

Abstract
Transplantation of stem cell derived β (SC-β) cells represents a promising new therapy for type 1 diabetes (T1D). However, the delivery of these cells, ideally without immunosuppression and in a retrievable device that is both functional and safe, remains a challenge. Indeed, prioritizing safety in cell delivery devices often compromises cell viability and function. Herein, we report the design of a safe and functional device composed of a highly porous, durable nanofibrous skin and an immunoprotective hydrogel core. The device made by electrospinning a medical grade thermoplastic silicone-polycarbonate-urethane is soft but tough (~15 MPa at a rupture strain of > 2). Tuning the nanofiber size to < ~500 nm prevented cell penetration while maintaining maximum mass transfer and interestingly decreased the cellular overgrowth on blank devices to as low as a single cell layer (~3 μm thick) in peritoneal cavity of mice. Using bioluminescence imaging (BLI), we confirmed the safety, indicated as continuous containment of even proliferative cells, within the device for 5 months. With either syngeneic, allogeneic or xenogeneic rodent islets, the device corrected chemically induced diabetes in mice and remained functional for up to 200 days. Most importantly, the device supported the function of human SC-β cells and reversed diabetes almost immediately (within a week) in both immunodeficient mice (for up to 120 days) and immunocompetent mice (for up to 60 days). Lastly, we demonstrated the scalability and retrievability of the device in dogs where we also observed viable human SC-β cells despite xenogeneic immune responses. The nanofibrous device design may therefore provide a translatable solution to the balance between safety and functionality in developing stem cell-based therapies for T1D.
One Sentence Summary:
A nanofibrous cell encapsulation design may provide a translatable solution to the balance between safety and functionality in developing stem cell-based therapies for type 1 diabetes.
Introduction
The replacement of missing β cells has been proposed as a promising new therapy for type 1 diabetes (T1D) (1, 2). Clinical trials with intrahepatic allogeneic islet transplantation have shown insulin independence in diabetic patients (3–5), but factors including the instant blood mediated inflammatory reaction (IBMIR), the side effects of immunosuppressive drugs and the shortage of human islets from cadavers limit the wide application to patients. Stem cell derived β (SC-β) cells could provide a nearly unlimited supply of cells and therefore holds great promise for the whole T1D population (6–11). However, the potential risks of immunosuppression and teratoma formation by undifferentiated stem cells remain significant concerns (12–14). Therefore, within the foreseeable future, the delivery of SC-β cells in a retrievable, immunoprotective encapsulation device that is both safe (i.e. prevents any potential cell escape) and functional (i.e. maintains facile mass transfer) may be critical to the clinical success of stem cell-based therapies for T1D.
Alginate microcapsule-based encapsulation systems have been extensively investigated and proven functional in numerous animal models (15–21). However, it is becoming increasingly recognized that the impossibility to ensure complete graft retrieval will hinder their application for SC-β cell delivery in clinical settings. In order to take advantage of the biocompatibility and immunoprotective property of alginate hydrogels while endowing retrievability, our lab has recently developed a Thread-Reinforced Alginate Fiber For Islet enCapsulation (TRAFFIC) device (22). Similar to alginate microcapsules, TRAFFIC showed great performance in maintaining cell viability and reversing diabetes in mouse models. Unlike microcapsules, TRAFFIC was completely retrievable using a laparoscopic procedure. However, alginate and hydrogels in general are intrinsically weak relative to other materials such as elastomers, prone to swelling and even breakage over time. This increases the risk of exposing transplanted cells to the host immune system and allowing undifferentiated cells to escape from the device.
In comparison, polymer-based encapsulation devices such as those made of semi-permeable polytetrafluoroethylene (PTFE) membranes (e.g. TheraCyte device or the ViaCyte device) (23–28) are sturdy and can prevent any potential cell escape, representing a much safer and more translatable strategy for developing cell replacement therapies. However, their small pore sizes (on the order of ~100 nm) and foreign body reaction (FBR)-induced fibrotic deposition diminishes mass transfer that is critical for long-term function. Similarly, a polycaprolactone (PCL) nanoporous membrane formed around sacrificial nanorods was used to form durable encapsulation devices (e.g. the Encellin device) (29, 30), but the small pore size (on the order of ~10 nm) and biodegradability of PCL makes the long-term, reliable membrane function less certain, posing a risk for clinical applications. In addition, in many of these devices, extra layers of more rigid membranes, such as polyethylene terephthalate (PET) mesh, are often required to give mechanical support to the devices and maintain their planar shape in vivo. These additional rigid layers may increase the severity of FBR and negatively impact the function. Lastly, these devices have been designed for subcutaneous implantation which has been proven to be challenging due to the low degree of vasculature and limited oxygen supply in the subcutaneous space (31).
Herein, we report an intraperitoneal, tubular, Nanofiber Integrated Cell Encapsulation (NICE) device that is both safe and functional in the long term for delivery of insulin-producing cells including human SC-β cells. The device consists of a highly porous and durable nanofibrous skin made by electrospinning a biocompatible medical grade thermoplastic silicone-polycarbonate-urethane (TSPU), and an alginate hydrogel core, which provides additional immunological protection and maintains the separation of islets or SC-β cells within the device. The nanofibrous skin is soft, stretchable and tough with a rupture strain of > 2 and an ultimate strength of ~15 MPa. The whole device is easy to handle, implant and retrieve using laparoscopic procedures. The pore structures of the nanofibrous skin are interconnected and tunable by controlling the nanofiber size. Interestingly, when the TSPU nanofiber skin was ~100 μm thick with nanofiber sizes less than ~ 500 nm, the device prevented cell penetration and induced only minimal cellular overgrowth as low as one cell layer of ~3 μm in the intraperitoneal space of mice. The pores of the device, although difficult to precisely define, were on the order of ~1 μm, much larger than those used in previous encapsulation devices, providing superb mass transfer. Live imaging and histological analysis confirmed the continuous containment of even proliferative cells for 5 months. After confirmation of safety, we showed that the device protected islets from diverse sources (syngeneic/allogeneic/xenogeneic) and restored normoglycemia in diabetic mice for up to 200 days. Most importantly, we further showed the NICE device supported the function of human SC-β cells and reversed diabetes almost immediately in both immunodeficient and immunocompetent mice for up to 120 days and 60 days respectively, which to our knowledge represents the first report of diabetes correction using human SC-β cells without any maturation period in vivo in a retrievable device. Lastly, to demonstrate the scalability and retrievability, we implanted the NICE device with human SC-β cells in dogs, a model that may be more challenging than human-to-human allogeneic transplantation. Despite xenogeneic immune responses during the short (2-week) transplantation, we successfully retrieved the device using a laparoscopic procedure and observed viable cells within the retrieved device. The NICE design may therefore provide a translatable solution to the balance between safety and functionality in developing a stem cell-based cell replacement therapy for T1D.
Results
Design, fabrication and characterization of the NICE device
We designed the NICE device based on several criteria: 1) mechanical toughness to ensure long-term stability and safety, 2) tubular form factor for high surface area and minimally invasive implantation and retrieval, 3) biocompatibility with host tissue (minimal foreign body reaction), 4) sufficiently small pores to prevent cell escape but high porosity to ensure facile mass transfer and 5) low complexity of design to facilitate translation (Fig. 1A). Based on these criteria, we chose a biocompatible medical grade thermoplastic silicone-polycarbonate-urethane (TSPU, CarboSil® from DSM Biomedical) as the polymer and used electrospinning to fabricate the device due to its ability to produce tough, nanofibrous membranes with tunable and interconnected pore structures.
Fig. 1.
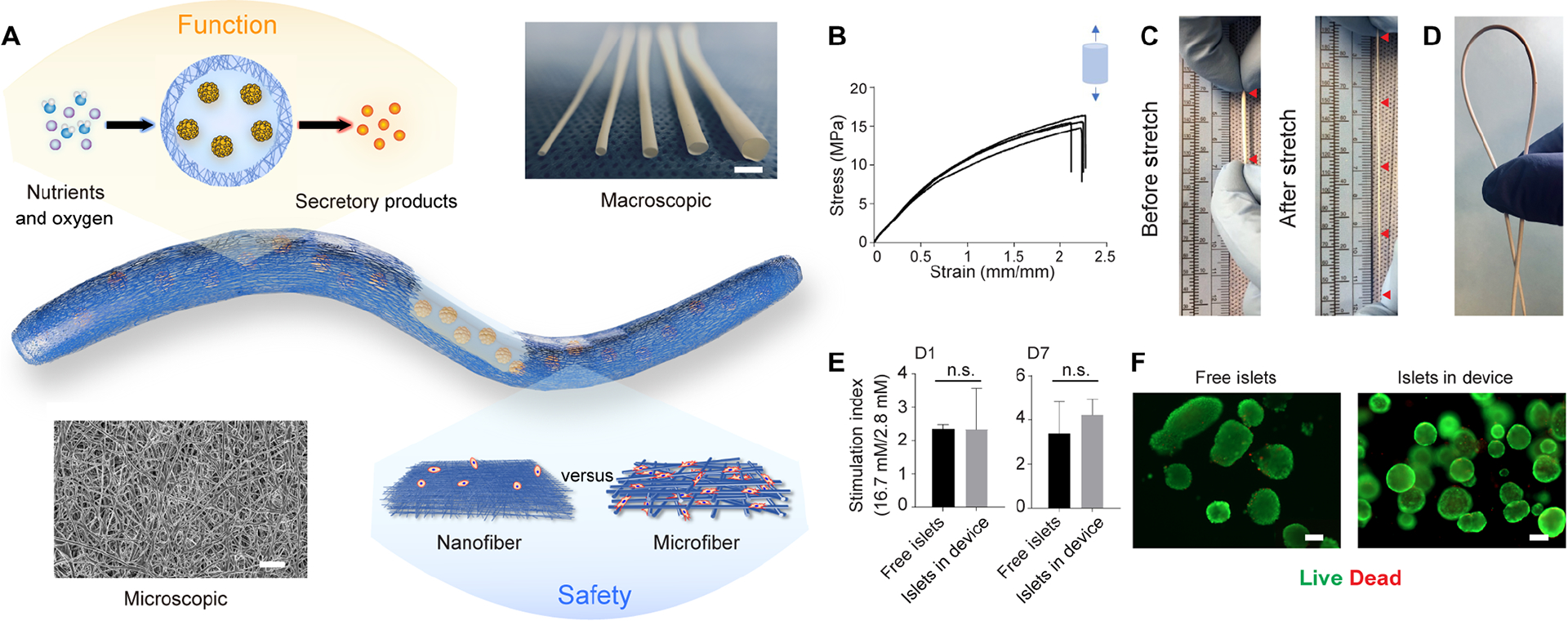
Design and characterization of the NICE device including mechanical properties, permeability and cell compatibility. (A) Schematics of the device showing the islet-laden hydrogel core surrounded by the nanofibrous skin that prevents cell penetration while allowing maximum mass transfer, along with a photo of nanofibrous tubes with different diameters (from left to right: 0.5 mm, 1 mm, 1.5 mm, 2 mm and 3 mm) and a SEM image of the nanofibers. (B) Tensile test (Stress-strain curves) of the nanofibrous tubes (n = 4). (C and D) Digital images showing the device being stretched more than three times (C) and bent without kink (D). (E) Stimulation index of islets (the ratio of insulin secretion in the buffers of high and low glucose concentrations) encapsulated in the device, compared to that of free-floating islets after 1-day and 7-days culture, mean ± SEM (n = 3). (F) Live (green) and dead (red) staining of free-floating islets and islets encapsulated in device after 1-day culture. The data was compared using the two-tailed Student’s t-test. The level of significance was labeled by n.s., denoting non-significant. Scale bars: 3 mm (A) macroscopic image, 100 μm (F) and 5 μm (A) microscopic image.
To fabricate the device, nanofibers were collected on a rotating, sugar-coated mandrel template (fig. S1A) and nanofibrous tubes were obtained after dissolution of the sugar coating. The tubes had inner diameters ranging from 500 μm to 3 mm and a wall thickness of ~100 μm (Fig. 1A). Examination using a scanning electron microscope (SEM) verified the non-woven fibrous nature of the NICE membrane with an average fiber diameter of 270 nm (Fig. 1A). Tensile tests demonstrated that the nanofibrous tube remained elastically deformable up to approximately 5 MPa under a 0.5 strain. The ultimate tensile strength was approximately 15 MPa at a device strain greater than 2. The Young’s modulus calculated from 10%−20% of the curve is ~12.6 ± 0.6 MPa (n = 4) (Fig. 1B). Qualitatively, the device can be stretched more than three times its original length (Fig. 1C and Movies S1), bent without kinks (Fig. 1D and Movies S2) and handled using forceps without damage.
Knowing the mechanical strength of the device, we next investigated the mass transfer properties and compatibility with islets. A static glucose stimulated insulin secretion (GSIS) test showed that mouse islets encapsulated in the NICE device were capable of responding to low and high glucose, with a mean stimulation index (SI) (insulin secretion at high glucose to that at low glucose) of 2.33 and 4.23 after 1 day and 7 days of culture, respectively (Fig. 1E). The SI of islets cultured inside the device was not significantly different from that of freely floating islets after 1 day (2.33 ± 1.23 vs. 2.34 ± 0.13) or 7 days (4.23 ± 0.72 vs. 3.39 ± 1.44) of culture (Fig. 1E). The live/dead staining and corresponding quantitative analysis indicated similar viability (~ 95%) of islets inside the device to free islets after 24h culture (Fig. 1F and fig. S2). These in vitro experiments therefore demonstrated that the NICE device met the basic requirements as a cell delivery vehicle, providing superb mass transfer and cell compatibility to ensure the function of islets.
Biocompatibility of the NICE device in the intraperitoneal (I.P.) space of mice
The in vivo biocompatibility of an encapsulation device is an important factor; foreign body reaction (FBR)-induced fibrotic deposition can diminish the mass transfer and affect the function of encapsulated cells. We implanted and compared the FBR to the blank NICE devices in three clinically relevant transplant sites: the intraperitoneal (I.P.) space, the epididymal fat pad (E.F.P.) (as a model of the omentum in large mammals) and the subcutaneous (S.C.) space in C57BL/6 mice. The results, as indicated by H&E and Masson’s Trichrome staining, demonstrated that the devices implanted in either the ventral S.C. space or dorsal S.C. space formed a thick fibrotic capsule (over 40 μm) (Fig. 2, A–D and I). The thickness of the fibrotic layer at the 4 week-time point was significantly increased compared to that at 2 weeks in both the dorsal S.C. space (87.7 ± 13.2 vs. 47.3 ± 4.1 μm) and ventral S.C. space (143.5 ± 11.3 vs. 73.6 ± 5.4 μm) (Fig. 2I). In contrast, devices implanted in the I.P. space or the E.F.P. had much thinner cellular overgrowth (<~10 μm) than in the S.C. space (Fig. 2, E–H and I). Although there was no statistically significant difference between the I.P. space and E.F.P. at either 2 weeks or 4 weeks, the cellular overgrowth on the device in the I.P. space at 4 weeks was only a single layer of cells with thickness of 2.9 ± 0.6 μm. In addition, the devices could be retrieved freely from the I.P. space (data not shown), while those implanted in the E.F.P. or the S.C. space were wrapped tightly within tissue. It was noted that the nanofibrous membranes remained impenetrable to cells in all these locations.
Fig. 2.
