consistent with the hydrophobicity of each molecule [octanol/water partition coefficients (logP), as determined by USA EPA Epi Suite: methylenebisacrylamide = −1.52, N,N′-bis (acryloyl)cystamine = 0.51]. Both nanogel formulations were similar in zeta potential [−35.5 ± 7.4 mV for non-degradable, and − 32.4 ± 7.7 mV for degradable nanogels, Figure S1(b)], exhibited pH-responsive swelling with a similar critical transition (pH~4.8) [Figure S1(c); Supporting Information], and had indistinguishable FTIR spectra [Figure S1(d); Supporting Information]. These results indicated that methacrylic acid and acrylamide were incorporated similarly in the two formulations, and that the sole difference was the crosslinker identity.
Sensor Functionalization
All sensors were first coated with an amine-terminated monolayer of cysteamine hydrochloride (HCl). While the cysteamine HCl flowed over the sensor (10 mg/mL, 1× PBS, pH = 7.4), the resonance frequency of the sensor increased continually. We attributed this increase to a displacement of solvent, which was previously interacting with the gold sensor. We discontinued infusion of cysteamine HCl after the resonance frequency, normalized to the overtone number, increased by at least 100 Hz for all sample and control sensors.
Degradable or non-degradable nanogels, activated with EDC (5-fold mass excess), were then infused over the amine-coated sensors. The nanogels were covalently coupled via a stable amide bond. The nanogel-coated sensors were washed under a flow (0.2 mL/min) of 1× PBS running buffer for at least 30 min to remove any unreacted species or remaining catalyst, prior to conducting further experiments.
We calculated the mass of bound nanogels by determining the change in resonance frequency, as a result of coupling, and applying the Sauerbrey equation. Representative data for the resonance frequency throughout the sensor functionalization procedure are given in Figure 2(a). As shown in this example, we noted the change in resonance frequency as a result of nanogel coupling, and calculated the change in bound mass. It is important to remember that this quantity is both the mass of bound nanogels as well as any additional associated water and ions. In Figure 2(a), the change in coupled mass was 2420 ng/cm2. In a separate experiment, we tested the effect of nanogel concentration on conjugation efficiency. We infused EDC-activated nanogels of across a range of concentration (1–10 mg/mL in 10 mM MES, pH = 5.5, 5-fold mass excess EDC: nanogels, 10 mg total nanogels, variable volume). The nanogel concentration affected the rate of conjugation (more rapid as the concentration of nanogels increased), but did not affect the final conjugated mass. This indicated that the nanogels were forming a dense layer on the sensor, and that upon reaching saturation no further conjugation occurred. For all further experiments, the nanogel concentration during the conjugation step was 2 mg/mL (5 mL total volume).
Figure 2.
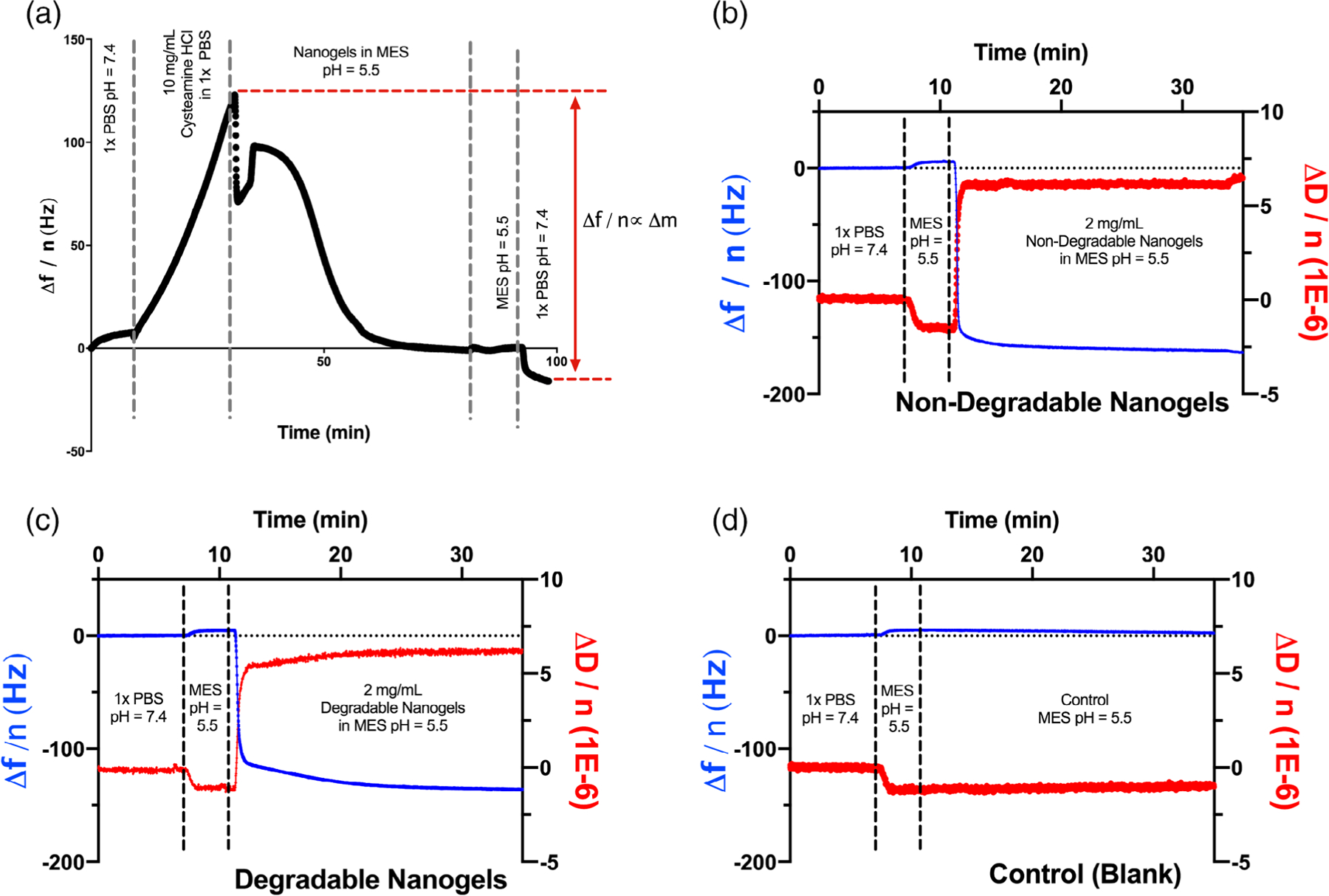
