

Abstract
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm caused by a reciprocal translocation [t(9;22)(q34;q11.2)] that leads to the fusion of ABL1 gene sequences (9q34) downstream of BCR gene sequences (22q11) and is cytogenetically visible as Philadelphia chromosome (Ph). The resulting BCR/ABL1 chimeric protein is a constitutively active tyrosine kinase that activates multiple signaling pathways, which collectively lead to malignant transformation. During the early (chronic) phase of CML (CP-CML), the myeloid cell compartment is expanded, but differentiation is maintained. Without effective therapy, CP-CML invariably progresses to blast phase (BP-CML), an acute leukemia of myeloid or lymphoid phenotype. The development of BCR-AB1 tyrosine kinase inhibitors (TKIs) revolutionized the treatment of CML and ignited the start of a new era in oncology. With three generations of BCR/ABL1 TKIs approved today, the majority of CML patients enjoy long term remissions and near normal life expectancy. However, only a minority of patients maintain remission after TKI discontinuation, a status termed treatment free remission (TFR). Unfortunately, 5–10% of patients fail TKIs due to resistance and are at risk of progression to BP-CML, which is curable only with hematopoietic stem cell transplantation. Overcoming TKI resistance, improving the prognosis of BP-CML and improving the rates of TFR are areas of active research in CML.
Keywords: Chronic myeloid leukemia, Philadelphia chromosome, BCR/ABL1, Tyrosine kinase inhibitor, Treatment free remission
1. Introduction
Chronic Myeloid Leukemia (CML) is a rare hematologic malignancy with an annual incidence of 1.0–1.5/105, without significant racial or geographic differences. In the last two decades, CML was the subject of transformational therapeutic advances that demonstrated the impact of clinical-translational biomedical research. CML patients treated with modern therapies have a life expectancy close to that of aged-matched controls(1). As a result, CML prevalence in the U.S. is predicted to rise from 70,000 in 2010 to 180,000 in 2050, and CML will become the most prevalent myeloid neoplasm(2). The only well-documented risk factor for development of CML is ionizing radiation(3–5).
The hallmark of CML is an acquired reciprocal translocation between the long arms of chromosomes 9 and 22 [t(9;22)(q34;q11.2)], cytogenetically visible as the Philadelphia chromosome (Ph). The t(9;22) translocation leads to the fusion of sequences from the ABL1 gene on 9q34 downstream of the BCR gene on 22q11 and formation of a BCR/ABL1 fusion gene. BCR is a ubiquitously expressed cytoplasmic protein with multiple functionalities. BCR−/− mice are viable and a defect in neutrophilic oxidative burst in phagocytic cells is the only known defect (6–9). ABL1 is also ubiquitously expressed and has several functions, including inhibition of cell cycle progression and proliferation, integrin signaling, and DNA repair (10–17). While ABL1 kinase activity is tightly controlled in physiological conditions, the chimeric BCR/ABL1 protein is constitutively active and re-localized to the cytoplasm. BCR/ABL1 activates numerous downstream pathways leading to increased proliferation, reduced apoptosis, abnormal adhesion and migration, and genetic instability. These signaling pathways were studied extensively, providing insights into the molecular machinery driving CML. Key downstream substrates and pathways include the phosphatidylinositol-3 kinase (PI3K) pathway, RAS/mitogen-activated protein kinase(MAPK) pathway, and Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathways, among others.
BCR/ABL1 is both necessary and sufficient for the development of CML. The introduction of BCR/ABL1 tyrosine kinase inhibitors (TKIs) revolutionized the treatment of CML and started the era of targeted therapies in oncology. With three successive generations of BCR/ABL1 TKIs approved today, the majority of CML patients enjoy long term remissions and near normal life expectancy(1). Moreover, ~50% of patients with deep molecular responses (>4-log reduction of BCR/ABL1 transcripts) maintain remission after discontinuation of TKIs, a state termed treatment free remission (TFR). Unfortunately, a subset of patients with CML develops resistance to TKIs leading to treatment failures and progression to blast phase CML (BP-CML). Despite the progress of BCR/ABL1 targeted therapies, stem cell transplant remains the only curative treatment of BP-CML. Current research frontiers in CML include overcoming TKI resistance, improving the prognosis of BP-CML and improving the rates of TFR which are all areas of active research in the field. Additionally, the success of TKIs has raised questions of a different nature, such as the exorbitant price of TKIs, late adverse effects of long-term TKI therapy, and the question of how we can make the success of TKIs available to less privileged parts of the world.
2. Current Management of CML
2.1. Diagnosis and Initial Work up
Patients with CML may present with constitutional symptoms including fatigue, weight loss and night sweats. Splenomegaly is fairly common and can result in abdominal pain and fullness. Hepatomegaly may be present and like splenomegaly reflects extramedullary hematopoiesis. In developed counties, more and more patients are diagnosed incidentally, when an abnormal complete count blood (CBC) obtained for an unrelated reason leads to a diagnostic workup. The CBC typically shows leukocytosis, and frequently thrombocytosis and mild anemia. Basophilia is a typical finding that should raise suspicion for CML. Mildly elevated eosinophils may be present, but significant eosinophilia raises the question of an alternative diagnosis(18). The peripheral blood smear usually shows left-shifted granulopoiesis encompassing the full spectrum of granulocytic precursors including promyelocytes and blasts. Cellular function is mostly normal and CP-CML patients are not at increased risk of infection or bleeding. As all risk stratification scores are based on pre-therapeutic values, it is important to document blood counts and spleen size (in cm below costal margin) before any therapy is initiated. Flow cytometry is indicated if blast phase or accelerated phase is suspected. In the case of discrepancies between morphological and immunophenotypic blast counts based on CD34 expression, morphological blast counts should take precedence over flow cytometry.
Morphologic examination of the bone marrow usually shows a hypercellular marrow with myeloid predominance, left shift and frequently basophilia. Abnormally small, nonlobated megakaryocytes (micromegakaryocytes) are typical for CML and there may be pseudo-Gaucher cells (large macrophages with foamy cytoplasm and a lobated eccentric nucleus) and harlequin cells (dysplastic eosinophils with metachromatic granules). Significant dysplasia raises the question of alternative diagnoses, such as atypical CML(19). Grade 1–2 reticulin fibrosis may be present, but severe fibrosis is uncommon(20). We routinely perform a bone marrow biopsy at diagnosis to identify the occasional patient with nests of blasts undetected by cytology, especially if the aspirate is suboptimal or the presentation is aggressive.
Metaphase karyotyping of bone marrow cells is indicated to confirm the diagnosis of CML. A minimum of 20 metaphase spreads should be analyzed. More than 95% of CML patients are Ph+. In ~3–5% of patients the Ph chromosome is part of complex translocations involving additional chromosomes. This must not be confused with clonal cytogenetic abnormalities in Ph+ cells (CCA/Ph+, also referred to as clonal cytogenetic evolution, discussed in depth below)(21). While patients with CCA/Ph+ have inferior progression-free and overall survival on TKI therapy, the complex translocations at diagnosis do not impact prognosis on TKIs (21, 22). Additionally, clonal cytogenetic abnormalities are detected in Ph-negative cells (CCA/Ph-) in 5–10% of CML patients with a cytogenetic response to TKI therapy and do not seem to alter prognosis (23). Rare (<5%) of patients have a silent Ph that cannot be identified by karyotyping, requiring molecular testing to establish the diagnosis of CML.
Fluorescence in situ hybridization (FISH) is usually performed on interphase nuclei, which increases sensitivity as 200–500 nuclei can be analyzed routinely. FISH is an excellent test to establish the diagnosis of CML in cases of silent Ph, but it should not routinely replace karyotyping, as it misses CCA/Ph+. Modern FISH probes also identify deletions in ABL1 and BCR adjacent to the translocation breakpoint. Deletions in the derivative chromosome 9 adjacent to the breakpoint or spanning the breakpoint confer an adverse prognosis to patients treated with interferon-α (IFN-α) and second-line imatinib, but not to patients on frontline TKI therapy(24–27). FISH is useful for monitoring patients with atypical BCR/ABL1 transcripts that are not detected by standard quantitative PCR assays (28). While other myeloproliferative diseases such as chronic myelomonocytic leukemia (CMML) and chronic neutrophilic leukemia can mimic CML clinically and morphologically, demonstration of Ph or BCR/ABL1 establishes a CML diagnosis in a patient with a myeloproliferative neoplasm, irrespective of other features.
Qualitative reverse transcription polymerase chain reaction (RT-PCR) is an alternative to FISH to identify a BCR/ABL1 fusion mRNA in patients with suspected CML who are Ph- by metaphase karyotyping. The breaks in chromosome 22 localize to one of three breakpoint cluster regions (BCRs) and determine how much of BCR is retained in the BCR/ABL1 fusion mRNA and protein. In the vast majority of CML patients, the breakpoint in BCR generates e13a2 or e14a2 fusion mRNAs (formerly referred to as b2a2 and b3a2) and a p210BCR/ABL1 fusion protein. Breakpoints in the minor BCR (m-BCR) generate an e1a2 fusion mRNA and p190BCR/ABL1, which is found in two-thirds of Ph+ acute lymphoblastic leukemia (ALL) cases, but very rarely in CML (29, 30). There is a number of rare BCR/ABL1 variants that cannot be detected with standard RT-PCR assays. Therefore, ruling out CML by RT-PCR requires multiplex assays that cover all possible BCR/ABL1 variants. (31–33). Importantly, in the case of rare variants, it is not possible to follow disease status using standard quantitative PCR (qPCR) assays(33–35). Finally, about two-thirds of CML cases also express the reciprocal ABL1-BCR mRNA (36, 37). Absence of ABL1-BCR mRNA indicates the presence of deletions flanking the breakpoints in BCR, ABL1, or both.
Next generation sequencing (NGS) to screen for mutations in genes involved in myeloid malignancies is increasingly used in CML but does not yet provide actionable information. The prognostic impact of such mutations is an area of active research(38, 39). Branford et al. recently presented data demonstrating that presence of mutations associated with myeloid malignancies predicted for inferior progression free survival and molecular response rates(40). Prospective data is needed to confirm the prognostic importance of mutations, before incorporating NGS into the routine diagnostic work-up and clinical decision-making. In our practice we consider myeloid NGS in patients with high-risk CML.
Imaging studies are not required for diagnosis and are mandatory only if palpation of spleen and liver is not possible or if extramedullary involvement in sites other than spleen and liver is suspected. Notably, there is no established algorithm to convert spleen size on imaging to the sizing measurements obtained from physical exam and used in the Sokal and other risk scores.
2.2. Risk Stratification and Disease Phases
The Sokal score, the first prognostication system in CML, was developed in the era of busulfan-based chemotherapy but was later applied successfully to risk-stratify patients treated with IFN-α or TKIs(41, 42). The Sokal score formula is based on age, blast count in the blood, platelet count and spleen size. Scores of <0.8, 0.8–1.2 and >1.2 define low, intermediate and high risk categories respectively(43). When IFN-α was the standard drug therapy for CML, Hasford developed a score optimized for patients treated with IFN-α(18). More recently, the European Treatment and Outcome Study for CML long-term survival score (EUTOS-LTSS, ELTS) was developed. The ELTS takes into account the fact that most CML patients with access to TKIs die from non-CML causes(44). ELTS was superior to all other scores in predicting CML-related death in CP-CML patients treated with imatinib, and its use is recommended in patients treated with TKIs.