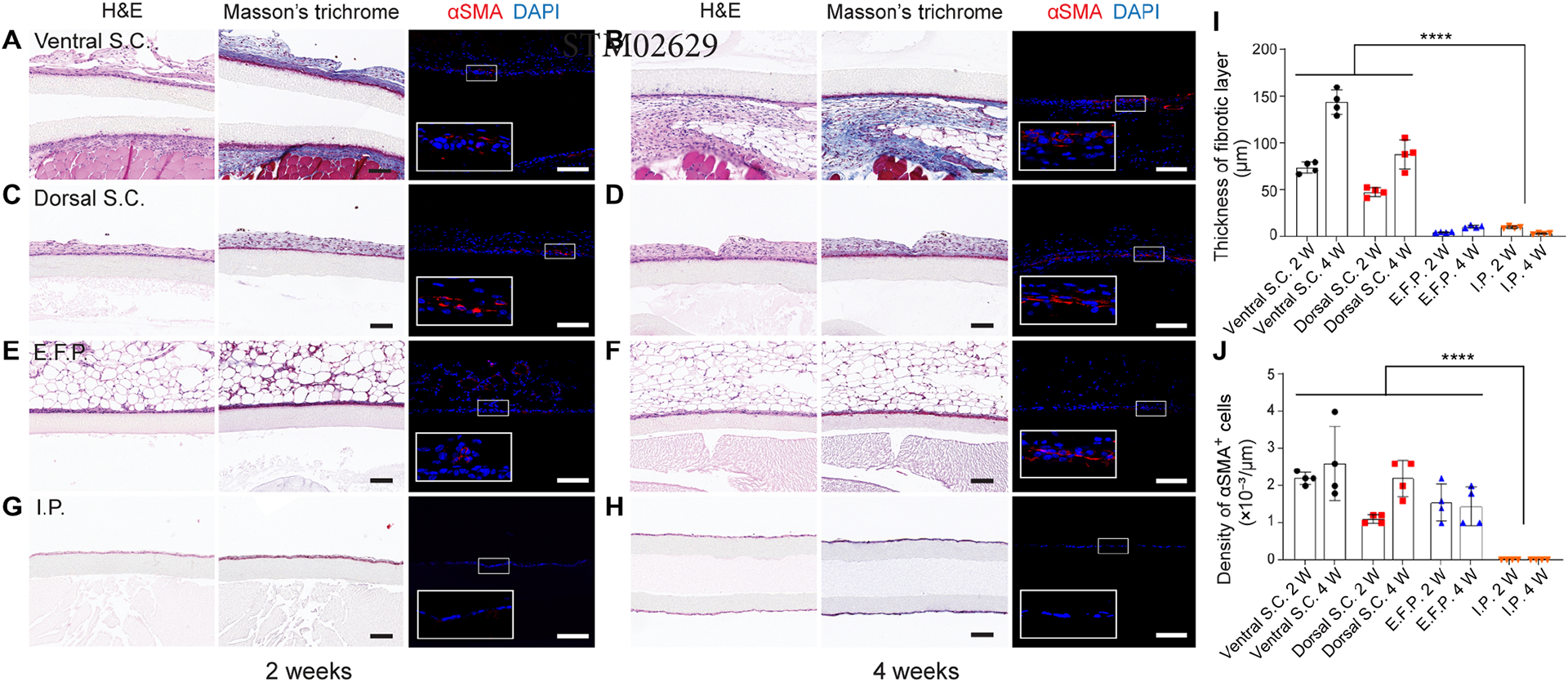
Biocompatibility of blank devices in different implantation sites in mice for 2 weeks and 4 weeks. (A-H) Blank devices implanted in the ventral S.C. space for two weeks (A) and four weeks (B), in the dorsal S.C. space for two weeks (C) and four weeks (D), in the E.F.P. for two weeks (E) and four weeks (F), and the in I.P. space for two weeks (G) and four weeks (H). In each panel, representative images from left to right are H&E staining, Masson’s Trichrome staining and immunofluorescent staining. Myofibroblasts were stained with αSMA (shown in red) and DAPI (shown in blue). (I) Analysis of the thickness of fibrotic layer measured from Masson’s Trichrome staining images, mean ± SD (n = 4). (J) Analysis of the αSMA positive cells measured from immunofluorescent staining images, mean ± SD (n = 4). The data was compared using one-way ANOVA followed by Tukey’s test. The level of significance was labeled by ****, denoting the p value of < 0.0001. Scale bars: 100 μm (A-H).
Myofibroblasts that are present in fibrotic capsules are identifiable by their expression of alpha smooth muscle actin (αSMA). These cells can arise from cell populations that enter the vicinity of a foreign material such as macrophages (32) and may be of bone marrow origin (33). The number and density of myofibroblasts trended with the fibrotic layer thickness (Fig. 2, I and J). More αSMA positive cells were observed in the S.C. space than in the I.P. space. Interestingly, there was no expression of αSMA in the I.P. group and no foreign body giant cells (FBGCs) were found adjacent to the implant surface. Thus, the FBR was determined to be the least severe in the I.P. space, motivating the choice to investigate this site in this study. Not only would the very thin cellular overgrowth facilitate facile mass transfer, but I.P. implantation was also generally adhesion free, allowing easy retrieval of the graft.
We hypothesized that the low FBR was due to unique properties of TSPU and the small fiber size. To test this hypothesis, we first investigated fiber sizes ranging from 270 nm to 1.04 μm by changing the concentration of TSPU from 8% to 14% during electrospinning (fig. S1, B and C). Histological examination of devices with different fiber sizes implanted in I.P. space for four weeks revealed that devices with fiber size below ~500 nm had no cell penetration and induced only thin (<10 μm) cellular overgrowth, while those above allowed significant cell penetration (fig. S1D). Then, we tested devices made from a different material (Nylon) for comparison. With a similar fiber size (~250 nm), the Nylon devices also prevented cell penetration after four weeks of I.P. implantation (fig. S1, B and D), but the delamination of different layers in the nanofiber membrane was observed (fig. S1D). These results confirmed the TSPU nanofibrous device with proper nanofiber sizes was uniquely suitable for cell encapsulation.
Protection of syngeneic/allogenic cells and confinement of proliferative cells
The ability of a cell encapsulation device to prevent any cell escape is of paramount importance in SC-β cell delivery due to risks of teratoma formation from undifferentiated cells. However, too tight pore structures will diminish the mass transfer that is necessary for cell viability and function. To test whether the NICE device can provide a solution to the safety/function balance, we encapsulated and implanted luciferase expressing cells and continuously monitored them over time in C57CL/6 mice. First, syngeneic mesenchymal stem cell (MSC) aggregates with GFP/luciferase expression (~100–150 μm in diameter) (fig. S3A) were encapsulated in NICE devices and implanted in the I.P. space. The bioluminescent signals were localized to the implantation site during the duration (5 months) of implantation and remined steady after an initial drop. (Fig. 3, A and B). The recipient animals showed no signals after device retrieval (Fig. 3A) and the retrieved devices were bioluminescent (Fig. 3C), suggesting that the cells were confined and viable in the NICE device for 5 months. To confirm these imaging results, we also performed histological analysis on retrieved samples. H&E images showed the confinement and viability of the MSC aggregates within the device (Fig. 3D).
Fig. 3.
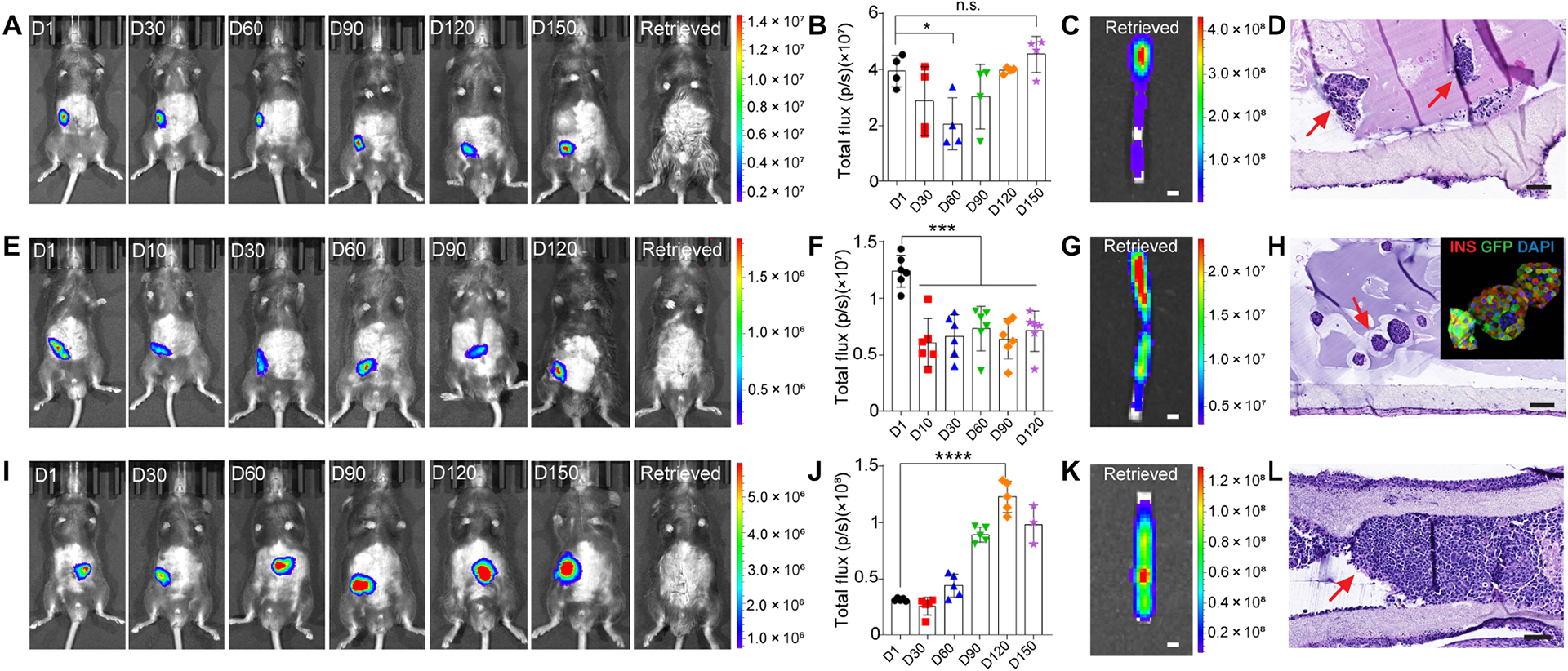
Survival and confinement of different types of cells within the device. (A) Representative bioluminescent images of healthy C57BL/6 mice transplanted with NICE devices encapsulating GFP/luciferase MSC spheroids for up to 150 days (from left to right are Day 1, 30, 60, 90, 120 and 150) and after retrieval. (B) Quantitative analysis of the bioluminescence intensity of the device. Data was presented as mean ± SD (n = 4). (C) A representative bioluminescent image of retrieved device. (D) H&E staining of a retrieved device at D150. Arrows point to the MSC spheroids. (E) Representative bioluminescent images of healthy C57BL/6 mice transplanted with NICE devices encapsulating FVB GFP/luciferase mouse islets for up to 120 days (from left to right are Day 1, 10, 30, 60, 90 and 120) and after retrieval. (F) Quantitative analysis of the bioluminescence intensity of the device. Data was presented as mean ± SD (n = 6). (G) A bioluminescent image of a retrieved device. (H) H&E staining of a retrieved device. Arrow points to the mouse islets. The top-right corner shows immunofluorescent staining of mouse islets transplanted for 120 days (green, GFP; red, insulin; blue, DAPI). (I) Representative bioluminescent images of healthy C57BL/6 mice transplanted with NICE devices encapsulating BALB/c GFP/luciferase 4T1 spheroids for up to 150 days (from left to right are Day 1, 30, 60, 90, 120 and 150) and after retrieval. (J) Quantitative analysis of the bioluminescence intensity of the device. Data was presented as mean ± SD (n = 5). (K) A bioluminescent image of a retrieved device. (L) H&E staining of a retrieved device. Arrows point to the 4T1 cells encapsulated in device for 120 days. The data was compared using one-way ANOVA followed by Tukey’s multiple comparisons test. The level of significance was labeled by n.s., *, *** and ****, denoting non-significant, p value of < 0.05, < 0.001 and < 0.0001, respectively. Scale bars: 1 mm (C, G and K), 100 μm (D, H and L).
Next, allogeneic GFP/luciferase islets with a size of around 100–150 μm (fig. S3B) were monitored within the device over time. It was found that 48.9% radiance was detected on day 10 compared to day 1 (Fig. 3E), similar to the initial signal drop in the MSC case. The acute loss of encapsulated cells following implantation was considered common and most likely caused by hypoxia (34, 35). However, there was no significant difference among day 10, 30, 60, 90 and 120, which demonstrated that the islets remained stable after 10 days (Fig. 3, E and F). In contrast, allogeneic islets transplanted in kidney capsules without the protection of the device were all rejected in 2 weeks (data not shown). Post-retrieval imaging of the recipients and devices (Fig. 3, E and G) as well as H&E and insulin/glucagon staining confirmed the robust function of the islets after 120 days (Fig. 3H and fig. S3, D–F).
Lastly, to challenge our device, we tested whether it could protect invasive cells and prevent them from escaping. Allogeneic cancer cell spheroids (4T1) with GFP/luciferase expression (fig. S3C) were encapsulated in the NICE device. In 2 out of the 6 mice, the bioluminescent signal disappeared after 2 weeks suggesting early failure possibly due to unintentional defects of the device and/or biological variation (Fig. 3J). However, in the other 4 mice the signal increased significantly after 60 days and lasted for up to 150 days indicating the cells were protected (i.e. no immune cell penetration) and proliferated in the device (Fig. 3, I and J). In all cases, no bioluminescence outside the implantation site or device was observed. The imaging of retrieved device and H&E staining confirmed that the NICE device was able to support cell viability and prevent them from escape (Fig. 3, K and L). Another GFP/luciferase-expressing, allogeneic cell line, NIT-1 β cell spheroids, were also confined and protected by the NICE device for 2 months (fig. S3G). All the data taken together suggested that the NICE device was protective and safe.
Diabetes correction in mice with diverse rodent islet sources
To investigate the function of the NICE device in reversing diabetes, different types of rodent islets (syngeneic, allogenic and xenogeneic) were encapsulated in the device and implanted in diabetic C57BL/6 mice. Rodent islets were dispersed evenly and embedded in alginate in the NICE device before transplantation. With 600–700 IEQ syngeneic islets, the NICE device corrected diabetes in 13 out of 17 mice. The engrafted mice maintained blood glucose levels within a normal range until device retrieval for as long as 120 days (Fig. 4A). Interestingly, the device with allogenic BALB/c islets (600–700 IEQ) engrafted in 16 out of 24 mice, with function for up to 200 days (Fig. 4B). Increasing immunological incompatibility of the transplanted cells by using rat islets (600–700 IEQ) demonstrated function for longer than 100 days (Fig. 4C). In all engrafted animals, the blood glucose levels dropped within a week of transplantation (Fig. 4, A–C) and increased after graft retrieval at different time points, confirming that the glucose levels were controlled by the grafts (Fig. 4, A–C). The devices were easily retrievable and there was no tissue adhesion in most cases. The increased body weight of all the engrafted mice similar to healthy mice showed metabolic recovery, while that of diabetic mice dropped continuously (Fig. 4D).
Fig. 4.