QCM-D sensor functionalization. (a) Cysteamine HCl forms a monolayer of free amines on the planar gold sensor through gold-thiol interactions. Nanogels were coupled to the amine monolayer via EDC-catalyzed crosslinking. The mass of nanogels coupled is linearly related to the reduction in resonance frequency during the coupling reaction. (b) Raw data for sensor functionalization with non-degradable poly(acrylamide-co-methacrylic acid) nanogels. Nanogel coupling increased the sensor bound mass by 3040 ng/cm2 as well as increased the viscosity of the sensor-associated layer. (c) A total of 2580 ng/cm2 degradable nanogels was conjugated to a parallel sensor. The viscosity of the sensor-associated layer increased in a manner similar to the non-degradable nanogels. (d) As expected, control sensors, without nanogels, exhibited no change in resonance frequency or dissipation.
Consistent with the similar methacrylic acid content in the degradable and non-degradable nanogels, there was no discernable difference in sensor conjugation efficiency between the two formulations [Figure 2(b–c)]. The final bound mass was 3040 ng/cm2 for the non-degradable nanogels and 2580 ng/cm2 for the degradable nanogels. Nanogel coupling led to an increase in dissipation, indicating that coupling increased the viscosity of the sensor-bound layer by increasing the amount of associated water. As shown in Figure 2(d), no polymer mass bound to the control sensor, as it was coated with cysteamine HCl but not exposed to nanogels.
Nanogel Swelling
Nanogel swelling analysis is given in Figure 3. In Figure 3(a), we show the change in sensor frequency and dissipation as buffers of varying ionic strength (0.01× PBS to 5× PBS) were infused over the non-degradable nanogel coated sensor. Swelling transitions were evident both in the frequency and dissipation measurements. As the nanogels swelled, they imbibed water, leading to increases in both the sensor-associated mass and the nanogel layer viscosity. We defined a physical transition as a position within the dataset where three consecutive frequency measurements trended in a similar (i.e., swelling or collapse) direction. We concluded that a transition was complete when the variation in frequency measurements returned to random fluctuation around an equilibrium value. The average noise in the frequency signal in the equilibrium state was 0.323 Hz. There was no difference in noise between sensors coated with non-degradable or degradable nanogels (0.324 and 0.321 Hz, respectively). The average swelling transition lasted 43 ± 8 s, before reaching a new equilibrium condition. It is worth noting that this methodology overestimates the duration of a swelling transition, as the QCM-D flow module exchanges buffer by diluting and/or displacing the previous medium (15 μL over the sensor). Therefore, a 43 s buffer infusion represents approximately a 1 to 11 dilution of the previous buffer into the new buffer, assuming that the sample buffer is continuously and uniformly diluted with running buffer. No significant differences were observed in the swelling behavior of non-degradable and degradable nanogels [Figure S2(a,b); Supporting Information].
Figure 3.
