

Abstract
Elevated infiltration of immunosuppressive alternatively polarized (M2) macrophages is associated with poor prognosis in cancer patients. The tumor microenvironment remarkably orchestrates molecular mechanisms that program these macrophages. Here we identify a novel role for oncogenic Hedgehog (Hh) signaling in programming signature metabolic circuitries that regulate alternative polarization of tumor-associated macrophages. Two immunocompetent orthotopic mouse models of mammary tumors were used to test the effect of inhibiting Hh signaling on tumor-associated macrophages. Treatment with the pharmacological Hh inhibitor Vismodegib induced a significant shift in the profile of tumor-infiltrating macrophages. Mass spectrometry-based metabolomic analysis showed Hh inhibition induced significant alterations in metabolic processes, including metabolic sensing, mitochondrial adaptations, and lipid metabolism. In particular, inhibition of Hh in M2 macrophages reduced flux through the UDP-GlcNAc biosynthesis pathway. Consequently, O-GlcNAc-modification of STAT6 decreased, mitigating the immune suppressive program of M2 macrophages, and the metabolically demanding M2 macrophages shifted their metabolism and bioenergetics from fatty acid oxidation to glycolysis. M2 macrophages enriched from Vismodegib-treated mammary tumors showed characteristically decreased O-GlcNAcylation and altered mitochondrial dynamics. These Hh-inhibited macrophages are reminiscent of inflammatory (M1) macrophages, phenotypically characterized by fragmented mitochondria. This is the first report highlighting the relevance of Hh signaling in controlling a complex metabolic network in immune cells. These data describe a novel immunometabolic function of Hh signaling that can be clinically exploited.
Keywords: Hedgehog signaling, lipid metabolism, mitochondrial bioenergetics, O-GlcNAcylation, tumor-associated macrophages
Introduction
In the tumor microenvironment (TME) tumor cells and stromal cells co-exist in a metabolically complex milieu. Immune cells are responsive to their surrounding environment and can readily react to perturbations by altering their metabolic profiles. Changes in the metabolic profile are noted in macrophages during the early phase of tumor evolution. Macrophages exist in a continuum of functional states, although, they are simplistically characterized as classically activated, pro-inflammatory (M1) macrophages or alternatively activated, immunosuppressive (M2) macrophages. While these represent the polar extremes of their functions, tumor-associated macrophages (TAMs) exist in a spectrum of intermediate activation states in response to the heterogeneous conditions they are exposed to in the TME (1).
Cytokines in the TME, including IL-12, TNF-α, and IFN-γ, promote macrophage polarization to an M1 state, whereas IL-4, IL-10, and IL-13 promote M2 macrophage polarization. These two states have distinct metabolic circuitries that contribute to survival, and subtype specific functions (2). The metabolism of M1 macrophages is predominantly governed by glycolysis, while M2 macrophages utilize oxidative phosphorylation (OXPHOS) (3). The mechanisms controlling these bioenergetic fates are unclear but are likely due to responses to signaling cues from the TME. Complex crosstalk between tumor and immune cells programs macrophages to a tumor-fostering M2 state. As such, the feed-forward loop between tumor and immune cells circumvents normalizing cues from the TME and evolves over time to sustain the tumor (2).
The Hedgehog (Hh) signaling pathway is aberrantly activated in multiple cancer types, including breast cancer, and promotes tumor invasion, metastasis, and multi-drug resistance (4–6). Hh signaling cascade is initiated through the binding of Hh ligands to Patched1 (Ptch1), relieving Ptch1’s inhibitory effect on the signal transducer, Smoothened (SMO). The ensuing regulatory circuit permits transcriptional activation by GLI transcription factors (7–9).
While Hh signaling in tumor cells has been studied extensively, the impact of Hh signaling on the immune TME is now being realized. We previously characterized a determinative role for Hh signaling in significantly altering the immune components of the TME (10). Since then, it has been confirmed that Shh promotes M2 polarization and reduces effector CD8+ T cell recruitment to the tumor (11,12). A high percentage of M2 TAMs is associated with poor patient outcomes (13). Approaches previously employed to ablate TAMs in breast and lung cancer have yielded little success (14,15). This is, in part, due to the fact that immunotherapeutic strategies have been successfully employed for highly heterogeneous tumor types with high T cell infiltration but remain less effective for tumor types with limited T cell infiltration.
Thus, inhibition of Hh signaling could present with the dual benefit of directly targeting tumor cells and re-configuring the tumor immune microenvironment to an immune-activated state. We investigated the possibility that Hh signaling orchestrates changes in macrophage metabolism that enable the immunosuppressive M2 polarization state. We demonstrate that Hh signaling critically controls metabolic regulation of M2 TAMs by impinging upon the Hexosamine Biosynthetic Pathway (HBP) and altering metabolic messaging that converges upon overlapping circuits of fatty acid oxidation (FAO), bioenergetics, and mitochondrial dysfunction.
Materials and Methods
Supplementary Table 1 includes a comprehensive list of all reagents used throughout this study.
Cell Culture
RAW 264.7 cells were cultured in DMEM (Thermo Fisher; Waltham, MA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher). 4T1 murine mammary carcinoma cell line was cultured in RPMI-1640 medium (Thermo Fisher) supplemented with 5% heat-inactivated FBS and 100μg/ml G418 (Thermo Fisher). EMT6 murine mammary carcinoma cell line was cultured in Dulbecco’s modified minimum essential medium supplemented with 5% heat-inactivated FBS, 1% Glutamax (Life Technologies; Carlsbad, CA), 1% Sodium Pyruvate (Thermo Fisher) and 1% Non-essential amino acids (Thermo Fisher). RAW264.7 and 4T1 cells were obtained from ATCC, while the EMT6 cells were a gift from Dr. Sophia Ran, Southern Illinois University. 4T1 cells have been validated by STR profiling from ATCC. Cell lines are typically used in their early passages. We expand cells at low passages and freeze multiple stocks. After about 10-15 passages, a new vial is thawed. All cell lines used are routinely tested for Mycoplasma.
Bone Marrow-derived Macrophage Isolation
Bone marrow from femurs and tibias of 6-8-week-old mice was flushed in MEMα medium (Life Technologies) supplemented with 10% FBS and Penicillin-Streptomycin. M-CSF (25ng/ml) was added to cells for 7 days to differentiate monocytes into macrophages (BMDMs).
In vivo Experiments
Luciferase-expressing 4T1 cells (5 x 105) or luciferase expressing EMT6 cells (2 x 105) were suspended in HBSS and injected into the inguinal mammary fat pad of eight-week-old female Balb/c mice. Tumor growth was documented by caliper measurements and bioluminescent imaging (BLI) (Xenogen Corp.; Alameda, CA). Once tumors were palpable, mice were orally gavaged with 100μl (2mg/mouse) of Vismodegib or DMSO as a vehicle control thrice weekly for three weeks. Animal studies were conducted in accordance with UAB IACUC.