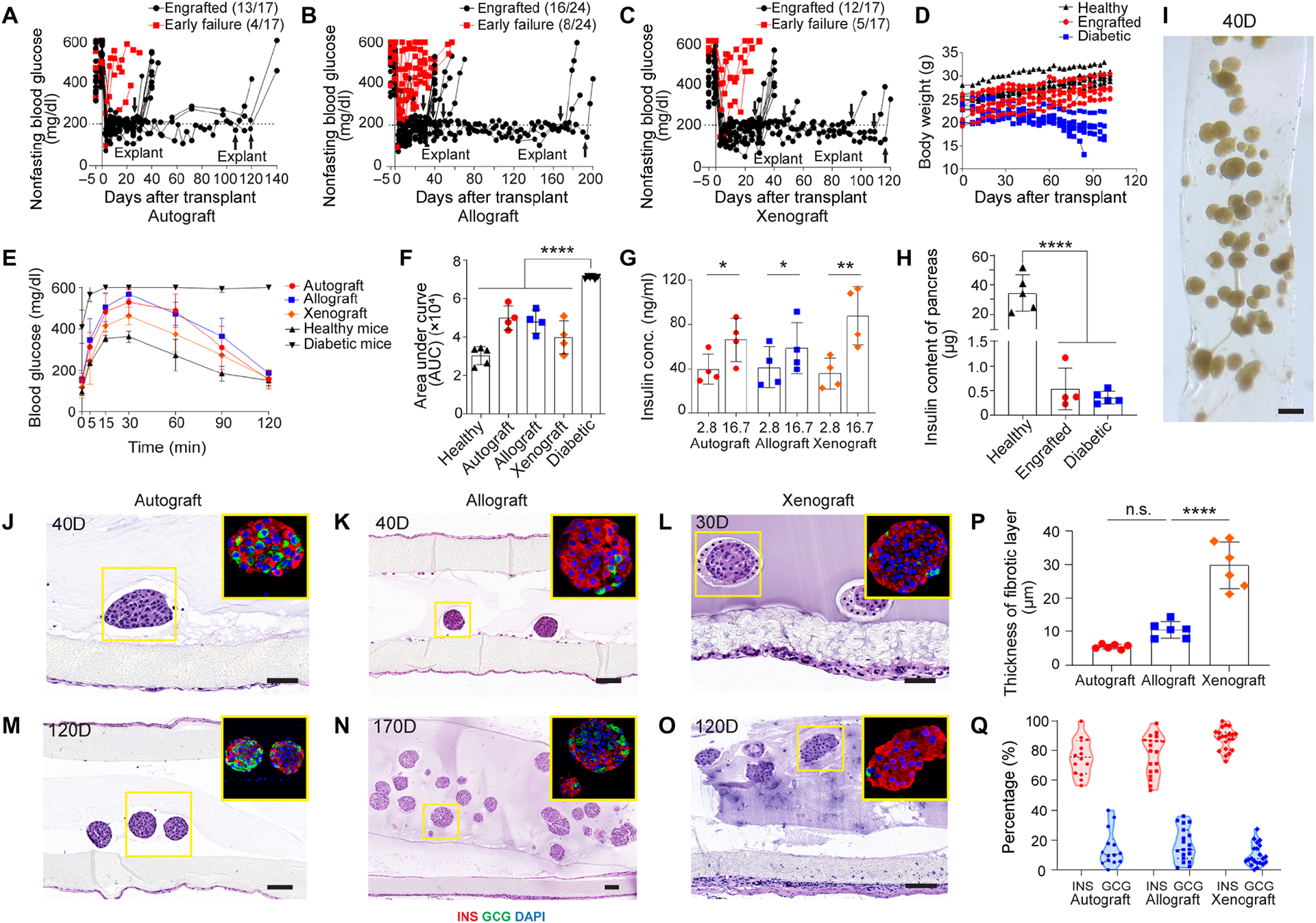
Device function with syngeneic, allogeneic and xenogeneic islets in diabetic immunocompetent C57BL/6 mice. (A-C) Non-fasting blood glucose levels of the recipients transplanted with syngeneic mouse islets (n = 17) (A), allogeneic mouse islets (n = 24) (B) and xenogeneic rat islets (n = 17) (C). Arrows indicate the time points when implants were retrieved from recipients. (D) Body weights of healthy, engrafted and diabetic mice measured for up to 105 days, (n = 5 in each group). (E) Measurement of blood glucose in IPGTT test in different groups on day 30. The data was presented as mean ± SD (n = 5 for healthy mouse group, n = 4 for engrafted groups, n = 6 for diabetic mouse group). (F) Quantification of area under curve (AUC) from (E) for different groups on day 30, mean ± SD. (G) Measurement of insulin concentration of retrieved devices from engrafted mice in different groups following ex vivo glucose stimulation insulin secretion (GSIS) test, mean ± SD (n = 4 for each group). (H) Measurement of total insulin content of the pancreas in different groups, mean ± SD (n = 4–5). (I) Bright field image of encapsulated mouse islets retrieved from allogeneic transplantation model on day 40. (J and K) H&E staining and immunofluorescent staining (shown in inset) of syngeneic islets (J) and allogeneic islets (K) from retrieved device after 40 days. (L) H&E staining and immunofluorescent staining (shown in inset) of xenogeneic rat islets from retrieved device after 30 days. (M and O) H&E staining and immunofluorescent staining (shown in inset) of syngeneic islets (M) and xenogeneic islets (O) from retrieved device after 120 days. (N) H&E staining and immunofluorescent staining (shown in inset) of allogeneic islets from retrieved device after 170 days (green, glucagon; red, insulin; blue, DAPI). (P) Percentage of hormone expressions (insulin and glucagon) quantified from immunofluorescent staining images of encapsulated islets retrieved from three different groups: autograft, allograft and xenograft. One dot represents one islet. (Q) Quantification of the thickness of fibrotic layer in three different groups: autograft, allograft and xenograft. The one-way ANOVA followed by Tukey’s test was performed for comparing the multi-group data. The two-tailed Student’s t-test was performed when the data consisted of only two groups. The level of significance was labeled by n.s., *, ** and ****, denoting non-significant, p value of < 0.05, < 0.01 and < 0.0001, respectively. Scale bars: 100 μm (K, M, N and O) and 50 μm (J and L).
To evaluate the metabolic control of blood glucose, an intraperitoneal glucose tolerance test (IPGTT) was performed on day 30. The IPGTT measurements indicated that all engrafted mice cleared glucose 2 hours post bolus similar to healthy mice, while the peak value of blood glucose post stimulation in engrafted mice was higher than that of healthy mice. The rate of glucose clearance in all the engrafted mice was significantly faster than that of the diabetic group (Fig. 4E). Quantitatively, the area under curve (AUC) for glucose clearance in the engrafted mice receiving syngeneic, allogeneic and xenogeneic islets were similar to the normal mice (Fig. 4F). Notably, there was no difference in the AUC among the engrafted mice implanted with different sources of islets. The IPGTT carried out on the engrafted mice with grafts lasting over 4 months also indicated long-term function (fig. S4A). To provide additional proof that the transplanted islets were responsible for glucose control, an ex vivo GSIS test was carried out (Fig. 4G). The responsiveness of islets inside the device indicated their viability and normal function. Lastly, an approximately 60X decrease of insulin content in the pancreas of engrafted mice compared to healthy mice confirmed again that the STZ induction has successfully depleted the β cells in pancreas and the transplanted grafts were responsible for the observed metabolic control (Fig. 4H).
Allogeneic islets in retrieved devices after 40 days appeared extremely healthy, similar to those before transplantation (Fig. 4I). The histological studies showed no tissue adhesion around the device and no cell penetration into the device (Fig. 4, J–O). Quantitative analysis of the cellular overgrowth in H&E staining images (Fig. 4, J–O) showed that while there was a relatively thicker fibrotic layer (~ 30 μm) for xenografts, the fibrotic deposition was minimal (around 10 μm in thickness) for autografts and allografts (Fig. 4Q). Islets from different sources within the device were viable in the short term (< 40 days) (Fig. 4, J–L and fig. S5, A–C) and in the long term (> 100 days) (Fig. 4, M–O). Moreover, islets inside the device were morphologically normal with a round shape, even at long time points (170 and 200 days) with a high density in an allogeneic environment (Fig. 4N). Immunostaining for insulin and glucagon demonstrated that cells maintained their individual hormone identities, many of which were insulin-expressing β cells (Fig. 4, J–O). Quantitative analysis of the expression of insulin and glucagon after transplantation showed that in average around 80% of the cells within one islet were insulin positive β cells and 15% of cells were glucagon expressing α cells in autografts and allografts (Fig. 4P), which were similar to the percentages of α and β cells before transplantation (fig. S4B). About 88% insulin positive cells and ~10% glucagon positive cells were observed in rat islets after transplantation, also similar to those before transplantation (Fig. 4P). Together these data demonstrated that the NICE device was biocompatible and long-term functional in correcting diabetes in mice with diverse rodent islets.
Immunoprotective function of NICE device in autografts, allografts and xenografts
To analyze the immune responses and understand the immunoprotective function of the device under different transplantation scenarios (i.e. autografts, allografts and xenografts), devices were retrieved after one month of implantation and analyzed for the cellular composition surrounding the device and the antibodies within the device. Flow cytometry analysis of the cells deposited on the device showed that 25% of the cells around the encapsulation device were lymphocytes in autograft group (Fig. 5A). In contrast, 80% of the cells deposited on the device were lymphocytes in allografts and xenografts, most of which were CD3+ T cells and macrophages (Fig. 5, A, B, fig. S6A and S7). Majority of the T cells in allografts and xenografts were CD4+ T cells (Fig. 5C). Importantly, less than 1.5% cells were CD8+ T cells, which suggested that CD8+ T cells were not recruited to the graft (Fig. 5D). Other types of immune cells found in the fibrotic layer included B cells (around 5%), neutrophils (less than 5%) and dendritic cells (around 15%) (fig. S6, B–D). These data showed that, despite the presence of various types of immune cells, there were very few CD8+ cells around the device even in xenografts.
Fig. 5.
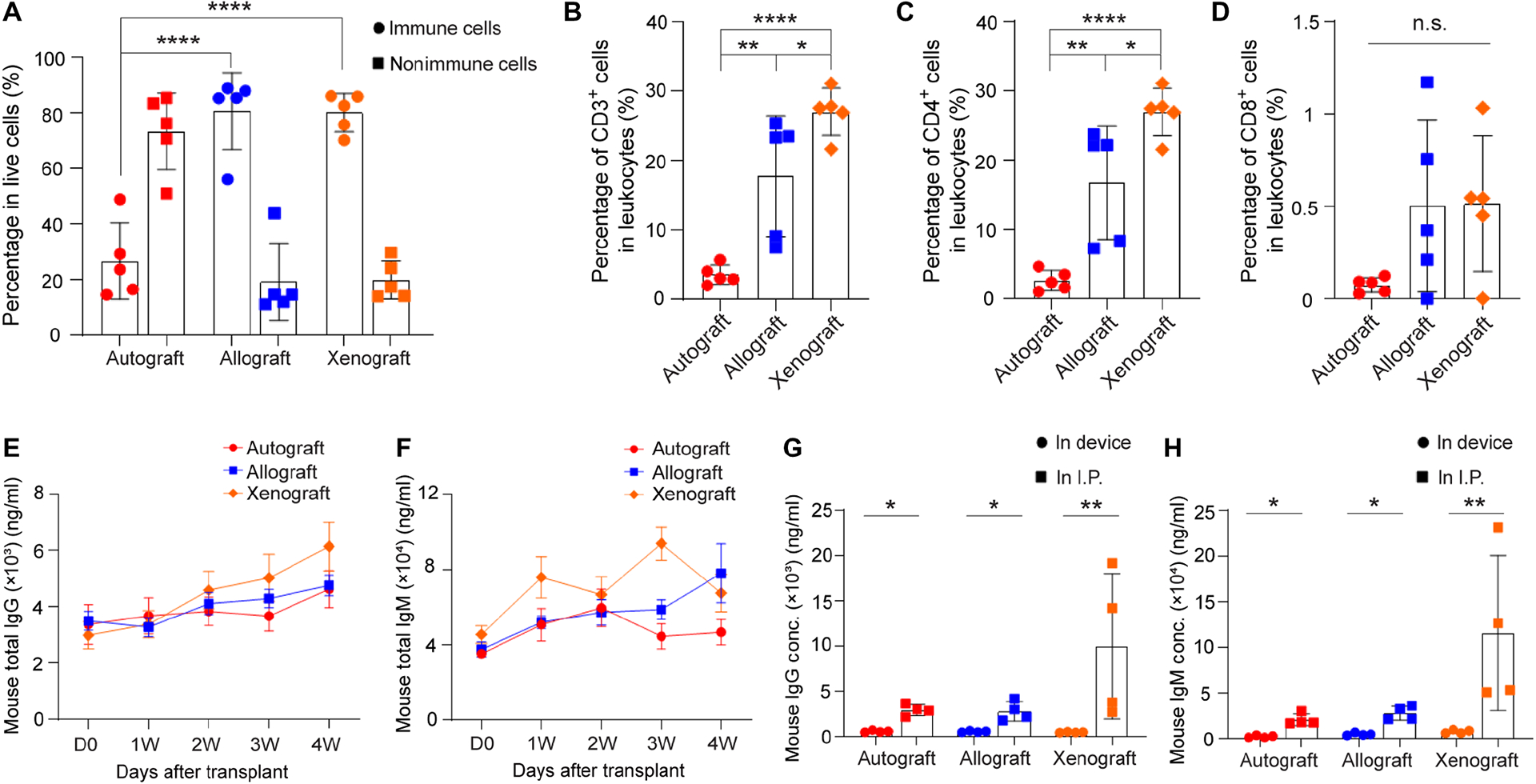
Analysis of immune cells in the fibrotic layer around the NICE device (A-D) and antibody levels (E-H) in three different groups: autograft, allograft and xenograft. (A) Percentage of lymphocytes and non-lymphocytes in live cells, mean ± SD (n = 5). (B) Percentage of CD3 positive cells in lymphocytes, mean ± SD (n = 5). (C) Percentage of CD4 positive cells in lymphocytes, mean ± SD (n = 5). (D) Percentage of CD8 positive cells in lymphocytes, mean ± SD (n = 5). (E) Measurement of mouse total IgG in serum extracted from recipients with autografts (red line), allografts (blue line) and xenografts (green line) before transplantation and at 1w, 2w, 3w and 4w post-transplantation, mean ± SD (n = 5). (F) Measurement of mouse total IgM in serum extracted from recipients with autografts (red line), allografts (blue line) and xenografts (green line) before transplantation and at 1w, 2w, 3w and 4w post-transplantation, mean ± SD (n = 5). (G) Measurement of mouse IgG in retrieved device and in I.P. fluid extracted from recipients after device retrieval, mean ± SD (n = 4). (H) Measurement of mouse IgM in retrieved device and in I.P. fluid extracted from recipients after device retrieval, mean ± SD (n = 4). The one-way ANOVA followed by Tukey’s test was performed for comparing the multi-group data. The two-tailed Student’s t-test was performed when the data consisted of only two groups. The level of significance was labeled by n.s., *, ** and ****, denoting non-significant, p value of < 0.05, < 0.01 and < 0.0001, respectively.
To study the humoral immune response to the encapsulated cells, mouse total antibodies (IgG and IgM) were analyzed in serum at different time points post-transplantation, in I.P. fluid and inside the device after retrieval. Mouse IgG antibody in xenografts increased significantly over the 4-week period, while it changed slightly in autografts and allografts (Fig. 5E). Elevated IgM antibodies were observed in the mouse serum in xenografts as early as 7 days after transplantation, with slight decrease at 2 weeks and another peak at 3 weeks (Fig. 5F). Different from xenografts, IgM increased over time in allografts (Fig. 5F). Moreover, it appeared that a stronger antibody response was found in the mice with devices encapsulating xenogeneic islets than in those with devices encapsulating syngeneic or allogeneic islets (Fig. 5, E and F). This response could be elicited by antigens shed from inside the device. More interestingly, data showed that the mouse total antibody (IgG and IgM) levels in the devices were significantly lower than those in I.P. fluids in all grafts including xenograft (Fig. 5, G and H). To determine whether the IgG and IgM detected in allografts were donor specific alloantibodies, the BALB/c T cells were incubated with sera withdrawn from the recipients with allografts and labeled with FITC anti-mouse IgG or anti-mouse IgM. Sera extracted from sensitized C57BL/6 mice with BALB/c islets transplanted in kidney capsules at 4 weeks post-transplantation was used as positive control. The flow cytometry data showed that there were anti-BALB/c alloantibodies (IgG and IgM) detected in C57BL/6 sera with allografts at 28 days post-transplantation although with much lower concentration than in the sera of sensitized mice (fig. S8). However, almost no alloantibodies were present inside the device (fig. S8). All these data taken together confirmed the immunoprotective function of the device.
The NICE device supports the function of human islets in immunodeficient mice
To demonstrate the potential of the NICE device for human islet encapsulation, we performed transplantation in diabetic, immunodeficient SCID-beige mice. Human islets with variable sizes but acceptable viability (70–90%) (Fig. 6A and fig. S9A) were received, encapsulated (~1700 IEQ per mouse) and transplanted. From immunostaining studies, the human islets before transplantation were positive for β cell markers such as insulin, C-peptide and NKX6.1 (fig. S9, B and C). Eight out of eleven recipients recovered from diabetes immediately (within a week) and maintained normoglycemia for up to 100 days before retrieval (Fig. 6B). Human C-peptide after IPGTT at 2-, 8- and 14-weeks’ time points was detectable in the serum and the concentration was significantly increased at 90 mins compared to 0 min (Fig. 6C), suggesting that human islets inside the device were responsive to glucose and secreted insulin. Blood glucose measurements during an IPGTT at week 8, representative of other time points, showed that the engrafted mice cleared glucose within 2 hours (Fig. 6D). The retrieved device had minimal cellular overgrowth and healthy islets were observed in the device in both the short term (Fig. 6E) and long term (Fig. 6H). Immunostaining showed the positive insulin and glucagon expression (Fig. 6, F and I) and C-peptide and NKX6.1 expression (Fig. 6, G and J) at 40 days and 105 days, even though some small necrotic areas existed in larger islets. Quantitative analysis of the expression of insulin and glucagon after transplantation showed that around 64% of cells within one islet were insulin positive β cells and 31% of cells were glucagon expressing α cells (Fig. 6K), which were similar to the percentages of α and β cells of human islets before transplantation (fig. S9D). Also, no polyhormonal cells were observed either before or after transplantation. Further analysis revealed that around 67.2% of cells within one islet were C-peptide expressing cells, 55.1% of cells were NKX6.1 positive and 53.6% were C-peptide/NKX6.1 double positive β cells (Fig. 6L), which were also similar to those before transplantation (fig. S9E). To further verify that the transplanted islets were responsible for glucose control, the engrafted devices were retrieved at one month and an ex vivo GSIS test was carried out. The retrieved devices secreted significantly more human C-peptide in high glucose solution than that in low glucose solution (Fig. 6M). In addition, dynamic calcium imaging was conducted on extracted, dissociated single cells. The cells responded with increased calcium influx when exposed to high glucose solution or KCl, an agent to depolarize the cell membrane (Fig. 6N), confirming their functions.
Fig. 6.
