

Abstract
Background
Pituitary neuroendocrine tumors (PitNETs) are the second most common intracranial tumor. We lacked a comprehensive understanding of the pathogenesis and heterogeneity of these tumors.
Methods
We performed high-precision single-cell RNA sequencing for 2679 individual cells obtained from 23 surgically resected samples of the major subtypes of PitNETs from 21 patients. We also performed single-cell multi-omics sequencing for 238 cells from 5 patients.
Results
Unsupervised clustering analysis distinguished all tumor subtypes, which was in accordance with the classification based on immunohistochemistry and provided additional information. We identified 3 normal endocrine cell types: somatotrophs, lactotrophs, and gonadotrophs. Comparisons of tumor and matched normal cells showed that differentially expressed genes of gonadotroph tumors were predominantly downregulated, while those of somatotroph and lactotroph tumors were mainly upregulated. We identified novel tumor-related genes, such as AMIGO2, ZFP36, BTG1, and DLG5. Tumors expressing multiple hormone genes showed little transcriptomic heterogeneity. Furthermore, single-cell multi-omics analysis demonstrated that the tumor had a relatively uniform pattern of genome with slight heterogeneity in copy number variations.
Conclusions
Our single-cell transcriptome and single-cell multi-omics analyses provide novel insights into the characteristics and heterogeneity of these complex neoplasms for the identification of biomarkers and therapeutic targets.
Keywords: intra-tumoral heterogeneity, pituitary neuroendocrine tumors, single-cell sequencing
Key Points.
Tumor-related genes in 3 types of PitNETs were identified for the first time.
Intra-tumoral transcriptomic heterogeneity of PitNETs was comprehensively examined.
Slight but clear intra-tumoral genomic heterogeneity of PitNETs was revealed.
Importance of the Study.
PitNETs are the second most common intracranial tumor. However, the clinical classification and diagnosis of these tumors are still not optimized. This is the first single-cell sequencing study and should provide a more comprehensive understanding of these complex neoplasms, serving as an invaluable resource for the field. One notable analysis was the comparison of tumor and matched normal endocrine cells, which has not previously been achieved. We identified novel tumor-related genes (eg, AMIGO2, ZFP36, BTG1, and DLG5), which will prompt further investigations to identify novel therapeutic targets. Additionally, intra-tumoral heterogeneity in the transcriptome and genome provides novel insights into the cellular origins and pathogenesis of PitNETs.
Pituitary neuroendocrine tumors (PitNETs, also named pituitary adenomas) are the second most common intracranial tumor and occur in approximately 1 in 1000 individuals.1,2 The classification of PitNETs includes defining the cell type by characterizing hormone genes and cell lineage-specific transcription factors (TFs), which includes the PIT-1 (also known as POU1F1) lineage (somatotroph, lactotroph, and thyrotroph), T-PIT (also known as TBX19) lineage (corticotroph), SF-1 (also known as NR5A1) lineage (gonadotroph), and null-cell and plurihormonal tumors.3–6 The PIT-1 lineage and corticotroph tumors often secrete excess amounts of hormones, which lead to hyperprolactinemia (prolactin-secreting lactotroph tumors), acromegaly (growth hormone-secreting somatotroph tumors), hyperthyroidism (thyrotropin-secreting thyrotroph tumors), and Cushing’s disease (corticotropin-secreting corticotroph tumors). Gonadotroph tumors are typically nonsecretory and cause local mass effects or hypogonadism. Plurihormonal tumors express multiple hormones, and null-cell tumors express none of the hormone genes or lineage-specific TFs. The cellular origin of these tumors has not been completely clarified.
The majority of PitNETs are sporadic. Early studies have established that these tumors are generally monoclonal in origin by X-inactivation analysis.7 Genetic studies have identified 2 main recurrent somatic mutations: one affecting the GNAS gene in 40%-60% of somatotroph tumors and the other affecting the USP8 gene in 40%-60% of corticotroph tumors.8–10 However, for approximately 60% of tumors, no recurrent somatic mutations can be found.11 Copy number variations (CNVs) are common in PitNETs, but a mechanistic link between CNVs and tumorigenesis has not been established. PitNETs are thought to usually be indolent due to their low proliferation rate and because they rarely metastasize. However, the pathogenesis of a large proportion of PitNETs remains unclear.
Single-cell sequencing technologies have emerged as powerful tools for comprehensively understanding genetic and functional heterogeneity at a single-cell resolution.12,13 For intracranial tumors, single-cell RNA sequencing (scRNA-seq) has been applied for malignant cancers, including gliomas and medulloblastoma.14,15 Here, we performed high-precision scRNA-seq to dissect the transcriptomes of 23 PitNETs that included most subtypes and characterized inter-tumoral and intra-tumoral transcriptomic heterogeneity. We identified normal pituitary cell types and compared tumor and matched normal pituitary endocrine cells, and investigated genomic heterogeneity with single-cell multi-omics sequencing.
Materials and Methods
Ethics Approval and Consent to Participate
This study was approved by the Institutional Review Boards of Beijing Tiantan Hospital, Capital Medical University (KY2017-049-03). All patients were enrolled after providing informed consent.
Sample Collection
We performed scRNA-seq for fresh specimens obtained from surgeries of 21 PitNET patients (patient information in Supplementary Table S1). For 2 patients (P1 and P2), we have analyzed samples for both the primary surgery and the secondary surgery for complete dissection of the tumor, making a total of 23 samples. Five of 23 samples were subjected to single-cell multi-omics sequencing; 9 of 23 samples to whole-genome sequencing (WGS); 2 of 23 samples (P4 and P6) to bulk RNA sequencing (RNA-seq). We also performed bulk RNA-seq for independent 18 tumor specimens (PB1-PB18) which were frozen in liquid nitrogen (patient information in Supplementary Table S1). Tumor invasion was assessed according to the Knosp classification based on magnetic resonance imaging (MRI) before surgery.16 PitNETs were clinically classified according to the World Health Organization (WHO) 2017 Classification based on the immunohistochemistry (IHC) results of 6 hormone genes and 3 lineage-specific TFs (PIT-1, SF-1, and T-PIT).3
Library Construction for scRNA-seq and Single-Cell Multi-Omics Sequencing
Fresh surgical specimens were cut into pieces and digested into single cells by collagenase type II and type IV (Gibco) at 37°C with shaking for 15-30 minutes.
