

Abstract
Adenosine-to-inosine (A-to-I) editing is a conserved eukaryotic RNA modification that contributes to development, immune response, and overall cellular function. RNA editing patterns can vary significantly between different cell and tissue types, and hyperactive A-to-I signatures are indicative of several diseases, including cancer and autoimmune disorders. Because of the biological and clinical importance of these differences, there is significant need for efficient methods to measure overall A-to-I editing levels in cellular RNA. The current standard approach relies on RNA-seq to indirectly detect editing sites, which requires significant investments in time and material as well as extensive computational analysis. Here, we utilize Endonuclease V (EndoV), which binds specifically to inosine in RNA, to develop a protein-based chemiluminescent bioassay to directly profile A-to-I RNA editing activity. We previously showed that EndoV can bind and enrich A-to-I edited transcripts prior to RNA-seq, and we now leverage this activity to construct an EndoV-linked immunosorbency assay (EndoVLISA) as a rapid, plate-based chemiluminescent method for measuring global A-to-I editing signatures in cellular RNA. We first optimize and validate our assay with chemically synthesized oligonucleotides, illustrating highly selective and sensitive detection of inosine in RNA. We then demonstrate rapid detection of inosine content in treated cell lines, demonstrating equivalent performance against current standard RNA-seq approaches. Lastly, we deploy our EndoVLISA for profiling differential A-to-I RNA editing signatures in normal and diseased human tissue, illustrating the utility of our platform as a diagnostic bioassay. Together, the EndoVLISA method is cost-effective, straightforward, and utilizes common laboratory equipment, offering a highly accessible new approach for studying A-to-I editing. Moreover, the multi-well plate format makes this the first assay amenable for direct high-throughput quantification of A-to-I editing for applications in disease detection and drug development.
Graphical Abstract
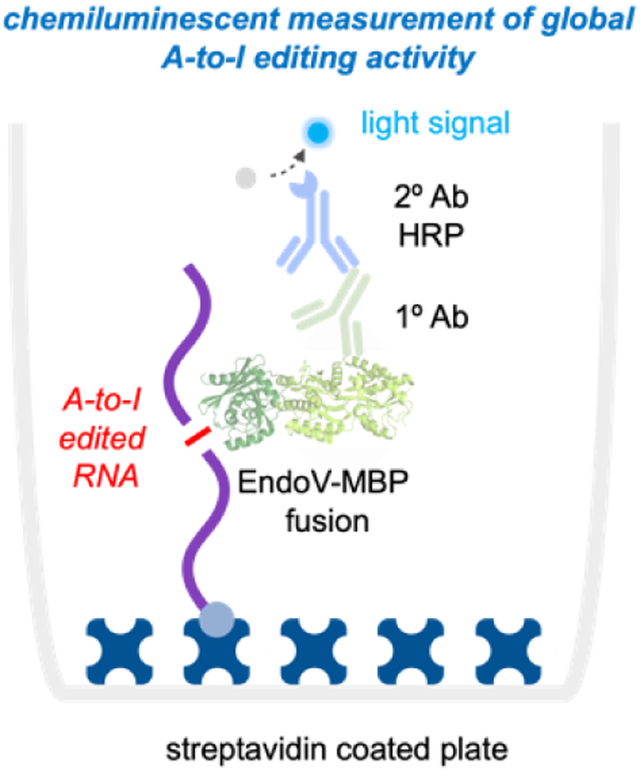
Adenosine-to-inosine (A-to-I) editing is a critical RNA modification and controls many cellular pathways. Dysregulation of this process is also linked with autoimmune disease, neurodegenerative disorders, and several types of cancer. Despite this importance, detecting this modification is both costly and time consuming. Here, we develop a protein-based chemiluminescent bioassay to rapidly characterize global A-to-I editing signatures in normal and diseased human tissue, addressing a significant technological need for studying RNA editing and its relationship with disease.
Introduction
Adenosine-to-inosine (A-to-I) RNA editing is catalyzed by adenosine deaminases acting on RNA (ADARs), and is a critical and widespread RNA modification in eukaryotes.1 Deamination changes the hydrogen bonding pattern of the nucleobase, and inosines are effectively decoded as guanine by cellular machinery (Fig. 1a). Editing sites within protein-coding regions can thus directly alter amino acid sequences and produce different protein isoforms.2 However, the vast majority of A-to-I editing occurs within repetitive Alu elements, which are embedded throughout the human transcriptome and form long (~300 bp) inverted dsRNA repeats that are recognized and edited by ADAR1.3 Millions of these A-to-I sites have been identified,3–6 and editing within these Alu regions is now recognized as an essential mechanism to regulate immune system activation and differentiate “host” RNA from pathogenic transcripts.7 In the absence of ADAR1, unmodified dsRNA accumulates within the cell and activates cytotoxic interferon responses.8–10 Genetic knockout of ADAR1 is also a lethal phenotype in mice, causing developmental defects arising from uncontrolled immune responses in specific tissues.8, 11 In humans, dysregulated A-to-I editing is also linked with autoimmune disorders,12–14 and most cancers display ADAR1 overexpression and hyperediting signatures as a potential means of immune system evasion.15–16 Interestingly, deletion of ADAR1 has been shown to overcome immune checkpoint blockade resistance in tumors,17 and there is significant interest in developing treatment methods to inhibit ADAR enzymes and decrease global editing activity. Beyond these critical roles in immune system function and embryogenesis, A-to-I editing is also broadly implicated in stem cell differentiation and neurological activity,2, 7, 18 and editing malfunctions in the nervous system can cause epilepsy, amyotrophic lateral sclerosis, glioblastoma, schizophrenia, autism, and Alzheimer’s disease.19–23
Figure 1. Designing a chemiluminescent bioassay for inosine detection.
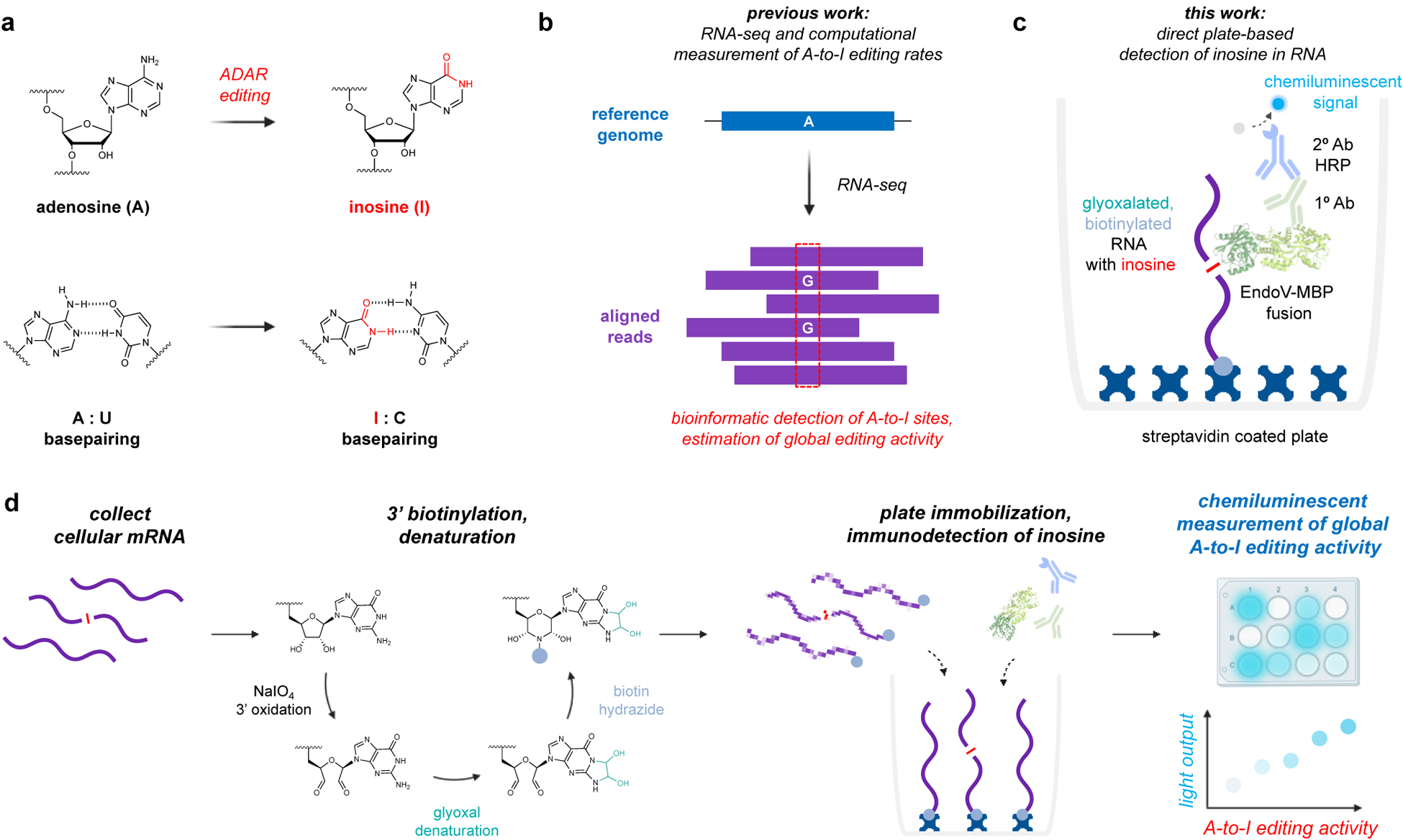
a) Adenosines are converted to inosine by ADAR enzymes. b) A-to-I sites are typically identified as A-G transitions between RNA-seq reads and a reference genome. c) We propose direct detection of inosine with an Endonuclease V – Linked Immunosorbency Assay (EndoVLISA). Cellular RNA is first glyoxal denatured, biotinylated, and immobilized in a streptavidin-coated well. Inosine is then specifically recognized by EndoV, which is fused to a maltose-binding protein (MBP) affinity tag. Wells are then probed with a mouse anti-MBP primary (1°) antibody and a goat anti-mouse secondary (2°) antibody conjugated to horse-radish peroxidase (HRP) to generate a chemiluminescent signal. d) Chemical strategy to prepare RNA for analysis using sodium meta-periodate (NaIO4 to oxidize terminal 3’ OH groups followed by covalent denaturation with glyoxal. Biotin-PEG4-hydrazide is then reacted with 3’ dialdehyde groups to immobilize RNA onto a streptavidin-coated surface.