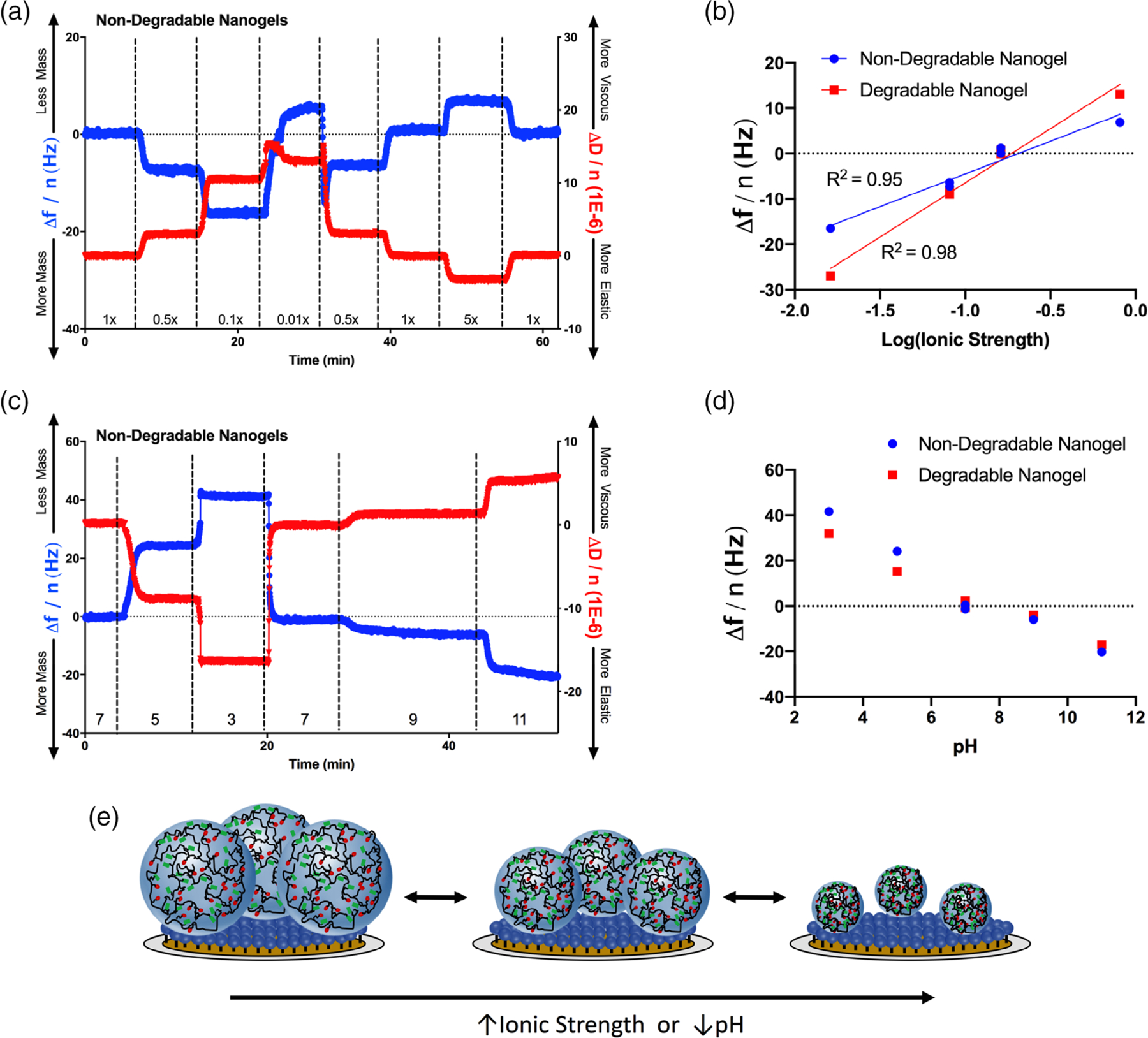
Nanogel swelling. (a) Non-degradable poly(acrylamide-co-methacrylic acid) nanogels were swollen or collapsed in a series of phosphate buffers of varying ionic strength. The frequency and dissipation of nanogel-conjugated quartz sensors was monitored. Reduced resonance frequencies corresponded with increases in dissipation, consistent with the expectation that nanogel swelling increases both the absorbed mass and the viscosity of the nanogel coating. The fold dilution of PBS (1× = 0.162 M) is given along the x axis. Corresponding data for the degradable nanogels are given in Figure S2 in Supporting Information. (b) From 0.1 to 5× PBS, nanogel swelling (degradable and non-degradable) scaled approximately linearly with the log of ionic strength. (c) Non-degradable nanogels were also pH-responsive, swelling in basic buffers and collapsing in acidic buffers. This swelling/hysteresis behavior is explained by protonation of the methacrylic acid moieties in the linear polymer chains. The buffer (1× PBS) pH is given along the x axis. Corresponding data for the degradable nanogels are given in Figure S2 in Supporting Information. (d) Degradable and non-degradable nanogels exhibited similar pH-responsive swelling behavior. (e) Schematic depiction of nanogel swelling, when coupled to a planar gold QCM sensor.
