

Abstract
Background
Oncolytic adenoviruses are promising new treatments against solid tumors, particularly for glioblastoma (GBM), and preclinical models are required to evaluate the mechanisms of efficacy. However, due to the species selectivity of adenovirus, there is currently no single animal model that supports viral replication, tumor oncolysis, and a virus-mediated immune response. To address this gap, we took advantage of the Syrian hamster to develop the first intracranial glioma model that is both adenovirus replication-permissive and immunocompetent.
Methods
We generated hamster glioma stem-like cells (hamGSCs) by transforming hamster neural stem cells with hTERT, simian virus 40 large T antigen, and h-RasV12. Using a guide-screw system, we generated an intracranial tumor model in the hamster. The efficacy of the oncolytic adenovirus Delta-24-RGD was assessed by survival studies, and tumor-infiltrating lymphocytes (TILs) were evaluated by flow cytometry.
Results
In vitro, hamGSCs supported viral replication and were susceptible to Delta-24-RGD mediated cell death. In vivo, hamGSCs consistently developed into highly proliferative tumors resembling high-grade glioma. Flow cytometric analysis of hamster gliomas revealed significantly increased T-cell infiltration in Delta-24-RGD infected tumors, indicative of immune activation. Treating tumor-bearing hamsters with Delta-24-RGD led to significantly increased survival compared to hamsters treated with phosphate buffered saline (PBS).
Conclusions
This adenovirus-permissive, immunocompetent hamster glioma model overcomes the limitations of previous model systems and provides a novel platform to study the interactions between tumor cells, the host immune system, and oncolytic adenoviral therapy; understanding of which will be critical to implementing oncolytic adenovirus in the clinic.
Keywords: glioma, immune-competent model, oncolytic adenovirus
Key Points.
First immunocompetent glioma model to support oncolytic adenovirus replication.
Delta-24-RGD induces tumor infiltration by T cells and prolongs survival.
Dexamethasone may dampen the response to oncolytic adenovirus.
Importance of the Study.
Clinical trials evaluating oncolytic adenoviruses in GBM patients have shown early promising results; however, critical questions remain surrounding the relative contributions of tumor cell oncolysis and antiviral and antitumor immune responses in the efficacy of these therapeutic strategies. Preclinical models provide a system to systematically evaluate these aspects that may have critical implications for patient stratification on clinical trials. Unfortunately, to date, there is not a single small animal model that is both immune-competent and adenovirus replication-permissive in which to model GBM. Here we describe the first intracranial, immune-competent, adenovirus-permissive animal model of glioma and demonstrate the preclinical value in using such a model to interrogate oncolytic adenovirus mechanisms of efficacy.
Oncolytic viruses are increasingly gaining traction as a therapeutic strategy against solid tumors, including glioblastoma (GBM), which has not benefitted from new or improved treatments in the past three decades. Several groups have reported favorable results in clinical trials using oncolytic viruses against GBM.1–3 Similarly, in a Phase I clinical trial evaluating Delta-24-RGD (DNX-2401, tasadenoturev), a tumor selective oncolytic adenovirus,4 20% of recurrent GBM patients receiving the virus experienced long-term survival of more than three years.5 Patients receiving Delta-24-RGD showed increased evidence of T cells in the tumors and alterations in the expression of immune checkpoint molecules.5 The results of this clinical trial demonstrated that Delta-24-RGD acts against tumor cells not only by direct oncolysis of tumor cells, but also by activating an antitumor immune response, implicating oncolytic adenovirus as a type of immunotherapy.6 Understanding the interplay between the direct oncolytic effects of adenoviruses and the virus-mediated antitumor immune response is critical for maximizing the efficacy of these promising agents. Additionally, the radiation and chemotherapy used as standard of care for GBM patients and the corticosteroids commonly administered to reduce symptoms associated with edema each may affect the immune response and therefore, should be evaluated in the context of oncolytic adenoviral therapy.
To date, animal models to evaluate oncolytic adenovirus efficacy against GBM have relied upon intracranial xenograft models, in which human glioma cell lines or human glioma stem-like cells (GSCs) are implanted into immunocompromised mice. These studies clearly demonstrated the ability of Delta-24-RGD to extend the survival of tumor-bearing mice due to direct tumor cell oncolysis4,7; however, the contribution of the host immune system could not be evaluated. To address this issue, Jiang and colleagues evaluated the efficacy of Delta-24-RGD in an immune-competent murine syngeneic model of glioma. Consistent with the observations from the clinical trial, Delta-24-RGD treatment of mouse GL261 glioma tumors promoted T-cell tumor infiltration.8 Further, Delta-24-RGD treatment enhanced antigen presentation of a tumor-specific antigen, induced T-cell activation against both the virus and the tumor, and significantly increased survival, indicating that the oncolytic adenovirus activity includes the activation of an antitumor immune response.8 Although this immune-competent model provided valuable evidence in support of Delta-24-RGD activating an antitumor immune response, adenoviruses replicate 1000-fold lower in mouse cells than in human cells,9,10 limiting the oncolytic effects of the virus and the contributions of oncolysis to direct tumor cell killing and activation of the immune system. In order to fully evaluate the efficacy and mechanisms of oncolytic adenoviruses against GBM, an animal model that is both immune-competent and permissive to adenovirus replication is required.
We hypothesized that an immunocompetent, adenovirus replication-permissive GBM model would address the gap in knowledge surrounding the mechanisms behind the efficacy of oncolytic adenoviral therapeutics and would allow more relevant preclinical strategies for translation to patients. The Syrian (Golden) hamster is one of only two small mammals that support efficient adenovirus replication and has been used as a model to evaluate antiviral immune responses.10–13 Here we describe the creation and characterization of a syngeneic, intracranial glioma model in the immune-competent Syrian hamster. We developed hamster glioma stem-like cells (hamGSCs) and determined that Delta-24-RGD efficiently infects, replicates in, and kills hamGSCs in vitro. Delta-24-RGD treatment of tumor-bearing hamsters increased tumor infiltration of immune cells and significantly prolonged survival. This model provides further support for the dual mechanisms of oncolytic adenovirus efficacy and provides an improved model in which to study such therapies. To this end, we found that dexamethasone treatment attenuated the tumor-associated immune response and impaired Delta-24-RGD efficacy. This model will be invaluable for further studies measuring the efficacy and mechanisms of oncolytic virotherapy, particularly in the context of clinical management strategies that may affect patient immune functions.
Methods
Cell Lines and Viruses
Delta-24-RGD4 was amplified and titered as previously described.8 UV-inactivated virus was generated by subjecting the virus to 7 cycles of 125 mJ UV light in a GS Gene Linker UV Chamber (BioRad).
