

Summary
Structural maintenance of chromosomes (SMC) complexes organize genome topology in all kingdoms of life and have been proposed to perform this function by DNA loop extrusion. How this process works is unknown. Here, we have analyzed how loop extrusion is mediated by human cohesin-NIPBL complexes, which enable chromatin folding in interphase cells. We have identified DNA binding sites and large-scale conformational changes that are required for loop extrusion and have determined how these are coordinated. Our results suggest that DNA is translocated by a spontaneous 50 nm-swing of cohesin’s hinge, which hands DNA over to the ATPase head of SMC3, where upon binding of ATP, DNA is clamped by NIPBL. During this process, NIPBL “jumps ship” from the hinge toward the SMC3 head and might thereby couple the spontaneous hinge swing to ATP-dependent DNA clamping. These results reveal mechanistic principles of how cohesin-NIPBL and possibly other SMC complexes mediate loop extrusion.
Keywords: loop extrusion, SMC complexes, cohesin, NIPBL, genome architecture, high speed AFM, single molecule FRET
Graphical abstract
Highlights
-
•
Identification of cohesin’s DNA binding sites and movements needed for loop extrusion
-
•
A DNA binding site on the hinge might translocate DNA by ATP-independent swinging
-
•
DNA clamping on the ATPase heads is regulated by ATP-dependent engagement cycles
-
•
NIPBL couples ATP-independent and -dependent DNA translocation events
Cohesin hands DNA over long distances from one binding site to another, explaining how SMC complexes might fold genomes in all kingdoms of life.
Introduction
The loop extrusion hypothesis posits that “structural maintenance of chromosomes” (SMC) complexes form chromosomal cis-interactions by reeling genomic DNA into loops. These have important functions, ranging from the local separation of replicated DNA molecules in bacteria to the formation of long-range enhancer-promoter contacts in eukaryotes (reviewed in Davidson and Peters, 2021). Direct evidence for the loop extrusion hypothesis has been obtained in biochemical reconstitution experiments. These experiments showed that condensin, a eukaryotic SMC complex that folds DNA in mitotic chromosomes, and cohesin, a related complex that mediates chromatin looping in interphase, can extrude DNA at up to 2.1 kb/s in a process that depends on the DNA-stimulated ATPase activity of these complexes (Davidson et al., 2019; Ganji et al., 2018; Golfier et al., 2020; Kim et al., 2019).
The mechanism of DNA loop extrusion is unknown but has been subject to numerous speculations. These are based on the observation that SMC complexes contain a dimer of elongated SMC subunits, which can adopt different conformations (reviewed in Yatskevich et al., 2019). SMC proteins form up to 50 nm long coiled coils. These contain a “hinge” at one end, via which the two SMC subunits dimerize, and ATPase “heads” at their other end. These are related to the ATPase domains of ATP binding cassette (ABC) transporters (Hopfner et al., 2000) and can engage with each other to form two composite ATP binding sites. In addition, the ATPase heads are connected by a kleisin subunit, resulting in the formation of tripartite ring structures. By electron microscopy (EM) and atomic force microscopy (AFM), SMC complexes can be seen in open ring conformations (in which the coiled coils are separate), in rod conformations (in which the coiled coils are aligned), and in bent conformations (in which the coiled coils are folded and the hinge is close to the ATPase heads) (for review, see Davidson and Peters, 2021). The kleisin subunit is associated with additional subunits, which are required for loop extrusion, called STAG1/STAG2 and NIPBL in cohesin (Davidson et al., 2019; Kim et al., 2019).
The “walking” and “inchworm models” posit that SMC complexes use movements between the ATPase heads to “walk” along DNA, analogous to how cytoskeletal motor proteins move along microtubules (Fudenberg et al., 2016; Nichols and Corces, 2018). The “pumping model” proposes that DNA is translocated from the hinge toward the kleisin by alignment of the coiled coils and disengagement of the ATPase heads into a juxtaposed state (Diebold-Durand et al., 2017; Marko et al., 2019). The “scrunching model” speculates that alternations of the coiled coils between straight and bent conformations translocate DNA (Ryu et al., 2020). Except for the observations mentioned above, there is limited experimental support for these models and none of them has been rigorously tested. It is therefore unknown, which of them, if any, is correct.
Here, we have used protein engineering and mutagenesis in conjunction with single-molecule experiments to analyze how DNA is translocated by human cohesin and NIPBL. Our results indicate that loop extrusion depends on five DNA binding sites on cohesin-NIPBL and on three large-scale movements between these sites that are partially coupled to each other and to ATP binding-hydrolysis cycles. We provide evidence that the upper coiled coils of the SMC subunits align and bend spontaneously, leading to a 50-nm movement of a high-affinity DNA binding site on the hinge toward the ATPase head of SMC3. In the absence of ATP, NIPBL interacts with the hinge, but in the presence of ATP, NIPBL separates from the hinge and clamps DNA onto the head of SMC3. We propose that loop extrusion depends on alternating DNA translocation and clamping steps, which are mediated by the 50 nm-hinge swing and interactions between NIPBL and the SMC3 head, respectively. Our data further suggest that NIPBL can participate only in hinge or SMC3-head interactions, but not in both simultaneously, making the ATP-dependent clamping step dependent on the spontaneous hinge swing and providing directionality to DNA extrusion.
Results
Identification of DNA binding sites on cohesin and NIPBL that are required for loop extrusion
Several cohesin subunits can bind DNA in vitro and structures of the ATPase heads, STAG1, and NIPBL bound to DNA have been solved (Figures 1A–1E and S1A) (for review, see Davidson and Peters, 2021). However, it is unknown if these DNA binding domains are required for loop extrusion. We therefore generated human cohesin complexes (containing SMC1, SMC3, SCC1, and STAG1) bound to NIPBL, in which individual DNA binding sites were mutated, and analyzed these complexes for their ability to mediate loop extrusion in an assay, in which this process can be visualized by total internal reflection fluorescence (TIRF) microscopy (Davidson et al., 2019).
Figure 1.

DNA binding sites on cohesin-NIPBL required for loop extrusion
(A) Cartoon illustration of human cohesin-NIPBL. DNA binding sites are indicated with black dots.
(B–E) Structures of the SMC1-SMC3 ATPase heads (B, PDB: 6WG3), the SMC1-SMC3 hinge (C, PDB: 2WD5), STAG1 (D; PDB: 6WG3), and NIPBL (E; PDB: 6WG3). Mutated residues are indicated as spheres.
(F–J) Electrophoretic mobility shift assays (EMSAs) of the SMC1 head (F), the SMC3 head (G), the hinge (H), ΔN-STAG1 (I), and ΔN-NIPBL (J). Increasing concentrations of wild-type (wt) or mutant proteins were incubated with a 40 base pair double stranded DNA and separated by native PAGE. U, unbound DNA; B, bound DNA.
(K) Time-lapse recordings of loop extrusion events catalyzed by wt and mutant cohesin complexes. DNA was stained with Sytox Orange. Scale bar, 2 μm.
(L) Frequencies of loop extrusion events. Shown are means ± SD and values of individual replicates.
(M) Rates of loop extrusion in kilobases per second (kb/s). Shown are rates of individual loop extrusion events and a violin plot of their distribution.
(N) NIPBL- and DNA-stimulated ATPase rates of the indicated cohesin complexes. Hydrolysis of ATP was detected by thin layer chromatography. Shown are means ± SD and values of individual replicates.
See also Figure S1.
Figure S1.

DNA binding sites in cohesin-NIPBL, related to Figure 1
A.) Structure of the DNA clamp formed by the SMC1-SMC3 heads and NIPBL. Shown is a space filling model of PDB: 6WG3.
B.) Design of the SMC1 head domain (a.), SMC3 head domain (b.), ΔN−STAG1 (c.), ΔN−NIPBL (d.) and the hinge domain (e.). With exception of ΔN−NIPBL, constructs were expressed with fragments of SCC1 to facilitate purification. Numbers correspond to the amino acid positions in the respective wild-type (wt) proteins. The cartoon illustration on the top right indicates the location of the individual constructs within the cohesin complex.
C.) Purity of the SMC1 head domain, SMC3 head domain, hinge, ΔN−STAG1, ΔN−NIPBL. Proteins were separated by SDS-PAGE and stained by Coomassie.
D.) Mass photometry analysis of the SMC1 head in presence of ATP and in absence or presence of tenfold molar excess of a 40 base pair (bp) double stranded (ds) DNA. Negative masses correspond to events, where protein-DNA complexes had dissociated from the surface between two frames. The theoretical masses of the SMC1 head and 40 bp DNA are indicated. The expected masses of SMC1 head dimers and complexes of SMC1 head dimers and one DNA molecule are indicated with arrows (“2xHD1” and “2xHD1 + DNA,” respectively). Note that the molecular mass of the 40 bp DNA is below the detection limit of mass photometry. For this reason, DNA alone is not detected.
E.) As in D.), but with the SMC3 head. Note that a small fraction of the SMC3 head preparation contains dimeric head domains, which are a result of mispairing of the fused SCC1 fragments. The expected masses of SMC3 head dimers and complexes between SMC3 head dimers and one DNA molecule are indicated with arrows (“2xHD3” and “2xHD3 + DNA,” respectively).
F.) Structure of the hinge domain (PDB: 2WD5), as viewed from the side. Residues substituted by para-benzoyl-phenylalanine (BPA) via amber suppression are indicated in blue. Residues mutated to alanine in hinge4A are shown in red. The cartoon shows the definition of the “South” and “North” interfaces of the hinge, as viewed from the top. The organic structure of BPA is indicated.
J.) Photo crosslinking of a 40 base single (ss) or double stranded (ds) DNA to the South interface of the hinge. Hinge domains with the indicated residues replaced by BPA were incubated in presence or absence of biotinylated DNA and treated with UV light. DNA was immobilized on streptavidin beads and washed under denaturing conditions. After heat-elution, DNA-hinge adducts were visualized by SDS-PAGE and silver staining. Note that crosslinking residues (550, 557) face toward the South interface whereas the non-crosslinking residue (555) faces away from it. Similar to what has been observed before (Shi et al., 2020), we found higher cross-linking efficiencies for ssDNA compared to dsDNA, possibly due to the increased flexibility of ssDNA at this length, but there is also crosslinking with dsDNA, visible as a smear.
K.) EMSAs of STAG1patch2 and STAG1patch3, as described in Figure 1I.
Electrophoretic mobility shift assays (EMSAs) performed with isolated recombinant domains and subunits of cohesin showed that the hinge, both ATPase heads, and N-terminal deletion mutants of STAG1 and NIPBL (ΔN-STAG1 and ΔN-NIPBL) are able to bind DNA (Figures 1F–1J, S1B, and S1C). For the SMC1 and the SMC3 heads, we confirmed these results by mass photometry (Sonn-Segev et al., 2020), which revealed that these domains can each bind a single DNA molecule as a monomer (Figures S1D and S1E). For the hinge, we confirmed DNA binding in photo-crosslinking experiments, which indicated that residues at the “South” interface of SMC1 and SMC3 can contact DNA (Figures S1F and S1G).
Mutation or deletion of surface exposed basic amino acid patches in the cohesin domains and NIPBL reduced (SMC1Δ58–62, SMC32E, hinge4A, ΔN-STAG1patch1, ΔN-STAG1patch2, and ΔN-STAG1patch3) or abrogated detectable DNA binding (SMC14E, SMC34E, and ΔN-NIPBL2A) (Figures 1F–1J and S1H), indicating that these patches are DNA binding sites. These results are consistent with partial cryoelectron microscopy (cryo-EM) structures of cohesin-NIPBL bound to DNA, which revealed that NIPBL clamps DNA onto the ATPase heads when these are engaged (Figures 1B, 1D, 1E, and S1A) (Collier et al., 2020; Higashi et al., 2020; Shi et al., 2020).
We next tested cohesin-NIPBL complexes containing mutated DNA binding sites for their ability to mediate loop extrusion. Cohesin containing SMC14E, SMC34E, hinge4A, ΔN-STAG1patch1, ΔN-STAG1patch2, ΔN-STAG1patch3, and NIPBL2A were unable to support loop extrusion, whereas cohesin containing SMC1Δ58–62 or SMC32E enabled loop extrusion (Figure 1K) but at reduced frequencies and rates (Figures 1L and 1M).
All mutant complexes were able to hydrolyze ATP, although at reduced rates, but the ATPase activities of complexes containing SMC1Δ58–62, SMC14E, SMC32E, and hinge4A were still stimulated by DNA to a similar extent as wild-type cohesin. In contrast, complexes containing NIPBL2A or versions of SMC3’s head domain, which carry mutations in the acetylatable residues K105 and K106 (SMC34E), could only be stimulated by DNA to a small extent (Figure 1N), consistent with observations reported for yeast cohesin (Murayama and Uhlmann, 2015; Petela et al., 2018).
These results indicate that cohesin-NIPBL contains at least five DNA binding sites that are required for loop extrusion (indicated by black dots in Figure 1A). The sites on the SMC3 head and NIPBL become part of the DNA clamp in the engaged state (Figure S1A) and are required for stimulation of cohesin’s ATPase activity by DNA. Their interaction with DNA might therefore trigger ATP hydrolysis. The sites on the SMC1 head, the hinge and STAG1 are dispensable for this effect of DNA. Complexes containing mutations in these sites can hydrolyze ATP at rates, which are sufficient to allow loop extrusion, because they are comparable to the ATP hydrolysis rates achieved by cohesin containing SMC1Δ58–62 and SMC32E, which can perform loop extrusion. The DNA binding sites on the SMC1 head, the hinge and STAG1 must therefore have a role in loop extrusion other than enabling cohesin’s ATPase activity, such as the translocation of DNA into loop structures. The DNA binding sites that stimulate cohesin’s ATPase could of course also contribute to the movement of DNA since these functions may not be mutually exclusive.
Visualization of conformational changes in cohesin by high-speed atomic force microscopy
If the DNA binding sites that we identified have roles in translocating DNA, one would expect that at least some of these can move relative to each other, to transport DNA and possibly to “hand over” DNA from one site to another. Cohesin has been observed in different conformations by EM (Anderson et al., 2002; Bürmann et al., 2019; Collier et al., 2020; Higashi et al., 2020; Hons et al., 2016; Huis in ’t Veld et al., 2014; Shi et al., 2020), but it is unknown which of these exist under native conditions and how cohesin changes from one conformation to another. We therefore imaged cohesin by high-speed atomic force microscopy (HS-AFM) (Ando et al., 2003) in real time.
To determine which subunits and domains can be visualized by this technique, we first analyzed individual subunits and sub-complexes of cohesin-NIPBL (Figures S2A–S2D). The hinge (hi), coiled coils, and head domains (he) could be seen in trimeric complexes containing SMC1, SMC3, and SCC1, but SMC1 and SMC3 could not be distinguished from each other (Figure 2A). SCC1 could not be visualized clearly, possibly because this subunit is predicted to be partially unstructured, but mass photometry confirmed that these complexes contained SCC1 (Figure S2E). In isolation, STAG1 (S) and NIPBL (N) appeared as spherical particles (Figure S2F) and when bound to cohesin trimers, as additional densities in the vicinity of the ATPase heads (Figure 2A). When STAG1 and NIPBL were simultaneously bound to trimers the size of the structure near the SMC heads increased, but in most complexes the SMC heads, STAG1 and NIPBL could not be clearly distinguished (Figure 2A). Volumetric measurements yielded data that were consistent with the predicted masses of all subunits and complexes and indicated that NIPBL forms dimers in isolation but binds to cohesin as a monomer (Figure S2G).
Figure S2.

Conformational changes of cohesin-NIPBL visualized by HS-AFM, related to Figure 2
A-D.) Purity of trimeric cohesin (A), tetrameric cohesin (B), trimeric cohesin bound to NIPBL (C), and NIPBL alone (D). Molecular weights are indicated in kilo Daltons (kDa). Note that cohesin holocomplexes were formed by mixing cohesinSTAG1 with NIPBL before imaging.
E.) Mass distributions of preparations of dimeric and trimeric cohesin, as determined by mass photometry. Note that the theoretical masses of dimers (286 kDa) and trimers (399 kDa) were in close agreement with the measured values.
F.) Examples of NIPBL (top) and STAG1 (bottom) in isolation. The sequence on the right shows morphological alterations of NIPBL that might represent changes in a monomer-dimer equilibrium.
G.) Volumetric analysis of different cohesin complexes and subunits. Shown are means, standard deviations and the values measured for individual molecules.
H.) Major conformations observed for cohesin tetramers in presence of ATP. Heads (he), hinge (hi) and STAG1 (S) are indicated.
I.) Distribution of head-head and head-hinge distances of trimeric cohesin bound to NIPBL and cohesin tetramers. For trimer-NIPBL, heads were distinguished based on the binding behavior of NIPBL (“N-bound head” versus “head 2”; note that NIPBL typically does not switch between heads). The total number of molecules (n) and frames imaged are indicated.
J.) Frequencies of different conformational changes. “hinge preference for head 1” was quantified as the fraction of frames with head 1-hinge distances smaller than 10 nm over the total number of frames with head-to-hinge distances smaller than 10 nm (for both head 1 and head 2). “hinge-head w/ engaged head-head” corresponds to the fraction of frames with both hinge-head and head-head distances smaller than 10 nm over the total number of frames with head-head distances smaller than 10 nm.
K.) Example of a trimer bound to NIPBL in presence of ATP showing head engagements and coiled coil alignment followed by hinge bending toward the NIPBL-bound head. The cartoons shown below illustrate the conformations observed in the individual stills. The position of NIPBL is indicated with a black ball.
L.) Image sequence of a cohesin trimer in presence of ATP undergoing sequential coiled coil alignment and hinge bending. The cartoons shown below illustrate the conformations observed in the individual stills.
Figure 2.