CML progresses in stages termed chronic phase (CP-CML), accelerated phase (AP-CML) and blast phase (BP-CML). While ≥20% blasts are used by the WHO classification of hematologic neoplasms to establish a diagnosis of blast phase, National Comprehensive Cancer Network (NCCN), and Center for International Blood and Marrow Transplant Research (CIBMTR) use a cutoff of ≥30%, which is also the definition employed by major prospective TKI trials, which form the basis for current treatment algorithms(45, 46). BP-CML can exhibit a pre B-lymphoid (25%), myeloid (70%) or indeterminate (5%) phenotype(47). BP-CM may develop suddenly or over a period of time through the intermediary stage of AP-CML. In addition to a blast count of ≥30% in the blood or bone marrow, extramedullary myeloblasts (chloroma) in tissues other than liver or spleen define BP-CML, and any proportion of pre-B lymphoblasts strongly suggests lymphoid blast phase. The acquisition of CCA/Ph+ in a patient on TKI therapy is diagnostic for AP-CML irrespective of other disease features(21, 48, 49). CML presenting in lymphoid blast crisis may be difficult to distinguish from de novo Ph+ ALL. The therapeutic approaches are almost identical though and the distinction is mainly of academic interest.
2.3. Parameters of Response
Response to therapy is assessed by clinical, hematological, cytogenetic and molecular parameters that reflect the leukemic burden in patients (Figure 1). Response to TKI therapy is highly predictive of long term outcomes, and milestones of response have been identified by the National Comprehensive Cancer Network (NCCN) and European Leukemia Net (ELN) consensus panels(45, 48). Complete Hematologic response (CHR)is defined as the resolution of all CML-related signs and symptoms (including palpable splenomegaly) and normalization of white blood cell count, white blood cell differential and platelet count. Normalization of hemoglobin is not part of the CHR definition. Complete Cytogenetic Response (CCyR) denotes the absence of Ph+ metaphases in at least 20 bone marrow cells analyzed by G- or R-banding. CCyR correlates strongly with overall survival (50).Partial cytogenetic responsedenotes 1–35% Ph+ metaphases, andmajor cytogenetic response (MCyR)combines CCyR and PCyR. Molecular Response refers to response assessed by PCR. Quantitative RT-PCR for BCR/ABL1 has emerged as the assay of choice for monitoring patients with CML on therapy. The introduction of the international scale (IS) as a universal metric to express BCR/ABL1 transcript levels helped establish the standard for reporting the results of molecular testing(51). Compared to cytogenetics, qPCR has a much greater dynamic range, which can span up to 5 orders of magnitude. However, this sensitivity is achieved only if the sample is of optimal quality. Thus, laboratories must report the sensitivity reached for a given sample to allow for correct interpretation of results. A housekeeping gene is used as a control for normalization of RNA input. Acceptable choices include ABL1, BCR, andβ-glucuronidase (GUS). It is often necessary to decide whether a rise of BCR/ABL1 mRNA is ‘real’ and should trigger a more extensive workup, or is only a variation of the assay. The most important factor is the quality of the test, which determines the extent of inter-test variation(52). Current recommendation is that generally a 5–10 - fold rise in transcript levels be regarded as significant (51). It is important to know the performance of the laboratory and to adjust standard operating procedures accordingly. It is also good practice to repeat the test in case of rising BCR/ABL1 mRNA levels before rushing to far-reaching conclusions, particularly if the changes occur at a low level, where the coefficient of variation is largest. It is also important to understand the meaning of the IS scale. A value of 100% on the IS is equivalent to the average BCR/ABL1 mRNA expression of 30 patients treated on the International Randomized Study of Interferon and STI571 (IRIS) study (53). Therefore, the fold reductions of BCR/ABL1 expressed on IS refer to a standardized baseline rather than the patient’s baseline, representing an absolute measurement of leukemic mRNA. A value of 0.1% corresponds to a 3-log reduction compared to this baseline and is referred to as major molecular response (MMR). Achievement of MMR on TKI therapy is an important milestone that portends a good prognosis. Further reductions of BCR/ABL1 mRNA by 4 or 4.5 logs are referred to as MR4 or MR4.5, respectively. MR4.0 or better is frequently referred to as deep molecular response (DMR) and is the minimum depth of response required to consider a trial of TFR. The first defined prognostic milestone for CP-CML is at 3 months after initiating TKI therapy (Table 1). An IS level of less than 10% at 3 months is referred to as an early molecular response, and highly correlated with progression free and overall survival(54). Subsequent studies showed that 6 months for achieving less than 10% BCR/ABL1 IS are also acceptable(55).
Figure 1. Response to treatment and residual leukemia in chronic myeloid leukemia.

CHR –complete hematologic response; CCyR –complete cytogenetic response; MMR –major molecular response.
Table 1.
Treatment response milestones as defined by the NCCN guidelines. If resistance is suspected (possible resistance), the physician has the option of increasing imatinib to 800 mg, switching to a different TKI or continuing the same 2G TKI. Switching to an alternate TKI is recommended for TKI resistant disease.
| BCR-ABL1 (IS) | 3 months | 6 months | 12 months |
|---|---|---|---|
| >10% | Possible Resistance | Resistance | |
| >1–10% | TKI-sensitive | Possible Resistance | |
| >0.1–1% | TKI-sensitive | TKI-sensitive* | |
| <0.1% | TKI-sensitive | ||
Shared decision making: consider switching to an alternate TKI to increase the chances for TFR.
2.4. Current Therapies in the Treatment of CML
2.4.1. Tyrosine kinase inhibitors.
TKIs are the standard of care for newly diagnosed CML patients in all disease phases. Six TKIs are currently approved –imatinib, dasatinib, nilotinib, bosutinib, ponatinib and radotinib (only in South Korea). Imatinib is referred to as a first-generation (1G) TKI, dasatinib, nilotinib, bosutinib and radotinib are second-generation (2G) TKIs, and ponatinib is a third generation TKI. The TKIs differ in their potency, activity against BCR/ABL1 mutants, activity against kinases other than BCR/ABL1, pharmacokinetics and adverse effect profiles.
2.4.1.1. Imatinib.
The introduction of Imatinib initially as salvage therapy in 2001 and subsequently as front line therapy revolutionized CML therapy, transforming this disease from a fatal condition into a manageable chronic ailment (56–59). The International Randomized Study of Interferon and STI571 (IRIS) study compared imatinib 400mg daily versus a combination of IFN-α and cytarabine and found imatinib to be vastly superior in all major endpoints. Crossover was permitted and the fact that many patients who failed IFN-α were effectively salvaged by imatinib may explain why no difference in overall survival between the two arms was observed (60). Subsequent phase 3 studies of imatinib mainly addressed dose optimization. Two prospective randomized studies compared 400mg vs. 800mg imatinib in patients with newly diagnosed CP-CML. Both studies failed in their primary endpoints, MMR or CCyR at 12 months, respectively(61, 62). Later the German CML IV study reported superior rates of MMR at 12 months for 800mg compared to 400 mg imatinib daily, without a difference in progression-free survival(63, 64). In this study the median dose of imatinib was 627mg daily due to intolerances and dose reductions. Similarly, the French SPIRIT study reported higher rates of MMR for patients treated with 600 vs. 400mg imatinib daily (65). Hence, the optimal dose of imatinib may be approximately 600mg daily, which is also the recommended dose for AP/BP (66).
2.4.1.2. Dasatinib.
Dasatinib was initially studied in the second line after imatinib failure, including several phase 2 studies that tested different doses and dosing schedules (67–72). Dasatinib at a dose of 100mg daily for CML-CP, and 140mg daily for AP/BP-CML emerged as the recommended doses. Collaborative group (Southwestern Working Group SWOG S0325) and industry-sponsored (Dasatinib versus Imatinib Study In treatment-Naive CML patients, DASISION) phase 3 studies compared dasatinib 100mg daily with imatinib 400mg daily in newly diagnosed CML-CP patients. Dasatinib proved superior in the primary endpoints, MMR and CCyR at 12 months, respectively (71, 73). No differences in overall survival were observed. Based on the results of DASISION, dasatinib was approved for use in newly diagnosed CML patients.
2.4.1.3. Nilotinib.
Nilotinib first showed impressive results in patients with CP-CML and AP-CML who had failed imatinib(74). It was later approved for second line therapy based on the confirmatory results in phase 2 studies at a dose of 400mg twice daily(75, 76). Although the phase 2 results for BP-CML were comparable to those of dasatinib, nilotinib is not currently approved for this indication (77). Based on the The Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed (ENESTnd) results, nilotinib (300mg twice daily) received approval for frontline therapy of CML-CP. At the doses of 300 or 400 mg twice daily, both experimental arms were superior to the standard arm in the major endpoint, MMR at 12 months (78). Unlike any other 2G TKI, nilotinib significantly improved progression-free survival when compared to imatinib in the first line setting, but overall survival was improved only for the nilotinib 400mg twice daily arm, which is not on the label as frontline therapy (79). Unfortunately, nilotinib was found to cause hyperglycemia and hyperlipidemia along with an increase in cardiovascular adverse events compared to imatinib. These cardiovascular events included peripheral arterial occlusive disease, cerebral ischemic events and ischemic heart disease (80). The cardiovascular toxicity of TKIs will be discussed in depth in a later section.
2.4.1.4. Bosutinib.
Bosutinib was initially approved at a dose of 500 mg daily for third-line therapy of CML in all phases (81, 82). In the frontline setting, bosutinib failed to improve CCyR at 12 months when compared to imatinib, the primary endpoint of the study, and consequently, was denied regulatory approval despite superior rates of MMR(83, 84). A high rate of discontinuation in the experimental arm prior to response assessment due to inadequate management of adverse events may have led to an underestimation of the true difference. Subsequently, the Bosutinib Trial in First Line Chronic Myelogenous Leukemia Treatment (BFORE) trial used a reduced dose of bosutinib (400mg daily) and demonstrated superiority for MMR at 12 months, but no difference in overall and progression free survival(85). Based on this data, bosutinib became the 4th TKI approved for the first line therapy of CP-CML.
2.4.1.5. Ponatinib.
First and second generation TKIs do not cover the BCR/ABL1T315I mutant (86). Ponatinib was designed to address this specific problem. Unlike other TKIs, ponatinib skirts isoleucine 315 through a carbon-carbon triple bond (87). Ponatinib is considered the most potent BCR/ABL1 TKI currently approved and also has a long half-life and long target occupancy. However, this potency comes at the cost of reduced selectivity leading to inhibition of multiple additional kinases (80). Ponatinib at an initial dose of 45mg daily was tested in relapsed/refractory (R/R) CML in all phases as well as relapsed/refractory Ph+ ALL (88, 89). Responses were durable in CP-CML, less stable in AP-CML and transient only in BP-CML and in Ph+ ALL. Cardiovascular events emerged with longer follow-up of the Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia (PACE) study and this lead to premature termination of an ongoing phase 3 frontline trial(90). The vascular toxicity was attributed to off-target inhibition of vascular system kinases such as vascular endothelial growth factor (VEGF), platelet-derived growth factor receptors (PDGFR) and tyrosine kinase with immunoglobulin like and EGF like domains 2 (TIE2). Recent animal models also revealed involvement of Van Willebrand Factor mediated platelet adhesion and a secondary microvascular angiopathy (91). There is evidence that ponatinib toxicity is dose-dependent, and dose reductions are recommended, once the desired response has been achieved. Recently presented data from the prospective randomized OPTIC trial showed that a response-adjusted dosing regimen of ponatinib (starting at 45 mg and reducing to 15 mg upon response) preserves efficacy, and reduces toxicity (92).