Although a critically important cellular process, our overall understanding of A-to-I editing regulation and its broader functional roles remains limited. Similarly, despite its clear relationship with several diseases,12–13, 16, 20, 24–28 both ADAR activity and global A-to-I editing signatures have yet to be effectively leveraged as biomarkers for disease diagnosis. These limitations are the direct result of technical challenges associated with detecting and measuring editing activity. One of the earliest methods employed enzymatic digestion of isolated mRNA followed by P32 radiolabeling and thin-layer chromatography.29
While this study provided a rough estimate of iinosine content in rat mRNA (~0.01 – <0.001% of all nucleotides) and identified tissue-level differences in editing activity, this method is laborious, hazardous, and suffers from low precision, and thus has not been adopted beyond these initial experiments. Because inosine is decoded as guanine during reverse transcription, the current standard method utilizes high-throughput RNA sequencing (RNA-seq) to identify editing sites from A-G transitions (Fig. 1b).30 This approach offers high-resolution mapping of A-to-I sites throughout the transcriptome, and when focused solely on Alu elements, enables computational estimation of global ADAR1 RNA editing levels.31 While these bioinformatic methods are effective and allow post-hoc measurement of A-to-I activity from published RNA-seq experiments, generating new datasets still requires high-cost consumables, specialized instrumentation, and data turnaround times can often extend past weeks or even months. RNA-seq is also limited to capturing small “windows” of RNA, making this technique inconsistent and prone to random sampling errors. Editing sites are also quite rare in the context of total RNA3, 29 and so RNA-seq is less suited for comprehensive measurement of editing levels from cellular samples. Because of these challenges, RNA-seq requires excessive amounts of RNA input material, very high numbers of sequencing reads, and customized computational pipelines to achieve sufficient depth and coverage for accurate indexing of global A-to-I levels.31–33 Modified bases can also be quantified by digesting cellular RNA and analyzing by LC/MS,34–37 and while capable of measuring inosine content in biological samples,38–40 these methods require impractically high amounts of RNA sample and utilize costly instrumentation. Additionally, LC/MS typically lacks the resolution needed to detect rare modifications or distinguish structurally similar nucleotides. Several commercial suppliers have also developed enzyme-based fluorometric kits for detecting free inosine, but these exhibit low detection sensitivity and cannot reliable measure inosine below ~0.1 nmol. A bioluminescent system was also recently developed, in which A-to-I editing of a UAG stop codon embedded in a luciferase mRNA reporter produces a measurable signal for inferring ADAR activity levels in immortalized cells.41 While this method enabled the first high-throughput screen for potential ADAR-modulating small molecules, this approach estimates A-to-I activity at a single editing site and does not directly assay actual inosine content in cellular RNA. Additionally, this approach requires genetic manipulation for establishing a stable reporter cell line, and thus cannot be extended beyond in vitro contexts or used as a diagnostic tool in primary cells or tissues.
Despite these advances, RNA-seq is still by far the most widely adopted and current method-of-choice for detecting A-to-I editing, and there remains a significant need for technology that can rapidly and inexpensively profile global editing signatures in a variety of biological contexts. This would not only provide faster and more accurate information regarding the regulation of global A-to-I editing activity in humans, but would also leverage differential A-to-I signatures as a diagnostic biomarker in several disease contexts. Additionally, an assay platform capable of detecting global changes in editing levels would facilitate the design and testing of pharmacological inhibitors of ADAR activity in both immortalized cells and primary tissues.
Toward these goals, we were inspired by our previous explorations of Endonuclease V (EndoV), a conserved nucleic acid repair enzyme that can specifically recognize inosine in nucleic acids.42–44 EndoV is present across all domains of life,45 and naturally utilizes Mg2+ to cleave inosine-containing substrates. In prokaryotes, this activity appears to have evolved for repair of inosine lesions in DNA,42 while in humans and other higher eukaryotes, EndoV exhibits specific activity toward inosine in RNA and is speculated to have roles in degrading A-to-I edited transcripts.43–44 Interestingly, enzyme activity can be modulated by replacing Mg2+ with Ca2+, enabling EndoV to bind instead of cleave inosine-containing nucleic acid substrates.46 Inspired by this, we recently demonstrated that E. coli EndoV (eEndoV) has high affinity and selectivity for A-to-I edited transcripts and can be repurposed to act as an “antibody” to bind and enrich edited transcripts prior to RNA-seq.47–48 Here, we leverage these properties to construct an EndoV-linked immunosorbency assay (EndoVLISA) to directly measure global A-to-I RNA editing signatures using a simple chemiluminescent plate-based bioassay (Fig. 1c). We first design an assay workflow to denature, biotinylate, and immobilize RNA into streptavidin-coated plates for subsequent immunodetection and chemiluminescent measurement (Fig. 1d). We then systematically optimize key assay parameters to validate its accuracy and sensitivity for detecting inosine in RNA, and further benchmark its performance against LC/MS and RNA-seq. We then demonstrate EndoVLISA as a facile bioassay for rapidly profiling A-to-I RNA editing in several human cell and tissue samples, addressing a significant gap in RNA-based analysis and improving our ability to profile global epitranscriptomic changes.
Results and Discussion
In designing a biosensing platform for measuring global changes in A-to-I RNA editing, we were inspired by the “antibody-like” qualities of EndoV and hypothesized that these could be applied in other established immunodetection approaches. In particular, we identified enzyme-linked immunosorbency assay (ELISA) as a potential platform due to its well-characterized performance, high sensitivity, and overall versatility for quantifying low abundance analytes in highly complex mixtures.49–50 These qualities have positioned ELISA as a routine research technique and a powerful diagnostic tool, and have enabled sensitive detection of human antibody titers,51 cytokine and small-molecule analytes in serum,52 and viral particles in different biological fluids.53 Moreover, primary antibodies identified for other rare nucleic acid modifications, including N6-methyladenosine (m6A),54–55 5-methylcytosine (5mC),56 5-hydroxymethylcytosine (5hmC), and 5-carboxylcytosine (5caC),57 have now also been successfully deployed in commercial ELISA formats for sensitively detecting global editing rates in RNA and DNA. ELISA also utilizes inexpensive consumables and commonplace equipment.