Lung carcinoma A549 cells (ATCC, CCL-185), 293T/17 (ATCC, CRL-11268), U-251MG (RRID: CVCL_0021), and murine GL261 (RRID: CVCL_Y003) cells were cultured in DMEM/F12 with 10% fetal bovine serum (FBS). Hamster neural stem cells (hamNSCs) were isolated from the cortex of 3–4 day old Syrian hamsters (Mesocricetus auratus, HsdHan®:AURA, Envigo) and cultured under neurosphere conditions: DMEM/F12 supplemented with B27 (ThermoFisher), EGF (20 ng/mL), FGF (20 ng/mL), and 100 µg/mL PenStrep. HamGSCs were isolated from tumors arising after intracranial implantation of transformed hamNSCs and cultured under neurosphere conditions. HamGSC1-4 was generated by serial dilution of hamGSC-1. Subclones were established by expanding spheres that formed in wells containing a single cell. All cells were maintained in 37°C, 5% CO2 incubator. All cell lines were tested mycoplasma-free. All cells were used for experiments within 10 passages after thawing, and hamGSCs were implanted for in vivo experiments within 4 passages after thawing.
Western Blot
HamNSCs and hamGSCs were lysed in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors and separated on a 4–20% Tris-Glycine gel (Invitrogen). Antibodies: Pan Ras (Invitrogen, MA1-012X, RRID: AB_2536664), hTERT (Abcam ab32020, cloneY182), Large T-antigen (Santa Cruz Biotechnology sc-147, clone Pab101), and α-Tubulin (Invitrogen, #32–2500, RRID: AB_2533071).
Animal Experiments
One thousand five hundred (hamGSC-1 or hamGSC-2) or 1.25 × 104 (hamGSC1-4) hamGSCs were implanted into the right hemisphere of 6–8 week old male Syrian hamsters (Harlan) using a guide screw placed 3.5 mm lateral and 1 mm anterior to the bregma.14 Animals were randomly assigned to treatment groups and Delta-24-RGD (1 × 108–5 × 108 pfu in 5 µL) or an equal volume of phosphate buffered saline (PBS) was injected intratumorally through the guide screw. Cells and virus were injected through the guide screw to a depth of 5 mm using a 26-guage Hamilton syringe and a microinfusion syringe pump (Harvard Apparatus). Animals were monitored daily and euthanized when moribund, except for time point experiments noted in the text. Dexamethasone-water soluble (Sigma D2915) or Dexamethasone injection 2 mg/mL (Patterson Veterinary, #07-808-8194) was administered daily at a dose of 10 mg/kg. Hematology was performed by the MD Anderson Cancer Center Department of Veterinary Medicine and Surgery Clinical Pathology core on whole blood collected by cardiac puncture using K2EDTA as an anticoagulant with the Advia 2120i automated hematology analyzer (Siemens). Athymic nude mice (nu/nu) were obtained from the Department of Experimental Radiation Oncology, University of Texas MD Anderson Cancer Center (Houston, TX). The guide screw was placed 2 mm lateral and 1 mm anterior to the bregma, and hamGSCs were implanted to a depth of 3 mm. All animal experiments were performed according to animal protocols approved by the Institutional Animal Care and Use Committee at MD Anderson Cancer Center. Power analysis was performed using α = 0.05, 90% power to select sample size for survival studies.
Electron Microscopy
Cells were infected with 10 MOI (U251 cells) or 100 MOI (GL261 and hamGSCs) Delta-24-RGD or UV-inactivated virus. After 48 and 72 hours of infection, cells were fixed with 2% paraformaldehyde and 3% glutaraldehyde and imaged with JOEL JSM-1010 transmission electron microscope.
Viral Replication and In Vitro Infection
To assess viral replication, 1 × 105 cells were infected with Delta-24-RGD-GFP at 10 MOI and cultured for 48 hours. Cells were subjected to three freeze-thaw cycles to release virus. Virus-containing supernatant was titered by end-point dilution with an antibody against viral hexon protein (Millipore AB1056). To determine IC50, 5 × 104 target cells were infected with serial dilutions of Delta-24-RGD. Cell viability was assessed on day 6 by Trypan-blue exclusion.
Immunohistochemistry
Brains were fixed in formalin and paraffin-embedded. Following Citrate antigen retrieval, sections were stained with Hexon (Millipore AB1056) and detected with 3,3’Diaminobenzidine.
Detection of Anti-adenovirus Antibodies in Serum
The Adenovirus ELISA assay was adapted from Crosby et al.15 Briefly, ELISA plates were coated with 1 × 107 pfu UV-inactivated adenovirus per well. Plates were blocked in 0.25% BSA, 0.05% Tween-20 in PBS and incubated with hamster serum (1:400 in blocking buffer). Antibodies were detected with goat anti-syrian hamster IgG-HRP (Santa Cruz, sc-2493) using substrate and stop solutions (R&D Systems) according to manufacturer’s instructions. Absorbance was read at 450 nm using 540 nm wavelength correction. Neutralizing antibody luciferase assay was adapted from Dhar et al.16 Briefly, 293T/17 cells were plated at 2 × 103 cells/well in opaque 96 well plates. Serum samples were incubated at 56°C for 30 minutes to inactivate complement and diluted in media in a round-bottom 96 well plate. 1 × 104 pfu of Ad-CMV-Luciferase (Vector Biolabs) was added to each well and incubated for 1 hour at 37°C. 293T/17 cells were infected with the serum/Ad-CMV-Luciferase mixture for 1 hour. Cells were washed and incubated for 24 hours before measuring luciferase activity by Dual-Glo Luciferase Assay (Promega) using a ClarioStar Luminometer.
Preparation of Splenocytes and Brain-infiltrating Lymphocytes
Hamster spleens were mechanically dissociated through a 100 µm cell strainer, then filtered through a 40 µm cell strainer. Cells were treated with Red Blood Cell Lysing Buffer Hybri-Max (Sigma), washed with PBS, and resuspended in flow cytometry staining buffer (FACS buffer) (PBS + 2 mM EDTA + 2% FBS) or splenocyte media (RPMI-1640 + 10% FBS + 50 µM β-mercaptoethanol). To isolate brain-infiltrating lymphocytes, tumor-bearing brain hemispheres were minced and mechanically dissociated through a 100 µm cell strainer, followed by filtering through a 40 µm cell strainer. Lymphocytes were separated from myelin debris using a Percoll gradient as described in Pino et al.17 Cells were washed with Hanks’ balanced salt solution (HBSS) and resuspended in FACS buffer.
Flow Cytometry
5 × 106 cells were blocked with FACS buffer + 2% mouse serum + 2% rat serum. Cells were then incubated with antibodies for 30 minutes. The antibodies were: Rat-anti-mouse CD4 clone GK1.5 eFlour 450 (eBioscience #48-0041-82, RRID:AB_10718983) 1:250, Mouse-anti-rat CD8b clone eBio341 PE (eBioscience #12-0080-82, RRID:AB_657776) 1:150, Mouse-anti-hamster PanT clone HAB2A Alexa-647 (Washington State University Monoclonal Antibody Core) 1:250, Mouse-anti-hamster CD45 clone HASA25A Alexa-488 (Washington State University Monoclonal Antibody Core) 1:500, and LIVE/DEAD Fixable Aqua-Dead Cell Stain Kit (405nM) (Thermo-Fisher #L34965). Stained cells were analyzed by flow cytometry using the BD LSRFortessa X-20 and data analyzed using BD FACSDiva 8.0.1 (Becton Dickinson) and FlowJo V10.