Library Preparation with polyA selection and HiSeq Sequencing for Transcriptomics Profiling
RNA library preparations and sequencing were conducted at GENEWIZ, LLC. (South Plainfield, NJ, USA). Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit using manufacturer’s instructions. The sequencing library was validated on the Agilent TapeStation and quantified using Qubit 2.0 Fluorometer and quantitative PCR. Samples were sequenced using a 2x150bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). FASTQ files were generated using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
RNA-seq Data Analysis
All raw data from sequencing in FASTq format were processed by removing adaptor sequences using Trimmomatic tool, alignment with HISAT2 algorithm, and removal of PCR replicates using SAMtools before expression level quantification. The resulting BAM or SAM files were analyzed through a workflow of Partek Genomics Suite. RPKM normalization was performed before any statistical analysis to select genes by fold changes and statistical p values. The resulting gene lists were used for pathway analyses using IPA (Redwood City, CA USA), GESTALT (15–18) NetworkAnalyst (19–21), and GSEA (22,23). Accession numbers for sequencing data are GSE171168 (whole tumor) and GSE146223 (RAW 264.7 Cells).
Chromatin Immunoprecipitation (ChIP)
RAW 264.7 cells were pretreated with 100nM recombinant SHH (R&D systems) in the presence of 20ng/ml M-CSF. Next, cells were treated with 10μM GANT61 and 20ng/mL IL-4. Cells were processed using the SimpleChip Plus Enzymatic kit. 10μg of cross-linked chromatin was immunoprecipitated with 5μg of anti-Gli1 (Novus Biologicals, Centennial, CO) or isotype control antibody. Chromatin was eluted, cross-links were reversed, and purified DNA was subjected to real-time quantitative PCR. CT values of input DNA were used to calculate percent immunoprecipitation. Each reaction was done in triplicate using Applied Biosystems Step One Plus.
Confocal Imaging
Cells (BMDMs or RAW 264.7) were plated on poly-L-lysine coated coverslips and polarized to M0, M2 + DMSO (10μM), M2 + GANT61 (10μM), M2 + Vismodegib (20μM), or M2 + BMS-833923 (2μM) for 24 hours and stained with 100ng of Mitotracker Green for 25 minutes. After fixation, nuclei were stained with DAPI and imaged (100x magnification) using Nikon A1R Confocal Microscope. Quantification was done using Nikon Elements software.
Flow Cytometry and FACS Sorting
Tumors were dissociated with the heated Miltenyi gentleMACS Octo Dissociator (Miltenyi Biotec). Cells were stained with viability dye eFluor 450 (Thermo Fisher), incubated with primary conjugated antibody cocktail (Biolegend, San Diego, CA), and processed using a BD LSRII Analyzer for flow cytometric analysis or ARIA Analyzer for sorting. FMO controls were utilized. Analysis was done using FlowJo v 10.
Gating strategy - M1 macrophages: Live cells, Cd11b+, F4/80+, Ly6C−, CD80+ CD86+; M2 macrophages: Live cells, Cd11b+, F4/80+, Ly6C−, Cd206+, Arg1+; and pan macrophages: Live, Cd11b+, F4/80+.
Fluorescence Imaging of Tumor Sections
Primary antibodies [anti-F4/80, anti-Cd206, or anti-Patched-1 (Novus Biologicals)] were applied to tumor sections (4.5μm). After washing with 1x PBS, secondary antibodies [Alexa Fluro 488-labeled goat anti-rabbit IgG antibody or Alexa Fluor 594-labeled donkey anti-rat antibody or Alexa Fluor 488-labeled donkey anti-goat antibody or Alexa Fluor 594-labeled goat anti-rabbit antibody] were applied to the sections. Autofluorescence was quenched using TrueVIEW Autofluorescence Quenching Kit with DAPI (Vector Labs; Burlingame, CA). F4/80 Cd206 double-positive staining signified M2 macrophages, and Cd206 Ptch1 double-positive staining marked M2 macrophages expressing Ptch1. Images were captured using a Nikon Eclipse Ti-U microscope (Nikon; Tokyo, Japan) and analyzed using NIS Elements.
Western Blotting
BMDMs were polarized to M0, M2 + DMSO (10μM), M2 + GANT61 (10μM), or M2 + SHH (100nM) for 24 hours. Protein lysates were immunoblotted for MFN, OPA1, DRP1, P-DRP1, STAT6, P-STAT6, RL2, CTD110.6, Cd206, OGA, or OGT and imaged using chemiluminescence. For Dot Blots, 0.1μg of protein lysate was spotted on a nitrocellulose membrane, allowed to dry, and immunoblotted with RL2. Beta-Actin was used as control.
Immunoprecipitation
RAW 264.7 cells were polarized using 20ng/ml IL-4 and 20ng/ml M-CSF with 20μM GANT61 or DMSO for 24 hours. 1 mg of protein lysate was pre-cleared using Protein A/G Plus Agarose bead slurry (50μl; Santa Cruz) for 1 hour at 4°C. Pre-cleared lysates were incubated with 2μg RL2 antibody for 2 hours at 4°C and 50μl of Protein A/G Plus Agarose bead slurry was added at 4°C overnight. Beads were resuspended in 2X Laemmli Buffer and heated at 95°C; supernatant was resolved on SDS PAGE gel.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Real time PCR was performed using TaqMan Fast Advanced Master Mix and Taqman gene expression assay probes for Arginase 1, Cd206, Acadl, Acadm, Cpt1b, Pparg, Ppargc1b, Ogt, Mgea5, Fasn, and Acaca (Thermo Fisher). Actb served as an endorse control gene. Each reaction was done in triplicate using an Applied Biosystems Step One Plus kit.
Luciferase Assay
Cells (RAW 264.7) were plated in triplicate and transfected with PPRE-X3-TK-luc (100ng) or p4xSTAT6-Luc2P (100ng) using Lipofectamine 2000 for 6 hours. Cells were polarized to M0, M2 + DMSO, M2 + GANT61 (10μM), M2 + Vismodegib (20μM), or M2 + OSMI-1 (5μM, 10μM, or 20μM). Luminescence was quantified using a luminometer (Promega) and normalized to total protein concentration.
Phagocytosis Assay
Cells (BMDMs) were polarized to M0, M1, or M2, followed by treatment with Hh inhibitors. Media was aspirated and 100μl of reconstituted pHrodo™ Red E. coli BioParticles were added to each dish in MEM alpha. Two hours later, cells were washed and imaged using the Nikon Eclipse Ti-U microscope and quantified using ImageJ.
JC1 Staining
Differentiated BMDMs were plated in the presence of 25ng/ml of M-CSF. Cells were polarized to M2, then treated with DMSO, GANT61 (10μM), or BMS-833923 (3μM) for 24 hours. BMDMs were stained with JC1 (2μM) for 30 minutes at 37°C. Data was acquired using a BD LSRII Analyzer and analyzed with FlowJo v10.
ATP Quantification
RAW 264.7 cells were polarized to M0, M1, or M2 followed by treatment with DMSO or GANT61 (10μM). ATP was assessed using the ATPLite 1 step Assay Kit (Promega).