To apply this assay format for quantifying inosine, we envisioned that RNA would first be biotinylated and immobilized into a streptavidin-coated well to enable the binding, washing, and detection steps that are typical of other ELISA approaches (Fig. 1c, d). We identified sodium metaperiodate (NaIO4) as a suitable approach for 3’ biotinylation of transcripts (Fig. 1d),58 and we reasoned that end-labeling and immobilizing each RNA strand would in theory maximize our potential sensitivity in detecting inosine by enabling multiple EndoV binding events per transcript while only occupying one streptavidin site on the two-dimensional surface of the well. In our initial demonstration using eEndoV for capturing A-to-I edited RNAs, we also observed that EndoV had a strong preference for binding inosine in unstructured, single-stranded RNA.47 This was especially problematic because ADAR targets structured duplexes for A-to-I editing,1, 59 and thus inosines are highly likely to reside in double-stranded RNAs. To circumvent this challenge, we identified glyoxal, a covalent denaturant that reacts with guanosine, adenosine, and cytidine, as an effective means for disrupting RNA structure (Fig S1a).60 Importantly, glyoxal does not react with inosine, and we found that this treatment step was not only compatible with EndoV-RNA pulldown, but enhanced the ability of EndoV to bind A-to-I edited transcripts regardless of structure (Fig. S1b).47
Incorporating this glyoxalation step, we first sequentially treated our test RNA I strand with NaIO4, glyoxal, and biotin-PEG3-hydrazide. We then fluorescently tagged this RNA with Cy5 and loaded increasing amounts into each well of a streptavidin-coated 96-well plate. After extensively washing each well, we then measured fluorescent signal across the plate. As shown in Figure S2, signal increased proportionally from 0 to 5 pmol of the treated RNA and plateaued at higher loading amounts, suggesting a maximum capacity of ~5 pmol biotinylated RNA. Considering the median length of human mRNA (~1400 nt),61 this capacity would conveniently enable us to measure inosine content in up to ~2–3 μg mRNA per well. Additionally, we observed no binding in a non-biotinylated RNA control strand, indicating that non-specific interactions in this step were minimal.
One of the key reasons that ELISA offers such high sensitivity is that sequential binding of analytes to both a primary (1°) and secondary (2°) antibody enables signal amplification. This is further enhanced by conjugating an enzyme to the 2° antibody, such as horse-radish peroxidase (HRP), which catalytically generates multiple detection signals for every initial analyte molecule bound. While powerful, this design also presents assay complexity, and ELISA platforms are known for requiring optimization to balance sensitivity with background signal. Our proposed system shares this potential complexity, wherein we first bind inosine in RNA with a recombinant eEndoV fused to a maltose-binding protein (MBP) affinity tag, followed by probing with a mouse anti-MBP 1° antibody and an HRP-conjugated, goat-anti-mouse 2° antibody (Fig 1c).
Each of these components are key to robustly measuring global changes in A-to-I editing, so we first sought to systematically optimize our workflow. Toward this end, we also identified mRNA as an analyte of choice, as the vast majority of ADAR1 editing events occur within repetitive Alu elements in these transcripts.3–6 Importantly, this would also eliminate RNA species that might interfere with our assay, including several transfer RNAs (tRNA) that contain inosine in the anticodon loop.62 While this modification is essential for protein translation, tRNA editing is performed by a different enzyme family (ADAT) and is unrelated to A-to-I editing activity catalyzed by ADAR1. Using the rough estimate of rat inosine content (~50–200 fmol/μg mRNA)29 as well as RNA-seq analysis showing that global human editing rates are ~5-fold higher,4 we wanted to first optimize our EndoVLISA approach to target this range (~0.1–2 pmol/μg mRNA). We also designed a combinatorial screen to systematically test each component (EndoV-MBP, 1° and 2° Ab-HRP) so that each EndoVLISA mixture could be assayed with 1) no RNA (blank), 2) “low on-target” (100 fmol/well RNA I), 3) “high on-target” (2 pmol/well RNA I) and 4) “high off-target” (2 pmol/well RNA A) (Fig. S9a). We also stringently measured assay linearity from 0 – 2 pmol RNA I using Pearson correlation coefficients (r) and associated p values for all EndoVLISA combinations. Interestingly, a clear trend emerged in that higher amounts of 1° MBP-targeting antibody (1:1000 and 1:2000 dilutions) and lower amounts of 2° antibody-HRP (1:20,000 and 1:40,000) yielded excellent linearity (p < 0.05, Fig. 9b). Conversely, lower 1° and higher 2° antibody concentrations were less linear and displayed both higher variability (Figs. S3–S5), which we suspect is due to use of a monoclonal 1° antibody which binds to MBP in a 1:1 ratio, whereas multiple 2° HRP (polyclonal) antibodies can subsequently attach to this complex.
Background signal is also a major consideration with ELISA development, so we evaluated performance with high amounts of both RNA A and RNA I (2 pmol/well). Better overall selectivity was again observed with higher 1° antibody concentrations (1:1000 and 1:2000, Figs. S9c, S6–S8), and there was a clear performance optimum when using 2° HRP at a dilution of 1:20,000. From this screen, we ultimately identified several EndoVLISA reagent combinations that produced comparable performance and robustness (Table S1), suggesting overall assay flexibility in a variety of conditions and indicating that minor adjustments in these components would not significantly alter assay quality.
Using optimized conditions, (EndoV 1:1000, 1° antibody 1:1000, and 2° antibody-HRP 1:20,000), we next confirmed that our method was specific and that each component was necessary for signal generation. As shown in Fig. 2a, signal was only observed in wells receiving all 3 detection components. Importantly, no response was generated without EndoV, indicating low non-specific binding of either the 1° or 2° HRP antibody. We also began testing a larger range of inosine concentrations in RNA, and found that our assay was both linear and sensitive, with an estimated lower detection limit of ~100 fmol (Fig. S10). Seeking to maximize this sensitivity, we lastly explored different chemiluminescent substrates. In particular, we employed SuperSignal™ Pico substrate (Thermo Fisher) for our initial optimization assays, and next tested the SuperSignal™ Atto variation, which is reported as the most sensitive commercially available substrate. Using slightly modified conditions to accommodate this substrate (2° HRP to 1:40,000) we observed excellent linearity below 0.5 pmol inosine and a lower limit of detection approaching ~30 fmol, representing a 2–3-fold improvement over the Pico substrate (Fig. S11–S12).
Figure 2. Characterizing EndoVLISA performance.