Results
Hamster GSCs Produce Tumors In Vivo
In order to take full advantage of the immune-competent and adenovirus replication-permissive nature of the Syrian hamster, we developed a syngeneic hamster glioma model. We isolated hamster neural stem cells and transformed them by simultaneously transducing the cells with retroviruses containing three oncogenes – hTERT, SV40 large T antigen (Tag), and H-RasV12. This strategy transforms rodent and human cells18 and targets major pathways commonly altered in GBM: the PI3K/MAPK pathway via H-RasV12, the TP53 and Rb pathways via Tag,19 and the telomerase pathway.20 The transformed cells were cultured under NSC conditions and in selective antibiotics to isolate NSCs expressing all three oncogenes, referred to as hamNSC-TTR cells. Using our established guide-screw system,14 we implanted hamNSC-TTR cells into hamster brains, where they developed into tumors. We collected tumors from two separate hamsters and isolated GSCs (referred to as hamGSCs) to establish two cell lines: hamGSC-1 and hamGSC-2 (Figure 1A). HamGSCs expressed the transduced oncogenes hTERT, Tag, and H-RasV12, as determined by Western blot (Figure 1B).
Fig. 1.

Hamster glioma stem-like cells GSCs (hamGSCs) produce tumors in vivo. (A) Generation of hamGSC lines. (B) Western blot of hamster neural stem cells (hamNSCs) and hamGSCs for transduced oncogenes. (C) Survival curve of hamsters implanted with increasing number of hamGSC-2 cells. (D) H&E of representative tumors arising from hamGSC-1 or hamGSC-2. Scale bars = 1 mm (left panels) and 50 µm (right panels).
GSCs can be thought of as glioma-initiating cells and therefore, can induce tumors even at low cell numbers.21 We evaluated hamGSC-1 and hamGSC-2 to determine an optimal cell number that would result in a high tumor penetrance, yet allow hamsters to survive long enough for treatment and survival analysis. When we implanted as few as 1500 GSCs, hamsters succumbed to tumors within 3–4 weeks (Figure 1C). In contrast, establishing GL261 murine brain tumors at a similar rate in immune-competent mice typically requires implanting 5 × 104 GL261 cells.22 Both hamGSC-1 and hamGSC-2 produced tumors when implanted into the brains of hamsters (Figure 1D). The tumors demonstrated hypercellularity and regions of necrosis, consistent with features of human gliomas. Injection of 2 × 106 nontransformed hamNSCs did not induce tumors in the hamster brain [not shown].
Hamster GSCs Are Susceptible to Delta-24-RGD Infection
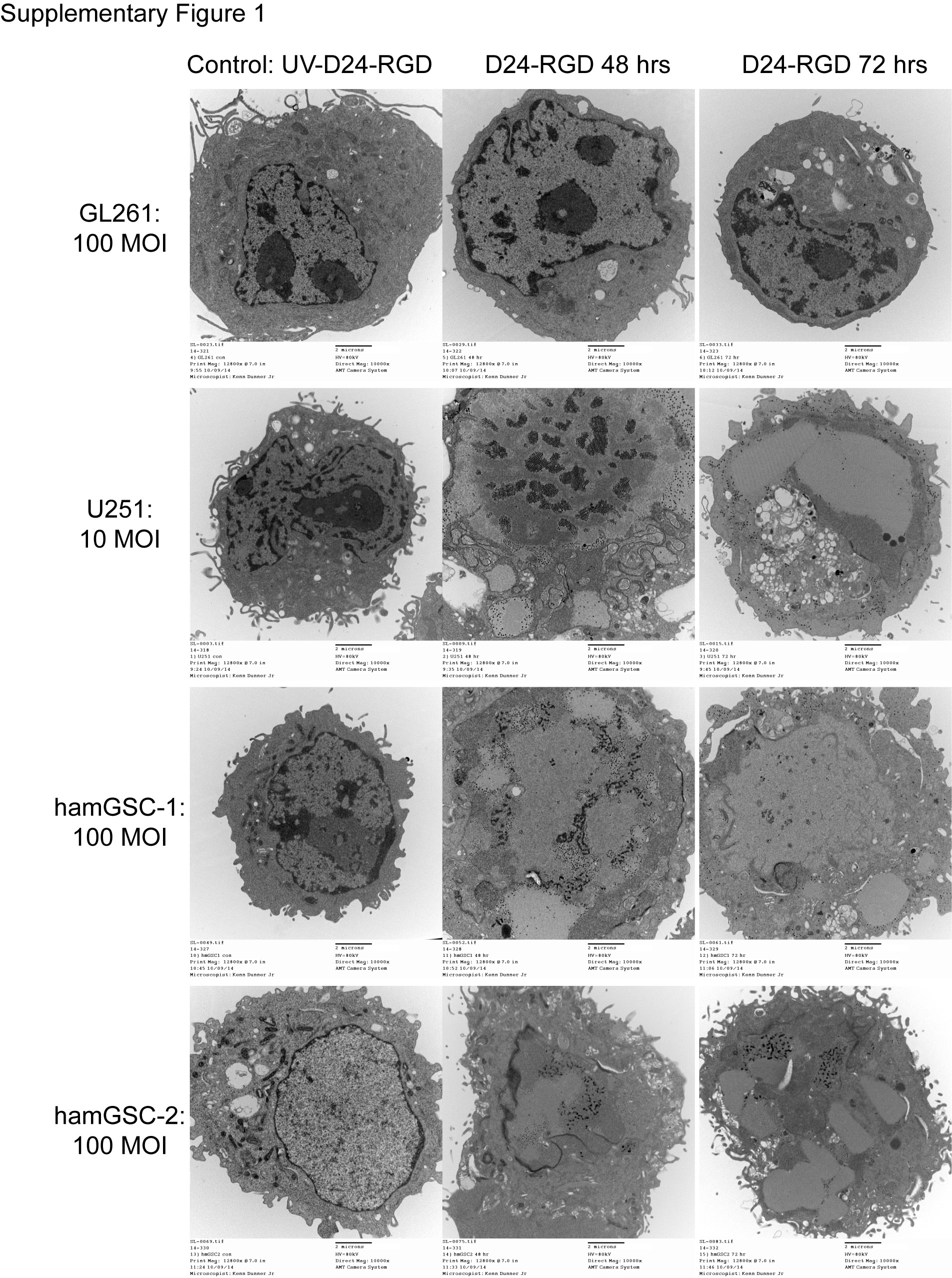
Previous studies have shown that, in contrast to mouse cells, hamster cells are permissive for adenovirus replication.23 The 24-basepair deletion within the E1A region of Delta-24-RGD further restricts replication to tumor cells with a disrupted Rb pathway.4 To verify the ability of Delta-24-RGD to replicate in hamGSCs, we measured the viral titer in virus-infected hamGSCs 48 hours following infection. Delta-24-RGD replicated efficiently in hamGSCs, although viral titers in hamGSCs were one log lower than in human glioma cells, whereas viral titers in mouse GL261 glioma cells did not increase beyond the input virus (Figure 2A). We next examined infected cells by electron microscopy. At 48 hours postinfection, viral particles were detected in hamGSC-1 and hamGSC-2 at varying levels. Viral particles were not identified in mock-infected hamGSCs or in mouse GL261 cells (Figure 2B). Cell morphology changes were also noted in hamGSCs and U251, but not GL261 cells, by 72 hours postinfection (Supplementary Figure S1). To evaluate the ability of Delta-24-RGD to kill hamGSCs, we infected hamGSC-1 and hamGSC-2 with a series of viral dilutions and evaluated cell viability after 6 days. Delta-24-RGD effectively killed infected hamGSCs with IC50 of 23 MOI for hamGSC-1 and 43 MOI for hamGSC-2 (Figure 2C). The IC50 for Delta-24-RGD in human glioma cells ranges from 0.5 to 20 MOI,7 and we observed an IC50 of 0.7 MOI for the U251 human glioma cell line. In contrast, GL261 is relatively resistant to Delta-24-RGD infection and killing in vitro, with an IC50 of 120 MOI, and requires infections as high as 100–200 MOI to elicit cell death.8,22 To evaluate Delta-24-RGD viral infection and replication in vivo, we implanted 1500 hamGSC-2 cells into hamster brains and delivered Delta-24-RGD intratumorally via the guide screw. We observed extensive expression of viral hexon protein within the tumor 5 days after virus treatment (Figure 2D).