Conformational changes of cohesin measured by hsAFM
(A) High speed atomic force microscopy recordings (HS-AFM) of isolated trimers, trimers bound to NIPBL, cohesin tetramers, or tetramers bound to NIPBL. Densities corresponding to the hinge (hi), heads (he), NIPBL (N), and STAG1 (S) are indicated.
(B) Major conformations observed in trimeric cohesin in the presence of ATP. The conformations are illustrated by cartoons.
(C) Cohesin trimer bound to NIPBL with twisted coiled coils. Closed and open hinge domains are indicated with arrows.
(D) Trimeric cohesin undergoing head engagements (top), coiled coil alignment (middle), and hinge bending (bottom).
(E) Distribution of head-head and head-hinge distances in trimeric cohesin. n, number of molecules analyzed.
In the presence of ATP, trimeric complexes alternated between ring-shaped, rod-shaped, and bent conformations (Figure 2B). In ring-shaped complexes, the coiled coils were outstretched but separate from each other, whereas the heads were either contacting each other, possibly representing an ATP-engaged state, or the heads were disengaged. The latter conformation resembles the state that is predominantly observed for vertebrate cohesin by rotary shadowing EM (rs-EM) (Anderson et al., 2002; Huis in ’t Veld et al., 2014). In rod-shaped complexes, the heads were disengaged, the coiled coils aligned, and in many cases, twisted around each other, as observed by negative staining EM (Hons et al., 2016). In this state, the hinge sometimes transiently opened (Figure 2C). Although interesting, because DNA has been proposed to enter cohesin via hinge opening (Gruber et al., 2006), we did not further analyze this phenomenon here because cohesin in which the two halves of the hinge have been cross-linked can still perform loop extrusion (Davidson et al., 2019).
In the bent state, the coiled coils were folded so that the hinge was close to the heads, which were always disengaged when bending occurred (Figure 2B). In this condition, the coiled coils were often, although not always, aligned near the hinge. These states are similar to conformations observed by negative staining (Bürmann et al., 2019; Hons et al., 2016) and cryo-EM (Collier et al., 2020; Higashi et al., 2020; Shi et al., 2020), in which the hinge and heads are separated by ∼15 nm, whereas HS-AFM imaging indicated that the hinge can come even closer to the heads (Figure 2B).
Trimeric rings oscillated between engaged and disengaged states and could, in the latter conformation, align and bend their coiled coils (Figure 2D; Videos S1, S2, and S3). Similar changes were seen when STAG1 and NIPBL bound to trimers (Figures S2H and S2K; Video S4). Folding of the coiled coils into the bent state occurred asymmetrically, with the hinge reaching close proximity to one head but not the other (Figures 2B and 2D). In some complexes, only one coiled coil was bending, also resulting in proximity between the hinge and one head (Figure S2L; Video S5), and possibly reflecting situations in which one coiled coil had been immobilized by adsorption on the mica surface used for imaging. Frame-by-frame measurements of distances between the two heads and the hinge confirmed the asymmetry of bending (Figures 2E and S2I), and in case of trimer-NIPBL, showed that in 80% of all bent conformations the hinge approached the NIPBL-bound head (Figures S2I and S2J). These analyses also confirmed that hinge bending occurred only in the absence of head engagements and vice versa, suggesting that the two conformational changes are mutually exclusive (Figures 2E, S2I, and S2J).
These results indicate that cohesin performs three types of reversible movements, (1) head engagements and disengagements, (2) coiled coil alignment and separation, and (3) reversible folding of the coiled coils, which leads to bending of the hinge toward one of the ATPase heads (“hinge bending”). Importantly, the HS-AFM movies show that the DNA binding sites that we identified as being essential for loop extrusion can move relative to each other, with the site on the hinge moving up to ∼50 nm. DNA could therefore be translocated over long distances by these movements.
ATP binding promotes engagement of the ATPase heads
To characterize these conformational changes kinetically and to analyze their regulation by ATP, NIPBL, and DNA, we generated a panel of 23 single molecule fluorescence resonance energy transfer (smFRET) “sensor” complexes, with which conformational changes can be measured with high spatial (∼1 nm) and temporal (50 ms/frame) resolution and for many molecules in parallel, which cannot be achieved by HS-AFM.
Because DNA loop extrusion depends on continuous ATP hydrolysis by cohesin-NIPBL (Davidson et al., 2019), we first characterized how the ATPase heads undergo engagement-disengagement cycles, which are thought to be required for ATPase activity. For this purpose, we fused YBBR-tags (Yin et al., 2005) to SMC1’s and SMC3’s C-termini, which are located in the ATPase heads and which are in close proximity in the engaged state (Shi et al., 2020). We labeled biotinylated cohesin complexes on these tags with Cy3 and Cy5 as donor and acceptor fluorophores, respectively (Figures 3A, S3A, and S3B). The labeled cohesin molecules were immobilized via neutravidin on passivated glass slides and imaged using TIRF microscopy and alternating laser excitation (ALEX) (Kapanidis et al., 2004), which enabled the selection of single molecules containing donor and acceptor fluorophores with 1:1 stoichiometry and to distinguish large conformational changes from blinking or bleaching events (Figure S3C).
Figure 3.

Dynamics of SMC head engagements visualized by smFRET
(A) Experimental setup of single molecule FRET (smFRET) experiments.
(B) Example smFRET traces of head engagement. Dex, direct donor excitation; Aex, direct acceptor excitation; S, stoichiometry; FRET, FRET efficiency; Dem, Aem, donor and acceptor emission in artificial units (A.U.); HMM, hidden Markov model fits of the raw FRET traces. Black arrows indicate donor or acceptor bleaching.
(C) Fraction of wild-type (wt) and mutant cohesin molecules undergoing dynamic head engagements. Shown are means ± SD and values of individual replicates.
(D) Normalized FRET distributions (top row) and transition density plots (bottom row) of head engagements of different cohesin complexes. N, total number of molecules quantified. The FRET distributions show means ± SD of 3 replicates, with exception of cohesinKA (2 replicates).
(E) Fraction of cohesinEQ/EQ complexes with stably engaged ATPase heads (“locked heads”). Shown are means ± SD and values of individual replicates.
(F) Example traces of disengaged ATP-heads (left) and locked ATPase heads (right).
(G) Cartoon illustration of cohesinSMC1-FKBP/SMC3-FRB.
(H) Distribution of head distances in HS-AFM recordings of cohesinSMC1-FKBP/SMC3-FRB. Shown are distances measured in individual frames. n, number of molecules analyzed. Example stills of cohesinSMC1-FKBP/SMC3-FRB are shown on the right. Heads (he), hinges (hi), and STAG1 (S) are indicated.
(I) Example stills of loop extrusion events by cohesinSMC1-FKBP/SMC3-FRB in presence of DMSO or rapamycin. DNA was stained with Sytox Orange. Scale bar, 2 μm.
(J) Frequencies of loop extrusion events catalyzed by cohesinSMC1-FKBP/SMC3-FRB or cohesinSMC1-FKBP. Shown are means ± SD (3 replicates).
(K) Rates of individual loop extrusion events catalyzed by cohesinSMC1-FKBP/SMC3-FRB and violin plot of their distribution.
(L) ATPase rates of wt cohesin or cohesinSMC1-FKBP/SMC3-FRB. Shown are means ± SD (2 replicates). ATP hydrolysis was determined by thin layer chromatography.
See also Figure S3.
Figure S3.

Kinetics of head engagement visualized by smFRET, related to Figure 3
A.) Positions of the head sensor probes in the cryo-EM model of human cohesin-NIPBL (PDB: 6WG3). The positions of the YBBR-tags are indicated with purple spheres.
B.) Purity and labeling of cohesin with donor and acceptor fluorophores (sulfo-Cy3 and sulfo-Cy5, respectively). CY3: donor fluorescence. CY5: acceptor fluorescence. Molecular weights are indicated in kilo Daltons (kDa). Note that the YBBR-tags are labeled randomly with donor and acceptor molecules.
C.) Example field of view of FRET-labeled cohesin molecules immobilized on glass slides and imaged by TIRF microscopy and alternating laser excitation. Top: First frame of a movie imaged upon direct donor excitation (Dex). Bottom: Second frame imaged upon direct acceptor excitation (Aex). Left: Image of donor emission (Dem). Right: Image of acceptor emission (Aem).
D.) Effect of DNA on the rates of head engagement. Shown are transition density plots of cohesin-NIPBL in presence of ATP and ds DNA. Transition density plots from the two conditions without DNA were taken from Figure 3D for direct comparison. N, total number of molecules.
E.) Dwell time distributions of low and high FRET states (left and right columns, respectively) of cohesin in presence of ATP alone or ATP and NIPBL (top and bottom, respectively). Single exponential decay fits are indicated with a red line. The half time (t1/2) of each fit is shown.
F.) Estimated head engagement rates under different experimental conditions. Engagement rates were quantified as the inverse sum of the half times of the low and high FRET states (as determined in E.).
G.) Kinetics of ATP hydrolysis of wild-type and head sensor cohesin complexes, as measured by a coupled ATPase assay. The assay measures the conversion NADH to NAD by a decrease in the absorbance at 340nm (A340nm), which is coupled to the regeneration of ATP hydrolyzed by cohesin-NIPBL.
H.) Quantification of ATPase rates as measured in G.) under different conditions. A 75-base ds DNA fragment was used as the DNA substrate. Shown are means ± SD and values of individual replicates.
I.) FRET distributions of the experiments shown in Figure 3E. Shown are means ± SD (3 replicates).
J.) Example smFRET trace of cohesinEQ/EQ undergoing reversible head engagement cycles in presence of NIPBL and ATP. Note that the observation of reversible head engagements by cohesinEQ/EQ indicates that head disengagements can occur without prior ATP-hydrolysis.
K.) Model of the head-engagement/disengagement cycle. In the apo-state (1), the heads are disengaged. In the presence of NIPBL (omitted for clarity), the binding of ATP induces the approach and subsequent engagement of the heads (2). Once engaged, in presence of DNA (3), ATP hydrolysis and release allows disengagement (4). In the absence of DNA (3′), the heads can disengage without ATP-hydrolysis (4’). Note that in this case, it is not clear if head disengagement occurs with or without prior ATP dissociation.
L.) Quantification of head distances in hsAFM recordings of cohesinSMC1-FKBP/SMC3-FRB in presence of DMSO or rapamycin for individual cohesin molecules. Each spot corresponds to one frame. The panel on the right shows example stills of the indicated molecules.
M.) Rotary shadowing EM images of cohesinSMC1-FKBP/SMC3-FRB in the presence of DMSO or rapamycin.
Analyses of these complexes revealed low FRET in the absence of either NIPBL or ATP (Figure 3B, left panel), with only a few molecules showing infrequent transitions into high FRET states (Figure 3C). In contrast, simultaneous addition of NIPBL and ATP induced rapid and frequent transitions into high FRET states in 38% of all complexes, indicating interactions between the ATPase heads (Figures 3B, right panel, and 3C).
We used hidden Markov modeling (van de Meent et al., 2014) to determine the major conformational states of the ATPase heads and the frequencies of transitions between them. Plotting the initial and final FRET values of these transitions in “heatmaps” (transition density plots) (McKinney et al., 2006) revealed that the majority of transitions occurred between FRET efficiency values (E-values) close to E∼0 and E∼0.8 (Figure 3D). These data indicate that the donor and acceptor fluorophores transiently come into close proximity of ∼4.3 nm (assuming a Förster radius of ∼5.4 nm) (Son et al., 2020), which is consistent with the ATPase heads being engaged in the high-FRET state.
Next, we used this assay to analyze the roles of ATP and DNA in engagement-disengagement cycles of the ATPase heads. An ATP hydrolysis deficient “Walker B” mutant of both SMC1 and SMC3 (SMC1E1157Q, SMC3E1144Q, and abbreviated cohesinEQ/EQ) showed oscillations between low and high FRET states similar to wild-type, whereas an ATP binding deficient “Walker A” mutant of SMC3 (SMC3K38A and abbreviated cohesinKA) showed only a few transitions, indicating that head engagements require ATP binding but not hydrolysis (Figures 3C and 3D). The addition of DNA to wild-type cohesin did not further increase the frequency of FRET transitions (Figures S3D–S3F), even though DNA stimulated the ATPase activity of this FRET sensor (Figures S3G and S3H), indicating that DNA is not rate limiting for head engagement but for ATP hydrolysis. Consistently, dwell times of the engaged and disengaged states were not affected by DNA in the case of wild-type cohesin (Figures S3E and S3F). In contrast, when added to the ATP hydrolysis-deficient Walker B mutant, DNA locked approximately half of the molecules in an engaged state (Figures 3E, 3F, S3I, and S3J), consistent with the finding that such complexes were amenable to structural analyses by cryo-EM and with bulk FRET data obtained for fission yeast cohesin (Collier et al., 2020; Higashi et al., 2020; Shi et al., 2020). These observations indicate that, in the absence of DNA, the ATPase heads of cohesin can disengage without ATP hydrolysis, but this process depends on ATP hydrolysis in the presence of DNA (Figure S3K).
Movements between the ATPase heads are required for DNA loop extrusion
To test if the ATPase head movements observed by smFRET are required for loop extrusion, we engineered cohesin in which these movements can be blocked. For this purpose, we fused FK506 binding protein 12 (FKBP12; hereafter called FKBP) and FKBP-rapamycin binding (FRB) domains to the C termini of SMC1 and SMC3, respectively, and tested if the resulting cohesinSMC1-FKBP/SMC3-FRB complexes could support loop extrusion in the presence of rapamycin, which induces FRB-FKBP dimerization (Chen et al., 1995; Gruber et al., 2006) and should therefore lock the ATPase heads in close proximity (Figure 3G). HS-AFM and rs-EM confirmed that ATPase head movements were restricted by rapamycin (Figures 3H, S3L, and S3M).
In extrusion assays without rapamycin, cohesinSMC1-FKBP/SMC3-FRB formed DNA loops with frequencies and at rates comparable to those of wild-type cohesin. However, in rapamycin’s presence, cohesinSMC1-FKBP/SMC3-FRB formed very few loops, and these loops were formed at reduced rates (Figures 3I–3K). This effect was dependent on the presence of both FRB and FKBP (Figure 3J). Unexpectedly, however, rapamycin did not inhibit the ATPase activity of cohesinSMC1-FKBP/SMC3-FRB, indicating that the heads of these complexes can still move to the extent that is necessary to enable ATP binding-hydrolysis cycles (Figure 3L). This implies that the deficiency of these complexes in mediating loop extrusion is not caused by defects in ATP hydrolysis but may be a result of their inability to undergo large-scale conformational changes (see below for why these complexes might be unable to extrude DNA).
Alignment of the upper coiled coils facilitates DNA loop extrusion
To analyze interactions between the coiled coils of SMC1 and SMC3 that occur when cohesin adopts a rod conformation, we introduced smFRET probes after amino acid residues 785 and 786 in SMC1 and SMC3, respectively, on the hinge-proximal side of the flexible “elbow” regions (Bürmann et al., 2019) (Figures 4A and S4A). The resulting smFRET sensor retained wild-type levels of DNA-stimulated ATPase activity (Figures S4B and S4C). As predicted by the finding that these regions can be chemically cross-linked (Huis in ’t Veld et al., 2014), cohesin-containing acceptor and donor fluorophores at these sites showed spontaneous transitions between low and high FRET states close to E∼0 and E∼0.8, respectively, which were reached via an intermediate FRET state at E∼0.4 (Figures 4B and S4D–S4F). This indicates that these regions of the coiled coils can interact dynamically, presumably reflecting the alignment of the upper coiled coils observed by HS-AFM (Figure 2B).
Figure 4.

Dynamics of coiled coil alignments visualized by smFRET
(A) Design of the coiled coil smFRET sensor.
(B) Example smFRET traces of the coiled coil sensor.
(C) FRET distributions of the coiled coil sensor. Shown are means ± SD from 3 replicates, with exception of –ATP/+NIPBL (2 replicates). N, total number of molecules analyzed.
(D) FRET distributions of cohesinR711Q and cohesinEEEG. Shown are means ± SD from 3 replicates.
(E) HS-AFM recordings of wt cohesin and cohesinEEEG. Heads (he), hinges (hi), and STAG1 (S) are indicated.
(F) ATPase rates of wt cohesin, cohesinR711Q and cohesinEEEG. Shown are means ± SD and values of individual replicates.
(G) Example stills of loop extrusion events by wt cohesin, cohesinR711Q, and cohesinEEEG. DNA was stained with Sytox Orange. Scale bar, 2 μm.
(H) Frequencies of loop extrusion events. Shown are means ± SD and values of individual replicates.
See also Figure S4.
Figure S4.