2.4.1.6. Choosing a TKI in the front-line setting.
Four different TKIs are currently approved for the frontline therapy of CP-CML. All three 2G TKIs are superior to imatinib in terms of MMR and DMR and these differences tend to be largest in Sokal high risk patients (Table 2). Similarly, there are trends for lower rates of progression to AP/BP with 2G TKIs compared to imatinib, although statistical significance is typically not reached, reflecting the overall low rates of events or avoidance of unplanned post hoc analyses. As an example, progression to AP/BP in the ENESTnd study was not different for low, but favored nilotinib 300 mg twice daily for intermediate (2% vs. 9.9%) and high (9% vs. 14.1%) Sokal risk, with similar trends for improved survival(79). Reflecting the ongoing discussion, the NCCN guidelines recommend a 2G TKI for Sokal high and intermediate risk patients, noting that imatinib is also an acceptable choice(45). The ELN 2020 recommendations though do not include such a recommendation, noting that imatinib is a safe and cost-effective TKI for patients with CP-CML(48). Another important consideration when selecting TKIs is their variable side effect profile and whether a TKI is likely to interfere with a patient’s comorbid conditions. Nilotinib is associated with arterial occlusive events, impaired glucose tolerance, and dyslipidemia, therefore it is relatively contraindicated in patients with diabetes and hyperlipidemia. Cardiovascular disease is a contraindication to nilotinib use. Dasatinib is associated with a high risk for pleural effusions and occasionally with pulmonary hypertension and is contraindicated in patients with pulmonary disease. Bosutinib is associated with liver toxicity and renal impairment. In patients with reduced glomerular filtration, nilotinib is the preferred TKI with the least evidence of renal toxicity (93). Imatinib is associated with many low-grade toxicities that can significantly impair quality of life but has demonstrated remarkable safety over many years of clinical use. Additional considerations include dosing schedules (Table 3), drug interactions, accessibility to drug and out of pocket expenses. Generic imatinib is available around the world. Unfortunately, imatinib prices in the U.S. have been slow to decline despite the availability of generic drug(94)(95). In our practice, although we give strong consideration for a 2G TKI in patients with intermediate and high Sokal risk, we recognize the importance of joint decision making, taking into account other factors such as treatment goals and toxicity, including financial toxicity.
Table 2.
Summary of phase 3 trials comparing second generation tyrosine kinase inhibitors to imatinib as first line therapy for chronic phase CML.
| Trial Name Reference | Second Generation TKI/Dose | Progression Free Survival | Overall Survival | CCyR (12 months) | MMR (12 months) |
|---|---|---|---|---|---|
| ENESTnd Hochhaus A et al (79) | Nilotinib 300 mg twice daily | 5 years: 96.5% | 5 years: 93.7% | 93%* | 44%* |
| Nilotinib 400 mg twice daily | 5 years: 98.3%* | 5 years: 96.2%* | 93%* | 43%* | |
| Imatinib 400 mg once daily | 5 years: 94.7% | 5 years: 91.7% | 76% | 22% | |
| BELA Brummendorf TH et al (83) | Bosutinib 500 mg once daily | 24 months: 92% | 24 months: 97% | 70% | 41%* |
| Imatinib 400 mg once daily | 24 months: 88% | 24 months: 95% | 68% | 27% | |
| BFORE Cortes et al (85) | Bosutinib 500 mg once daily | 12 months: 96.3 | 12 months: 99.6% | 77.2%* | 47.2%* |
| Imatinib 400 mg once daily | 12 months: 93.6 | 12 months: 97.9% | 66.4% | 36.9% | |
| DASISION Cortes et al (163) | Dasatinib 100 mg once daily | 5 year: 85% | 5 year: 91% | 77%* | 46%* |
| Imatinib 400 mg once daily | 5 year: 86% | 5 year: 910% | 66% | 28% |
CCyR - complete cytogenetic response; MMR - major molecular response.
p<0.05 compared to imatinib.
Table 3.
Molecular formulas, half-lives and dosing schedules for BCR-ABL1 TKIs.
| TKI | Structural Formula | Half Life | Recommended Starting Dose |
|---|---|---|---|
| Imatinib |
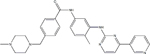
|
18 hours | 400 mg daily |
| Dasatinib |
|
3–5 hours | 100 mg once daily |
| Nilotinib |
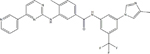
|
17 hours | 300 mg twice daily |
| Ponatinib |
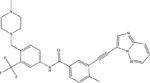
|
12–66 hours | 45 mg once daily |
| Bosutinib |
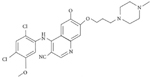
|
22 hours (oral) | 400 mg once daily |
| Asciminib |
|
8 hours | 40 mg BID (estimated) |
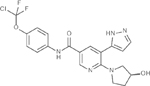
After starting a TKI in the first line setting, response must be assessed at 3 and 6 months as shown in Table 1. An early molecular response indicates TKI sensitive disease and is a favorable prognostic finding. It is important to first review the patient’s compliance before switching TKIs for lack of early molecular response. TKIs have significant adverse effects that may reduce adherence to therapy. Drug-drug interactions should also be assessed as multiple drugs and herbal preparations interfere with TKI absorption and metabolism, and a change of TKIs may not be required to correct the situation. A switch in TKIs should also be considered if the IS ration remains above 1% at 12 months post therapy(45).
2.4.2. Interferon-α (IFN-α)
IFN was that standard of care drug therapy prior to the introduction of TKIs, and continues to be an option for the rare patients who are intolerant of all TKIs. It remains the safest option to maintain responses in women who plan to become pregnant(96). There is also data showing that combining IFN-α with TKIs can improve the depth of molecular responses(65).
2.4.3. Cytotoxic agents
Cytototoxic agents are rarely used in CML nowadays. Hydrea is useful to reduce leukocytosis in patients with signs and symptoms of leukostasis such as retinal ischemia, hypoxia or priapism. To avoid TKI therapy interruptions, care must be taken to prevent the severe myelosuppression that can ensue when TKIs are started immediately after or simultaneously with high doses of hydrea.
2.4.4. Hematopoietic stem cell transplant (HSCT)
HSCT played a major rule in the treatment of CML during the pre-TKI era (97). Donor leukocyte infusions (DLIs) were first used in CML patients, proving the existence of a graft-versus-leukemia effect(98). While HSCT is no longer justified in newly diagnosed patients with CP-CML, there is consensus that HSCT should be offered to all patients with progression to AP/BP(45, 48, 99). In this setting, HSCT should be pursued as soon as the patient achieves a second chronic phase, as transplant in chronic phase yields better outcomes than transplant in AP/BP. In the CML IV study 3-year overall survival was 91% for patients transplanted in CP after failing imatinib vs. 59% for patients transplanted in AP/BP(100). Another situation in which transplant should be considered is at the emergence of primary or secondary TKI resistance. TKI resistance confers a poor prognosis and will be discussed in detail in subsequent sections. CML is exquisitely sensitive to the graft versus leukemia effect. The consensus is that peripheral blood is preferred over bone marrow as the source of donor stem cells in patients with AP/BP CML, while the opposite is the case in patients with CML-CP, where graft-versus-host disease is of greater concern than disease control(101). Treatment with imatinib before HSCT seems to reduce the risk of chronic graft-versus-host disease and possibly the risk of relapse as well(102–104). Our approach is to keep patients with high relapse risk on a TKI post-transplant for at least one year. This recommendation is based on limited data and must be balanced against the considerable myelosuppression caused by TKIs in the post-transplant setting.
2.4.5. Omacetaxine
Omacetaxine (previously homoharringtonine) is a subcutaneously administered protein synthesis inhibitor approved only in the United States for the treatment of CP-CML and AP-CML with resistance to at least two TKIs(105). Omacetaxine is used infrequently due to inconvenient route of administration and significant myelosuppression but it remains a palliative option for patients with intolerance of or resistance to all TKIs. In CP-CML, the rate of MCyR was 18.4%, with a median duration of 12.5 months(105).
2.5. Treatment free remission (TFR)
Treatment free remission is emerging as a therapeutic goal for patients with CML to avoid the long-term complications of TKIs, especially in younger patients(48). The first study exploring TFR was a small pilot study on 12 patients with undetectable residual disease on imatinib (106). Subsequently the Stop Imatinib Trial (STIM-1) clinical trial explored TFR in patients with sustained complete molecular response (CMR)onimatinib. Sustained CMR was defined as undetectable CML by quantitative RT-PCR for at least two consecutive years, irrespective of the sensitivity achieved in an individual sample(107). Patients were taken off imatinib and closely monitored with monthly RT-PCR testing(107). Molecular recurrence was defined by two consecutive positive PCR results or loss of MMR in a single test. STIM-1 results showed a projected recurrence free survival of 41% at 12 months of follow up. Fortunately, patients who experienced recurrence achieved MMR with resumption of imatinib. Longer TKI exposure and longer duration of DMR were associated with a higher probability of successful TFR, while patients with high Sokal were at higher risk of recurrence. Multiple independent studies confirmed TFR rates between 40 and 60%(108). Subsequent trials assessed TFR for patients on 2G TKIs and showed very similar results(109–114). Patients treated with a 2G TKI have higher rates of DMR in a shorter time frame which theoretically translates into higher rates of TFR. This could argue for the use of 2G generation TKIs as first line therapy as a means to increase overall TFR rates. However, 2G TKIs are not without risks and adverse effects and the risk benefit assessment should be individualized to each patient(115, 116). At our center, we consider a trial of TFR only for patients with sustained DMR for more than 2 years, total length of TKI treatment of at least 3 years, and no history of AP/BP-CML. Compliance and access to reliable qPCR monitoring are also important (Table 4). It is worth noting that the criteria for discontinuation reflect a reasonable compromise between clinical experience, eligibility criteria of clinical trials, and the dearth of prospective comparative studies. Numerous studies have identified associations between TFR and cellular immune parameters, including higher NK cells, lower T regulatory cells and lower numbers of CD86+ plasmacytoid dendritic cells compared to patients who experienced recurrence(117–119). These data suggest that TFR is based on immunological control, but thus far the mechanisms remain elusive. Of note, about one-quarter of patients who discontinue TKIs develop a TKI withdrawal syndrome. This syndrome consists of musculoskeletal pain and sometimes flushing(120). Patients with preexisting arthritis are more susceptible to developing TKI syndrome. Non-steroidal anti-inflammatories or a short course of oral steroids are typically effective, but some patients may require opiates or even resumption of TKI therapy. Cytokine release, regrowth of mast cells and reactivation of an underlying inflammatory state have been proposed as possible causes of TKI withdrawal syndrome. Interestingly, patients who experience TKI withdrawal syndrome seem to have a higher likelihood of successful TFR.
Table 4.