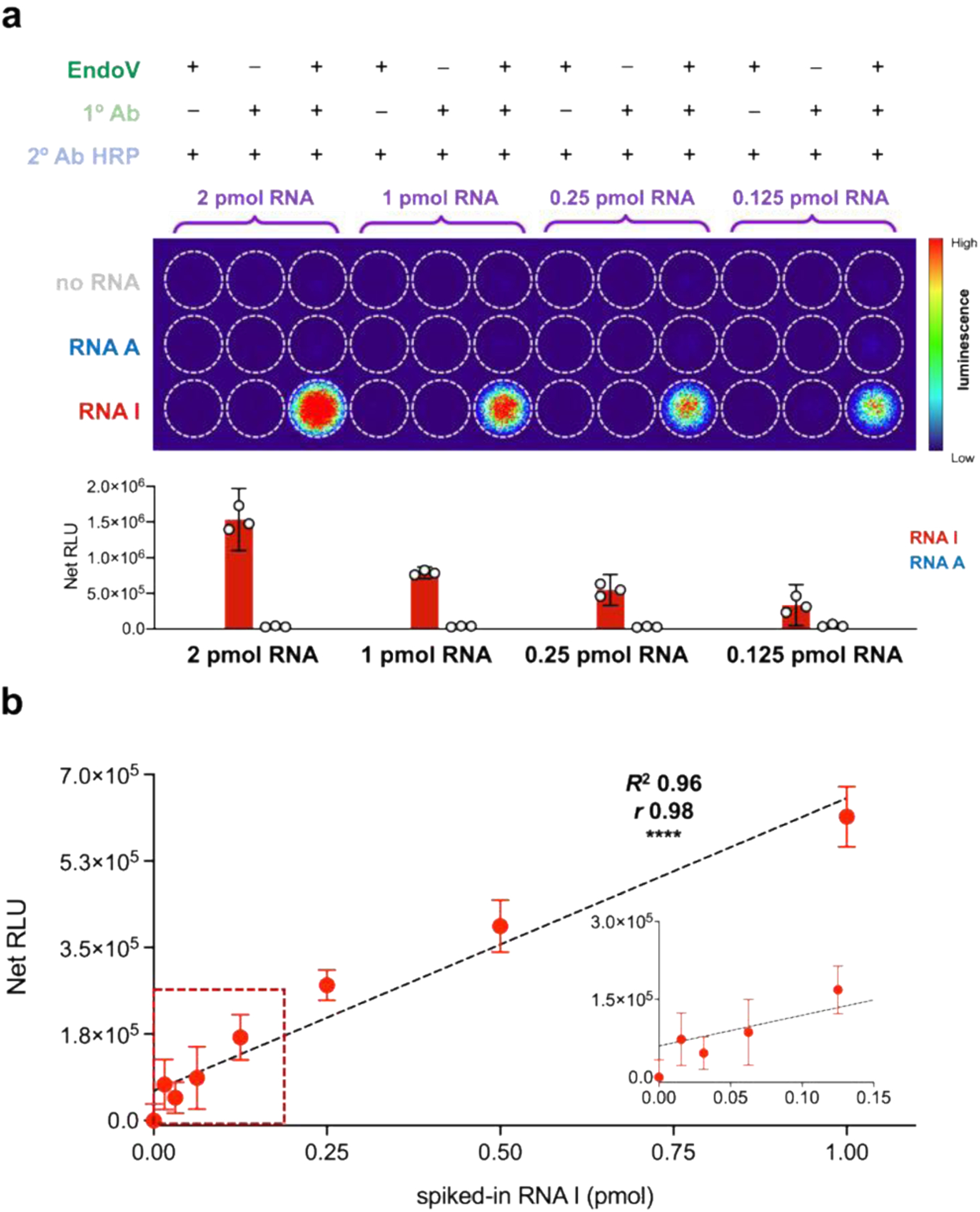
a) Representative image of chemiluminescent detection of inosine. Different RNA samples were immobilized into a 96-well streptavidin coated plate, followed by immunodetection with the indicated reagents and incubated with substrate for 1 minute. Reactions were then transferred to a clear 96-well plate and chemiluminescent signal was captured using a Typhoon biomolecular imager. White outlines indicate relative position of each well. Heat map values represent signal intensity using the acquisition software. Bar graph denotes signals generated from different RNA A and RNA I loading amounts (2, 1, 0.25, and 0.125 pmol per well). b) Linearity and sensitivity of detecting inosine in complex mixtures. Decreasing amounts of RNA I strand were spiked into 1 μg of in vitro transcribed mRNA and detected using optimized EndoVLISA workflow (EndoV 1:1000, 1° 1:1000, 2° HRP 1:40,000, and SuperSignal™ Atto Substrate). Inset displays a zoomed-in portion of the curve illustrating a lower limit of detection ~62.5 fmol of inosine per μg RNA. Values represent mean (n = 3) and error bars denote 95% confidence intervals. Linear regression (black dashed line), R2 and pearson (r) correlation (**** denotes p < 0.0001) were computed in Prism.
While these results were encouraging, detecting small amounts of A-to-I editing in complex samples is pivotal to our eventual goals, and so we next tested EndoVLISA performance in the presence of off-target mRNA. We first synthesized an ~800 nt mRNA by in vitro transcription, importantly using only the canonical ribonucleosides (A, U, C, G) to enable precise control over nucleobase content. We first combined this mRNA (1 μg) with different ratios of our RNA I strand, and subjected these samples to both glyoxal denaturation and 3’ biotinylation prior to EndoVLISA. While we did observe an overall signal decrease in the presence of off-target mRNA (Figure S13), these samples are more complex than our previous tests using oligonucleotides, and it is likely that EndoV and antibody binding is less efficient. Despite this, EndoVLISA displayed excellent linearity from 0–1 pmol (Fig. 2b). Additionally, we were able to reliably detect ~100 fmol inosine per μg mRNA, indicating that our method can sense 1 inosine molecule for every ~30,000 nucleotides (0.003%), which is in the range of the estimated lower levels of inosine content in cellular mRNA.4, 29
Although LC/MS is not typically used to quantify inosine content in cellular RNA due to practical limitations, we were curious to benchmark our assay relative to this approach. We first analyzed decreasing amounts of inosine ribonucleoside, and found that MS detection was only reliable above ~5 pmol (Figs S14, S15). To directly compare with our previous EndoVLISA selectivity test (Fig. 2b), we also spiked inosine into 1 μg of an equimolar mixture containing each of the four canonical nucleosides (A, U, C, G) as well as the two other major modified bases found in RNA (m6A and pseudouridine) (Fig. S16, S17). Given the upstream chromatography separation step, LC/MS was predictably unaffected by this sample complexity, and yielded similar detection performance when mixed with other ribonucleosides (Fig. S17). This lower limit of detection (~5–10 pmol using an accurate mass TOF-ESI instrument) was comparable to other studies employing LC/MS for inosine quantification,38–39 but still represented >100-fold decrease in sensitivity compared to EndoVLISA (Fig 2b). While specialized triple-quadrupole instruments can provide enhanced sensitivity for detecting inosine (<20 fmol),40 these approaches can require large amounts of RNA (50–100 μg) to achieve this level of performance. These limitations may explain why LC/MS methods have found wide use for detecting more abundant RNA modifications such as m6A but have not been widely adopted for detecting A-to-I editing.
After demonstrating feasibility and robustness of our method, we next sought to test our approach in a cellular context for measuring global changes in A-to-I editing. We were also curious how EndoVLISA would compare to inosine quantification with RNA-seq. Because the majority of editing sites are found in mRNA Alu elements,3–6, 63 the “Alu Editing Index” (AEI) was developed to computationally measure editing frequencies at these sites and provide a global estimate of ADAR1 editing activity.31, 64–65 To compare this approach to EndoVLISA, we first selected HEK293T as a suitable cell line for overexpressing ADAR1. Interestingly, most immortalized cell lines exhibit very low editing activity compared to human primary cells and tissues, which is partly explained by low ADAR1 expression levels.65 AEI profiling of different cell lines also showed that 293T cells display particularly low editing levels (Fig. S18), and this would allow us to simulate and detect ADAR1 increases that are characteristic of different developmental and disease-specific changes. We first induced overexpression using increasing amounts of a plasmid encoding a GFP-tagged ADAR1 p110 isoform.66 We then harvested whole cell lysate and isolated mRNA by two rounds of oligo d(T)25 purification (New England Biolabs). After validating mRNA purity and content in this workflow (Fig. S19), we then tested these materials by western blotting, RNA-seq, and EndoVLISA (Fig. 3). We confirmed very low ADAR1 levels in untreated 293T cells, and observed a clear increase in ADAR1 expression with increasing amounts of transfected plasmid by both western blot (Fig. 3a,b) and GFP signal in collected lysates (Fig. S20).
Figure 3. EndoVLISA detects global cellular changes in A-to-I editing.
