

Abstract
Immune evasion is a hallmark of cancer, and therapies that restore immune surveillance have proven highly effective in cancers with high tumor mutation burden (TMB) (e.g., those with microsatellite instability (MSI)). Whether low TMB cancers, which are largely refractory to immunotherapy, harbor potentially immunogenic neoantigens remains unclear. Here, we show that tumors from all patients with microsatellite stable (MSS) colorectal cancer (CRC) express clonal predicted neoantigens despite low TMB. Unexpectedly, these neoantigens are broadly expressed at lower levels compared to those in MSI CRC. Using a versatile platform for modulating neoantigen expression in CRC organoids and transplantation into the distal colon of mice, we show that low expression precludes productive cross priming and drives immediate T cell dysfunction. Strikingly, experimental or therapeutic rescue of priming rendered T cells capable of controlling tumors with low neoantigen expression. These findings underscore a critical role of neoantigen expression level in immune evasion and therapy response.
Approximately 12% of CRC has defects in DNA mismatch repair (MMR) resulting in MSI1, with a high burden of mutation-derived tumor-specific antigens (neoantigens) that underlies favorable response to immune checkpoint blockade (ICB)2–4. The remaining majority of CRC is MSS with lower TMB. However, MSS CRC has on average more mutations than some cancers that respond favorably to ICB1,5. Presentation of neoantigen-derived epitopes (neoepitopes) on Human Leukocyte Antigen class I (HLA-I) has also been observed in a small study of MSS CRC6. This suggests that other factors, both tumor intrinsic and microenvironmental, likely contribute to the poor immunogenicity of these and other immune “cold” cancers. Indeed, the intestinal microenvironment is tolerant to commensal bacteria and food-derived antigens, and these mechanisms may be co-opted by tumors to undermine immune responses7,8. In addition, the vast majority of CRC is associated with aberrant WNT/β-catenin signaling1, which can promote exclusion of dendritic cells and failure to prime productive T cell responses in melanoma and liver cancer9,10. To rigorously study processes underlying T cell dysfunction and immunotherapy resistance, it is critical that models faithfully recapitulate the tissue microenvironment and genetics of the human disease. Models should also enable isolation of defined antigen-specific T cells. To our knowledge, no single model of CRC meets all these criteria. Therefore, we adapted a technique employing endoscope-guided submucosal injection11,12 to induce genetically-defined tumors in the mouse colon harboring model CD8+ T cell antigens.
An additional feature we sought to model is the role of neoantigen expression level in modulating the anti-tumor immune response. While mutations in cancer are generally enriched in poorly expressed genes due to reduced transcription-coupled repair13, this pattern is absent in MMR-deficient tumors14. This raises the possibility that poor immunogenicity of MSS CRC and other MMR-proficient cancers is not only due to lower burden but also lower expression of neoantigens. It is appreciated that antigen expression is a central determinant of the magnitude of T cell response in viral infection15, and low neoantigen expression or affinity for MHC-I results in tumor immune evasion in flank transplant models16,17. Clinical studies have also shown that tumors frequently evade immune recognition via loss of heterozygosity of HLA alleles and dysregulation of antigen processing and presentation18. Despite these results, there remains some controversy surrounding the role of initial T cell receptor (TCR) signaling strength in shaping T cell fate and function19. In cancer specifically, the impact of low neoantigen expression on T cell dysfunction is poorly characterized.
Results
MSS CRC has lower burden and expression of neoantigens.
To guide development of a mouse model of CRC enabling tracking of tumor-specific T cell responses, we first developed a neoantigen prediction pipeline integrating HLA haplotype calling20 and affinity prediction algorithms21–25 and applied it to whole-exome and RNA sequencing data from The Cancer Genome Atlas (TCGA) colorectal adenocarcinoma (COADREAD) cohort. Tumors from all MSS CRC patients (excluding rare cases with hypermutation) expressed at least 14 (median 121) single nucleotide variant (SNV)- or insertion/deletion (indel)-derived neoantigens with predicted affinity to their respective HLA-I of IC50 ≤ 500 nM, despite a lower TMB compared to MSI high (MSI-H) CRC patients (Fig. 1a–b, Extended Data Fig. 1a). Interestingly, average expression of genes encoding predicted neoantigens was also significantly lower in MSS versus MSI-H tumors (Fig 1b, Extended Data Fig. 1b), consistent with more mutations in highly expressed genes in MMR-deficient tumors14.
Figure 1.
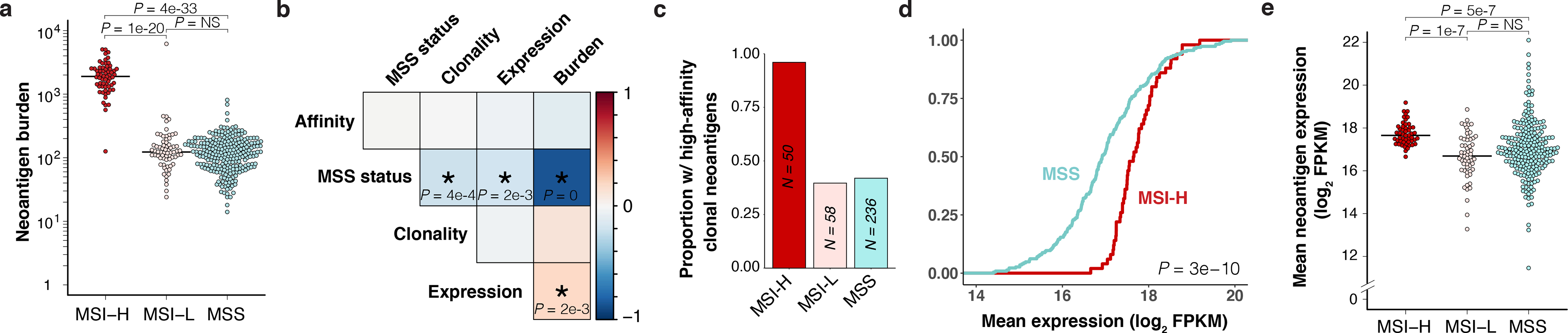
MSS CRC harbors both lower burden and expression of predicted neoantigens. Analysis of predicted neoantigens in human CRC (TCGA COADREAD) with high MSI (MSI-H), low MSI (MSI-L), and MSS. (a) Total expressed neoantigens with strong predicted HLA-I binding (IC50 ≤ 500 nM) by patient. N = 62 MSI-H, 68 MSI-L, and 266 non-hypermutant MSS patients. (b-e) Analysis of patients with available ABSOLUTE purity for estimation of clonality (adjVAF). N = 50 MSI-H, 58 MSI-L, and 236 non-hypermutant MSS patients. (b) Spearman rank correlation matrix of MSS status (MSS versus MSI-H) and mean neoantigen expression, predicted affinity, burden, and clonality by patient. Strength of correlation is represented by color scale (red = positive, blue = negative), and significance is indicated by asterisk with P-values displayed. (c) Proportion of patients expressing at least one clonal (adjVAF ≥ 0.5) neoantigen with very strong predicted binding affinity (IC50 ≤ 10 nM). (d) Empirical cumulative distribution function of mean neoantigen expression by patient, showing enrichment of lower expression in MSS patients. Significance assessed by two-sided Kolmogorov-Smirnov test. (e) Mean expression of clonal neoantigens by patient (FPKM, upper quartile-normalized). Significance in (a) and (e) was assessed by two-tailed Wilcoxon Rank Sum test with Holm’s correction for multiple comparisons.
We also observed significantly lower average predicted neoantigen clonality (ABSOLUTE26 tumor purity-adjusted variant allele frequency (adjVAF) ≥ 0.5, see Methods) in MSS tumors (Fig. 1b, Extended Data Fig. 1c), which may contribute to lower expression. Given this and the fact that clonal neoantigens are more likely to elicit productive immune responses following immunotherapy27,28, we repeated our analysis focusing only on clonal predicted neoantigens. Surprisingly, all tumors from MSS patients still expressed at least two clonal predicted neoantigens (median 42) (Extended Data Fig. 1d), and 42% of these tumors expressed at least one clonal neoantigen with predicted affinity as high or higher than the commonly used model CD8+ T cell antigen SIINFEKL (H-2Kb IC50 ≤ 10 nM)29 (Fig. 1c). Additionally, average expression of genes encoding clonal predicted neoantigens was still significantly lower in tumors from MSS versus MSI-H patients (Fig. 1d–e). Allele-specific expression of all SNV-derived clonal neoantigens, while limited by sparse coverage, recapitulated these results (Extended Data Fig. 1e).
Finally, a published immunopeptidomics study of human MSS CRC identified three clonal HLA-I neoepitopes in two of five patient-derived organoids (PDOs) analyzed by mass spectrometry (MS)6. While this study is small and the number of neoepitopes validated is substantially lower than predicted, the results suggest a lower limit of ~40% of MSS CRC presenting bona fide neoepitopes. Our re-analysis of this dataset located the neoepitopes in the 6th, 31st, and 45th percentiles of abundance of all detected HLA-I epitopes (Extended Data Fig. 1f–g). While peptide-specific properties can influence the efficiency of separation and ionization in MS, this qualitative analysis suggests lower surface abundance than most self-epitopes. Given that the detection limit of MS in these types of experiments is poorly defined and the recovery rate between replicates limited30, it is highly likely that a number of bona fide neoepitopes were not recovered, particularly those of lowest abundance. Indeed, some neoepitopes that went undetected by MS have been shown to be presented by tumor cells via immunogenicity assays31. Altogether, our integrated analysis of the TCGA and literature argue that many MSS CRC patients may harbor therapeutically actionable neoantigens. However, it remains poorly understood how low expression of neoantigens shapes resulting T cell responses in MSS CRC.
Neoantigen expression level is a critical determinant of immunity.
We first developed an autochthonous model in Apcflox/flox mice initiated by lentivirus expressing Cre-recombinase and the chicken ovalbumin antigen linked to luciferase (LucOS) (Extended Data Fig. 2a), as we have previously done in models of lung cancer32 and soft-tissue sarcoma33. Injection with LucOS dramatically reduced tumor incidence in a T cell-mediated manner, and tumors that did arise invariably lost antigen expression (Extended Data Fig. 2b–e). To assess effects of antigen expression in established tumors, T cells were continuously depleted for 5 weeks, at which point tumors retained antigen expression. However, 7 weeks after withdrawal of depleting antibodies, tumors had grown and lost antigen expression (Extended Data Fig. 2f–g). Given this potent immune editing and variability of antigen expression (Extended Data Fig. 2h), we developed a colon organoid model that maintains distinct levels of antigen expression throughout tumorigenesis. Organoid engineering also enables the use of a Kras mutant allele to model metastatic colon adenocarcinoma, which is confounded in the autochthonous model by concomitant Kras-driven fibrosarcoma formation (unpublished observations).
To enforce stable and continuous expression of antigen, we generated CRC organoids with SIINFEKL directly linked to Apc knockdown, an essential transforming event. Specifically, we transformed normal colon organoids from C57Bl/6 KrasLSL-G12D; Trp53flox/flox (KP) mice with adenoviral Cre, followed by lentivirus expressing miR-30 shRNA against Apc (shApc)34 and SIINFEKL fused to the fluorophore mScarlet (mScarletSIIN) (Fig. 2a). Given that relief from Apc knockdown in shApc-transformed tumors results in regression34, dependence on shApc provides powerful selection against antigen loss (Extended Data Fig. 2i). Finally, deletion of Smad4, commonly mutated in CRC35, was achieved by CRISPR/Cas9 editing. Selection of organoids harboring complete mutation of all genes was performed following published protocols36,37. This resulted in isogenic quadruple-mutant (shAKPS) organoids modeling some of the most common genetic mutations in MSS CRC35 (Extended Data Fig. 2j), which are co-mutated with high frequency in metastatic disease and associated with poor prognosis38. To investigate the importance of neoantigen expression level, we generated organoids with 400-fold range of mScarletSIIN expression via modifications to the shApc-expressing lentivirus, including placement in reverse orientation to the promoter (EF1α initiates bidirectional transcription), removal of the Kozak consensus sequence, and replacing 25% of codons with rare variants (Fig. 2a–b, Extended Data Fig. 2k). This flexible system is broadly applicable to other cancers via linkage to relevant essential events (e.g., knockdown of Trp53) and is easily adapted to the study of other immune epitopes.
Figure 2.
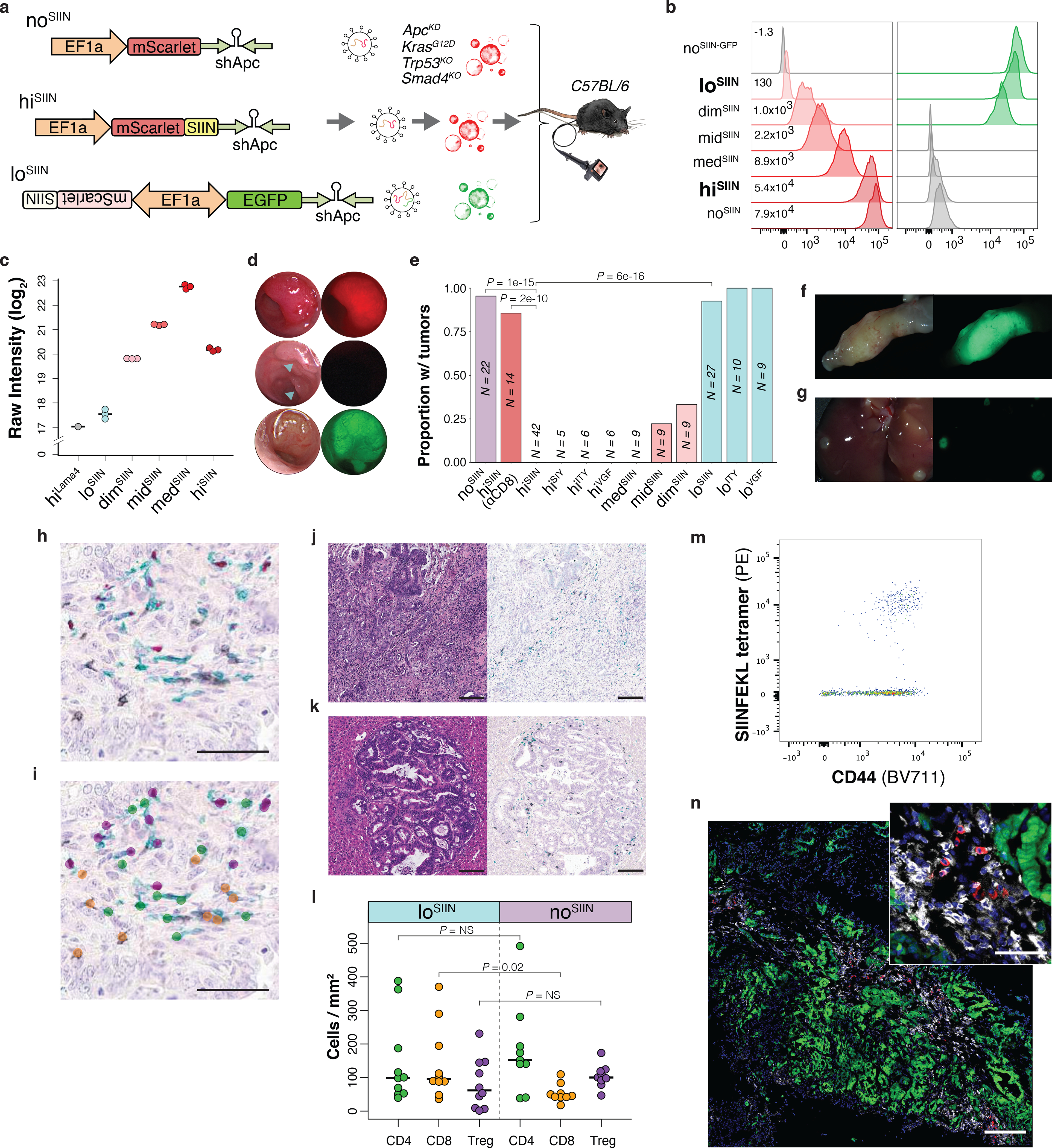
Development of an organoid system to interrogate neoantigen expression level in CRC. (a) shApc-expressing lentiviruses used to transform KP organoids, with no (noSIIN), high (hiSIIN) and low (loSIIN) expression of SIINFEKL. Resulting shAKPS organoids were orthotopically-transplanted into the colons of syngeneic mice. (b) Expression of mScarlet/mScarletSIIN and EGFP in expression variant organoids by flow cytometry. Experiment was performed three times with consistent results. (c) TMT-MS quantification of MHC-I bound SIINFEKL across three independent preparations of each line. (d) Colonoscopy images of noSIIN tumor (RFP channel), hiSIIN scars (RFP channel, blue arrows indicate injection sites), and loSIIN tumor (GFP channel) 28 days post-transplant. Representative of animals in (e). (e) Efficiency of tumor formation 42 days post-transplant with the lines indicated. αCD8 = continuous antibody depletion of CD8+ T cells. Significance assessed by 2×2 Fisher’s exact test with Holm’s correction for multiple comparisons. N = independent animals. (f-g) Stereoscopic brightfield and fluorescent images of loSIIN colon tumor (f) and liver metastases (g) 42 days post-transplant. Representative of animals transplanted in (e). (h-i) Three color IHC (black = CD8, green = CD4, red = FOXP3) (h) and automated annotation by convolutional neural network (i). Scale bars = 50 μM. (j-k) H&E and three color IHC of loSIIN primary colon tumor (j) and liver metastasis (k) 42 days post-transplant. Scale bars = 100 μM. Representative of animals in (l). (l) Quantification of CD8, CD4, and regulatory T cells infiltrating loSIIN and noSIIN tumors by convoluted neural network analysis. Each point represents at least one tumor from a single animal. N = 10 loSIIN and 9 noSIIN transplanted animals. Significance assessed by two-tailed Wilcoxon Rank Sum with Holm’s correction for multiple comparisons. (m-n) Identification of SIINFEKL tetramer-specific CD8+ T cells infiltrating 42-day loSIIN tumors by flow cytometry (m) and immunofluorescence (n) with in situ tetramer staining (green = tumor, white = CD8, red = SIINFEKL tetramer, blue = DAPI). Scale bar = 200 μM in main image, and 50 μM in zoom inset. Representative of N = 10 independent animals.