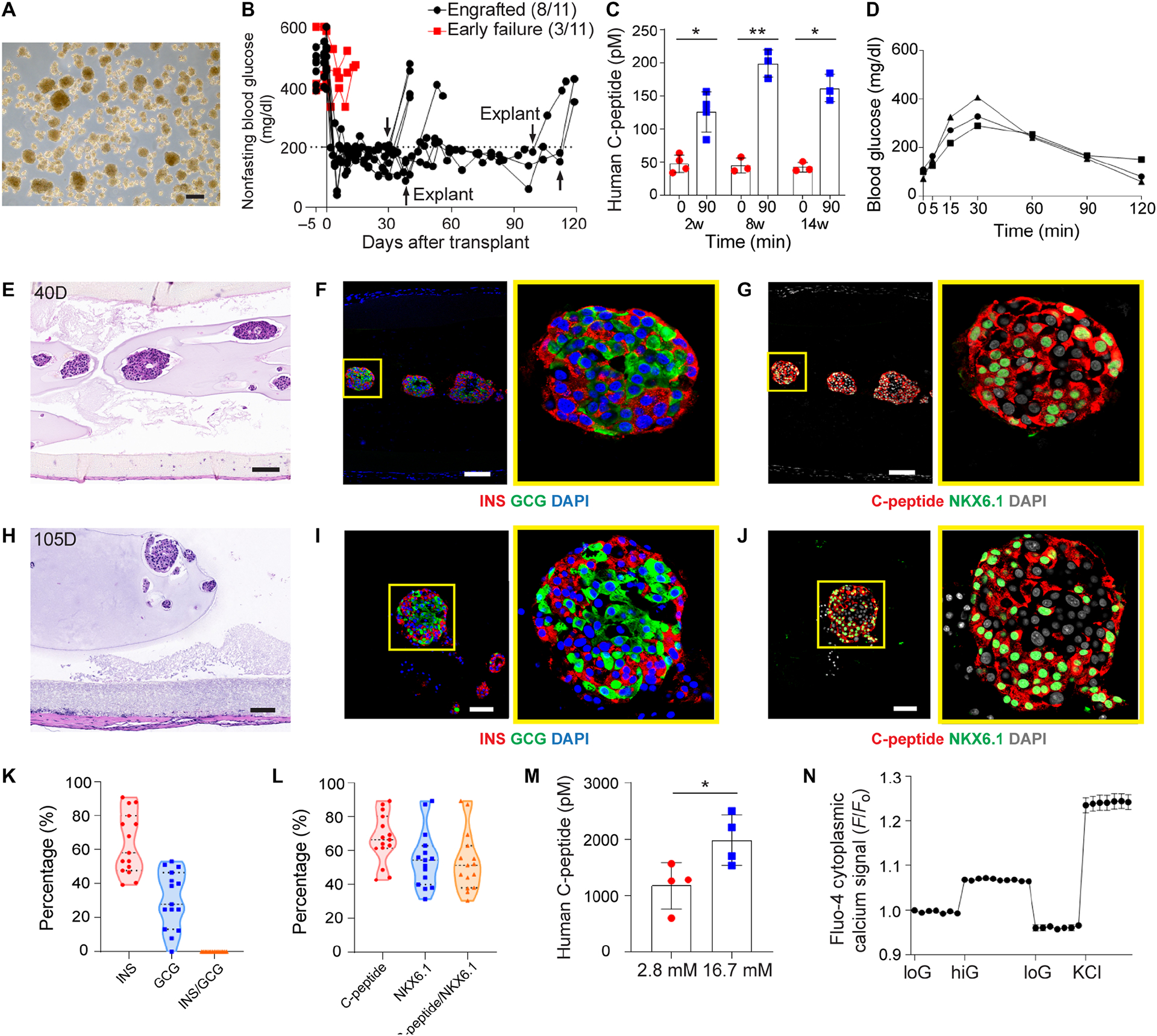
Device function with human islets in diabetic immunodeficient SCID-beige mice. (A) Bright field image of human islets before transplantation. (B) Measurement of non-fasting blood glucose levels (n = 11). Arrows indicate the time points when implants were retrieved from recipients. (C) Measurement of human C-peptide in serum of the mice at 0 min and 90 mins following IPGTT test after 2, 8 and 14 weeks of transplantation, mean ± SD (n = 3–4). (D) Typical blood glucose measurement in IPGTT test of engrafted recipients (week 8). (E-G) H&E staining (E) and immunofluorescent staining (F and G) of human islets from retrieved device after 40 days (higher magnifications on the right). (H-J) H&E staining (H) and immunofluorescent staining (I and J) of human islets from retrieved device after 105 days (higher magnifications on the right). (F and I) Co-immunostaining of insulin (red), glucagon (green) and DAPI (blue). (G and J) Co-immunostaining of C-peptide (red), NKX6.1 (green) and DAPI (gray). (K) Percentage of hormone expressions (insulin, glucagon and insulin/glucagon) quantified from immunofluorescent staining images. One dot represents one islet (n = 15). (L) Percentage of β cell marker expressions (C-peptide, NKX6.1 and C-peptide/NKX6.1) quantified from immunofluorescent staining images. One dot represents one islet (n = 15). (M) Measurement of C-peptide concentration of retrieved devices from engrafted mice following ex vivo glucose stimulation insulin secretion (GSIS) test, mean ± SD (n = 4). (N) Measurements of dynamic normalized Fluo-4 fluorescence intensity for retrieved human islets challenged sequentially with 2, 20, 2 mM glucose and 30 mM KCl, mean ± SD (n = 11). The two-tailed Student’s t-test was performed when the data consisted of two groups. The level of significance was labeled by * and **, denoting the p value of < 0.05 and < 0.01, respectively. Scale bars: 200 μm (A), 100 μm (E-H) and 50 μm (I and J).
The NICE device supports the function of human SC-β cells in immunodeficient mice
From a broad point of view, the highest impact of a cell encapsulation device that is both safe and functional would be its use for delivery of human SC-β cells. To provide evidence that the NICE device has this potential, we first encapsulated SC-β cells (6, 9) and implanted them in immunodeficient NSG diabetic mice (~2500 clusters per mouse). The SC-β cells had a more uniform size (~100–150 μm in diameter) compared to the human islets (Fig. 6A and 7A). From immunostaining studies, the SC-β cells before transplantation were positive for β cell markers such as insulin, C-peptide and NKX6.1 (fig. S10, A and B). About 60% of cells were positive for both C-peptide and NKX6.1 as measured by flow cytometry (Fig. 7B) and 12% were polyhormonal cells with expression of both C-peptide and glucagon (Fig. 7C). Slightly lower C-peptide expression (~55%) was obtained by quantifying the immunostaining images (fig. S10, C and D). In a dynamic GSIS assay, SC-β cells secreted higher amounts of insulin with a clear first- and second-phase response when exposed to increased glucose (Fig. 7D). Static GSIS also demonstrated that SC-β cells before transplantation can secrete more insulin in high glucose solution than that in low glucose solution (fig. S10E). Furthermore, oxygen consumption rate (OCR) measurement showed that SC-β cells had a higher OCR normalized to DNA than human islets, indicating SC-β cells might have a higher metabolism than human islets (Fig. 7E).
Fig. 7.
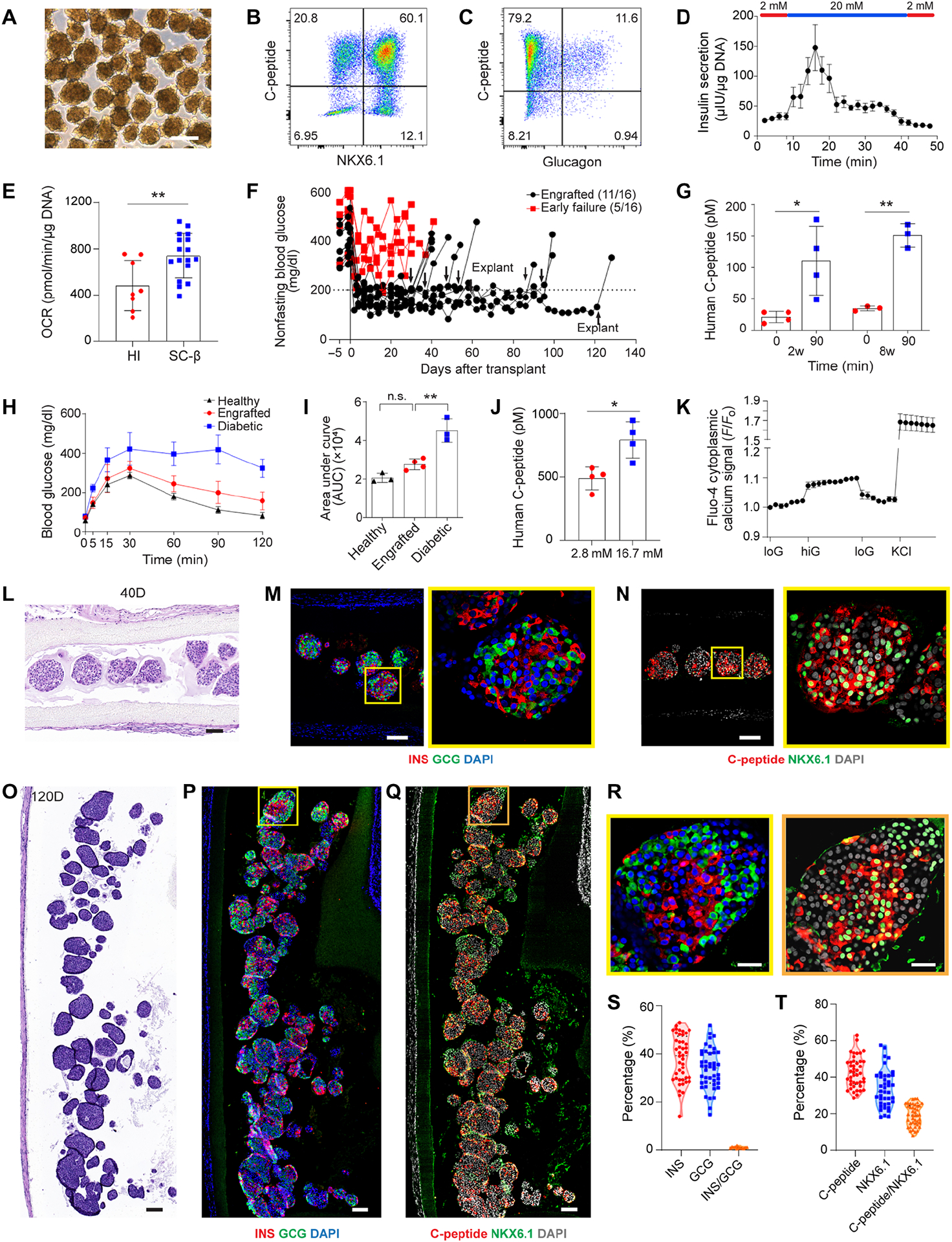
Device function with SC-β cells in diabetic immunodeficient NSG mice. (A) Bright field image of SC-β cell aggregates before transplantation. (B and C) Representative flow cytometric dot plots of dispersed stage 6 SC-β cells immunostained for the indicated markers. (D) Dynamic glucose stimulated human insulin secretion of cells in stage 6 in a perfusion GSIS assay. Data for each individual time point is shown as mean ± SEM (n = 3). Cells are perfused with low glucose (2 mM) and high glucose (20 mM) as indicated. (E) Oxygen consumption rate of human islets (n = 8) and SC-β cells (n = 17) at high glucose (20 mM) normalized to DNA content, mean ± SD. (F) Measurement of non-fasting blood glucose level of the mice transplanted with SC-β cells in the device (n = 16). Arrows indicate the time points when implants were retrieved from recipients. (G) Measurement of human C-peptide in mouse serum at 0 min and 90 mins following IPGTT test after 2 and 8 weeks of transplantation, mean ± SD (n = 3–4). (H) Representative blood glucose measurement in IPGTT test of engrafted recipients (n = 3–4). (I) Quantification of AUC from (H) for different groups, mean ± SD. (J) Measurement of C-peptide concentration of retrieved devices from engrafted mice following ex vivo glucose stimulation insulin secretion (GSIS) test, mean ± SD (n = 4). (K) Measurements of dynamic normalized Fluo-4 fluorescence intensity for retrieved SC-β cells challenged sequentially with 2, 20, 2 mM glucose and 30 mM KCl, mean ± SD (n = 14). (L-N) H&E staining (L) and immunofluorescent staining (M and N) of SC-β cells from retrieved device after 40 days (higher magnification on the right). (O-R) H&E staining (O) and immunofluorescent staining (P-R) of SC-β cells from retrieved device after 120 days. (M and P) Co-immunostaining of insulin (red), glucagon (green) and DAPI (blue). (N and Q) Co-immunostaining of C-peptide (red), NKX6.1 (green) and DAPI (gray). (R) High magnification images of cell aggregates from (P) and (Q). (S) Percentage of hormone expressions (insulin, glucagon and insulin/glucagon) in one aggregate quantified from immunofluorescent staining images. One dot represents one aggregate (n = 44). Samples were collected from four animals. (T) Percentage of β cell marker expressions (C-peptide, NKX6.1 and C-peptide/NKX6.1) per cell aggregate quantified from immunofluorescent staining images. One dot represents one aggregate (n = 43). Samples were collected from four animals. The two-tailed Student’s t-test was performed when the data consisted of two groups. The level of significance was labeled by n.s., * and **, denoting non-significant, p value of < 0.05 and < 0.01, respectively. Scale bars: 250 μm (A), 100 μm (L-Q), 25 μm (R).
The device with SC-β cells corrected glucose levels immediately (within a week) in 11 out of 16 mice, which was maintained for up to 120 days (Fig. 7F). Recipients became diabetic again after explanting the devices, an evidence that the blood glucose was controlled by the NICE device with SC-β cells (Fig. 7F). Human C-peptide in the blood at 90 minutes post glucose challenge (111 ± 21.3 pM) was significantly higher than the fasting value (21.4 ± 7.7 pM) at 2 weeks after implantation (Fig. 7G). At 8 weeks, there was also glucose responsive C-peptide secretion at 90 min (151.2 ± 15.4 pM) compared to that at 0 min (34.8 ± 3.1 pM) (Fig. 7G). There was no significant difference in terms of the stimulation index at 2 weeks (5.4 ± 1.9) and that at 8 weeks (4.3 ± 0.1), suggesting stable device function during that period (fig. S10F). For the engrafted mice studied at 30 days, glucose clearance after IPGTT was completed by 120 minutes (Fig. 7H); the AUC of the glucose concentration curve was similar to that of healthy mice, while significantly less than that of diabetic mice (Fig. 7, H and I). An IPGTT study carried out at 8 weeks also demonstrated the long-term function of SC-β cells in the device (fig. S10G). In addition, the total insulin content in the pancreas of the engrafted mice was significantly lower than that of healthy mice, which again confirmed that the transplanted grafts were responsible for the observed metabolic control (fig. S10H).
Ex vivo GSIS test showed that devices retrieved at one month secreted significantly more human C-peptide in high glucose solution than that in low glucose solution (Fig. 7J). Dynamic calcium imaging conducted on dissociated cells corroborated the GSIS results: the retrieved SC-β cells exhibited glucose- and KCl-responsive calcium influx (Fig. 7K). Explanted SC-β cells at 40 days and 120 days remained viable with a healthy and round morphology, even though the cells were packed at a high density within the device (Fig. 7, L and O). The positive expression of insulin and glucagon (Fig. 7, M, P and R) and C-peptide and NKX6.1 (Fig. 7, N, Q and R) of the cells retrieved at different time points (up to 120 days) indicated device function in both short and long terms (fig. S11). Quantitative analysis of the hormone expression within each cell aggregate post transplantation averaged over four recipients showed that around 38.1% of cells were insulin positive and 33.7% of cells were glucagon positive α cells (Fig. 7S and fig. S10I). Interestingly, while polyhormonal cells were observed prior to transplantation (Fig. 7C, fig. S10, A and C), less than 1% of cells expressed both insulin and glucagon after retrieval (Fig. 7S and fig. S10I). About 42.9% of cells within one aggregate expressed C-peptide, 36.9% of cells expressed NKX6.1 and 19.4% were double positive β cells (Fig. 7T and fig. S10J). More importantly, around 32% of cells within the device expressed MAFA, a maturation marker of β cells, after transplantation (fig. S12, A, B and D). The decline of C-peptide or insulin expressing cells after transplantation may be due to the fact that the polyhormonal cells observed before transplant tend to mature into α cells in vivo (6, 7). Despite a lower percentage of β cells, the percentage of α cells in SC-β cells was similar to that in human islets; α cells are important in the crosstalk with β cells and controlling glucose levels (36, 37). All these results showed that the NICE device supported the function of the human SC-β cells in immunodeficient mice.