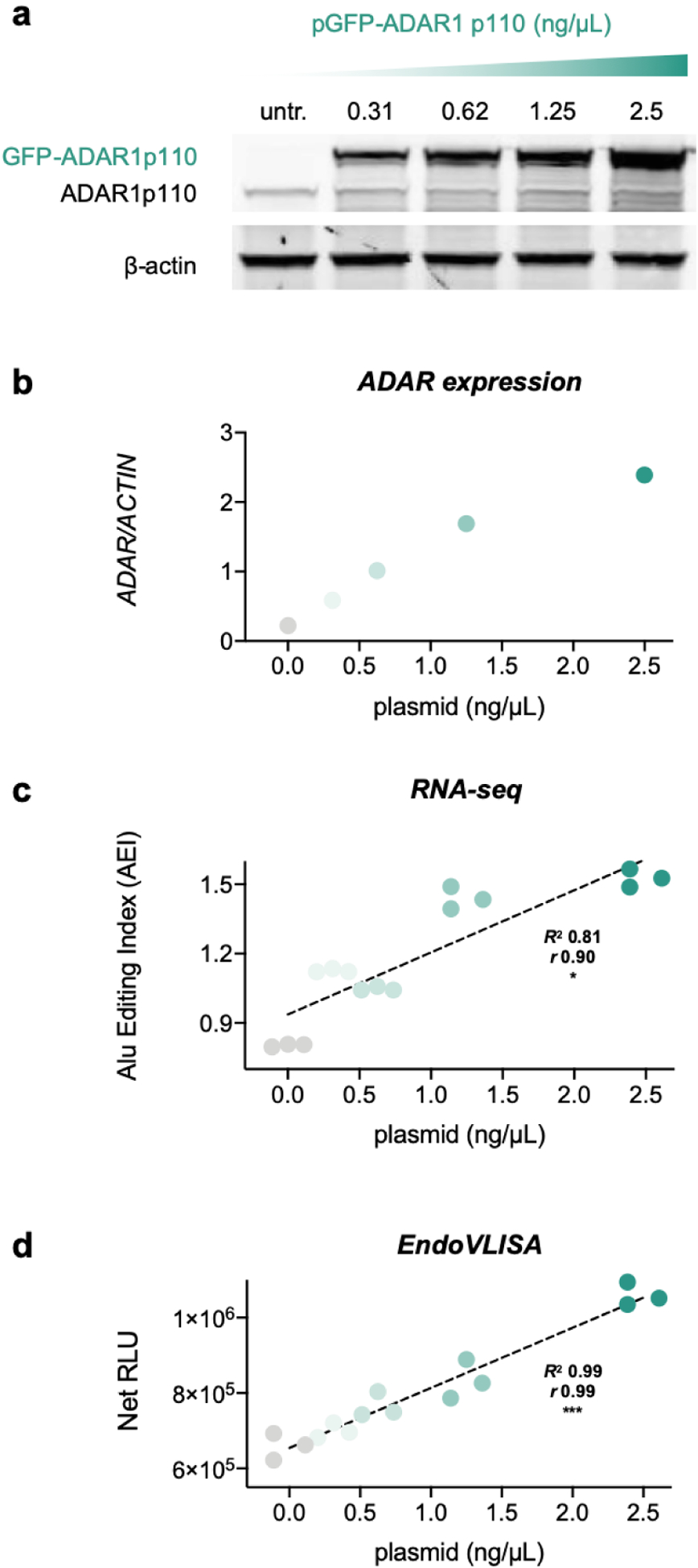
HEK293T cells were transfected with increasing amounts of GFP-tagged ADAR1 p110. a) Western blotting of lysates from untreated (untr.) and transfected 293T cells, illustrating higher ADAR expression. b) Densitometric estimation of ADAR1 expression relative to β-actin control. c) A-to-I editing activity calculated using the Alu Editing Index (AEI). Values represent individual samples for each group (n = 3). d) mRNA material was also tested using EndoVLISA. Individual values (n = 3 for each treatment group) represent net relative luminescent units (RLU, arbitrary units) by subtracting blank well values (no RNA). Linear regression (black dashed lines), R2 and pearson (r) correlation (* denotes p < 0.05, *** p < 0.001) were computed in Prism.
Using isolated mRNA from these treated cells, we performed RNA-seq to calculate global A-to-I editing activity and observed a proportional rise in AEI values with overall ADAR expression (Fig. 3c). In parallel, we tested this same mRNA material using EndoVLISA, and were delighted to see a commensurate rise in chemiluminescent response that correlated with ADAR1 transfection (Fig. 3d). Using a standard curve comprised of RNA I control strand added to transcribed mRNA, we also estimated inosine concentrations and saw a comparable increase in these l levels (Fig. S21). Both RNA-seq and EndoVLISA showed statistically significant linearity (Figs. 3c,d) and were also in good agreement with each other (Fig. S22), indicating our method was reliable for detecting global increases in A-to-I editing. Although the AEI was more sensitive in detecting smaller changes in ADAR1 activity, particular between untreated cells and those receiving lower plasmid amounts (<1 ng/ul), this approach was less responsive in higher transfection ranges. As shown in Fig. 3c–d, similar index values were observed in these samples, whereas EndoVLISA yielded more proportional responses. Overall, our results demonstrated that EndoVLISA is feasible and effective for accurately detecting global cellular changes in A-to-I RNA editing. Additionally, our method produced highly similar responses compared to RNA-seq while offering significant time and cost-savings per sample, (~200-fold reduction, Tables S2, S3) positioning this method as a highly accessible and robust tool for characterizing RNA editing signatures.
We were next interested in testing our method in mRNA from primary human samples, as A-to-I editing can vary between different organs, tissues, and cell types within the body.3, 31 These editing changes are key drivers of stem cell differentiation, embryogenesis, and immune activation, and large-scale editing fluctuations can also be indicative of a variety of developmental diseases, autoimmune illnesses, neurodegenerative disorders, and cancer types.7, 12, 21–27 Towards our goal of profiling these changes, we first measured relative inosine levels in a panel of normal human tissues. Although most organs exhibit similar A-to-I activity, higher editing has been observed in both brain and aorta, while skeletal muscle and pancreatic mRNA display uniquely low editing rates.3, 31 Excitingly, we were able to detect these large-scale differences using EndoVLISA (Fig. 4b). Additionally, these global trends are highly similar to large-scale Alu indexing of >9,000 RNA-seq datasets from the Genotype-Tissue Expression (GTEx) project (Fig. 4a).31, 67 In particular, while brain and aorta mRNA displayed the highest mean response among all tissues in both RNA-seq and EndoVLISA, these were also similar to either breast (mammary gland) or kidney mRNA (Figs. 4a,b). However, skeletal muscle and pancreas mRNA were both substantially lower than other tissues and reproduced previous observations of these differences.3, 31
Figure 4. EndoVLISA detects tissue-specific A-to-I editing signatures.
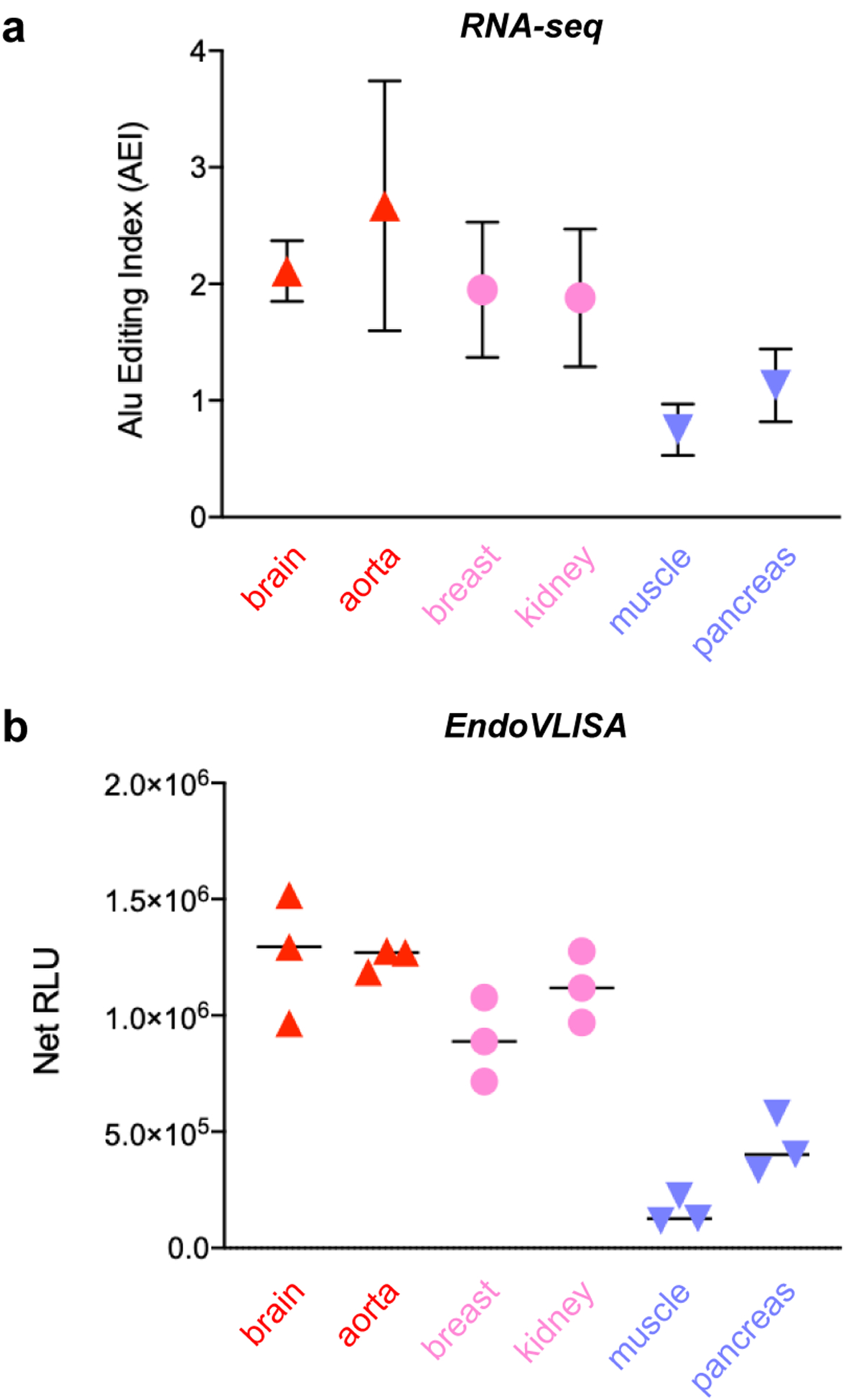
a) RNA-seq AEI analysis of A-to-I editing in different human tissues from the GTEx project.67 Reproduced from compiled data,31 and values represent mean and SD (brain n = 103 individuals, aorta n = 246, breast n = 219, kidney n = 38, muscle n = 450, pancreas n = 192). b) Purified mRNA (500 ng) from the indicated human tissues was tested using EndoVLISA. Values represent net relative luminescent units (RLU, arbitrary units) calculated by subtracting appropriate blank wells (no RNA) from each set. Data points represent individual values from each well (n = 3 for each sample).