Kinetics of coiled coil alignment visualized by smFRET, related to Figure 4
A.) Purity and labeling of the cohesin coiled coil sensor with donor and acceptor fluorophores. YBBR-tags were introduced into the ‘elbow’ region of the SMC1 and SMC3 coiled coils. This flexible region comprises residues 392-393 and 781-803 in SMC1 and 375-392 and 790-791 in SMC3. CY3: donor emission. CY5: acceptor emission.
B.) Coupled ATPase assay of wild-type and coiled coil sensor cohesin complexes (as described in Figure S3G).
C.) Quantification of ATPase rates as measured in B.) under different conditions. A 75 base pair DNA fragment was used as the DNA substrate. Shown are means ± SD and values of individual replicates.
D.) Example smFRET traces of coiled coil alignments in presence of ATP and NIPBL.
E.) Example smFRET traces of coiled coil alignments in the absence of ATP but in the presence of NIPBL.
F.) Transition density plots of wt cohesin, cohesinR711Q and cohesinEEEG in presence of ATP and in presence or absence of NIPBL. N: total number of molecules.
G.) FRET distributions of the coiled coil sensor in presence of ATP and NIPBL and in presence or absence of either 75 bp DNA or puc19 plasmid DNA. N: total number of molecules. Shown are means ± SD from three replicates (‘- DNA’ and ‘+ 75 bp DNA’) and two replicates (‘+ puc19’ DNA).
H.) Effect of high salt concentrations on the behavior of the coiled coil sensor. Cohesin was imaged in the presence of ATP and in the presence of 50 mM (blue bars) or 750 mM (red bars) potassium chloride (KCl). Shown are FRET distributions of the two conditions.
I.) Examples of Cornelia de Lange mutations found in the coiled coil domain of SMC1.
J.) Further HS-AFM stills of cohesinEEEG. Heads (he), hinge (hi) and STAG1 (S) are indicated.
K.) Frequencies of frames with aligned coiled coils for wt cohesin and cohesinEEEG (left) and frequencies of hinge bending (right), as observed by HS-AFM. Shown are the frequencies determined from movies of individual molecules. The number of molecules analyzed is indicated (n). Shown are values of individual molecules and violin plots of their distributions.
In the presence of ATP alone, transitions into the high FRET states occurred occasionally, but at steady state, most cohesin complexes were in the low FRET state. Addition of NIPBL caused rapid oscillations between low and high FRET states and increased the occupancy of the high FRET state (Figures 4B and 4C, right panel), indicating that NIPBL promotes alignment of the upper coiled coils. Stimulation of ATP hydrolysis by DNA caused only a small decrease in the high FRET states (Figure S4G), whereas omission of ATP in either the presence or absence of NIPBL had no detectable effect (Figures 4C, left panel, and S4E). These results suggest that the association of the coiled coils does not depend on ATP binding and hydrolysis and might instead be a movement that occurs spontaneously and is driven by Brownian motion.
To test whether coiled coil interactions are required for loop extrusion, we searched for mutations that reduce these interactions. We observed that high salt concentrations reduced the occupancy of the high FRET state in the coiled coil sensor complexes, indicating that the coiled coils might align by forming electrostatic interactions (Figure S4H). We noticed further that several Cornelia de Lange syndrome (CdLS) mutations in the coiled coil of SMC1 cause the neutralization of positively charged residues, which might normally form such electrostatic interactions (Figure S4I). We therefore generated coiled coil sensors, in which some of these residues are mutated, either singly (SMC1R711Q) or in combination (R693E, R711E, R790E, and R816G, referred to here as SMC1EEEG). smFRET showed that the SMC1R711Q mutation reduced the occupancy of high FRET states, but in these complexes coiled coil alignments were still stimulated to some extent by NIPBL (Figure 4D, left panel). By contrast, complexes containing SMC1EEEG failed to stably align their coiled coils in either the absence or presence of NIPBL (Figures 4D, right panel, and S4F). HS-AFM indicated that this defect was not caused by misfolding or aggregation of cohesinSMC1-EEEG because cohesin containing these mutations formed ring-shaped complexes that were indistinguishable from wild-type cohesin in this conformation. No rod-shaped conformations of cohesinSMC1-EEEG were observed, supporting the conclusion that these complexes are unable to align their coiled coils (Figures 4E, S4J, and S4K).
The ATPase activities of cohesin complexes containing SMC1R711Q and SMC1EEEG were similarly stimulated by NIPBL and DNA as wild-type cohesin (Figure 4F). In extrusion assays, cohesin containing SMC1R711Q formed DNA loops, but at reduced frequencies compared to wild-type cohesin, whereas complexes containing SMC1EEEG were unable to form any loops (Figures 4G and 4H).
These results suggest that alignment of the upper coiled coils facilitates loop extrusion even though this interaction is not needed for ATPase head engagement and ATP hydrolysis and it does not depend on these processes. Interactions between the coiled coils might therefore be required for loop extrusion because they enable conformational changes in cohesin that are needed for the translocation of DNA.
Bending of the hinge toward the SMC3 head is required for DNA loop extrusion
To analyze the bending of cohesin complexes by smFRET, we introduced one FRET probe into the hinge by inserting a YBBR-tag at amino acid residue 594 in SMC3 and varied the position of the second between regions in the SMC1 and SMC3 heads (Figures 5A and S5A–S5C). Dynamic FRET traces could be obtained with three smFRET sensor complexes carrying a fluorophore on or adjacent to the SMC3 head (sensor pairs 1, 2, and 3) (Figures 5B–5D, 5E, left panel, and S5D), but not with two complexes containing a fluorophore on the SMC1 head (sensor pairs 4 and 5) (Figures 5E, right panel, and S5E).
Figure 5.

Dynamics of hinge bending visualized by smFRET
(A) Design of the hinge bending sensors.
(B) Example smFRET trace of sensor pair 1.
(C) FRET distributions of hinge bending (sensor pair 1). Shown are means ± SD (3 replicates). N, number of molecules analyzed.
(D) Transition density plots of sensor pair 1.
(E) Transition density plots of sensor pairs 2–5. Pooled data from 3 replicates (sensors 2 and 3) and 2 replicates (sensors 4 and 5).
(F) Rotary shadowed electron micrographs of cohesinFKBP-SCC1/SMC3-intFRB. Top cartoon: design of cohesinFKBP-SCC1/SMC3-intFRB. Bottom cartoons: observed conformations.
(G) Time-lapse recordings of loop extrusion events by cohesinFKBP-SCC1/SMC3-intFRB in presence of DMSO or rapamycin. DNA was stained with Sytox Orange. Scale bar, 2 μm.
(H) Loop maintenance in presence of DMSO or rapamycin. Loops were first formed by cohesinFKBP-SCC1/SMC3-intFRB (step 1) followed by flow-in of DMSO or rapamycin (step 2). Bottom: example stills of the three major fates of loops upon rapamycin flow-in. Scale bar, 2 μm.
(I) Loop extrusion frequencies of cohesinSMC3-intFRB and cohesinFKBP-SCC1/SMC3-intFRB. Shown are means ± SD and values of individual replicates.
(J) Fraction of loops formed before (1) and after (2) flow-in of DMSO or rapamycin. Shown are means ± SD (3 replicates).
See also Figure S5.
Figure S5.

Kinetics of hinge bending visualized by smFRET, related to Figure 5
A.) Positions of YBBR-tag and FRB-insertions in the hinge domain. The X-ray structure of the mouse hinge domain is shown (PDB: 2WD5), with SMC3 in green and SMC1 in yellow. Residues in between which the YBBR-tag and the FRB-domain were inserted are shown as red and blue spheres, respectively.
B.) Cartoon illustration showing the positions of the YBBR-tags in the hinge-bending sensor constructs used in Figure 5.
C.) Purity and labeling of the hinge-bending sensor constructs used in Figure 5. CY3: donor fluorescence. CY5: acceptor fluorescence.
D.) Example FRET traces of hinge-bending sensor pairs 1, 2 and 3 in the presence of ATP and NIPBL.
E.) Example FRET traces of hinge bending sensor pairs 4 and 5 in the presence of ATP and NIPBL.
F.) Coupled ATPase assay of wild-type and hinge-bending sensor cohesin complexes (sensor pair 1).
G.) Quantification of ATPase rates under different conditions as performed under F.). A 75 base pair (bp) DNA fragment was used as the DNA substrate. Shown are means ± SD and values of individual replicates.
H.) FRET distributions of hinge-bending sensor pair 1 in presence of ATP and NIPBL and in presence or absence of either 75 bp DNA or puc19 plasmid DNA. N: total number of molecules. Shown are means ± SD (3 replicates).
I.) Purity of cohesinSMC3-intFRB/FKBP-SCC1 and cohesinSMC3-intFRB. Shown is a Coomassie stained gel.
J.) Examples of cohesinSMC3-intFRB/FKBP-SCC1 in presence of DMSO or rapamycin as visualized by rotary shadowing EM. Note that complexes locked with rapamycin appear to be in conformations with aligned upper coiled coils, even though the harsh specimen preparation required for rotary shadowing EM separated the coiled coils in the absence of rapamycin. This observation suggests that the coiled coils are very stably aligned in the bent conformation, perhaps because they are twisted around each other as we observed by HS-AFM (Figures 2B and 2C).
K.) Loop maintenance in presence of rapamycin but absence of NIPBL. Loops were first formed by cohesinFKBP-SCC1/SMC3-intFRB in presence of ATP and NIPBL (step 1) followed by flow-in of rapamycin in presence of ATP but in absence of NIPBL (step 2). Shown are means ± SD (2 replicates).
These results confirm the asymmetric bending seen by HS-AFM, show that the hinge bends toward the head of SMC3, and indicate that the hinge and the SMC3 head come into close proximity of up to ∼4 nm. As mentioned, this distance is shorter than the head-hinge distance observed by cryo-EM (Shi et al., 2020; Petela et al., 2021), indicating that the hinge can come closer to the SMC3 head than can be seen in these cryo-EM structures.
In subsequent experiments, we analyzed hinge bending by using sensor pair 1 (FRET probes in the SMC3 hinge and the N terminus of SCC1, which is located next to the SMC3 head) because these displayed the highest FRET efficiencies of up to E∼0.8. Dynamic traces were also observed in the absence of ATP and NIPBL, suggesting that hinge bending—similar to coiled coil alignment—occurs spontaneously and is driven by Brownian motion (Figure 5C). In contrast to the data obtained for the ATPase heads and the coiled coils, the observed FRET distributions did not center on discrete states but were spread across the FRET spectrum, indicating that the movements that bring the hinge and SMC3 head into proximity have many degrees of freedom (Figure 5C). Although we did not observe major effects of ATP and NIPBL on the steady state FRET distributions (Figure 5C), addition of ATP and NIPBL separately or in combination affected the frequency and to a lesser extent also the FRET values of the transitions counted in density plots (Figure 5D), suggesting that ATP and NIPBL modify the geometry of the bending movements. Addition of DNA in presence of NIPBL and ATP did not significantly change the FRET distributions, even though DNA stimulated the ATPase activity of the hinge bending sensor (Figures S5F–S5H).
To address whether bending is required for loop extrusion, we integrated FRB into the SMC3 hinge and fused FKBP to the N terminus of SCC1 to generate cohesin that could be trapped in a bent conformation by rapamycin (Figures 5F and S5I). rs-EM of the resulting cohesinSMC3-intFRB/FKBP-SCC1 complexes revealed predominantly ring-like conformations in the absence of rapamycin, as described for wild-type cohesin (Anderson et al., 2002; Huis in ’t Veld et al., 2014). By contrast, complexes with a sharp bend in the center of the coiled coils were observed in the presence of rapamycin (Figures 5F and S5J), confirming that these complexes can be locked in this conformation.
In the absence of rapamycin, cohesinSMC3-intFRB/FKBP-SCC1 extruded DNA into loops similarly frequently as wild-type cohesin, but rapamycin inhibited this activity almost completely, an effect that was dependent on the presence of both FRB and FKBP (Figures 5G and 5I). Hinge bending was also essential for loop maintenance because flow-in of rapamycin after loop establishment caused full or partial disassembly of most loops (Figures 5H, 5J, and S5K). These results indicate that hinge bending is required for both the initiation and elongation phases of loop extrusion.
Alignment of the upper coiled coils is required for hinge bending
Our results so far indicated that all three conformational changes that we had observed by HS-AFM—head movements, coiled coil alignment, and reversible hinge bending—are required for loop extrusion, consistent with the possibility that they contribute to the translocation of DNA. For this to be the case, one would expect that these conformational changes are coordinated with each other and with cohesin’s ATP binding-hydrolysis cycle. We therefore analyzed interdependencies between these conformational changes.
Because we had observed by HS-AFM that the upper coiled coils were often aligned when bending occurred (Figures 2B and 2D), we tested first whether upper coiled coil alignment is required for hinge bending. We therefore introduced FRET probes for bending into cohesin complexes containing SMC1EEEG, which are defective in coiled coil interactions. These complexes were unable to transit into high FRET states even in the presence of NIPBL and ATP and instead displayed FRET efficiencies close to E∼0 (Figures 6A and S6A). These results suggest that hinge bending occurs preferentially when the coiled coils are aligned. This conclusion was supported by HS-AFM imaging of cohesinSMC1-EEEG complexes because these did neither align their coiled coils nor show any bending movements (Figure S4K).
Figure 6.

Coupling of head engagement, coiled coil alignment, and hinge bending
(A) Hinge bending in cohesinEEEG. Left: FRET distributions of wt cohesin (blue, 3 replicates) and cohesinEEEG (orange, 5 replicates). N, total number of molecules. Shown are means ± SD. Right: quantification of the probability of hinge bending. Shown are means ± SD and values of individual replicates.
(B) FRET distributions of coiled coil alignment in cohesinFKBP-SCC1/SMC3-intFRB. Shown are means ± SD (3 replicates).
(C) ATPase activity of cohesinFKBP-SCC1/SMC3-intFRB. Shown are means ± SD and individual replicates. ATP hydrolysis was measured by thin layer chromatography.
(D) Frequencies of head-engagements of cohesinFKBP-SCC1/SMC3-intFRB. Pooled data of 4 replicates per condition.
(E) Example smFRET traces of hinge bending in cohesinEQ/EQ.
(F) Quantification of hinge bending of cohesinEQ/EQ. Shown are means ± SD and values of individual replicates.
(G) FRET distribution and probabilities of hinge bending in cohesinSMC1-FKBP/SMC3-FRB (a) and cohesinSMC1-FKBP (b). Shown are means ± SD and values of individual replicates.
(H) FRET distributions of coiled coil alignment in cohesinEQ/EQ. Shown are means ± SD (3 replicates).
(I) Model of cohesin’s conformational cycle. NIPBL is omitted for clarity. In absence of ATP (“X”), cohesin cycles between separated and aligned states (1 to 2). When aligned, cohesin can cycle between bent and outstretched conformations (2 to 3). Binding of ATP (“A”) promotes head-engagement and coiled coil separation (4). These events promote the displacement of the hinge from the SMC3 head (4 to 6). Upon full head engagement, ATP hydrolysis separates the heads and resets the cycle (6 to 1).
See also Figure S6.
Figure S6.