Testing of the NICE device with human SC-β cells in immunocompetent models
After confirming the function of encapsulated human SC-β cells in immunodeficient mice, we explored the use of the NICE device for delivering them in immunocompetent diabetic mice and healthy dogs. It is noted that these xenotransplantation models may be more challenging than allogenic, human SC-β cells-to-human transplantation and the explorations here are intended to test the robustness, safety, scalability and retrievability in the setting of SC-β cell delivery but not necessarily the full, long-term function of the device. Nevertheless, when devices with the human SC-β cells (~2500 clusters) were implanted in diabetic C57BL/6 mice, 10 out of 17 became normoglycemic immediately (within a week) after transplantation and lasted for up to 8 weeks (Fig. 8A), a result better than we anticipated. Human C-peptide in the blood of mice with engrafted SC-β cells at 90 minutes post glucose challenge (86.7 ± 10.4 pM) was significantly higher than the fasting value (12.4 ± 1.1 pM) at 2 weeks after implantation (Fig. 8B). However, at 4 weeks, there was no significant difference of C-peptide secretion before and after glucose injection, which indicated that the function of cells declined after 4 weeks (Fig. 8B). Blood glucose measurement during IPGTT at 2 weeks showed that the engrafted mice cleared glucose within 2 hours, while the early failed mice remained at high glucose levels (Fig. 8C). The mice engrafted with SC-β cells showed similar blood glucose clearance following IPGTT with those engrafted with human islets (Fig. 8C), which were included as a control (fig. S13). The SC-β cells in retrieved devices secreted significantly more human C-peptide in high glucose solution than that in low glucose solution following ex vivo GSIS test (Fig. 8D). In addition, the dynamic calcium imaging conducted on retrieved cells showed glucose-responsive calcium influx (Fig. 8E). Histological studies showed that the SC-β cells were reasonably healthy after 35 days, despite that the FBR was more severe than that observed in immunodeficient mice and there were some cells that penetrated from outside into the membrane (Fig. 8F and fig. S14). Immunostaining also confirmed the existence of insulin, glucagon, C-peptide, NKX6.1 (Fig. 8G and fig. S15) and MAFA (fig. S12, A, C and D).
Fig. 8.
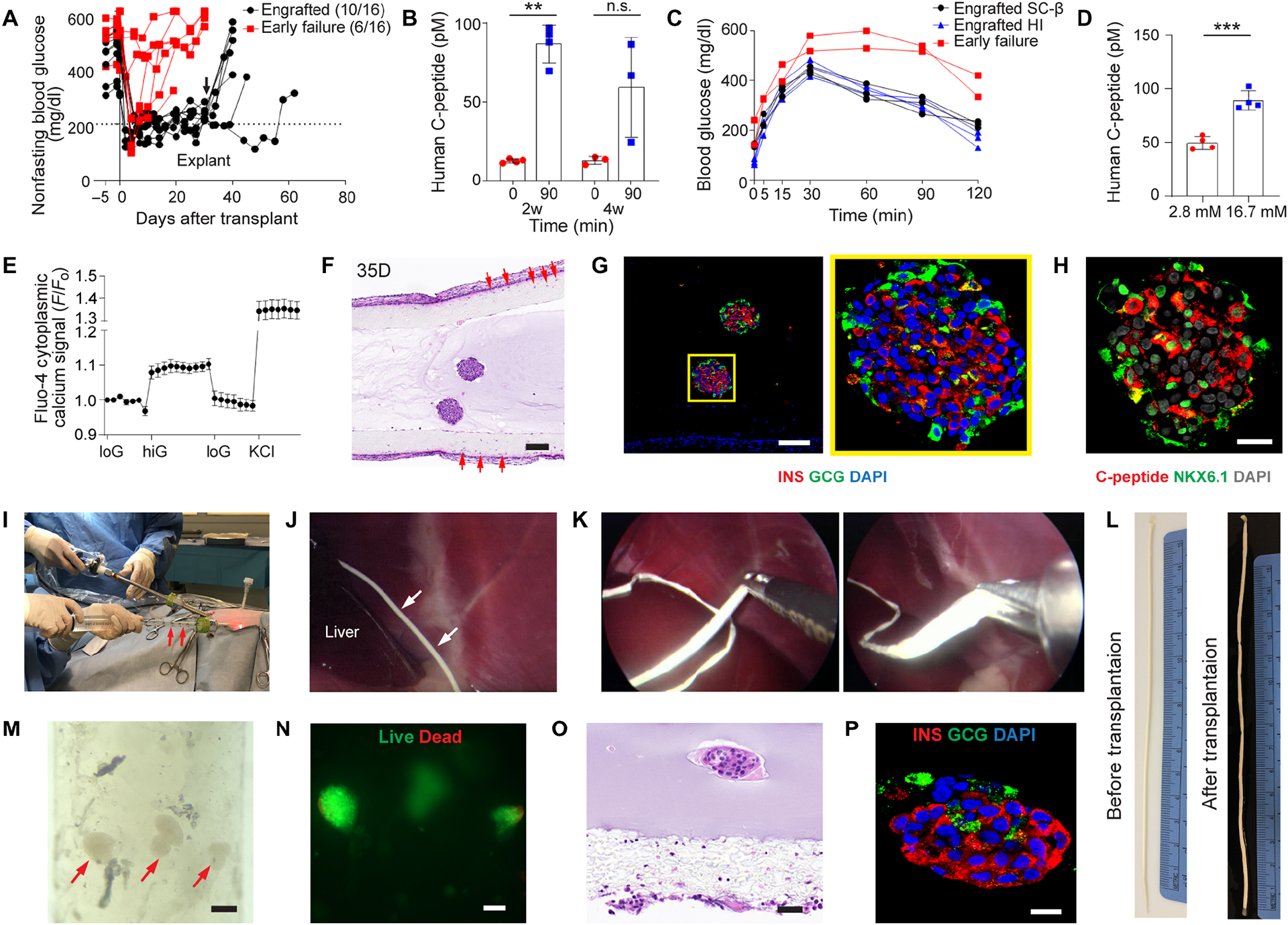
Device function with SC-β cells in diabetic immunocompetent C57BL/6 mice (A-G), and scalability, retrievability and SC-β cell survival in healthy dogs (H-P). (A) Measurement of non-fasting blood glucose level of the mice transplanted with SC-β cells in device (n = 17). Arrow indicates the time points when implants were retrieved from recipients. (B) Measurement of human C-peptide in mouse serum at 0 min and 90 mins following IPGTT test after 2 and 4 weeks of transplantation, mean ± SD (n = 3–4). (C) Representative blood glucose measurement in IPGTT test of engrafted recipients with SC-β cells (black line), human islets (blue line) and failed ones (red line) on 14 days post-transplant. (D) Measurement of C-peptide concentration of retrieved devices from engrafted mice following ex vivo glucose stimulation insulin secretion (GSIS) test, mean ± SD (n = 4). (E) Measurements of dynamic normalized Fluo-4 fluorescence intensity for retrieved SC-β cells challenged sequentially with 2, 20, 2 mM glucose and 30 mM KCl, mean ± SD (n = 6). (F and G) H&E staining (F) and immunofluorescent staining (G) of SC-β cells from retrieved device after 35 days (higher magnification on the right). Co-immunostaining of insulin (red), glucagon (green) and DAPI (blue). Co-immunostaining of C-peptide (red), NKX6.1 (green) and DAPI (gray). Red arrows in (F) indicated cells penetrating from outside to inside of the outer surface of the nanofibrous membrane. (H) Digital image showing the laparoscopic implantation of device (n = 3). Red arrows point to the device in a 10 mL pipette during implantation. (I) Laparoscopic image showing the device implanted in I.P. near the liver in a dog. White arrows point to the device. (J) Laparoscopic images showing the device being pulled out from a dog during retrieval. (K) Digital image of a device before transplantation. (L) Digital image of a device retrieved from a dog after 2 weeks. (M) Bright field image of encapsulated SC-β cells after retrieval (red arrows point to SC-β cells). (N) Live (green) and dead (red) staining of the SC-β cells in (M). (O and P) H&E staining (O) and immunofluorescent staining (P) of SC-β cells from retrieved device after 2 weeks. (P) Co-immunostaining of insulin (red), glucagon (green) and DAPI (blue). The two-tailed Student’s t-test was performed. The level of significance was labeled by n.s., ** and ***, denoting non-significant, p value of < 0.01 and < 0.001, respectively. Scale bar: 100 μm (F, M and N), 50 μm (O) and 25 μm (P).
To prove the concept that the NICE device is scalable, easy to handle and implant, and retrievable, we implanted devices with human SC-β cells into healthy dogs without administering immunosuppressive drugs (n = 3). We loaded a sub-therapeutic dose (2500 clusters) of SC-β cells into 1 mm-diameter, 17 cm-long devices (Fig. 8K). In a typical procedure, the device was placed in a 10 mL pipette, which was then inserted through a trocar (Fig. 8H and fig. S16A). Under the laparoscopic visualization, the device was implanted into the dog’s peritoneal cavity by flushing the device with saline through the trocar (fig. S16B and Movies S3). The device was implanted freely in the I.P. cavity without anchoring and placed in the cranial abdomen near the liver (Fig. 8I). Two weeks later, the device was retrieved by grasping one end of the device using laparoscopic forceps and pulling the device through a trocar (Fig. 8J). In two dogs, minor adhesions occurred between the peritoneal wall and a short segment of the device. In another dog, there was a point adhesion to the liver. In all 3 dogs, adhesions were considered minor and easy to separate, and the devices were all retrievable in their entirety (Fig. 8L, fig. S16C and Movies S4, S5). The SC-β cells were still encapsulated in intact alginate hydrogel within retrieved devices (Fig. 8M). In addition, while many cells were dead (fig. S16D), likely due to the severe immune responses in the challenging xenogeneic setting, it was surprising to observe viable cells through live/dead staining (Fig. 8N). The H&E staining and immunostaining of insulin and glucagon also confirmed the viability and function of some cells (Fig. 8, O and P). These stringent, exploratory studies in immunocompetent mice and dogs, which may be more immunologically challenging than the scenario of human trial, demonstrated procedure-wise the feasibility of the NICE device for SC-β cell delivery.
Discussion
In this study, we developed a nanofibrous-skin, hydrogel-core encapsulation device for the safe delivery of insulin-producing cells, especially human SC-β cells. The design takes advantage of the unique, tunable nanofibrous structure of electrospun nonwoven membranes and the biocompatible, tough and easy-to-process TSPU polymer. With proper nanofiber size (< 500 nm) and membrane thickness (~100 μm), the nanofibrous skin prevented cell escape or penetration while maintaining superb mass transfer. Inside the device, we use a common, easy-to-crosslink alginate hydrogel to disperse the islets and avoid cell clumping for better access to nutrients, notably oxygen (38). In addition, alginate hydrogel materials have been shown to be immunoprotective (39), support the survival of islets via high hydration (40) and reduce diffusion of particular antibodies (41). The whole device is soft but tough, easy to fabricate and does not require any extra internal or external support to maintain its shape, leaving a unique nanofibrous surface as the host interface.
The long-term efficacy of cell encapsulation devices is highly dependent on the biocompatibility of the material and the transplantation site. We explored three clinically relevant transplant sites, the I.P. space, the E.F.P. (42) and the S.C. space (43) for the NICE device. Subcutaneous implantation was the least invasive transplantation site but caused the thickest FBR-induced fibrotic deposition around the device; modification of the site to induce a supporting vasculature will likely be needed for future development (44–48). Implantation in the E.F.P. (as a surrogate for the omentum) would provide access to vasculature (49), but the device-tissue adhesion observed during retrieval may become a challenge for clinical applications. On the other hand, the I.P. space was accessible, allowed facile retrieval of the device without significant adhesion and importantly caused the least cellular overgrowth. We attribute the minimal fibrotic reaction against the NICE device to both the biocompatibility of the TSPU polymer as well as the softness and nanofibrous structures of the electrospun nanomembranes. Previous studies have demonstrated that nanofibers may more closely mimic the natural ECM and provide unique contact cues to modulate macrophage cells toward anti-inflammatory phenotypes in vitro and in vivo compared to microfibers (50, 51).
In addition to good biocompatibility, the function of the islets in the encapsulation device also highly depends on mass transfer under the constraint that the pores should be sufficiently small to at least prevent direct contacts between the host immune cells and the encapsulated ones. While it is difficult to measure the exact pore sizes of an electrospun nonwoven membrane, the average pore size for the NICE device was estimated to be around ~1 μm which was shown in most cases to be effective in preventing the invasion of immune cells into the device, perhaps also partly due to the unique nonwoven, multilayer nature of the membrane. Indeed, research has been directed at modifying the structure of electrospun membranes to increase cell infiltration when that is desirable (52). It is important to note that the range of pore sizes for the NICE device is much larger than the ones in other previously reported devices which are on the order of 10 or 100 nm. The relatively large pore size allows better mass transfer of nutrients, insulin and metabolic wastes. It should also be noted that alginate hydrogel core of the device likely played a role in the immunoprotection. Our results with proliferative cells confirmed that the NICE device was both safe and functional, preventing cell infiltration or escape while allowing cell viability for 5 months.
Besides a feasible implantation site and a safe and functional device, the islet source is another factor that determines the clinical success of a cell replacement therapy. Thus, in this study, we established the performance of our device using various islet sources, to mimic different clinical scenarios. Currently, both islet auto-transplantation after total pancreatectomy for the indication of pancreatitis and allotransplantation with immunosuppression for the treatment of T1D have been demonstrated clinically feasible (53, 54). We demonstrated that the NICE device protected syngeneic and allogeneic islets to correct diabetes in immunocompetent mice for up to 200 days. Even though there are promising results with these cell sources, the search for a more ideal cell source continues. There is much research interest in xenotransplantation of porcine islets, due to donor tissue abundance, economic considerations (55–57) and particularly the recent advancement of genome editing to inactivate the porcine endogenous retrovirus (58). However, it is likely challenging for devices alone, without any immunosuppression, to protect and support the function of porcine islets in humans. Indeed, comparisons of diabetes correction results in mice using rat islets to that using mouse ones, or the functional outcome of human SC-β cells in immunodeficient mice to that in immunocompetent ones, suggest that the NICE device will be more suited for allogeneic than xenogeneic transplantation.
To better understand the immunoprotective function of the device with the different islet sources, we examined the composition of the cells surrounding the device following transplantation and the antibody levels in circulation, in the I.P. fluid surrounding the device and within the device. While there were various types of immune cells surrounding the device including CD4+ T cells, B cells, macrophages, dendritic cells and neutrophils, almost no CD8+ T cells were present even in xenografts. This may be important because CD8+ T cells are known to be recruited in non-immunoprotected β cell transplantation and can directly destroy allogeneic islets by releasing granzyme B and perforins (28, 59). Moreover, antibody analysis showed there were elevated IgG and IgM concentrations and existence of alloantibodies in the sera of recipient mice and that a stronger antibody response was found in the mice with devices encapsulating xenogeneic islets than in those with devices encapsulating syngeneic or allogeneic islets. The antibody response is not unexpected since the device can prevent direct immune recognition but small molecular weight shed antigens can still diffuse out of the device and elicit immune responses by indirect recognition. However, results showed that almost no alloantibodies were present inside the device. This is likely because the nanofiber membrane and the alginate core combinedly present diffusion barriers for the relatively large antibodies. Lastly, we expect small molecule cytokines such as IFN-γ, IL-1β, and TNF-α with molecular weight of 17 to 51 kDa and hydrodynamic radii of 2 to 3 nm can likely diffuse into the device and may cause harmful effects on islets over time (28, 60, 61). Nevertheless, these immunological analyses confirm the immunoprotective function of the NICE device as supported by the functional data with multiple islet sources.
Above all, human SC-β cells are one of the most promising alternative cell sources for diabetes cell replacement therapy. A critical challenge, however, is the delivery of these cells ideally in a safe and retrievable device without immunosuppression. In particular, since the auto-immune disease process that originally destroyed β cells in the pancreas may be re-invigorated by the introduction of new insulin producing cells (62), new methods including development of safe encapsulation devices to prevent both allo- and auto-immunity without systemic immunosuppression are needed (63–70). We showed the NICE device was effective in supporting the function of relatively mature human SC-β cells and reversing diabetes in both immunodeficient mice (for up to 120 days) and immunocompetent mice (for up to 60 days). It is also noted that diabetes correction occurred within a week after transplantation. These results to our knowledge represent the first report of immediate diabetes correction without any in vivo maturation period in a retrievable encapsulation device. Lastly, immunosuppression such as treatment of rapamycin which suppresses T cell activation and proliferation led to improved engraftment of encapsulated SC-β cells in immunocompetent mice (fig. S17).