Overall signal was also much higher than in our experiments using 293T cells (Fig. 3), supporting previous analyses showing lower overall editing levels in immortalized cell lines.65 This higher signal did require us to use 500 ng instead of 1 μg mRNA per well as used in our previous tests, and we were able to estimate inosine content using an appropriate standard curve (Fig. S23). However, some of these samples produced very high signal (especially aorta and brain) and were slightly above the maximum amount of RNA I used in our standard curve (2 pmol), so actual inosine concentrations may be slightly higher. Regardless, EndoVLISA consistently detected tissue-level epitranscriptomic signatures and was highly comparable to large-scale RNA-seq analyses (Fig. 4) while requiring a fraction of the time and cost, further verifying our method as an effective means for profiling global A-to-I editing signatures.
ADAR1 overexpression is rapidly emerging as a potential molecular mechanism for cancer progression, and the majority of cancer types display significantly upregulated A-to-I RNA editing levels.15–16, 26 Because of this potential as a disease biomarker, we were interested in using EndoVLISA to identify epitranscriptomic differences between healthy and diseased tissue. We first obtained total RNA from several normal human tissues as well as samples from breast cancer, kidney renal cell carcinoma, lung adenocarcinoma, and liver hepatocellular carcinoma. We purified mRNA from each source through two rounds of polyT selection, and then performed EndoVLISA.
As shown in Fig 5, we detected significantly higher chemiluminescent signal in all 4 cancer types, reflecting large overall increases in global editing activity. While these tumor RNA samples were actually too high for accurate quantification using our typical standard curve, Net RLU values were used to roughly estimate fold-increases in inosine content (Fig. 5). In particular, breast, liver, and lung cancer samples exhibited the largest increases, displaying ~8, ~6, and ~5-fold upregulated activity, respectively. These observations were in close agreement with prior studies of RNA editing in all three cancer types,25, 68–70 and additionally support large-scale bioinformatic profiling of editing changes from the Cancer Genome Atlas,71 which identified significant upregulation of A-to-I activity in breast, liver, and lung cancer progression.16, 26 Interestingly, different kidney carcinoma types can exhibit hypo- or hyperediting signatures,16, 24 and while we observed ~4-fold increase in EndoVLISA response in our sample, RNA-seq analysis of a similar renal cancer found no significant difference in average Alu editing rates between normal and tumorigenic tissue.26 However, this study also noted variability between individuals and observed both over- and underediting phenotypes in renal carcinoma datasets (62 patients),26 and so our results may simply reflect the composition of our own sample. Additionally, Cancer Genome Atlas datasets revealed that while overall editing differentials were higher in kidney tumors compared to matched normal tissue, Alu-specific editing was significantly depleted in these samples.16 These results suggest additional molecular mechanisms that regulate editing patterns in these cancer types and may point to a major limitation in relying solely on Alu editing signatures for inferring global cellular A-to-I editing activity. Expanding EndoVLISA profiling of tumor-specific editing signatures in high numbers of individuals will be a worthy pursuit for resolving this heterogeneity and will likely enable identification of other important editing regulation mechanisms and drivers of cancer progression. Overall, these experiments demonstrated rapid, effective, and consistent EndoVLISA detection of upregulated A-to-I editing activity in several known cancer types, highlighting the bioanalytical potential of our method.
Figure 5. EndoVLISA detects upregulated A-to-I editing in several cancer types.
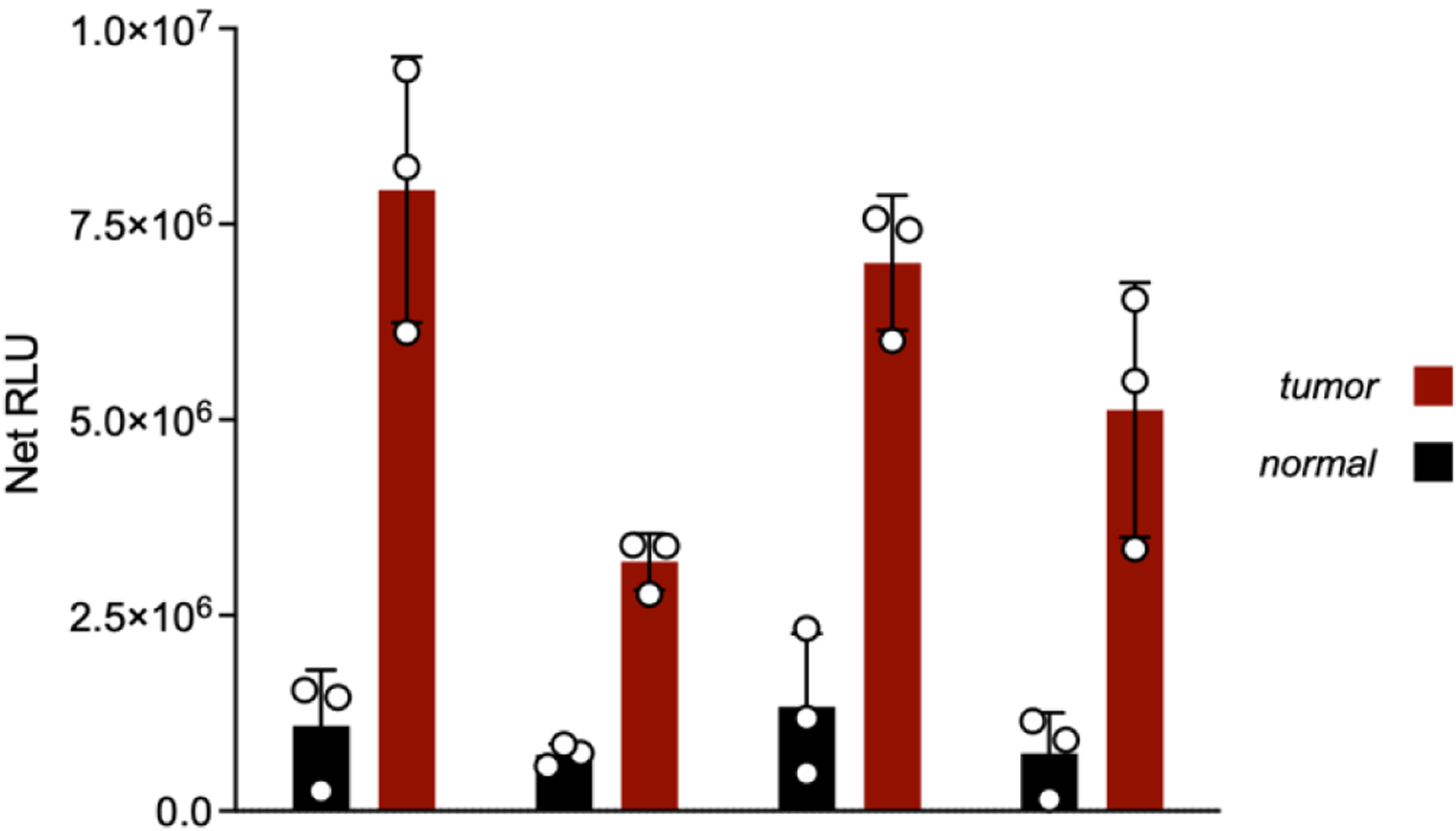
mRNA from the indicated human tissues (normal = black and tumor = red) was tested using EndoVLISA. Values represent net relative luminescent units (RLU, arbitrary units) calculated by subtracting appropriate blank wells (no RNA) from each set. Data points represent individual values from each well (n = 3 for each sample). Unpaired t-tests were computed in Prism between normal and tumor samples for all tissue types (* denotes p < 0.05, ** p < 0.05, *** p < 0.001).