Oil Red O Staining
Cells were polarized to M0, M1, or M2 with DMSO or GANT61 (10μM), and assessed for lipid accumulation using Oil Red O Staining following manufacturer’s protocol. ImageJ was used to quantify accumulated lipid droplets.
Metabolomics Data Analysis
Metabolite extraction was performed as described in previous study (16). LC-MS peak extraction and integration were performed using commercially available software Sieve 2.2 (Thermo Scientific). The peak area was used to represent the relative abundance of each metabolite. The missing values were handled as described in previous study (17).
FAO Seahorse Palmitate oxidation assay
BMDMs polarized to M2 or M2 + GANT61 [10μM] (5 x 104) were plated in MEM alpha with inhibitors for 24 hours. Media was changed to DMEM supplemented with 2mM Glucose and 0.5mM Carnitine pH 7.4. Cells were pretreated with Etomoxir (40μM) 15 min prior to assay. Immediately before assay, BSA or BSA/Palmitate was added to plates. Respiration was determined using the XF96 Seahorse extracellular flux analyzer (Seahorse Bioscience, MA, USA). BMDMs were sequentially treated with inhibitors: 1.5 μg/mL oligomycin, 3μM FCCP, 10μM AntimycinA, and 1μM Rotenone. Protein concentration was determined to be equal across all wells.
Metabolic Extracellular Flux Analyzer
BMDMs were polarized to M0, M1, or M2 followed by treatment with DMSO or Hh inhibitors for 12 hours and incubated in unbuffered DMEM containing 5.55mM glucose, 2mM L-Glutamine, and 1mM pyruvate at pH 7.4 for 1 hour at 37°C in a non-CO2 incubator. BMDMs were sequentially treated with inhibitors at a final port concentration of 0.8μM Oligomycin, 0.45μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and 0.4μM Antimycin A. OCR and ECAR analyses were performed according to the Seahorse Biosciences manual.
Statistical Analysis
We used the student’s t-test or ANOVA analysis, as appropriate, and plotted using GraphPad Prism 8 software (La Jolla, CA). Statistical significance was determined for p ≤ 0.05 (indicated with a *). All data shown as mean +/− SEM.
Results
Hedgehog signaling blockade alters metabolic processes in macrophages
To elucidate the effect of Hh activity on macrophage polarization, we utilized two immunocompetent mammary tumor mouse models (Figure 1A) - the 4T1-Balb/c model and the EMT6-Balb/c model. We targeted Hh signaling using the pharmacological FDA-approved, orally available inhibitor Vismodegib (Smo-i) that targets the signal transducer, SMO, of the Hh pathway. We initiated administration of Smo-i once the tumors were ~ 3-4 mm in diameter. Smo-i did not significantly impact tumor growth (Supplementary Figure 1A) or the portfolio of chemokines that direct macrophage recruitment (Figure 1B). However, Smo-i-treated tumors had significantly reduced levels of IL-4 and IL-13 cytokines that are instrumental in M2 macrophage polarization with a concomitant increase in IFN-γ, the M1 programming cytokine (Figure 1B). While there were no appreciable changes in the tumor infiltrating pan-macrophage population (Cd11bhi F4/80hi) (Supplementary Figure 1B), we registered a significant decrease in the abundance of tumor infiltrating M2 macrophages concurrent with increased M1 macrophage infiltration (Figures 1C–F, Supplementary Figure 1C, 1D). We also evaluated mammary tumor sections from both models for Ptch. Tumor infiltrating Cd206-positive macrophages express the Hh receptor Ptch1 (Supplementary Figures 1E–1H), which is expectedly decreased in Smo-i-treated tumors. M2 polarization integrally involves activation of STAT6, which potentiates M2-specific polarization by transcriptionally upregulating arginase 1 (Arg1), mannose receptor 1 (Mrc1, Cd206), and resistin-like α (Fizz1). To determine the effect of Hh on STAT6 activity within the M2 TAMs, we evaluated the activation status of STAT6 in F4/80hi Cd206hi M2 TAMs. There was a significant reduction in active p-STAT6 in M2 TAMs from Smo-i tumors, indicating that Hh blockade mitigates STAT6 activity within the M2 TAMs (Supplementary Figure 1I). In order to evaluate a direct role for the activity of the Gli transcription factors, we used the small molecule inhibitor, GANT61 which directly targets and disables DNA binding of Gli transcription factors at the terminal end of the Hh pathway. To understand the functional ramifications of Hh signaling on macrophages, we evaluated their phagocytic activity. While activation of Hh with exogenous recombinant SHH decreased the phagocytic capability of M2-polarized macrophages, inhibiting Gli activity remarkably elevated their phagocytic activity, indicating that blockade of Hh signaling promotes the effector functions of immunosuppressive macrophages (Supplementary Figure 1J).
Figure 1. Hedgehog signaling blockade alters the tumor infiltrating macrophage polarization and metabolic processes.
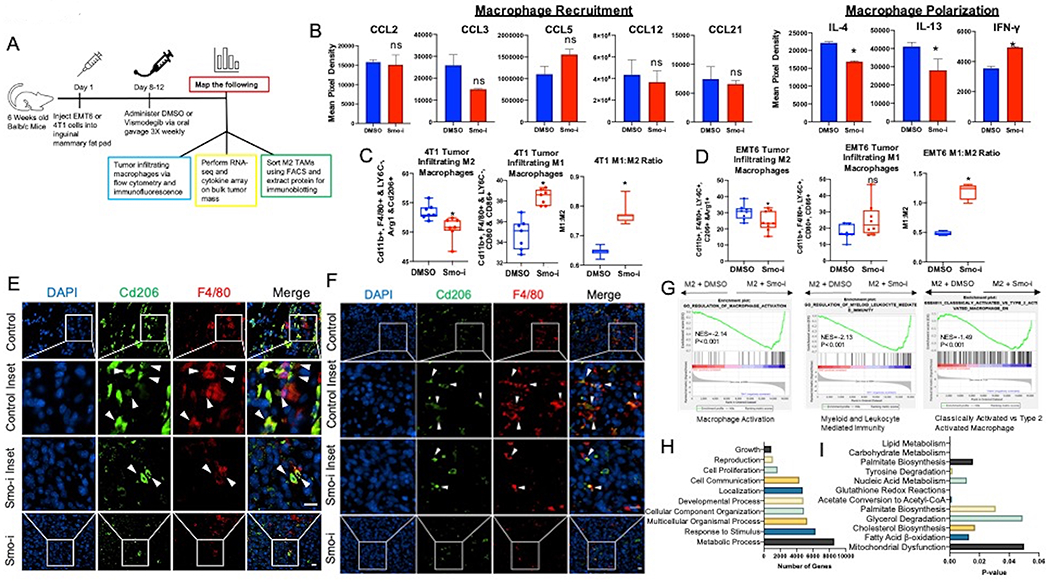
A. Schematic of in vivo experimental timeline. B. Quantitative analysis of chemiluminescent intensity from the cytokine array measurements. (n = 4 per group). C,D. Quantitation of flow cytometric data from 4T1 (C) and EMT6 (D) tumors represented as % of tumor infiltrating TAMs. M2 macrophages are defined as Cd11b+, F4/80+ LY6C−, Arg1+ and Cd206+ populations, and M1 macrophages are defined as Cd11b+, F4/80+, LY6C−, Cd80+, Cd86+ populations (n = 7 per group). E, F. Immunofluorescent staining from 4T1 (E) and EMT6 (F) tumors for Cd206 (green), F4/80 (red), and DAPI (blue). White arrows point to M2 macrophages (F4/80+ Cd206+). Magnification bar is 15μm. G. GSEA Enrichment plots from RNA-seq of bulk 4T1 tumor mass depicting changes in macrophage related signatures. H,I. Pathway analysis of RNA-sequencing data shows top pathways based on number of relevant genes in (H) and top pathways based on significant p-value in (I). *P<0.05
We analyzed the transcriptome of bulk tumors resected from vehicle control or Smo-i-treated mice. Enrichment plots from RNA-seq data indicate that genes involved in macrophage activation are enriched in Smo-i-treated tumors (Figure 1G). Analysis of RNA-sequencing data based on KEGG, Wikipathways, Panther (Figure 1H), NetworkAnalyst, and ingenuity pathway analysis (IPA) reveal remarkable alterations in metabolic processes with Smo-i treatment, and out of the top metabolic pathways that are significantly altered, carbohydrate and lipid metabolism present with high significance (Figure 1I, Supplementary Figure 1K). These changes are also captured in RNA seq analysis of M2 macrophages (skewed from RAW264.7 cells, Supplementary Figure 1L) inhibited for Hh signaling, highlighting alterations in FAO, PPAR signaling, O-linked glycosylation, and glycolysis (Supplementary Figure 1M, 1N,).