Limitations exist in this study. For example, early failures in engraftments were observed and might be due to various factors including sealing defects (fig. S18A), material defects (fig. S18B), unintentional inconsistencies in encapsulation and surgical procedures, number and quality of islets, and biological variations of recipients. Quality control of cells and devices will be critical to minimizing the earlier failures and eventually making the cell encapsulation a clinical reality. More specifically, for our device, quality control strategies include standardizing the fabrication and examining representative devices from every batch including scanning electron microscopy, tensile test, porosity measurement, in vitro biocompatibility with islets and in vivo biocompatibility; only after passing these assessments could that batch of devices be used in islet and β cell encapsulation (fig. S19). Another limitation is the lack of ideal animal models used in our study. The true potential of the NICE device for human SC-β cell-based T1D therapy would be best and ideally tested in human patients in an allogenic setting; human SC-β cells elicited much stronger immune responses than rodent islets in mice (fig. S20). As a preclinical proof of concept, we tested the scalability, safety and procedural feasibility (i.e. handling, implantation and retrieval) of the device with human SC-β cells in healthy dogs. Despite the challenging xenogeneic environment and only a short 2-week transplantation, the fact that we observed viable and functional SC-β cells in retrieved devices was encouraging and more importantly the device was safe, relatively easy to implant and completely retrievable with minimally invasive laparoscopic procedures. Longer-term implantation will likely lead to more tissue adhesions and make the retrieval less smooth. The fibrotic cellular overgrowth after long-term implantation in large animal models or human patients is another major challenge for clinical applications. Given our mouse studies which seem to suggest that the NICE device supported long-term function of allogeneic cells even in presence of cellular overgrowth, it remains to be investigated what level of fibrotic deposition may be permissible for cellular function in higher order animals or human patients and whether additional fibrosis-mitigating coatings (18) may be applicable to improve the device function.
In conclusion, rapid stem cell technology advancements in recent years (6–10) have fueled a great hope that stem cell-derived β cells may be used one day to replace the missing cell type in patients with diabetes and functionally cure T1D. To enable these therapies, encapsulation represents a promising, immunosuppression-free method to protect the cells from the autoimmune and allo-/xeno- immune attack while mitigating potential risks such as teratoma formation by undifferentiated stem cells (22). In this work, we report the NICE device as a translatable strategy in the delivery of insulin producing cells especially human SC-β cells. The low complexity design, the relatively easy fabrication and importantly the balance of safety and function make the NICE device an ideal candidate for future development for eventual clinical applications.
Materials and Methods
Study design:
The purpose of this study was to design a translational device to deliver insulin-producing cells, especially human SC-β cells for T1D treatment. Animals were handled and cared by trained scientists and approved by Cornell Institutional Animal Care and Use Committee. We carried out islet encapsulation experiments using different types of islets in the I.P. space in mice to examine the islet viability and function in this device and scaled up the device with SC-β cells in healthy dogs to test its safety, retrievability and biocompatibility. Rodent islets were transplanted in immunocompetent diabetic mouse models, human islets were transplanted into immunodeficient diabetic mouse models, and SC-β cells were transplanted into immunodeficient and immunocompetent diabetic mouse models as well as immunocompetent healthy dogs. The optimal number of islets used in each study was determined based on previous experience. Sample size, including number of mice per group, was chosen to ensure adequate power and was based on historical data. All mice used were males to eliminate any potential confounding influences of gender differences. All mice were randomly assigned to treatment groups, and all analyses were performed blinded to treatment conditions. No animals were excluded from analysis, and no outliers were excluded. The number of biologic replicates is specified in the figure legends.
Chemicals:
Calcium chloride (CaCl2), barium chloride (BaCl2), sodium chloride (NaCl), tetrahydrofuran (THF) and N, N-Dimethylformamide (DMF) were purchased from Sigma-Aldrich Co. (St. Louis, MO). Glucose was purchased from Mallinckrodt Pharmaceuticals (Dublin, Ireland). Ultrapure, sterile sodium alginate (SLG100) was purchased from FMC BioPolymer Co. (Philadelphia, PA). Thermoplastic silicone polycarbonate urethane (TSPU), a product of DSM Biomedical (CarboSil®, Exton, PA, USA) was received as a gift.
Animals:
8 weeks- old male C57BL/6, BALB/c, FVB-Tg(CAG-luc,-GFP)L2G85Chco/J (L2G85) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). SCID-Beige mice were obtained from Taconic Farms (Hudson, NY). Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). Human islets were provided by Prof. Yong Wang from Division of Transplant Surgery, University of Virginia (Charlottesville, VA), Prof. Chengyang Liu and Prof. Ali Naji from Department of Surgery, Hospital of the University of Pennsylvania. Beagle dogs were obtained from Marshall Bioresources (Clyde, NY). All animal procedures were approved by the Cornell Institutional Animal Care and Use Committee.
Stem cell culture:
The nondiabetic HUES8 hESC line was used to generate SC-β cells in this study, which has been previously published (6, 9). Undifferentiated hESCs were cultured in 30-mL spinner flasks (Reprocell; ABBWVS03A) on a rotator spinning plate (Chemglass) at 60 rpm in mTeSR1 media (StemCell Technologies; 05850). The flask remained in a humidified 5% CO2 incubator at 37° C. The cells were passaged every 3 days by single cell dispersion with Accutase (StemCell Technologies; 07920) and counted with a Vi-Cell XR (Beckman Coulter). 18 × 106 viable cells were seeded for propagation in mTeSR1 with 10 μM Y27632 (Abcam; ab120129).
Stem cell differentiation:
18 × 106 viable cells are seeded into a 30-mL spinner flask. The cells are cultured in mTeSR1 for 72 hr and then differentiation begins. The table below details duration, basal media, and added supplements for each stage of differentiation. On the first day of stage 6, the cells are resized by single cell dispersion with TrypLE at 37° C. The cells are then replanted in S6 media, replenished every other day, and placed on an orbital shaker (Benchmark) set at 100 RPM in 6 well plates (6, 9).
Static GSIS of SC-β cells:
Clusters are collected and washed twice in KRB buffer, made from 128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2, 1.2 mM MgSO4, 1 mM Na2HPO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES (Gibco; 15630-080), and 0.1% BSA. The clusters are incubated in transwells (Corning; 431752) in a 24-well plate at 2 mM glucose KRB for a 1-hour equilibration. The transwells are drained and transferred to 2 mM glucose and then 20 mM glucose for 1-hour incubations. Supernatant is collected and insulin secretion is quantified with a Human Insulin Elisa kit (ALPCO; 80-INSHU-E10.1). The cells are then single cell dispersed with TrypLE and viable cells are counted on a Vi-Cell XR to normalize insulin secretion.
Dynamic GSIS of SC-β cells:
An 8-channel dispenser pump (ISMATEC; ISM931C) connected to 0.015” inlet and outlet two-stop tubing (ISMATEC; 070602-04i-ND) with a 275-μl cell chamber (BioRep; Peri-Chamber) in between and a dispensing nozzle (BioRep; PERI-NOZZLE) attached at the end through 0.04” connection tubing (BioRep; Peri-TUB-040). The system was maintained in a water bath at 37 °C. Clusters are washed twice in KRB buffer and loaded into the cell chamber between Bio-Gel P-4 polyacrylamide beads (Bio-Rad; 150-4124). The chamber is perfused with 2 mM glucose KRB for 90 minutes prior to collecting effluent for 8 minutes. Next, 24 minutes with 20 mM glucose KRB and then 12 additional minutes with 2 mM KRB. The flow rate is 100 μL/min with 2–4 min collections. Last, clusters within the cell chamber were lysed in 10 mM TRIS (MilliporeSigma; T6066), 0.2% Triton-X 100 solution, and 1 mM EDTA. DNA from the lysed solution and insulin secretion were quantified with Quant-iT Picogreen dsDNA (Invitrogen; P7589) and Human Insulin Elisa kits, respectively.
Oxygen consumption rate (OCR):
15–30 clusters of primary human islets or SC-β cells were placed in XF24 islet capture microplates (Agilent; 101122-100) with Seahorse media (7.4 pH RPMI-1640 (Sigma; R6504)) at 20 mM glucose. The Seahorse XFe24 flux analyzer (Agilent) measured OCR of the clusters. Measurements were normalized to DNA through Quant-iT Picogreen dsDNA assay kit (Invitrogen; P7589).
Flow cytometry:
Clusters were single cell dispersed with TrypLE in a water bath at 37 °C. The cells were washed with PBS once and then fixed with 4% paraformaldehyde overnight at 4 °C. The cells are blocked with staining buffer (5% donkey serum (Jackson Immunoresearch; 017-000-121) and 0.1% Triton-X 100 (Acros Organics; 327371000) in PBS), incubated overnight at 4 °C with primary antibodies (list below), washed with staining buffer, stained with secondary antibodies (list below) for 2 hr room temperature, and resuspended in staining buffer to analyze on a LSRII (BD Biosciences). FlowJo was used to generate dot plots and percentages.
Device fabrication:
8%, 10%, 12% and 14% (w/v) TSPU solutions were prepared by dissolving TSPU in a mixture of THF and DMF (3/2). The custom-built electrospinning device was equipped with a syringe pump (Harvard Apparatus, MA), a 10-mL syringe, a stainless-steel blunt needle, a constantly moving needle holder, a rotating collector and a high voltage supply (Gamma High Voltage, Ormond Beach, FL). The nanofibers were spun at 13 kV with a pumping rate of 0.5 mL/h and with a 23G blunt needle as the spinneret. The spinneret was mounted on a sliding table moving back and forth with a speed of 5 cm/s. Working distance was fixed at 10 cm. A rotating target (i.e. stainless-steel rods with diameters ranging from 0.5 mm to 3 mm) was placed in the path of the polymer solution jet. The rod was connected to an AV motor controlled by rheostat (VWR) and rotated at 400–500 rpm. The rod template was pre-coated with a thin layer of sucrose. The nanofibrous tubes were removed from the template by soaking in water. The tubes were cut into desired length and sealed at one end by hand impulse sealer (Impulse Sealer Supply, CA). The devices were soaked in 70% ethanol and sterilized using UV light for further study.
Characterizations of device:
To observe the morphology of nanofibers, membrane samples were gold sputter-coated and examined by the scanning electron microscope (SEM) (LEO 1550 FESEM). ImageJ (NIH, Bethesda, USA) was applied to quantitatively characterize the fiber diameter. 10 random fibers were selected and measured in each image, and a total of 10 images were counted. To study the tensile strength of the electro-spun fibers, the tube was mounted on the dynamic mechanical analysis instrument (DMA Q800) with a distance of ~1.5 cm between the holders. The tensile testing was conducted at a rate of 0.5 N/min at room temperature. Stress (MPa) and strain (%) were automatically calculated by the software. The Young’s modulus was obtained by measuring the slope of the stress-strain curve in the elastic region between 10% and 20%. Four samples were tested for the experiment.
Isolation of rodent pancreatic islets:
Mouse pancreatic islets were isolated from 8-week-old male C57BL/6 mice, BALB/c mice or L2G85 mice. For mice, one bottle of Collagenase (CIzyme RI 005-1030, Vitacyte, Indiana, USA) was reconstituted in 30 mL M199 media (Gibco, USA). The bile duct was cannulated with a 27 G needle as described previously (71) and the pancreas distended with cold Collagenase. The perfused pancreases were then removed and digested in a 37 °C water bath for 17 min. Sprague Dawley rats from Charles River Laboratories weighing approximately 300 g were used for harvesting islets. For rat islet isolation, one bottle of Liberase (Research Grade, Roche) was reconstituted in 33 mL M199 media (Gibco, USA). Briefly, the bile duct was cannulated, and the pancreas was distended by an injection of 0.15% Liberase solution. The pancreas was digested a 37 °C water bath for 28 min. The digestion was stopped by adding 20–25 mL of cold M199 media with 10% heat-inactivated fetal bovine serum (FBS) and a slight shaking. For both mouse and rat islets, digested pancreases were washed twice in the same M199 media, filtered through a 450 mm sieve, and then suspended in an LSM (Lymphocyte Separation Medium, Corning, 25-072-CV) / M199 media gradient and centrifuged at 1750 RCF for 20 min at 4 °C. This gradient centrifugation step was repeated when desired for higher purity islets. Finally, the islets were collected from the gradient and further isolated by a series of gravity sedimentations, in which each supernatant was discarded after 4 min of settling. Purified islets were hand-counted by aliquot under a stereomicroscope (Olympus SZ61). Islets were then cultured in RPMI 1640 media with 10% FBS and 1% penicillin/streptomycin (P/S) for further use.
Isolation of human pancreatic islets:
The pancreatic organs were obtained from the organ procurement organization under the United Network for Organ Sharing (UNOS). The islets were isolated in the Human Islet Core at the University of Pennsylvania following the guidelines of Clinical Islet Transplantation (CIT) consortium protocols (72). Briefly, the pancreas was digested following intraductal injection of collagenase and neutral protease enzyme in Hanks’ balanced salt solution (HBSS). Liberated islets were then purified on continuous density gradients (Cellgro/Mediatech) using the COBE 2991 centrifuge, cultured in CIT culture media and kept in a humidified 5% CO2 incubator.
Cell encapsulation for mouse study:
For a typical mouse study, the electrospun nanofiber device with a diameter of 1 mm was cut into 2.5 cm lengths. Immediately prior to encapsulation, the cultured islets (300–350 IEQ) were centrifuged at 1000 rpm for 1 min and all supernatant was aspirated. 20 μl 2% (w/v) solution of SLG100 alginate dissolved in 0.9% (w/v) NaCl solution was added into the islets pellet. The cell-loaded alginate solution was filled into the device. For human islets, ~800 IEQ were collected and loaded with alginate as described above. For SC-β cells, ~1250 clusters were used. The device was further submerged in a crosslinking buffer containing 100 mM CaCl2 and 5 mM BaCl2. The device was taken out and extra solution on the surface of the device was removed with autoclaved tissue. The end of the tube was thermally sealed with hand impulse sealer (Impulse Sealer Supply). The device was washed with 0.9% (w/v) NaCl solution and ready to use.
In vitro and ex vivo GSIS of islets inside device:
Islet-laden devices and the same number of free-floating islets were cultured in 2 mL RPMI 1640 complete media for 1 day or 7 days in nonadherent 25 mm2 culture dishes. After culture, the devices or free-floating islets were incubated in pre-warmed Krebs Ringer Buffer (KRB) solution (125 mM NaCl, 5.9 mM KCl, 2.56 mM CaCl2, 1.2 mM MgCl2) supplemented with 25 mM HEPES, 1mM L-glutamax, 0.1% BSA and 2.8 mM D-glucose for 30 min at 37 °C, 5% CO2, and then incubated for 1 h with 2.8 mM or 16.7 mM D-glucose under the same condition. The supernatant was collected and frozen for future analysis. The insulin content in the supernatant was quantified by mouse insulin ELISA kit (ALPCO) according to the manufacturer’s specifications. Absorbance of reaction solution at 450 nm was measured in the Synergy plate reader (Biotek). For devices retrieved from mice, the devices were put in KRB buffer supplemented with 2.8 mM D-glucose for 30 min and incubated in KRB buffer supplemented with 2.8 mM or 16.7 mM D-glucose for 90 min. The stimulation index (SI) was calculated as the ratio of the insulin value after high glucose (16.7 mM) stimulation divided by insulin value after low glucose (2.8 mM) solution.
Live and dead staining:
Islet-laden devices and a same number of free-floating islets were cultured as described above for 1 day in nonadherent culture dish. After culture, the nanofiber skin of the device was peeled off. The islet-laden alginate inside the device and the free-floating islets were stained by calcein-AM (green, live) and ethidium homodimer (red, dead) according to the manufacture’s protocol (R37601, Thermo fisher). Fluorescent microscopic images were taken using a digital inverted microscope (EVOS FL Cell Imaging System). Quantification of the percentage of live cells in islets was carried out by calculating the intensity of fluorescence using ImageJ.
Generation of GFP/luciferase expressing cell line:
Plasmid containing enhanced green fluorescent protein (EGFP) gene (720 bp) and humanized firefly luciferase (Luc2) gene (1653 bp) was constructed by Vector Builder. 293T cell line and 4T1 cell line were received as gifts. Strain C57BL/6 Mouse Mesenchymal Stem Cells (MSCs) (MUBMX-01001) were purchased from Cyagen (Santa Clara, CA). NIT-1 cells (ATCC® CRL-2055™) were purchased from ATCC. 293T cells were cultured in DMEM (#2051526, Gibco) supplemented with 10% FBS and 1% P/S. 4T1 cells were cultured in RPMI 1640 media with 10% FBS and 1% P/S. MSCs were cultured in Mesenchymal Stem Cell Growth Medium (GUXMX-90011, Cyagen). NIT-1 cells were cultured in F-12K medium with 10% FBS and 1% P/S. Lentivirus vectors were produced by transfecting 293T cells with the designed plasmid using ViraPower™ Bsd Lentiviral Support Kit (K497000, Thermofisher). Media containing lentivirus vectors were collected and stored at 4 °C. MSC single cell solutions were prepared and 3 mL media containing lentivirus vectors were used to infect MSCs in 6-well plate. After 48h infection, lentivirus medium was discarded and fresh MSC growth medium was added. GFP/luciferase positive MSCs were verified under a fluorescent microscope. For the formation of MSC spheroids, around 2.5 mL solution containing 1 million MSCs were added in a non-adherent 25 mm2 petri dish. Then the cells were cultured on an orbital shaker with a speed of 50 rpm overnight. The spheroids were imaged with a fluorescent microscope mentioned above. The GFP/luciferase 4T1 spheroids were generated in the same manner as GFP/luciferase MSC spheroids.