Fig. 2.

Hamster GSCs are susceptible to Delta-24-RGD infection. (A) Viral replication in human, hamster, and mouse cells. 48 hours after infection with Delta-24-RGD (10 MOI), cells were lysed and the supernatant titered on A549 cells. Dashed line indicates input viral particles (1 × 106 pfu/105 cells). Data are shown as mean ± SEM, *adjusted P <.05, and ** adjusted P <.005. Dunnett’s test was used to compare each of the experimental arms with the control, adjusting for multiple comparisons. (B) Transmission electron microscopy 48 hours after virus infection demonstrates visible viral particles in human glioma cells (10 MOI) and hamster GSCs (100 MOI), but not in the mouse GL261 cell line (100 MOI). Scale bar = 500 nm. (C) IC50 assays demonstrate that Delta-24-RGD kills human glioma cell lines and hamGSCs in a dose-dependent manner. (D) Representative H&E and immunohistochemistry for viral hexon protein in hamGSC-2 tumors 5 days after Delta-24-RGD treatment. Scale bars = 1 mm (1.25× objective), 200 µm (4× objective), and 100 µm (10× objective).
Delta-24-RGD Prolongs Survival in Tumor-bearing Hamsters
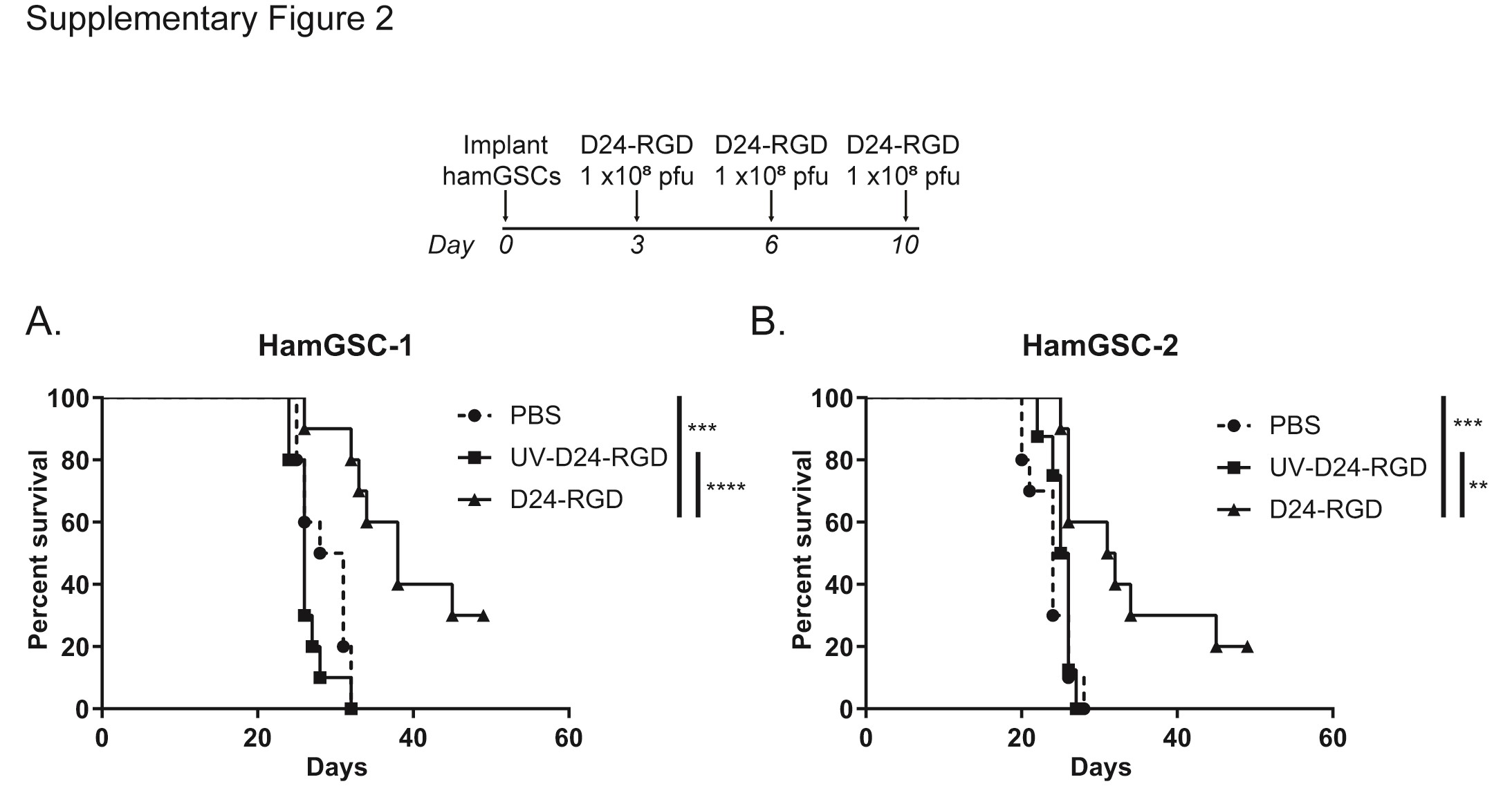
To determine the ability of Delta-24-RGD to eradicate tumor cells in vivo, we implanted hamGSC-1 and hamGSC-2 into the brains of nude mice and treated them with Delta-24-RGD using a dosing strategy previously evaluated in nude mice implanted with human glioma cells. Consistent with our observations in human glioma cells,4,7 Delta-24-RGD treatment significantly prolonged survival (Supplementary Figure S2) in immunocompromised mice. We next implanted 1500 hamGSC-1 or hamGSC-2 cells into the brains of immunocompetent hamsters. We administered 5 × 108 pfu of Delta-24-RGD on days 3 and 7 following tumor cell implantation. As controls, we treated hamsters with PBS or UV-inactivated Delta-24-RGD. Delta-24-RGD treatment significantly increased the median survival to 40 days compared with 22 days in PBS and 24 days in UV-inactivated virus treated controls (GSC-1: P = .032, GSC-2:P = .0005 log-rank test of D24-RGD vs UV-D24-RGD) (Figure 3A and B).