For single-cell transcriptome library construction, we used the single-cell tagged reverse transcription (STRT)-seq method that we previously described.17 For single-cell multi-omics sequencing, the cell nucleus was captured by Dynabeads MyOne Streptavidin C1 beads, and mRNAs were released in lysis buffer, and then DNA and RNA libraries were separately constructed.18,19 In brief, a single cell was put into a 2.5 µl lysis buffer with Streptavidin C1 beads. After cell lysis, the tube was placed on a magnetic stand, and the supernatant which contained the released mRNAs was transferred to another tube containing 1 µl barcoded primer at a concentration of 10 µM. Then, DNA cell lysis buffer was added to the tube, and the tube was incubated in a PCR thermal cycler to release genomic DNA. Genomic DNA library construction methods were performed using multiple annealing and looping-based amplification cycle (MALBAC) with modifications.20 After pre-amplification, products with different cell barcodes were pooled and processed similarly to scRNA-seq.
The libraries were purified with AMPure XP beads and quantified by the Qubit High-Sensitivity DNA kit (Invitrogen). Then, the purified libraries were sequenced on an Illumina HiSeq 4000 platform with paired-end 150 base reads.
Bulk RNA Sequencing and WGS
Specimens frozen in liquid nitrogen were ground to a homogenate with pestles. Then, genomic DNA and/or total RNA were extracted using the DNA/RNA mini kits (QIAGEN) and quantified by the Qubit High-Sensitivity DNA kit (Invitrogen). 100 ng DNA was ultrasonically sheared by a Covaris S2 before library construction with the Kapa Hyper Prep Kit (Kapa Biosystems). 2 µg total RNA was used to construct a library with the RNA-seq kit (New England Biolabs).
Immunostaining Assays
Fresh samples were fixed and dehydrated and embedded in paraffin. Approximately 10-µm paraffin sections were prepared. After deparaffinization, antigen retrieval, and blocking, the sections were stained with anti-PIT-1 antibody (Santa Cruz) and anti-collagen III antibody (Abcam). The secondary antibody was marked with the Opal 7-Color Manual IHC Kit (NEL811001KT, AKOYA). Finally, the section images were captured by the Vectra Polaris multispectral imaging system (PerkinElmer).
scRNA-seq Reads Processing
First, we split cells by cell barcodes in Reads2, and added the barcode and unique molecular identifiers (UMIs) to Reads1. Next, we removed sequences of the template switch oligo (TSO) and the ploy-A tail and trimmed low-quality bases to obtain clean reads from Reads1. Then, we aligned the clean reads to the hg19 human genome reference (downloaded from UCSC) with TopHat (version 2.0.12).21 For uniquely mapped reads, quantification was counted with HTSeq (version 0.6.0),22 and duplicates were removed based on UMIs to obtain the single-cell gene expression count matrix. In the subsequent quality control step, we removed low-quality cells with less than 2000 positive gene counts and the genes with positive counts in less than three cells.
Single-Cell Expression Matrix Dimension Reduction and Identification of the Differentially Expressed Genes (DEGs)
The count expression matrix was generated by Seurat2 (version 2.3.3),23 in which feature gene selection, principal component analysis (PCA), and t-distributed stochastic neighbor embedding (t-SNE) dimension reduction were performed. To identify DEGs, we used the FindMarkers function in the Seurat2 package and the Wilcoxon test. Genes with adjusted P values less than .05 were considered significant DEGs.
Analysis of Intra-Tumoral Heterogeneity and Cell Cycle Phase Projection
For the tumor cells from each patient, Seurat2 was used for cell clustering by using the genes of the top 5 principal components (PCs), and gene ontology (GO) analysis (ToppGene: https://toppgene.cchmc.org) was performed on the DEGs to identify significantly enriched functions with false discovery rate (FDR) less than 0.05. A cell cluster was regarded with biological function only if it enriched upregulated DEGs with significant GO terms.
To make a 2-dimensional single-cell projection of the cell cycle, we calculated the average expression of cell cycle marker genes in G2/M and S phases.12
CNV Analysis by Single-Cell MALBAC (scMALBAC) and Bulk WGS
For the scMALBAC raw data, we first assigned paired-end reads to single cells based on barcodes and trimmed low-quality bases and adapters by seqtk (https://github.com/lh3/seqtk). Then, FASTQ files with clean reads were aligned to the hg19 human genome reference with Burrows-Wheeler Aligner (BWA) (version 0.7.17).24 Subsequent alignment quality control (mapQ > 30) and duplicates deletion were conducted with SAMtools (version 1.9).25 Cells with more than 100 000 mapped read pairs were considered qualified with sufficient sequencing depth. Finally, the qualified BAM files of cells were used to conduct CNV analysis.
To obtain a low-noise single-cell CNV profile, we used Control-FREEC26 CNV analysis, which normalized the copy ratio by considering the GC content. Running FREEC without control set, we first got 10 euploid cells and made them as a noise control. Subsequent analysis exploited this control set to eliminate copy ratio estimation noise. We used the default parameters except that the bin size was set to 1 Mb. Bins possessing an NA copy ratio caused by insufficient read counts in cells were also removed. Thus, we generated a single-cell copy ratio matrix. Finally, Pheatmap (version 1.0.12) was used to visualize the matrix as a single-cell CNV heatmap.
FREEC was also applied for calling CNVs in bulk WGS, except that GC-content normalization was performed without a control set. We eliminated samples with less than 1 000 000 mapped reads, annotated and visualized the CNV fragments, and finally plotted the copy ratio scatters along the chromosome axis.
Data Availability
Raw sequencing data in this study are available in the Genome Sequence Archive (GSA) of number PRJCA002946.
Results
scRNA-seq Analysis of PitNETs
To generate a transcriptome map of PitNETs, we used a modified single-cell STRT-seq method to analyze a total of 2926 cells obtained from surgically resected tumor tissue samples from 21 patients, including somatotroph tumors (n = 4), lactotroph tumors (n = 1), thyrotroph tumors (n = 1), silent corticotroph tumors (n = 4), gonadotroph tumors (n = 8), PIT-1-positive plurihormonal tumors (n = 2), and 1 tumor with unusual immunohistochemical combinations (PAwUIC, n = 1) (Figure 1A, B; Supplementary Table S1). For 2 patients (P1 and P2), samples were obtained from both the primary surgery and the secondary surgery; therefore, there were a total of 23 samples for scRNA-seq. After quality controls, we retained 2679 (91.6%) high-quality cells, with a median of 5616 genes detected in an individual cell. We also performed single-cell multi-omics (genome and transcriptome) sequencing of 409 cells for 5 patients (P17, P18, P19, P20, and P21) and bulk WGS for 9 patients (Figure 1B).