Bioluminescence imaging:
The GFP/luciferase expressing aggregates were collected and centrifuged at 1000 rpm for 3 min. The cells were washed with saline and centrifuged again to form a pellet. Then 300–400 GFP/luciferase MSC or 4T1 spheroids were loaded with alginate into the NICE device as described above. The GFP/luciferase MSCs- or 4T1-laden devices were implanted into healthy C57BL/6 mice. The GFP/luciferase islet-laden device was prepared as described above and implanted into healthy C57BL/6 mice. The mice were injected with 150 mg luciferin/kg body weight (#122799, PerkinElmer) and imaged with IVIS Spectrum System (PerkinElmer) at the Biotechnology Resource Center at Cornell.
Transplantation and retrieval of the device in mice:
For diabetes study, immunocompetent male C57BL/6 or immunodeficient male SCID-beige or male NSG mice were utilized for transplantation. To create diabetic mice, mice were injected I.P. with (130 mg/kg body weight) freshly prepared streptozotocin (STZ) (Sigma Aldrich) solution (13 mg/mL in 5 mM sodium citrate buffer solution). Only mice whose non-fasted blood glucose levels were above 300 mg/dL with two consecutive measurements were considered diabetic and underwent transplantation. The mice were anesthetized using 3% isoflurane in oxygen and maintained at the same rate throughout the procedure. The abdomens of the mice were shaved and alternately scrubbed with betadine and isopropyl alcohol to create a sterile field before being transferred to the surgical field. A ~1 cm incision was made along the midline of the abdomen and the peritoneum was exposed using blunt dissection. The peritoneum was then grasped with forceps and a ~1 cm incision was made. Two devices with 600–700 IEQ in total were then inserted into the peritoneal cavity through the incision. The incision was closed using 6–0 silk sutures (DemeTECH, FL). The skin was then closed over the incision using wound clips. Glucose was monitored twice a week. A small drop of blood was collected from the tail vein using a lancet and tested using a commercial glucometer (Contour next, Ascensia Diabetes Care, NJ). Mice changing from a diabetic state (blood glucose >~350 mg/dl for at least two consecutive measurements) to a non-diabetic state (blood glucose <~200 mg/dl for at least two consecutive measurements were considered as engrafted mice. For all the engrafted mice, devices were explanted in a survival surgery after a certain time and the mice remained alive after retrieval. Blood glucose was monitored after explanting the device to further confirm the function of the device. Some retrieved samples were examined by ex vivo GSIS test (described above) and fixed in 10% formalin for further staining. For the other devices, the nanofibrous skin was peeled off and the islet-laden alginate inside the device was imaged using the stereomicroscope.
To measure the total insulin content of the pancreas of the engrafted mice, diabetic mice and healthy mice, the homogenized tissue was placed into acid-ethanol (1.5% HCl in 70% ethanol), cut into small pieces using scissors and digested overnight at −20 °C. Then the acid-ethanol extract solution was neutralized with pH 7.5 TRIS buffer. The samples were further diluted, and the insulin content was quantified as mentioned before.
To analyze the in vivo biocompatibility of the device, immunocompetent male C57BL/6 mice were used, and blank devices were implanted in the peritoneum, E.F.P., ventral S.C. space or the dorsal S.C. space of normal C57BL/6 mice. After 2 weeks or 1 month, the implants were harvested, fixed in 10% buffered formalin and embedded in paraffin. Cross-sections were analyzed as described in the Histological Analysis section.
To build an immunosuppressed mouse model, 50 mg/ml Rapamycin (LC Laboratories) stock solution in 100% ethanol was prepared and stored in −80°C. The stock solution was diluted in 10 ml mixture (5 ml of 10% PEG 400 in DI water and 5 ml of 10% Tween 80 in DI water) to make a final concentration of 0.05 mg/ml. Diabetic recipients were injected with 0.5 mg/kg body weight in I.P. daily from −1 to 14 days post-transplantation. The immunosuppressed mice were transplanted with NICE devices encapsulating SC-β cells as described previously.
IPGTT:
Mice were fasted overnight before receiving an intraperitoneal glucose bolus (2 g/kg body weight). The healthy mice and diabetic mice were used as positive control and negative control respectively. Blood glucose levels were monitored at regular intervals (time 0, 5, 15, 30, 60, 90, and 120 min) after injection, allowing for the AUC to be calculated and analyzed between groups.
In vivo GSIS:
The SCID-beige mice implanted with human islet-laden devices or NSG mice implanted with SC-β cells were fasted overnight before receiving an intraperitoneal glucose bolus (2 g/kg body weight). Blood was collected from the orbital vein before injection and 90 min after injection using a tube with clotting activator (Microvette® 300 Z, Sarstedt). The tubes were then centrifuged at 2000 g for 10 min. Serum was collected from the tube and frozen for further analysis. Human C-peptide concentration in the serum was detected using ultra-sensitive human C-peptide ELISA kit (Mercodia).
Cellular analysis of fibrotic layer by flow cytometry:
After the engrafted devices were retrieved from the recipients, the fibrotic layer surrounding the encapsulation device was carefully peeled off using tweezers. The fibrotic layer was cut into small pieces and digested with 1 mg/ml Type I collagenase (Worthington Biochemical Corporation; LS004194) for 1 h in incubator. The digestion was stopped by adding cell culture medium containing 10% FBS. The cell solution was filtered through Falcon 40 μm strainer (Corning; 431750) to get single cell solution. Cells were centrifuged at 1000 rpm for 5 min. Supernatant was discard and cells were washed with PBS solution to remove the remaining FBS. The cells were stained with Zombie Yellow Fixable Viability Kit (BioLegend; 423103) following manufacture’s instruction. Cells were washed one time with 2 ml Cell Staining Buffer (BioLegend; 420201) and centrifuged into a pellet. Fc receptors were blocked by pre-incubating cells with TruStain FcX™ PLUS (anti-mouse CD16/32) Antibody (BioLegend; 156603) in 100 μl Cell Staining Buffer for 5 min on ice. Then cells were labeled with Alexa Fluor® 488 anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody (BioLegend; 108419), PerCP anti-mouse CD45 Antibody (BioLegend; 103129), APC anti-mouse CD11c Antibody (BioLegend; 117309), Alexa Fluor® 700 anti-mouse/human CD45R/B220 Antibody (BioLegend; 103231), APC/Cyanine7 anti-mouse/human CD11b Antibody (BioLegend; 101225), Pacific Blue™ anti-mouse CD3 Antibody (BioLegend; 100213), PE anti-mouse F4/80 Antibody (BioLegend; 123109), Alexa Fluor® 594 anti-mouse CD4 Antibody (BioLegend; 100446) and PE/Cyanine7 anti-mouse CD8a Antibody (BioLegend; 100721) at 1:100 dilution on ice for 15 min. The cells were washed twice with 2 ml of Cell Staining Buffer by centrifugation at 350 g for 5 min. The cell pellet was resuspended in 0.5 ml of Cell Staining Buffer. UltraComp eBeads™ Compensation Beads (Thermofisher; 01-2222-41) incubated with each antibody following manufactures’ instruction were used for compensation. Finally, stained cells were analyzed using Attune NxT flow cytometer (Thermo Fisher). The data were analyzed by FlowJo software v10.7.
Mouse total antibody analysis:
C57BL/6 mice were transplanted with NICE devices encapsulating syngeneic, allogeneic and xenogeneic islets (rat islets or human SC-β cells) for a month. Blood was withdrawn from retro-orbital sinus using capillary at D0, 1w, 2w, 3w and 4w post-transplantation and collected in tubes with clotting activator (Sarstedt; Microvette® 300 Z). The blood samples were centrifuged at 2000 g for 15 min. The serum was transferred in a new tube and stored at −80°C. After devices were retrieved from the recipients, 100 μl saline was added into the I.P. space. The I.P. fluids were collected after gently shaking the belly. The I.P. fluid was centrifuged at 1000 rpm for 5 min to remove cellular debris. The supernatant was transferred into a new tube and store at −80°C. The alginate core inside retrieved devices was collected and degraded with 100 μl 1 mg/ml alginate lyase (Sigma; A1603) and 1X Halt™ Protease Inhibitor Cocktail (Thermofisher; 87786) in saline in incubator for 1 h. Then the tube was centrifuged at 1000 rpm for 5 min to separate the alginate and the encapsulated islets. The supernatant was collected and stored at −80°C. Mouse total IgG (Thermofisher; 88-50400-22) and IgM (Thermofisher; 88-50470-22) were analyzed in all the samples (serum, I.P. fluids and degraded alginate) by using ELISA kit following manufacturer’s instruction.
Donor specific alloantibody analysis:
BALB/c mouse spleen was harvested and placed in 6 well plate. 5 ml RPMI1640 culture medium (Fisher; 11875-119) with 10% FBS was added in each dish. The spleen was smashed using 3 ml syringe head. The spleen cell suspension was filtered through Falcon 40 μm strainer (Corning; 431750) to get single cell solution. The cells were pelleted by centrifugation at 1000 rpm for 5 min and the supernatant was aspirated. Red blood cells were removed by adding RBC lysis buffer (BioLegend; 420301) and cells were incubated on ice for 5 min. The reaction was stopped by adding 20 ml PBS. Cells were centrifuged at 1000 rpm for 5 min and the supernatant was discarded. T cells were isolated using EasySep™ Mouse T Cell Isolation Kit (Stem cell technologies; 19851) following manufacture’s instruction. 105 T cells in 70 μl cell staining buffer were incubated with 30 μl samples (serum collected from recipients with allografts at D0 and 4w, and degraded alginate) at 4 °C for 30 min. Serum collected from sensitized mice with BALB/c mouse islets transplanted in kidney capsule at 4w post-transplantation was used as positive control. After incubation, cells were washed with PBS and centrifuged at 1000 rpm for 5 min. Supernatant was removed and cells were stained with Zombie Yellow Fixable Viability Kit (BioLegend; 423103) following manufacture’s instruction. Cells were washed one time with 2 ml Cell Staining Buffer (BioLegend; 420201) and centrifuged into a pellet. Fc receptors were blocked by pre-incubating cells with TruStain FcX™ PLUS (anti-mouse CD16/32) Antibody (BioLegend; 156603) in 100 μl Cell Staining Buffer for 5 min on ice. Cells were labeled with APC anti-mouse CD3 antibody (BioLegend; 100235) and FITC anti-mouse IgG Antibody (BioLegend; 406001) or FITC anti-mouse IgM antibody (Sigma; F9259) at 1:100 dilution on ice for 15 min. Cells were washed twice with 2 ml of Cell Staining Buffer by centrifugation at 1000 rpm for 5 min. Finally, stained cells were analyzed using Attune NxT flow cytometer (Thermo Fisher). The data were analyzed by FlowJo software v10.7.
Ca2+ imaging:
SC-β cells and human islets were retrieved from the engrafted devices as mentioned above. The SC-β cells or human islets were extracted as mentioned before, shipped overnight, and cultured in S6 or human islet media (CMRLS with 10% FBS) for recovery of SC-islets or human islets, respectively. The clusters were single-cell dispersed with TrypLE for 10 min and seeded in Matrigel-coated 96 well plates (Cellvis, 963-1.5H-N) overnight for attachment in their respective recovery media. Cells were washed with 2 mM glucose KRB and incubated with 20 μM Fluo-4 AM (Invitrogen; F14201) in 2mM glucose for 45 min at 37°C in incubator. Next, the cells were washed with 2 mM glucose KRB and challenged with 2 mM glucose KRB, 20 mM glucose KRB, and 20mM glucose 30 mM KCl KRB, sequentially. Images were taken every minute using a Leica DMI4000 fluorescence microscope and calcium flux was calculated with ImageJ.
Cell encapsulation for dog experiment:
Three devices about 17 cm long were prepared and sterilized as mentioned above. 2500 clusters SC-β cells were suspended in 140 μl 2% SLG100 solution and loaded into one device using PE50 tubing. The device was crosslinked and sealed as mentioned above. The devices were submerged in 0.9% saline and ready for implantation.
Laparoscopic implantation and retrieval in dogs:
Dogs were premedicated with glycopyrrolate and butorphanol, induced with propofol, and anesthetized with isoflurane and oxygen. The abdomen was clipped and prepared for sterile surgery. A 10-mm laparoscopic camera port and two 5-mm instrument ports were percutaneously inserted into the abdomen. The abdomen was insufflated to 12 mm Hg pressure with CO2. The device in a 10 mL pipette was inserted into the abdomen through the left-side instrument port. And then the device was flushed out from the pipette using sterile saline. A laparoscopic probe was introduced through the right-sided 5-mm port and was used to manipulate the device so that it was placed between the liver and the diaphragm. The remaining ports were then removed, and the port sites were closed with 3–0 polydioxanone suture material. For retrieval of the devices, the procedure was similar using one 10-mm camera port and one or two 5-mm instrument ports. The previously implanted device was located and photographed. The device was grasped with laparoscopic Kelly forceps.
Histological analysis:
The implants were harvested from the mice and fixed in 10% formalin, dehydrated with graded ethanol solutions, embedded in paraffin, and sectioned by Cornell Histology Core Facility. The samples were sliced on a microtome at a thickness of 5 μm. The sections were stained with H&E and then imaged by a microscope (IN200TC, Amscope). To conduct immunofluorescence staining, the histological slides were deparaffinized followed by antigen retrieval as described before (42). Non-specific binding was blocked via incubation with 5% donkey serum (S30-M, Sigma) for 1 h at room temperature. Sections were decanted and incubated with primary antibodies overnight at 4 °C. The sections were then washed and incubated with the fluorescently conjugated secondary antibodies for 1 h at room temperature. Nuclei were labeled with DAPI and slides were covered with fluorescent mounting medium (F6057, Sigma). Finally, the sections were imaged through confocal microscopy (FV1000, Olympus, Japan). The primary antibodies used here were: rat anti-C-peptide (1:100, GN-ID4, University of Iowa Developmental Hybridoma Bank; staining for both C-peptide and pro-insulin), mouse anti-NKX6.1 (1:300, F55A12, University of Iowa Developmental Hybridoma Bank), rabbit anti-alpha smooth muscle actin antibody (1:200, ab5694, Abcam), rabbit anti-insulin (1:200, ab63820, Abcam), mouse anti-glucagon (1:200, G2654, Sigma), goat anti-GFP (1:200, 600-101-215S, Rockland), rabbit anti-MAFA (1:200, LP9872, LifeSpan Bioscience), rabbit anti-CD3 (1:100, ab5690, Abcam) and rat anti-F4/80 (1:50, 14-4801-82, Thermofisher). The secondary antibodies used here are: goat anti-rat IgG AF555 (1:400, A21434, Thermofisher), donkey anti-mouse IgG AF488 (1:400, A21202, Thermofisher), goat anti-rabbit IgG AF594 (1:400, A11037, Thermofisher), donkey anti-goat IgG AF488 (1:400, A11055, Thermofisher), donkey anti-rabbit IgG AF568 (1:400, A10042, Thermofisher) and donkey anti-rat IgG AF488 (1:400, A21208, Thermofisher). The thickness of fibrotic layer, the density of α-SMA positive cells and the percentage of hormone positive cells were analyzed using ImageJ software.
Statistical analysis:
Unless otherwise stated, data were expressed as mean ± standard deviation of the mean (SD). For comparisons between two groups, means were compared using unpaired two-tailed Student’s t-tests. Comparisons between multiple groups were performed by ANOVA followed by Tukey’s post-hoc analysis. Samples size, including number of mice per group, was chosen to ensure adequate power and was based on historical data. No exclusion criteria were applied for all analyses. All statistical analyses were performed using GraphPad Prism v.8 software (GraphPad Software Inc.). The level of significance was labeled by n.s., *, **, *** and ****, denoting non- significant and p value of < 0.05, < 0.01, < 0.001 and < 0.0001, respectively.
Supplementary Material
Acknowledgments:
We thank Prof. Nikolaos Bouklas (Cornell University) for discussions on the mechanical properties of the device. We thank Cornell Center for Materials Research Facility for the SEM images, which is supported by the National Science Foundation under Award Number DMR-1719875. We thank Cornell Histology Core Facility for histological cross-sectioning and staining. We thank Dr. Kevin D’Amour (ViaCyte, Inc.) for the MAFA antibody.
Funding:
This work was mainly sponsored by the Novo Nordisk Company and partially supported by the Juvenile Diabetes Research Foundation (JDRF, 2-SRA-2018-472-S-B), the Hartwell Foundation and the National Institutes of Health (NIH, 1R01DK105967-01A1). J.R.M. was supported by the NIH (5R01DK114233) and JDRF (5-CDA-2017-391-A-N). K.G.M. was supported by the NIH (T32DK108742). The NIDDK IIDP (Integrated Islet Distribution Program) for the grant (Beckman Research Center, no. 10028044) was awarded to A.N..