Following this lead, we adopted a mass spectrometry-based untargeted metabolomics analysis approach with BMDMs skewed to M0, M2, or M2 inhibited for Gli activity (Supplementary Figure 2A). This analysis revealed that 98 metabolites were significantly altered by a fold change of two or greater (Figure 2A). IPA of metabolomics presented the UDP-N-acetyl-D-glucosamine Biosynthesis II pathway as a top significantly altered pathway in M2 macrophages inhibited for Hh signaling (Figure 2B). One metabolite that is enhanced in M2 macrophages and possibly contributes to their immune-suppressive phenotype is Uridine diphosphate N-acetyl-alpha-D-glucosamine (UDP-GlcNAc) (5), an end product of the HBP. Relative to M0 macrophages, UDP-GlcNAc levels are significantly increased in M2 macrophages (Supplementary Figure 2B). Inhibiting Hh activity significantly reduced the levels of UDP-GlcNAc and the majority of metabolites in the UDP-GlcNAc synthesis pathway (Figure 2C, Supplementary Figure 2C). Thus, Hh signaling blockade alters the UDP-GlcNAc metabolic signature in M2 macrophages.
Figure 2. Hedgehog signaling blockade alters UDP-GlcNAc related metabolic processes in macrophages.
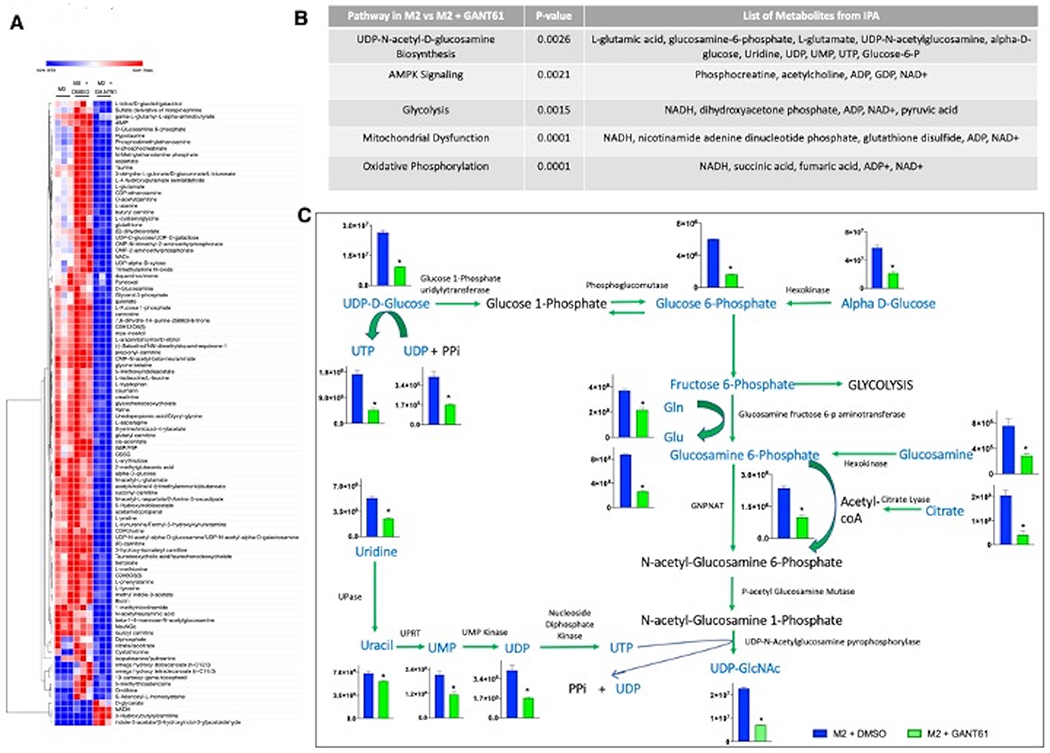
A. Heatmap of top metabolites significantly altered in M2 macrophages treated with GANT61 compared with vehicle control. All changes are statistically significant and represent alterations by greater than two-fold (DMSO vs GANT61 comparison). B. IPA analysis of metabolomics data showing top significant metabolic pathways altered. C. Metabolites of the UDP-GlcNAc biosynthesis pathway are significantly altered by blockade of Hh signaling. Metabolomic data for each respective metabolite in the pathway is plotted adjacent to its metabolite. Metabolites are plotted as Metabolite Concentration (AU) +/− SEM (Y-axis) for M2 + DMSO and M2 + GANT61 groups. Metabolites in blue font are significantly downregulated in M2 macrophages inhibited for Hh signaling (n=3) *P<0.05.
Hedgehog signaling enhances O-GlcNAcylation to potentiate the M2 phenotype
UDP-GlcNAc is the substrate for O-GlcNAcylation, a post translational modification which culminates in the addition of O-GlcNAc moieties to serine and threonine residues on proteins (Figure 3A). Elevated O-GlcNAcylation in the TME contributes to increased tumor growth and invasion (18)(19). Hh blockade diminished the cellular pool of O-GlcNAcylated proteins (Figure 3B, Supplementary Figures 3A, 3B, 3C). This is accompanied by a significant decrease in the levels of O-GlcNAc Transferase (OGT), the enzyme that adds O-GlcNAc to target proteins (Figure 3C, Supplementary Figure 3D). No changes in transcript or protein levels were evident in O-GlcNAcase (OGA, Mgea5), the enzyme that removes O-GlcNAc residues (Supplementary Figure 3E, 3F).