Fig. 1.

Workflow and clinical information for single-cell sequencing analysis of PitNETs. (A) The workflow for the scRNA-seq and single-cell multi-omics sequencing analysis of PitNETs. (B) Clinical details of the patients, who are ordered by tumor type. Somato, somatotroph tumors; Lacto, lactotroph tumors; Thyro, thyrotroph tumors; PIT-1+Pluri, plurihormonal PIT-1-positive tumors; Cortico, corticotroph tumors; Gonado, gonadotroph tumors; PAwUIC, pituitary adenoma with unusual immunohistochemical combinations. * indicates the patient had 2 surgical samples. (C) Heatmap showing clinical immunohistochemical results and scRNA-seq results with the scaled average expression of hormone genes and transcription factors (TFs). Samples are arranged in the same order as Figure 1B. Abbreviations: PitNETs, pituitary neuroendocrine tumors; scRNA-seq, single-cell RNA sequencing.
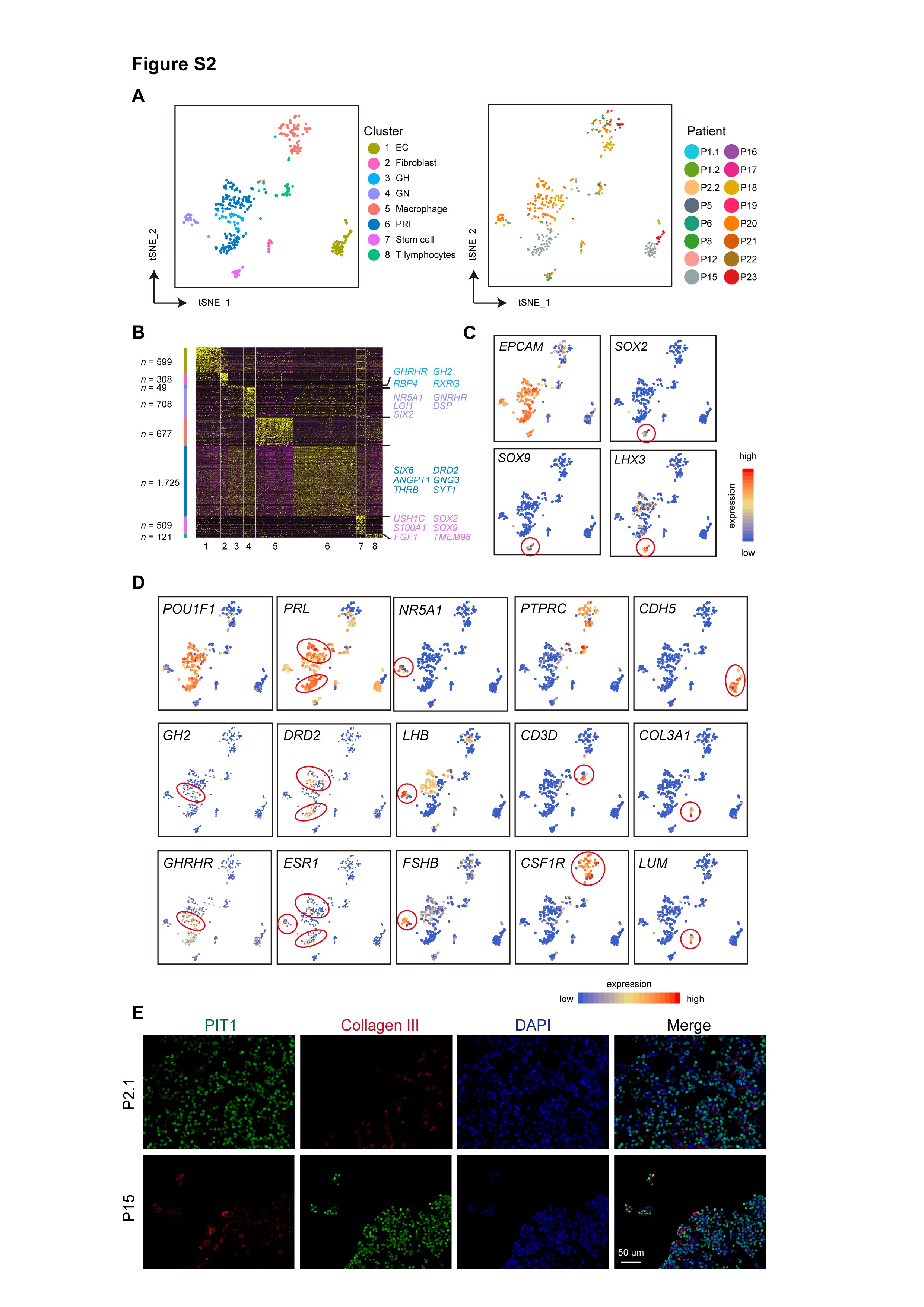
We distinguished tumor cells from non-tumoral cells by combining 2 complementary approaches. In the first approach, we inferred CNVs from scRNA-seq data with InferCNV methods and distinguished cells with and without CNVs27 (Supplementary Figure S1A, B). In the second approach, different cell types were identified by unsupervised clustering (Supplementary Figure S1C). Finally, we identified 2311 tumor cells and 368 normal cells. The normal cell types included lactotroph cells (n = 125, marked by PIT-1, PRL, ESR1, and D2R), somatotroph cells (n = 30, marked by PIT-1, GH2, and GHRHR), gonadotroph cells (n = 24, marked by SF-1, GATA2, ESR1, and LHB), and pituitary stem cells (n = 17, marked by SOX2, SOX9, and LHX3) (Supplementary Figure S2A–D). We also identified endothelial cells (n = 49, marked by CDH5), fibroblasts (n = 14, marked by LUM and COL3A1), macrophages (n = 74, marked by PTPRC and CSF1R), and T lymphocytes (n = 35, marked by PTPRC and CD3D) (Supplementary Figure S2D). Immunostaining results showed that although the majority of cells were tumor cells, there were many non-tumor cells, including fibroblasts in tumor tissues, emphasizing the need for single-cell analysis (Supplementary Figure S2E). It is noteworthy that we used the mouth pipette for picking individual cells. We found that the tumor cell was generally larger than the normal cell, so this contributed to enrichment of the tumor cell, and result in the potential reduction of the proportion of the non-tumor cellular composition.