Coupling of head engagement, coiled coil alignment, and hinge bending, related to Figure 6
A-C.) Purity and labeling of the hinge bending (A), coiled coil alignment (B) and head engagement sensors (C) used in Figure 6.
D.) FRET distributions of the data shown in Figure 6D. Shown are means and standard deviations from four replicates per condition. The total number of molecules analyzed is indicated (N). Shown are means ± SD (4 replicates).
E.) FRET distributions of the data shown in Figures 6E and 6F. Shown are means ± SD for +ATP/+NIPBL/+DNA and +NIPBL/+DNA (3 replicates) and +ATP/+NIPBL (2 replicates). The total number of molecules analyzed is indicated (N).
To further test whether coiled coil alignment and hinge bending are linked, we integrated the FRET sensor for coiled coil interactions into cohesin containing SMC3intFRB and SCC1FKBP (i.e., into complexes that can be locked in the bent state) (Figure 5F). Without rapamycin, these complexes behaved similarly as the coiled coil FRET sensor complexes not containing FRB and FKBP, i.e., they showed predominantly low FRET efficiencies in the absence of NIPBL but an increase in high FRET in the presence of NIPBL. Interestingly, addition of rapamycin mimicked the effect of NIPBL, i.e., led to a strong increase in high FRET efficiencies, indicating that locking cohesin in a bent conformation traps the upper coiled coils in an aligned state (Figures 6B and S6B), as can also be observed by rotary shadowing EM (Figure 5F). These results support the hypothesis that hinge bending occurs preferentially when the coiled coils are aligned. Because hinge bending is required for loop extrusion (Figures 5G–5J), these results also provide additional support for the notion that coiled coil alignment facilitates loop extrusion (Figures 4G and 4H).
Hinge bending and head engagement are mutually exclusive
Because HS-AFM had shown that hinge bending occurs only when the ATPase heads are separated (Figures 2B, 2D, and 2E), we tested whether hinge bending is also coupled to head engagement-disengagement cycles. We therefore analyzed whether locking cohesin in the bent state affects its ability to hydrolyze ATP. For this purpose, we measured the effect of rapamycin on the ATPase activity of cohesinSMC3-intFRB/FKBP-SCC1. Without rapamycin, these complexes (supplemented with NIPBL and DNA) hydrolyzed ATP at rates similar to those achieved by wild-type cohesin, but in the presence of rapamycin, the ATPase activity of cohesin containing SMC3intFRB and SCC1FKBP was strongly inhibited, whereas wild-type cohesin was much less affected (Figure 6C). Measurement of head movements in cohesinSMC3-intFRB/FKBP-SCC1 by smFRET showed that this defect in ATP hydrolysis upon rapamycin addition was caused by an inability of the ATPase heads to engage and did not arise at a later stage of the ATP hydrolysis cycle (Figures 6D and S6D). These results indicate that bending inhibits cohesin’s ATPase activity by preventing head engagement, in line with our HS-AFM data, in which bending could only be observed when the heads were disengaged (Figures 2B, 2D, and 2E).
To be able to test whether the opposite is also true (i.e., whether head engagement prevents hinge bending), we introduced the FRET probes for bending into cohesinEQ/EQ, in which the ATPase heads can be trapped in an engaged pre-hydrolysis state by addition of ATP, NIPBL, and DNA (Figures 3E and 3F). In the absence of either ATP or DNA, these complexes behaved like cohesin containing wild-type ATPase heads (i.e., these complexes oscillated rapidly between high and low FRET states), indicative of dynamic bending and straightening events, and spent ∼50% of their time in high FRET states with E > 0.3. By contrast, simultaneous addition of ATP, NIPBL, and DNA reduced this time by more than half (Figures 6E, 6F, and S6E), suggesting that locking the ATPase heads in an engaged state strongly reduces hinge bending.
The inability of cohesin to bend when the ATPase heads are engaged could be directly caused by the engaged pre-hydrolysis state of the ATPase cycle or by the mere proximity of the ATPase heads. To distinguish between these possibilities, we introduced the FRET probes for bending into cohesin containing SMC3FRB and SMC1FKBP, i.e., into complexes that can be locked in a conformation, in which the heads are in close proximity but ATP binding and hydrolysis can still occur (Figure S6A). In the presence of NIPBL and ATP, these complexes showed FRET distributions similar to those seen for the corresponding FRET sensor complexes not containing FRB and FKBP, with a substantial occupancy of high FRET states. Strikingly, the addition of rapamycin drastically shifted the FRET distributions toward FRET efficiencies close to E∼0, indicating that the mere physical proximity of the ATPase heads prevented bending of cohesin (Figure 6G, left panel). This effect of rapamycin depended on the presence of both FRB and FKBP on the SMC3 and SMC1 heads, respectively (Figure 6G, right panel). These results indicate that hinge bending and head engagement occur in a mutually exclusive manner, possibly because these conformational changes are mechanically and structurally coupled in a way that makes them incompatible with each other.
Importantly, these findings provide a potential explanation for our observation that locking the ATPase heads in proximity by rapamycin prevents loop extrusion without inhibiting the ATPase activity of these complexes (Figures 3G–3L). The smFRET experiments above indicate that it may be the inability of these complexes to bend the hinge toward the SMC3 head that prevents loop extrusion. If so, these results also argue against the possibility that locking cohesin in the bent conformation prevents loop extrusion solely by inhibiting its ATPase activity (Figures 5H and 6C) and instead support the notion that hinge bending has an important direct role in loop extrusion.
These results, combined with the observation that bending usually occurs in a conformation, in which the upper coiled coils are aligned, predicts that head engagement also triggers the separation of the upper coiled coils. To test this, we introduced the coiled coil FRET sensor into cohesinEQ/EQ, which can be locked in the head-engaged conformation by addition of ATP, NIPBL, and DNA (Figure S6B). In the presence of ATP and NIPBL, but in the absence of DNA, cohesinEQ/EQ showed a similar distribution of FRET efficiencies as cohesin containing wild-type ATPase heads. However, addition of DNA resulted in a strong decrease in the high FRET states and a concomitant increase in the low FRET state (Figure 6H), indicating that head engagement promotes separation of the coiled coils.
These results indicate that in cohesin, coiled coil alignment and hinge bending are mutually exclusive with head engagement. This mutual exclusivity might “synchronize” the ATP-independent movements of the coiled coils and the hinge with the ATP-dependent movements of the ATPase heads and by doing so, create oscillations between two different conformations: ATP binding would lead to head engagement, coiled coil opening, and straightening, whereas subsequent ATP hydrolysis in the presence of DNA would cause head disengagement and allow coiled coil alignment and hinge bending (Figure 6I).
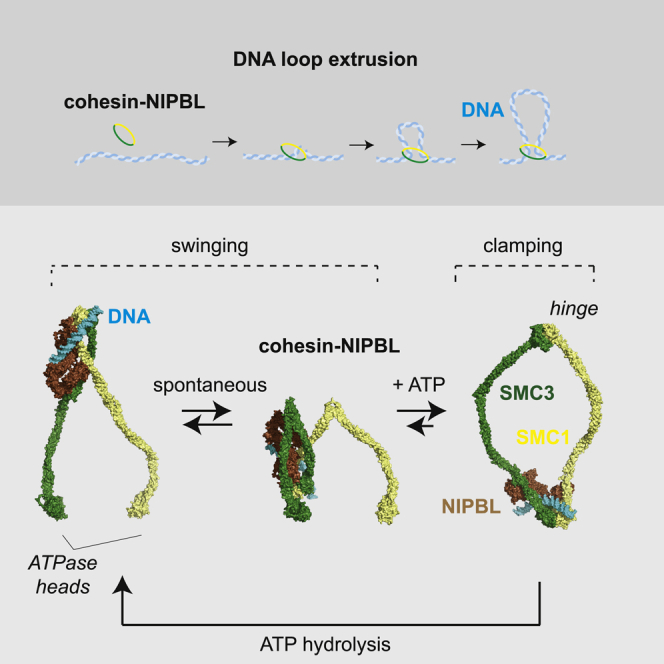
How could oscillations between these two conformations move DNA? Our observations that DNA binding sites on the hinge and the heads and movements between these are required for loop extrusion raise the possibility that DNA is translocated from the hinge to the heads or vice versa. According to this idea, DNA would be translocated by the hinge in the absence of ATP but would be clamped onto the ATPase heads by NIPBL in the presence of ATP. Cycles of ATP binding and hydrolysis would then drive oscillations between these states and thereby translocate DNA. We call this the “swing-and-clamp” model of loop extrusion.
The DNA clamp formed by the ATPase heads and NIPBL is assembled and disassembled during each ATP binding-hydrolysis cycle
The swing-and-clamp hypothesis makes several testable predictions. One of these predictions is that the hinge should bind DNA with high affinity to be able to translocate DNA. Fluorescence polarization experiments revealed that this is indeed the case because, depending on the experimental conditions, the hinge bound DNA with apparent affinities of 200–400 nM (for comparison, we found that the isolated SMC1 and SMC3 heads bound DNA with 1–2 and 2–8 μM, respectively, and NIPBL with 1–2 μM) (Figures S7A–S7C).
Figure S7.

Nucleotide-dependent interactions between STAG1, NIPBL, and cohesin domains, related to Figure 7
A.) Design and purity of a hinge construct containing the upper coiled coils. The coiled coils emanate from the hinge up until the elbow region and are directly fused to one another with a peptide linker that can be cleaved by 3C-protease. In the uncleaved form, the upper coiled coils are in a closed, but upon cleavage in an open conformation, thus mimicking the rod and ring conformations observed in cohesin.
B.) Rotary shadowing EM images of hinge constructs as in (A), before and after cleavage of the peptide linker (‘closed’ and ‘open’, respectively).
C.) Affinities of different cohesin domains and ΔN-NIPBL to DNA as measured by fluorescence polarization. Proteins were incubated at increasing concentrations with a 75 bp fluorescine-labeled DNA probe under the indicated conditions. DNA binding was monitored by measuring the change in polarization (in milli-polarization units, ‘mP’) of the DNA probe upon protein binding. For the hinge containing short coiled coils, the inlet shows an expanded view of the binding data in the low concentration range and binding data for a construct containing the hinge4A mutations in the closed (black interrupted line) and open conformations (black continuous line). For the ATPase heads, experiments were performed in the presence and absence of 2 mM ATP. Note that the presence of ATP increases the apparent affinities of the ATPase heads for DNA. This indicates that nucleotide-dependent conformational changes affect DNA-binding and that the ATPase heads can bind ATP without forming heterodimers.
D.) Binding of TNP-ATP to the ATPase heads. The fluorescence of TNP-ATP was measured at increasing concentrations in the presence and absence of a constant amount of the SMC3 or SMC1 head domains. Plotted is the change in fluorescence due to binding to protein as a function of the concentration of TNP-ATP (three replicates per condition). Note that we could not detect significant binding of TNP-ATP to the SMC1 head using this assay. However, the effect of addition of 2 mM ATP on the DNA-binding affinity of the SMC1 head (C) would indicate that also the SMC1 head binds ATP at high concentrations, which we could not test using TNP-ATP. Furthermore, it is interesting to note that the opposite behavior has been observed for the ATPase heads of yeast condensin, in which the SMC2 head (most similar to SMC1) binds ATP with high affinity and binding of ATP to the SMC4 head (most similar to SMC3) could not be detected (Hassler et al., 2019).
E.) Cartoon illustration of the FRET sensor used to detect interactions between STAG1 and the hinge. CohesinEQ/EQ was used in these experiments to compare engaged and disengaged head conformations.
F.) Example smFRET traces of the hinge-STAG1 sensor, showing transient oscillations between low and high FRET states in the presence but not in absence of ATP.
G.) Left column: FRET distributions of STAG1-hinge interactions. Shown are means ± SD of 3 replicates, with exception of –NIPBL/+ATP/+DNA (2 replicates). The total number of molecules analyzed (N) is indicated. Right column: Transition density plots of conditions shown in the first column.
H.) Cartoon illustration of the FRET sensor pair used to measure interactions between the cohesin hinge and the NIPBL nose. The YBBR-tag was positioned on the N terminus of SCC1, which is close to the SMC3 head.
I.) FRET distributions of nose-SMC3 head interactions in the engaged (top), pre-engaged (middle) and apo conformations (bottom). Means and standard deviations from three replicates.
J.) Example smFRET traces of the hinge-nose sensor in cohesinEQ/EQ (top) and wild-type cohesin (bottom) in the presence of ATP and DNA. Note that the ability to hydrolyze ATP allows cohesin to undergo cycles of binding and dissociation between the hinge and the nose of NIPBL.
K.) Cartoon illustration of the hinge-nose sensor in wild-type cohesin.
L.) FRET distributions of the hinge-nose sensor in wild-type cohesin in different nucleotide conditions (orange). Shown are means ± SD (3 replicates). The corresponding distributions recorded for cohesinEQ/EQ (Figure 7) are outlined in black.
M.) Estimated rates of hinge-nose interactions in cohesinEQ/EQ and wild-type cohesin under different conditions. Half times of low and high FRET states were determined by fitting the distribution of their dwell times to single exponential decay curves. The rates were estimated as the inverse sum of the half times of the low and high FRET states.
Another prediction is that the DNA clamp is assembled and disassembled during each ATP-binding-hydrolysis cycle. To test this, we generated smFRET sensors with which interactions between NIPBL and the ATPase heads (i.e., the integrity of the DNA clamp) could be analyzed. We introduced pairs of FRET probes into the N-terminal part of NIPBL’s U-shaped “body” (at residue 1860; “body-N”) and the N-terminal helix of SCC1 (next to the SMC3 head at residue 92; therefore referred to as “SMC3 head”) and into the C-terminal part of NIPBL’s body (at residue 2154; “body-C”) and the C-terminal ATPase lobe of SMC1 (at residue 1092; referred to as “SMC1 head”) to monitor interactions between NIPBL and the SMC3 head and between NIPBL and the SMC1 head, respectively (Figure 7A). We used cohesinEQ/EQ to be able to compare the binding of NIPBL between the engaged and disengaged conformations of the ATPase heads and ΔN-NIPBL to facilitate protein labeling.
Figure 7.