To compare surface MHC-I presentation of SIINFEKL across the expression series of organoids quantitatively, we performed H-2Kb immunoprecipitation and multiplexed tandem mass tag mass spectrometry (TMT-MS) on eluted peptides. While loSIIN through medSIIN showed near perfect correlation between SIINFEKL abundance and mScarletSIIN protein expression, hiSIIN unexpectedly showed intermediate abundance (Fig. 2c, Extended Data Fig. 2l). Given that antigen expression in midSIIN and medSIIN was adjusted by altering translation efficiency, it is possible that proteasomal degradation of mScarletSIIN is enhanced in these lines, resulting in greater surface presentation. Alternatively, high expression of mScarletSIIN in hiSIIN may lead to insoluble protein aggregates that are sequestered from antigen processing machinery. Regardless, these results validate the flexibility of our system to modulate surface presentation levels of neoantigen. While the multiplexed format of TMT-MS precludes meaningful intrasample comparisons of the abundance of SIINFEKL versus other epitopes, SIINFEKL abundance in loSIIN was only 1.4-fold above background in hiVGF (which lacks SIINFEKL), consistent with a low level of surface presentation.
Endoscope-guided transplant of shAKPS organoids without antigen (noSIIN) efficiently induced tumors (Fig. 2d–e) and spontaneous metastases to liver and lung, with histology remarkably like human CRC (Extended Data Fig. 2m). In contrast, transplant of the highest expression variant (hiSIIN) resulted in CD8+ T cell-mediated rejection in all animals (Fig. 1h–i). While transplant of medSIIN also resulted in complete rejection, midSIIN and dimSIIN formed tumors with intermediate efficiency (Fig. 2e, Extended Data Fig. 2n–o), suggesting a non-discrete effect of neoantigen expression level in tumorigenesis. We also generated shAKPS organoids harboring different epitopes with high affinity for MHC-I, including SIYRYYGL (hiSIY), ITYTWTRL (hiITY), and VGFNFRTL (hiVGF) (Extended Data Fig. 2p). The latter two are mutant epitopes of Alg8 (A506T) and Lama4 (G1254V) that arose in a methylcholanthrene-induced mouse sarcoma, which were reported to be insufficient for tumor rejection but critical for ICB response in a syngeneic flank transplant model39. Here, high expression of all three epitopes resulted in tumor rejection (Fig. 2e), demonstrating that immunogenicity is not idiosyncratic to SIINFEKL, but a general feature associated with high expression of high-affinity epitopes. This also argues that the major genetic features of MSS CRC do not confer cell-autonomous resistance to T cell killing.
Strikingly, transplant of the lowest expression variant (loSIIN) induced tumors with similar efficiency, histology, and infiltration as noSIIN organoids (Fig. 2d–l). This was also true of organoids with low expression of ITYTWTRL (loITY) and VGFNFRTL (loVGF), which formed tumors with high efficiency (Fig. 2e, Extended Data Fig. 2p). CD8+ T cells were sparse and only modestly increased in loSIIN tumors, while helper and regulatory T cell infiltration was not significantly different (Fig. 2h–l, Extended Data Fig. 2m). This is characteristic of the immune “cold” landscape of MSS CRC in humans40. Importantly, immune escape in loSIIN, loITY, and loVGF tumors did not result from neoantigen ignorance, as advanced tumors were infiltrated by antigen-experienced (CD44+) and specific (H-2Kb tetramer+) CD8+ T cells (Fig. 2m–n, Extended Data Fig. 2q). Altogether, these results demonstrate that MSS tumors can harbor high affinity neoantigens despite poor T cell infiltration, and low neoantigen expression is an important mechanism of tumor immune evasion.
Low neoantigen expression drives early T cell dysfunction.
To investigate why loSIIN tumors escaped immune rejection, we first compared the kinetics of the antigen-specific T cell response in loSIIN versus hiSIIN lesions. Low neoantigen expression resulted in both delayed and lower magnitude response (Fig. 3a). Interestingly, this difference was far less pronounced in the caudal and iliac draining lymph nodes (DLNs) (Extended Data Fig. 3a), suggesting that early T cells in loSIIN animals are either impaired in their ability to traffic to or proliferate within the tumor. The latter is unlikely, however, as loSIIN and hiSIIN T cells within tumors and DLNs showed no difference in proliferation (Extended Data Fig. 3b). Alternatively, T cells arriving at the tumor may have undergone deletional tolerance41. A critical step in the early maturation of functional T cell responses is effector differentiation, characterized by production of cytokines and cytolytic granzymes, particularly Granzyme B (GZMB), and loss of progenitor potential. TCF1 is a marker of progenitor potential and is expressed in naïve, memory precursor and memory T cells42–44. Consistent with impaired effector differentiation, at 8 days significantly more antigen-specific T cells from loSIIN tumors and DLNs were TCF1+/GZMB-, and significantly fewer were TCF1-/GZMB+ (Fig. 3b, d–e, Extended Data Fig. 3c). This is unlikely a result of delayed kinetics, as the percentage of TCF1-/GZMB+ T cells in loSIIN tumors at peak response (14 days) remained significantly lower (Fig. 3c, f, Extended Data Fig. 3d). Unexpectedly, the percentage of antigen-specific T cells capable of secreting both TNFα and IFNγ effector cytokines was higher in loSIIN versus hiSIIN DLNs and not different in tumors at 8 days (Extended Data Fig. 3e). However, this cytokine-proficient population also showed higher TCF1 and lower GZMB (Extended Data Fig. 3f), suggesting similar lack of effector differentiation. Strikingly, the percentage of double-negative (TCF1-/GZMB-) T cells was greater in loSIIN versus hiSIIN animals at 8 days (Fig. 3b, g), and became even more pronounced by 14 days (Fig. 3c, h). Absence of TCF1 and GZMB implies lack of progenitor and effector functionality and indicates dysfunction. Indeed, by 14 days TCF1-/GZMB- antigen-specific T cells in loSIIN versus hiSIIN tumors showed higher expression of co-inhibitory receptors PD-1, TIM3, LAG3, and 2B4 (Extended Data Fig. 3g), and an increased fraction co-expressing three or all four (Fig. 3i, Extended Data Fig. 3h). To further interrogate functionality of the loSIIN T cell response, we performed an in vivo killing assay45 by transferring SIINFEKL-loaded “target” splenocytes into tumor-bearing mice. Targets were eliminated in the DLNs and spleen 6 hours post-transfer in hiSIIN, but not loSIIN, animals at 8 days (Fig. 3j–k, Extended Data Fig. 3i). Even at the peak of the loSIIN response (14 days), killing was incomplete (Fig. 3l–m, Extended Data Fig. 3j), with fewer targets killed per antigen-specific T cell (Fig. 3n). Finally, to assess the TCR repertoire breadth of antigen-specific T cells infiltrating early loSIIN and hiSIIN lesions, we performed TCRβ sequencing. Consistent with an attenuated response, antigen-specific T cells infiltrating loSIIN tumors were more clonal (Extended Data Fig. 3k–l). Altogether, these results demonstrate that low neoantigen expression drives an immediately dysfunctional T cell response with reduced magnitude, diversity, and per cell functionality.
Figure 3.
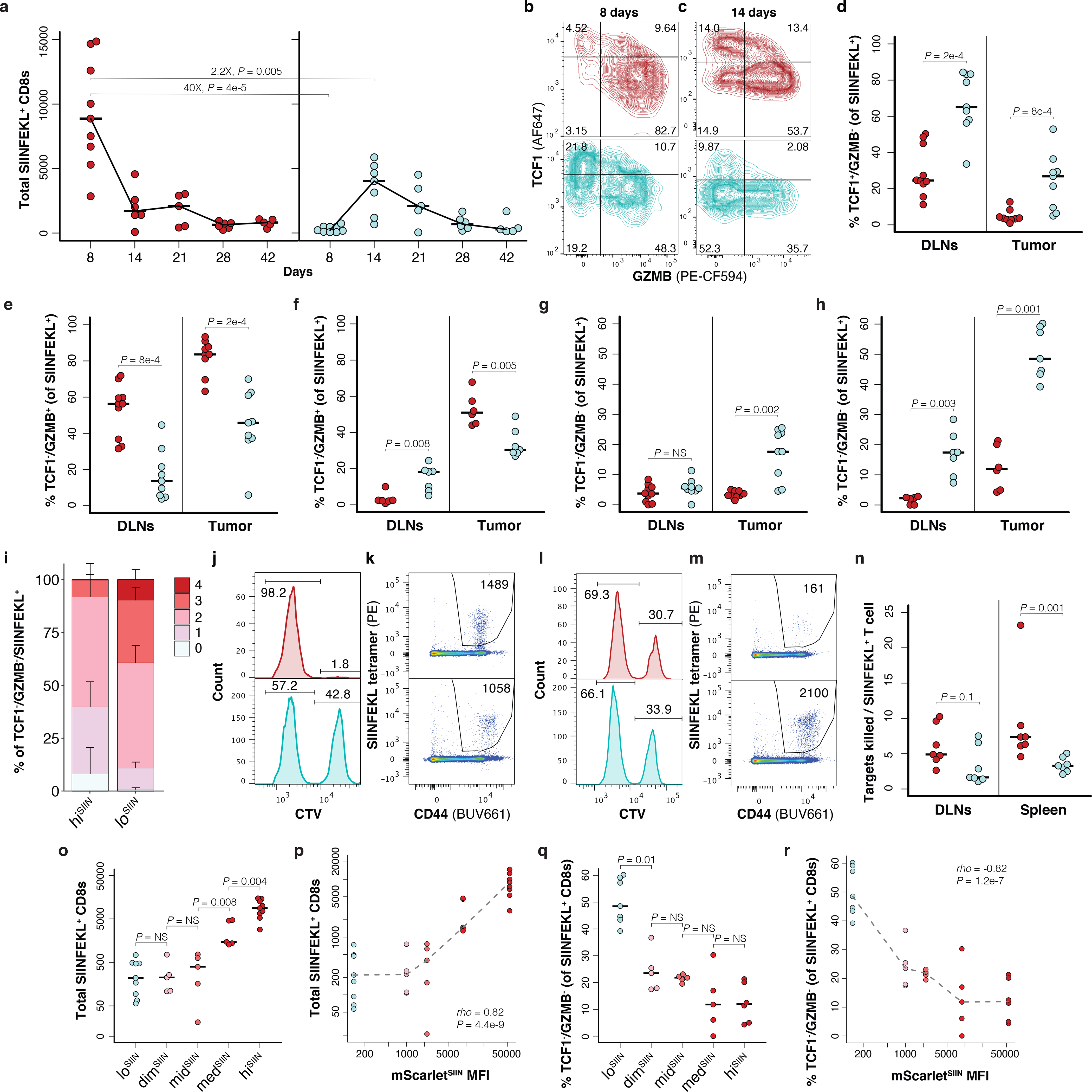
Low neoantigen expression drives impaired T cell effector commitment and dysfunction. (a) Total CD44+/CD8+ antigen-specific T cells isolated from lesions at indicated days post-transplant of hiSIIN (red) and loSIIN (blue) organoids by flow cytometry. N = 5–9 independent animals per line and timepoint. (b-c) Antigen-specific T cell expression of TCF1 versus GZMB in tumors at 8 (b) and 14 (c) days. Representative of animals in (a). (d-e, g) Percent of antigen-specific T cells from DLNs and tumors at 8 days positive for TCF1 and negative for GZMB (d), negative for TCF1 and positive for GZMB (e), and double-negative for TCF1 and GZMB (g). N = 10 hiSIIN and 9 loSIIN-transplanted animals. (f,h) Percent of antigen-specific T cells from DLNs and tumors at 14 days negative for TCF1 and positive for GZMB (f), and double-negative for TCF1 and GZMB (h). N = 6 hiSIIN and 7 loSIIN-transplanted animals. (i) Median percentage of TCF1-/GZMB- antigen-specific T cells from tumors at 14 days co-expressing 0 through 4 inhibitory receptors (PD-1, TIM3, LAG3, and 2B4). N = 6 hiSIIN and 7 loSIIN-transplanted mice. Bars = standard deviation. (j-m) In vivo killing assay of transferred control (weak CTV stain) and SIINFEKL-loaded “target” (strong CTV stain) splenocytes and flow plots of antigen-specific T cells recovered from DLNs at 8 (j-k) and 14 (l-m) days post-transplant of hiSIIN (red) and loSIIN (blue) organoids. Representative of N = 6–7 independent animals per line and timepoint. (n) Target killing normalized to total antigen-specific T cells recovered in 14-day killing assay. N = 7 independent animals per line. (o-p) Total antigen-specific T cells isolated from lesions at 8 days across all expression variant lines (o) and versus mScarletSIIN MFI (p). (q-r) Percent of TCF1-/GZMB- antigen-specific T cells from 14-day lesions across expression variants (q) and versus mScarletSIIN MFI (r). Dashed lines connect medians. Significance assessed by Spearman’s rank correlation. N = 5–9 independent animals per line in (o-r). Significance in (a), (d-h), (n-o), and (q) assessed by two-tailed Wilcoxon Rank Sum. Holm’s correction applied in (o) and (q).
To determine if these effects are mediated by discrete or continuous levels of neoantigen expression, we characterized the T cell responses to dimSIIN, midSIIN, and medSIIN organoids at 8 and 14 days. Consistent with the tumor efficiency data (Fig. 2e) and a continuous model, SIINFEKL expression correlated strongly and positively with the magnitude of the initial T cell response (8 days) (Fig. 3o–p). While no correlation was seen at 14 days (Extended Data Fig. 3m–n), likely due to T cell contraction in the intermediate expressing lines, SIINFEKL expression correlated strongly and negatively with the percentage of TCF1-/GZMB- antigen-specific T cells at this time point (Fig. 3q–r, Extended Data Fig. 3o–p). These results demonstrate a continuous positive effect of neoantigen expression level on the magnitude of the T cell response and effector differentiation.
Lowly expressed neoantigen-specific T cells undergo exhaustion.