Fig. 3.

Delta-24-RGD prolongs survival in tumor-bearing hamsters. Kaplan–Meier survival curves for hamGSC-1 (n = 9 per group) (A) and hamGSC-2 (PBS n = 10, D24-RGD and UV-D24-RGD n = 11) (B). 1500 hamGSCs were implanted on day 0 and animals were treated with PBS, UV-inactivated virus, or 5 × 108 pfu Delta-24-RGD on days 3 and 7. *P < .05, ***P < .005 log-rank test.
Immune-competent Hamsters Develop an Immune Response Against Delta-24-RGD
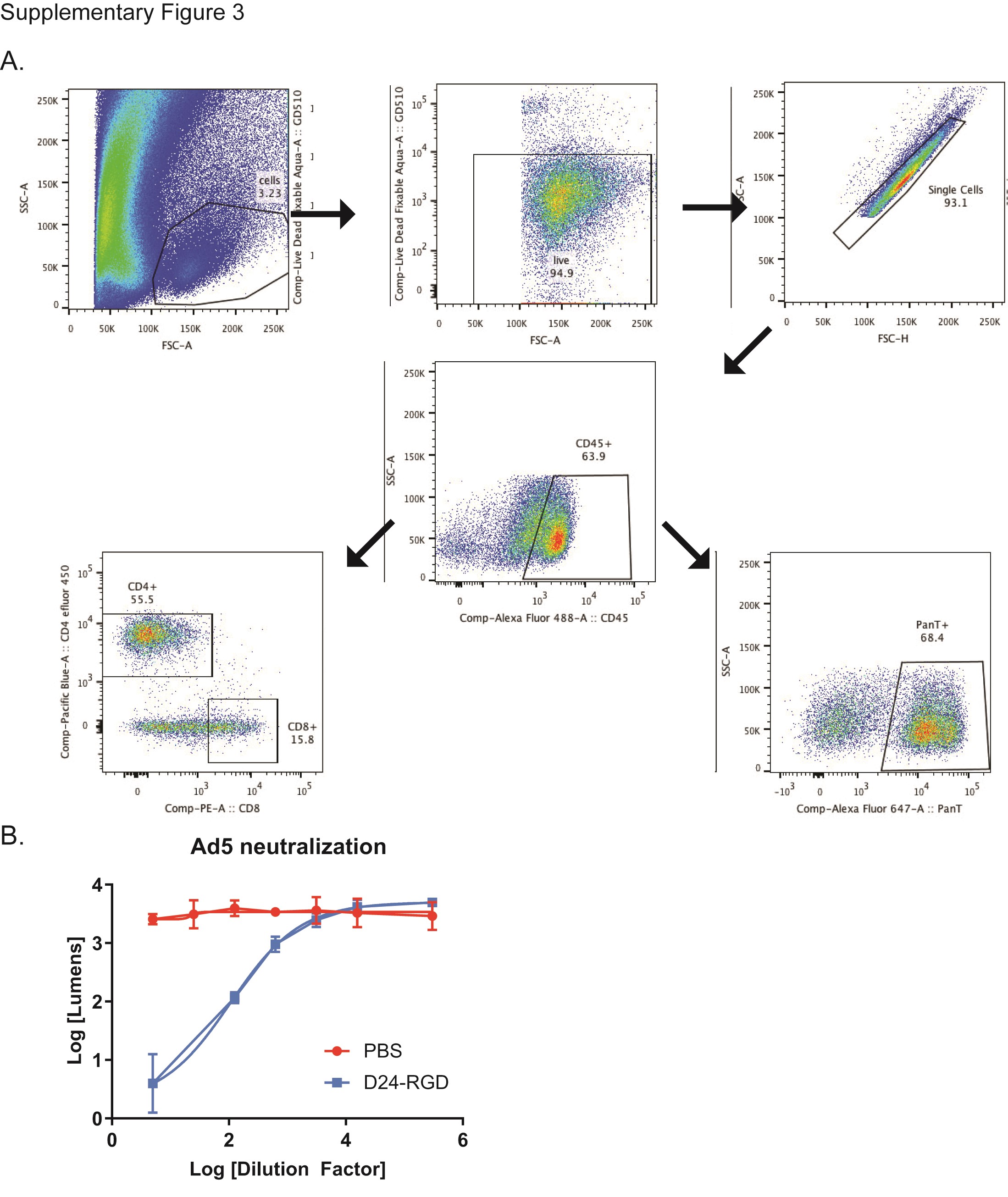
Having established an immune-competent hamster glioma model and demonstrated the efficacy of the Delta-24-RGD oncolytic adenovirus in this model, we sought to evaluate the hamster immune response to virus treatment. We evaluated tumor-infiltrating lymphocytes (TILs) in hamGSC-2 tumors by flow cytometry 1- and 2-weeks following treatment with PBS, UV-inactivated virus, or Delta-24-RGD. Using commercially available antibodies against rat CD8 and mouse CD4, which have been previously demonstrated to cross-react with hamster CD8 and CD4, in combination with a hamster PanT antibody and an antibody against hamster CD45,24 we analyzed T cell populations as a percentage of CD45+ cells within the GSC-2 tumors (Supplementary Figure S3A). One week after Delta-24-RGD treatment, there was a significant increase in CD4+ and CD8+ T cells within the tumors as compared with tumors treated with PBS or UV-inactivated virus (Figure 4A). At 2 weeks following treatment, the percentage of CD45+ cells expressing T cell markers was increased in all groups, with the Delta-24-RGD treated tumors containing the highest numbers of T cells, with significantly elevated CD8+ T cells (Figure 4B). These results collectively demonstrate that Delta-24-RGD treatment activates the hamster immune system and recruits T cells to the tumor microenvironment.
Fig. 4.

Immune-competent hamsters develop an immune response against Delta-24-RGD and hamGSC tumors. (A, B) Flow cytometric analysis of brain-infiltrating lymphocytes (n = 3 hamsters per group) 1 week (A) and 2 weeks (B) after Delta-24-RGD treatment. Data are shown as mean ± SEM, * adjusted P < .05, ** adjusted P < .005, t tests corrected for multiple comparisons using the Holm–Sidak method and α = 0.05. (C) ELISA demonstrating anti-adenovirus antibodies in hamster serum collected 10 days after Delta-24-RGD treatment.
To further evaluate the immune response to Delta-24-RGD administration, we assessed the serum of tumor-bearing hamsters treated with PBS or Delta-24-RGD for the presence of neutralizing IgG antibodies against adenovirus by ELISA. Anti-adenovirus antibodies were consistently detected in the serum of Delta-24-RGD treated hamsters 10 days after treatment, but not in PBS treated animals (Figure 4C). Anti-adenovirus antibodies were neutralizing, as they inhibited adenoviral infection of target cells (Supplementary Figure S3B).