Recommended clinical criteria to consider a trial of treatment free remission in patients with chronic phase CML (45,48).
| Criteria for discontinuation of TKI therapy |
|---|
| Age ≥ 18 years |
| History of good compliance |
| TKI therapy for ≥ 3 years |
| Quantifiable BCR-ABL1 transcript |
| Deep molecular response for ≥ 2 years |
| Chronic phase CML with no prior accelerated or blast phase CML |
| Access to reliable quantitative PCR testing |
| Monthly testing for months 1–6, bimonthly for month 7–12, and every three months afterwards |
| Prompt resumption of TKI therapy upon loss of major molecular response |
3. Challenging Situations in the Management of CML
3.1. TKI resistance
Primary resistance to TKIs implies the failure to achieve a desired level of response while secondary or acquired resistance implies the loss of response while on therapy. On a molecular level, TKI resistance can be the result of BCR/ABL1-dependent mechanisms (reactivation of BCR/ABL1 kinase activity) or BCR/ABL1-independent mechanisms (resistance despite continued suppression of BCR/ABL1 kinase activity). TKI resistance represents a serious turn in a CML patient’s history, as it is associated with impaired survival outcomes.
3.1.1. BCR/ABL1-dependent TKI resistance
BCR/ABL1 kinase domain mutations (Figure 2).
Fig. 2.

BCR-ABL1 kinase domain mutations confer resistance to first and second generation TKIs. The crystal structure of the ABL1 kinase domain in complex with the indicated TKI (indicated in green) is shown. The P-loop is shown in yellow and the activation loop in green. Mutations at the highlighted residues confer resistance to the indicated TKI in vitro, with orange (moderate) and red (high) spheres indicating the level of resistance. Compared with imatinib, the newer inhibitors have less vulnerability to kinase domain mutations. Imatinib has a broad range of resistance mutations and a switch to a 2G TKI is indicated at in imatinib resistant disease. From O’Hare et al. Nat Rev Cancer. 2012;12(8):513–26.
The most common mechanism of BCR/ABL1 dependent resistance is mutations in the kinase domain of BCR/ABL1 that impair drug binding(9, 121, 122). Kinase domain mutations are generally more common in acquired resistance and in AP/BP-CML (123). Guidelines call for BCR/ABL1 mutation testing in case of failure to achieve therapeutic milestones, at relapse, and at progression to AP/BP-CML on therapy. NGS increases sensitivity of detection to between 1 and 5% of variant allele frequency (VAF), depending on sequencing depth. Recent data using NGS points to the presence of small BCR/ABL1 resistance mutations as early as 3 months in some patients taking imatinib. These clones were shown to expand due to their survival advantage under an ineffective TKI(124, 125). Sanger sequencing is unable to detect these small but clinically significant clones, making NGS the preferred modality for detection of TKI resistance mutations. In the future, error-corrected NGS may allow for ultra-sensitive mutation detection, but the clinical significance of very small mutant clones will need to be validated first (126). In some cases, subclones may acquire a second mutation in the same BCR/ABL1 allele, a situation termed compound mutation(116). Many compound mutations, particularly those including BCR/ABL1T315I confer high level resistance to most or all approved TKIs (127, 128). Imatinib, the TKI with the lowest potency, has the broadest spectrum of resistance mutations including hotspots at the ATP binding loop (P-loop), activation loop, and threonine 315. Threonine 315, frequently referred to as the gatekeeper residue, controls access to a hydrophobic pocket in the catalytic site that is visited by first and second generation TKIs. In aggregate 2G TKIs cover all clinical mutants except T315I (Table 5). Except for ponatinib, all first and second generation TKIs make a hydrogen bond with T315 and require access to the hydrophobic pocket. Ponatinib abuts T315I through a rigid triple carbon bond (87).
Table 5.
The most common BCR-ABL1 resistance mutations are listed here along with the 2G TKIs contraindicated to use in these scenarios. Figure 3 shows the crystal structure of ABL1 kinase domain with the annotated resistance mutations and 2G TKIs in complex. Imatinib is contraindicated for all these mutations.
| BCR-ABL1 Mutation | Contraindicated TKIs |
|---|---|
| T315I | dasatinib, nilotinib, bosutinib |
| V299L | dasatinib, bosutinib |
| G250E | bosutinib, nilotinib |
| F317L | bosutinib, dasatinib |
| Y253H | nilotinib |
| E255K/V | nilotinib bosutinib |
| F359V/C/I | nilotinib |
| L248V | nilotinib, bosutinib |
Increased BCR/ABL1 expression through gene amplification or transcriptional upregulation is implicated in TKI resistance in cell lines, but the clinical importance of these findings is less clear (129).
3.1.1.2. Drug efflux and influx.
Organic-cation transporter-1 (OCT-1) is required for imatinib uptake, and low OCT-1 activity has been implicated in BCR/ABL1 dependent imatinib resistance(130). TKIs are substrates for various efflux pumps, including ABCB1 (MDR1). Several polymorphisms of ABCB1 and ABCG2 (BCRP) have been correlated with the depth of response to imatinib (131, 132). Overexpression of MDR1 may be sufficient to protect CML stem and progenitor cells, until they acquire a BCR/ABL1 kinase domain mutation leading to overt resistance (133).
3.1.2. BCR/ABL1-independent resistance
Multiple pathways have been implicated in activation of growth and survival mechanisms independent of BCR/ABL1 kinase activity, including several downstream of BCR/ABL1, such as PI3K and MAPK(9). This mechanistic heterogeneity with its lack of universal vulnerabilities poses a challenge for the development of therapies to overcome BCR/ABL1 independent resistance. Interestingly, extensive overlap is seen in the pathways involved in transformation to AP/BP-CML and BCR/ABL1-independent resistance, including imbalanced activity of polycomb repressive complex 1 (PRC1) and PRC2 that lead to global epigenetic aberrancies(134). The fact that there is pathway conversion at the epigenetic level suggests that targeting universal epigenetic regulators such as enhancer of zeste homolog 2 (EZH2) may have broad applicability to BCR/ABL1-independent resistance. Moreover, specific additional chromosomal abnormalities (ACA) are deemed high risk and herald TKI resistance. These chromosomal abnormalities likely result in amplification or deletions of cancer associated genes which in turn lead to TKI resistance. Three high risk ACAs were associated with poor prognosis, including i(17)(q10), −7/del7q, and 3q26.2 rearrangements. Isochromosome 17 (i(17)(q10)) leads to loss of one copy of the TP53 tumor suppressor gene(135). Finally, many cases of clinical TKI resistance seem to combine BCR/ABL1-dependent and independent mechanisms. Single cell analysis will be required to achieve the resolution needed to map these complex myeloid neoplasms and devise rational treatment approaches.
3.1.3. Approach to the patient with TKI resistance
The first step in addressing this issue is investigating the cause of a patient’s loss of response or inadequate response. A careful history must be taken to exclude noncompliance and drug interactions. Once non-compliance and drug interactions are ruled out, a complete resistance workup is indicated. At the minimum this includes a physical exam, CBC, bone marrow aspirate and biopsy, bone marrow metaphase karyotyping and BCR/ABL1 mutation screen. Management of TKI resistance depends on whether other TKIs are expected to be effective. Resistance to first generation TKIs can be addressed by 2G TKIs as long as the patient remains in chronic phase and does not have the BCR/ABL1T315I mutation, with TKI selection dependent on the BCR/ABL1 genotype if there is a BCR/ABL1 mutation (Table 5). Ponatinib is the only option for patients with BCR/ABL1T315I. Patients on 2G or 3G TKIs due to resistance require close monitoring to ensure adequate responses are achieved. Although data are limited, we generally follow the consensus response milestones used for first line TKI therapy in this situation. When patients fail a 2G TKI, switching to another 2G produces CCyR rates of only ~20%. We use ponatinib in these cases, which produces an estimated 5-year overall survival of 73% in CP-CML(136, 137). Close monitoring for patients who switched to ponatinib from 2G TKIs is important. Only 7% of patients who failed to achieve a minor cytogenetic response (≤65% Ph+ metaphases) to 2G TKIs at three months were in MCyR at 12 months after starting ponatinib, suggesting that alternative approaches need to be considered early (138). Patients who fail 2G TKIs are candidates for a clinical trial, and should undergo a work-up for HSCT. We consider ponatinib as the last non-transplant option capable of inducing a durable remission and recommend transplant, unless a convincing response is achieved. Weighing the risk of HSCT vs. the risk of progression to AP/BP-CML can be extremely challenging, as outcomes are far inferior if HSCT is performed after progression. The available armamentarium to address TKI resistance may soon be expanded by the availability of asciminib (ABL001), an investigational allosteric ABL1 TKI that targets the myristate binding pocket of BCR/ABL1, stabilizing an inactive kinase conformation (139, 140). A phase 1 study of asciminib showed significant clinical activity in patients who failed multiple TKIs. Toxicity was low, mirroring high selectivity and suggesting that high doses may be tolerated, achieving deep suppression of BCR/ABL1 kinase activity (140–142). Results from a phase 3 study comparing asciminib versus bosutinib in the third line in patients without the T315I and V299L mutation (ASCEMBL) were recently reported in abstract form. The MMR rate at 24 weeks was 25.5% with asciminib and 13.2% with bosutinib, meeting the primary objective of the study (143).
3.2. Accelerated and Blast Phase CML
Transformation from CP-CML to BP-CML was common in the pre-TKI era but nowadays is rare in patients with access to appropriate management. The most prominent feature of BP-CML is the loss of terminal differentiation capacity, which leads to an acute leukemia with myeloid (M-BP) or pre-B cell phenotype (L-BP).
3.2.1.1. Cytogenetic abnormalities.
CCA/Ph+ is present in 70–80% of BP-CML cases. In fact, the acquisition CCA/Ph+ on TKI therapy is diagnostic of AP-CML, even in the absence of other features. CCA/Ph+ includes a number of nonrandom abnormalities. Classically, trisomy 8, +Ph, i(17q), and trisomy 19 were considered as major route abnormalities and are included in the ELN classification of AP-CML. Monosomy 7 and 3q26 rearrangements are minor route abnormalities but also confer a poor prognosis (20, 46). The major and minor route designation was based on the frequency of these abnormalities in advanced disease. Subsequent evidence showed that a number of minor route abnormalities signify high risk disease as mentioned in the section on TKI resistance (135). Somatic mutations in multiple genes associated with myeloid malignancies have been also been reported at progression to BP-AML(144). In contrast to AML, the reciprocal translocations involving core binding factors and retinoic acid receptor α are rare.
3.2.1.2. Molecular mechanisms of transformation.
The pathogenesis of BP-CML is complex and involves both somatic mutations and epigenetic dysregulation. As a comprehensive review of BP-CML biology is beyond the scope of this manuscript, we will focus on selected mechanisms that are well supported by data. In CP-CML the myeloid cell compartment is expanded, but differentiation is mostly maintained. In contrast, BP-CML is characterized by loss of differentiation and accumulation of immature blasts. It is thought that BCR/ABL1 first arises in a leukemia stem cell with the ability to differentiate into myeloid or lymphoid progeny. This may explain the existence of both a lymphoid and myeloid BP-CML. Increased BCR/ABL1 expression contributes to the differentiation arrest in M-BP. In some patients, rising BCR/ABL1 levels are associated with duplication of Ph, but in the majority this seems to result from transcriptional upregulation due to as yet unknown mechanisms(145). High levels of BCR/ABL1 were shown to impair translation of CAAT enhancer binding protein α (C/EBPα), a positive regulator of granulocytic differentiation. Suppression of C/EBPα is seen in the majority of patients with M-BP(146, 147). M-BP also commonly displays inactivating RUNX1 mutations similar to those seen in AML that lead to a block of neutrophil differentiation (38, 134, 148, 149). Disruption of other myeloid transcription factors such as C/EBPβ and over-expression of MECOM (EVI-1) have also been implicated in M-BP(150–152).