We plotted the change in frequency (relative to nanogels in 1× PBS) of degradable and non-degradable nanogels in response to the ionic strength. These data are presented in Figure 3(b), with the ionic strength given on a log scale. With the 0.01× PBS swelling measurement omitted, the data approximately fit the function:
where n is the overtone number, C is a constant (17.7 ng cm−2 Hz−1), and I is the solution ionic strength. We believe that in 0.01× PBS, the leaching of adsorbed ions from the nanogel-sensor layer into the buffer leads to behavior which outside of the above trend. This hypothesis was supported by that, in 0.01× PBS, the sensor frequency and dissipation both increased. This combination indicated that the 0.01× condition promoted a simultaneous decrease in adsorbed mass and increase in viscosity, consistent with the combination of nanogel swelling and adsorbed ion release.
We also measured nanogel swelling in response to the pH environment. It is worth noting that the experiment was conducted in 1× PBS adjusted the various pH values. Therefore, the solution ionic strength varied simultaneously as the ionization state of the phosphate ions within the buffer changed. The kinetic frequency and dissipation changes, as a function of buffer and time, for non-degradable nanogels are presented in Figure 3(c), and degradable nanogels in Figure S3(d) in Supporting Information. In the baseline state (pH = 7), the nanogels were swollen [pKa of poly(methacrylic acid)~4.8]. As the pH was decreased, the nanogels collapsed, as indicated by a decrease in adsorbed mass and increase in layer elasticity. As the pH was increased above 7, the adsorbed mass and layer viscosity both increased, consistent with a greater extent of swelling. The change in sensor resonance frequency for degradable and non-degradable nanogels, as a function of pH, is given in Figure 3(d).
Our results were consistent with the Brannon–Peppas theory for swelling of ionizable gels, which predicts that swelling is a function of the solution pH, ionizable monomer pKa, and ionic strength of the medium.26 Theoretical considerations and biomedical applications of the swelling of ionizable hydrogels have also been discussed extensively.27,28 Swelling increased as the extent of gel ionization increased (pH > pKa) and as the ionic strength of the medium decreased. Gel layer viscosity and sensor adsorbed mass both correlated with nanogel swelling, while increases in elasticity indicated hysteresis [Figure 3(e)].
Model Protein Adsorption
For the purpose of this study, lysozyme was selected as a model ligand. We knew from previous studies that lysozyme binds in significant quantity to poly(methacrylic acid-co-acrylamide) copolymers in aqueous buffer.29,30 We infused lysozyme dissolved in PBS at up to 5 mg/mL over the nanogel-coated and uncoated sensors. Significantly more lysozyme bound to the nanogel-coated sensors than the uncoated controls [Figure 4(a)], and no significant differences were observed between degradable and non-degradable nanogels. This suggested that, in this case, interaction of lysozyme with the components of the linear polymer within the nanogels drove protein-nanogel affinity, and that the two crosslinkers exhibited similar size exclusion properties. The majority of the bound lysozyme desorbed when running buffer (1× PBS) without protein was infused.
Figure 4.