T cell dysfunction in human cancer is often attributed to upregulation of co-inhibitory receptors, terminal differentiation and loss of effector function, or “exhaustion”46,47. More recently, it has been shown that terminally-differentiated T cells in cancer are characterized by low TCF1 and high TIM3 expression, and are regenerated by “progenitor exhausted” T cells that have high TCF1 and low TIM3 expression48–50. Consistent with progressive dysfunction, terminally differentiated T cells (TCF1-/TIM3+) made up a greater proportion of the response at 42 compared to 8 days in loSIIN tumors, but not in hiSIIN rejected lesions (Fig. 4a–b, Extended Data Fig. 4a). In contrast, progenitors (TCF1+/TIM3-) were substantially depleted in loSIIN tumors by 42 days (Fig. 4a–b, Extended Data Fig. 4b). Antigen-specific T cells in loSIIN tumors at 42 days also showed increased expression of PD-1, LAG3, and TIGIT (Fig. 4c). In loSIIN tumors, T cells negative for TCF1 and triple-positive for TIM3, PD-1, and LAG3 peaked at 42 days (Fig. 4d), consistent with progressive dysfunction. A similar trend was observed in TCF1+/TIM3- progenitor exhausted T cells double-positive for PD-1 and LAG3 (Fig. 4e). Finally, loSIIN tumors and DLNs at 42 days showed a reduced percentage of TNFα and IFNγ double-positive antigen-specific T cells (Extended Data Fig. 4c–d), indicating loss of effector functionality. Therefore, in addition to immediate dysfunction, T cells responding to low neoantigen expression undergo progressive exhaustion.
Figure 4.
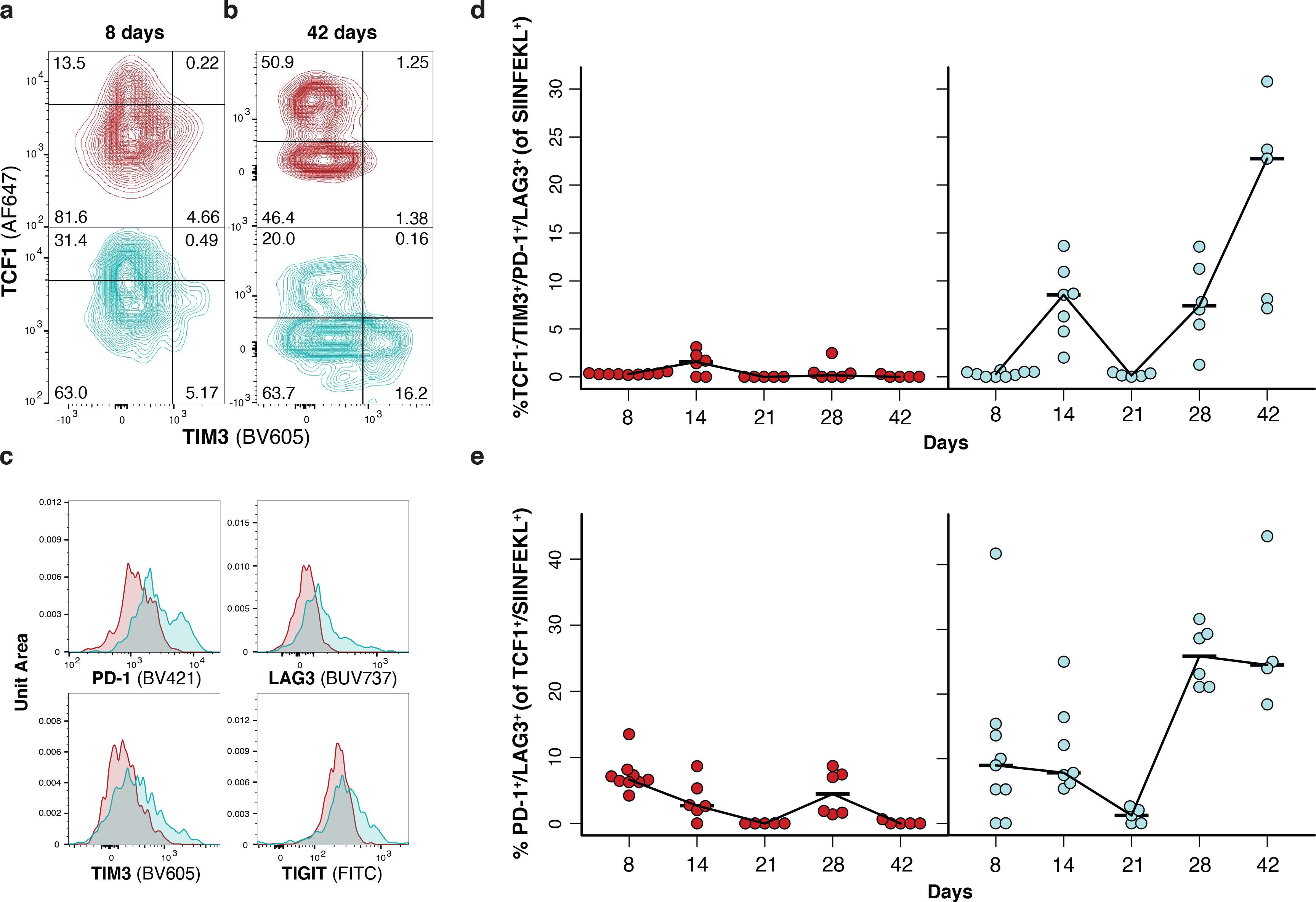
T cells in tumors with low neoantigen expression become progressively dysfunctional. (a-b) Antigen-specific T cell expression of TCF1 versus TIM3 in tumors at 8 (a) and 42 (b) days post-transplant, and (c) inhibitory receptor expression on antigen-specific T cells from tumors at 42 days post-transplant by flow cytometry. Representative of N = 9 hiSIIN and 9 loSIIN-transplanted animals at 8 days, and N = 5 hiSIIN and 5 loSIIN-transplanted animals at 42 days. (d-e) Percent TCF1-/TIM3+/PD-1+/LAG3+ (d) and TCF1+/PD-1+/LAG3+ (e) antigen-specific T cells isolated from tumors at 8-, 14-, 21-, 28-, and 42-days post-transplant. Red = hiSIIN, blue = loSIIN. N = 4–9 independent animals per line and timepoint.
Neoantigen expression level is limiting for cross priming.
Impaired effector differentiation and early dysfunction are indicative of poor priming, such as occurs in the absence of CD4+ T cell “help”51. While loSIIN and hiSIIN organoids lack a defined MHC-II-restricted model neoantigen, depletion of CD4+ T cells completely rescued formation of hiSIIN tumors (Fig. 5a). Therefore, absence of help is unlikely the mechanism of dysfunction in the loSIIN model. It is possible that CD4+ T cells are primed against uncharacterized neoantigens in mScarlet, tumor-associated self-antigens, or microbial antigens in the colon microenvironment. Consistent with the importance of neoantigen expression level in priming, hiSIIN tumor formation was partially rescued in Batf3−/− mice, which lack conventional cross-presenting dendritic cells (DC1s) (Fig. 5a). To directly test the role of neoantigen expression level in cross presentation, we co-cultured bone marrow-derived dendritic cells (BM-DCs)—comprised of ~13% CD103+ DC1s—with naïve TCR-transgenic T cells specific to SIINFEKL (OT-1) (Extended Data Fig. 5a–b). Compared to BM-DCs loaded with hiSIIN organoids, those loaded with loSIIN were markedly less capable of promoting OT-1 proliferation and effector differentiation (Fig. 5b–e, Extended Data Fig. 5c). On the other hand, in vitro-primed OT-1 cells were equally capable of killing loSIIN as hiSIIN organoids when co-cultured (Fig. 5f–g). These results argue that neoantigen expression is limiting for T cell cross priming, but not tumor cell recognition by effector T cells.
Figure 5.
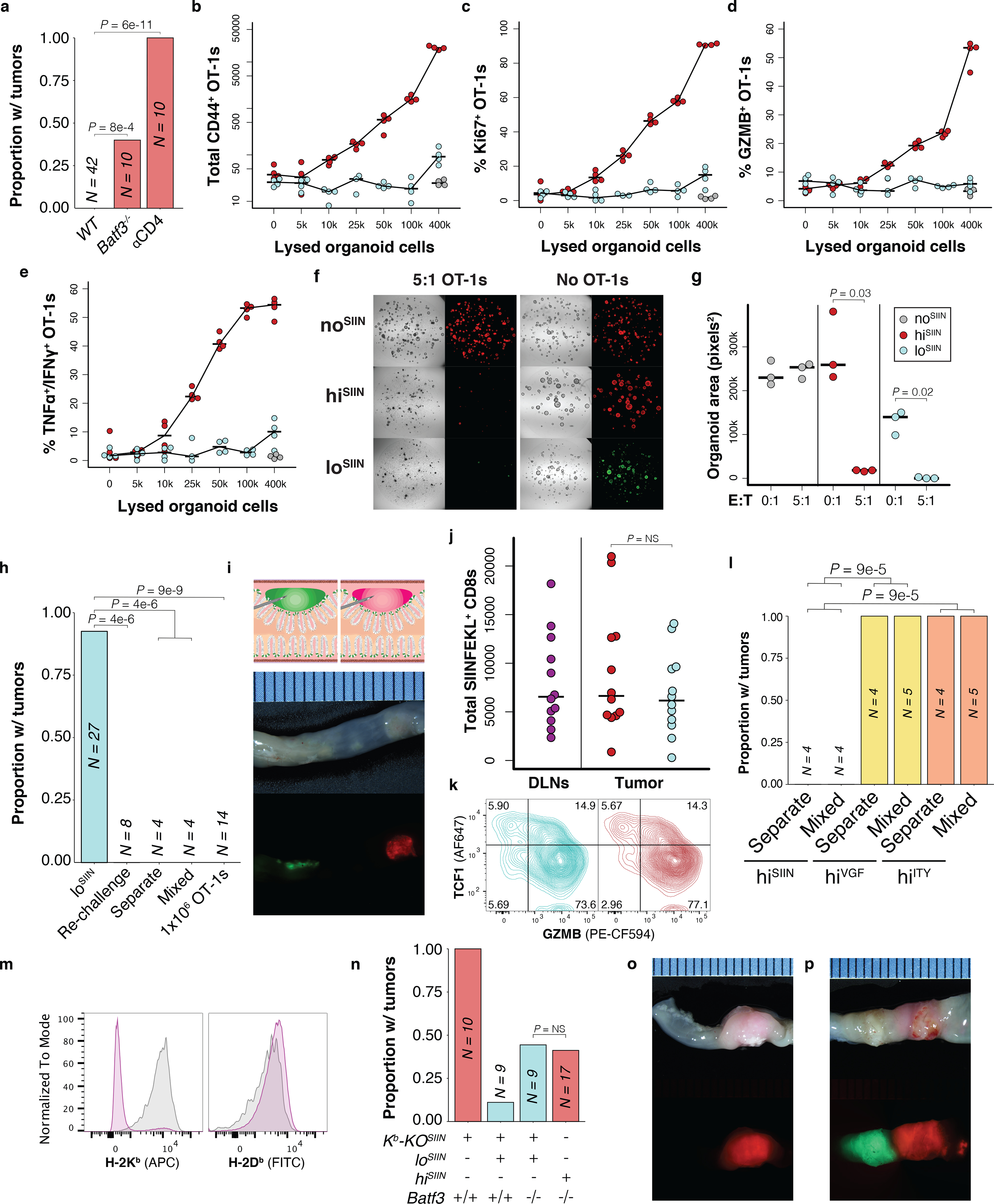
Low neoantigen expression limits T cell cross priming. (a) Efficiency of tumor formation 6 weeks post-transplant of hiSIIN organoids into WT, Batf3−/−, and WT mice with continuous anti-CD4 treatment. N = independent animals. (b-e) Cross priming of 10,000 naïve OT-1s co-cultured with 50,000 activated BM-DCs loaded with lysed noSIIN (grey), loSIIN (blue), and hiSIIN (red) cells. Flow cytometric quantification of CD44+, Ki67+, GZMB+, and TNFα+/IFNγ+ OT-1s. N = 4 co-cultures per line and condition. (f) Images of day 4 organoid and activated OT-1 co-cultures, representative of (g). (g) Quantification of organoid confluence. E:T = effector-to-target ratio. N = 3 co-cultures per line and condition. Significance assessed by two-tailed t-test. (h) Efficiency of tumor formation 6 weeks post-transplant of loSIIN, loSIIN 4 weeks post-transplant of hiSIIN (Re-challenge), loSIIN and hiSIIN at separate sites or mixed, and loSIIN concurrent with retro-orbital injection of activated OT-1s. N = independent animals. (i) Schematic of co-transplant of loSIIN (green) and hiSIIN (red) organoids (top), and stereoscopic images 8 days post-transplant (bottom) representative of N = 12 animals. (j-k) Flow cytometric analysis of antigen-specific T cells from DLNs and lesions 8 days post-co-transplant of hiSIIN (red) and loSIIN (blue) organoids at separate sites. N = 12 independent animals. (j) Total antigen-specific T cells. Significance assessed by two-tailed Wilcoxon Rank Sum. (k) Expression of GZMB and TCF1, representative of animals in (j). (l) Efficiency of tumor formation 6 weeks post co-transplant of loSIIN with indicated organoids mixed or at separate sites. N = independent animals. (m) Expression of H-2Kb/H-2Db on hiSIIN organoids after electroporation with Cas9 complexes targeting H2-k1 (pre-sorting). Purple: targeted; grey: untargeted. Organoids pre-treated with IFNγ. N = 1 experiment. (n) Efficiency of tumor formation 6 weeks post-transplant of Kb-KOSIIN, hiSIIN or co-transplant of loSIIN and Kb-KOSIIN in WT and Batf3−/− mice. N = independent animals. (o-p) Stereoscopic images of tumors 6 weeks post co-transplant of Kb-KOSIIN and loSIIN organoids in WT (o) and Batf3−/− mice (p), representative of animals transplanted in (n). Significance in (a), (h), (l) and (n) assessed by 2×2 Fisher’s exact test, with Holm’s correction applied in (h).
To interrogate priming in vivo, we performed: 1) re-challenge with loSIIN organoids 28 days after transplant of hiSIIN organoids, 2) co-injection of loSIIN and hiSIIN organoids in the same animals, and 3) transfer of in vitro-activated OT-1 cells concurrent with transplant of loSIIN organoids. All these approaches resulted in complete rejection of loSIIN organoids (Fig. 5h), demonstrating unequivocally that efficiently primed T cells are capable of killing tumors with low neoantigen expression in vivo. Priming in the context of high neoantigen expression also rescued the phenotype of T cells infiltrating loSIIN lesions. Notably, antigen-specific T cells infiltrating loSIIN and hiSIIN lesions from the same animals (Fig. 5i) showed similar abundance (Fig. 5j) and overlapping expression of TCF1 and GZMB that is indistinguishable from that of animals transplanted with only hiSIIN organoids (Fig. 5k, Extended Data Fig. 5d–f). To determine if these effects were SIINFEKL specific, we performed co-injection of loSIIN and hiVGF or hiITY organoids in the same animals. While hiVGF and hiITY did not form tumors in any animals, 100% of animals developed loSIIN tumors (Fig. 5l, Extended Data Fig. 5g), demonstrating that rescue of priming by hiSIIN is mediated through increased SIINFEKL expression.
The incomplete penetrance of hiSIIN tumor formation in Batf3−/− animals implicates additional mechanisms of priming, such as direct priming by tumor cells or cross priming by non-DC1s. Consistent with this, we detected activated antigen-specific T cells in the colons and DLNs of most of these animals 6 weeks post-transplantation, albeit at very low numbers (Extended Data Fig. 5h–j). To assess the role of direct priming, we used CRISPR/Cas9 to generate hiSIIN organoids lacking H-2kb (Kb-KOSIIN) (Fig. 5m, Extended Data Fig. 5k). Consistent with failure to present SIINFEKL, these organoids formed tumors with 100% efficiency in immunocompetent hosts. However, co-transplantation of loSIIN with Kb-KOSIIN resulted in complete rejection of loSIIN in all but one animal (Fig. 5n–o), despite outgrowth of Kb-KOSIIN in all animals, demonstrating that SIINFEKL from Kb-KOSIIN is efficiently cross presented and that this is the dominant mechanism of priming in our model. To further interrogate any potential contribution of direct tumor cell priming, we repeated the co-transplantation experiments in Batf3−/− animals. In this DC1 deficient context, loSIIN formed tumors with ~40% efficiency when co-transplanted with Kb-KOSIIN (Fig. 5n, p), indistinguishable from the efficiency of tumor formation with hiSIIN transplanted alone. These results strongly suggest that direct priming by tumor cells is not operative, and that non-DC1s play an important role in anti-tumor T cell cross priming in the colon.
Therapeutic priming rescues the poorly primed T cell response.