Another consequence of increased BCR/ABL1 expression is elevated expression of SET, an inhibitor of the tumor suppressor protein phosphatase 2A (PP2A). Reduced PP2A de-activates Src homology region 2 domain-containing phosphatase-1 (SHP-1), resulting in increased phosphorylation of BCR/ABL1 and other substrates (153). Reduced SHP-1 activity is also associated with resistance to TKIs, highlighting the importance of this pathway(154). Cancerous inhibitor of PP2A (CIP2A) contributes to PP2A inhibition in CML, with high CIP2A activity predicting a high risk of transformation to BP-CML(155). Transformation to L-BP is caused by inactivation of critical B cell transcription factors, such as IKAROS Family Zinc Finger 1 (IKZF1) or paired Box 5 (PAX-5)(156).
Recent work reported that heterogeneous upstream pathways converge on a relatively uniform pattern epigenetic dysregulation, specifically reduced activity of polycomb repressive complex 2 (PRC2) and upregulation of PRC1 (134). Although, expectedly, monotherapy with hypomethylating agents does not produce durable responses, the future development of therapies in BP-CML should include correcting epigenetic dysregulation rather than targeting individual upstream pathways only(157, 158). For instance, the combination and PRC1 inhibitor and decitabine showed activity against AP-CML in cell lines. A phase I/II study combining decitabine with dasatinib in advanced CML showed promising clinical activity providing further evidence that epigenetic dysregulation contributes to this phase of disease(159). The goal of therapy for patients who progress to AP/BP CML is to establish a second chronic phase, then proceed to HSCT if the patient is eligible. Second and third generation TKIs, specifically ponatinib, are a cornerstone of therapy for AP/BP CML following the same strategy described above for CP-CML. Some patients have no TKI options left, necessitating the use of intensive cytotoxic chemotherapy, but in general TKIs are often combined with cytotoxic agents, despite lack of randomized data showing an advantage for the addition of chemotherapy. For patients who have a TKI option, the risk of chemotherapy-induced toxicity needs to be considered as this may delay transplant and increase non-relapse mortality. Less toxic regimens such as hypomethylating agents in myeloid BP and vincristine/prednisone in lymphoid BP provide reasonable options while avoiding excessive toxicity, but prospective, randomized studies are lacking (160).
3.3. Cardiovascular TKI Toxicity
Aside from imatinib, all TKIs are associated with cardiovascular toxicity including associations with life threatening events such as arterial thrombosis and pulmonary hypertension. The incidence of cardiovascular toxicity is highest with ponatinib and nilotinib but is also increased with dasatinib and to a lesser extent bosutinib(80, 83, 161–164). There is consensus that imatinib has no significant cardiovascular toxicity. Several mechanisms were proposed to explain cardiovascular adverse events associated with ABL1 TKIs, and it is likely that the underlying pathophysiology differs between the different TKIs. For example, nilotinib reduces glucose tolerance and may cause accelerated atherosclerosis(78, 116). Ponatinib was shown to induce an angiopathy caused by excessive endothelial-associated von Willebrand factor (VWF) and secondary platelet adhesion(91). It remains possible that inhibition of ABL1 itself contributes to toxicity. If confirmed this on-target effect would represent the natural limitation of BCR/ABL1 kinase inhibitor therapy. As there is no uniform preventive strategy, the priority is prevention through minimizing CV risk and selecting the most appropriate TKI for a given patient. In our practice, we avoid nilotinib in patients with higher Framingham risk scores and consider imatinib for high-risk patients. We have a low threshold in obtaining a cardiology consult. Patients are followed using a set of practice guidelines tailored to the specific TKI and the patient’s comorbidities. A strategy worth exploring for patients who experience severe toxicities is dose reduction below the doses recommended in the current labels, with careful monitoring of BCR/ABL1 qPCR. For instance, in a single-arm study of CP-CML patients, a lower starting dose of dasatinib (50 mg daily) was shown to be well-tolerated and at least as effective as 100 mg daily in historical controls(165–167).
4. Future Directions
What are the next steps? From a clinical perspective, the most important frontiers are increasing the rate of TFR, improving the prognosis of BP-CML and overcoming BCR/ABL1-independent TKI resistance. Increasing the rate of TFR has proven surprisingly difficult, and as of now the majority of patients require lifelong TKI therapy (Figure 3). An important motivation to achieve higher rates of TFR is reduction of the financial toxicity associated with long term TKI use (95). Rational strategies to improve our understanding of TFR and increase the rates of successful TFR are of particular interest. Drug combinations are a possible strategy to improve TFR by increasing the rates of deep molecular response. Current clinical studies focus on combinations of IFNα with imatinib or 2G TKIs but toxicity is a challenge in a setting where long-term survival is all but assured(65, 168–171). Pre-clinical models have implicated multiple pathways that promote the survival of primitive CML cells in the presence or in the absence of TKIs, but surprisingly few have led to clinical trials, and ever fewer were tested in the most relevant clinical setting: residual CML in TKI responders. Addition of the autophagy inhibitor chloroquine to imatinib moderately improved molecular response in patients with major cytogenetic response, but residual CML detectable by qPCR (172). In a phase 1 trial combining nilotinib and the JAK kinase inhibitor ruxulitinib improved molecular responses, and a phase 2 study is currently underway(173). A complete review of this subject is beyond the scope of our overview article and the reader is referred to a recent publication(174). An alternative strategy for increasing TFR may be to optimize TKIs to achieve complete inhibition of BCR/ABL1 kinase activity. The abandoned EPIC study of ponatinib 45 mg daily may provide some clues into whether we have exploited the full potential of inhibiting BCR/ABL1. Only 73 patients were evaluable for response at 6 months, but the DMR rate was 32% compared to ~5% with nilotinib 300mg twice daily in the ENESTnd study(90). This may suggest that a very potent yet well-tolerated TKI, such as asciminib is the first step to maximizing TFR rates(90). Enhancing immune response and surveillance may be another strategy to improve the rates of DMR and reduce recurrence. Correlative studies showed decreased natural killer (NK) cell numbers and functionality in patients with recurrence in comparison to those who continued in TFR(117, 175). Myeloid-derived suppressor cells were shown to be increased in patients with recurrence, while enhanced cytotoxic T lymphocyte responses were decreased(176). While these immune mediated mechanisms present exciting insights into TFR, they have not translated into therapeutic interventions yet. Clinical approaches utilizing immune surveillance to eradicate residual leukemic cells are potential future research directions to improve TFR rates. In patients who are unable to achieve TFR, prevention of cardiovascular side effects associated with prolonged TKI therapy is an important focus for future studies to address.
Fig. 3.

Only a small subset of patients with newly diagnosed CML will be able to achieve treatment free remission. Among 100 patients with newly diagnosed CML about 10 percent will have accelerated or blast phase at diagnosis and are not eligible for a trial of treatment free remission per current guidelines. Of the 90% who have chronic phase CML, two thirds will achieve sustained DMR. Half of the patients who qualify for a TKI discontinuation trial will experience recurrence necessitating reintroduction of TKIs.
Progression to BP-CML is becoming an increasingly rare scenario in the age of TKIs, but the prognosis remains poor despite the advances made in CP-CML. Current therapies rely on TKIs, conventional cytotoxic chemotherapy and SCT. The lack of randomized trials in BP-CML is a major impediment, and given its rarity, progress will require international collaboration. The improved understanding of the molecular pathogenesis BP-CML has not yet led to new therapeutic modalities, a reflection of the complexity nature of this disease. BCR/ABL1 independent resistance continues to be a unique challenge due to the heterogenous underlying mechanisms converging on the resistance phenotype. Current efforts continue to uncover new pathways involved in BCR/ABL1 independent TKI resistance. Interestingly, a number of these pathways have clinical inhibitors under use in solid tumors and other hematologic malignancies. Inhibition of BCL2 and MEK for example both showed promise in pre-clinical models and await testing in clinical trials(177, 178). That being said, only a few of these preclinical discoveries lead to clinically relevant interventions so far. Establishing pre-clinical models that more faithfully reflect human CML and its progression to BP, along with simplifying clinical trials will also be critical for progress in this area.
Finally, while TKIs revolutionized the treatment of CML, access to TKIs remains challenging in the developing world and sometimes also in the United States due to their high costs. This demonstrates the importance of socioeconomic factors in the treatment of CML and hematologic malignancies in general. At an individual patient level, TKI side effects can be debilitating and life altering. A strong therapeutic alliance between the patient and treating hematologist is needed to navigate side effects and assure compliance. As the number of CML patients increases, a nuanced knowledge of the BCR/ABL1 TKIs and their side effects, resistance patterns and molecular monitoring of CML is of increasing importance for the practicing hematologist.
Practice Points
Demonstration of Ph or BCR/ABL1 using karyotyping, FISH or RT-PCR establishes the diagnosis of CML in a patient with a myeloproliferative neoplasm, irrespective of other features.
The choice of a BCR/ABL1 TKI for newly diagnosed chronic phase CML depends on the patient’s comorbidities, Sokal or ELTS risk score, cost and drug availability.
Patients with a total duration of TKI therapy of at least 3 years and no history of progression to AP/BP-CML who maintain a deep molecular response (BCR/ABL1<0.01% IS) for more than 2 years are potential candidates for a trial of treatment free remission. Adherence to contemporary management guidelines is strongly recommended, including BCR/ABL1 monitoring (monthly for the first 6 months, 2-monthly for month 7–12, and quarterly thereafter). Confirmed loss of MMR is the indication for re-initiation of TKI therapy.
Ponatinib is the only TKI with activity against the BCR/ABL1T315I mutant. Ponatinib is associated with significant cardiovascular toxicity and should be used only after careful assessment and discussion of risks and benefits.
Blast phase CML has a poor prognosis. 2G TKIs or ponatinib with or without chemotherapy are used to induce a second chronic phase as a bridge to allogeneic stem cell transplant.
Research Agenda
Increasing the rate of treatment free remission.
Overcoming TKI resistance in patients with multi-TKI failure.
Improving outcomes for patients with accelerated and blast phase CML
Developing better animal models that more faithfully recapitulate the clinic-pathologic features of human CML.
Preventing cardiovascular toxicities associated with second and third generation TKIs.