Figure 3. Hedgehog signaling regulates O-GlcNAcylation in M2 polarized macrophages.
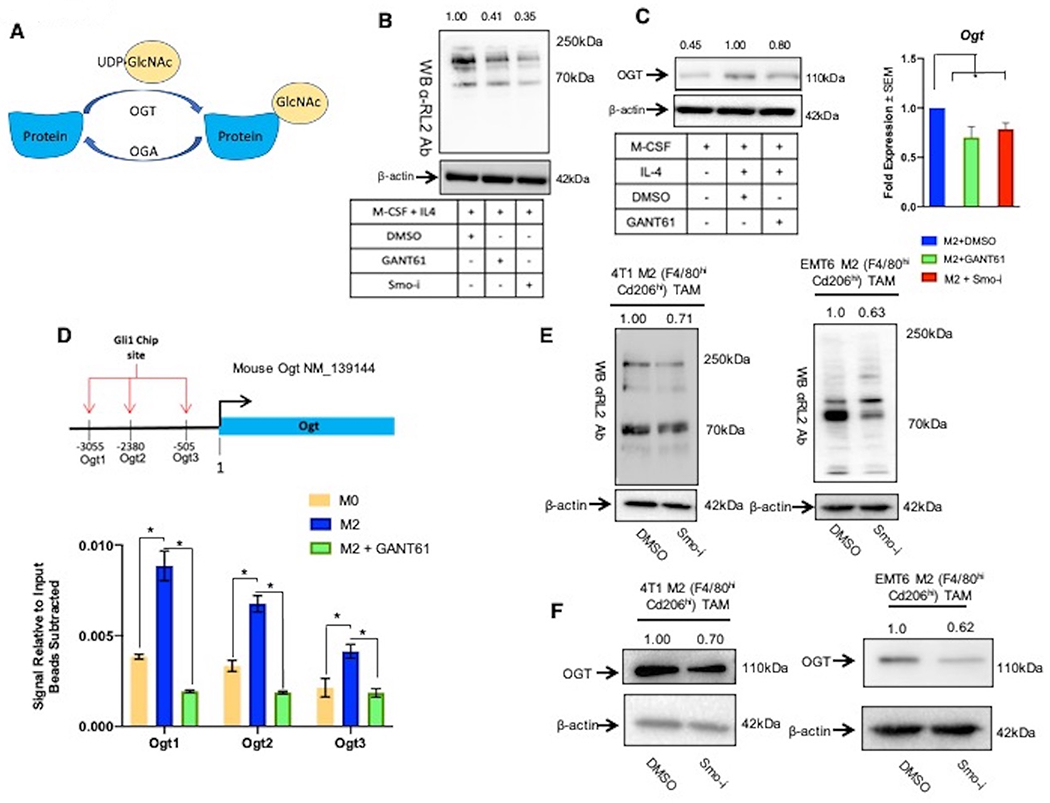
A. Schematic of O-GlcNAcylation. Ogt adds O-GlcNAc to target proteins from the substrate UDP-GlcNAc, and Oga removes O-GlcNAc moieties. B. Immunoblot for all cellular O-GlcNAcylated proteins (using RL2 antibody) in M2 macrophages (BMDMs) +/− Hhi. C. Protein (Immunoblot) and transcript (qPCR) levels for Ogt in M2 macrophages (BMDMs) +/− Hhi. D. ChIP was performed in macrophages (skewed from RAW264.7 cells) for Gli1 binding to the Ogt promoter followed by qPCR with primers specific for Ogt promoter. Graph shows qPCR results with signal relative to input with beads subtracted. E. Immunoblots for cellular O-GlcNAcylated proteins (using RL2 antibody) in M2 macrophages sorted from 4T1 tumors and EMT6 tumors of mice treated with DMSO or Smo-i. F. Immunoblots for Ogt in tumor infiltrating M2 macrophages sorted from 4T1 and EMT6 tumors of mice treated with DMSO or Smo-i *P<0.05. These lysates were also used for data depicted in Figure 8C. As such the same image for β-actin loading control is used.
We next explored if Hh/Gli activity transcriptionally regulates Ogt. We identified three putative Gli1 binding sites in the promoter sequence of Ogt. By employing a ChIP assay, we registered that Gli1 binding to all three predicted sites is significantly enriched in M2 skewed macrophages compared to resting (M0) macrophages (Figure 3D). GANT61 caused a significant decrease in Gli1 binding to the Ogt promoter (Figure 3D). Furthermore, exogenous Shh ligand elevated cellular O-GlcNAcylation, establishing a heretofore unknown direct link between Hh signaling and O-GlcNAcylation in M2-polarized macrophages (Supplementary Figure 3G).
In order to evaluate the translational relevance of this finding, we assessed O-GlcNAcylation in M2 macrophages (F4/80hi Cd206hi) sorted from mammary tumors (Supplementary Figure 3H). F4/80hi Cd206hi macrophages from tumors of Smo-i-treated mice demonstrate an overall decrease in the landscape of O-GlcNAcylated proteins and reduced Ogt protein levels (Figure 3E, 3F, Supplementary Figure 3I). Aligned with this, we registered decreased O-GlcNAcylation in the F4/80hi TAM population sorted from Smo-i mammary tumors (Supplementary Figure 3J), indicating that even though the overall abundance of F4/80hi TAMs is unchanged (Figure 1B, Supplementary Figure 1C), their O-GlcNAc landscape is altered. Cumulatively these data establish a role for Hh signaling in directing metabolic changes and altering cellular O-GlcNAcylation in M2 TAMs.
Cd206 is a critical marker of M2-macrophages that is modified by posttranslational N-glycosylation (20)(21). Inhibiting Hh notably decreased Cd206 protein levels in M2 macrophages (Supplementary Figure 4A). We explored if Hh activity might impact its O-GlcNAcylation, based on predictions of several possible O-GlcNAc sites in Cd206 protein (Supplementary Figure 4B). Hh blockade remarkably decreased the pool of O-GlcNAc-modified Cd206 (Supplementary Figure 4C). This likely reflects a cumulative decrease in Cd206 and its O-GlcNAcylation.
STAT6 has a prime role in orchestrating the M2 transcriptional program, in part by transcriptionally regulating Cd206. In addition to phosphorylation, O-GlcNAc-modification of STAT6 enhances its transcriptional activity (22,23). The Yin-O-Yang1.2 engine predicts three robust sites of O-GlcNAcylation in STAT6 protein (Figure 4A). Inhibiting Hh activity significantly decreased O-GlcNAc modification of STAT6 without impacting total STAT6 levels (Figure 4B, 4C; Supplementary Figure 4D). In order to test the impact of O-GlcNAc-modification on the transcriptional activity of STAT6 we inhibited Ogt using the small molecule inhibitor, OSMI-1, in M2 macrophages. Ogt blockade diminished STAT6 activity in a dose-dependent manner (Figure 4D) and significantly reduced the expression of STAT6 transcriptional targets, Arg1 and Cd206, suggesting that O-GlcNAc-modification of STAT6 critically impacts M2 gene expression (Figure 4E, Supplementary Figure 4E). These outcomes collectively substantiate a role for Hh signaling in impacting STAT6 activity through enhancing its O-GlcNAcylation.