Thus, we performed scRNA-seq for PitNETs that included most subtypes and captured major normal pituitary cell types. The acquisition of normal cells provides us with an excellent opportunity to compare differences with corresponding tumor cells.
Classification of PitNETs by scRNA-seq
To investigate global transcriptional interrelationships, we performed PCA on tumor cells. The results distinguished 3 major cell groups corresponding to 3 adenohypophyseal cell lineages6 (Figure 2A, B). The first group was the PIT-1 lineage of 10 PIT-1-positive tumors. The second group was the corticotroph lineage of 4 T-PIT-positive tumors, and the third group was 8 gonadotroph tumors and 1 tumor with the unusual coexpression of both T-PIT and SF-1 (Figure 2B). Unsupervised clustering analysis separated the tumor cells into the same 3 major groups. The results further showed that the cells from 4 somatotroph tumors (P2, P5, P15, and P16) clustered together (Figure 2C). We also performed bulk RNA-seq to 20 samples, 18 of which were independent specimens frozen in liquid nitrogen (PB1-PB18), and 2 were from the patients (P4 and P6) subjected to scRNA-seq. The PCA results were consistent with the scRNA-seq results (Figure 2D). We also identified DEGs from these 3 PitNET lineages by scRNA-seq and bulk RNA-seq (Figure 2E; Supplementary Table S2).
Fig. 2.

Clustering of PitNETs by scRNA-seq. (A) PCA plot showing the principal component 1 (PC1) vs PC2 + PC3 projection of all tumor cells. (B) PCA plots showing the expression of 4 lineage-specific TFs. (C) Unsupervised clustering of all tumor cells and the lineage information of PitNETs. (D) PCA plot showing the PC1 vs PC2 projection of tumors at the bulk RNA-seq level. (E) Heatmap showing the expression of DEGs in 3 adenohypophyseal cell lineages of 20 samples performed bulk RNA-seq (left), and 23 samples performed scRNA-seq, which are arranged in the same order as Figure 1B (right). (F) Violin plots showing the expression of SOX2 and SOX9 in tumor and normal cells, including pituitary stem cells. Samples are arranged in the same order as Figure 1B. Somato, somatotrophs; Lacto, lactotrophs; Gonado, gonadotrophs; Stem, stem cells; Fibro, fibroblasts; EC, endothelial cells; IMM, immune cells. Abbreviations: DEGs, differentially expressed genes; PCA, principal component analysis; PitNETs, pituitary neuroendocrine tumors; scRNA-seq, single-cell RNA sequencing; TFs, transcription factors.
The tumor from P14 was clinically diagnosed as a “null-cell” tumor with negative IHC results for all 6 hormones; scRNA-seq showed that even SF-1 was not expressed in this tumor (Figure 1C). PCA and clustering analysis clearly showed that the tumor cells were of the gonadotroph origin (Figure 2A). The examination of other TFs showed that GATA2, GATA3, and ISL1 were expressed in the tumor cells from P14 (Figure 1C). These results supported the use of a set of TFs for the diagnosis of a “null-cell” PitNET. Interestingly, the PAwUIC tumor (P11) showed the unusual coexpression of both T-PIT and SF-1, and the cells were positioned between the corticotroph and gonadotroph lineages, suggesting that they represented an intermediate cell state.6 In addition, these cells were clustered with the gonadotroph lineage, suggesting that they were of the gonadotroph origin.
We examined the expression of a set of lineage and stem cell-specific TFs (Figures 1C and 2F). PIT-1, T-PIT, and SF-1 were the most specific markers for the classification of tumors; other genes, such as ESR1, GATA2, and GATA3, also added useful information. Stem cell-specific SOX2 and SOX9 were expressed in a minor fraction of cells in some tumors (Figure 2F). SOX9 was prominently expressed in the tumor expressing both T-PIT and SF-1 (P11), reflecting the transcriptional dysregulation of the tumor (Figure 2F).
Together, the results showed that scRNA-seq classification is in accordance with the classification of PitNETs based on IHC. At the same time, it provides more information for understanding the cellular origin and the molecular characteristics of tumors.
Identification of Tumor-Related Genes by Comparing Tumor and Matched Normal Cells
We captured 3 normal endocrine cell types: somatotrophs, lactotrophs, and gonadotrophs. This provided an excellent chance for identifying tumor-related genes with high accuracy that was not previously possible due to the lack of transcriptome information on normal endocrine cells.