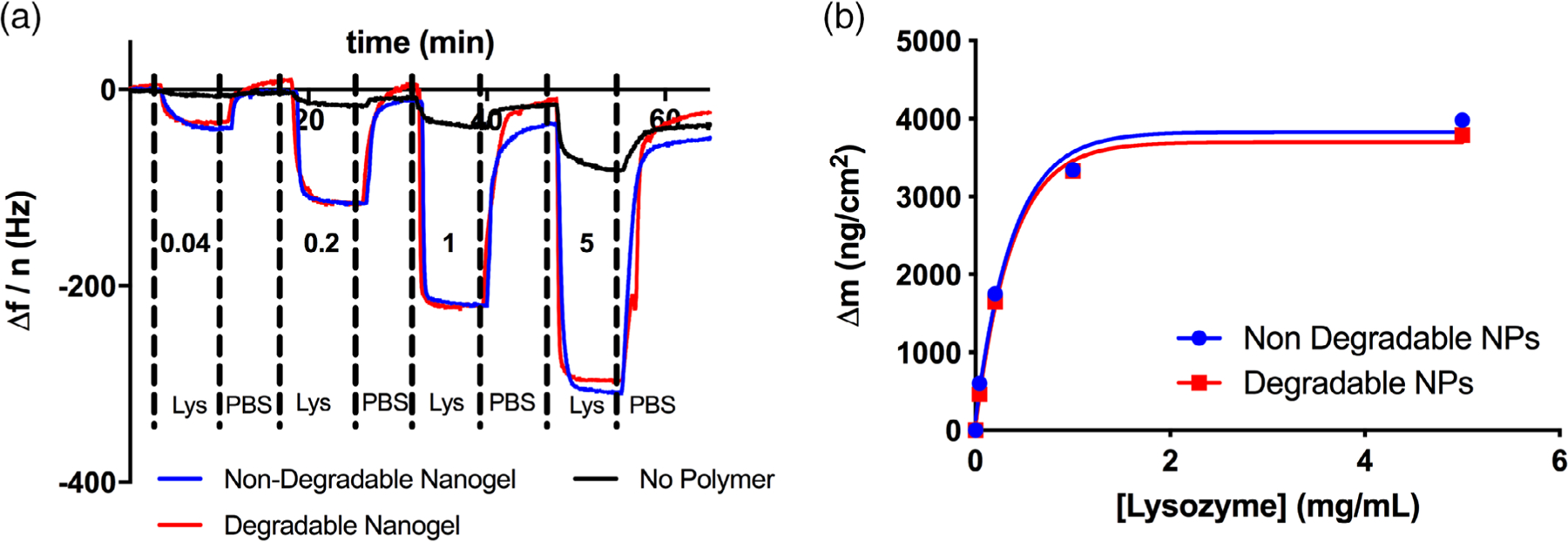
Lysozyme-nanogel affinity. (a) Raw data of lysozyme (Lys) adsorption to degradable or non-degradable poly(acrylamide-co-methacrylic acid) nanogels. The values written above/below each peak are the equilibrium lysozyme concentrations. (b) Corresponding plot of the lysozyme adsorption isotherm. No significant differences were observed in lysozyme adsorption between degradable and non-degradable nanogels. Substantial quantities of lysozyme bound to both formulations, exceeding the nanogel dry weight at equilibrium. Fit of a Langmuir isotherm estimated the dissociation constant of lysozyme for either formulation as 15.5 μM.
We calculated a Langmuir adsorption isotherm for ligand-polymer binding data.31 To calculate this isotherm, we converted each mass bound value to fractional occupancy:
where θ is the fractional occupancy. Then, we plotted the fractional occupancy as a function of concentration, and fit the Langmuir isotherm:
where Ka is the association constant and Ce is the equilibrium ligand (lysozyme) concentration. We converted Ka to the dissociation constant, Kd, as this is the value typically used to compare different extents of ligand-receptor affinity.
the Kd value for both the degradable and non-degradable nanogels was approximately 15.5 μM.
In future iterations of this method, it will be necessary to test the adsorption of more than one protein. To do this, we recommend testing the full range of concentration for the first protein, then ensuring complete extraction before moving on to the next protein. We recommend the following steps to extract protein from the nanogels, which were the most successful in our optimization studies. As a first step, flow running buffer (1× PBS, etc.). For proteins with low affinity for the nanogels, this will be sufficient to promote desorption. If pure buffer does not extract the bound protein, add solutes to the buffer that will compete with the protein-nanogel interaction. We recommend the addition of Tween-20 (hydrophobic interactions), glycine (compete with hydrogen bonds), or ethylenediaminetetraacetic acid (EDTA) (compete with anion-cation interaction). Acidic or basic buffers, such as dilute hydrochloric acid or sodium hydroxide, may be necessary to break ionic interactions between the protein and polymer.
If none of these buffers are sufficient to extract all of the bound protein, infusion of Trypsin–EDTA solution (0.25 wt %) will digest the absorbed protein. In the case of lysozyme adsorption to our degradable and nondegradable nanogels, this was the case, and a digestion step was necessary. Wash the sensor with plenty of running buffer to remove any bound trypsin, and establish a stable baseline reading before moving on to subsequent protein adsorption experiments.
Biodegradation
The novelty of our QCM-D method is the ability to quantify each of these biologically relevant characteristics (rheology, swelling, protein adsorption, and degradation) in a single experiment. Degradation is a terminal measurement, so one must ensure that no further experiments are necessary before initiating degradation steps. In our experiments, we measured the biodegradation kinetics of nanogels with a disulfide crosslinker in the presence of a reducing agent (DTT).
During degradation, an increase in resonance frequency indicates mass loss, and the change in dissipation provides information about the degradation mechanism. For surface eroding gels, the sensor will lose mass without exhibiting a substantial change in dissipation (i.e., neither more viscous nor elastic). On the other hand, bulk-eroding gels will increase in viscosity (i.e., due to loss of crosslinks and subsequent swelling). An increase in resonance frequency (i.e., mass loss) may or may not accompany this change in viscosity, as polymer loss is simultaneous with water uptake. Frequency and dissipation data for the non-degradable and degradable nanogels in the presence of a reducing agent (10 mM DTT in PBS, pH = 7.4) are given in Figure 5.