Figure 4. Hedgehog signaling regulated O-GlcNAcylation potentiates STAT6 signaling in M2 polarized macrophages.
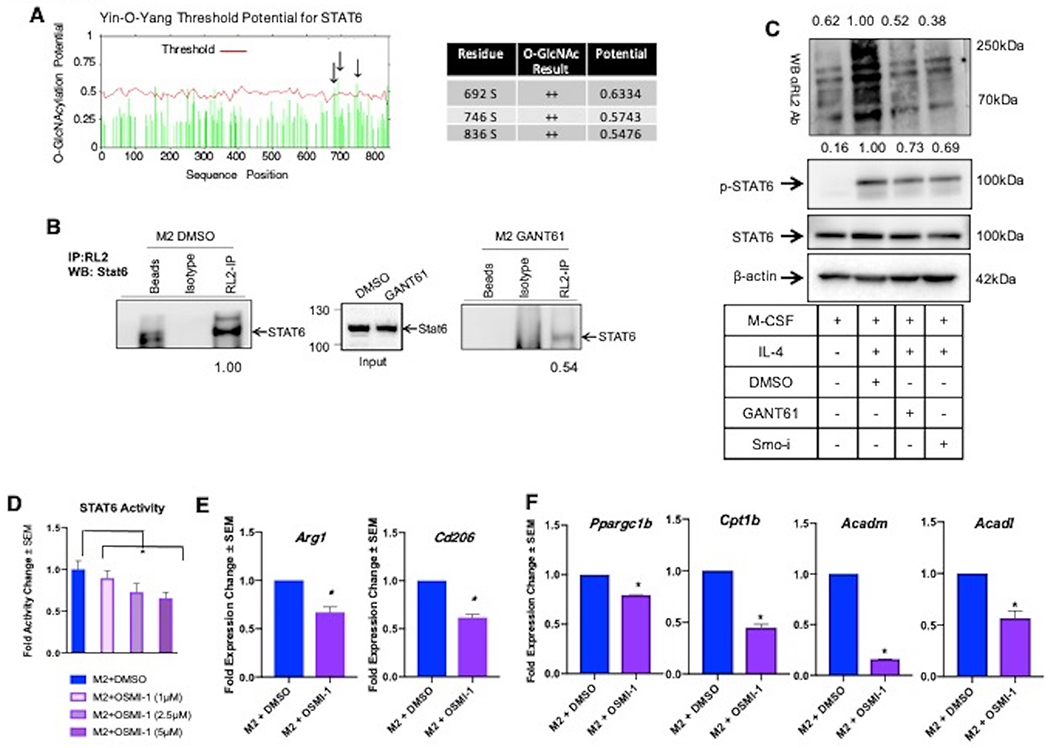
A. O-GlcNAc potential for murine STAT6 as determined by the Yin-O-Yang search engine. Black arrows point to sites of high likelihood for Cd206 O-GlcNAcylation that are specified in the adjacent table. B. Macrophages (RAW 264.7 cells) polarized to M2 + DMSO, M2 + GANT61, were immunoprecipitated for O-GlcNAcylated proteins with anti-RL2, then immunoblotted for STAT6. Densitometry values were determined by normalizing by the average of empty beads and isotype control lanes. C. Controls for IP depicting total O-GlcNAc levels (anti-RL2), STAT6 levels, and phospho-STAT6 levels in M2 macrophages inhibited for Hh signaling. D. Luciferase assay for STAT6 activity was performed on M2 macrophages inhibited for Ogt with OSMI-1. E. qRT-PCR analysis of steady-state transcript levels of M2 markers Arg1 and Cd206 in M2 macrophages (BMDMs) inhibited for Ogt using OSMI-1. F. Transcript levels of FAO genes in M2 BMDMs treated with OSMI-1. *P<0.05
As an essential aspect of immunosuppressive programming of macrophages, STAT6 acts as a co-factor for peroxisome proliferator-activator receptor gamma coactivator 1-beta (PGC-1β) to activate transcription of genes that regulate lipid metabolism. Accordingly, we tested the effects of O-GlcNAc manipulation on the expression of these genes. Inhibition of Ogt significantly decreased expression of Ppargc1b, and of fatty acid oxidation (FAO) genes Cpt1b, Acadm, and Acadl (Figure 4F). Collectively, these findings support a role for Hh/Gli signaling in intersecting with the UDP-GlcNAc biosynthesis pathway impinging upon critical metabolic programs that regulate alternative polarization of macrophages.
Hedgehog signaling alters FAO in immunosuppressive macrophages
In agreement with STAT6 regulating genes critical for FAO, global transcriptomics analyses of the bulk mammary tumor and skewed M2 macrophages, and our metabolomics analysis, independently underscore a role for Hh/Gli signaling in orchestrating lipid oxidation in macrophages (Figures 5A–D). Blockade of Hh in mammary tumors diminished several fatty acid metabolism signatures (Figure 5A). Inhibiting Hh signaling in skewed M2 macrophages resulted in a metabolome signature of lipid oxidation of short, long, and branched chain fatty acids (Figure 5B), and highlighted signatures of lipid metabolic processes within the bulk tumor mass (KEGG pathways) (Figure 5C, Supplementary Tables 2–5). GSEA from macrophage RNA-seq highlights alterations consistent with fatty acid metabolic processes and nuclear receptor activity (Figure 5D). STAT6 functions as a cofactor with peroxisome proliferator-activated receptor gamma (PPARγ) and promotes lipid uptake, and enhances FAO as a means for generating energy for the metabolically demanding M2 macrophages, potentiating a switch to OXPHOS (24). Hh inhibition in M2 macrophages reduced the transcript levels of Pparg and Ppargcb1 (Figure 5E; Supplementary Figure 5A, 5B) and blunted the activity of PPARγ (Figure 5F, Supplementary Figure 5C). Concordantly, we registered decreased expression of genes involved in FAO (Figure 5G, Supplementary Figure 5D, 5E) with a simultaneous increase in fatty acid synthesis genes (Supplementary Figure 5F). Silencing Gli1 expression phenocopied the effects of GANT61 on these alterations in FAO (Supplementary Figure 5G).
Figure 5. Hedgehog signaling blockade reduces transcriptional signatures of fatty acid oxidation.