Conclusion
A-to-I RNA editing is a key molecular event that significantly influences cellular function and disease progression. Despite this importance, our understanding of RNA editing regulation and its precise role in different biological pathways remains limited. Much of this uncertainty results from a lack of accessible tools to measure large-scale differences in A-to-I editing activity between different cell and tissue types. Gaining a deeper understanding of these and other RNA modification patterns would provide invaluable information on necessary cellular mechanisms and could potentially lead to the identification of novel druggable protein and RNA targets. Overall A-to-I editing activity is also a key biosignature for a number of disorders, and the ability to rapidly detect these changes could be powerfully leveraged for disease diagnosis.
Here, we have addressed several of these needs by demonstrating a novel A-to-I editing bioassay based on the well-characterized ELISA format. Our method is the first assay of this type specifically designed to quantify A-to-I RNA editing, and enables direct immunodetection of global epitranscriptomic activity without sequencing. EndoVLISA is straightforward and exclusively uses commercially available components, positioning it as a highly accessible approach for measuring changes in A-to-I RNA editing. After carefully optimizing key parameters in our assay, we successfully validated its performance and demonstrated its use for detecting global cellular changes in RNA editing activity in both treated cell lines and primary tissue samples.
Looking to the future, we aim to deploy EndoVLISA for answering other long-standing questions in the field, and in particular envision that multiplexing our assay with existing ELISA kits specific for other RNA and DNA modifications, including N6-methyladenosine (m6A),54–55 5-methylcytosine (5mC),56 5-hydroxymethylcytosine (5hmC), and 5-carboxylcytosine (5caC),57 will provide an unprecedented opportunity to elucidate functional relationships between different nucleic acid modifications. EndoVLISA is also well-poised for implementation in CRISPR-based knockout screens,72 and will likely find utility in identifying regulators of ADAR activity that influence A-to-I signatures. Building off of our initial results, we anticipate that additional developmental efforts will yield increased benefit in both assay performance and versatility. Moreover, streamlining and automating our overall workflow for high-throughput drug screening could provide a significant technological advance for identifying ADAR-modulating pharmaceuticals, especially in non-engineered cell lines or primary tissues. Together, this report details a simple yet powerful new tool to complement existing epitranscriptomic sequencing technologies. Our assay has broad versatility across many research disciplines, and we anticipate that EndoVLISA will dramatically improve our ability to rapidly measure global changes in A-to-I RNA editing across a diverse range of biological contexts.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R21GM134564, and R01GM140657 to J.M.H.). This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. We would also like to thank Christopher Wojewodzki for his helpful advice and technical expertise in ELISA development, Heather Hundley for her expertise in epitranscriptomics, and Viren Patel for his help in computational analysis.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- (1).Bass BL Annu Rev Biochem 2002, 71, 817–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nishikura K Nat Rev Mol Cell Biol 2016, 17 (2), 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tan MH; Li Q; Shanmugam R; Piskol R; Kohler J; Young AN; Liu KI; Zhang R; Ramaswami G; Ariyoshi K Nature 2017, 550 (7675), 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Porath HT; Knisbacher BA; Eisenberg E; Levanon EY Genome biology 2017, 18 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ramaswami G; Li JB Nucleic acids research 2013, 42 (D1), D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bazak L; Haviv A; Barak M; Jacob-Hirsch J; Deng P; Zhang R; Isaacs FJ; Rechavi G; Li JB; Eisenberg E Genome research 2014, 24 (3), 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Eisenberg E; Levanon EY Nature Reviews Genetics 2018, 19 (8), 473–490. [DOI] [PubMed] [Google Scholar]
- (8).Liddicoat BJ; Piskol R; Chalk AM; Ramaswami G; Higuchi M; Hartner JC; Li JB; Seeburg PH; Walkley CR Science 2015, 349 (6252), 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pestal K; Funk CC; Snyder JM; Price ND; Treuting PM; Stetson DB Immunity 2015, 43 (5), 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mannion NM; Greenwood SM; Young R; Cox S; Brindle J; Read D; Nellåker C; Vesely C; Ponting CP; McLaughlin PJ Cell reports 2014, 9 (4), 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang Q; Miyakoda M; Yang W; Khillan J; Stachura DL; Weiss MJ; Nishikura K Journal of Biological Chemistry 2004, 279 (6), 4952–4961. [DOI] [PubMed] [Google Scholar]
- (12).Shallev L; Kopel E; Feiglin A; Leichner GS; Avni D; Sidi Y; Eisenberg E; Barzilai A; Levanon EY; Greenberger S RNA 2018, 24 (6), 828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Roth SH; Danan-Gotthold M; Ben-Izhak M; Rechavi G; Cohen CJ; Louzoun Y; Levanon EY Cell reports 2018, 23 (1), 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rice GI; Kasher PR; Forte GM; Mannion NM; Greenwood SM; Szynkiewicz M; Dickerson JE; Bhaskar SS; Zampini M; Briggs TA Nature genetics 2012, 44 (11), 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Maas S; Kawahara Y; Tamburro KM; Nishikura K RNA biology 2006, 3 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Han L; Diao L; Yu S; Xu X; Li J; Zhang R; Yang Y; Werner HMJ; Eterovic AK; Yuan Y; Li J; Nair N; Minelli R; Tsang YH; Cheung LWT; Jeong KJ; Roszik J; Ju Z; Woodman SE; Lu Y; Scott KL; Li JB; Mills GB; Liang H Cancer Cell 2015, 28 (4), 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ishizuka JJ; Manguso RT; Cheruiyot CK; Bi K; Panda A; Iracheta-Vellve A; Miller BC; Du PP; Yates KB; Dubrot J Nature 2018, 565 (7737), 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nishikura K Annual review of biochemistry 2010, 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hwang T; Park C-K; Leung AK; Gao Y; Hyde TM; Kleinman JE; Rajpurohit A; Tao R; Shin JH; Weinberger DR Nature neuroscience 2016, 19 (8), 1093. [DOI] [PubMed] [Google Scholar]
- (20).Tran SS; Jun H-I; Bahn JH; Azghadi A; Ramaswami G; Van Nostrand EL; Nguyen TB; Hsiao Y-HE; Lee C; Pratt GA Nature neuroscience 2019, 22 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Breen MS; Dobbyn A; Li Q; Roussos P; Hoffman GE; Stahl E; Chess A; Sklar P; Li JB; Devlin B Nature neuroscience 2019, 22 (9), 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Khermesh K; D’Erchia AM; Barak M; Annese A; Wachtel C; Levanon EY; Picardi E; Eisenberg E Rna 2016, 22 (2), 290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Silvestris DA; Picardi E; Cesarini V; Fosso B; Mangraviti N; Massimi L; Martini M; Pesole G; Locatelli F; Gallo A Genome biology 2019, 20 (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Paz N; Levanon EY; Amariglio N; Heimberger AB; Ram Z; Constantini S; Barbash ZS; Adamsky K; Safran M; Hirschberg A; Krupsky M; Ben-Dov I; Cazacu S; Mikkelsen T; Brodie C; Eisenberg E; Rechavi G Genome Res 2007, 17 (11), 1586–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chan TH; Lin CH; Qi L; Fei J; Li Y; Yong KJ; Liu M; Song Y; Chow RK; Ng VH; Yuan YF; Tenen DG; Guan XY; Chen L Gut 2014, 63 (5), 832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Paz-Yaacov N; Bazak L; Buchumenski I; Porath HT; Danan-Gotthold M; Knisbacher BA; Eisenberg E; Levanon EY Cell Rep 2015, 13 (2), 267–76. [DOI] [PubMed] [Google Scholar]