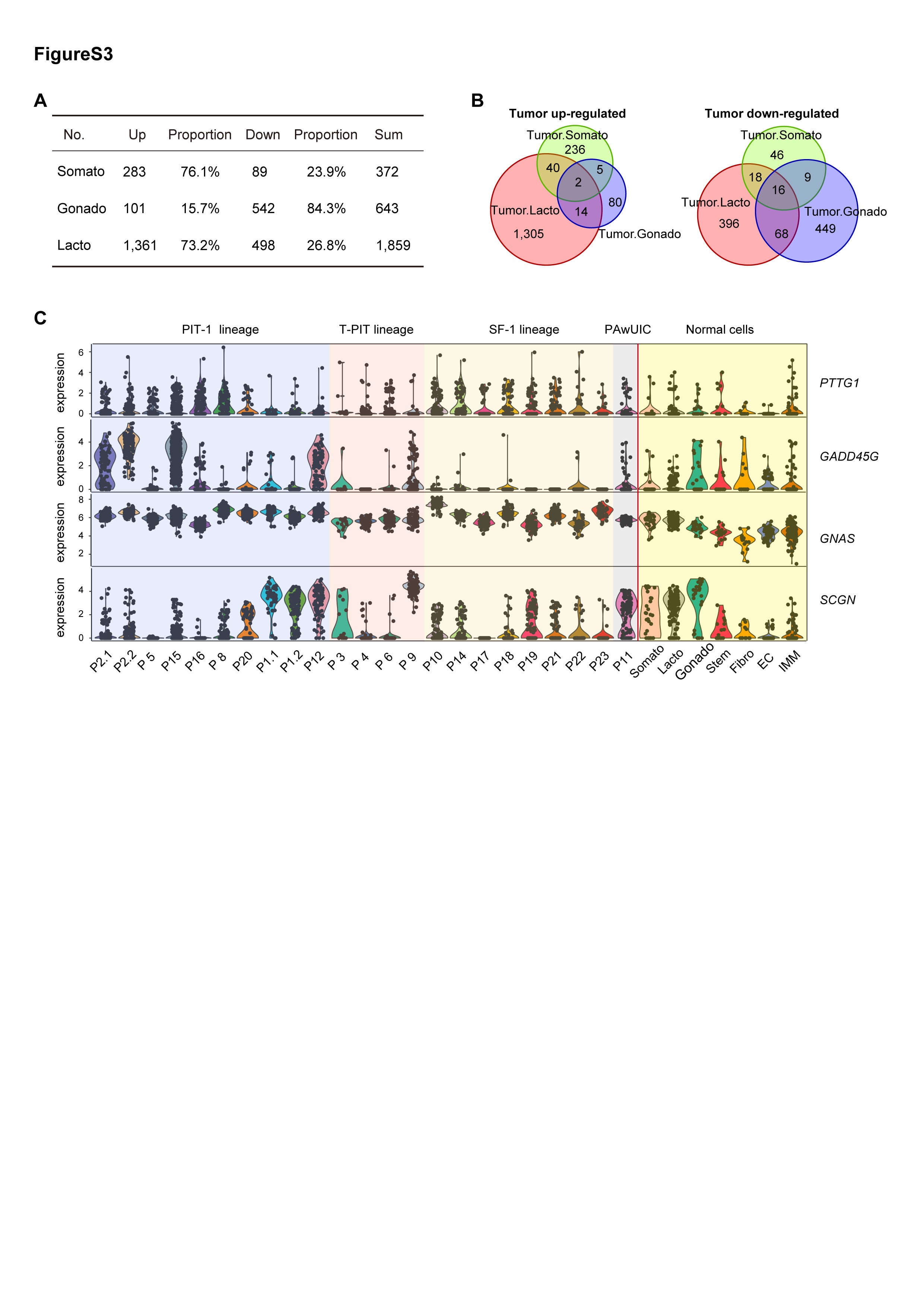
Comparison of somatotroph, gonadotroph, and lactotroph tumor cells and their matched normal cells identified 372, 643, and 1859 DEGs, respectively (Figure 3A–C; Supplementary Figure S3A, B; Supplementary Table S3). Notably, among gonadotroph DEGs, the majority of them (84.3%, 542 of 643) were downregulated, while in contrast, the majority of the somatotroph and lactotroph DEGs were upregulated (76.1%, 283 of 372 for somatotrophs, and 73.2%, 1361 of 1859 for lactotrophs, Supplementary Figure S3A). The higher number of DEGs for the lactotroph tumor may be due to individual differences as there was only one lactotroph tumor sample.
Fig. 3.

Identification of tumor-related genes by comparing tumor and matched normal cells. (A–C) Volcano plots showing DEGs between the tumor and matched normal of somatotrophs (A), gonadotrophs (B), and lactotrophs (C). The number of DEGs is labeled. (D) Representative GO terms of downregulated genes in gonadotroph tumors and upregulated genes in somatotroph tumors. (E) Violin plots showing the expression of representative known and novel tumor-related genes in normal pituitary cell types and tumor cells from each patient. Samples are arranged in the same order as Figure 1B. Somato, somatotrophs; Lacto, lactotrophs; Gonado, gonadotrophs; Stem, stem cells; Fibro, fibroblasts; EC, endothelial cells; IMM, immune cells. (F) Immunostaining results showing the expression of AMIGO2 (green) and PIT-1 (red) in tumor cells from 1 somatotroph tumor. Scale bar, 50 µm. Abbreviations: DEGs, differentially expressed genes; GO, gene ontology.
LHB, GNRHR, ESR1, and PGR were remarkably downregulated in gonadotroph tumors compared with normal gonadotrophs, which was consistent with the nonfunctioning nature of this tumor subtype (Figure 3B). Downregulated genes of gonadotroph tumors enriched for GO terms such as “regulation of cell population proliferation” (eg, CDKN1A, CDKN2A, ZFP36, BTG2, DLG5, and ZBTB16), “epithelium development” (eg, KRT8, KRT18, and KLF4), and “hormone metabolic process” (eg, LHB, GAL, and ESR1), which suggested abnormalities in cell proliferation, hormone production, and epithelium function of these tumors (Figure 3D). For somatotroph tumors, high expression levels GHRHR, GH1, and GH2 were maintained, which was consistent with the functioning nature of this tumor type. Consistently, upregulated genes of somatotroph tumors are mainly enriched for GO terms “regulated exocytosis” (eg, SCG3, ANXA2, CLU, and GAA) and “secretion by cell” (eg, A1BG, HEXB, ATP6V0A1, ATP6AP1, PSAP, and PSMA5) (Figure 3D).
We examined previously reported PitNET-related genes including PTTG1, GADD45G, MEG3, CDKN2A, and CCND1. Consistent with previous reports of the selective silencing of MEG3 in gonadotroph tumors, the expression levels of MEG3 were remarkably decreased in all (8 of 8) gonadotroph tumors compared with normal gonadotrophs28 (Figure 3E). CDKN2A was markedly downregulated in 6 of 8 gonadotroph tumors, and 2 of 8 PIT-1 tumors, a pattern that was consistent with previous studies.29CCND1 was upregulated in all gonadotroph tumors and 2 of 4 somatotroph tumors (Figure 3E). Interestingly, GADD45G was prominently downregulated in gonadotroph tumors but upregulated in somatotroph tumors, which was not previously recognized.30,31PTTG1 was significantly upregulated in the lactotroph tumor and a portion of somatotroph and gonadotroph tumors (Supplementary Figure S3C).
Two genes (AMIGO2 and SERF2) were consistently upregulated in all 3 tumor types, and 5 genes (CLU, BEX1, C4orf48, NDUFA1, and TSPAN3) were upregulated in both somatotroph and gonadotroph tumors (Figure 3A and Supplementary Figure S3B; Supplementary Table S3). The high expression level of CLU in PitNETs has been reported.32AMIGO2 was not upregulated in the corticotroph tumors and thus was tumor type-specific. An association between AMIGO2 and PitNETs has not been reported, and this gene has recently been reported to play roles in the proliferation or metastasis of several malignant cancer types.33,34 To confirm the expression of AMIGO2 in PitNETs, we performed immunostaining in 2 somatotroph tumors. The results showed that AMIGO2 was strongly expressed in the cytoplasm and membrane of PIT-1-positive tumor cells (Figure 3F).