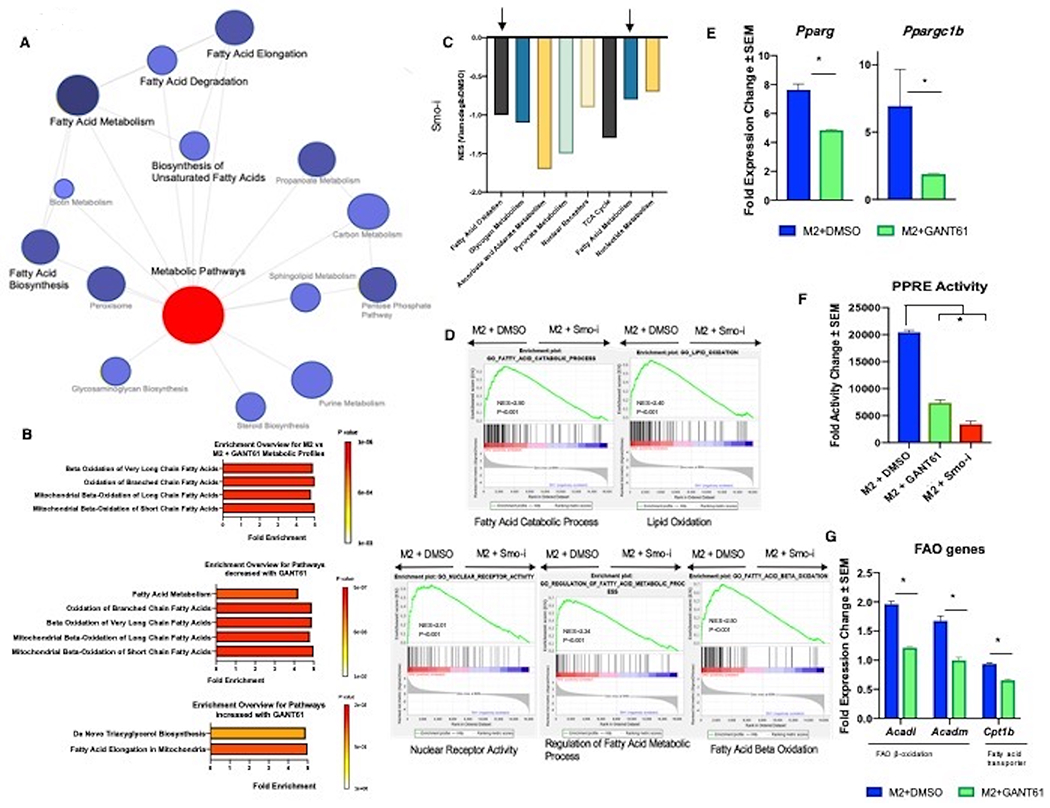
A. PANTHER Network analyst yields a network of metabolic pathways that are differentially regulated in 4T1 tumors from mice treated with DMSO or Smo-i. B. Pathway enrichment score of metabolomics data. The graphs depict (Top) all pathways, (Middle) pathways that are downregulated with Hh inhibition, and (bottom) pathways that are upregulated with Hh inhibition. C. KEGG pathway analysis depicting metabolic pathways that are significantly downregulated in tumors from mice treated with DMSO or Smo-i. D. GSEA Enrichment plots from RNA-seq of RAW 264.7 cells skewed to M2 macrophages and inhibited for Hh signaling with GANT61. Normalized Enrichment Score (NES) and P-Value are depicted. E. Transcript levels of Pparg and Ppargc1b in BMDMs polarized to M2 macrophages +/− Hhi. F. PPARγ activity in RAW 264.7 cells polarized to M2 macrophages +/− Hhi. G. Transcript levels of FAO genes in BMDMs polarized to M2 +/− Hhi. *P<0.05
In order to assign functional relevance to transcriptional alterations in lipid metabolism, we utilized the Seahorse XF Palmitate-BSA FAO assay to quantify oxidation of exogenous fatty acids. In brief, M2 macrophages were pretreated with Etomoxir, BSA, and/or BSA-Palmitate prior to the assay. Oligomycin was introduced first to inhibit ATP synthase, followed by FCCP to uncouple the mitochondrial inner membrane, and finally Antimycin A/Rotenone to inhibit complex III of the electron transport chain (Figure 6A). Etomoxir was used as a FAO inhibitor and elicited effective inhibition of FAO. Concordant with the transcriptional FAO profile, GANT61 significantly reduced FAO at basal and maximal levels in M2 polarized macrophages (Figure 6B, 6C). At the cellular level, this manifested as increased accumulation of lipid droplets (Figures 6D; Supplementary Figure 6A). These observations are consistent with findings linking the family of nuclear receptors to induce a metabolic shift contributing to M2 polarization (25). Such metabolic signatures involve activation of PPARγ in controlling homeostasis by regulating FAO (Figure 5A, 5B and Supplementary Tables 2–5) to ensure a supply of ATP to sustain cellular functions. As such, reduced FAO in Hh inhibited M2 macrophages is reflected in significantly diminished levels of ATP (Figure 6E, Supplementary Figure 6B). Collectively, our studies establish an important role for Hh signaling in altering FAO in immunosuppressive macrophages.
Figure 6. Inhibiting Hedgehog signaling significantly decreases lipid oxidation in M2 macrophages.
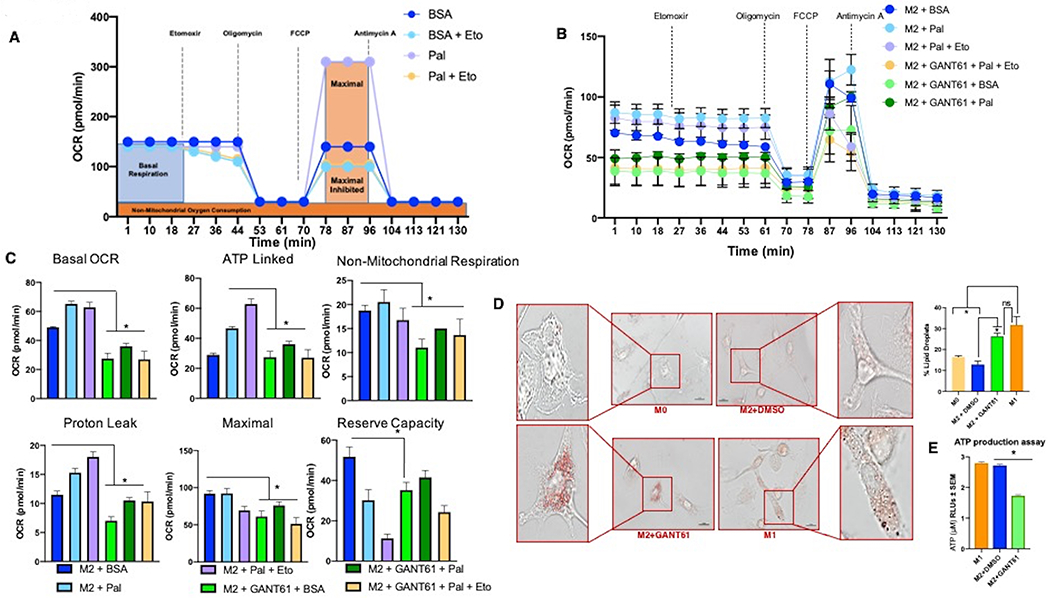
BMDMs skewed to M2 macrophages were used for all panels. The figure legends detail the specific treatments applied to M2 macrophages. A. Panel outlining experimental setup and depicting idealized FAO Seahorse Palmitate assay results. Seahorse XFe96 extracellular flux analyzer was used to calculate: B. Oxygen consumption rate (OCR), C. Basal, maximal, non-mitochondrial, ATP-linked respiration, proton leak, and reserve capacity. D. BMDMs [M0 (resting macrophages), polarized to M1 or M2 macrophages] were stained for lipid droplets by Oil Red-O. GANT61 was used to inhibit Gli activity. Scale bar represents 10 μm. E. Cellular ATP content in M2 macrophages (skewed from BMDMs) +/− Hhi. *P<0.05
Inhibition of Hh/Gli signaling alters mitochondrial bioenergetic activity and morphology
Mitochondria are the main site for FAO and function of nuclear receptors PGC-1β and PPARγ (26). We surmised that changes in ATP levels reflect a switch in the bioenergetic profile of the macrophages. M2 macrophages principally rely on OXPHOS, whereas M1 macrophages depend on glycolysis for energy production (5,18,19,27). For M1 macrophages, this reliance on glycolysis results in enhanced Krebs cycle intermediates, such as succinate, which activate HIF-1α to stimulate the production of inflammation-related genes. For M2 macrophages, their reliance on oxidative metabolism is important for prolonged energy generation to maintain wound healing and tissue repair functions (28). Our untargeted metabolomics assessment has identified significant changes in OXPHOS, mitochondrial dysfunction, and glycolysis (Figure 2B), suggestive of a bioenergetic re-calibration when Hh signaling is impeded in macrophages.