Footnotes
Conflict of interest statement
No conflicts of interest, financial or other, exist for Afaf Osman. Michael Deininger’s potential conflicts of interest includes serving on the advisory board of Blueprint, Takeda, Novartis, Incyte, Sangamo and Pfizer, paid consultation at Blueprint, Fusion Pharma, Medscape, Novartis, Sangamo, DisperSol and the NCCN, research funding from Blueprint, Takeda, Novartis, Incyte, SPARC, Leukemia and Lymphoma Society and Pfizer and study management committees at Blueprint and Takeda. He is a case author at Medscape.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M, Fava C, et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst. 2011;103(7):553–61. [DOI] [PubMed] [Google Scholar]
- 2.Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118(12):3123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichimaru M, Tomonaga M, Amenomori T, Matsuo T. Atomic bomb and leukemia. J Radiat Res. 1991;32 Suppl:162–7. [DOI] [PubMed] [Google Scholar]
- 4.Little MP, Weiss HA, Boice JD, Jr., Darby SC, Day NE, Muirhead CR. Risks of leukemia in Japanese atomic bomb survivors, in women treated for cervical cancer, and in patients treated for ankylosing spondylitis. Radiat Res. 1999;152(3):280–92. [PubMed] [Google Scholar]
- 5.Van Kaick G, Wesch H, Luhrs H, Liebermann D, Kaul A. Neoplastic diseases induced by chronic alpha-irradiation-- epidemiological, biophysical and clinical results of the German Thorotrast Study. JRadiatResTokyo. 1991;32 Suppl 2:20–33. [DOI] [PubMed] [Google Scholar]
- 6.Reckel S, Gehin C, Tardivon D, Georgeon S, Kukenshoner T, Lohr F, et al. Structural and functional dissection of the DH and PH domains of oncogenic Bcr-Abl tyrosine kinase. Nat Commun. 2017;8(1):2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reckel S, Hamelin R, Georgeon S, Armand F, Jolliet Q, Chiappe D, et al. Differential signaling networks of Bcr-Abl p210 and p190 kinases in leukemia cells defined by functional proteomics. Leukemia. 2017;31(7):1502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aceves-Luquero CI, Agarwal A, Callejas-Valera JL, Arias-Gonzalez L, Esparis-Ogando A, del Peso Ovalle L, et al. ERK2, but not ERK1, mediates acquired and “de novo” resistance to imatinib mesylate: implication for CML therapy. PLoS One. 2009;4(7):e6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12(8):513–26. [DOI] [PubMed] [Google Scholar]
- 10.Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. ProcNatlAcadSciUSA. 1996;93(26):15174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kipreos ET, Wang JY. Differential phosphorylation of c-Abl in cell cycle determined by cdc2 kinase and phosphatase activity. Science. 1990;248(4952):217–20. [DOI] [PubMed] [Google Scholar]
- 12.Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77(1):121–31. [DOI] [PubMed] [Google Scholar]
- 13.Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387(6632):516–9. [DOI] [PubMed] [Google Scholar]
- 14.Yuan ZM, Utsugisawa T, Huang Y, Ishiko T, Nakada S, Kharbanda S, et al. Inhibition of phosphatidylinositol 3-kinase by c-Abl in the genotoxic stress response. JBiolChem. 1997;272(38):23485–8. [DOI] [PubMed] [Google Scholar]
- 15.Kharbanda S, Pandey P, Jin S, Inoue S, Bharti A, Yuan ZM, et al. Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature. 1997;386(6626):732–5. [DOI] [PubMed] [Google Scholar]
- 16.Yuan ZM, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Kharbanda S, et al. Regulation of Rad51 function by c-Abl in response to DNA damage. JBiolChem. 1998;273(7):3799–802. [DOI] [PubMed] [Google Scholar]
- 17.Yuan ZM, Utsugisawa T, Ishiko T, Nakada S, Huang Y, Kharbanda S, et al. Activation of protein kinase C delta by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene. 1998;16(13):1643–8. [DOI] [PubMed] [Google Scholar]
- 18.Hasford J, Pfirrmann M, Hehlmann R, Allan NC, Baccarani M, Kluin-Nelemans JC, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML Prognostic Factors Project Group. JNatlCancer Inst. 1998;90(11):850–8. [DOI] [PubMed] [Google Scholar]
- 19.Vardiman JW. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: An overview with emphasis on the myeloid neoplasms. Chemico-Biological Interactions. 2010;184(1):16–20. [DOI] [PubMed] [Google Scholar]
- 20.Thiele J, Kvasnicka HM, Schmitt-Graeff A, Zirbes TK, Birnbaum F, Kressmann C, et al. Bone marrow features and clinical findings in chronic myeloid leukemia--a comparative, multicenter, immunohistological and morphometric study on 614 patients. LeukLymphoma. 2000;36(3–4):295–308. [DOI] [PubMed] [Google Scholar]
- 21.Fabarius A, Leitner A, Hochhaus A, Muller MC, Hanfstein B, Haferlach C, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760–8. [DOI] [PubMed] [Google Scholar]
- 22.Luatti S, Castagnetti F, Marzocchi G, Baldazzi C, Gugliotta G, Iacobucci I, et al. Additional chromosomal abnormalities in Philadelphia-positive clone: adverse prognostic influence on frontline imatinib therapy: a GIMEMA Working Party on CML analysis. Blood. 2012;120(4):761–7. [DOI] [PubMed] [Google Scholar]
- 23.Bumm T, Muller C, Al Ali HK, Krohn K, Shepherd P, Schmidt E, et al. Emergence of clonal cytogenetic abnormalities in Ph- cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood. 2003;101(5):1941–9. [DOI] [PubMed] [Google Scholar]
- 24.Huntly BJ, Guilhot F, Reid AG, Vassiliou G, Hennig E, Franke C, et al. Imatinib improves but may not fully reverse the poor prognosis of patients with CML with derivative chromosome 9 deletions. Blood. 2003;102(6):2205–12. [DOI] [PubMed] [Google Scholar]
- 25.Sinclair PB, Nacheva EP, Leversha M, Telford N, Chang J, Reid A, et al. Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood. 2000;95(3):738–43. [PubMed] [Google Scholar]
- 26.Quintas-Cardama A, Kantarjian H, Talpaz M, O’Brien S, Garcia-Manero G, Verstovsek S, et al. Imatinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood. 2005;105(6):2281–6. [DOI] [PubMed] [Google Scholar]
- 27.Kreil S, Pfirrmann M, Haferlach C, Waghorn K, Chase A, Hehlmann R, et al. Heterogeneous prognostic impact of derivative chromosome 9 deletions in chronic myelogenous leukemia. Blood. 2007;110(4):1283–90. [DOI] [PubMed] [Google Scholar]
- 28.Testoni N, Marzocchi G, Luatti S, Amabile M, Baldazzi C, Stacchini M, et al. Chronic myeloid leukemia: a prospective comparison of interphase fluorescence in situ hybridization and chromosome banding analysis for the definition of complete cytogenetic response: a study of the GIMEMA CML WP. Blood. 2009;114(24):4939–43. [DOI] [PubMed] [Google Scholar]
- 29.Verma D, Kantarjian HM, Jones D, Luthra R, Borthakur G, Verstovsek S, et al. Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 2009;114(11):2232–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melo JV, Myint H, Galton DA, Goldman JM. P190BCR-ABL chronic myeloid leukaemia: the missing link with chronic myelomonocytic leukaemia? Leukemia. 1994;8(1):208–11. [PubMed] [Google Scholar]
- 31.Cross NC, Melo JV, Feng L, Goldman JM. An optimized multiplex polymerase chain reaction (PCR) for detection of BCR-ABL fusion mRNAs in haematological disorders. Leukemia. 1994;8(1):186–9. [PubMed] [Google Scholar]
- 32.Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC, et al. beta-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2015;29(12):2328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hochhaus A, Reiter A, Skladny H, Melo JV, Sick C, Berger U, et al. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood. 1996;88(6):2236–40. [PubMed] [Google Scholar]
- 34.Demehri S, Paschka P, Schultheis B, Lange T, Koizumi T, Sugimoto T, et al. e8a2 BCR-ABL: more frequent than other atypical BCR-ABL variants? Leukemia. 2005;19(4):681–4. [DOI] [PubMed] [Google Scholar]
- 35.Al Ali HK, Leiblein S, Kovacs I, Hennig E, Niederwieser D, Deininger MW. CML with an e1a3 BCR-ABL fusion: rare, benign, and a potential diagnostic pitfall. Blood. 2002;100(3):1092–3. [DOI] [PubMed] [Google Scholar]
- 36.Melo JV, Gordon DE, Cross NC, Goldman JM. The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood. 1993;81(1):158–65. [PubMed] [Google Scholar]
- 37.Huntly BJ, Bench AJ, Delabesse E, Reid AG, Li J, Scott MA, et al. Derivative chromosome 9 deletions in chronic myeloid leukemia: poor prognosis is not associated with loss of ABL-BCR expression, elevated BCR-ABL levels, or karyotypic instability. Blood. 2002;99(12):4547–53. [DOI] [PubMed] [Google Scholar]
- 38.Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948–61. [DOI] [PubMed] [Google Scholar]
- 39.Hehlmann R, Voskanyan A, Lauseker M, Pfirrmann M, Kalmanti L, Rinaldetti S, et al. High-risk additional chromosomal abnormalities at low blast counts herald death by CML. Leukemia. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shanmuganathan N, Wadham C, Shahrin NH, Thomson D, Feng J, Saunders VA, et al. Mutated Cancer-Related Genes Detected at Diagnosis of CML and a Novel Class of Variant Associated with the Philadelphia Translocation Are Both Independent Predictors of Inferior Outcomes. Blood. 2020;136(Supplement 1):46–7. [Google Scholar]
- 41.Bonifazi F, de Vivo A, Rosti G, Guilhot F, Guilhot J, Trabacchi E, et al. Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood. 2001;98(10):3074–81. [DOI] [PubMed] [Google Scholar]
- 42.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. NEnglJMed. 2006;355(23):2408–17. [DOI] [PubMed] [Google Scholar]
- 43.Sokal JE, Cox EB, Baccarani M, Tura S, Gomez GA, Robertson JE, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789–99. [PubMed] [Google Scholar]
- 44.Pfirrmann M, Baccarani M, Saussele S, Guilhot J, Cervantes F, Ossenkoppele G, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. 2016;30(1):48–56. [DOI] [PubMed] [Google Scholar]
- 45.Radich JP, Deininger M, Abboud CN, Altman JK, Berman E, Bhatia R, et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16(9):1108–35. [DOI] [PubMed] [Google Scholar]
- 46.Kantarjian HM, Talpaz M. Definition of the accelerated phase of chronic myelogenous leukemia. J ClinOncol. 1988;6(1):180–2. [DOI] [PubMed] [Google Scholar]
- 47.Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737–47. [DOI] [PubMed] [Google Scholar]
- 48.Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76–94. [DOI] [PubMed] [Google Scholar]
- 50.Hasford J, Baccarani M, Hoffmann V, Guilhot J, Saussele S, Rosti G, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118(3):686–92. [DOI] [PubMed] [Google Scholar]
- 51.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Press RD, Willis SG, Laudadio J, Mauro MJ, Deininger MW. Determining the rise in BCR-ABL RNA that optimally predicts a kinase domain mutation in patients with chronic myeloid leukemia on imatinib. Blood. 2009;114(13):2598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. NEnglJMed. 2003;349(15):1423–32. [DOI] [PubMed] [Google Scholar]
- 54.Marin D, Ibrahim AR, Lucas C, Gerrard G, Wang L, Szydlo RM, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012;30(3):232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nazha A, Kantarjian H, Jain P, Romo C, Jabbour E, Quintas-Cardama A, et al. Assessment at 6 months may be warranted for patients with chronic myeloid leukemia with no major cytogenetic response at 3 months. Haematologica. 2013;98(11):1686–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. NEnglJMed. 2001;344(14):1038–42. [DOI] [PubMed] [Google Scholar]
- 57.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. NEnglJMed. 2001;344(14):1031–7. [DOI] [PubMed] [Google Scholar]
- 58.O’Brien SG, Deininger MW. Imatinib in patients with newly diagnosed chronic-phase chronic myeloid leukemia. SeminHematol. 2003;40(2 Suppl 3):26–30. [DOI] [PubMed] [Google Scholar]