We did not capture the normal corticotroph. For identifying potential corticotroph tumor-related genes, we compared the corticotroph tumor cell with the normal human fetal corticotroph (Corticotroph2) published in our recent study using the same scRNA-seq platform.35 We identified tumor-upregulated genes such as CLU, BEX1, C4orf48, and NDUFA1 which also upregulated in somatotroph and gonadotroph tumors, and downregulated genes such as POMC (Supplementary Table S3).
Thus, the comparison of tumor and matched normal cells at a single-cell resolution uncovered distinct characteristics between gonadotroph and the PIT-1 lineage tumors and identified known and novel tumor-related genes.
Intra-Tumor Heterogeneity in Transcriptome
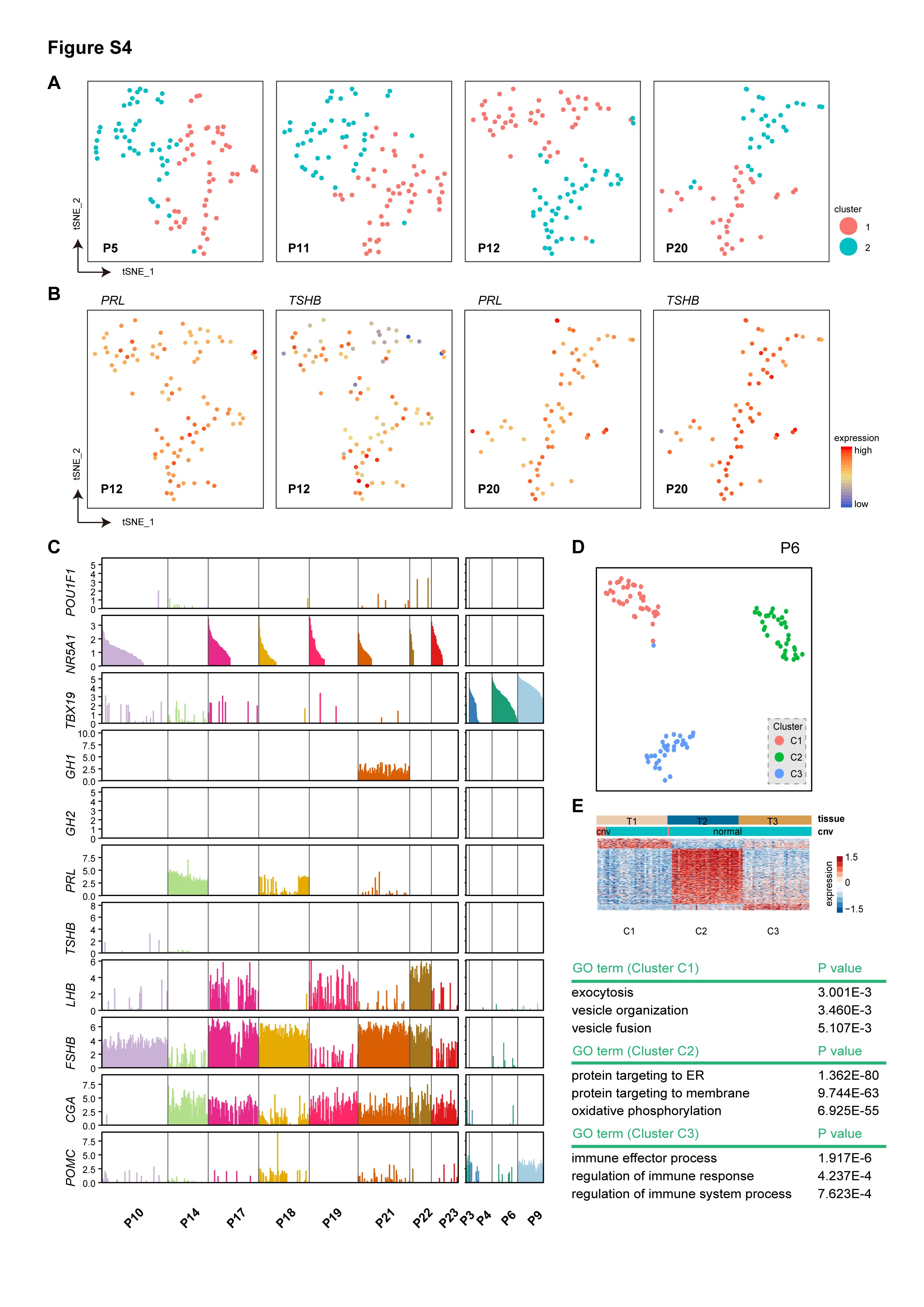
We next explored the intra-tumor transcriptomic heterogeneity of PitNETs. Cell cycle analysis showed that a small portion of tumor cells (0.7%) were actively proliferating, which is consistent with the notion that PitNETs have a low proliferation rate (Figure 4A). Correlation analysis showed that these tumors were generally homogeneous (Figure 4B). PitNETs usually expressed more than one hormone gene, eg, P1, P5, P11, P12, and P15 in this cohort. We performed clustering analysis to examine the heterogeneity of these multiple hormone tumors. The results showed that tumor cells expressing multiple hormone genes or TFs were not segregated in all cases (Figure 4C, D; Supplementary Figure S4A–C). In the PAwUIC tumor from P11, SF1 (NR5A1) and T-PIT (TBX19) were clearly co-expressed in the same individual cells (Figure 4C, D). Additionally, in the somatotroph tumor from P5, cells expressing GHRH and GHRHR were not segregated into subclusters, suggesting the presence of an autocrine loop (Figure 4C; Supplementary Figure S4A). Clustering analysis of other tumors identified a cell subcluster expressing the cell cycle program including TOP2A, UBE2C, AURKB, and CCNB2, in 1 invasive tumor (P16, Figure 4E, F). This tumor indeed gave the highest Ki-67 staining (8%) in the cohort (Supplementary Table S1). Another invasive tumor (P6) showed mass-associated subpopulations with unexplained functions (Supplementary Figure S4D, E). We also identified cell subclusters enriched for genes involved in extracellular matrix organization and epithelial-to-mesenchymal transition in 4 tumors (P1.2, P2.2, P10, and P15); however, we failed to verify the presence of these cells in vivo using immunostaining (data not shown).
Fig. 4.