Dexamethasone Suppresses Antiviral Immune Response and Impairs Delta-24-RGD Efficacy
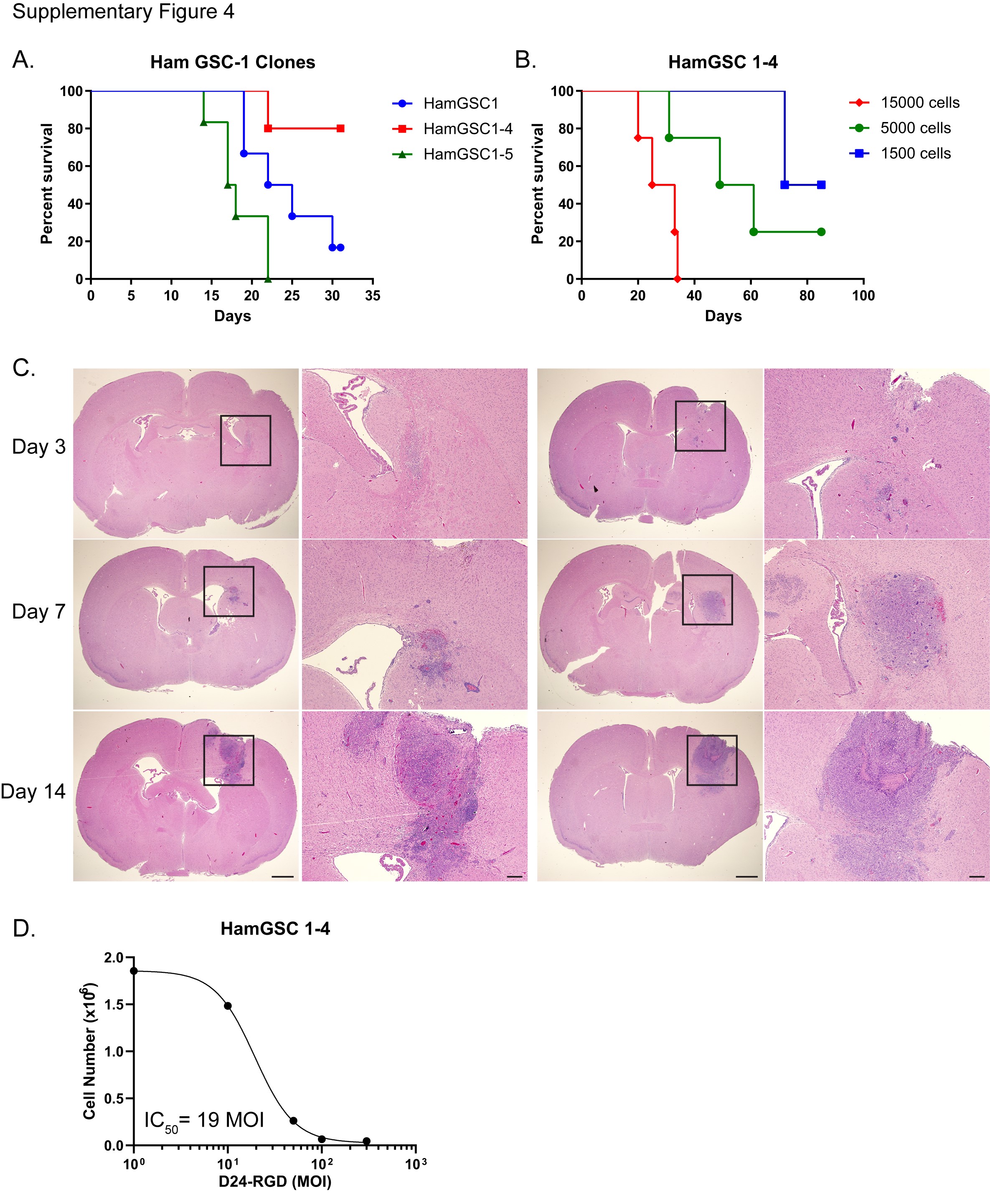
We reasoned that implanting a larger cell number of a slower-growing hamGSC line would ensure the presence of tumor at the time of Delta-24-RGD treatment and produce a median survival of sufficient length to allow the hamster immune response to develop in response to treatment. The hamGSC-1 subclone, hamGSC1-4, produced a longer survival compared with the parental hamGSC-1 (Supplementary Figure S4A). The median survival of hamGSC1-4 after implanting 15 000 cells (10 times the number implanted for hamGSC-1) was 29 days (Supplementary Figure S4B). Tumors were visible in the hamster brain within 3 days of implanting 10 000 hamGSC1-4 cells (Supplementary Figure S4C) and the IC50 after Delta-24-RGD treatment was 19 MOI, similar to the parental line (23 MOI) (Supplementary Figure S4D, Figure 2C).
To demonstrate how this model might be used to study the effects of immune-modulating factors on the efficacy of oncolytic viruses, we evaluated the effects of the commonly administered corticosteroid, dexamethasone, on the hamster immune response and Delta-24-RGD efficacy. We hypothesized that if Delta-24-RGD efficacy is partly mediated by the host immune response, the immune-suppressive effects of corticosteroids would impair Delta-24-RGD efficacy in our model. For this study, we implanted 12,500 hamGSC1-4 cells. As observed in patients receiving dexamethasone to relieve symptoms associated with edema, dexamethasone was associated with significant lymphopenia (Figure 5A). This was accompanied by an increase in neutrophil to lymphocyte ratio (NLR) (Figure 5B and C), a phenomenon also observed in patients treated with dexamethasone.25,26 Consistent with the studies above using hamGSC-2, in the absence of dexamethasone, we observed significant increases in TILs upon Delta-24-RGD treatment compared with PBS. Dexamethasone treatment significantly impaired the infiltration of CD4+ and CD8+ T cells to the tumor (Figure 5D). The TILs in tumors from hamsters treated with Delta-24-RGD plus dexamethasone were not significantly different from those of hamsters treated with PBS.
Fig.5.

Dexamethasone suppresses antiviral immune response and impairs Delta-24-RGD efficacy. (A–C) Hematology analysis of hamster blood following a 2-week course of dexamethasone treatment indicating changes in lymphocytes (A), neutrophils (B), and neutrophil to lymphocyte ratio (NLR) (C) in response to dexamethasone treatment. Data are shown as mean ± SD and evaluated with two-tailed, two-sample t tests. (D) Tumor-infiltrating lymphocytes (TILs) from hamsters treated with PBS or Delta-24-RGD with and without dexamethasone treatment (PBS and PBS + Dex: N = 5, Delta-24-RGD and Delta-24-RGD + Dex: N = 6). TILs were isolated on day 19 (following a 2-week course of dexamethasone and 14 days after the first virus injection) and analyzed by flow cytometry. Data are shown as mean ± SEM, ** adjusted P < .005, *** adjusted P < .0005, two-tailed, two sample t tests corrected for multiple comparisons using the Holm–Sidak method and α = 0.05. (E) Survival plot of GSC 1–4 tumor-bearing hamsters treated with Delta-24-RGD in the presence or absence of dexamethasone (N = 10 per group). **P-.004, log rank test. (F–H) Levels of anti-adenoviral antibodies in serum from the animals in the survival study (E) on day 19; 24 h following the final dexamethasone dose for those animals receiving a 2-week course (F), on day 70; surviving animals evaluated after completion of the 10-week course of dexamethasone (G), and at time of death or at the end of the survival study (day 120) (H). Colored symbols indicate animals euthanized due to tumor development on or before day 30 (red), between day 31 and day 55 (pink), on day 65–66 (blue), and long-term survivors ≥120 days (black). Data are shown as mean ± SD and evaluated with two-tailed, two-sample t tests corrected for multiple comparisons using the Holm–Sidak method and α = 0.05. ** adjusted P < .05, *** adjusted P < .005, **** adjusted P < .001.