Figure 5.
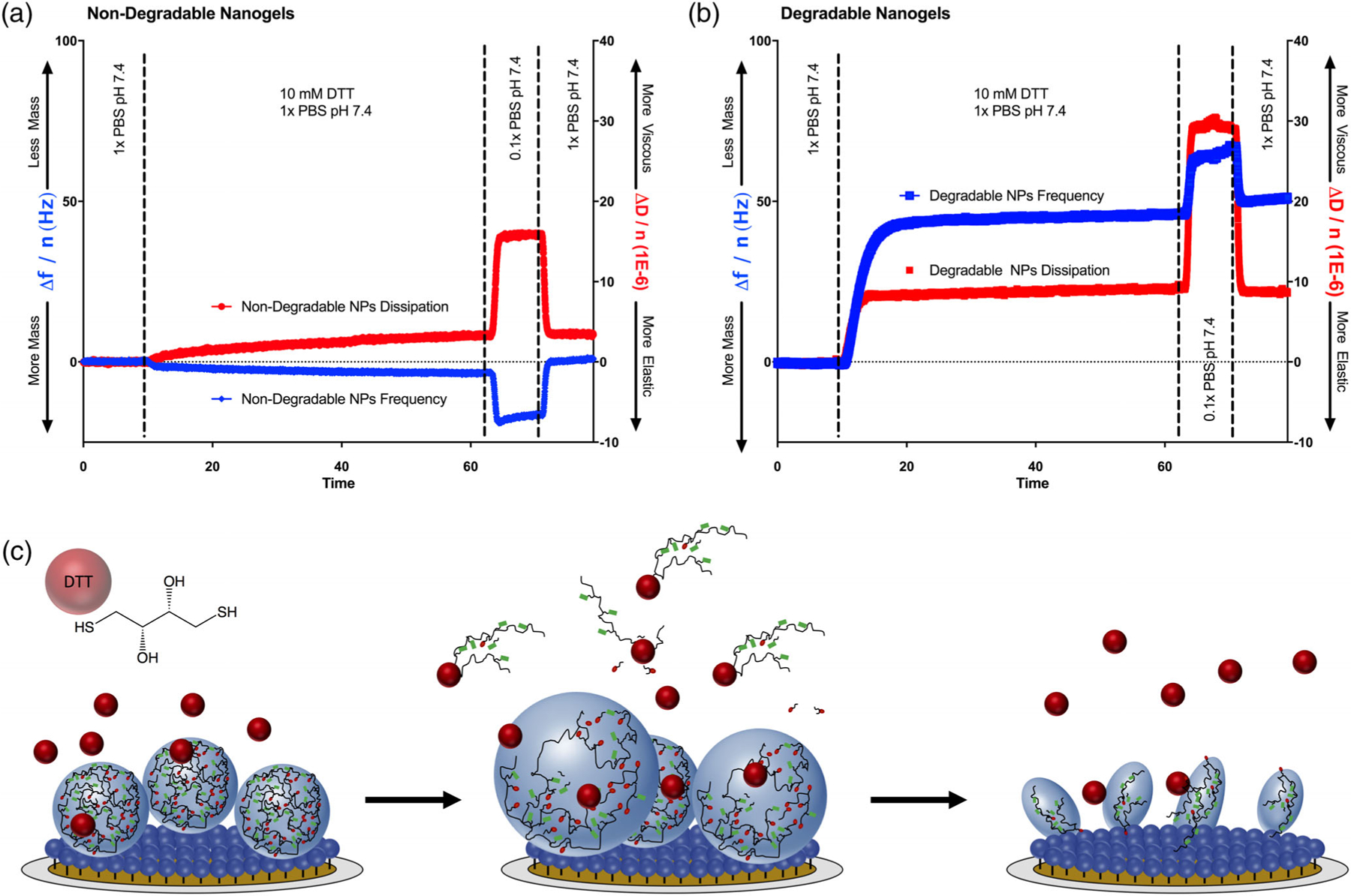
Nanogel degradation. (a) Non-degradable nanogels were subjected to reducing conditions (10 mM DTT). No evidence of degradation was observed. The nanogels instead swelled slightly, as indicated by a decrease in frequency and increase in dissipation. The nanogels swelled further in a subsequent 0.1x PBS infusion, indicating that intact nanogels were present. (b) The degradable nanogels rapidly lost mass and increased in viscosity when exposed to 10 mM DTT. This indicated that the degradable nanogels were decomposing by bulk degradation, and were simultaneously losing crosslinks and imbibing water. Upon exposure to 0.1x PBS, the sensor further lost mass and increased in viscosity, consistent with the behavior of a completely degraded polymer brush. (c) Schematic depiction of nanogel degradation upon exposure to DTT. First, DTT competes with disulfide crosslinks and leads to simultaneous leaching of linear / branched polymer and water uptake. Once degradation is complete, a brush of covalently bound linear polymer remains.