Given the central role of T cell priming in our model, we tested the therapeutic potential of neoantigen vaccination in animals with established tumors. Mice with GFP-expressing tumors, as determined by colonoscopy, were randomly enrolled to receive SIINFEKL containing OVA250–270 (CGLEQLESIINFEKLTEWTSS) or non-specific mutant gp10020–39 (CAVGALEGPRNQDWLGVPRQL)52 peptide-based vaccines consisting of a peptide-amphiphile and adjuvant amphiphile-CpG53 administered at 14- and 21-days post-transplant (Extended Data Fig. 6a). Vaccination with OVA250–270, but not non-specific peptide, induced profound expansion of tumor-specific T cells in peripheral blood (Fig. 6a–b, Extended Data Fig. 6b–c). One week following the second dose, tumors in the OVA250–270 vaccine arm trended towards greater reduction in size (Fig. 6c) and at termination (6 weeks), tumor burden was significantly reduced, with four complete regressions (Fig. 6d). These proof-of-principle results suggest that it may be therapeutically tractable to vaccinate against poorly expressed—albeit clonal—neoantigens, and that strict neoantigen expression cutoffs in anti-tumor vaccine pipelines should be re-evaluated.
Figure 6.
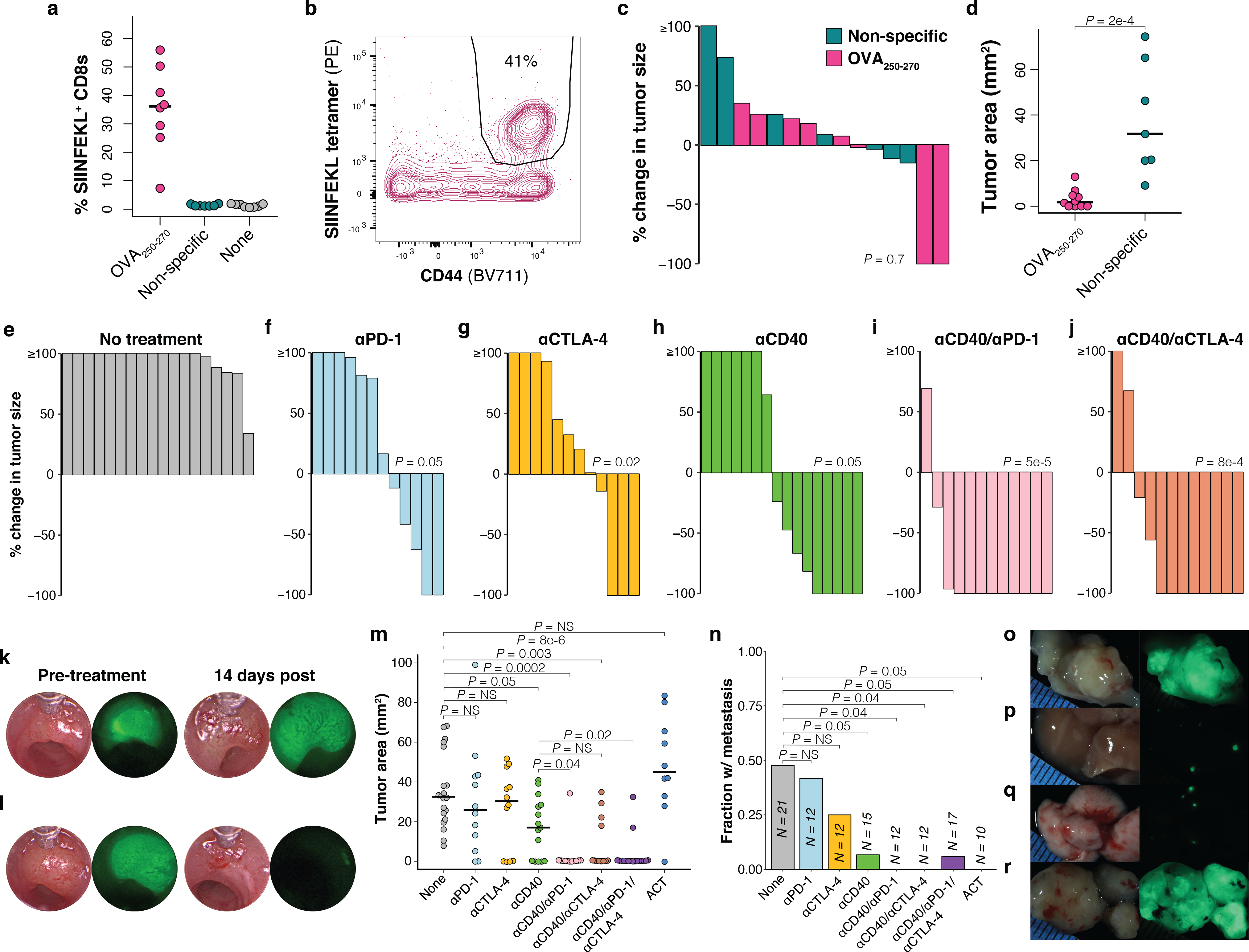
Therapeutic vaccination and agonistic anti-CD40 are efficacious in low neoantigen expressing tumors. (a) Percent of total peripheral blood CD8+ T cells that are antigen specific (CD44+/SIINFEKL tetramer+) in loSIIN tumor-bearing mice following two weeks (two doses) of OVA250–270 (N = 8 animals), non-specific (N = 7 animals), or no peptide-based vaccination (N = 8 animals). (b) Flow plot of peripheral blood antigen-specific CD8+ T cells from OVA250–270 vaccinated mouse, representative of N = 8 animals. (c) Change in loSIIN tumor size as measured by longitudinal colonoscopy following 14 days (two doses) of OVA250–270 (N = 8 animals) or non-specific vaccination (N = 7 animals). Significance assessed by Wilcoxon Rank Sum of percent change in tumor size. (d) Primary tumor sizes at necropsy 28 days post-vaccine regimen initiation. N = 10 OVA250–270 and 7 non-specific vaccine treated animals. Significance assessed by Wilcoxon Rank Sum. (e-j) Immunotherapy preclinical trial of mice bearing loSIIN tumors showing change in tumor size after 14 days of treatment, as determined by colonoscopy. N = 18 (e), 12 (f), 12 (g), 16 (h), 12 (i), and 12 (j) independent animals. Significance assessed by Wilcoxon Rank Sum of percent change in tumor size of treatment groups versus no treatment, with Holm’s correction. (k-l) Colonoscopy images of tumors pre- and post-treatment from mice receiving no treatment (k) and αCD40/αPD-1/αCTLA-4 (l), representative of N = 18 and 17 animals, respectively. (m) Primary tumor sizes at necropsy 28 days post-treatment initiation. ACT = adoptive cell transfer of OT-1s. N = 21 no treatment, 12 αPD-1, 12 αCTLA-4, 15 αCD40, 12 αCD40/αPD-1, 12 αCD40/αCTLA-4, 17 αCD40/αPD-1/αCTLA-4, and 10 ACT arm animals. Significance assessed by Wilcoxon Rank Sum with Holm’s correction. (n) Fraction of mice with any metastases (liver, lung, or omentum). N = same as (m). Significance assessed by 2×2 Fisher’s exact test with Holm’s correction. (o-r) Stereoscopic images of primary colon tumor (o), liver (p), lung (q), and omental (r) metastases from an αPD-1-treated mouse 28 days post-treatment initiation, representative of N = 12 animals.
We next asked whether more readily deployable antibody-based immunotherapies are efficacious in our model. Agonistic antibodies against the CD40 receptor (αCD40) enhance priming by potentiating the co-stimulatory function of antigen-presenting cells54. αCD40 is efficacious in preclinical mouse models of pancreatic ductal adenocarcinoma (PDAC), particularly when combined with ICB and immunogenic chemotherapy55. This is notable in light of the low TMB and immunogenicity of PDAC, which, like MSS CRC, is refractory to ICB56. Therapeutic combinations with αCD40 may be able to rescue or generate new T cell responses against weak affinity or poorly expressed neoantigens, or against tumor-associated self-antigens that lack high affinity T cell clones due to central tolerance. However, clinical studies in CRC are lacking.
We performed preclinical trials in mice bearing loSIIN colon tumors starting 14 days post-transplant (Extended Data Fig. 6a) with single agents αCD40, αPD-1, and αCTLA-4, and combinations αCD40/αPD-1, αCD40/αCTLA-4, and αCD40/αPD-1/αCTLA-4. Response was evaluated by colonoscopy at 28 days post-transplant following Response Evaluation Criteria in Solid Tumors (RECIST). All animals in the no treatment arm presented with progressive disease, while 2 of 12 (17%), 3 of 12 (25%), and 5 of 16 (31%) showed complete responses in the αPD-1, αCTLA-4, and αCD40 arms, respectively. Response was notably better in all combination arms, with 9 of 12 (75%), 8 of 12 (67%), and 12 of 17 (71%) complete responses in the αCD40/αPD-1, αCD40/αCTLA-4, and αCD40/αPD-1/αCTLA-4 arms, respectively (Fig. 6e–m, Extended Data Fig. 6d). Comparing all combination arms against single agent αCD40 showed significantly more objective responses (P = 0.02) and complete responses (P = 0.01, Fisher’s exact test). Interestingly, adoptive cell transfer (ACT) of one million ex vivo-activated OT-1 T cells at 14 days post-transplant significantly delayed tumor growth but only resulted in one complete response (Fig. 6m, Extended Data Fig. 6e), suggesting that transferred T cells rapidly become dysfunctional.
Despite initially delayed tumor growth in the single ICB arms, no significant difference in final tumor burden was observed at necropsy, suggesting only transient effect in most tumors (Fig. 6m). In addition, incidence of metastasis was not significantly decreased in single ICB arms (Fig. 6n–r). These results are reminiscent of the poor response to ICB seen in MSS CRC and demonstrate that ICB is only modestly effective at rescuing a poorly primed T cell response. In contrast, single agent αCD40 significantly decreased primary tumor size at endpoint, while combination with ICB significantly reduced tumor size further (Fig. 6m). All treatment arms with αCD40 resulted in significantly reduced rates of metastasis (Fig. 6n), although this could reflect the absence of primary tumors in many of these animals. However, the combined rate of metastasis in animals with progressive disease across all αCD40 arms was still significantly reduced (Extended Data Fig. 6f). Interestingly, while ACT had no effect on reducing primary tumor size at endpoint, it resulted in complete control of metastatic tumor burden (Fig. 6n).
To determine if therapy resistance is mediated by down-regulation of antigen expression or MHC-I, we isolated ex vivo loSIIN tumor-derived organoids from two αCD40/αPD-1 escapers, three αPD-1 escapers, and three untreated mice. All lines showed comparable sensitivity to IFNγ stimulation, expression of H-2Kb and mScarletSIIN, and were similarly sensitive to killing when co-cultured with activated OT-1s (Fig. 7a–g). These results suggest that low neoantigen expression obviates the need for dysregulation of antigen presentation, and are consistent with the lower frequency of such events in MSS versus MSI cancers18. Critically, MSS tumors may remain sensitive to T cell killing if priming against poorly expressed neoantigens can be rescued (Fig. 7h).
Figure 7.
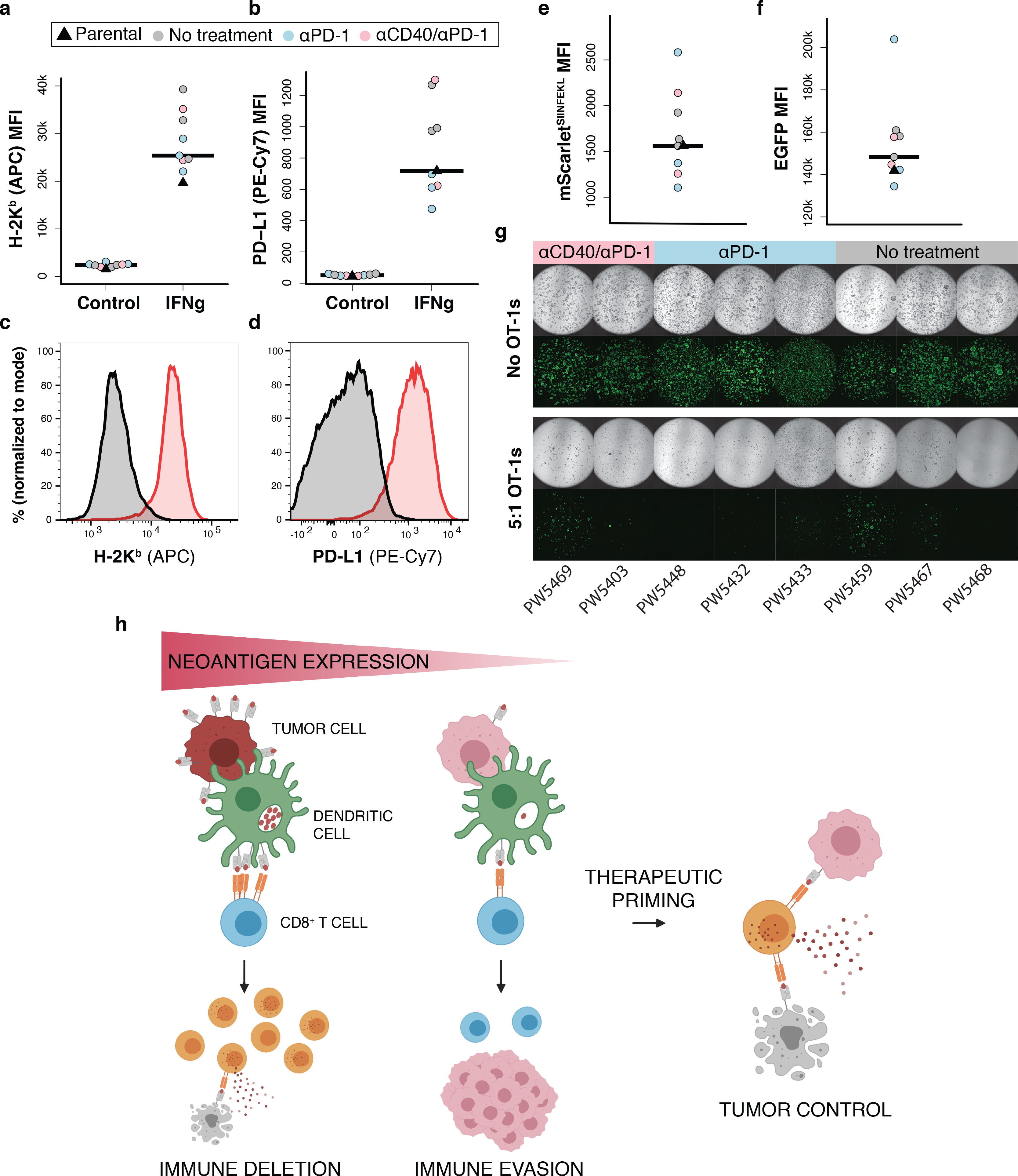
Immunotherapy refractory low neoantigen expressing tumors remain vulnerable to antigen-specific T cell killing. (a-d) Flow cytometric analysis of H-2Kb and PD-L1 MFI (a-b) and representative histograms of expression (c-d) following 24 hours of IFNγ stimulation (10 ng/mL) in ex vivo loSIIN tumor-derived organoids. Each organoid line was derived from a treatment refractory tumor taken from an independent animal in the indicated treatment arms in Fig. 6. Parental = un-transplanted loSIIN organoids. N = 3 no treatment, 3 αPD-1, and 2 αCD40/αPD-1 independent organoid lines. (e-f) Flow cytometric analysis of mScarletSIIN (e) and EGFP (f) expression in ex vivo tumor-derived organoids. N = same as above. (g) Images of co-cultures with ex vivo tumor-derived organoids and activated OT-1s at an effector-to-target ratio of 5:1 at day 4. (h) Schematic representation of the role of neoantigen expression level in immune evasion and response to therapeutic priming.
Discussion
The poor response of most CRC to immunotherapy represents a major unmet clinical need. Mouse models have provided invaluable insights into T cell dysfunction in cancer, but none to our knowledge recapitulate essential features of human CRC while facilitating detailed study of antigen-specific T cells. Here, we developed colonoscopy-guided models enabling comparison of functional versus dysfunctional tumor-specific T cell responses in a context highly faithful to the microenvironment, genetics, histopathology, and metastatic progression of the human disease.