Nucleotide dependent interface switching of NIPBL
(A) FRET sensor pairs for NIPBL-head and NIPBL-hinge interactions. Body and nose of NIPBL are indicated.
(B) FRET distributions of the SMC3 head-body-N sensor in the engaged, the pre-engaged, and the apo states (top to bottom). N, number of molecules. Shown are means ± SD (3 replicates).
(C) As in (B) but for the SMC1 head-body-C sensor. Shown are means ± SD (3 replicates).
(D) As in (B) but for the hinge-nose sensor. Shown are means ± SD (3 replicates).
(E) Fraction of stably bound complexes. Shown are means ± SD and values of individual replicates.
(D) “Swing and clamp” model of DNA translocation. In the nucleotide free state (“X”), NIPBL interacts via its nose with the hinge. Bending moves the hinge together with NIPBL to the SMC3-head. Upon ATP-binding (“A”), the body of NIPBL forms a clamp with the SMC3 head, inducing the engagement of the heads. The hinge dissociates from the nose and the coiled coils stretch. The engagement of the heads triggers ATP hydrolysis and results in dissociation of the clamp and disengagement of the heads, resetting cohesin for the next cycle. The proposed pathway of a DNA stretch from the hinge to the SMC3 head is indicated. At the end of one cycle, the DNA segment may be transferred to the SMC1 head.
See also Figure S7.
In the presence of ATP and DNA (i.e., under conditions that promote head engagement), addition of ΔN-NIPBLbody-N-Cy5 to cohesin containing SMC3head-Cy3 resulted in traces with high FRET efficiencies (E∼0.8), consistent with an alignment of the DNA-binding regions of NIPBL and SMC3 as part of the DNA clamp (Figure 7B, top panel). Most traces were static, indicating that ΔN-NIPBL was bound to the SMC3 head in a single stable conformation (Figure 7E, top panel). A similar distribution of static high FRET traces was observed when we monitored the binding of ΔN-NIPBLbody-C-Cy5 to cohesin containing SMC1head-Cy3, indicating that NIPBL is simultaneously bound to the SMC1 and the SMC3 heads when these are engaged, as has been observed by cryo-EM (Shi et al., 2020) (Figures 7C and 7E, top panels).
In the presence of ATP, but in the absence of DNA, i.e., under conditions in which the ATPase heads interact only transiently (i.e., the “pre-engaged state”) (Figure 3E), smFRET between the SMC3 head and the NIPBL body remained high and static (Figures 7B and 7E, middle panels). By contrast, the static high-FRET interactions between the SMC1 head and the NIPBL body seen in the presence of DNA were almost completely lost, with the majority of traces showing FRET efficiencies close to E∼0 (Figures 7C and 7E, middle panels).
Omission of ATP (in presence of DNA, i.e., the “apo” state) resulted in a complete loss of FRET for both sensor pairs, suggesting that under these conditions, NIPBL had dissociated from the ATPase heads. However, NIPBL could still be detected by direct acceptor excitation, indicating that NIPBL remained bound to other sites on cohesin (Figures 7B, 7C, and 7E, bottom panels).
These results indicate that the DNA clamp that is formed by cohesin and NIPBL is indeed assembled and disassembled during each ATP binding-hydrolysis cycle. Unexpectedly, however, our data show that ATP binding already promotes an interaction between NIPBL and the SMC3 head before the heads are engaged. We could not directly test if the formation of the NIPBL-SMC3 head interface can capture a DNA segment before head engagement occurs, but the existence of similar static FRET states in the presence and absence of DNA is consistent with this possibility. Using fluorescent ATP-analogs, we could further show that the SMC3 head can bind ATP in the absence of head engagement (Figure S7D). Our results therefore suggest that the SMC3 head and NIPBL clamp DNA already upon ATP binding to SMC3 and before head engagement. In this pre-engaged clamp, DNA would contact the two binding sites on the SMC3 head and on NIPBL that are required for stimulation of cohesin’s ATPase activity. However, ATP hydrolysis could only occur once, in a second step, the pre-engaged clamp and the SMC1 head approached each other and engaged, leading to the formation of two composite ATP binding sites. Upon engagement, DNA would trigger ATP hydrolysis, causing disassembly of the clamp and disengagement of the two ATPase heads, thereby resetting cohesin for the next ATP binding-hydrolysis cycle.
Cohesin’s nucleotide state determines whether NIPBL forms part of the DNA clamp or interacts with the hinge
In the cryo-EM structures of cohesin, the hinge is located in proximity to STAG1 and to NIPBL’s N-terminal HEAT repeats (Collier et al., 2020; Higashi et al., 2020; Shi et al., 2020). In this conformation, STAG1 and parts of NIPBL are located between the hinge and the SMC3 head. Another prediction of the swing-and-clamp hypothesis is therefore that these two subunits have to undergo conformational changes or change their positions for the hinge to be able to bend toward the SMC3 head. We therefore analyzed by smFRET under which conditions the hinge is located in proximity to STAG1 and NIPBL.
Analyses of cohesinEQ/EQ complexes containing FRET sensors in the hinge (introduced at SMC3 residue 594) and STAG1 (introduced at residue 714) revealed dynamic interactions that depended on the presence of NIPBL and ATP, suggesting that the hinge comes close to STAG1 only transiently in the presence of these factors and might therefore not affect the passage of the hinge (Figures S7E–S7G).
Interestingly, we obtained very different results for interactions between NIPBL and the hinge. In the cryo-EM structures of cohesin-NIPBL in its ATP-bound engaged state, the hinge is located next to NIPBL’s N-terminal HEAT-repeats. These are located in a domain called “the nose” (Yatskevich et al., 2019), whereas the DNA binding site that is part of the clamp is located in the U-shaped body of NIPBL (Figure 1E). Contrary to what we had expected based on the cryo-EM structure, FRET probes introduced into the hinge (at SMC1 residue 579) and NIPBL’s nose (residue 1217) (Figure 7A) showed no interactions when cohesinEQ/EQ was locked in the engaged state in the presence of ATP and DNA (Figures 7D and 7E, top panels), and only a few high FRET traces were observed in the presence of ATP alone (i.e., when the heads interact transiently) (Figures 7D and 7E, middle panels). However, the apo state resulted in strong interactions between NIPBL and the hinge (Figure 7D, bottom panel). Most of these traces were static (Figure 7E, bottom panel), indicating that NIPBL’s nose is stably interacting with the hinge in the nucleotide-free state. Because the coiled coils bend frequently under these conditions (Figure 5), leading to movement of the hinge, these results suggest that NIPBL’s nose “travels” with the hinge in the nucleotide-free state. To test this idea further, we measured interactions between FRET sensors placed in the nose of NIPBL and the N terminus of SCC1, which is close to the SMC3 head (Figure S7H). Consistent with the observations that the hinge comes into close proximity to the SMC3 head, and the hinge interacts with NIPBLs nose only in the nucleotide-free state, we found interactions between the nose and the SMC3 head only in the apo but not in the engaged state (Figure S7I, bottom versus top panels).
Because these experiments had been performed with cohesinEQ/EQ, which does not undergo cycles of ATP hydrolysis, we next tested if hinge-nose interactions also occur in cycling cohesin complexes. In contrast to cohesinEQ/EQ, in which hinge-nose interactions could only be seen in the absence of ATP, wild-type cohesin showed dynamic and rapid oscillations between low and high FRET states in the presence of ATP and mostly static traces in the absence of nucleotide (Figures S7J–S7M). Strikingly, the rates of these transitions were strongly stimulated by the presence of DNA (Figure S7M). Because DNA stimulates cohesin’s ATPase activity, these findings indicate that in cycling cohesin, hinge-nose interactions occur upon ATP hydrolysis and persist until the SMC3 head binds to the next ATP molecule.
These results support the hypothesis that NIPBL’s nose travels at least some distance with the hinge (i.e., moves toward the SMC3 head and away from it during coiled coil bending and straightening). The proximity that has been observed between the hinge and NIPBL’s nose in the engaged state by cryo-EM might therefore represent a rare or intermediate conformation, possibly because a subset of only 10% of the particles (6,857 out of 68,161) were selected to resolve the locations of the hinge (Shi et al., 2020). Consistent with this possibility, “time resolved” cryo-EM studies have revealed a high degree of conformational flexibility in macromolecular complexes and shown that even sparsely populated conformations can be detected by cryo-EM (Fischer et al., 2010).
Together with the cyclic DNA clamping described above, these results indicate that not only SMC1 and SMC3, but also NIPBL, undergoes major conformational changes and/or changes in its position relative to other subunits during cohesin’s ATP binding-hydrolysis cycle (Figure 7F; note that conformational changes in fungal NIPBL orthologs have also been suggested based on comparisons of crystal and cryo-EM structures) (Higashi et al., 2021). In the absence of ATP (i.e., when cohesin’s ATPase heads are disengaged and the DNA clamp is disassembled), NIPBL’s nose is located in close proximity to the hinge; in the presence of ATP (i.e., when the heads can engage), NIPBL’s body clamps DNA onto the ATPase heads, but the hinge and NIPBL’s nose separate from each other. Depending on the nucleotide state, different domains of NIPBL therefore interact with the DNA binding sites on the ATPase heads or the hinge (Figure 7F).
Discussion
A swing and clamp model for DNA translocation
How DNA is folded by loop extruding SMC complexes has remained a mystery, although this process is thought to have important structural and regulatory functions and to determine genome architecture in all kingdoms of life (for review, see Davidson and Peters, 2021; Yatskevich et al., 2019). We have therefore analyzed at the single-molecule level how loop extrusion is mediated by human cohesin-NIPBL.
Based on the results of these experiments, we propose that DNA is translocated by movements of the hinge toward the ATPase head of SMC3 and handed-over there from the hinge to the ATPase heads in a manner that is controlled by NIPBL (Figure 7F). According to this hypothesis, cohesin-NIPBL would bind DNA in the nucleotide-free apo state at the hinge, which under these conditions is interacting with NIPBL’s nose. Cohesin’s coiled coils would spontaneously align and fold in a process driven by thermal motion and thereby translocate the DNA segment bound to the hinge over a distance of ∼50 nm toward the SMC3 head. We refer to this step as “the hinge swing.” If in this conformation ATP bound to the ATPase heads, this would cause separation of NIPBL’s nose from the hinge and would enable transfer of the DNA segment from the hinge to the SMC3 head where NIPBL’s body would clamp it. Subsequent engagement of the ATPase heads would force the hinge away from the SMC3 head because coiled coil bending and head engagement are incompatible. Because the DNA has been clamped onto the ATPase heads by NIPBL at this stage, the hinge could not move the DNA back but would have to dissociate from it, possibly in a step that is facilitated by the separation of NIPBL’s nose from the hinge. The large degree of freedom with which the hinge moves during this process might enable the hinge to search for the next DNA segment. ATP hydrolysis triggered by head engagement and DNA inside the clamp would recreate the apo state by leading to disengagement of the heads and disassembly of the clamp. These events would enable the hinge to interact again with NIPBL’s nose for the next round of DNA translocation and would unclamp DNA bound to the ATPase heads so that it could be pushed beyond them by the next hinge swing.
This model elegantly explains how DNA translocation could be synchronized with the head-engagement and ATPase cycle: because NIPBL is associated with the hinge in the nucleotide-free state, ATP binding might induce head engagement and/or ATP hydrolysis only once the spontaneous bending of the coiled coils has moved the hinge-NIPBL interface into proximity of the SMC3 head, so that NIPBL can clamp DNA onto the SMC3 head and DNA-NIPBL contacts inside this clamp can (together with DNA-SMC3 head contacts) trigger ATP hydrolysis. If such a coupling mechanism did not exist, head engagement could occur at any time, even when the coiled coils are outstretched, which would result in non-productive engagement-ATP hydrolysis cycles without DNA translocation. Because cohesin-NIPBL is able to extrude DNA at high rates of up to 2.1 kb/s (Davidson et al., 2019; Golfier et al., 2020; Kim et al., 2019), we suspect that such non-productive head engagement events are rare. The ability of NIPBL to interact in a mutually exclusive manner with the hinge and the SMC3 head, and by doing so to “jump ship” from one DNA binding site to another, may therefore be a key element of the DNA translocation process. Importantly, this model could also explain how cohesin might be able to move DNA in the presence of nucleosomes or other DNA bound proteins (Pradhan et al., 2021). Because chromatin fibers would be translocated beyond the ATPase heads when these are disengaged and could be 30–50 nm apart from each other, there would be space for DNA bound proteins to pass by the ATPase heads.
It is also possible that DNA is translocated into the opposite direction (i.e., from the ATPase heads toward the hinge), as has been proposed based on modeling and the speculation that STAG1 moves with the hinge (Higashi et al., 2021). Note that our smFRET experiments failed to detect stable interactions between the hinge and STAG1 and thus do not support this speculation (Figures S7E–S7G). We also consider this possibility to be less plausible based on our observation that the hinge can only come close to the SMC3 head when this is disengaged from the SMC1 head. For the hinge to be able to pick up DNA from the ATPase heads these would therefore first have to disengage, which could lead to dissociation of DNA from the heads before the hinge would be in their proximity and could bind DNA. As a result, DNA might neither be bound to the heads nor to the hinge at this stage and would have to be held in place and the correct orientation by other sites. Although possible, we consider this scenario less plausible.
In summary, our results provide first insights into the mechanistic principles through which human cohesin-NIPBL complexes translocate DNA to mediate DNA loop extrusion and may have broader implications for understanding how other SMC complexes perform this function.
Limitations of the study
Although essential for loop extrusion, we have not been able yet to determine the molecular function of STAG1 in this process. For technical reasons, the conformational changes described here could so far only be observed by HS-AFM for trimeric and tetrameric cohesin complexes in the absence of DNA, but not for active loop extruding cohesin-NIPBL molecules on DNA. FRET can only measure proximity between labeled protein domains over a range of up to ∼10 nm but cannot determine how far beyond this the domains are separating during conformational changes. How these changes translocate DNA has been inferred from a combination of results, the identification of DNA binding sites that are required for loop extrusion, the movement of these sites during conformational changes, and the regulation and interdependency of these movements, but DNA translocation has not been measured or visualized directly yet. Further studies will be needed to achieve these aims and to resolve how DNA translocation leads to loop extrusion.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| BL21(DE3) | in house | N/A |
| DH10MultiBAC DH10EmBacY | in house | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Toyopearl AF-Chelate-650M | Tosoh | 0014475 |
| Flag M2 agarose beads | Sigma | A2220-25ML |
| Para-benzoyl phenylalanine | BACHEM | 4017646.0001 |
| Pierce Ultralink Streptavidin Resin | Thermo Scientific | 53113 |
| Culture Well Reusable Gaskets 3x1mm | Grace Biolabs/ Sigma | GBL103250 |
| Hellmanex III | Hellma | 9-307-011-4-507 |
| Gamma-32-ATP | Hartmann Analytic | SCP-501 |
| lambda DNA | New England Biolabs | N3011S |
| polyethylene imide | Sigma | 1055790001 |
| NADH | Sigma | N8129 |
| Phosphoenolpyruvate | Sigma | N0564 |
| lactate dehydrogenase/ pyruvate kinase | Sigma | P0294 |
| avidin DN | Vectorlabs | A-3100-1 |
| BSA (loop extrusion assays) | Thermo Scientific | AM2616 |
| BSA fraction V (others) | VWR | 422351S |
| ATP (loop extrusion assays) | Jena Biosciences | NU-1010 |
| ATP (others) | Roche/ Sigma | ATPD-RO |
| Sytox Orange | Thermo Scientific | S11368 |
| glucose oxidase | Sigma | G2133 |
| Catalase | Sigma | C- 40 |
| Trolox (loop extrusion assays) | Cayman Chemical | 10011659 |
| Trolox (smFRET) | Sigma | 238813 |
| Rapamycin | Sigma | R8781 |
| glass rods | Hilgenberg GmbH | 7001854 |
| mica sheets | NanoAndMore GmbH | 50-S-25-50-1 |
| uhu two component epoxy | Conrad Electronics | 478703 |
| Cantilevers | Oxford Instruments | 803.OLY.BL-AC10DS |
| sulfo Cyanine 3 maleimide | Lumiprobe | 11380 |
| sulfo Cyanine 5 maleimide | Lumiprobe | 13380 |
| Coenzyme A trilithium salt | Sigma | C3019 |
| EZ-Link biotin-PEG11-maleimide | Thermo Scientific | 21911 |
| Dichloromethylsilane | Sigma | 440272 |
| Hexane | Sigma | 139386 |
| Sulfuric acid (for Piranha solution) | Sigma | 258105 |
| Hydrogen peroxide (for Prinanha solution) | Sigma | H1009 |
| Permanent double sided sticking tape 3m | Scotch | N/A |
| Pluronic F-127 | Sigma | P2443 |
| Neutravidin | Thermo Scientific | 31000 |
| beta casein | Sigma | C6905 |
| 4-nitrobenzyl alcohol (NBA) | Sigma | N12821 |
| Cyclooctotetraene (COT) | Sigma | 138924 |
| Protocatechuic acid (PCA) | Sigma | 37580 |
| Protocatechuic acid-3,4-Dioxygenase (PCD) | Sigma | P8279 |
| 2’,3’-O-Trinitrophenyl-adenosine-5’triphosphate | Jena Biosciences | NU-221 |