Non degradable nanogels [Figure 5(a)] increased in adsorbed mass slightly (Δf = −4.15 Hz, Δm = 73 ng/cm2) while also exhibiting a slight increase in dissipation (ΔD = 3.2 × 10−6), indicating that the nanogels swelled slightly in the reducing medium. No mass loss was observed following 40 min exposure to 10 mM DTT. On the other hand, the degradable nanogels lost mass rapidly in 10 mM DTT, which was accompanied with an increase in dissipation, consistent with a bulk degradation mechanism. The majority of the nanogel degradation was observed within the first 3 min, with complete degradation observed in 15 min. Following 40 min exposure to 10 mM DTT, the total mass loss was 832 ng/cm2, which was a 32% reduction, relative to the initial bound mass. As shown in Figure 5(c), this mass remainder is because many of the linear poly(acrylamide-co-methacrylic acid) chains remain covalently immobilized following degradation of the crosslinks. When each polymer-coated sensor was transitioned from 1 to 0.1× PBS, the non-degradable nanogels imbibed water but the degraded nanogels lost mass. This suggested that the non-degradable nanogels were still acting as an environmentally responsive nanogel, whereas the degraded nanogels were acting as a polymer brush.
Protocol Validation and Discussion
Nanogel swelling results were consistent with our pH-responsive swelling data (via DLS) shown in Figure S1 in Supporting Information. The pH-responsive swelling transition was sharper in DLS measurements, but the observed pH critical point was similar via both DLS and QCM. Previous studies looking at lysozyme adsorption to poly(acrylamide-co-methacrylic acid-co-diethylaminoethyl methacrylate) microgels estimated a dissociation constant of 0.80 ± 0.18 μM.29 Our measured dissociation constant from QCM-D for similar poly(acrylamide-co-methacrylic acid) nanogels was 15.5 μM, which is of a similar order of magnitude. Further, it is reasonable that additional electrostatic and hydrophobic interactions between lysozyme and the terpolymer synthesized by Culver et al.,29 as opposed to the copolymer in the present work, would lead to an increase in affinity.
Our degradation kinetics via QCM was more rapid than those measured by DLS (Figure S3 in Supporting Information). The nanogels degraded to 50% of the initial count rate in 6.75 min (DLS), as compared to 50% of the total mass loss (QCM) in 2.25 min. Complete degradation was achieved in approximately 30 min (DLS) as opposed to 15 min (QCM). However, this was be expected, as the DLS experiment was static (i.e., no fluid flow, no mixing, and no exchange of fresh buffer) whereas in QCM, the flow module was continually flushed with fresh buffer at 0.2 mL/min. Further, in the DLS experiment, linear polymers were retained in the cuvette, whereas in QCM the released linear and branched fragments were continually removed.
Finally, it is worth noting that we were able to directly apply the Sauerbrey equation in our analyses because of the thickness (80 nm) and relative homogeneity of our nanogel layer. However, for larger species, such as hydrogel microparticles, these assumptions would not be valid. In order to properly study these thick, viscoelastic films with QCM-D,25,32 one would need to apply a continuum mechanics model that includes viscous and elastic contributions, such as the Voigt model.33
In summary, the QCM-D method reveals both the magnitude and kinetics of physical gel transitions, association/dissociation of ligands, and decomposition of the nanogel layer. The results collected are of similar magnitude to other widely accepted techniques, but QCM-D has the added benefit of being able to conduct the diverse physical tests in series, within a single experiment. Therefore, it is a useful platform for comparing a limited number of formulations (as shown here with degradable and non-degradable nanogels of similar composition) with a high degree of precision and biological rigor.
CONCLUSIONS
In this work, we developed a new QCM-D assay for measuring nanogel swelling, adsorption of ligands, and degradation, in a single experiment. By analyzing the modulation in sensor resonance frequency and dissipation, we elucidated the extents and mechanisms of the pH- and ionic strength-responsiveness, lysozyme affinity, and biodegradation kinetics of poly(acrylamide-co- methacrylic acid) nanogels. Further, by detailing our practical guidelines and lessons learned, we sought to facilitate the future development of related QCM-D assays for quantifying the biologically relevant characteristics of other soft nanomaterials.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from the UT-Portugal Collaborative Research Program (CoLAB), the Cockrell Family Chair Foundation, and the National Institutes of Health (NIH) (EB022025 to NAP). J.R.C. was supported by an NSF Graduate Research Fellowship (DGE-1610403). C.M.L. was supported by an undergraduate research fellowship from the UT Austin Office of Undergraduate Research.