Intra-tumoral heterogeneity of the cell cycle and multiple hormone genes. (A) Classification of all tumor cells as noncycling (blue) and cycling cells (red) based on cell cycle scores. (B) Pairwise correlations between expression profiles of single cells across all tumor cells. Cells are hierarchically clustered by patients. (C) t-SNE plot showing coexpression of TBX19 and NR5A1 in individual tumor cells from P11 and coexpression of GHRH and GHRHR in the tumor from P5. (D) Bar plot showing the expression of TFs and hormone genes in single cells of PIT-1 lineage tumors and the tumor from P11. Samples are arranged in the same order as Figure 1B. (E) t-SNE plot showing clustering results of tumor cells from P16. (F) Heatmap showing the expression of DEGs in each of 3 clusters from P16, with the expression of representative genes and the main GO term of cluster C3 shown on the right panel. Abbreviations: GO, gene ontology, TFs, transcription factors, t-SNE, t-distributed stochastic neighbor embedding.
Together, the results indicated that PitNETs were generally had homogeneous transcriptomes, even among tumors expressing multiple hormone genes.
Single-Cell CNV Analysis
Genomic CNVs occur frequently in PitNETs.36,37 However, this has not been analyzed at a single-cell resolution. We deduced CNVs from scRNA-seq data as reported in previous studies.38 The results showed that 62% (13 of 21) of tumors had CNVs; 5 tumors (P1, P8, P10, P12, and P20) displayed severe genomic disruption as defined by genomic alterations on 4 or more chromosomes, and 8 tumors displayed CNVs on 1-3 chromosomes (Figure 5A and Supplementary Figure S1A). We performed WGS of bulk samples from 7 tumors, and the results generally verified the deduced CNVs from single-cell data (Figure 5A and Supplementary Figure S1B). In all cases, including 5 tumors displaying genomic disruptions, individual tumor cells from the same patient showed similar CNV patterns, suggesting minor intra-tumoral genomic heterogeneity.
Fig. 5.

Single-cell multi-omics sequencing of PitNETs. (A) CNV profiles inferred from scRNA-seq in individual cells from each patient, verified by bulk WGS in selected cases. (B–D) CNV profiles detected by MALBAC at a 1-Mb resolution in 4 single tumor cells from P20 (B), P21 (C), and P18 (D). Cells are in cluster C2 in Supplementary Figure S5C–E. Abbreviations: CNV, copy number variations; MALBAC, multiple annealing and looping-based amplification cycle; PitNETs, pituitary neuroendocrine tumors; scRNA-seq, single-cell RNA sequencing; WGS, whole-genome sequencing.
The scRNA-seq based CNV detection method had a low resolution. To investigate the intra-tumoral genomic heterogeneity at a higher resolution, we performed single-cell multi-omics sequencing, which simultaneously detected the transcriptome and the genome in an individual cell. In this analysis, the identity of cells could be clearly determined by transcriptome, while genomic alterations can be accurately determined by the single-cell genome sequencing MALBAC method.20 We analyzed a total of 409 cells from 5 patients (P17-P21) with 238 cells passing the quality criteria (Supplementary Figure S5A–F). Tumor cells from P20 showed genomic disruptions with CNVs on 4 chromosomes; all 8 analyzed tumor cells showed the same CNV pattern, confirming that there was no significant intra-tumoral heterogeneity (Figure 5B and Supplementary Figure S5G). Interestingly, tumor cells from P21 and P18 showed slight but clear intra-tumoral heterogeneity in genome. In P21, except for the gain of chromosome 7 in all tumor cells, 5 of these 80 tumor cells showed a gain of a 61-Mb portion of chromosome 1q (Figure 5C). In P18, most tumor cells showed a gain of chromosome X, while a small portion of tumor cells (6 of 86) showed no gain of chromosome X (Figure 5D).
Thus, single-cell sequencing analysis indicated that PitNETs had a relatively uniform pattern of genomes, but slight intra-tumoral CNV heterogeneity did exist in some tumors.
Discussion
For the first time, we conducted high-precision scRNA-seq and single-cell multi-omics analyses of PitNETs. First, we compared tumor and matched normal endocrine cells at a single-cell resolution and comprehensively examined tumor-related transcriptome alterations. Previous studies have compared tumor and normal pituitary tissue. This approach is not accurate since it is complicated by mixed cell types in normal pituitary tissue.39–41 One notable finding of this study is that gonadotroph tumors predominantly exhibited downregulated genes, while somatotroph and lactotroph tumors mainly exhibited upregulated genes. This likely reflects the different characteristics and pathogenesis of gonadotroph tumors in comparison with the other 2 tumor types. The vast majority of gonadotroph tumors are nonfunctioning, while somatotroph and lactotroph tumors are typically functioning.42 Consistently, the downregulated genes in the gonadotroph tumors included LHB and GNRHR, while in the somatotroph tumors, GH1 and GHRHR were not downregulated. Furthermore, the pathogenesis differs between somatotroph tumors and gonadotroph tumors. GNAS mutations, which stimulate GH secretion, occur in 40%-60% of somatotroph tumors.8,37 In contrast, no recurrent mutations have been identified in gonadotroph tumors, and this tumor type is thought to be driven by epigenetic mechanisms including DNA methylation.4,5 Thus, many downregulated genes in gonadotroph tumors may be silenced by DNA methylation as has been reported for the tumor suppressors MEG3, CDKN2A, and GADD45G. Among the newly identified genes, there were some novel putative tumor suppressors, including ZFP36, BTG1, DLG5, and ZBTB16, were downregulated in gonadotroph tumors. AMIGO2 is an interesting tumor-associated gene that was upregulated in most gonadotroph, somatotroph, and lactotroph tumors, but not corticotroph tumors in our cohort. Further studies should investigate whether these genes are required for the survival of PitNET cells.
Second, we investigated inter- and intra-tumoral heterogeneity in the transcriptome and genome of PitNETs. Our data failed to reveal intra-tumoral heterogeneity in several multiple hormone tumors including P11; the TFs or hormone genes of different lineages were clearly co-expressed in individual cells. These results thus suggested that these tumors originated from a single “ancestor” cell with a dysregulated epigenome and transcriptome.
Previous studies have shown that approximately 30% of PitNETs display genomic disruptions involving up to 99% of the genome, a level similar to most malignant cancers.36,37 For 5 such tumors, individual cells showed the same CNV patterns, indicating that they were derived from a single tumor “ancestor” cell suffering all genomic alterations; this further suggested that CNVs were one of the causes of these PitNETs. We identified intra-tumoral genomic heterogeneity in 2 PitNETs. However, the frequency and extent of the heterogeneity are much lower than those of colorectal cancers.18
This study has limitations. The scRNA-seq landscape of 21 tumors for 6 subtypes was still preliminary, and there was no functional corticotroph adenoma case during the period of sampling; less cases were subjected for multi-omics analysis. The number of cells for each tumor was limited and thus we may not fully capture intra-tumoral heterogeneity. We have also not captured the normal adult corticotroph and thyrotroph.