- 59.Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med. 2017;376(10):917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guilhot F, Druker B, Larson RA, Gathmann I, So C, Waltzman R, et al. High rates of durable response are achieved with imatinib after treatment with interferon alpha plus cytarabine: results from the International Randomized Study of Interferon and STI571 (IRIS) trial. Haematologica. 2009;94(12):1669–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cortes JE, Baccarani M, Guilhot F, Druker BJ, Branford S, Kim DW, et al. Phase III, randomized, open-label study of daily imatinib mesylate 400 mg versus 800 mg in patients with newly diagnosed, previously untreated chronic myeloid leukemia in chronic phase using molecular end points: tyrosine kinase inhibitor optimization and selectivity study. J Clin Oncol. 2010;28(3):424–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castagnetti F, Palandri F, Amabile M, Testoni N, Luatti S, Soverini S, et al. Results of high-dose imatinib mesylate in intermediate Sokal risk chronic myeloid leukemia patients in early chronic phase: a phase 2 trial of the GIMEMA CML Working Party. Blood. 2009;113(15):3428–34. [DOI] [PubMed] [Google Scholar]
- 63.Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Muller MC, Pletsch N, et al. Tolerability-Adapted Imatinib 800 mg/d Versus 400 mg/d Versus 400 mg/d Plus Interferon-{alpha} in Newly Diagnosed Chronic Myeloid Leukemia. JClinOncol. 2011;29(12):1634–42. [DOI] [PubMed] [Google Scholar]
- 64.Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ, et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia. 2017;31(11):2398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Preudhomme C, Guilhot J, Nicolini FE, Guerci-Bresler A, Rigal-Huguet F, Maloisel F, et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. NEnglJMed. 2010;363(26):2511–21. [DOI] [PubMed] [Google Scholar]
- 66.Hoffmann VS, Hasford J, Deininger M, Cortes J, Baccarani M, Hehlmann R. Systematic review and meta-analysis of standard-dose imatinib vs. high-dose imatinib and second generation tyrosine kinase inhibitors for chronic myeloid leukemia. J Cancer Res Clin Oncol. 2017;143(7):1311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109(6):2303–9. [DOI] [PubMed] [Google Scholar]
- 68.Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109(8):3207–13. [DOI] [PubMed] [Google Scholar]
- 69.Guilhot F, Apperley J, Kim DW, Bullorsky EO, Baccarani M, Roboz GJ, et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood. 2007;109(10):4143–50. [DOI] [PubMed] [Google Scholar]
- 70.Shah NP, Kantarjian HM, Kim DW, Rea D, Dorlhiac-Llacer PE, Milone JH, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and -intolerant chronic-phase chronic myeloid leukemia. JClinOncol. 2008;26(19):3204–12. [DOI] [PubMed] [Google Scholar]
- 71.Kantarjian H, Cortes J, Kim DW, Dorlhiac-Llacer P, Pasquini R, DiPersio J, et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood. 2009;113(25):6322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41. [DOI] [PubMed] [Google Scholar]
- 73.Deininger MW, Kopecky KJ, Radich JP, Kamel-Reid S, Stock W, Paietta E, et al. Imatinib 800 mg daily induces deeper molecular responses than imatinib 400 mg daily: results of SWOG S0325, an intergroup randomized PHASE II trial in newly diagnosed chronic phase chronic myeloid leukaemia. Br J Haematol. 2014;164(2):223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–51. [DOI] [PubMed] [Google Scholar]
- 75.Le CP, Ottmann OG, Giles F, Kim DW, Cortes J, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–9. [DOI] [PubMed] [Google Scholar]
- 76.Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110(10):3540–6. [DOI] [PubMed] [Google Scholar]
- 77.Giles FJ, Kantarjian HM, le Coutre PD, Baccarani M, Mahon FX, Blakesley RE, et al. Nilotinib is effective in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26(5):959–62. [DOI] [PubMed] [Google Scholar]
- 78.Saglio G, Kim DW, Issaragrisil S, Le CP, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. NEnglJMed. 2010;362(24):2251–9. [DOI] [PubMed] [Google Scholar]
- 79.Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moslehi JJ, Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol. 2015;33(35):4210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khoury HJ, Cortes JE, Kantarjian HM, Gambacorti-Passerini C, Baccarani M, Kim DW, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cortes JE, Khoury HJ, Kantarjian HM, Lipton JH, Kim DW, Schafhausen P, et al. Long-term bosutinib for chronic phase chronic myeloid leukemia after failure of imatinib plus dasatinib and/or nilotinib. Am J Hematol. 2016;91(12):1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brummendorf TH, Cortes JE, de Souza CA, Guilhot F, Duvillie L, Pavlov D, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia: results from the 24-month follow-up of the BELA trial. Br J Haematol. 2015;168(1):69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cortes JE, Kim DW, Kantarjian HM, Brummendorf TH, Dyagil I, Griskevicius L, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30(28):3486–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cortes JE, Gambacorti-Passerini C, Deininger MW, Mauro MJ, Chuah C, Kim DW, et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J Clin Oncol. 2018;36(3):231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–9. [DOI] [PubMed] [Google Scholar]
- 87.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cortes J, Talpaz M, Bixby D, Deininger M, Shah N, Flinn IW, et al. A Phase 1 Trial of Oral Ponatinib (AP24534) In Patients with Refractory Chronic Myelogenous Leukemia (CML) and Other Hematologic Malignancies: Emerging Safety and Clinical Response Findings. Blood. 2010;116(21):210.20304810 [Google Scholar]
- 89.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(5):612–21. [DOI] [PubMed] [Google Scholar]
- 91.Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood. 2019;133(14):1597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kantarjian HM, Deininger MW, Abruzzese E, Apperley J, Cortes JE, Chuah C, et al. Efficacy and Safety of Ponatinib (PON) in Patients with Chronic-Phase Chronic Myeloid Leukemia (CP-CML) Who Failed One or More Second-Generation (2G) Tyrosine Kinase Inhibitors (TKIs): Analyses Based on PACE and Optic. Blood. 2020;136(Supplement 1):43–4. [Google Scholar]
- 93.Steegmann JL, Baccarani M, Breccia M, Casado LF, Garcia-Gutierrez V, Hochhaus A, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016;30(8):1648–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dusetzina SB, Muluneh B, Keating NL, Huskamp HA. Broken Promises - How Medicare Part D Has Failed to Deliver Savings to Older Adults. N Engl J Med. 2020;383(24):2299–301. [DOI] [PubMed] [Google Scholar]
- 95.The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. Blood. 2013;121(22):4439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lasica M, Willcox A, Burbury K, Ross DM, Branford S, Butler J, et al. The effect of tyrosine kinase inhibitor interruption and interferon use on pregnancy outcomes and long-term disease control in chronic myeloid leukemia. Leuk Lymphoma. 2019;60(7):1796–802. [DOI] [PubMed] [Google Scholar]
- 97.Fefer A, Cheever MA, Thomas ED, Boyd C, Ramberg R, Glucksberg H, et al. Disappearance of Ph1-positive cells in four patients with chronic granulocytic leukemia after chemotherapy, irradiation and marrow transplantation from an identical twin. N Engl J Med. 1979;300(7):333–7. [DOI] [PubMed] [Google Scholar]
- 98.Kolb HJ, Mittermuller J, Clemm C, Holler E, Ledderose G, Brehm G, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76(12):2462–5. [PubMed] [Google Scholar]
- 99.Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. JClinOncol. 2009;27(35):6041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saussele S, Lauseker M, Gratwohl A, Beelen DW, Bunjes D, Schwerdtfeger R, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115(10):1880–5. [DOI] [PubMed] [Google Scholar]
- 101.Zhang H, Chen J, Que W. Allogeneic peripheral blood stem cell and bone marrow transplantation for hematologic malignancies: meta-analysis of randomized controlled trials. Leuk Res. 2012;36(4):431–7. [DOI] [PubMed] [Google Scholar]
- 102.Deininger M, Schleuning M, Greinix H, Sayer HG, Fischer T, Martinez J, et al. The effect of prior exposure to imatinib on transplant-related mortality. Haematologica. 2006;91(4):452–9. [PubMed] [Google Scholar]
- 103.Oehler VG, Gooley T, Snyder DS, Johnston L, Lin A, Cummings CC, et al. The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood. 2007;109(4):1782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee SJ, Kukreja M, Wang T, Giralt SA, Szer J, Arora M, et al. Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood. 2008;112(8):3500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cortes JE, Kantarjian HM, Rea D, Wetzler M, Lipton JH, Akard L, et al. Final analysis of the efficacy and safety of omacetaxine mepesuccinate in patients with chronic- or accelerated-phase chronic myeloid leukemia: Results with 24 months of follow-up. Cancer. 2015;121(10):1637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007;109(1):58–60. [DOI] [PubMed] [Google Scholar]
- 107.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–35. [DOI] [PubMed] [Google Scholar]
- 108.Rousselot P, Charbonnier A, Cony-Makhoul P, Agape P, Nicolini FE, Varet B, et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J Clin Oncol. 2014;32(5):424–30. [DOI] [PubMed] [Google Scholar]
- 109.Kimura S, Imagawa J, Murai K, Hino M, Kitawaki T, Okada M, et al. Treatment-free remission after first-line dasatinib discontinuation in patients with chronic myeloid leukaemia (first-line DADI trial): a single-arm, multicentre, phase 2 trial. Lancet Haematol. 2020;7(3):e218–e25. [DOI] [PubMed] [Google Scholar]
- 110.Okada M, Imagawa J, Tanaka H, Nakamae H, Hino M, Murai K, et al. Final 3-year Results of the Dasatinib Discontinuation Trial in Patients With Chronic Myeloid Leukemia Who Received Dasatinib as a Second-line Treatment. Clin Lymphoma Myeloma Leuk. 2018;18(5):353–60.e1. [DOI] [PubMed] [Google Scholar]
- 111.Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2(12):e528–35. [DOI] [PubMed] [Google Scholar]
- 112.Rea D, Nicolini FE, Tulliez M, Guilhot F, Guilhot J, Guerci-Bresler A, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: interim analysis of the STOP 2G-TKI study. Blood. 2017;129(7):846–54. [DOI] [PubMed] [Google Scholar]
- 113.Shah NP, Garcia-Gutierrez V, Jimenez-Velasco A, Larson S, Saussele S, Rea D, et al. Dasatinib discontinuation in patients with chronic-phase chronic myeloid leukemia and stable deep molecular response: the DASFREE study. Leuk Lymphoma. 2020;61(3):650–9. [DOI] [PubMed] [Google Scholar]
- 114.Ross DM, Masszi T, Gomez Casares MT, Hellmann A, Stentoft J, Conneally E, et al. Durable treatment-free remission in patients with chronic myeloid leukemia in chronic phase following frontline nilotinib: 96-week update of the ENESTfreedom study. J Cancer Res Clin Oncol. 2018;144(5):945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aichberger KJ, Herndlhofer S, Schernthaner GH, Schillinger M, Mitterbauer-Hohendanner G, Sillaber C, et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am J Hematol. 2011;86(7):533–9. [DOI] [PubMed] [Google Scholar]
- 116.Kim TD, Rea D, Schwarz M, Grille P, Nicolini FE, Rosti G, et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia. 2013;27(6):1316–21. [DOI] [PubMed] [Google Scholar]