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Koetting MC; Peppas NA Int. J. Pharm 2014, 471, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steichen S; O’Connor C; Peppas NA Macromol. Biosci 2017, 17, 1600266. [DOI] [PubMed] [Google Scholar]
- 3.Marek SR; Peppas NA AIChE J 2013, 59, 3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forbes DC; Peppas NA ACS Nano 2014, 8, 2908. [DOI] [PubMed] [Google Scholar]
- 5.Liechty WB; Scheuerle RL; Peppas NA Polymer 2013, 54, 3784. [Google Scholar]
- 6.Liechty WB; Scheuerle RL; Vela Ramirez JE; Peppas NA Bioeng. Transl. Med 2019, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marx KA Biomacromolecules 2003, 4, 1099. [DOI] [PubMed] [Google Scholar]
- 8.Sadman K; Wiener CG; Weiss RA; White CC; Shull KR; Vogt BD Anal. Chem 2018, 90, 4079. [DOI] [PubMed] [Google Scholar]
- 9.O’Neal JT; Dai EY; Zhang Y; Clark KB; Wilcox KG; George IM; Ramasamy NE; Enriquez D; Batys P; Sammalkorpi M; Lutkenhaus JL Langmuir 2018, 34, 999. [DOI] [PubMed] [Google Scholar]
- 10.Silva JM; García JR; Reis RL; García AJ; Mano JF Acta Biomater 2017, 51, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagasawa D; Azuma T; Noguchi H; Uosaki K; Takai MJ Phys. Chem. C 2015, 119, 17193. [Google Scholar]
- 12.Siow KS; Britcher L; Kumar S; Griesser HJ Colloids Surf. B Biointerfaces 2019, 173, 447. [DOI] [PubMed] [Google Scholar]
- 13.Contreras AE; Steiner Z; Miao J; Kasher R; Li Q Environ. Sci. Technol 2011, 45, 6309. [DOI] [PubMed] [Google Scholar]
- 14.Glasmästar K; Larsson C; Höök F; Kasemo BJ Colloid Interface Sci 2002, 246, 40. [DOI] [PubMed] [Google Scholar]
- 15.Hu M; Zheng S; Mi B Environ. Sci. Technol 2016, 50, 685. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S; Qiu G; Ting YP; Chung T-S Colloids Surf. A Physicochem. Eng. Asp 2013, 436, 207. [Google Scholar]
- 17.Goor OJGM; Brouns JEP; Dankers PYW Polym. Chem 2017, 8, 5228. [Google Scholar]
- 18.EL-Sharif HF; Aizawa H; Reddy SM Sens. Actuators B 2015, 206, 239. [Google Scholar]
- 19.Hussain M; Northoff H; Gehring FK Biosens. Bioelectron 2015, 66, 579. [DOI] [PubMed] [Google Scholar]
- 20.Yilmaz E; Majidi D; Ozgur E; Denizli A Sens. Actuators B 2015, 209, 714. [Google Scholar]
- 21.Zhong JX; Clegg JR; Ander EW; Peppas NA J. Biomed. Mater. Res. A 2018, 106, 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clegg JR; Irani AS; Ander EW; Ludolph CM; Venkataraman AK; Zhong JX; Peppas NA Sci. Adv 2019, 5, eaax7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denison DJ Vac. Sci. Technol 1973, 10, 126. [Google Scholar]
- 24.Behrndt KH J. Vac. Sci. Technol 1971, 8, 622. [Google Scholar]
- 25.Dunér G; Thormann E; Dėdinaitė AJ Colloid Interface Sci 2013, 408, 229. [DOI] [PubMed] [Google Scholar]
- 26.Brannon-Peppas L; Peppas NA Chem. Eng. Sci 1991, 46, 715. [Google Scholar]
- 27.Slaughter BV; Khurshid SS; Fisher OZ; Khademhosseini A; Peppas NA Adv. Mater 2009, 21, 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koetting MC; Peters JT; Steichen SD; Peppas NA Mater. Sci. Eng. R Rep 2015, 93, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culver HR; Steichen SD; Peppas NA Biomacromolecules 2016, 17, 4045. [DOI] [PubMed] [Google Scholar]
- 30.Clegg JR; Zhong JX; Irani AS; Gu J; Spencer DS; Peppas NJ Biomed. Mater. Res. A 2017, 105, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latour RA J. Biomed. Mater. Res. A 2015, 103, 949. [DOI] [PubMed] [Google Scholar]
- 32.Malmström J; Agheli H; Kingshott P; Sutherland DS Langmuir 2007, 23, 9760. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Z; Ji X; Dimova R; Lipowsky R; Liu Y Macromolecules 2015, 48, 1824. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.