We found that tumors from all MSS CRC patients analyzed harbored clonal neoantigens with high predicted HLA-I affinity, but these were broadly expressed at lower levels compared to those from MSI CRC. This raises the intriguing possibility that poor immunogenicity in MSS CRC and other immune cold cancers is driven by both lower burden and lower expression of neoantigens. Indeed, our low neoantigen-expressing model, like MSS CRC, demonstrated poor T cell infiltration and ICB response. We showed that neoantigen expression is an analog input that tunes the quality of anti-tumor T cell cross priming. Low expression shifts priming towards a tolerogenic response characterized by reduced magnitude, diversity, effector commitment, and per T cell functionality. A general feature of early immune evasion in cancer may be that T cell dysfunction begins as a tolerogenic program initiated during priming with insufficient antigen stimulation, in addition to a lack of local inflammatory and/or co-stimulatory cues. By extension, it is likely that immune responses against clonal neoantigens, at least those acquired early in tumorigenesis, are poorly primed and tolerogenic—axiomatic to their failure to restrain tumor outgrowth. We showed that rescuing priming is sufficient to prevent tumor initiation in our model, suggesting that early neoplasia lack immunosuppressive mechanisms to evade functional T cells. We also showed that poorly primed T cells undergo progressive exhaustion, in line with prevailing literature and suggesting that T cell dysfunction is a heterogeneous state shaped by multiple processes operative early and late in tumorigenesis.
Therapeutically targeting priming via αCD40 was highly efficacious in our model, particularly in combination with ICB. While ICB alone had no effect on the rate of metastasis, αCD40 and ACT almost completely prevented metastases, even in mice with progressive primary disease. Therefore, targeting priming may be efficacious against early metastatic lesions that may not be detected at time of treatment. These results establish the preclinical utility of our model and highlight the therapeutic promise of combined αCD40 and ICB for MSS CRC and other immune cold cancers. Given that no adequately powered clinical trials of αCD40 in CRC have been initiated to date, these results warrant clinical evaluation. Our demonstration that anti-tumor immunity against a poorly expressed neoantigen can be rescued by therapeutic vaccination is particularly relevant to analogous efforts ongoing in humans57,58. In a recent consortium study integrating neoantigen prediction pipelines from 28 research teams, it was concluded that expression, among other variables, is an important predictor of neoepitope immunogenicity, and a minimum threshold of >33 transcripts per million was imposed57. However, neoepitope immunogenicity in this and other studies was validated by experimentally measuring reactivity of existing T cells in patient blood or tumor57–59. Given that tumor-specific T cells in advanced tumors are dysfunctional, it is likely that functional readouts of these assays are limited by low sensitivity. Additionally, it is possible that therapeutic priming against neoantigens overlooked by these assays could unleash productive T cell responses from naïve T cells or reservoirs of clonally expanded precursors in lymphoid tissues. Indeed, a recent phase 1b trial (NEO-PV-01) found that while personalized vaccines elicited de novo T cell responses in all patients, the vast majority showed no detectable responses in peripheral blood prior to vaccination58. Our findings argue that RNA sequencing in these trials should be performed at sufficient depth to distinguish lack of expression from dropout due to poor coverage, and that any detectable allele-specific expression of clonal neoantigens is sufficient to nominate them for vaccination. It has been shown in vitro that effector T cells can lyse targets presenting only three cognate MHC-I:epitope complexes60. Therefore, tumor cells likely must undergo near absolute loss of neoepitope presentation to render responding T cells truly ignorant.
Altogether, the results of our study describe a model in which tumors harboring poorly expressed neoantigens elicit tolerogenic responses and evade deletion, whilst remaining vulnerable to those same T cell responses following therapeutic priming (Fig. 7h). It will be important to determine if therapies that potentiate priming mediate their effects predominantly through naïve T cells or reservoirs of antigen experienced T cells in the tumor bed or lymphoid tissues. The flexible organoid-based system developed here should facilitate a broad range of future studies in faithful models of cancer. Our use of single clonal neoantigens is a simplification of most human tumors, which are mutationally heterogeneous. It will therefore be important to interrogate the impact of intratumoral heterogeneity of neoantigens in future studies. Importantly, organoids can be rapidly engineered to express multiple antigens with a range of affinities and mixed to varying degrees of clonality. Finally, our model should provide a powerful preclinical platform for future studies of emerging immunotherapies.
Methods
TCGA neoantigen prediction analysis
350 colon adenocarcinoma patients and 76 rectum adenocarcinoma patients were analyzed from The Cancer Genome Atlas COAD and READ studies. These represented all samples with tumor (−01A) and matched-normal whole-exome sequencing (WES), RNA-Seq, and mutation annotation format (MAF) files available. Sequencing data were obtained as Binary Alignment Maps (BAMs) files aligned to GRCh38. HLA-A, -B, and -C alleles were called using OptiType, v1.3.120. Tumor/normal WES BAMs were used to create inputs to OptiType. Reads were filtered to those mapping to the HLA region (chr6:28510120–33480577 in GRCh38) with Samtools v1.1061, converted to FASTQ and filtered with RazerS 3 v3.5.862, as recommended in the OptiType documentation. OptiType was run with default parameters.
A custom Python v2.7.13 script was employed to evaluate concordance between normal and tumor HLA allele calls. 1917/2100 alleles (91.3%) in the COAD cohort and 428/456 (93.9%) alleles in the READ cohort were consistent between tumor and normal WES-based calls; the tumor allele was accepted as the final call to resolve discrepancies between calls from tumor and normal sample sequencing data. Patient MAFs were converted to Variant Call Format (VCF) and filtered to SNVs only. All non-PASS variants were removed except for some in KRAS and TP53 that had been marked as either panel_of_normals, clustered_events, or homologous_mapping_event in the TCGA MAF files.
Indels were called using Strelka2 v2.9.263 and Scalpel v0.5.464. Scalpel was run with default parameters, with a bed file derived from the CGHub bitbucket account (https://cghub.ucsc.edu; whole_exome_agilent_1.1_refseq_plus_3_boosters.targetIntervals.bed), with coordinates converted to GRCh38. Scalpel failed to call variants for 7/426 patients due to excessive read buildup at some loci; these samples were excluded from downstream analysis. VCF files containing the union of PASS variants from Strelka2/Scalpel were annotated with variant allele frequencies (VAF) from Strelka2 output and merged with corresponding SNV VCFs.
Variant consequence was annotated using Ensembl Variant Effect Predictor (VEP) v9965 with Wildtype and Downstream plugins and the following parameters: --symbol, --terms=SO, --cache, --offline, --transcript_version, --pick. The --pick parameter was reordered from default to report transcript with most extreme consequence for each variant: rank, canonical, appris, tsl, biotype, ccds, length, mane. Neoepitopes were predicted with HLA allele calls and variant effect predictions using pVACtools v1.5.725. Mutant peptides were generated for lengths 8- through 11- amino acids. MHC:peptide binding affinity was predicted for all peptide:MHC allele pairs with NetMHC-4.0, NetMHCpan-4.0, SMM v1.0, and SMMPMBEC v1.021–24, and the median value across all affinity predictions was taken.
Only neoantigens with evidence of expression (RNA-seq FPKM Upper Quartile Normalized (FPKM-UQ) > 0) were included in analyses. Tumor purity estimates (ABSOLUTE algorithm26) for TCGA COADREAD were acquired from a previous publication66. Neoantigen clonality was estimated by dividing WES level VAF by ABSOLUTE purity (adjVAF), with adjVAF ≥ 0.5 considered clonal. This is an estimate of clonality only, as other factors not considered here can also influence VAF.
Tumor RNA-Seq BAMs were analyzed to detect and quantify SNV expression at the transcriptional level. For all SNVs corresponding to predicted neoepitopes, the corresponding chromosomal coordinates were used as inputs to bam-readcount v0.8.0, which was run with default parameters (-b 20) to obtain the sequencing depth and read counts for reference and alternative alleles at each position. A custom Python v2.7.13 script was constructed to parse the output from bam-readcount and to quantify RNA VAF. Allele-specific expression was calculated as the product of RNA VAF and corresponding gene expression (FPKM-UQ). While neoantigens with no gene level expression were excluded from analysis, those with RNA VAF = 0 were included due to poor RNA-Seq coverage at many SNVs, and the high likelihood of detection failure versus true lack of expression.
Mice
Mice were housed in the animal facility at the Koch Institute for Integrative Cancer Research at MIT with a 12-hour light/12-hour dark cycle with temperatures within 68–72°F and 30–70% humidity. All animal use was approved by the Department of Comparative Medicine (DCM) at MIT and the Institutional Animal Care and Use Committee (IACUC). Apcflox/flox67, KrasLSL-G12D68, Trp53flox/flox69, Rag2−/− 70, OT-171, R26Cas9−2A-EGFP72 and Batf3−/− 73 mice were maintained on a pure C57BL/6 background. Approximately equal numbers of male and female mice between 6 to 12 weeks of age were used for all experiments. Organoids were derived from female C57BL/6 mice, allowing transplant into male recipients without minor histocompatibility antigen-driven responses. loSIIN, dimSIIN, midSIIN, medSIIN, hiSIIN, loVGF, and loITY organoids were transplanted into R26Cas9−2A-EGFP mice, which are tolerant to EGFP. In some instances, hiSIIN organoids were transplanted into wild-type mice, with no differences in phenotype observed between host genotypes. hiVGF, hiITY, and Kb-KOSIIN organoids were transplanted into wild-type or R26Cas9−2A-EGFP mice, whereas co-injection experiments with loSIIN were performed only in R26Cas9−2A-EGFP mice.
Organoid isolation and transformation
Normal colon crypts were isolated from wild-type female C57BL/6 mice as previously described11. Crypts and organoids were cultured in 60 μL domes comprised of 10 μL conditioned L-WRN (for primary crypts and wild-type organoids) or minimal media (for Apc knockdown organoids) and 50 μL of growth-factor reduced phenol-red free Matrigel Matrix (Corning) in 24-well TC-treated Olympus plates (Genesee Scientific). Conditioned L-WRN media was produced as previously described74. Minimal media is comprised of 50X B-27 Serum-Free Supplement (Thermo Fisher), Penicillin/Streptomycin (Corning), and 100x GlutaMAX (Thermo Fisher).
Lentivirus was produced in HEK-293 cells (ATCC) and concentrated as previously described75, and functional titers (Cre activity, mScarlet/EGFP fluorescence) measured as previously described76. Confluent organoids were dissociated to single cells using TrypLE Express (Thermo Fisher), diluted, and washed with PBS, and resuspended in 1.5 mL of appropriate media with 10 μM of Y-27632 (Sigma-Aldrich). Organoids were divided into three wells of a 24-well plate and transduced with 10k to 100k transduction units (TU) of virus. The plate was then spun at 600g for 1 hour and incubated for 4 hours at 37 °C, after which organoids were plated in Matrigel. Adeno-Cre was used to recombine KrasLSL-G12D and Trp53flox/flox in normal KP organoids, after which organoids were grown in L-WRN + Y-27632 for 4 days and then selected for 1 week with 10 μM NUTLIN-3A (Sigma-Aldrich). Complete recombination of KrasLSL-G12D and Trp53floxl/flox was confirmed by PCR using published primers68,69. Next, organoids were infected with shApc-expressing lentiviruses. To select for stable integration, organoids were grown in minimal media lacking WNT (described above) one week after infection. Organoids with lowest multiplicity of infection (~0.5), as determined by mScarlet or GFP fluorescence, were chosen. Finally, organoids were infected with integration-deficient lentivirus expressing Cas9 (lenti CRISPR v2)77 and sgRNA against Smad4 (5’-GATGTGTCATAGACAAGGT-3’)37, and selected by addition of TGFβ. Integration deficient lentivirus was generated using a D64V mutant psPax2 packaging vector78, and absence of integration was confirmed by absence of Cas9 protein by Western blot, and sensitivity to puromycin killing.
MHC-I immunoprecipitation and peptide isolation
MHC-I (H-2Kb) peptide isolation was performed on x210 20 μl plugs per triplicate for each organoid line using a modified immunoprecipitation and protein filtration protocol, as described previously79. Organoids were grown to confluence over 3 days before stimulation with 10 ng/mL murine IFNγ (PeproTech) for 18 hours prior to harvest. Organoids were washed with PBS and mechanically liberated by vigorous pipetting in PBS. Cells were washed twice in 50 mL PBS and pellets snap frozen in liquid nitrogen. Pellets were lysed in 2 mL of lysis buffer containing 50 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA, 1% Triton X-100, 60 mM octylglucopyranoside (Sigma), 20 mM iodoacetamide, 10 U DNase, and 1x Halt protease inhibitors (Pierce). Isolations were performed with 40 μL (bed volume) of rProtein A Sepharose beads (GE Healthcare) preloaded with 1 mg anti-H-2Kb antibody (Y3, BioXcell). Peptides were eluted in 500 μL of 10% acetic acid and purified with 10 kDa MWCO spin filters (PALL Life Science).
Tandem mass tag mass spectrometry (TMT-MS)
Dried down MHC-I eluted peptides were resuspended in 100 μL triethylammonium bicarbonate buffer and labeled with TMT16plex (Pierce, Rockford, IL, USA). Samples were then mixed and cleaned with C18 ZipTip (Millipore Sigma). 1/5 sample was used for one LC/MS/MS analysis. Samples were again dried and reconstituted in 2% formic acid (FA) for MS analysis. Peptides were loaded with the autosampler directly onto a 50cm EASY-Spray C18 column (ES803a, Thermo Scientific). Peptides were eluted from the column using a Dionex Ultimate 3000 Nano LC system with a 5 min gradient from 1% buffer B to 5 % buffer B (100 % acetonitrile, 0.1 % formic acid), followed by 84.8 min gradient to 25%, and a 15.2 min gradient to 35%B, followed by a 12min gradient to 60%B, followed by a 4 min gradient to 80%B, and held constant for 4 min. Finally, the gradient was changed from 80% buffer B to 99% buffer A (0.1% formic acid in water) over 0.1 min and held constant at 99% buffer A for 19.9 more minutes. The application of a 2.2 kV distal voltage electrosprayed the eluting peptides directly into the Thermo Exploris480 mass spectrometer equipped with a FAIMS and an EASY-Spray source (Thermo Scientific). Mass spectrometer-scanning functions and HPLC gradients were controlled by the Xcalibur data system (Thermo Scientific). MS1 scans parameters were 60,000 resolution, scan range m/z 390–1500, AGC at 300%, IT at 50ms. MS2 scan parameters were either at 45,000 or 60,000 resolution, isolation width at 0.7, HCD collision energy at 30%, AGC target at 300% and IT set to 300ms. Cycle time for MS2 was 1sec for each MS1 scan. The scan cycle MS1/MS2 was repeated for FAIMS voltages at −40V, −60V and −80V.
Tandem mass spectra were searched with Sequest (Thermo Fisher Scientific, San Jose, CA, USA; version IseNode in Proteome Discoverer 2.5.0.400). Sequest was set up to search a mouse uniprot database (database version July 3, 2020; 55650 entries containing common contaminants and the three proteins mScarlet-SIINFEKL, mScarlet-VGFNFRTL and EGFP) assuming no digestion enzyme (unspecific). Sequest was searched with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 10.0 PPM. TMTpro was added as a fixed modification on K and N-terminus of peptides. Oxidation of methionine was specified in Sequest as a variable modification. Resulting peptides were filtered to exclude peptides with an isolation interference of >30% and ppm error >+/−3ppm of the median ppm error of all PSMs. SIINFEKL intensity across samples was normalized by the overall abundance of all peptides detected in each sample.
Organoid CRISPR/Cas9 RNP electroporation
Confluent hiSIIN organoids were dissociated to single cells as described above and resuspended in 100 μL OPTI-MEM. Ribonucleoprotein (RNP) complexes were formed by mixing 1.64 μL (0.1 nmol) Alt-R Cas9 (IDT) with 3 μL (0.3 nmol) synthetic sgRNA (Synthego) and incubating for 10–20 minutes at room temperature. Cells were then added to the RNP mix, 100 μL transferred to a 2 mm gap cuvette (Bulldog Bio), and electroporated using a NEPA21 electroporator (Bulldog Bio) with the following poring pulse parameters: 175 V, 5 msec length, 50 msec interval, 2 pulses, 10% decay rate, + polarity; and transfer pulse parameters: 20 V, 50 msec length, 50 msec interval, 5 pulses, 40% decay rate, +/− polarity. Electroporated organoids were resuspended gently in pre-warmed minimal media and incubated at 37°C for 15 minutes before plating in Matrigel. sgRNAs used for electroporation are: H-2Kb: 5’-CAAUGAGCAGAGUUUCCGAG-3’; previously published B2m sequence79: 5’-UUGAAUUUGAGGGGUUUCUG-3’.