| Platinum | Baltic | BP2261 |
| Carbon | Balzers | B80100761 |
| Cu/pd grids | Agar Scientific | AGG2440PD |
| Experimental models: Cell lines | ||
| SF9 insect cells | Thermo Scientific | B82501 |
| Hela SCC1-Halo-Flag | Davidson et al. (2019) | N/A |
| Recombinant DNA | ||
| pET21a | Novagen | N/A |
| pEVOL-pBpf | Addgene | RRID:Addgene_31190 |
| Plib | Weissman et al. (2016) | RRID:Addgene_80610 |
| pBig1a | Weissman et al. (2016) | RRID:Addgene_80611 |
| pBig1b | Weissman et al. (2016) | RRID:Addgene_80612 |
| pBig1c | Weissman et al. (2016) | RRID:Addgene_80613 |
| pBig2ab | Weissman et al. (2016) | RRID:Addgene_80616 |
| Software and algorithms | ||
| Prism | Graphpad | Version 8 |
| Fiji | NIH | Version 2.0.0 |
| Gwyddion | http://gwyddion.net/ | Version 2.55 |
| Matlab | Mathworks | Version 2015b |
| iSMS | Preus et al. (2015) | Version 2.01 |
| ebFRET | Van de Meent et al. (2014) | N/A |
| plotTDP Matlab script | http://ebfret.github.io/ | N/A |
Resource availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan-Michael Peters (peters@imp.ac.at).
Materials availability
Requests for plasmids generated in this study should be directed to the lead contact.
Experimental model and subject details
Molecular cloning and mutagenesis
Homo sapiens (hs) SMC1, hs SMC3 with a C-terminal Flag tag, hs SCC1(TEV) with three internal tabacco etch virus (TEV) sites substituting the endogenous separase cleavage site and a C-terminal Halo-tag, and hs STAG1 with an N-terminal 10 histidine tag in pLib vectors were as described in Huis in ’t Veld et al. (2014) and Weissmann et al. (2016). Hs NIPBL with N-terminal FLAG and Halo-tags and a C-terminal 10 histidine tag alone or as a tandem construct with untagged hs MAU2 in pLib were as previously described (Davidson et al., 2019). These constructs were used as the starting points for site-directed mutagenesis to introduce point mutations, deletions and tags by polymerase chain reactions using Phusion Hotstart High Fidelity DNA Polymerase (NEB). Larger protein domain fusions, such as those involving FRB and FKBP domains (Chen et al., 1995) were introduced by Gibson Assembly (Gibson et al., 2009). In the case of STAG1 constructs bound to their cognate SCC1 sequence, the SCC1 fragment was fused to the C terminus of STAG1 via the self-cleaving P2A tag.
pLib constructs encoding cohesin subunits were combined into polycistronic expression cassettes using the bigBAC method (Weissmann et al., 2016). Unless stated otherwise, SCC1 and STAG1 genes were subcloned into Big1a vectors and SMC1 and SMC3 genes into Big1b vectors. For trimeric cohesin, SMC1, SMC3-FLAG and SCC1(TEV)-Halo-His14 were assembled into Big2ab.
Cohesin fragments were cloned into pET21a (Novagen) for bacterial expression using PCR and Gibson assembly. For the SMC3 head, the N-terminal SCC1 fragment was fused via a 39 amino acid linker to the C terminus of the head, as described in Davidson et al. (2019). For the SMC1 head, the C-terminal SCC1 fragment was co-expressed from the same plasmid using a Shine Dalgarno sequence (5′-gaattcgctagcaaaggaataagta-3′) between the coding sequences of the two proteins. For the hinge constructs, the SMC1 and SMC3 fragments were either co-expressed using a Shine Dalgarno sequence between the two coding sequences, or they were fused directly via a cleavable 3C-protease recognition site.
A list of all constructs is shown in Table S1.
Protein expression and purification from SF9 insect cell cultures
pLib, Big1a and Big1b vectors were transformed into DH10 Multibac cells to generate bacmid DNA as described in Weissmann et al. (2016). Baculoviruses were produced in SF9 cells transfected with bacmid DNA by lipofection and cultured for 72 h. A 50 mL V1 virus stock was cultured from this V0 stock for 48 h and used to directly infect expression cultures. Cohesin complexes were expressed by co-infection of SF9 cultures with equal titers of two viruses encoding SCC1/STAG1 and SMC1/SMC3, respectively, with the exception of cohesinSMC1(EQ)-hinge-YBBR, which was expressed by co-infection of cultures with viruses encoding SCC1/STAG1, SMC1(EQ)-hingeYBBR and SMC3(EQ), cohesinSMC1-hinge-YBBR, which was expressed by co-infection of cultures with viruses encoding SCC1/STAG1, SMC1-hingeYBBR and SMC3, and cohesinSMC3(EQ)-hingeYBBR, STAG1-intYBBR, which was expressed by co-infection of cultures with viruses encoding SMC1(EQ)/SMC3(EQ)-hingeYBBR, STAG1-intYBBR, and SCC1. NIPBL, NIPBL/MAU2, STAG1 and versions thereof and in some cases full or partial cohesin complexes were expressed after (co-)infection with viruses generated from pLib vectors. All expression cultures were infected with virus at a density of 1 million cells per milliliter and grown at 100 rpm and 27°C in Grace medium in baffled 2 L flasks filled to 750 ml. Cells were harvested after 48 h by centrifugation, washed once in phosphate buffered saline (PBS) and either processed immediately or snap frozen in liquid nitrogen and stored at −80°C.
All protein purifications were performed at 4°C. Purifications of trimeric and tetrameric cohesin constructs and of full length and ΔN- NIPBL constructs were performed essentially as in Davidson et al. (2019), with minor modifications. Cells were lysed by douncing 25 times in five pellet volumes of Lysis Buffer (50 mM NaPO4 pH 7.6, 500 mM NaCl, 5% glycerol, 0.1% TWEEN, 10 mM imidazole) supplemented with 3 mM beta mercapto ethanol (bME), 1 mM benzamidine, 1 mM phenyl-methyl-sulfonyl fluoride (PMSF) and 1x EDTA-free protease inhibitor cocktail (PIC, Roche). Insoluble material was removed by centrifugation (35 min, 40000 xg, 4°C) and the supernatant was incubated for 3 h at 4°C with 5 mL of Toyopearl AF-chelate-650M resin (Tosoh Bioscience) pre-charged with Ni2+ ions. Beads were collected by centrifugation, washed three times each with 50 mL Lysis-buffer supplemented with additional 5 mM imidazole but lacking bME and protease inhibitors and transferred to a glass column. Bound protein was eluted with 5 × 5 mL Ni-NTA Elution Buffer (50 mM NaPO4 pH 7.6, 150 mM NaCl, 5% glycerol, 300 mM imidazole). The eluate was incubated for further 3 h at 4°C with 5 mL of equilibrated FLAG-M2 agarose resin (Sigma). In the case of NIPBL, the incubations were supplemented with 1 tablet of protease inhibitor cocktail. Beads were collected by centrifugation and washed three times with FLAG buffer (50 mM NaPO4 pH 7.6, 100 mM NaCl, 5% glycerol, 50 mM imidazole), followed by elution of bound protein with 5 × 5 mL of FLAG-buffer supplemented with 0.25 mg/ml 3xFLAG peptide (DYKDHDGDYKDHDIDYKDDDDK). Eluates were concentrated to ∼1 mL in 100kDa molecular weigh cut off (MWCO) spin filters (Amicon), snap frozen in liquid nitrogen and stored at −80°C. Protein concentrations were analyzed by running samples next to BSA standards (Biorad) on SDS-PAGE followed by Coomassie staining and quantification of protein bands in ImageJ (NIH).
For dimeric cohesin, cells were lysed as described for trimeric and tetrameric cohesin. After centrifugation, lysates were incubated with 5 mL FLAG-resin for 3 h at 4°C, beads were collected by centrifugation and washed three times with FLAG buffer (50 mM NaPO4 pH 7.6, 100 mM NaCl, 5% glycerol, 50 mM imidazole), followed by elution of bound protein with five times 5 mL of FLAG-buffer supplemented with 0.25 mg/ml 3xFLAG peptide. Eluates were concentrated to ∼1 mL in 100kDa molecular weigh cut off (MWCO) spin filters (Amicon), snap frozen in liquid nitrogen and stored at −80°C. Protein concentrations were analyzed as described above.
For STAG1 purifications, cells were lysed by douncing with 25 strokes in 5 pellet volumes of Lysis Buffer (20 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole, 3 mM bME), supplemented with 1x PIC, 1 mM PMSF and 1 mM benzamidine. Insoluble material was removed by centrifugation (35 min, 40000 xg, 4°C) and the supernatant applied to 1.5 mL NiNTA-agarose (QIAGEN) for 1.5 h at 4 °C. Beads were collected by centrifugation, washed three times with 50 mL Lysis buffer and transferred to a glass column. Bound protein was eluted with 4 × 2.5 mL of NiNTA elution buffer (20 mM Tris pH 7.5, 300 mM NaCl, 300 mM imidazole, 3 mM bME). Eluates were concentrated to ∼500 μL in 100 kDa MWCO spin filters (Amicon), and subjected to size exclusion chromatography using a Superdex 200 Increase 10/300 GL column (GE Life Sciences), equilibrated in Gelfiltration Buffer (20 mM Tris pH 7.5, 300 mM NaCl, 1 mM DTT). Fractions were analyzed by SDS-PAGE. STAG1-containing fractions were pooled, concentrated, snap frozen in liquid nitrogen in small aliquots and stored at −80°C. Protein concentrations were determined by the Bradford assay.
Protein expression and purification from E. coli cell cultures
100 μL chemically competent BL21(DE3) cells were transformed with expression vectors by heat shock, outgrown at 37°C in 1 mL super optimal broth (SOC) for 1 h, plated on Lysogeny Broth (LB) agar plates supplemented with 100ug/ml ampicillin and grown to colonies over night at 37°C. The colonies were resuspended in LB and used to inoculate 3 × 2L of LB supplemented with 100 μg/ml ampicillin in 5 L baffled flasks. Cells were grown shaking at 180 rpm and 37°C until an optical density (OD) of ∼1.0 had been reached. At this point, the incubator temperature was lowered to 18°C, and protein expression was induced by addition of 0.5 mM isopropyl-beta-D-1-thiogalactopyranoside (IPTG). Cells were harvested after ∼18 h and either processed immediately or snap frozen in liquid nitrogen and stored at −80 °C.
For SMC1 head and SMC3 head constructs, cells were resuspended in five pellet volumes of Lysis Buffer (20 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole, 3 mM bME) supplemented 1 x PIC by douncing and lysed by sonication (Branson Digital Sonifier, 60% amplitude, 150 × 1 s cycles with 1 s pause in between) on ice. Insoluble material was removed by centrifugation (35 min, 40000 xg, 4°C) and the supernatant applied to 1.5 mL NiNTA-agarose for 1.5 h at 4°C. Beads were collected by centrifugation, washed two times with 50 mL Lysis buffer and transferred to a glass column. Bound protein was eluted with 4 × 2.5 mL of NiNTA elution buffer (20 mM Tris pH 7.5, 100 mM NaCl, 300 mM imidazole, 3 mM bME). Eluates were concentrated to ∼500 μL in 50 kDa MWCO spin filters (Amicon), and subjected to size exclusion chromatography using a Superdex 200 Increase 10/300 GL column, equilibrated in Gelfiltration Buffer (20 mM Tris pH 7.5, 100 mM NaCl, 1 mM dithiotreitol (DTT)). The SMC3 head constructs migrated as two major peaks during size exclusion chromatography, likely corresponding to monomeric and dimeric species. The dimeric species was a result of mispairing of SCC1 fragments between to SMC3 head domains. The monomeric species was enriched by pooling the fractions close to 12 ml. Fractions containing head domains were identified by SDS-PAGE, pooled, concentrated and snap frozen in liquid nitrogen in small aliquots. Protein concentrations were determined by the Bradford assay.
For preparations of hinge constructs lacking coiled-coils, cells were lysed by sonication in 10 pellet volumes of lysis buffer (50 mM NaPO4, pH 8.0, 300 mM NaCl, 20 mM imidazole, 3 mM bME) supplemented with 1x PIC. Insoluble material was removed by centrifugation and the supernatant applied to NiNTA agarose as above. Beads were washed with 2 × 50ml Lysis buffer and transferred to a glass column. Bound protein was eluted with 4 × 2.5ml of Elution buffer (50 mM NaPO4, pH 8.0, 100 mM NaCl, 300 mM imidazole, 3 mM bME). Protein was concentrated to ∼500 μL and subjected to size exclusion chromatography using a Superdex 75 Increase 10/300 GL column (GE Life Sciences), equilibrated in Gelfiltration Buffer (20 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM DTT). Fractions were analyzed by SDS-PAGE followed by Coomassie staining. Hinge-containing fractions were concentrated in 10 kDa MWCO spin filters, aliquoted, snap frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined using the Bradford assay.
For preparations of hinge constructs with elongated coiled-coils, cells were lysed by sonication in Lysis buffer (50 mM NaPO4, pH 7.6, 500 mM NaCl, 5% glycerol, 20 mM imidazole, 3 mM bME) supplemented with 1 x PIC. Insoluble material was removed by centrifugation and the supernatant applied to NiNTA agarose as above. Beads were washed with 2 × 50 mL Lysis buffer. Bound protein was eluted on column with 4 × 2.5 mL of Elution buffer (50 mM NaPO4, pH 7.6, 100 mM NaCl, 300 mM imidazole, 3 mM bME). Protein was concentrated to ∼500 μL and subjected to size exclusion chromatography using a Superdex 200 Increase 10/300 GL column, equilibrated in Gelfiltration Buffer (20 mM Tris pH 7.5, 100 mM NaCl, 1 mM DTT). Protein was concentrated, aliquoted, snap frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined using the Bradford assay.
Protein expression and purification from HeLa cell cultures
A HeLa cell line expressing cohesinSCC1-Halo-Flag (HeLaSCC1-Halo-Flag) was as described previously (Davidson et al., 2019). Based on this clone, a cell line expressing cohesinSMC1-FKBP/SMC3-FRB/SCC1-Halo-Flag was generated using CRISPR-Cas9 genomic engineering (Cong et al., 2013). For this purpose, we first introduced the FKBP-tag to the C terminus of SMC1 in HeLaSCC1-Halo-Flag, generating HeLaSMC1-FKBP/SCC1-Halo-Flag. To introduce FKBP, we used the gRNAs 5′-CACCGCTGCTAATCAAGCATGAAG-3′ and 5′-CACCGGACTACACCAAAAAGGGCC-3′ together with the repair template ‘CRISPR-SMC1-FKBP’. As a second step, the FRB-tag was introduced to the C terminus of all genomic loci encoding SMC3, resulting in HeLaSMC1-FKBP/SMC3-FRB/SCC1-Halo-Flag. FRB was introduced using the gRNA sequences 5′-CACCGATACATCTCCCAAACCAGT-3′ and 5′-CACCGTAATATGATTCTCATACCC-3′ together with the repair template ‘CRISPR-SMC3-FRB’. Clones were selected after verification of homozygous FRB- and FKBP- integration by PCR of genomic DNA (primers used for SMC1-FKBP: 5′-TGGACCTCTAGGTAGCTCGG-3′ and 5′-GAGGGTGTTCGGTATGTGTC-3′; primers used for SMC3-FRB: 5′-CATAGCTCTTCCTGGGTGTCTT-3′ and 5′-AGTAGCTCCTAACAAATTCTCCTG-3′). Protein expression was confirmed by western blotting.
Purification of HeLa cohesin constructs
cohesinSCC1-Halo-Flag, cohesinSMC1-FKBP/SCC1-Halo-Flag and cohesinSMC1-FKBP/SMC3-FRB/SCC1-Halo-Flag from HeLa cell cultures were purified essentially as described (Davidson et al., 2019). Cell pellets were resuspended in 10 pellet volumes of hypotonic buffer (20 mM Tris 7.5, 1.5 mM MgCl2, 10 mM KCl, 1 mM PMSF, 1 x PIC). Cells were lysed by douncing (10 strokes with a tight pestle), incubated for 10 min on ice and dounced with additional 15 strokes. Nuclei were isolated by centrifugation (2000 rpm, 15 min) and resuspended in 8 pellet volumes of hypotonic buffer by douncing with 3 strokes. Nuclei were lysed by adding a 5 M NaCl solution dropwise to a final concentration of 500 mM while stirring, followed by addition of TWEEN to a final concentration of 0.1%. The lysate was stirred for 10 min on ice and subsequently sonified (60% amplitude, 60 × 0.4 s pulses with 1.5 s pauses between pulses). Insoluble material was removed by centrifugation (40000 xg, 30 min) and the supernatant incubated with 1 mL of FLAG-M2 agarose (Sigma) for 3 h at 4°C. Beads were washed with 10 column volumes of high salt wash buffer (25 mM NaPO4 pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM EDTA) and three column volumes of low salt wash buffer (25 mM NaPO4 pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM EDTA). Bound protein was eluted with 5 bead volumes of low salt wash buffer supplemented with 0.5 mg/ml 3X FLAG peptide. Eluates were pooled, concentrated, aliquoted and snap frozen in liquid nitrogen. Protein concentrations were analyzed next to BSA standards (Biorad) by SDS-PAGE followed by Coomassie staining.
Method details
Amber suppression and UV-induced protein-DNA crosslinking
Amber stop codons were introduced by site-directed mutagenesis at the indicated positions in the hinge constructs (lacking coiled-coils). The constructs were transformed into electrocompetent BL21(DE3) cells harboring the pEVOL-pBpF plasmid for co-expression of an engineered aminoacyl t-RNA synthethase/tRNA pair, allowing the incorporation of p-benzoyl phenylalanine by amber suppression. pEVOL-pBpF was a kind gift from Peter Schulz (Addgene plasmid # 31190). The transformations were grown in 1 mL SOC for 1 h at 37°C, plated on LB agar plates supplemented with 100 μg/ml ampicillin and 17 μg/ml chloramphenicol and grown to colonies over night. The colonies were resuspended in LB and used to inoculate 500 mL LB cultures (supplemented with the respective antibiotics) in 2 L baffled flasks. Cells were grown at 180 rpm and 37°C to an OD of 0.7, at which point 1 mM pBpF (Bachem, dissolved to 1 M in 1 M NaOH), 0.5 mM IPTG and 0.2% L-arabinose were added. Cells were harvested after 3h expression and pellets processed immediately or snap frozen in liquid nitrogen and stored at −80°C. The proteins were prepared as described above, omitting bME in the buffers and omitting the final size exclusion chromatography step. Incorporation of pBpF was confirmed by mass-spectrometry.
10 μM hinge domains were incubated in reaction buffer (20 mM HEPES pH 7.6, 50 mM NaCl, 2 mM MgCl2, 5% glycerol) in 100 μL reactions with 5 μM double or single stranded DNA containing a biotinylated 5′end (sequence forward: 5′-[Biotin]GCGGGTAATCCAGAGCCAGACGAGCACTACGAACAACTAA-3′; sequence reverse 5’-TTAGTTGTTCGTAGTGCTCGTCTGGCTCTGGATTACCCGC-3′) for 20 min on ice. The reactions were transferred to the wells of a black bottom 96 well plate (Corning) and treated with UV-light for 15 min on ice using a Blackray UV-lamp at a distance of 5 cm. The reactions were combined with 25 μL of equilibrated streptavidin beads (Pierce Ultralink streptavidin resin) in 500 μL of binding buffer (10 mM Tris, pH 7.5, 10 mM EDTA, 2 M NaCl) and incubated for 40 min rotating at room temperature. The beads were collected by centrifugation and washed three times with 500 μL of wash buffer (10 mM Tris pH 7.5, 10 mM EDTA, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS)). Beads were collected by centrifugation, resuspended in 50 μL 1 x SDS-sample buffer and bound DNA was eluted at 95°C for 5 min. Beads were removed by centrifugation and the supernatants subjected to SDS-PAGE on 16% Tris-Glycine gels (Invitrogen) in Tris-Glycine running Buffer. Proteins and DNA were visualized by silver staining.