Footnotes
Competing interests: J.R.M. is an inventor on patent and patent application related to the differentiation approaches described in this manuscript. X.W., J.A.F. and M.-L.M. are inventors on a patent application related to the device described in this manuscript.
Data and materials availability:
All data needed to evaluate the conclusion in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References and notes
- 1.Latres E, Finan DA, Greenstein JL, Kowalski A, Kieffer TJ, Navigating two roads to glucose normalization in diabetes: automated insulin delivery devices and cell therapy, Cell Metab 29, 545–563 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Aguayo-Mazzucato C, Bonner-Weir S, Pancreatic β cell regeneration as a possible therapy for diabetes, Cell Metab 27, 57–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro AMJ, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, Cagliero E, Alejandro R, Ryan EA, DiMercurio B, Morel P, Polonsky KS, Reems J-A, Bretzel RG, Bertuzzi F, Froud T, Kandaswamy R, Sutherland DER, Eisenbarth G, Segal M, Preiksaitis J, Korbutt GS, Barton FB, Viviano L, Seyfert-Margolis V, Bluestone J, Lakey JRT, International trial of the Edmonton Protocol for islet transplantation, New Engl J Medicine 355, 1318–1330 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Shapiro AMJ, Lakey JRT, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV, Islet transplantation in seven patients with Type 1 Diabetes Mellitus using a glucocorticoid-free immunosuppressive regimen, New Engl J Medicine 343, 230–238 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Hering BJ, Clarke WR, Bridges ND, Eggerman TL, Alejandro R, Bellin MD, Chaloner K, Czarniecki CW, Goldstein JS, Hunsicker LG, Kaufman DB, Korsgren O, Larsen CP, Luo X, Markmann JF, Naji A, Oberholzer J, Posselt AM, Rickels MR, Ricordi C, Robien MA, Senior PA, Shapiro AMJ, Stock PG, Turgeon NA, Consortium CIT, Phase 3 Trial of transplantation of human islets in Type 1 Diabetes complicated by severe hypoglycemia, Diabetes Care 39, 1230–1240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagliuca FW, Millman JR, Gürtler M, Segel M, Van A Dervort, J. H. Ryu, Q. P. Peterson, D. Greiner, D. A. Melton, Generation of functional human pancreatic β cells in vitro, Cell 159, 428–439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YHC, Johnson JD, Kieffer TJ, Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells, Nat Biotechnol 32, 1121 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Millman JR, Xie C, Dervort AV, Gürtler M, Pagliuca FW, Melton DA, Generation of stem cell-derived β-cells from patients with type 1 diabetes, Nat Commun 7, 11463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, Millman JR, Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells, Stem Cell Rep 12, 012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Li M, Nguyen VQ, Giacometti S, Puri S, Xing Y, Wang Y, Szot GL, Oberholzer J, Bhushan A, Hebrok M, Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells, Nat Cell Biol 21, 263–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, Morikawa S, Urano F, Millman JR, Gene-edited human stem cell–derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice, Sci Transl Med 12, eaax9106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weir GC, Susan B-W, Scientific and Political Impediments to Successful Islet Transplantation, Diabetes 46, 1247–1256 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Zahr E, Molano RD, Pileggi A, Ichii H, Jose SS, Bocca N, An W, Gonzalez-Quintana J, Fraker C, Ricordi C, Inverardi L, Rapamycin impairs in vivo proliferation of islet beta-cells, Transplantation 84, 1576–1583 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR, Teratoma formation by human embryonic stem cells: Evaluation of essential parameters for future safety studies, Stem Cell Res 2, 198–210 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacík I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG, Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates, Nat Mater 14, 643–651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Lacík I, Brissová M, Anilkumar AV, Prokop A, Hunkeler D, Green R, Shahrokhi K, Powers AC, An encapsulation system for the immunoisolation of pancreatic islets, Nat Biotechnol 15, 358 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Graham AC, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R, Anderson DG, Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates, Nat Biotechnol 34, 345–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bray N, Biomaterials: Modified alginates provide a long-term disguise against the foreign body response, Nat Rev Drug Discov 15, nrd.2016.41 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, Chiu A, Wang L-H, An D, Zhong M, Smink AM, de Haan BJ, de Vos P, Keane K, Vegge A, Chen EY, Song W, Liu WF, Flanders J, Rescan C, Grunnet LG, Wang X, Ma M, Zwitterionically modified alginates mitigate cellular overgrowth for cell encapsulation., Nat Commun 10, 5262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vegas AJ, Veiseh O, Gürtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, Tam HH, Jhunjhunwala S, Langan E, Aresta-Dasilva S, Gandham S, McGarrigle JJ, Bochenek MA, Hollister-Lock J, Oberholzer J, Greiner DL, Weir GC, Melton DA, Langer R, Anderson DG, Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice, Nat Med 22, 306–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alagpulinsa DA, Cao JJL, Driscoll RK, Sîrbulescu RF, Penson MFE, Sremac M, Engquist EN, Brauns TA, Markmann JF, Melton DA, Poznansky MC, Alginate-microencapsulation of human stem cell-derived β cells with CXCL12 prolongs their survival and function in immunocompetent mice without systemic immunosuppression., Am J Transplant Official J Am Soc Transplant Am Soc Transpl Surg 19, 1930–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 22.An D, Chiu A, Flanders JA, Song W, Shou D, Lu Y-C, Grunnet LG, Winkel L, Ingvorsen C, Christophersen NS, Fels JJ, Sand FW, Ji Y, Qi L, Pardo Y, Luo D, Silberstein M, Fan J, Ma M, Designing a retrievable and scalable cell encapsulation device for potential treatment of type 1 diabetes, Proc National Acad Sci 115, E263–E272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haller C, Piccand J, Franceschi FD, Ohi Y, Bhoumik A, Boss C, Marchi UD, Jacot G, Metairon S, Descombes P, Wiederkehr A, Palini A, Bouche N, Steiner P, Kelly OG, Kraus MR-C, Macroencapsulated human iPSC-derived pancreatic progenitors protect against STZ-induced hyperglycemia in mice, Stem Cell Rep 12, 787–800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumagai-Braesch M, Jacobson S, Mori H, Jia X, Takahashi T, Wernerson A, Flodström-Tullberg Malin A Tibell, The TheraCyte™ device protects against islet allograft rejection in immunized hosts, Cell Transplant 22, 1137–1146(10) (2013). [DOI] [PubMed] [Google Scholar]
- 25.Bruin JE, Rezania A, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ, Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice, Diabetologia 56, 1987–1998 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Robert T, Mesmaeker ID, Stangé GM, Suenens KG, Ling Z, Kroon EJ, Pipeleers DG, Functional Beta Cell Mass from Device-encapsulated hESC-derived pancreatic endoderm achieving metabolic control, Stem Cell Rep 10, 739–750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezania A, Bruin JE, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ, Enrichment of human embryonic stem cell‐derived NKX6.1‐expressing pancreatic progenitor cells accelerates the maturation of insulin‐secreting cells in vivo, Stem Cells 31, 2432–2442 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Faleo G, Lee K, Nguyen V, Tang Q, Assessment of immune isolation of allogeneic mouse pancreatic progenitor cells by a macroencapsulation device, Transplantation 100, 1211–1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang R, Faleo G, Russ HA, Parent AV, Elledge SK, Bernards DA, Allen JL, Villanueva K, Hebrok M, Tang Q, Desai TA, Nanoporous immunoprotective device for stem cell derived β cell replacement therapy, Acs Nano 11, 7747–7757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nyitray CE, Chang R, Faleo G, Lance KD, Bernards DA, Tang Q, Desai TA, Polycaprolactone thin-film micro- and nanoporous cell-encapsulation devices, Acs Nano 9, 5675–5682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pepper AR, Gala-Lopez B, Pawlick R, Merani S, Kin T, Shapiro AMJ, A prevascularized subcutaneous device-less site for islet and cellular transplantation, Nat Biotechnol 33, 518–523 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Mooney JE, Rolfe BE, Osborne GW, Sester DP, van Rooijen N, Campbell GR, Hume DA, Campbell JH, Cellular plasticity of inflammatory myeloid cells in the peritoneal foreign body response, Am J Pathology 176, 369–380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell JH, Efendy JL, Han C-L, Girjes AA, Campbell GR, Haemopoietic origin of myofibroblasts formed in the peritoneal cavity in response to a foreign body, J Vasc Res 37, 364–371 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Carlsson P-O, Palm F, Andersson A, Liss P, Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site, Diabetes 50, 489–495 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Komatsu H, Kandeel F, Mullen Y, Impact of oxygen on pancreatic islet survival, Pancreas 47, 533–543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, Leibiger IB, Kenyon NS, Ricordi C, Pileggi A, Caicedo A, Berggren P-O, Paracrine interactions within the pancreatic islet determine the glycemic set point, Cell Metab 27, 549–558.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, Holst JJ, Insulin secretion depends on intra-islet glucagon signaling, Cell Reports 25, 1127–1134.e2 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Papas KK, Leon HD, Suszynski TM, Johnson RC, Oxygenation strategies for encapsulated islet and beta cell transplants., Adv Drug Deliver Rev 139, 139–156 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Omer A, Duvivier-Kali VF, Trivedi N, Wilmot K, Bonner-Weir S, Weir GC, Survival and maturation of microencapsulated porcine neonatal pancreatic cell clusters transplanted into immunocompetent diabetic mice, Diabetes 52, 69–75 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Hoffman AS, Hydrogels for biomedical applications, Adv Drug Deliver Rev 64, 18–23 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Juárez GAP, Spasojevic M, Faas MM, de Vos P, Immunological and technical considerations in application of alginate-based microencapsulation systems, Frontiers Bioeng Biotechnology 2, 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Wang K, Zhang W, Qiang M, Luo Y, A bilaminated decellularized scaffold for islet transplantation: Structure, properties and functions in diabetic mice, Biomaterials 138, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Merani S, Toso C, Emamaullee J, Shapiro AMJ, Optimal implantation site for pancreatic islet transplantation, Brit J Surg 95, 1449–1461 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Pepper AR, Gala-Lopez B, Pawlick R, Merani S, Kin T, Shapiro AMJ, A prevascularized subcutaneous device-less site for islet and cellular transplantation, Nat Biotechnol 33, 518–523 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Pepper AR, Pawlick R, Bruni A, Wink J, Rafiei Y, O’Gorman D, Yan-Do R, Gala-Lopez B, Kin T, MacDonald PE, Shapiro AMJ, Transplantation of human pancreatic endoderm cells reverses diabetes post transplantation in a prevascularized subcutaneous site, Stem Cell Rep 8, 1689–1700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song W, Chiu A, Wang L-H, Schwartz RE, Li B, Bouklas N, Bowers DT, An D, Cheong SH, Flanders JA, Pardo Y, Liu Q, Wang X, Lee VK, Dai G, Ma M, Engineering transferrable microvascular meshes for subcutaneous islet transplantation, Nat Commun 10, 4602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowers DT, Song W, Wang L-H, Ma M, Engineering the vasculature for islet transplantation, Acta Biomater 95, 131–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vlahos AE, Cober N, Sefton MV, Modular tissue engineering for the vascularization of subcutaneously transplanted pancreatic islets., P Natl Acad Sci Usa 114, 9337–9342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weaver JD, Headen DM, Aquart J, Johnson CT, Shea LD, Shirwan H, García AJ, Vasculogenic hydrogel enhances islet survival, engraftment, and function in leading extrahepatic sites, Sci Adv 3, e1700184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garg K, Pullen NA, Oskeritzian CA, Ryan JJ, Bowlin GL, Macrophage functional polarization (M1/M2) in response to varying fiber and pore dimensions of electrospun scaffolds, Biomaterials 34, 4439–4451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang K, Hou W-D, Wang X, Han C, Vuletic I, Su N, Zhang W-X, Ren Q-S, Chen L, Luo Y, Overcoming foreign-body reaction through nanotopography: Biocompatibility and immunoisolation properties of a nanofibrous membrane, Biomaterials 102, 249–258 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Chen S, Wang H, McCarthy A, Yan Z, Kim HJ, Carlson MA, Xia Y, Xie J, Three-Dimensional Objects Consisting of Hierarchically assembled nanofibers with controlled alignments for regenerative medicine, Nano Lett 19, 2059–2065 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Robertson RP, Lanz KJ, Sutherland DER, Kendall DM, Prevention of diabetes for up to 13 years by autoislet transplantation after pancreatectomy for chronic pancreatitis, Diabetes 50, 47–50 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Millman JR, Pagliuca FW, Autologous pluripotent stem cell–derived β-like cells for diabetes cellular therapy, Diabetes 66, 1111–1120 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Omer A, Duvivier-Kali VF, Trivedi N, Wilmot K, Bonner-Weir S, Weir GC, Survival and maturation of microencapsulated porcine neonatal pancreatic cell clusters transplanted into immunocompetent diabetic mice, Diabetes 52, 69–75 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Krishnan R, Arora RP, Alexander M, White SM, Lamb MW, Foster CE, Choi B, Lakey JRT, Noninvasive evaluation of the vascular response to transplantation of alginate encapsulated islets using the dorsal skin-fold model, Biomaterials 35, 891–898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mesmaeker ID, Robert T, Suenens KG, Stangé GM, Hulle FV, Ling Z, Tomme P, Jacobs-Tulleneers-Thevissen D, Keymeulen B, Pipeleers DG, Increase Functional Beta Cell Mass in Subcutaneous Alginate Capsules with Porcine Prenatal Islet Cells but Loss with Human Adult Islet Cells, Diabetes 67, db180709 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Niu D, Wei H-J, Lin L, George H, Wang T, Lee I-H, Zhao H-Y, Wang Y, Kan Y, Shrock E, Lesha E, Wang G, Luo Y, Qing Y, Jiao D, Zhao H, Zhou X, Wang S, Wei H, Güell M, Church GM, Yang L, Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9, Science 357, 1303–1307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harper SJF, Ali JM, Wlodek E, Negus MC, Harper IG, Chhabra M, Qureshi MS, Mallik M, Bolton E, Bradley JA, Pettigrew GJ, CD8 T-cell recognition of acquired alloantigen promotes acute allograft rejection, Proc National Acad Sci 112, 12788–12793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Åkerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, Laybutt DR, Cytokine-induced β-Cell death is independent of endoplasmic reticulum stress signaling, Diabetes 57, 3034–3044 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soldevila G, Buscema M, Doshi M, James RFL, Bottazzo GF, Pujol-Borrell R, Cytotoxic effect of IFN-γ plus TNF-α on human islet cells, J Autoimmun 4, 291–306 (1991). [DOI] [PubMed] [Google Scholar]
- 62.Wen L, Ley RE, Volchkov P. Yu., Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV, Innate immunity and intestinal microbiota in the development of Type 1 diabetes, Nature 455, 1109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ernst AU, Wang L-H, Ma M, Islet encapsulation, J Mater Chem B 6, 6705–6722 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Desai T, Shea LD, Advances in islet encapsulation technologies, Nat Rev Drug Discov 16, nrd.2016.232 (2016). [Google Scholar]
- 65.Dolgin E, Diabetes: Encapsulating the problem, Nature 540, S60 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Dolgin E, Encapsulate this, Nat Med 20, 9 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Orive G, Emerich D, Vos PD, Reply to Encapsulate this: the do’s and don’ts, Nat Med 20, nm.3491 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Fuchs S, Ernst AU, Wang L-H, Shariati K, Wang X, Liu Q, Ma M, Hydrogels in emerging technologies for Type 1 Diabetes, Chem Rev (2020). [DOI] [PubMed] [Google Scholar]
- 69.Ernst AU, Bowers DT, Wang L-H, Shariati K, Plesser MD, Brown NK, Mehrabyan T, Ma M, Nanotechnology in cell replacement therapies for type 1 diabetes, Adv Drug Deliver Rev 139, 116–138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Desai TA, Tang Q, Islet encapsulation therapy — racing towards the finish line, Nat Rev Endocrinol 14, 630–632 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Wang K, Wang X, Han C, Chen L, Luo Y, Scaffold-supported transplantation of islets in the epididymal fat pad of diabetic mice, J Vis Exp 125, 54995 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ricordi C, Goldstein JS, Balamurugan AN, Szot GL, Kin T, Liu C, Czarniecki CW, Barbaro B, Bridges ND, Cano J, Clarke WR, Eggerman TL, Hunsicker LG, Kaufman DB, Khan A, Lafontant D-E, Linetsky E, Luo X, Markmann JF, Naji A, Korsgren O, Oberholzer J, Turgeon NA, Brandhorst D, Chen X, Friberg AS, Lei J, Wang L, Wilhelm JJ, Willits J, Zhang X, Hering BJ, Posselt AM, Stock PG, Shapiro AMJ, National Institutes of Health–Sponsored Clinical Islet Transplantation Consortium Phase 3 Trial: Manufacture of a complex cellular Product at eight processing facilities, Diabetes 65, 3418–3428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusion in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.