Colonoscopy-guided injections
Orthotopic injection of lentivirus and organoids was performed similarly to previously described methods11,12. Intact organoids were always harvested two days post-passaging by washing in PBS and dissociating in Dispase. Matrigel was broken up by gentle scraping and pipetting four times using a 1 mL pipette and incubating at 37°C for 15 minutes. Organoids were washed thoroughly in PBS and resuspended in OPTI-MEM with 10% Matrigel at 50 organoids per μL. Intact organoids and lentivirus (20,000 or 100,000 TU/μL) were injected via Hamilton syringe (Hamilton, 7656–01) and custom injection needle (Hamilton, 33-gauge, small Hub RN NDL, 16 inches, point 4, 45-degree bevel, like 7803–05) fed through the working channel of the colonoscope and inserted into the colonic mucosa at ~30°. ~50 μL was delivered per injection, resulting in large “blebs” within the mucosa.
Tissue preparation and flow cytometry
Colon draining lymph nodes (DLNs, caudal and iliac) were harvested and mechanically dissociated in RPMI1640 (Corning) with 5% heat-inactivated fetal bovine serum (HI-FBS) (harvest media). Tumors were identified using a Dual Fluorescent Protein Flashlight, Model DFP-1 (Nightsea), dissected and placed in a digestion buffer containing 500 Units/mL Collagenase Type 1 (Worthington) and 20 μg/mL DNAse (Sigma-Aldrich) in harvest media, minced using surgical scissors and digested at 37 °C for 40 minutes with gentle agitation. Tumors were then further dissociated with a gentleMACS Octo Dissociator (Miltenyi Biotec) on the tumor_imp1.1 setting and filtered through a 100 μM filter. DLN and tumor preparations were divided for immediate staining or peptide stimulation. Intravenous CD45 staining prior to sacrifice of animals (to differentiate tissue-infiltrating versus circulating T cells) was not routinely performed, as this stained less than 1% of total SIINFEKL-specific T cells.
Live/dead staining (ghost ef780 (Corning), 1:500) was performed in PBS and surface stains in FACS buffer (1 mM EDTA, 25 mM HEPES, 0.5% HI-FBS in PBS). Cells were fixed for 1 hour at room temperature in Fixation/Permeabilization Concentrate (Thermo Scientific) diluted 1:3 in Fixation/Permeabilization diluent (Thermo Scientific) and washed in permeabilization buffer (Thermo Scientific). Intracellular staining was performed in permeabilization buffer overnight at 4 °C. Cells were washed and resuspended in FACS buffer for analysis on a BD LSRFortessa 4 laser, 18 color flow cytometer running BD FACSDiva v8.0 software. Results were analyzed in FlowJo v10.4.2. Single lymphocytes were gated first on FSC-A versus SSC-A and then FSC-A versus FSC-H. Then, live CD8+ T cells were gated on positive CD8α and negative ghost ef780 staining. Antigen-specific CD8+ T cells were further gated on CD44 and tetramer positivity. Expression of additional markers was analyzed specifically in this antigen-specific CD8+ T cell population in all flow cytometric experiments on T cells presented in this manuscript.
Antigen-specific in vivo killing assay
Splenocytes were prepared for in vivo transfer as described by Durward et. al, 201045. Briefly, spleens were harvested from female C57Bl/6 mice, red blood cells lysed using ACK Lysing Buffer (Thermo Fisher), and cells resuspended in PBS in a round bottom 96-well plate at 1×108 cells per mL. Half of the wells were pulsed with SIINFEKL peptide (Anaspec) at 1 μg/mL, followed by labeling with the membrane dye CellTrace Violet (CTV; Thermo Fisher) at 20 μM. The remaining wells were labeled with 2 μM CTV. Peptide loaded “target” and unloaded control splenocytes were then mixed 1:1 and 200 μL retro-orbitally injected (2×107 cells) into experimental animals 8- and 14-days post-transplant of organoids. DLNs and spleens were harvested 6 hours later and processed for flow cytometry as described above. Target and control splenocytes were identified by live/dead staining and CTV labeling intensity, and percent target killing determined relative to the control population. Targets killed per antigen-specific T cell was determined by dividing the total number of targets killed (control minus target splenocytes) by the total number of SIINFEKL tetramer+ CD8+ T cells. This metric was meaningful at 14 days when target killing was incomplete in both loSIIN and hiSIIN animals but precluded at 8 days by effectively complete target killing in hiSIIN animals.
Peptide stimulation for cytokine staining
Samples were prepared as described above, and prior to surface staining were stained with antibodies for Ly-6G (BioLegend, 1A8, 1:200), EpCAM (BioLegend, G8.8 1:80) and F4/80 (BioLegend, BM8, 1:80) in FACS buffer for 30 minutes at 4°C and depleted using Dynabeads Goat Anti-Mouse IgG kit (Thermo Fisher) following manufacturer recommendations. T cells were then stimulated in T-cell media (RPMI-1640 with 10% HI-FBS, 20 mM HEPES, 1 mM Sodium Pyruvate, 2 mM L-Glutamine, 50 μM β-mercaptoethanol, 1x non-essential amino acids, and 0.5x Penicillin/Streptomycin) with 1:1000 GolgiPlug (BD) and 2 μM Monensin Solution (BioLegend), and 1 μM SIINFEKL peptide (Anaspec) for 3 hours at 37 °C. Cells were washed and stained for surface and intracellular markers as described above.
Bone marrow-derived dendritic cell (BM-DC) isolation and OT-1 co-culture
Bone marrow from C57Bl/6 mouse femurs and tibias was isolated, red blood cells lysed, and cells plated at 1.5×106 cells per mL and cultured in T-cell media (described above) plus 600 ng/mL recombinant human Flt-3L-Ig (hum/hum, BioXcell) and 5 ng/mL recombinant mouse GM-CSF (BioLegend). After 1 week, BM-DCs were switched to fresh media and activated with 20 μg/mL of the mouse STING ligand DMXAA (InvivoGen). The following day, BM-DCs were plated in 96-well plates at 10,000 cells per well in fresh media (without DMXAA) and cultured with lysed organoids overnight. Organoids were first dissociated to single cells in TrypLE and counted, then lysed in water at 2×106 cells per mL for 10 minutes at 37 °C. Lysed cells were pelleted at 1000 G for 15 minutes and resuspended in BM-DC media at appropriate dilutions. The next day, loaded BM-DCs were washed and cultured with 50,000 naïve OT-1s per well. CD8+ T cells were purified from spleen and LNs of OT-1 mice using the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotech) following manufacturer specifications. Cells were harvested at 72 hours for staining and flow cytometric analysis. For cytokine stains, cells were treated with 1:1000 GolgiPlug and 2 μM Monensin Solution for 3 hours prior to harvest.
OT-1 T cell activation and organoid co-culture
Spleen and LNs from OT-1 mice were harvested in PBS, red blood cells lysed, cells resuspended in T cell media (described above) + 10 ng/mL hIL-2 (PeproTech) and 1 μM SIINFEKL peptide (Anaspec), counted, and plated at 1×106 cells per mL. Stimulation was performed for 24 hours at 37 °C. CD8+ T cells were then purified using the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotech), and expanded in T cell media + hIL-2 with daily splitting. T cells were used for ACT or co-culture assays at day 3 or 4.
Organoids and OT-1s were plated at 0:1 and 5:1 effector to target ratios at 2500 organoid single cells in 10 μl minimal media and 50 μl Matrigel. Co-cultures were plated in triplicate at 20 μl per dome, grown in minimal media, and imaged on day 4. Total fluorescent area of organoids within images was quantified in ImageJ v2.1.0/1.53c by setting left and right thresholds of greyscale images to 22 and 255 on B&W setting, respectively, and analyzing particles with size threshold set to >20 pixel^2 and circularity set to 0.1–1.0 with holes included.
Immunohistochemistry and automated quantification
Tissues were fixed in zinc formalin, washed in 70% ethanol and paraffin embedded. Antigen retrieval was performed in citrate buffer pH 6 in a pressure cooker at 125 °C for five minutes. Blocking was performed with BLOXALL Endogenous Peroxidase and Alkaline Phosphatase Blocking Solution (Vector) followed by Normal Horse Serum (2.5%) (Vector). Slides were stained with CD8α (ab217344, Abcam) 1:1000 overnight, incubated with Alkaline Phosphatase (AP) anti-Rabbit IgG (Vector) and developed with Vector Black substrate (Vector). Sections then underwent a second round of antigen retrieval in a pressure cooker at 110 °C for two minutes, followed by co-incubation with FOXP3 (FJK-16s, eBioscience) 1:125 and CD4 (ab183685, Abcam) 1:400 overnight. Sections were then sequentially incubated with AP anti-Rat IgG (Vector) and HRP anti-Rabbit IgG (Vector) and developed sequentially with Vector Red (Vector) and Vina Green (Biocare Medical). Slides were counterstained with Harris Acidified Hematoxylin and dehydrated. Aqueous wash steps following counterstain were shortened from 1 minute to 30 seconds to minimize loss of Vina Green stain.
Immune infiltration was calculated by a convolutional neural network (CNN) trained to identify the three cell types stained (black = CD8, green = CD4, green/red = Treg), using Aiforia’s cloud-based platform (Aiforia Technologies Oy). Whole slides were scanned with a Leica AT2 (Aperio) using the Rainbow color profile. First, the CNN was trained to identify a tissue layer. Within that layer, the CNN was trained to identify black, green, and green/red staining. Within each of these layers, an object counter was trained to quantify the number of cells with the stain. Training was performed by manual annotation of each layer and counting of objects within training regions across 20 separate slides, with roughly five training regions per layer per slide. Performance was validated against human counting and found to be highly accurate and consistent.
In situ SIINFEKL tetramer staining
Tissue was stained in situ with SIINFEKL tetramer as previously described80. Tissues were additionally stained with CD8β AF647 (YTS156.7.7, BioLegend) (1:100), and anti-human β2-microglobulin (β2M) PE (2M2, BioLegend) (1:50). Anti-β2M staining is specific to human β2M in the SIINFEKL tetramer and serves to amplify signal. Images were taken at 30X on an Olympus FV1200 Laser Scanning Confocal Microscope and analyzed in ImageJ v2.1.0/1.53c.
In vivo antibody and vaccine dosing
All antibody dosing was performed via intraperitoneal injection in PBS. αCD4 (GK1.5, BioXCell) and αCD8 (2.43, BioXCell) depleting antibodies were administered at 200 ug every 4 days. αPD-1 (29F.1A12, BioXCell) was administered at 200 μg three times a week. αCTLA (9H10, BioXCell) was administered at an initial dose of 200 μg, with all subsequent doses at 100 μg, three times a week. αCD40 (FGK4.5, BioXCell) was administered once at the beginning of treatment at 100 μg.
The adjuvant amphiphile-CpG (amph-CpG) and antigen amphiphile (amph-peptide) were produced as previously described53. Briefly, class B CpG 1826 oligonucleotide with a G2 spacer (5’-diacyl lipid-GGTCCATGACGTTCCTGACGTT- 3’) was conjugated via the 5’ end to an 18 carbon diacyl tail. Antigen peptide OVA250–270 (CGLEQLESIINFEKLTEWTSS) and non-specific mutant gp10020–39 (optimized S27P, EGP long52, CAVGALEGPRNQDWLGVPRQL) were conjugated via N’ cysteine residue to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethyleneglycol-2000] (Avanti Polar Lipids). Mice were vaccinated subcutaneously at the base of the tail with 1.24 nmol amph-CpG and 25 μg of amph-peptide, with half dose given to each side. Vaccination was performed once weekly starting 14 days post-transplant of loSIIN organoids.
Colonoscopy imaging
Tumor progression was monitored longitudinally using a Karl Storz colonoscopy system with white light, RFP and GFP fluorescence. This consists of Image 1 H3-Z Spies HD Camera System (part TH100), Image 1 HUB CCU (parts TC200, TC300), 175-Watt D-Light Cold Light Source (part 20133701–1), AIDA HD capture system, and fluorescent filters in the RFP and GFP channels (all from Karl Storz). The endoscope used for imaging was the Hopkins Telescope (Karl Storz, part 64301AA) with operating sheath (Karl Storz, part 64301AA). To consistently measure tumor area, biopsy forceps (Richard Wolf) were fed through the operating sheath and positioned consistently given two landmarks: widthwise grooves that appear as concentric semi-circles in the field of view, and a lengthwise groove at the forceps tip. Images were captured upon gentle contact of forceps with tumor. Tumor area in the field of view and length of the lengthwise forceps groove were calculated using ImageJ v2.1.0/1.53c. Tumor area was normalized to groove length.
TCR sequencing
Using a BD FACSAria flow cytometer, live SIINFEKL tetramer-positive CD8+ T cells were directly sorted into 50 μl lysis buffer with proteinase K, from the Arcturus PicoPure DNA Extraction kit (ThermoFisher), in low binding microcentrifuge tubes (Biotix), and genomic DNA extraction performed following manufacturer instructions. Mouse TCRβ sequencing was performed by Adaptive Biotechnologies. Analysis was performed in R v4.0.2, and Simpson diversity calculated using the ‘Vegan’ v2.5.7 package. To account for differences in total numbers of T cells surveyed in samples between groups, unique productive TCR sequences were randomly downsampled to match between groups. Down-sampled data is presented in Extended Data Fig. 3k–l, although down-sampling did not impact observed trends.
Statistics and reproducibility
Statistical analyses and figure generation were performed in R v4.0.2 using built in functions and ggplot2 v3.3.3, beeswarm v0.3.1, corrplot v0.88 and RColorBrewer v1.1.2. For statistical assessment of differences in proportionality, Fisher’s exact 2×2 test was performed. For continuous data, two-tailed Wilcoxon Rank Sum test was performed, apart from the organoid and OT-1 co-culture results, which were analyzed with two-tailed Student’s t-test. Multiple comparison corrections were performed using Holm’s method. No statistical method was used to predetermine sample size. Of animals transplanted with loSIIN organoids, only those that formed tumors were taken for flow cytometric analysis. No other data were excluded from analyses. Preclinical trials were randomized, and investigators blinded to allocation during dosing, colonoscopy imaging, and tumor quantification. Preclinical studies were performed across three independent cohorts with the aim of validating consistency and reaching 10 or more animals per treatment arm. All in vivo and co-culture experiments were repeated at least two times. No experiments presented in this manuscript failed to replicate. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1: Lower burden and expression of predicted neoantigens in MSS versus MSI-H CRC.