- 117.Ilander M, Olsson-Stromberg U, Schlums H, Guilhot J, Bruck O, Lahteenmaki H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31(5):1108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schutz C, Inselmann S, Saussele S, Dietz CT, Mu Ller MC, Eigendorff E, et al. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia. 2017;31(4):829–36. [DOI] [PubMed] [Google Scholar]
- 119.Irani YD, Hughes A, Clarson J, Kok CH, Shanmuganathan N, White DL, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. British Journal of Haematology. 2020;191(3):433–41. [DOI] [PubMed] [Google Scholar]
- 120.Richter J, Söderlund S, Lübking A, Dreimane A, Lotfi K, Markevärn B, et al. Musculoskeletal pain in patients with chronic myeloid leukemia after discontinuation of imatinib: a tyrosine kinase inhibitor withdrawal syndrome? J Clin Oncol. 2014;32(25):2821–3. [DOI] [PubMed] [Google Scholar]
- 121.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–80. [DOI] [PubMed] [Google Scholar]
- 122.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–25. [DOI] [PubMed] [Google Scholar]
- 123.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12(24):7374–9. [DOI] [PubMed] [Google Scholar]
- 124.Kizilors A, Crisa E, Lea N, Passera R, Mian S, Anwar J, et al. Effect of low-level BCR-ABL1 kinase domain mutations identified by next-generation sequencing in patients with chronic myeloid leukaemia: a population-based study. Lancet Haematol. 2019;6(5):e276–e84. [DOI] [PubMed] [Google Scholar]
- 125.Soverini S, Bavaro L, De Benedittis C, Martelli M, Iurlo A, Orofino N, et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: the NEXT-in-CML study. Blood. 2020;135(8):534–41. [DOI] [PubMed] [Google Scholar]
- 126.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell. 2019;36(4):431–43.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96(3):1070–9. [PubMed] [Google Scholar]
- 130.White DL, Saunders VA, Dang P, Engler J, Venables A, Zrim S, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007;110(12):4064–72. [DOI] [PubMed] [Google Scholar]
- 131.Dulucq S, Bouchet S, Turcq B, Lippert E, Etienne G, Reiffers J, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2008;112(5):2024–7. [DOI] [PubMed] [Google Scholar]
- 132.Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55(11):731–7. [DOI] [PubMed] [Google Scholar]
- 133.Eadie LN, Hughes TP, White DL. ABCB1 Overexpression Is a Key Initiator of Resistance to Tyrosine Kinase Inhibitors in CML Cell Lines. PLoS One. 2016;11(8):e0161470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ko TK, Javed A, Lee KL, Pathiraja TN, Liu X, Malik S, et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood. 2020;135(26):2337–53. [DOI] [PubMed] [Google Scholar]
- 135.Wang W, Cortes JE, Tang G, Khoury JD, Wang S, Bueso-Ramos CE, et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood. 2016;127(22):2742–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Garg RJ, Kantarjian H, O’Brien S, Quintas-Cardama A, Faderl S, Estrov Z, et al. The use of nilotinib or dasatinib after failure to 2 prior tyrosine kinase inhibitors: long-term follow-up. Blood. 2009;114(20):4361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132(4):393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tam CS, Kantarjian H, Garcia-Manero G, Borthakur G, O’Brien S, Ravandi F, et al. Failure to achieve a major cytogenetic response by 12 months defines inadequate response in patients receiving nilotinib or dasatinib as second or subsequent line therapy for chronic myeloid leukemia. Blood. 2008;112(3):516–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J Med Chem. 2018;61(18):8120–35. [DOI] [PubMed] [Google Scholar]
- 140.Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543(7647):733–7. [DOI] [PubMed] [Google Scholar]
- 141.Mauro MJ, Hughes TP. Asciminib in Relapsed Chronic Myeloid Leukemia. Reply. N Engl J Med. 2020;382(14):1379. [DOI] [PubMed] [Google Scholar]
- 142.Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ, et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N Engl J Med. 2019;381(24):2315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hochhaus A, Boquimpani C, Rea D, et al. Efficacy and Safety Results from ASCEMBL, a Multicenter, Open-Label, Phase 3 Study of Asciminib, a First-in-Class STAMP Inhibitor, vs Bosutinib (BOS) in Patients (Pts) with Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Previously Treated with ≥2 Tyrosine Kinase Inhibitors (TKIs). Abstract LBA-4 Presented at the 2020 American Society of Hematology Annual Meeting; December 8, 2020. [Google Scholar]
- 144.Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25(3):557–60. [DOI] [PubMed] [Google Scholar]
- 145.Elmaagacli AH, Beelen DW, Opalka B, Seeber S, Schaefer UW. The amount of BCR-ABL fusion transcripts detected by the real-time quantitative polymerase chain reaction method in patients with Philadelphia chromosome positive chronic myeloid leukemia correlates with the disease stage. AnnHematol. 2000;79(8):424–31. [DOI] [PubMed] [Google Scholar]
- 146.Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. NatGenet. 2002;30(1):48–58. [DOI] [PubMed] [Google Scholar]
- 147.Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140(5):652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gerritsen M, Yi G, Tijchon E, Kuster J, Schuringa JJ, Martens JHA, et al. RUNX1 mutations enhance self-renewal and block granulocytic differentiation in human in vitro models and primary AMLs. Blood Adv. 2019;3(3):320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mullighan CG, Williams RT, Downing JR, Sherr CJ. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008;22(11):1411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guerzoni C, Bardini M, Mariani SA, Ferrari-Amorotti G, Neviani P, Panno ML, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood. 2006;107(10):4080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cuenco GM, Ren R. Both AML1 and EVI1 oncogenic components are required for the cooperation of AML1/MDS1/EVI1 with BCR/ABL in the induction of acute myelogenous leukemia in mice. Oncogene. 2004;23(2):569–79. [DOI] [PubMed] [Google Scholar]
- 152.De Weer A, Poppe B, Cauwelier B, Carlier A, Dierick J, Verhasselt B, et al. EVI1 activation in blast crisis CML due to juxtaposition to the rare 17q22 partner region as part of a 4-way variant translocation t(9;22). BMC Cancer. 2008;8:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8(5):355–68. [DOI] [PubMed] [Google Scholar]
- 154.Esposito N, Colavita I, Quintarelli C, Sica AR, Peluso AL, Luciano L, et al. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood. 2011;118(13):3634–44. [DOI] [PubMed] [Google Scholar]
- 155.Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. 2011;117(24):6660–8. [DOI] [PubMed] [Google Scholar]
- 156.Thomson DW, Shahrin NH, Wang PPS, Wadham C, Shanmuganathan N, Scott HS, et al. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2020;34(8):2051–63. [DOI] [PubMed] [Google Scholar]
- 157.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103(5):1635–40. [DOI] [PubMed] [Google Scholar]
- 158.Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23(17):3948–56. [DOI] [PubMed] [Google Scholar]
- 159.Abaza Y, Kantarjian H, Alwash Y, Borthakur G, Champlin R, Kadia T, et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am J Hematol. 2020;95(11):1288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ruggiu M, Oberkampf F, Ghez D, Cony-Makhoul P, Beckeriche F, Cano I, et al. Azacytidine in combination with tyrosine kinase inhibitors induced durable responses in patients with advanced phase chronic myelogenous leukemia. Leuk Lymphoma. 2018;59(7):1659–65. [DOI] [PubMed] [Google Scholar]
- 161.Larson RA, Hochhaus A, Hughes TP, Clark RE, Etienne G, Kim DW, et al. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012;26(10):2197–203. [DOI] [PubMed] [Google Scholar]
- 162.Montani D, Bergot E, Gunther S, Savale L, Bergeron A, Bourdin A, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–37. [DOI] [PubMed] [Google Scholar]
- 163.Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boque C, et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients Trial. J Clin Oncol. 2016;34(20):2333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. The Lancet Oncology. 2016;17(5):612–21. [DOI] [PubMed] [Google Scholar]
- 165.Clark RE, Polydoros F, Apperley JF, Milojkovic D, Rothwell K, Pocock C, et al. De-escalation of tyrosine kinase inhibitor therapy before complete treatment discontinuation in patients with chronic myeloid leukaemia (DESTINY): a non-randomised, phase 2 trial. Lancet Haematol. 2019;6(7):e375–e83. [DOI] [PubMed] [Google Scholar]
- 166.Naqvi K, Jabbour E, Skinner J, Anderson K, Dellasala S, Yilmaz M, et al. Long-term follow-up of lower dose dasatinib (50 mg daily) as frontline therapy in newly diagnosed chronic-phase chronic myeloid leukemia. Cancer. 2020;126(1):67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Naqvi K, Jabbour E, Skinner J, Yilmaz M, Ferrajoli A, Bose P, et al. Early results of lower dose dasatinib (50 mg daily) as frontline therapy for newly diagnosed chronic-phase chronic myeloid leukemia. Cancer. 2018;124(13):2740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Burchert A, Wang Y, Cai D, von Bubnoff N, Paschka P, Muller-Brusselbach S, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–82. [DOI] [PubMed] [Google Scholar]
- 169.Hjorth-Hansen H, Stentoft J, Richter J, Koskenvesa P, Hoglund M, Dreimane A, et al. Safety and efficacy of the combination of pegylated interferon-alpha2b and dasatinib in newly diagnosed chronic-phase chronic myeloid leukemia patients. Leukemia. 2016;30(9):1853–60. [DOI] [PubMed] [Google Scholar]
- 170.Nicolini FE, Etienne G, Dubruille V, Roy L, Huguet F, Legros L, et al. Nilotinib and peginterferon alfa-2a for newly diagnosed chronic-phase chronic myeloid leukaemia (NiloPeg): a multicentre, non-randomised, open-label phase 2 study. Lancet Haematol. 2015;2(1):e37–46. [DOI] [PubMed] [Google Scholar]
- 171.Burchert A, Saussele S, Eigendorff E, Muller MC, Sohlbach K, Inselmann S, et al. Interferon alpha 2 maintenance therapy may enable high rates of treatment discontinuation in chronic myeloid leukemia. Leukemia. 2015;29(6):1331–5. [DOI] [PubMed] [Google Scholar]
- 172.Horne GA, Stobo J, Kelly C, Mukhopadhyay A, Latif AL, Dixon-Hughes J, et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia. 2020;34(7):1775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sweet K, Hazlehurst L, Sahakian E, Powers J, Nodzon L, Kayali F, et al. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk Res. 2018;74:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhao H, Deininger MW. Declaration of Bcr-Abl1 independence. Leukemia. 2020;34(11):2827–36. [DOI] [PubMed] [Google Scholar]
- 175.Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102(8):1368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hughes A, Clarson J, White DL, Ross DM, Hughes TP, Yong AS. Enhanced Natural Killer and Cytotoxic T Lymphocyte Responses, with Decreased Monocytic Myeloid Derived Suppressor Cells May Promote Treatment Free Remission in Chronic Myeloid Leukaemia Patients Following Tyrosine Kinase Inhibitor Cessation. Blood. 2016;128(22):1122-. [Google Scholar]
- 177.Carter BZ, Mak PY, Mu H, Wang X, Tao W, Mak DH, et al. Combined inhibition of MDM2 and BCR-ABL1 tyrosine kinase targets chronic myeloid leukemia stem/progenitor cells in a murine model. Haematologica. 2020;105(5):1274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6(252):252ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]