In summary, our single-cell sequencing analysis provides insights into the characteristics and pathogenesis of PitNETs. The data serve as an invaluable resource for the identification of diagnostic biomarkers and therapeutic targets for PitNETs.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the Core Facilities at School of Life Sciences, Peking University for assistance with confocal microscopy. We thank Yuqiong Hu, Lin Li, and other members of Tang laboratory for helpful advice and appreciate the patients who participated in this study.
Funding
This study was supported by grants from the National Natural Science Foundation of China (31671109 and 31871457). This work was also supported by the grants from Beijing Advanced Innovation Center for Genomics and the Open Research Fund of the National Center for Protein Sciences at Peking University. Part of the high-throughput sequencing data were processed at the Computing Platform of the Center for Life Science.
Conflict of interest statement. The authors declare that they have no competing interests.
Authorship statement. D.Z., L.Wen, and F.T. conceived and lead the project. Y.C. did the experiments with the help of C.L., R.L., L.Wei, and L.L. and Q.Z., S.Z., and Z.J. conducted the bioinformatics analyses. L.Wen and Y.C. wrote the manuscript with help from all of the authors. All authors read and approved the final manuscript.
References
- 1. Asa SL, Casar-Borota O, Chanson P, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club Proposal. Endocr Relat Cancer. 2017;24(4):C5–C8. [DOI] [PubMed] [Google Scholar]
- 2. Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72(3):377–382. [DOI] [PubMed] [Google Scholar]
- 3. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134(4):521–535. [DOI] [PubMed] [Google Scholar]
- 4. Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol. 2009;4:97–126. [DOI] [PubMed] [Google Scholar]
- 5. Melmed S. Pituitary-tumor endocrinopathies. N Engl J Med. 2020;382(10):937–950. [DOI] [PubMed] [Google Scholar]
- 6. Neou M, Villa C, Armignacco R, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. 2020;37(1):123–134.e5. [DOI] [PubMed] [Google Scholar]
- 7. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary-adenomas. J Clin Endocr Metab. 1990;71(6):1427–1433. [DOI] [PubMed] [Google Scholar]
- 8. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–696. [DOI] [PubMed] [Google Scholar]
- 9. Reincke M, Sbiera S, Hayakawa A, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47(1):31–38. [DOI] [PubMed] [Google Scholar]
- 10. Ma ZY, Song ZJ, Chen JH, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25(3):306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bi WL, Larsen AG, Dunn IF. Genomic alterations in sporadic pituitary tumors. Curr Neurol Neurosci Rep. 2018;18(1):4. [DOI] [PubMed] [Google Scholar]
- 12. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puram SV, Tirosh I, Parikh AS, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Venteicher AS, Tirosh I, Hebert C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355(6332):eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hovestadt V, Smith KS, Bihannic L, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572(7767):74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery. 1993;33(4):610–618; discussion 617. [DOI] [PubMed] [Google Scholar]
- 17. Cui Y, Zheng Y, Liu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26(7):1934–1950.e5. [DOI] [PubMed] [Google Scholar]
- 18. Bian SH, Hou Y, Zhou X, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362(6418):1060–1063 . [DOI] [PubMed] [Google Scholar]
- 19. Zhou Y, Bian S, Zhou X, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell. 2020;38(6):818–828.e5. [DOI] [PubMed] [Google Scholar]
- 20. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv:1303.3997 [q-bio.GN]. 2013;1303. [Google Scholar]
- 25. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boeva V, Popova T, Bleakley K, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28(3):423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tickle T, Ti GC, Brown M, Haas B. inferCNV of the Trinity CTAT Project.https://github.com/broadinstitute/inferCNV. Accessed May 31, 2017.
- 28. Zhao J, Dahle D, Zhou Y, Zhang X, Klibanski A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab. 2005;90(4):2179–2186. [DOI] [PubMed] [Google Scholar]
- 29. Simpson DJ, Bicknell JE, McNicol AM, Clayton RN, Farrell WE. Hypermethylation of the p16/CDKN2A/MTSI gene and loss of protein expression is associated with nonfunctional pituitary adenomas but not somatotrophinomas. Genes Chromosomes Cancer. 1999;24(4):328–336. [PubMed] [Google Scholar]
- 30. Bahar A, Bicknell JE, Simpson DJ, Clayton RN, Farrell WE. Loss of expression of the growth inhibitory gene GADD45γ, in human pituitary adenomas, is associated with CpG island methylation. Oncogene. 2004;23(4):936–944. [DOI] [PubMed] [Google Scholar]
- 31. Zhang X, Sun H, Danila DC, et al. Loss of expression of GADD45γ, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab. 2002;87(3):1262–1267. [DOI] [PubMed] [Google Scholar]
- 32. Chesnokova V, Zonis S, Zhou C, et al. Lineage-specific restraint of pituitary gonadotroph cell adenoma growth. PLoS One. 2011;6(3):e17924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fontanals-Cirera B, Hasson D, Vardabasso C, et al. Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol Cell. 2017;68(4):731–744.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanda Y, Osaki M, Onuma K, et al. Amigo2-upregulation in tumour cells facilitates their attachment to liver endothelial cells resulting in liver metastases. Sci Rep. 2017;7:43567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang S, Cui Y, Ma X, et al. Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat Commun. 2020;11(1):5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bi WL, Horowitz P, Greenwald NF, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. 2017;23(7):1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song ZJ, Reitman ZJ, Ma ZY, et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016;26(11):1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X, Zhou Y, Klibanski A. Isolation and characterization of novel pituitary tumor related genes: a cDNA representational difference approach. Mol Cell Endocrinol. 2010;326(1–2):40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morris DG, Musat M, Czirják S, et al. Differential gene expression in pituitary adenomas by oligonucleotide array analysis. Eur J Endocrinol. 2005;153(1):143–151. [DOI] [PubMed] [Google Scholar]
- 41. Kober P, Boresowicz J, Rusetska N, et al. The role of aberrant DNA methylation in misregulation of gene expression in gonadotroph nonfunctioning pituitary tumors. Cancers (Basel). 2019;11(11):1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7(5):257–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data in this study are available in the Genome Sequence Archive (GSA) of number PRJCA002946.