As steroid treatment courses vary in duration, we then evaluated the effects of a short-term (2-week) and long-term (10-week) course of dexamethasone on survival. Delta-24-RGD treatment of hamGSC1-4 bearing hamsters induced long-term survival in 70% of hamsters and significantly improved survival compared with PBS treated animals (P = .004, log-rank test) (Figure 5E). Despite the long-term (10-week) course of steroids, the addition of dexamethasone did not significantly reduce survival compared with virus alone. However the long-term survivors in the Delta-24-RGD + Dex (2 weeks) and Delta-24-RGD + Dex (10 weeks) groups were reduced to 50% (median survival 87 days) and 40% (median survival 50.5 days), respectively, as compared with Delta-24-RGD alone (70% long-term survival). We also quantified the serum levels of anti-adenovirus antibodies in the animals from this survival study. Anti-adenovirus antibodies could be detected in the serum of Delta-24-RGD treated hamsters by day 19 (14 days after the first dose of Delta-24-RGD), and even at this early time point, dexamethasone treatment significantly impaired the detection of anti-adenovirus antibodies (Figure 5F). The decreased level of neutralizing antibodies was even more pronounced when we evaluated the surviving animals at the end of the 10-week course of dexamethasone and at the end of the study (day 70, Figure 5G and H). These findings suggest that dexamethasone impairs the efficacy of Delta-24-RGD, at least in part by inducing systemic lymphopenia, which also results in an impaired antiviral antibody response and decreased TILs.
Discussion
We have developed a novel animal model that addresses several historical shortcomings to studying oncolytic adenovirus in animal models. We describe the first intracranial glioma animal model that is both immune competent and that supports adenoviral replication. Specifically, we used this model to demonstrate that Delta-24-RGD activated the host immune response, which led to increased survival in tumor-bearing hamsters. Finally, we showed the utility of this model in answering key clinical questions by evaluating the effects of a frequently administered corticosteroid on the efficacy of Delta-24-RGD.
Existing mouse models have demonstrated direct oncolytic effects of Delta-24-RGD on human glioma cell lines and patient-derived GSCs.4,7 These early studies provided a foundation for clinical implementation of Delta-24-RGD by demonstrating tumor specificity, viral replication, safety, and efficacy in an immunocompromised mouse model. Similarly, we demonstrated that Delta-24-RGD treatment prolongs survival in nude mice implanted with hamGSCs. However, the contributions of an intact host immune system could not be evaluated. In a syngeneic immunocompetent mouse model, Delta-24-RGD prolonged survival and induced an immune response against both the tumor and the virus.8 Importantly, these effects increased when the oncolytic virus was modified to encode for a T-cell costimulation molecule, OX40L.22 Together, these studies indicate that infecting tumor cells with Delta-24-RGD causes an initial phase of local inflammation, oncolysis, and the release of DAMPs and tumor antigens, which leads to the recruitment of immune cells and activation of the innate immune system against both viral and tumor antigens. Although Delta-24-RGD can infect and kill mouse cells, this occurs at a much lower rate than in human cells. Therefore, the syngeneic immunocompetent mouse models, which require high doses of virus yet do not result in high intratumoral viral load, may underestimate the contribution of viral replication and oncolysis to Delta-24-RGD efficacy.
The Syrian hamster has been used as a model for studying viral infections and oncolytic adenovirus activity against renal and pancreatic cancers.13This study demonstrated the ability of human adenovirus type 5 to infect and replicate in hamster cells, inhibit tumor growth, and suppress metastasis.13 In a hamster model of subcutaneous renal cancer, the broad-spectrum immunosuppressive agent, cyclophosphamide, cooperated with the oncolytic adenovirus to suppress tumor growth better than with oncolytic virus alone.27,28 In these studies, cyclophosphamide abrogated the production of adenovirus neutralizing antibodies and allowed persistent replication of the oncolytic virus within the tumor, suggesting that the primary antitumor effects of the virus were due to oncolysis and that a functional immune system primarily targets the virus. In contrast, antibody-mediated T cell depletion impaired the efficacy of an oncolytic adenovirus against subcutaneous hamster pancreatic tumors.29 In this case, the authors also demonstrated that treatment with oncolytic virus induced cytotoxic T cells directed against both viral and tumor antigens, and that this adaptive immune response played an important role in the antitumor efficacy of the oncolytic virus.29 These studies demonstrate the utility of the Syrian hamster as an immunocompetent and adenovirus replication-permissive model; however, the contribution of the immune system in mediating the efficacy of oncolytic adenovirus remains unclear and may have different effects depending on the specific tumor type, tumor location, and virus studied.
To address this gap, we took advantage of the immunocompetent, replication-permissive hamster model and our expertise with GSCs to develop the first intracranial, orthotopic GBM model in the hamster. This model has two additional advantages in that it more accurately represents the brain specific immune microenvironment and it uses GSCs that can be implanted as low numbers of tumor-initiating cells.
Corticosteroids are commonly administered to counteract symptoms associated with edema; however, the effects of corticosteroids on treatment response in the context of adenoviral therapy remain elusive. Corticosteroid dose and duration vary considerably among patients and are often not well reported in clinical studies, making it difficult to evaluate how such treatments might affect tumor growth and respond to standard therapies. Therefore, we utilized our novel hamster model to evaluate the immunosuppressive effects of the corticosteroid, dexamethasone, on the efficacy of Delta-24-RGD. When administered concurrent with oncolytic adenovirus, dexamethasone significantly reduced peripheral blood lymphocytes in the treated hamsters and impaired the production of neutralizing antibodies against Delta-24-RGD. Although dexamethasone did not significantly impair the survival of Delta-24-RGD treated hamster, the number of long-term survivors was reduced. Consistent with a study of Delta-24-RGD in the immunocompetent, but nonpermissive GL261 model,30 we observed a significant reduction in the TILs in Delta-24-RGD treated tumors from hamsters receiving dexamethasone. Recent studies have called into question the appropriateness of steroid use for GBM patients.31 Although further experiments will be required to fully evaluate the effects of corticosteroids on Delta-24-RGD efficacy, in our model, dexamethasone dampened virus-mediated host immune activation in response to Delta-24-RGD, suggesting that such immunosuppressive cotherapies be carefully considered prior to use in patients receiving oncolytic virotherapy.