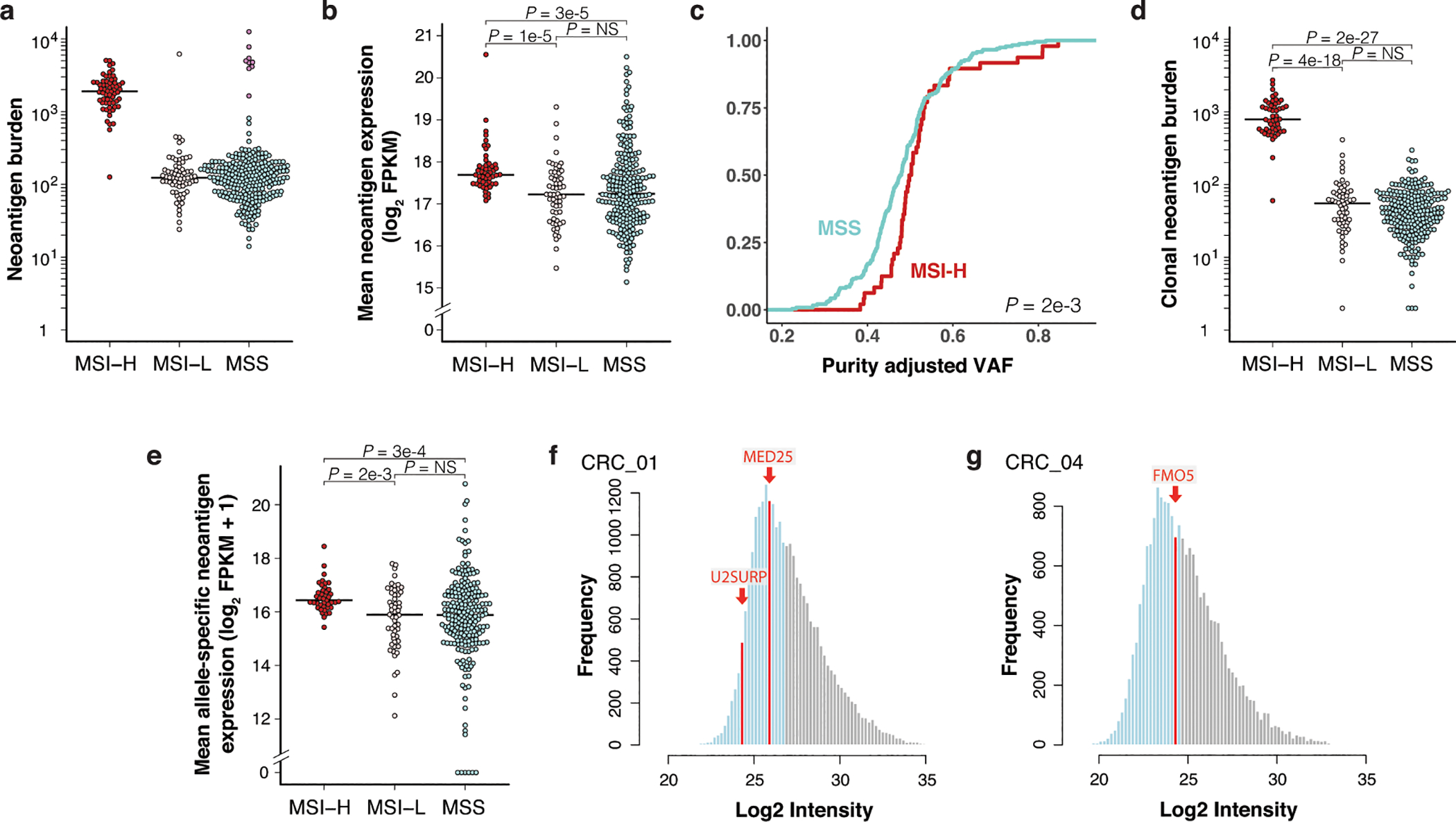
(a) Total expressed neoantigens by patient including hypermutant MSS cases (in purple). N = 62 MSI-H, 68 MSI-L, and 275 (including 9 hypermutant) MSS patients. All other plots exclude hypermutant MSS cases. (b) Mean expression of all neoantigens, regardless of clonality, by patient. N = 62 MSI-H, 68 MSI-L, and 266 MSS patients. (c-d) Analysis of patients with available ABSOLUTE purity for estimation of clonality (adjVAF). N = 50 MSI-H, 58 MSI-L, and 236 non-hypermutant MSS patients. (c) Empirical cumulative distribution function of mean neoantigen clonality (adjVAF) by patient. Significance was assessed by two-sided Kolmogorov-Smirnov test. (d) Total expressed clonal neoantigens with predicted HLA-I binding IC50 ≤ 500 nM by patient. (e) Mean allele-specific expression of clonal SNV-derived neoantigens by patient, excluding neoantigens with zero gene level expression but including those with zero allele-specific expression. N = 41 MSI-H, 53 MSI-L, and 219 MSS patients. (f-g) Abundance distributions of HLA-I ligandomes by MS in PDOs from MSS CRC patients CRC_01 (f) and CRC_04 (g) with epitope abundance above the median in grey, below the median in light blue, and neoantigens in red. Data from Newey, A, et al., 2019. Significance in (b), (d), and (e) was assessed by two-tailed Wilcoxon Rank Sum test with Holm’s correction for multiple comparisons. Source data for panels (a-e) can be found in Source Data Figure 1a–e.
Extended Data Fig. 2: Development of in vivo lentiviral and organoid models of CRC with neoantigen expression.
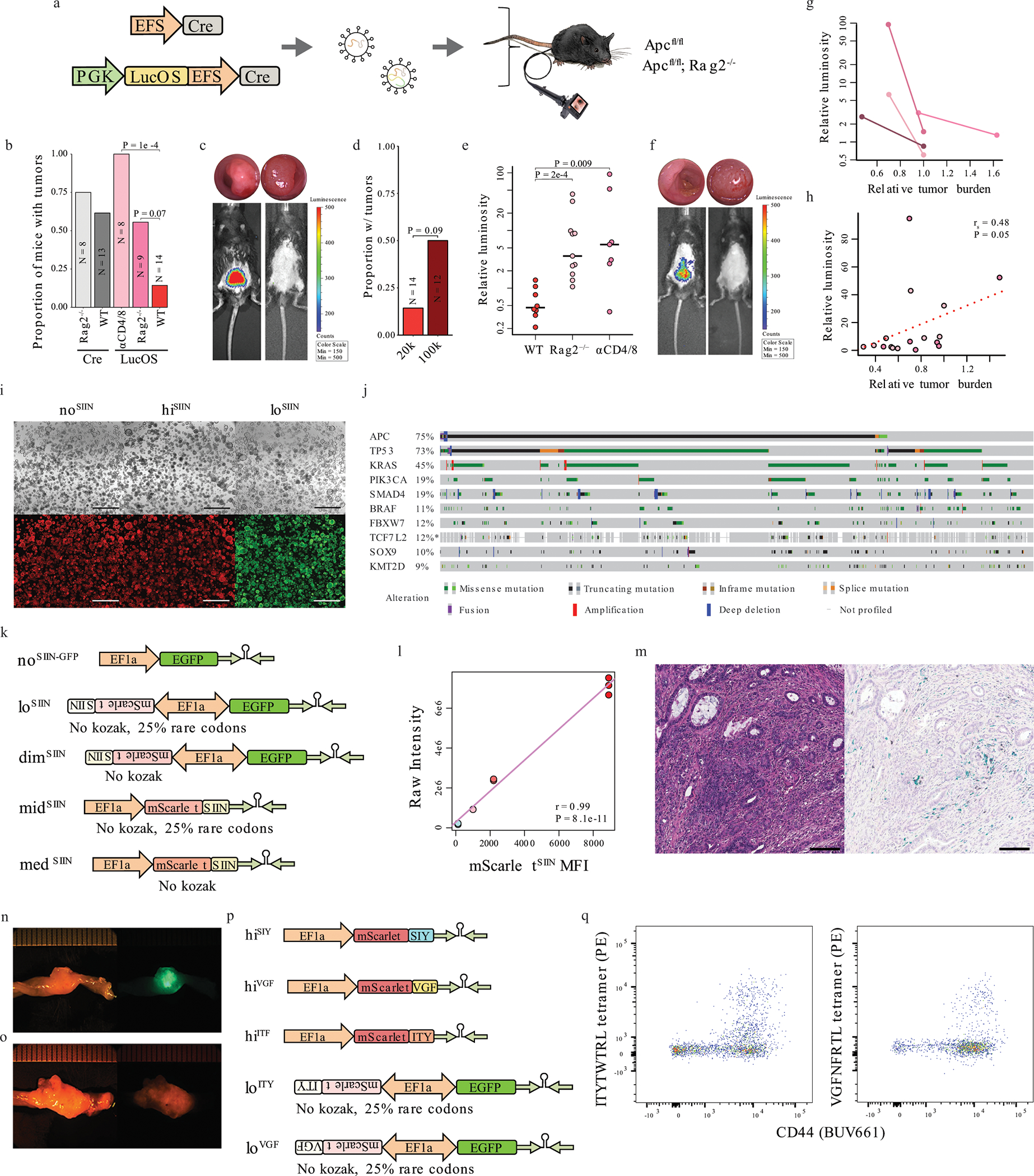
(a) Lentiviruses used to initiate colon tumors in Apcflox/flox and Apcflox/flox; Rag2−/− mice. (b) Efficiency of tumor formation 16 weeks post-injection. N = independent animals. Significance assessed by two-tailed Wilcoxon Rank Sum with Holm’s correction for multiple comparisons. (c) Antigen expression in LucOS-induced tumors in Rag2−/− (left) and wild-type (right) mice at 12 weeks (colonoscopy above, bioluminescence below). (d) Efficiency of tumor induction with LucOS lentivirus at 20,000 and 100,000 transduction units (TU)/μl. N = 26 independent animals. (e) Antigen expression (bioluminescence). N = 26 independent animals. Significance assessed by two-tailed Wilcoxon Rank Sum. (f) Antigen expression in LucOS-induced tumors with continuous T cell depletion at 5 weeks (left) and 7 weeks after T cell depletion (right), and colonoscopy (above). (g) Antigen expression versus relative tumor size (percent of colon occluded) following withdrawal of depleting antibodies. N = 4 independent animals. (h) Correlation of antigen expression and tumor burden in Rag2−/− (dark pink) and αCD4/8 (light pink)-treated mice 12 weeks post-injection with LucOS. N = 17 independent animals. Significance measured by Spearman’s rank-order correlation. (i) noSIIN, hiSIIN, and loSIIN organoids grown in the absence of WNT. Scale bars = 1 mm. Representative of N = 3 independent cultures. (j) Top 10 mutated genes in MSK-IMPACT colon adenocarcinoma (cBioPortal). (k) Lentiviral constructs used to generate organoids expressing only EGFP (noSIIN-GFP) and SIINFEKL expression variants. (l) Linear regression with Pearson correlation of SIINFEKL abundance (TMT-MS) versus mScarletSIIN MFI (flow cytometry). TMT-MS was performed on three independent preparations of each line. (m) H&E and IHC of noSIIN primary colon tumor 42 days post-transplant. Representative of N = 9 independent animals. Scale bar = 100 μM. (n-o) Images of dimSIIN (n) and midSIIN (o) tumors that formed in N = 2/9 and 1/9 transplanted animals, respectively. (p) Lentiviral constructs used to generate organoids expressing SIYRYYGL, ITYTWTRL, and VGFNFRTL at high and low levels. (q) ITYTWTRL and VGFNFRTL tetramer-specific CD8+ T cells infiltrating 42-day loITY and loVGF tumors by flow cytometry. Representative of N = 10 loITY and 9 loVGF transplanted animals.
Extended Data Fig. 3: Low neoantigen expression drives reduced T cell function and diversity.
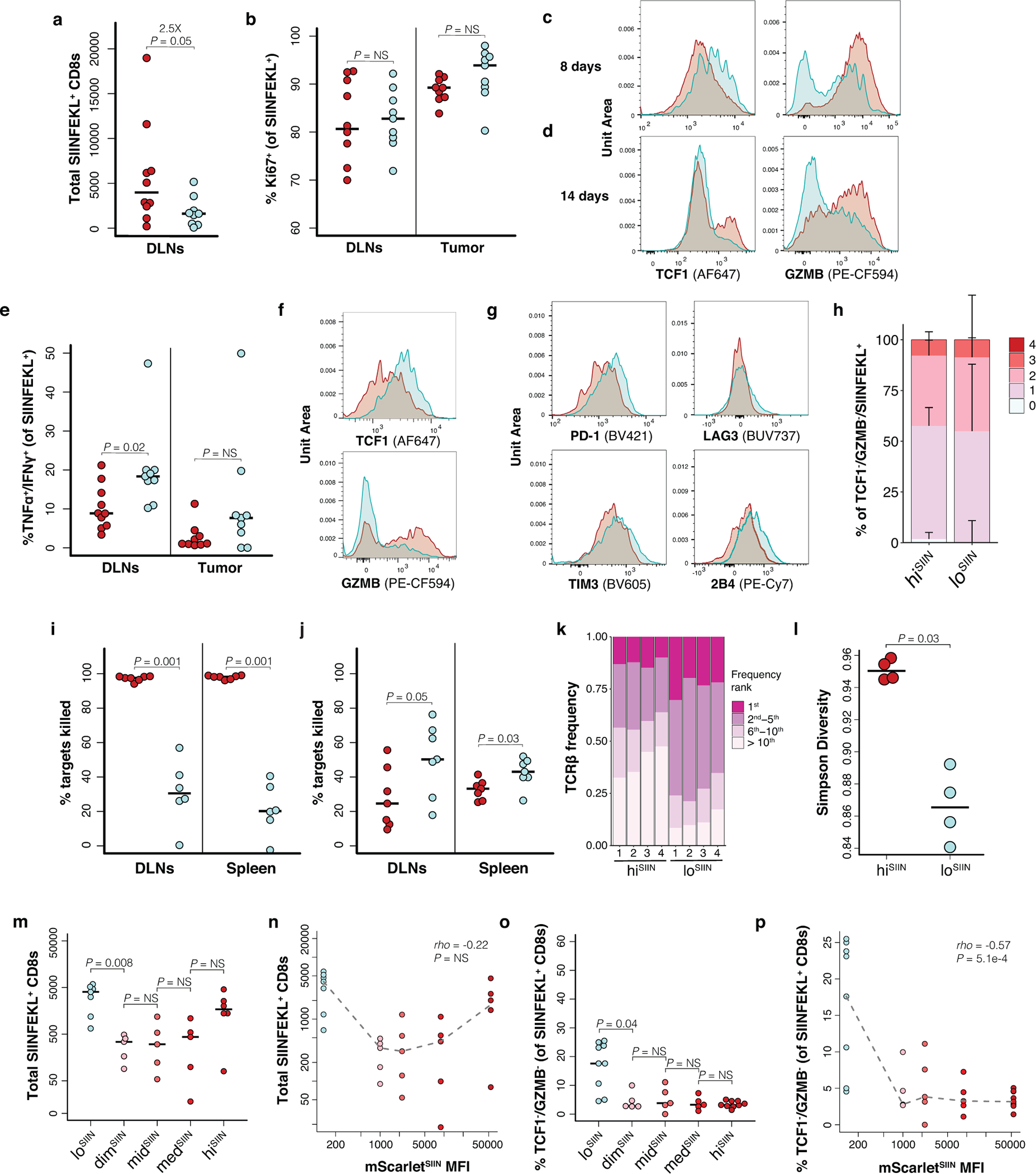
(a-m) Flow cytometry of CD44+/CD8+ antigen-specific T cells from lesions and DLNs post-transplant of hiSIIN (red) and loSIIN (blue) organoids. (a) Total antigen-specific T cells and (b) percent Ki67 positive in DLNs at 8 days. N = 10 hiSIIN and 9 loSIIN-transplanted animals. (c-d) TCF1 and GZMB expression in antigen-specific T cells in lesions at 8 (c) and 14 (d) days. Representative of N = 59 animals per line and timepoint. (e) Percent of antigen-specific T cells double-positive for TNFα and IFNγ, and (f) and representative expression of TCF1 and GZMB in this subset within DLNs at 8 days. N = 10 hiSIIN and 9 loSIIN-transplanted animals. (g) Inhibitory receptor expression on TCF1-/GZMB- antigen-specific T cells from tumors at 14 days. Representative of N = 6–7 animals per line. (h) Median percent of TCF1-/GZMB- antigen-specific T cells from tumors expressing 0 through 4 inhibitory receptors (PD-1, TIM3, LAG3, and 2B4) at 8 days. N = 9 hiSIIN and 9 loSIIN-transplanted animals. Bars = standard deviation. (i-j) Percent of SIINFEKL-loaded “target” splenocytes killed in DLNs and spleens from killing assay at 8 (i) and 14 (j) days post-transplant of hiSIIN and loSIIN organoids. N = 6–7 animals per line and timepoint. (k-l) Frequency of most common clonotypes (k) and Simpson diversity score (l) from TCRβ chain sequencing of antigen-specific T cells from hiSIIN (down-sampled) and loSIIN lesions at 8 days. N = 4 independent animals per line. (m-n) Total antigen-specific T cells isolated from lesions at 14 days across all lines (m) and versus mScarletSIIN MFI (n). (o-p) Percent of antigen-specific T cells from lesions at 8 days double-negative for TCF1 and GZMB across lines (o) and versus mScarletSIIN MFI (p). Dashed lines connect medians. Significance assessed by Spearman’s rank correlation. N = 5–9 independent animals per line in (m-p). Significance in (a-b), (e), (i-j), (l-m) and (o) assessed by two-tailed Wilcoxon Rank Sum. Holm’s correction applied in (m) and (o). Source data for panels (a, h-j, m-p) can be found in Source Data Figure 3a, i, n–r.
Extended Data Fig. 4: T cells in tumors with low neoantigen expression lose effector function over time.