Electrophoretic Mobility Shift Assays (EMSAs)
A double stranded (ds) 40bp DNA oligomer labeled at the 5′ end of one strand with fluorescine was used as the DNA probe (sequence forward: 5′-[fluorescine]GCGGGTAATCCAGAGCCAGACGAGCACTACGAACAACTAA-3′; sequence reverse: 5′-TTAGTTGTTCGTAGTGCTCGTCTGGCTCTGGATTACCCGC-3′). Increasing concentrations of proteins were incubated with 50 nM of the DNA-probe in 20 mM HEPES pH 7.6, 2 mM MgCl2, 50 mM NaCl, 10% glycerol and trace amounts of Orange G (Sigma) for 30 min on ice. Samples were loaded on a 6% polyacrylamide gel cast in 0.5 x tris-borate-EDTA (TBE) and protein-DNA complexes were separated from free DNA in 0.5 x TBE running buffer for 90 min at 4°C and 80 V constant voltage. DNA was visualized on a fluorescent gel scanner (Biorad). For EMSAs involving STAG1/SCC1int proteins, gels were cast and PAGE was performed in 1 x tris-acetate-EDTA (TAE) instead of 0.5 x TBE.
Mass photometry
Mass photometry experiments were performed essentially as described in Sonn-Segev et al. (2020). Cover slides (Menzel, 24x60mm, #1.5) were cleaned by sequential 5min sonications in 2% Hellmanex (Hellma), milliQ water, 100% isopropanol, two times milliQ water and subsequently dried under an air stream. Silicon buffer gaskets (Culture Well reusable gaskets 3 mm x 1 mm, Grace Biolabs) were attached to the glass slides by applying a mild pressure. The glass slides were then mounted on a Refeyn OneMP mass photometer (Refeyn Ltd.).
SMC1 and SMC3 head domains were incubated at 500 nM in MP buffer (20 mM HEPES pH 7.6, 50 mM NaCl, 5% glycerol, 2 mM MgCl2) supplemented with or without 2 mM ATP and/or 5 μM of the 40 bp dsDNA probe for at least 15 min at room temperature. A gasket was filled with buffer and the focal position of the glass surface determined and held constant using an autofocus system. The sample was then diluted 10-fold into the buffer-filled gasket. For mass photometry recordings of trimeric and dimeric cohesin, proteins were diluted directly 20-fold from stocks into MP buffer, yielding final concentrations of 110 nM and 135 nM, respectively. A sequence of 6000 frames was recorded immediately after dilution at 1 kHz. Each condition was recorded in a separate gasket and repeated at least twice. Data was acquired using AcquireMP and processed using DiscoverMP (Refeyn Ltd.). Contrast-to-mass calibration was performed by analyzing a native protein standard (NativeMark Unstained Protein Standard, Thermo Scientific) diluted in MP buffer. Contrast peaks corresponding to 66, 146, 480 and 1048 kDa were used for calibration. Mass distributions were plotted in the software and mean mass peaks determined by Gaussian fitting.
ATPase assays
Thin layer chromatography based ATPase assay
Recombinant cohesin was incubated at 60 nM in ATPase reaction buffer (final composition: 25 mM NaPO4 pH pH 7.5, 50 mM NaCl, 2.5 mM MgCl2, 1 mM DTT, 2% glycerol, 3.9 mM imidazole pH 7.5, 0.1 mg/ml BSA, 2 mM ATP and 10 nM [g-32P]ATP (Hartmann Analytic; SCP-501). Reactions were supplemented with 60 nM NIPBL-MAU2 and, where indicated, 10 ng/μl λ-DNA (NEB). For the experiments comparing NIPBL wild-type and 2A, recombinant cohesin was incubated at 60 nM in ATPase buffer supplemented with 60 nM NIPBL variants and, where indicated, 10 ng/μl λ-DNA. For FRB-FKBP dimerization experiments, rapamycin (1 μM) or an equal volume of DMSO was included in the ATPase reaction buffer. Reactions were incubated at 37°C for various times and stopped by adding SDS and EDTA to 1% and 10 mM final concentration, respectively. Reaction products were separated on polyethyleneimide plates (Sigma; 1055790001) by thin-layer-chromatography using 0.75 M KH2PO4 (pH 3.4), analyzed by phosphor imaging with a Typhoon Scanner (GE Healthcare) and quantified with ImageJ. ATPase rates were determined as previously described (Davidson et al., 2019).
Coupled ATPase assay
Coupled ATPase assays were performed based on the method described in Nørby (1988). 100 nM cohesin was incubated with 200 nM ΔN-NIPBL and 1 μM of a 75 bp ds DNA in ATPase reaction buffer (20 mM NaPO4 pH pH 7.6, 50 mM NaCl, 20mM KCl, 2.5 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA, 2 mM ATP), supplemented with 0.795 mM NADH (Sigma), 5.5mM phosphoenol pyruvate (Sigma), 16 U/ml lactate dehydrogenase and 14 U/ml pyruvate kinase (Sigma). Reactions were measured at 37°C in a volume of 100 μL in transparent 96 well (Corning) plates by monitoring the decrease in absorbance at 340 nm at 30 s intervals using a Pherastar FX plate reader (BMG Lab tech). The sequence of the 75 bp ds DNA was as follows: sequence forward: 5′-ACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAGTTAAGC-3′; sequence reverse 5’-GCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGT-3′.
The linear regions of the decay curves were fit to straight lines using PRISM (Graphpad). ATPase rates were determined from the slope of these lines, the molar extinction coefficient of NADH (6.23 mM-1 cm-1), a path length of 0.28 cm and a concentration of cohesin of 100 nM.
Loop extrusion assays
Microfluidic flow cells were prepared as previously described (Davidson et al., 2019). Flow cells were incubated with 1 mg/ml Avidin DN (Vector Laboratories) for 15 min and washed extensively with DNA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.25 mg/ml BSA (ThermoFisher Scientific; AM2616)). 40 μL of 22 pM λ-DNA (New England Biolabs; N3011S) biotinylated at both ends (Davidson et al., 2016) was introduced into flow cells at 5 μl/min in DNA buffer supplemented with 20 nM Sytox Orange (ThermoFisher Scientific; S11368). Flow cells were washed with 20 μL of wash buffer (50 mM Tris pH 7.5, 200 mM NaCl, 1 mM MgCl2, 5% glycerol, 1 mM DTT, 0.25 mg/ml BSA, 20 nM Sytox Orange) at 5 μl/min. Flow was then switched to perpendicular mode and a further 0.4 mL of wash buffer was introduced at 100 μl/min. 0.5 mL of imaging buffer (50 mM Tris pH 7.5, 50 mM NaCl, 2.5 mM MgCl2, 0.25 mg/ml BSA, 0.05% Tween-20) supplemented with 20 nM Sytox Orange was then introduced at 100 μl/min, followed by 0.1 mL of imaging buffer supplemented with an oxygen scavenging system (0.2 mg/ml glucose oxidase (Sigma; G2133), 35 μg/ml catalase (Sigma; C- 40), 9 mg/ml β-D-glucose, 2 mM Trolox (Cayman Chemical; 10011659)), 1 mM DTT, 220 nM Sytox Orange and 5 mM ATP (Jena Biosciences; NU-1010-SOL) at 100 μl/min. Stock solutions of glucose oxidase (20 mg/ml), catalase (3.5 mg/ml) and glucose (450 mg/ml) were prepared in 50 mM Tris pH 7.5, 50 mM NaCl, 50% glycerol. A stock solution of Trolox (100 mM) was prepared in 50 mM Tris pH 7.5, 50 mM NaCl, 10% methanol and adjusted to pH 7.5 with NaOH. Cohesin and NIPBL-MAU2 or NIPBL variants were then introduced together into flow cells at 30 μl/min in 0.25 mL imaging buffer supplemented with oxygen scavenging system, 1 mM DTT, 220 nM Sytox Orange and 5 mM ATP. For FRB-FKBP dimerization experiments, rapamycin (1 μM) or an equal volume of DMSO was included in the supplemented imaging buffer and in the cohesin/NIPBL flow in mixture.
Recombinant cohesin variants and NIPBL-MAU2 were introduced into flow cells at 0.25 nM and 2.5 nM, respectively, for all experiments with the following exceptions: for the experiments comparing wild-type NIPBL and NIPBL2A, cohesinSCC1-Halo-flag from HeLa cells and recombinant NIPBL were introduced into flow cells at a final concentration of 1.5 nM and 2.5 nM, respectively. For the experiments comparing the effect of FRB-FKBP dimerization of the ATPase heads, cohesinSMC1-FKBP/SMC3-FRB/SCC1-Halo-Flag and cohesinSMC1-FKBP/SCC1-Halo-Flag from HeLa cells were used at ∼3nM and 6nM, respectively, since they contained substoichiometric amounts of SCC1 and STAG1/2. The complexes were introduced together with 2.5 nM NIBPL-MAU2. For the experiments comparing STAG1 variants, recombinant trimeric cohesin was introduced into flow cells at 0.9 nM together with 5 nM NIBPL-MAU2 and 5.4 nM STAG1. For the experiments comparing the effect of rapamycin-induced dimerization of cohesinSMC3-intFRB/FKBP-SCC1, recombinant cohesinSMC3-intFRB/FKBP-SCC1 or recombinant cohesinSMC3-intFRB were introduced into flow cells at 2 nM together with 12 nM NIBPL-MAU2.
All experiments were performed at 37°C. Time-lapse microscopy images were acquired at 4 s intervals using a Zeiss TIRF 3 Axio Observer setup and a 561 nm laser (Davidson et al., 2016).
Loop maintenance assays
For loop maintenance experiments, flow cells were washed as above and 50 pM recombinant cohesinSMC3-intFRB/FKBP-SCC1 was introduced together with 1 nM NIPBL-MAU2 and 5 mM ATP in 0.25 mL imaging buffer supplemented with oxygen scavenging system components, 1 mM DTT, 250 nM Sytox Orange and 5 mM ATP at 30 μl/min (step 1). Further 0.25 mL of imaging buffer supplemented with oxygen scavenging system components, 1 mM DTT, 250 nM Sytox Orange and 5 mM ATP was then introduced in the presence or absence of NIPBL-MAU2 (1 nM) and either rapamycin (1 μM) or an equal volume of DMSO at 30 μl/min (step 2).
High Speed Atomic Force Microscopy
Glass rods (2 mm diameter, 2 mm length, Hilgenberg GmbH) were glued on the sample stage with acetone-diluted nail polish (Astor). At least 24 h before the beginning of each experiment mica discs with a diameter of 1.5 mm (V-1 grade Muscovite mica sheet; NanoAndMore GmbH) were cut with a mica cutter and glued to the opposite side of the glass rods using two-compound epoxy glue (UHU, Bolton Adhesives). Prior to imaging, cohesin preparations were thawn on ice and diluted to a concentration of 3nM in imaging buffer (10 mM HEPES pH 7.7, 150 mM KCl, 2.5 mM MgCl2, 2 mM ATP). The mica disc was cleaved and 1.5 μl of these dilutions were applied to the surface. After incubation for 5 min at room temperature the surface was rinsed with 10 μL of imaging buffer and the sample stage was mounted into the imaging bath of the HS-AFM (custom-built based on the design of Ando et al., 2003). CohesinSMC1-FKBP/SMC3-FRB was imaged following the same procedure as described above, with the exception that imaging buffers supplemented with either 1 μM DMSO or 1 μM rapamycin were used in the dilution, rinsing and imaging steps. NIPBL or Stag1 in isolation were imaged following the same procedure, with the exception that the imaging buffers did not contain ATP. Silicon nitride ultra-short cantilevers, (BL-AC10DS-A2, Olympus, Tokyo, Japan) with a nominal spring constant of 0.1 N/m, a resonance frequency of ∼500 kHz, and a quality factor of ∼2 in liquid were used for imaging. Laser alignment, surface approach and imaging were performed essentially as previously described (Uchihashi et al., 2012). Scanning areas of 300 × 300 nm were first imaged to identify regions containing single cohesin molecules, which were subsequently recorded on scan areas of 100 × 100 nm at a resolution of 200 × 200 pixels and a scan rate of 500 ms per frame. The imaging was performed in tapping mode, the free amplitude was set to ∼3 nm and the amplitude set point to 90%–85%. All experiments were performed at room temperature.
Labeling of proteins for smFRET experiments
Sulfo-Cyanine3-maleimide and Sulfo-Cyanine5-maleimide were coupled to Coenzyme A by mixing 333 μM Coenzyme-A Tri-lithium Salt (Sigma), 1.67 μM sulfo-Cyanine3-maleimide or sulfo-Cyanine5-maleimide (Lumiprobe) in 283 mM HEPES KOH pH 7.7, 100 mM NaCl, 33.3% dimethyl sulfoxide (DMSO) at room temperature for 60 min. Conjugates were subsequently purified by high performance liquid chromatography (HPLC). Fractions containing the conjugates were lyophilized, resuspended in 100% DMSO and stored in small aliquots at −20°C.
500 μL cohesin preparations (at ∼1 μM) were treated for 5min at room temperature with 100 μM biotin-PEG11-maleimide (Pierce). The labeling reactions were stopped by addition of 10 mM DTT. The reactions were supplemented with 10 mM MgCl2, 20 μM sulfo-Cy3-Coenzyme A, 20 μM sulfo-Cy5-Coenzyme A and 5 μM SfP-synthase, adjusted to 600 μL final volume with FLAG buffer and labeled over night at 4°C. For experiments involving SMC3head-Cy3 and SMC1head-Cy3, cohesin complexes were biotinylated and labeled with sulfo-Cy3-CoA only. Unreacted dye was removed by ultracentrifugation in 4 mL 5%–25% sucrose gradients (in FLAG buffer supplemented with 1 mM DTT) for 6 h at 55000 rpm in a SW60 Ti rotor (Beckmann). Gradients were fractionated in 500 μL steps and fractions containing cohesin were identified by SDS-PAGE. The peak fraction was aliquoted and frozen in liquid nitrogen.
ΔN-NIPBL containing internal YBBR tags was labeled similarly to cohesin, but with sCy5-CoA only and omitting the biotinylation step. After over night labeling, unreacted dye was removed by size exclusion chromatography using a Superdex200 10/300 GL column equilibrated in FLAG buffer containing 1 mM DTT. Fractions containing labeled NIPBL were pooled, concentrated, aliquoted and snap frozen in liquid nitrogen. Protein concentrations were determined using the Bradford assay.
Single-molecule FRET
Glass slides were cleaned by repeated sonication in acetone, ethanol and milliQ water, followed by boiling in Piranha solution for 1h. Glass slides were washed in milliQ water, dried and functionalized with dichloromethylsilane in hexane for 1h at room temperature. After washing in chloroform, milliQ water and chloroform, glass slides were stored dry and protected from dust. Coverslips were cleaned by sonication in a water bath for 15 min in 2% Hellmanex solution, washed in milliQ water and sonified for further 15 min in 100% ethanol. Coverslips were subsequently dried under an air stream and assembled into a flow cell with functionalized coverslips separated by two stripes of double-sided sticking tape (3M). 0.1 mg/ml biotinylated BSA (Pierce) in 20 mM HEPES, pH 7.6, 150 mM NaCl was injected into the dry flow cell and allowed to adhere for 5 min. The surface was further passivated using 1% Pluronic F127 (Sigma) in 20 mM HEPES, pH 7.6, 150 mM NaCl for 15 min. The flow cells were washed twice with 30 μL WB150 (20 mM HEPES, pH 7.6, 150 mM KCl, 2.5 mM MgCl2, 0.6 mg/ml BSA, 0.6 mg/ml b-casein) and incubated for at least 5 min with 0.05 mg/ml neutravidin (Pierce) diluted in WB150.
Before each experiment, flow cells were washed two times with WB750 (20 mM HEPES, pH 7.6, 750 mM KCl, 2.5 mM MgCl2, 0.6 mg/ml BSA, 0.6 mg/ml b-casein). Labeled and biotinylated cohesin was diluted into WB750 (20 mM HEPES, pH 7.6, 750 mM KCl, 2.5 mM MgCl2, 0.6 mg/ml BSA, 0.6 mg/ml b-casein) 5,000 to 20,000 fold (depending on the coverage), injected into the flow cell and allowed to bind for 2 min at room temperature. Flow cells were subsequently washed with two times 50 μL of WB750 and two times 50 μL of WB150. Before imaging, image buffer IB50 (20 mM HEPES, pH 7.6, 50 mM KCl, 2.5 mM MgCl2, 0.6 mg/ml BSA, 0.6 mg/ml b-casein, 3 mM Trolox, 1 mM nitrobenzaldehyde, 1 mM 1,3,5,7-cyclooctatetraene) was supplemented with 0.2 μM protecatechuic acid decarboxylase (Sigma) and the indicated additives and injected into the flow cell, followed by imaging. Experiments involving NIPBL-cohesin interactions were performed with ΔN-NIPBL instead of NIBPL-MAU2. Additives were added at the following concentrations: ATP: 2 mM; unlabeled NIBPL-MAU2: 60 nM, acceptor-labeled ΔN-NIPBL: 0.5 nM; 75 bp dsDNA: 1 μM; puc19 plasmid: 0.1 mg/ml; rapamycin: 1 μM.
For imaging, excitation lasers (Coherent OBIS, 532 nm, 150 mW and 647 nM, 140mW) were synchronized to the fire output of the camera by NI DAQ to produce an alternating laser excitation (ALEX) scheme of one frame donor excitation and one frame acceptor excitation. Molecules were imaged simultaneously in both wavelengths using an Andor iXon 897 EMCCD camera with a custom-made optical split with a 50 ms exposure time and saved as tiff stacks. A nanogrid (Miraloma Tech) was recorded on the day of the experiment to determine the transformation function between the donor and the acceptor channel. Channel registration was then performed using custom built ImageJ scripts.
Rotary Shadowing EM
Cohesin was diluted to a concentration of approximately 0.1 mg/mL in FLAG buffer in the absence or presence of 1 μM rapamycin. Hinge domains were diluted to 0.1 mg/ml in 20 mM Tris pH 7.5, 100 mM NaCl. Samples were subsequently diluted 1:1 in spraying buffer, containing 200 mM ammonium acetate and 60% (v/v) glycerol, pH adjusted to 7.6.
After dilution, the samples were sprayed onto freshly cleaved mica chips (Agar Scientific, UK) and immediately transferred into a BAL-TEC MED020 high vacuum evaporator (BAL-TEC, Liechtenstein) equipped with electron guns. While rotating, samples were coated with 0.7 nm Platinum (BALTIC, Germany) at an angle of 4-5°, followed by 8 nm Carbon (Balzers, Liechtenstein) at 90°. The obtained replicas were floated off from the mica chips, picked up on 400 mesh Cu/Pd grids (Agar Scientific, UK), and inspected in an FEI Morgagni 268D TEM (FEI, the Netherlands) operated at 80 kV. Positions on the grid were chosen randomly. Images were acquired using an 11 megapixel Morada CCD camera (Olympus-SIS, Germany).
Fluorescence polarization assays
Proteins were incubated at increasing concentrations with 25 nM of a 75bp ds DNA probe labeled at both 5′ ends with fluorescience (sequence forward: 5′-[Fluorescine]ACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAGTTAAGC-3′; sequence reverse 5’-[Fluorescine]GCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGT-3′) in reaction buffer (20 mM HEPES, pH 7.6, 50 mM NaCl, 5% glycerol, 2 mM MgCl2, 1 mM DTT) at room temperature for 30 min. For experiments involving hinge domains, aliquots of hinge preparations were thawn on ice, split and one half was treated with 3C protease for 1h on ice while the other half was treated with buffer only. The treated and untreated samples were then titrated on the DNA probe and incubated as above. For experiments involving ATPase heads, protein/DNA mixtures were incubated with either 2 mM ATP (diluted from a 100 mM stock in 500 mM HEPES pH 7.6) or with the same volume of 500 mM HEPES, pH 7.6). The reactions were transferred into black 96 well plates (Corning) and polarization measured using Pherastar FX plate reader (BMG Lab tech) using a 488 nm laser and a filter cube with parallel and perpendicular emission slits at 520 nm. The EM gain was adjusted so that the free DNA probe control had a polarization value of 200 mP. This reference value was subtracted from the polarization signal recorded in the presence of protein. Data was plotted in PRISM and fitted to a ‘one site - total binding’ model.