- (27).Vlachogiannis NI; Gatsiou A; Silvestris DA; Stamatelopoulos K; Tektonidou MG; Gallo A; Sfikakis PP; Stellos K Journal of autoimmunity 2020, 106, 102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hideyama T; Yamashita T; Aizawa H; Tsuji S; Kakita A; Takahashi H; Kwak S Neurobiology of disease 2012, 45 (3), 1121–1128. [DOI] [PubMed] [Google Scholar]
- (29).Paul MS; Bass BL The EMBO journal 1998, 17 (4), 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Oakes E; Vadlamani P; Hundley HA, Methods for the Detection of Adenosine-to-Inosine Editing Events in Cellular RNA. In mRNA Processing, Springer: 2017; pp 103–127. [DOI] [PubMed] [Google Scholar]
- (31).Roth SH; Levanon EY; Eisenberg E Nature methods 2019, 1–8. [DOI] [PubMed] [Google Scholar]
- (32).Zhang R; Li X; Ramaswami G; Smith KS; Turecki G; Montgomery SB; Li JB Nature methods 2014, 11 (1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Giudice CL; Silvestris DA; Roth SH; Eisenberg E; Pesole G; Gallo A; Picardi E Frontiers in Genetics 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Su D; Chan CT; Gu C; Lim KS; Chionh YH; McBee ME; Russell BS; Babu IR; Begley TJ; Dedon PC Nature protocols 2014, 9 (4), 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhang N; Shi S; Jia TZ; Ziegler A; Yoo B; Yuan X; Li W; Zhang S Nucleic acids research 2019, 47 (20), e125–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Addepalli B; Limbach PA Journal of the American Society for Mass Spectrometry 2011, 22 (8), 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Popova AM; Williamson JR Journal of the American Chemical Society 2014, 136 (5), 2058–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Laverdiere I; Caron P; Couture F; Guillemette C; Lévesque E. r., Analytical chemistry 2012, 84 (1), 216–223. [DOI] [PubMed] [Google Scholar]
- (39).Jimmerson LC; Bushman LR; Ray ML; Anderson PL; Kiser JJ Pharmaceutical research 2017, 34 (1), 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Prestwich EG; Mangerich A; Pang B; McFaline JL; Lonkar P; Sullivan MR; Trudel LJ; Taghizedeh K; Dedon PC Chemical research in toxicology 2013, 26 (4), 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fritzell K; Xu L-D; Otrocka M; Andréasson C; Öhman M Nucleic acids research 2019, 47 (4), e22–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dalhus B; Arvai AS; Rosnes I; Olsen ØE; Backe PH; Alseth I; Gao H; Cao W; Tainer JA; Bjørås M Nature structural & molecular biology 2009, 16 (2), 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Morita Y; Shibutani T; Nakanishi N; Nishikura K; Iwai S; Kuraoka I Nature communications 2013, 4, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Vik ES; Nawaz MS; Andersen PS; Fladeby C; Bjørås M; Dalhus B; Alseth I Nature communications 2013, 4, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wu J; Samara NL; Kuraoka I; Yang W Molecular cell 2019, 76 (1), 44–56. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hitchcock TM; Gao H; Cao W Nucleic acids research 2004, 32 (13), 4071–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Knutson SD; Arthur RA; Johnston HR; Heemstra JM Journal of the American Chemical Society 2020, 142 (11), 5241–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Knutson SD; Heemstra JM Current Protocols in Chemical Biology 2020, 12 (2), e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Clark MF; Lister RM; Bar-Joseph M Methods in enzymology 1986, 118, 742–766. [Google Scholar]
- (50).Engvall E; Perlmann P The Journal of Immunology 1972, 109 (1), 129–135. [PubMed] [Google Scholar]
- (51).GeurtsvanKessel CH; Okba NM; Igloi Z; Bogers S; Embregts CW; Laksono BM; Leijten L; Rokx C; Rijnders B; Rahamat-Langendoen J Nature communications 2020, 11 (1), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Baran P; Hansen S; Waetzig GH; Akbarzadeh M; Lamertz L; Huber HJ; Ahmadian MR; Moll JM; Scheller J Journal of Biological Chemistry 2018, 293 (18), 6762–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).De La Rica R; Stevens MM Nature nanotechnology 2012, 7 (12), 821–824. [DOI] [PubMed] [Google Scholar]
- (54).Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M Nature 2012, 485 (7397), 201. [DOI] [PubMed] [Google Scholar]
- (55).Matsuzawa S; Wakata Y; Ebi F; Isobe M; Kurosawa N PloS one 2019, 14 (10), e0223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Tobias IC; Kao M-MC; Parmentier T; Hunter H; LaMarre J; Betts DH Stem cell research & therapy 2020, 11 (1), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chowdhury B; Cho I-H; Hahn N; Irudayaraj J Analytica chimica acta 2014, 852, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Qiu C; Liu W-Y; Xu Y-Z, Fluorescence labeling of short RNA by oxidation at the 3′-end. In RNA Nanotechnology and Therapeutics, Springer: 2015; pp 113–120. [DOI] [PubMed] [Google Scholar]
- (59).Nishikura K Nature reviews Molecular cell biology 2016, 17 (2), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nakaya K; Takenaka O; Horinishi H; Shibata K Biochimica et Biophysica Acta (BBA)-Nucleic Acids and Protein Synthesis 1968, 161 (1), 23–31. [DOI] [PubMed] [Google Scholar]
- (61).Sommer SS; Cohen JE Journal of molecular evolution 1980, 15 (1), 37–57. [DOI] [PubMed] [Google Scholar]
- (62).Torres AG; Piñeyro D; Filonava L; Stracker TH; Batlle E; de Pouplana LR FEBS letters 2014, 588 (23), 4279–4286. [DOI] [PubMed] [Google Scholar]
- (63).Kim DD; Kim TT; Walsh T; Kobayashi Y; Matise TC; Buyske S; Gabriel A Genome research 2004, 14 (9), 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bazak L; Levanon EY; Eisenberg E Nucleic acids research 2014, 42 (11), 6876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Schaffer AA; Kopel E; Hendel A; Picardi E; Levanon EY; Eisenberg E Nucleic acids research 2020, 48 (11), 5849–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Galipon J; Ishii R; Suzuki Y; Tomita M; Ui-Tei K Genes 2017, 8 (2), 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Lonsdale J; Thomas J; Salvatore M; Phillips R; Lo E; Shad S; Hasz R; Walters G; Garcia F; Young N Nature genetics 2013, 45 (6), 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fumagalli D; Gacquer D; Rothé F; Lefort A; Libert F; Brown D; Kheddoumi N; Shlien A; Konopka T; Salgado R Cell reports 2015, 13 (2), 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Kang L; Liu X; Gong Z; Zheng H; Wang J; Li Y; Yang H; Hardwick J; Dai H; Poon RT; Lee NP; Mao M; Peng Z; Chen R Genomics 2015, 105 (2), 76–82. [DOI] [PubMed] [Google Scholar]
- (70).Anadón C; Guil S; Simó-Riudalbas L; Moutinho C; Setien F; Martínez-Cardús A; Moran S; Villanueva A; Calaf M; Vidal A Oncogene 2016, 35 (33), 4407–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Weinstein JN; Collisson EA; Mills GB; Shaw KRM; Ozenberger BA; Ellrott K; Shmulevich I; Sander C; Stuart JM Nature genetics 2013, 45 (10), 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Shalem O; Sanjana NE; Hartenian E; Shi X; Scott DA; Mikkelsen TS; Heckl D; Ebert BL; Root DE; Doench JG Science 2014, 343 (6166), 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.