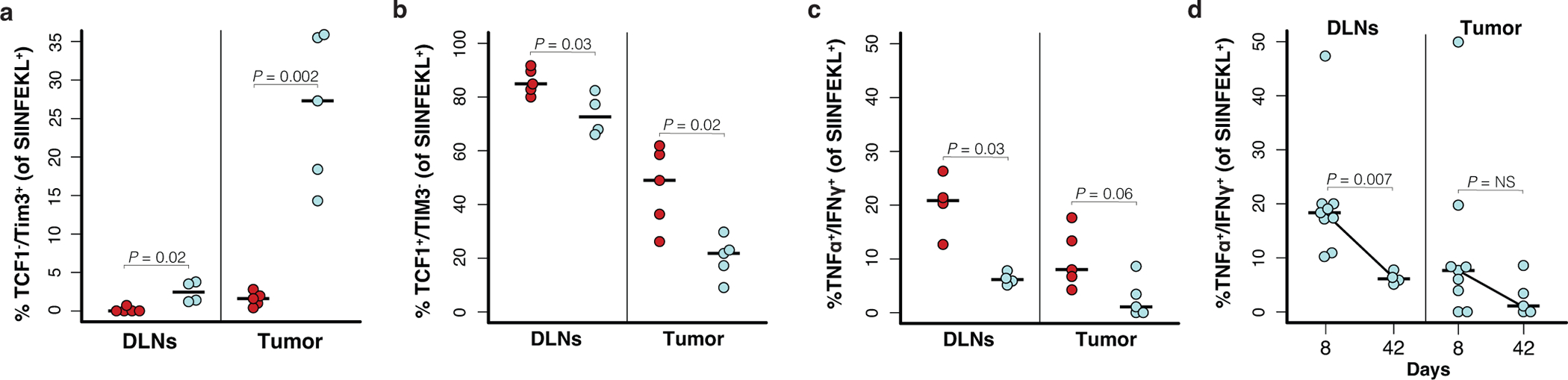
(a-b) Percent of antigen-specific T cells from DLNs and tumors at 42 days negative for TCF1 and positive for TIM3 (a), and positive for TCF1 and negative for TIM3 (b) by flow cytometry. N = 4–5 independent animals per line. (c-d) Percent of antigen-specific T cells from DLNs and tumors double-positive for TNFα and IFNγ at 42 days (N = 4–5 independent animals per line) (c) and both 8 and 42 days (d) (N = 4–9 independent animals per line). Red = hiSIIN, blue = loSIIN. Significance in (a-d) was assessed by Wilcoxon Rank Sum. Source data for panels (a-d) can be found in Source Data Figure 4d–e.
Extended Data Fig. 5: Neoantigen expression is limiting for cross priming by canonical and noncanonical antigen-presenting cells.
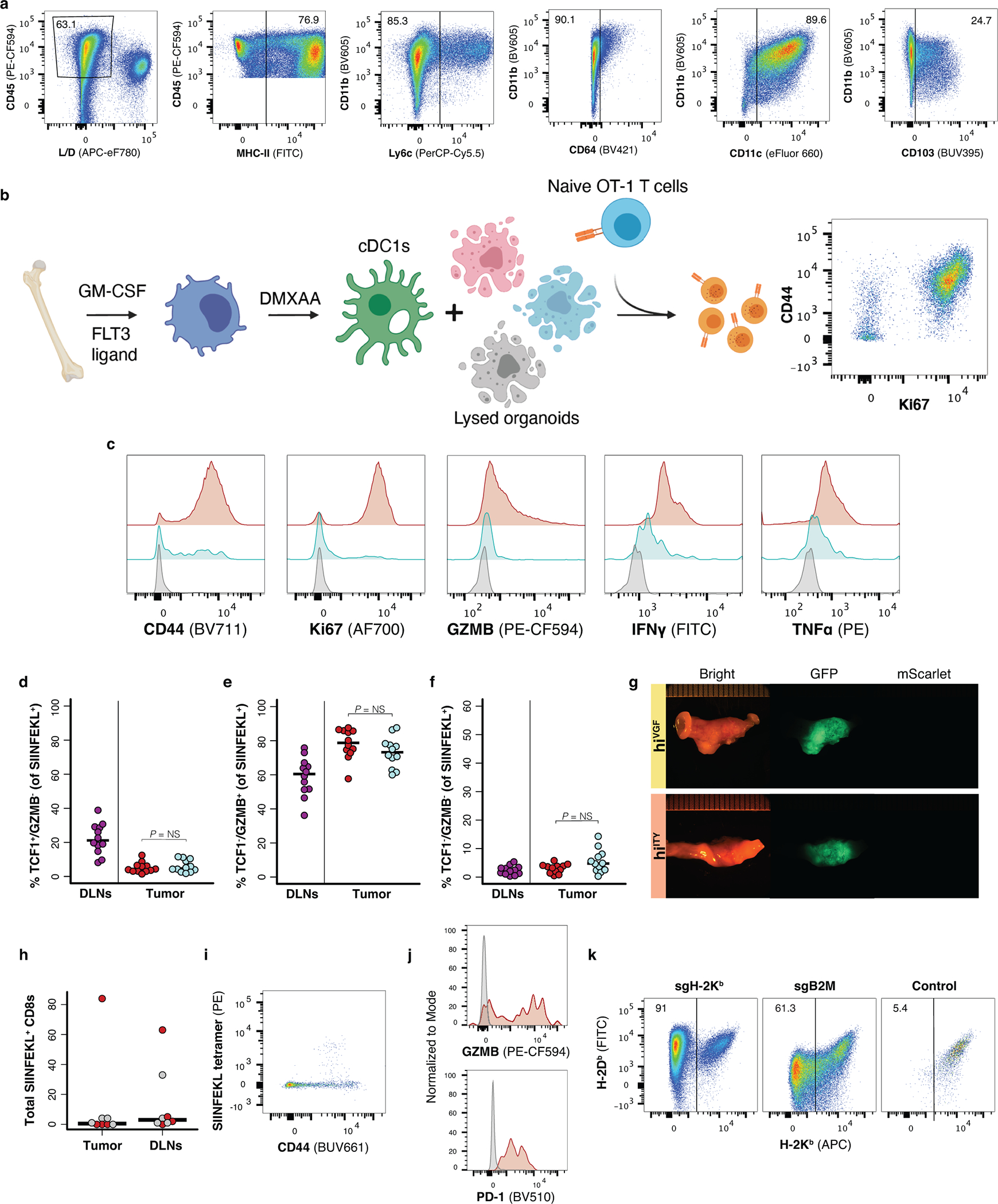
(a) Flow cytometry gating strategy to determine percentage of CD11c+/CD103+ DC1s in BM-DC culture. (b) Schematic of BM-DC isolation, activation, and co-culture with naïve OT-1s. (c) Histograms of CD44, Ki67, GZMB, TNFα, and IFNγ expression on OT-1s representative of N = 4 co-cultures in the 400,000 lysed organoid cells condition. (d-f) Flow cytometric analysis of antigen-specific T cells from DLNs and lesions 8 days post-co-transplant of hiSIIN (red) and loSIIN (blue) organoids at separate sites. Percent TCF1+/GZMB- (d), TCF1-/GZMB+ (e), and TCF1-/GZMB- (f). N = 12 animals. Significance assessed by two-tailed Wilcoxon Rank Sum. (g) Brightfield and fluorescent images of colons and tumors 6 weeks post co-transplant of loSIIN and hiVGF or hiITY, representative of N = 9 animals each. (h-j) Flow cytometric analysis of antigen-specific (CD44+/SIINFEKL+) CD8s in colon and DLNs 6 weeks post-transplant of hiSIIN in Batf3−/− mice. (h) Total SIINFEKL+ CD8s, with progressive tumors in grey (N = 4 animals) and rejected lesions in red (N = 4 animals). (i) Flow plot of SIINFEKL+ CD8s infiltrating rejected lesion, and (j) PD-1 and GZMB expression on CD44+/SIINFEKL+ CD8s (red) versus CD44- CD8s (grey) from rejected lesion representative of N = 4 animals. (k) Flow plots of H-2Kb/H-2Db expression on hiSIIN organoids post electroporation with Cas9 complexes targeting H2-k1 and B2m, or untargeted control (pre-sorting). Organoids were pre-treated with IFNγ. N = 1 experiment. Source data for panels (d-f) can be found in Source Data Figure 5j.
Extended Data Fig. 6: Design of preclinical trials to test therapies that rescue priming in low neoantigen expressing tumors.
(a) Schematic of vaccination and immunotherapy preclinical trial design and dosing schedule. (b-c) Flow plots of peripheral blood antigen-specific (CD44+/SIINFEKL tetramer+) CD8+ T cells from non-specific peptide-based vaccination (b) and no vaccination control (c) mice, representative of N = 7 and 8 independent animals, respectively. (d-e) Change in tumor size after 14 days of treatment, as determined by colonoscopy. ACT = adoptive cell transfer of OT-1s. N = 17 (d) and 10 (e) independent animals. Significance assessed by Wilcoxon Rank Sum of percent change in tumor size of treatment group versus no treatment, with Holm’s correction. (f) Fraction of mice with any metastases (liver, lung, or omentum), including only mice with progressive primary disease. N = independent animals. Significance assessed by 2×2 Fisher’s exact test of number of mice with metastases across all αCD40 treatment arms (with and without ICB) versus all other arms (no treatment and ICB single agent arms). Source data for panels (d-e) can be found in Source Data Figure 6e–j, m.
Supplementary Material
Acknowledgements
This work was supported by the NCI Cancer Center Support Grant P30-CA14051, R01 CA233983 and the Howard Hughes Medical Institute. P.M.K.W was supported by a Damon Runyon Fellowship Award. We thank K. Yee, J. Teixeira, K. Anderson, and M. Magendantz for administrative support, and our colleagues in the Jacks laboratory and the broader community at the Koch Institute of MIT for thoughtful discussions and technical advice, with special thanks to Dr. S. Spranger and members of her laboratory. We thank the Koch Institute Swanson Biotechnology Center for core support from the Flow Cytometry, Proteomics, Histology, and Microscopy facilities, with particular thanks to A. Koller for help with design and analysis of mass spectrometry experiments; M. Griffin, M. Jennings, and G. Paradis for flow cytometry support; and K. Cormier and C. Condon for histology support. We are also grateful for a fruitful collaboration with T. Westerling and Aiforia in developing an automated CNN for IHC quantification. Finally, we thank T. Tammela and J. Roper for early inspiration and mentorship in the colonoscopy-guided injection technique.
Competing interest declaration
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represents a conflict of interest with respect to any of the studies described in this manuscript. The Jacks laboratory also currently receives funding from Johnson & Johnson, but this did not support the research described in this manuscript. This work was supported by the Howard Hughes Medical Institute. The remaining authors declare no competing interests.
Footnotes
Code availability
Analyses were performed using open-source software and in-house scripts in R v4.0.2 and Python v2.7.13 that are available from the corresponding author on reasonable request. Quantification of CD4, CD8, and FOXP3 staining by IHC was performed using a custom convoluted neural network developed with Aiforia’s cloud-based image analysis platform. This is a commercial platform with proprietary technology and therefore did not generate any code. An interactive example of algorithm functionality can be provided free-of-charge upon request at https://www.aiforia.com.
Additional Information
Supplementary Information is available for this paper. All unique biological materials (e.g. organoid lines and lentiviral constructs used to generate them) derived in this work are available from the corresponding author (tjacks@mit.edu) upon reasonable request.
Data availability
Mass spectrometry data generated in this study have been deposited on MassIVE under accession code MSV000087648. TCRβ sequencing data generated in this study have been deposited on immuneACCESS under DOI: 10.21417/PMKW2021NC: clients.adaptivebiotech.com/pub/westcott-2021-nc. TCGA COADREAD data analyzed in this study are available for download on the NCI Genomics Data Commons. Numerical source data for Main and Extended Data Figures are provided as Source Data Figs. and Extended Data Figs. 1–6 and Source Data Fig. 7. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
References
- 1.Muzny DM et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Germano G et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Mandal R et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samstein RM et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newey A et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class i neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 7, 309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steimle A & Frick J-S Molecular Mechanisms of Induction of Tolerant and Tolerogenic Intestinal Dendritic Cells in Mice. J. Immunol. Res. 2016, 1958650 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim M et al. Critical Role for the Microbiota in CX3CR1+ Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49, 151–163.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Galarreta M. R. de et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) doi: 10.1158/2159-8290.CD-19-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roper J et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 35, 569–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roper J et al. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 13, 217–234 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence MS et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Supek F & Lehner B Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521, 81–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wherry EJ, Puorro KA, Porgador A & Eisenlohr LC The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J. Immunol. 163, 3735–45 (1999). [PubMed] [Google Scholar]
- 16.Spiotto MT et al. Increasing tumor antigen expression overcomes "ignorance" to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity 17, 737–47 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Engels B et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 23, 516–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jhunjhunwala S, Hammer C & Delamarre L Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 1–15 (2021) doi: 10.1038/s41568-021-00339-z. [DOI] [PubMed] [Google Scholar]
- 19.Richard AC et al. T cell cytolytic capacity is independent of initial stimulation strength. Nat. Immunol. 19, 849–858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szolek A et al. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurtz V et al. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreatta M & Nielsen M Gapped sequence alignment using artificial neural networks: Application to the MHC class i system. Bioinformatics 32, 511–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters B & Sette A Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, Sidney J, Pinilla C, Sette A & Peters B Derivation of an amino acid similarity matrix for peptide:MHC binding and its application as a Bayesian prior. BMC Bioinformatics 10, 394 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hundal J et al. PVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 8, 409–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (80-. ). 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf Y et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 179, 219–235.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karandikar SH et al. Identification of epitopes in ovalbumin that provide insights for cancer neoepitopes. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh M et al. Guidance document: Validation of a high-performance liquid chromatography-tandem mass spectrometry immunopeptidomics assay for the identification of HLA class I ligands suitable for pharmaceutical therapies. Molecular and Cellular Proteomics vol. 19 432–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebrahimi-Nik H et al. Mass spectrometry–driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DuPage M et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DuPage M, Mazumdar C, Schmidt LM, Cheung AF & Jacks T Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dow LE et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 161, 1539–1552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drost J et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015). [DOI] [PubMed] [Google Scholar]
- 37.de Sousa e Melo F et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543, 676–680 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Schell MJ et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 7, 11743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gubin MM et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mlecnik B et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 44, 698–711 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Parish IA et al. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood 113, 4575–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin W-HW et al. CD8+ T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep. 17, 1773–1782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danilo M, Chennupati V, Silva JG, Siegert S & Held W Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 22, 2107–2117 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Zhou X et al. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–40 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durward M, Harms J & Splitter G Antigen specific in vivo killing assay using CFSE labeled target cells. J. Vis. Exp. 45 (2010) doi: 10.3791/2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wherry EJ T cell exhaustion. Nat. Immunol. 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 47.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siddiqui I et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Kurtulus S et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunity 50, 181–194.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Provine NM et al. Immediate Dysfunction of Vaccine-Elicited CD8+ T Cells Primed in the Absence of CD4+ T Cells. J. Immunol. 197, 1809–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Stipdonk MJB et al. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer Res. 69, 7784–7792 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Liu H et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cella M et al. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 184, 747–752 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byrne KT & Vonderheide RH CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 15, 2719–2732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabacaoglu D, Ciecielski KJ, Ruess DA & Algül H Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 9, 1878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wells DK et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ott PA et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 183, 347–362.e24 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Krogsgaard M & Davis MM How T cells ‘see’ antigen. Nat. Immunol. 6, (2005). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 61.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weese D, Holtgrewe M & Reinert K RazerS 3: Faster, fully sensitive read mapping. Bioinformatics 28, 2592–2599 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Kim S et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Narzisi G et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aran D, Sirota M & Butte AJ Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuraguchi M et al. Adenomatous Polyposis Coli (APC) Is Required for Normal Development of Skin and Thymus. PLoS Genet. 2, e146 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson L et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–6 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Marino S, Vooijs M, van Der Gulden H, Jonkers J & Berns A Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004 (2000). [PMC free article] [PubMed] [Google Scholar]
- 70.Hao Z & Rajewsky K Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med. 194, 1151–64 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hogquist KA et al. T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27 (1994). [DOI] [PubMed] [Google Scholar]
- 72.Chu VT et al. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 16, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hildner K et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science (80-. ). 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miyoshi H & Stappenbeck TS In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DuPage M, Dooley AL & Jacks T Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 4, 1064–1072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sánchez-Rivera FJ et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Certo MT et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 8, 671–676 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jaeger AM et al. Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res. 25, 6392–6405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steinert EM et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data generated in this study have been deposited on MassIVE under accession code MSV000087648. TCRβ sequencing data generated in this study have been deposited on immuneACCESS under DOI: 10.21417/PMKW2021NC: clients.adaptivebiotech.com/pub/westcott-2021-nc. TCGA COADREAD data analyzed in this study are available for download on the NCI Genomics Data Commons. Numerical source data for Main and Extended Data Figures are provided as Source Data Figs. and Extended Data Figs. 1–6 and Source Data Fig. 7. All other data supporting the findings of this study are available from the corresponding author on reasonable request.