TNP-ATP binding assays
The isolated SMC3 and SMC1 head domains were incubated at 2 μM with increasing concentrations of 2′,3′-O-Trinitrophenyl-adenosine-5′triphosphate (TNP-ATP; Jena Biosciences) in binding buffer (20 mM HEPES pH 7.6, 50 mM NaCl, 2mM MgCl2, 5% glycerol) for 15 min at room temperature in 50 μL volumes. Samples were transferred to black 384 wells and fluorescence measured using a Synergy H1 plate reader (excitation 403 nm, emission 540 nm; BioTek). Fluorescence measurements of reactions in the absence of protein and/or in the absence of TNP-ATP were subtracted from the raw fluorescence counts to correct for intrinsic fluorescence of protein and to quantify the change in fluorescence upon head-domain binding to TNP-ATP. The binding data was fit to a ‘one site - total binding’ model in PRISM.
Quantification and statistical analysis
Analysis of loop extrusion experiments
Image analysis was performed in ImageJ. The frequency and rate of loop extrusion events were determined as previously described (Davidson et al., 2019).
For loop maintenance assays, DNA loops were classified by visually comparing the length of DNA remaining outside of the loop immediately after loop extrusion to that remaining at the end of step 1. Loops in which the length of DNA outside of the loop was unchanged were classed as ‘maintained’. Loops in which the length of DNA outside of the loop increased were classed as ‘partially released’. DNAs in which the loop was completely released were classed as ‘released’. This classification was then repeated in step 2 for all loops that had been formed in step 1. Statistical details for loop extrusion experiments (means, standard deviations and number of replicates) can be found in the figures and figure legends.
High Speed Atomic Force Microscopy data analysis
Volumetric analysis was performed in Gwyddion 2.55. Images were selected and processed to remove background and transient noise. Horizontal scars due to feedback instabilities or particles sticking to the AFM tip were selected and removed by Laplacian background substitution. A height threshold mask was used for selecting the background prior to correction of scanning artifacts, e.g., scan line artifacts and polynomial background. For volume quantification, the protein surface was selected using a height threshold mask defined from a minimum height of 0.25 – 0.35 nm to the maximum height of the protein structure. The same image treatment described for the volume analysis was applied to the images displayed in the figures, with the addition that a Gaussian filter was applied to reduce the remaining noise effect, increase smoothness and improve the visualization of the features of the molecules. Statistical details (means, standard deviations and number of replicates) can be found in the figures and figure legends.
Frame by frame distances between hinge and head-domains were quantified in MATLAB (Mathworks). The total number of molecules and frames analyzed are indicated in the figures. This data was further summarized in the following way: “hinge preference for head 1” was quantified as the fraction of frames with head 1-hinge distances smaller than 10 nm over the total number of frames with head-to-hinge distances smaller than 10 nm (for both head 1 and head 2). “hinge-head w/ engaged head-head” corresponds to the fraction of frames with both hinge-head and head-head distances smaller than 10 nm over the total number of frames with head-head distances smaller than 10 nm. The fraction of aligned conformations was quantified by categorizing each frame manually for the presence of aligned or separated coiled coils.
Analysis of smFRET data
smFRET traces were extracted using the iSMS software (version 2.01) (Preus et al., 2015) and default settings. Bona fide smFRET traces were selected based on the single step bleaching behavior of donor or acceptor, anti-correlation between donor and acceptor, and the stability of the stoichiometry traces over time. The acceptor signal was corrected in iSMS for donor- and acceptor-bleed through by analyzing the signals of multiple traces after acceptor and donor bleaching, respectively. This correction was performed for each flow channel. Multiple fields of view (typically containing 10-20 FRET pairs) were recorded so that one replicate had more than 100 FRET pairs. Replicates were performed by assembling the reactions in new channels on at least two different days.
Transition densities were analyzed by fitting the FRET traces to Hidden Markov Models (HMM) using the ebFRET software (van de Meent et al., 2014). Head engagement sensors, head-NIPBL, hinge-NIPBL and hinge-STAG1 sensors were fitted to 2 state models. Coiled-coil and bending sensors were fitted to 3 state models. Plots were generated by the plotTDP MATLAB script (http://ebfret.github.io/), using a variance of 0.003, and normalized for the total number of frames in each condition (‘normbyframes’).
‘Dynamic traces’ were quantified based on the HMM fits as traces that showed at least one reversible transition during the acquisition time. ‘locked heads’ (Figure 3) and ‘stably bound complexes’ (Figure 7) were quantified based on the HMM fits as traces that were in a high FRET state and static, i.e., did not show any transitions during the acquisition time. The probability of bending ‘p(head-hinge)’ (Figure 6) was defined as the fraction of FRET values above 0.3 in a given condition. Statistical details (means, standard deviations and number of replicates) can be found in the figures and figure legends.
FRET histograms were generated by counting the number of all FRET values in each replicate ranging from −0.1 to 1.1 FRET values in bin sizes of 0.025. For each bin, the mean and standard deviation from at least two replicates was plotted. The number of replicates for each condition is indicated in the figure legends.
For analysis of head engagement and hinge-nose engagement rates, dwell times of the low and high FRET states from all molecules were extracted from the HMM fits. Single molecule traces shorter than 15 frames were excluded from the analysis and the extracted dwell times were binned into 493.2 ms from 0 to 40 s. The data was then plotted in Prism (Graphpad) and fit to a single exponential decay function. In conditions with low engagement rates (e.g., in absence of either ATP or NIPBL for the head engagement sensor), the dwell time distributions displayed non-zero peaks. Here, the distributions were fit at the point were the distribution was starting to decay after the peak. Rates were determined by calculating the inverse sum of the half times determined from the low and high FRET states.
Acknowledgments
We thank Maxim Molodtsov, Alipasha Vaziri, Thomas Lendl, and Francisco Balzarotti for technical assistance on smFRET, Daniela Goetz for providing data on NIPBL-cohesin interactions, and Mathias Madalinski for help with HPLC. rs-EM experiments were performed by Marlene Brandstetter at the EM facility of the Vienna Biocenter Core Facilities GmbH (VBCF). pEVOL-pBpF was a kind gift from Peter Schultz. We thank Alexander Schleiffer, Anton Goloborodko, David Haselbach, Anton Meinhart, and Marco Catipovic for helpful discussions and Tom Rapoport and Kim Nasmyth for critical reading of the manuscript. B.W.B. was supported by long-term fellowships from EMBO and HFSP. D.C. was supported by a doctoral fellowship from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement number 721874 (SPM2.0) and from the Vienna Science and Technology Fund (LS19-029). Research in the laboratory of J.-M.P. is supported by Boehringer Ingelheim, the Austrian Research Promotion Agency (Headquarter grant FFG-852936), the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme (693949), the Human Frontier Science Program (RGP0057/2018), and the Vienna Science and Technology Fund (LS19-029). J.-M.P. is also an adjunct professor at the Medical University of Vienna.
Author contributions
B.W.B. designed research, generated constructs, performed DNA-binding, mass photometry, ATPase assays and smFRET experiments, analyzed data, and drafted the manuscript. I.F.D. performed and analyzed loop extrusion experiments and ATPase assays. D.C. performed and analyzed HS-AFM experiments. G.W. performed and analyzed loop extrusion experiments and purified cohesin constructs from HeLa cells. W.T. engineered HeLa cell lines for cohesin purification. S.H. supported work on FRB/FKBP dimerizable constructs. G.L. supported construct cloning and protein expression. P.H. supervised HS-AFM work. J.-M.P. supervised research and wrote the manuscript with critical input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: October 7, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.09.016.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Anderson D.E., Losada A., Erickson H.P., Hirano T. Condensin and cohesin display different arm conformations with characteristic hinge angles. J. Cell Biol. 2002;156:419–424. doi: 10.1083/jcb.200111002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T., Kodera N., Naito Y., Kinoshita T., Furuta K., Toyoshima Y.Y. A high-speed atomic force microscope for studying biological macromolecules in action. ChemPhysChem. 2003;4:1196–1202. doi: 10.1002/cphc.200300795. [DOI] [PubMed] [Google Scholar]
- Bürmann F., Lee B.G., Than T., Sinn L., O’Reilly F.J., Yatskevich S., Rappsilber J., Hu B., Nasmyth K., Löwe J. A folded conformation of MukBEF and cohesin. Nat. Struct. Mol. Biol. 2019;26:227–236. doi: 10.1038/s41594-019-0196-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zheng X.F., Brown E.J., Schreiber S.L. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. USA. 1995;92:4947–4951. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J.E., Lee B.G., Roig M.B., Yatskevich S., Petela N.J., Metson J., Voulgaris M., Gonzalez Llamazares A., Löwe J., Nasmyth K.A. Transport of DNA within cohesin involves clamping on top of engaged heads by Scc2 and entrapment within the ring by Scc3. eLife. 2020;9:1–36. doi: 10.7554/eLife.59560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson I.F., Peters J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021;22:445–464. doi: 10.1038/s41580-021-00349-7. [DOI] [PubMed] [Google Scholar]
- Davidson I.F., Goetz D., Zaczek M.P., Molodtsov M.I., Huis In ’t Veld P.J., Weissmann F., Litos G., Cisneros D.A., Ocampo-Hafalla M., Ladurner R. Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J. 2016;35:2671–2685. doi: 10.15252/embj.201695402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson I.F., Bauer B., Goetz D., Tang W., Wutz G., Peters J.M. DNA loop extrusion by human cohesin. Science. 2019;366:1338–1345. doi: 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- Diebold-Durand M.L., Lee H., Ruiz Avila L.B., Noh H., Shin H.C., Im H., Bock F.P., Bürmann F., Durand A., Basfeld A. Structure of Full-Length SMC and Rearrangements Required for Chromosome Organization. Mol. Cell. 2017;67:334–347.e5. doi: 10.1016/j.molcel.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N., Konevega A.L., Wintermeyer W., Rodnina M.V., Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016;15:2038–2049. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganji M., Shaltiel I.A., Bisht S., Kim E., Kalichava A., Haering C.H., Dekker C. Real-time imaging of DNA loop extrusion by condensin. Science. 2018;360:102–105. doi: 10.1126/science.aar7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Golfier S., Quail T., Kimura H., Brugués J. Cohesin and condensin extrude DNA loops in a cell cycle-dependent manner. eLife. 2020;9:1–34. doi: 10.7554/eLife.53885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S., Arumugam P., Katou Y., Kuglitsch D., Helmhart W., Shirahige K., Nasmyth K. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell. 2006;127:523–537. doi: 10.1016/j.cell.2006.08.048. [DOI] [PubMed] [Google Scholar]
- Hassler M., Shaltiel I.A., Kschonsak M., Simon B., Merkel F., Thärichen L., Bailey H.J., Macošek J., Bravo S., Metz J. Structural Basis of an Asymmetric Condensin ATPase Cycle. Mol. Cell. 2019;74:1175–1188.e9. doi: 10.1016/j.molcel.2019.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T.L., Eickhoff P., Sousa J.S., Locke J., Nans A., Flynn H.R., Snijders A.P., Papageorgiou G., O’Reilly N., Chen Z.A. A Structure-Based Mechanism for DNA Entry into the Cohesin Ring. Mol. Cell. 2020;79:917–933.e9. doi: 10.1016/j.molcel.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T.L., Pobegalov G., Molodtsov M., Uhlmann F. A Brownian ratchet model for DNA loop extrusion by the cohesin complex. eLife. 2021;10:e67530. doi: 10.7554/eLife.67530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hons M.T., Huis In ’t Veld P.J., Kaesler J., Rombaut P., Schleiffer A., Herzog F., Stark H., Peters J.M. Topology and structure of an engineered human cohesin complex bound to Pds5B. Nat. Commun. 2016;7:12523. doi: 10.1038/ncomms12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner K.P., Karcher A., Shin D.S., Craig L., Arthur L.M., Carney J.P., Tainer J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- Huis in ’t Veld P.J., Herzog F., Ladurner R., Davidson I.F., Piric S., Kreidl E., Bhaskara V., Aebersold R., Peters J.M. Characterization of a DNA exit gate in the human cohesin ring. Science. 2014;346:968–972. doi: 10.1126/science.1256904. [DOI] [PubMed] [Google Scholar]
- Kapanidis A.N., Lee N.K., Laurence T.A., Doose S., Margeat E., Weiss S. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA. 2004;101:8936–8941. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Shi Z., Zhang H., Finkelstein I.J., Yu H. Human cohesin compacts DNA by loop extrusion. Science. 2019;366:1345–1349. doi: 10.1126/science.aaz4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko J.F., De Los Rios P., Barducci A., Gruber S. DNA-segment-capture model for loop extrusion by structural maintenance of chromosome (SMC) protein complexes. Nucleic Acids Res. 2019;47:6956–6972. doi: 10.1093/nar/gkz497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney S.A., Joo C., Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y., Uhlmann F. DNA Entry into and Exit out of the Cohesin Ring by an Interlocking Gate Mechanism. Cell. 2015;163:1628–1640. doi: 10.1016/j.cell.2015.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols M.H., Corces V.G. A tethered-inchworm model of SMC DNA translocation. Nat. Struct. Mol. Biol. 2018;25:906–910. doi: 10.1038/s41594-018-0135-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørby J.G. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- Petela N.J., Gligoris T.G., Metson J., Lee B.G., Voulgaris M., Hu B., Kikuchi S., Chapard C., Chen W., Rajendra E. Scc2 Is a Potent Activator of Cohesin’s ATPase that Promotes Loading by Binding Scc1 without Pds5. Mol. Cell. 2018;70:1134–1148.e7. doi: 10.1016/j.molcel.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petela N.J., Gonzalez Llamazares A., Dixon S., Hu B., Lee B.-G., Metson J., Seo H., Ferrer-Harding A., Voulgaris M., Gligoris T. Folding of cohesin’s coiled coil is important for Scc2/4-induced association with chromosomes. eLife. 2021;10:e67268. doi: 10.7554/eLife.67268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan B., Barth R., Kim E., Davidson I.F., Bauer B., van Laar T., Yang W., Je-Kyung R., van de Torre J., Peters J.M., Dekker C. SMC complexes can traverse physical roadblocks bigger than their ring size. bioRxiv. 2021 doi: 10.1101/2021.07.15.452501. [DOI] [PubMed] [Google Scholar]
- Preus S., Noer S.L., Hildebrandt L.L., Gudnason D., Birkedal V. iSMS: single-molecule FRET microscopy software. Nat. Methods. 2015;12:593–594. doi: 10.1038/nmeth.3435. [DOI] [PubMed] [Google Scholar]
- Ryu J.K., Katan A.J., van der Sluis E.O., Wisse T., de Groot R., Haering C.H., Dekker C. The condensin holocomplex cycles dynamically between open and collapsed states. Nat. Struct. Mol. Biol. 2020;27:1134–1141. doi: 10.1038/s41594-020-0508-3. [DOI] [PubMed] [Google Scholar]
- Shi Z., Gao H., Bai X.C., Yu H. Cryo-EM structure of the human cohesin-NIPBL-DNA complex. Science. 2020;368:1454–1459. doi: 10.1126/science.abb0981. [DOI] [PubMed] [Google Scholar]
- Son H., Mo W., Park J., Lee J.W., Lee S. Single-Molecule FRET Detection of Sub-Nanometer Distance Changes in the Range below a 3-Nanometer Scale. Biosensors (Basel) 2020;10:3–9. doi: 10.3390/bios10110168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonn-Segev A., Belacic K., Bodrug T., Young G., VanderLinden R.T., Schulman B.A., Schimpf J., Friedrich T., Dip P.V., Schwartz T.U. Quantifying the heterogeneity of macromolecular machines by mass photometry. Nat. Commun. 2020;11:1772. doi: 10.1038/s41467-020-15642-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihashi T., Kodera N., Ando T. Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat. Protoc. 2012;7:1193–1206. doi: 10.1038/nprot.2012.047. [DOI] [PubMed] [Google Scholar]
- van de Meent J.W., Bronson J.E., Wiggins C.H., Gonzalez R.L., Jr. Empirical Bayes methods enable advanced population-level analyses of single-molecule FRET experiments. Biophys. J. 2014;106:1327–1337. doi: 10.1016/j.bpj.2013.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann F., Petzold G., VanderLinden R., Huis In ’t Veld P.J., Brown N.G., Lampert F., Westermann S., Stark H., Schulman B.A., Peters J.M. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl. Acad. Sci. USA. 2016;113:E2564–E2569. doi: 10.1073/pnas.1604935113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatskevich S., Rhodes J., Nasmyth K. Organization of Chromosomal DNA by SMC Complexes. Annu. Rev. Genet. 2019;53:445–482. doi: 10.1146/annurev-genet-112618-043633. [DOI] [PubMed] [Google Scholar]
- Yin J., Straight P.D., McLoughlin S.M., Zhou Z., Lin A.J., Golan D.E., Kelleher N.L., Kolter R., Walsh C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.