We demonstrated that Delta-24-RGD treatment of hamGSC tumors prolonged survival of both immunocompromised mice and immunocompetent hamsters. However, evaluating the contribution of the immune system based on direct comparison of the two species is complicated by differences in animal size, virus treatment schedule and dose, and other factors. Ultimately, understanding the mechanisms of oncolytic adenovirus efficacy will include determining the contributions and balance of several actions: direct tumor cell killing by viral oncolysis, immune-mediated tumor cell killing due to viral infection of tumor cells, immune-mediated viral clearance vs viral spread, and immune-mediated antitumor immunity. By generating syngeneic hamGSCs that support adenovirus replication, we have developed the first model that will allow evaluation of these factors in the context of intracranial tumors. Additionally, we demonstrate how this model might be used to evaluate the effects of other, coadministered treatments that may affect host immune function and therefore, oncolytic adenovirus efficacy. With this model, future studies should advance our understanding of the role of direct oncolysis in driving immune-mediated viral clearance vs driving an antiglioma immune response, understanding of which will be critical to implementing oncolytic adenovirus therapy in the clinic.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Select data from this manuscript were presented at the Society for Neuro-Oncology Annual Meeting, November 2020. This study was supported by grants from the National Cancer Institute and by generous philanthropic contributions to The University of Texas MD Anderson Cancer Center and to F.F.L.
Conflict of interest statement. C.G.-M. and J.F. report ownership interest (including patents) in and are consultants for DNAtrix, Inc. F.F.L. is patent holder for Delta-24-RGD.
Authorship statement. Experimental design, L.M.P., S.L., C.G.-M., J.F., and F.F.L.; Implementation, L.M.P., S.L., J.G., M.D., D.L., J.Y., and A.H.; Data analysis and interpretation, L.M.P., S.L., J.G., M.D., S.S., B.P.K., A.H., Y.Y., C.G.-M., J.F., and F.F.L.; Writing and revision of manuscript, all authors; Approval of manuscript, all authors.
Funding
This study was supported by the University of Texas MD Anderson Cancer Center Moon Shots Program™, the Broach Foundation for Brain Cancer Research, the Elias Family Fund, the Jim & Pam Harris Fund, the Chuanwei Lu Fund, the Bauman/CUREFEST Fund, the Sweet Family Fund, and the Ira Schneider Fund and National Institutes of Health/National Cancer Institute CA247970 and SPORE in Brain Cancer P50CA127001. Electron microscopy and flow cytometry were performed by the High-Resolution Electron Microscopy Facility and the Flow Cytometry Facility supported by the NIH/NCI Cancer Center Support Grant P30CA016672.
References
- 1. Desjardins A, Gromeier M, Herndon JE 2nd, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018; 379(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cloughesy TF, Landolfi J, Vogelbaum MA, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018; 20(10):1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wheeler LA, Manzanera AG, Bell SD, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016; 18(8):1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fueyo J, Alemany R, Gomez-Manzano C, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003; 95(9):652–660. [DOI] [PubMed] [Google Scholar]
- 5. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018; 36(14):1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forsyth PA, Abate-Daga D. Oncolytic Virotherapy for Malignant Gliomas. J Clin Oncol. 2018; 36(14):1440–1442. [DOI] [PubMed] [Google Scholar]
- 7. Jiang H, Gomez-Manzano C, Aoki H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007; 99(18):1410–1414. [DOI] [PubMed] [Google Scholar]
- 8. Jiang H, Clise-Dwyer K, Ruisaard KE, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014; 9(5):e97407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blair GE, Dixon SC, Griffiths SA, Zajdel ME. Restricted replication of human adenovirus type 5 in mouse cell lines. Virus Res. 1989; 14(4):339–346. [DOI] [PubMed] [Google Scholar]
- 10. Ying B, Toth K, Spencer JF, et al. INGN 007, an oncolytic adenovirus vector, replicates in Syrian hamsters but not mice: comparison of biodistribution studies. Cancer Gene Ther. 2009; 16(8):625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prescott J, Falzarano D, Feldmann H. Natural Immunity to Ebola Virus in the Syrian Hamster Requires Antibody Responses. J Infect Dis. 2015; 212Suppl 2:S271–S276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prescott J, Safronetz D, Haddock E, Robertson S, Scott D, Feldmann H. The adaptive immune response does not influence hantavirus disease or persistence in the Syrian hamster. Immunology. 2013; 140(2):168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas MA, Spencer JF, La Regina MC, et al. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res. 2006; 66(3):1270–1276. [DOI] [PubMed] [Google Scholar]
- 14. Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000; 92(2):326–333. [DOI] [PubMed] [Google Scholar]
- 15. Crosby CM, Nehete P, Sastry KJ, Barry MA. Amplified and persistent immune responses generated by single-cycle replicating adenovirus vaccines. J Virol. 2015; 89(1):669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhar D, Spencer JF, Toth K, Wold WS. Pre-existing immunity and passive immunity to adenovirus 5 prevents toxicity caused by an oncolytic adenovirus vector in the Syrian hamster model. Mol Ther. 2009; 17(10):1724–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pino PA, Cardona AE. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J Vis Exp. 2011; (48):2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005; 25(15):6464–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network . The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013; 110(15):6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004; 432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 22. Jiang H, Rivera-Molina Y, Gomez-Manzano C, et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017; 77(14):3894–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wold WS, Toth K. Chapter three–Syrian hamster as an animal model to study oncolytic adenoviruses and to evaluate the efficacy of antiviral compounds. Adv Cancer Res. 2012; 115:69–92. [DOI] [PubMed] [Google Scholar]
- 24. Rees J, Haig D, Mack V, Davis WC. Characterisation of monoclonal antibodies specific for hamster leukocyte differentiation molecules. Vet Immunol Immunopathol. 2017; 183:40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chitadze G, Flüh C, Quabius ES, et al. In-depth immunophenotyping of patients with glioblastoma multiforme: Impact of steroid treatment. Oncoimmunology. 2017; 6(11):e1358839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mason M, Maurice C, McNamara MG, et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J Neurooncol. 2017; 132(3):463–471. [DOI] [PubMed] [Google Scholar]
- 27. Dhar D, Toth K, Wold WS. Cycles of transient high-dose cyclophosphamide administration and intratumoral oncolytic adenovirus vector injection for long-term tumor suppression in Syrian hamsters. Cancer Gene Ther. 2014; 21(4):171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas MA, Spencer JF, Toth K, Sagartz JE, Phillips NJ, Wold WS. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther. 2008; 16(10):1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Wang P, Li H, et al. The Efficacy of Oncolytic Adenovirus Is Mediated by T-cell Responses against Virus and Tumor in Syrian Hamster Model. Clin Cancer Res. 2017; 23(1):239–249. [DOI] [PubMed] [Google Scholar]
- 30. Kleijn A, Kloezeman J, Treffers-Westerlaken E, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS One. 2014; 9(5):e97495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pitter KL, Tamagno I, Alikhanyan K, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016; 139(Pt 5):1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.