We evaluated mitochondrial respiration in macrophages using the Seahorse extracellular flux analyzer (Figure 7A) to assess changes in the mitochondrial oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Relative to M1 macrophages, the M2 macrophages show robust respiration characterized by elevated basal and maximal reserve capacity. Inhibiting Hh significantly decreased basal and maximal respiration of M2 macrophages (Figure 7B, 7C, 7D; Supplementary Figures 7A, 7B). In contrast to OCR, the ECAR of M2 macrophages is significantly reduced relative to that of M1 macrophages. Inhibiting Hh/Gli signaling results in a significant increase in ECAR, reminiscent of M1 macrophages (Figure 7E, Supplementary Figure 7C). In agreement with this, mitochondria from M2 macrophages displayed high membrane potential as indicated by greater numbers of macrophages with JC-1 stained red-fluorescent aggregates. In contrast, Hh inhibited macrophages had increased proportion mitochondria with low membrane potential (Figure 7F).
Figure 7. Hh/GLI signaling impacts metabolic reliance on glycolysis with reciprocal decrease in OXPHOS.
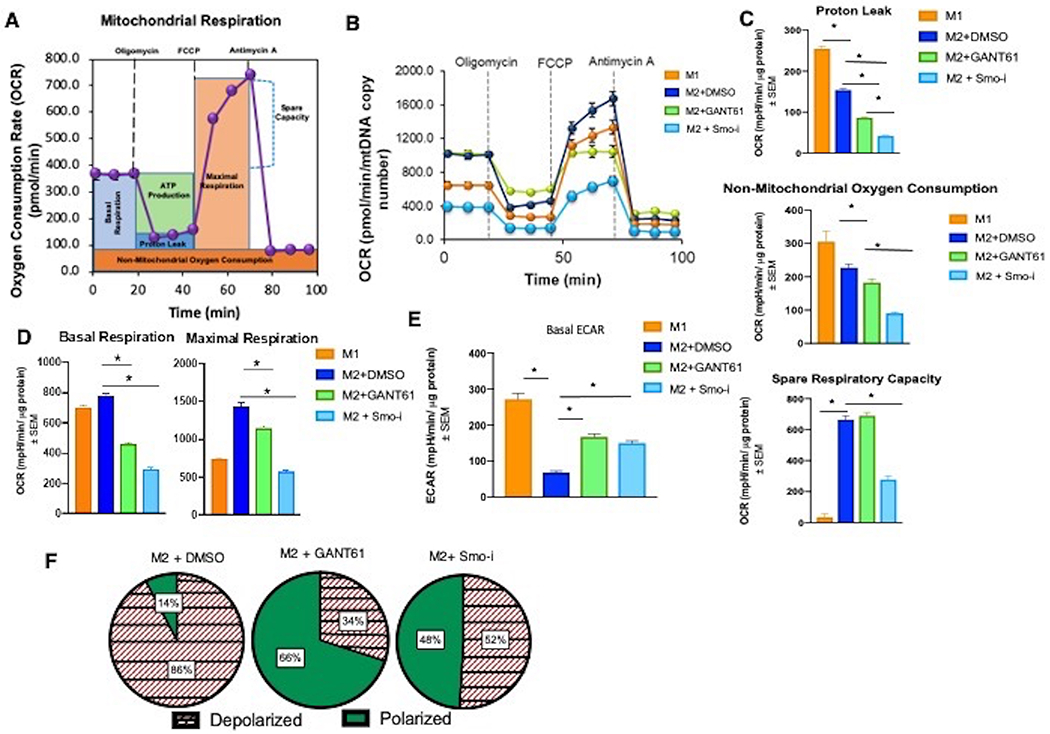
BMDMs skewed to macrophages were used for all panels. A. Schematic for Seahorse mitochondrial stress test. Seahorse XFe24 extracellular flux analyzer was used to calculate: B. Oxygen consumption rate (OCR), C. Proton leak, non-mitochondrial respiration, reserve capacity, D. Basal and maximal respiration, and E. Basal extracellular acidification rate (ECAR). F. Macrophages (BMDMs skewed to M2 +/− Hhi) stained with JC-1.
Cellular bioenergetics is also intricately associated with distinct mitochondrial morphology (29). Cells utilizing OXPHOS have mitochondria with a fused morphology, whereas cells reliant on upregulated glycolysis have characteristically fragmented mitochondria due to increased electron leakage (30). We utilized confocal imaging to visualize mitochondrial morphology of skewed macrophages inhibited for Hh signaling (Figure 8A, Supplementary Figure 8A). In contrast to M1 macrophages with fragmented mitochondria, M2 macrophages demonstrate a fused mitochondrial phenotype that is disrupted upon Hh inhibition. Moreover, mitochondria from macrophages inhibited for Hh/Gli signaling resemble those of M1 macrophages (Figure 8A, Supplementary Figure 8A). Such morphological changes are orchestrated by proteins including MFN1, OPA1, and p-DRP that regulate fusion (MFN1 and OPA1) or fission (p-DRP). Agreeably, inhibition of Hh/Gli signaling registers as diminished expression of MFN1 and OPA1. There is concurrent upregulation in the expression of p-DRP1 (Figure 8B, Supplementary Figures 8B, 8C, 8D).
Figure 8. Inhibition of Hh/GLI signaling alters mitochondrial morphology in M2 macrophages.
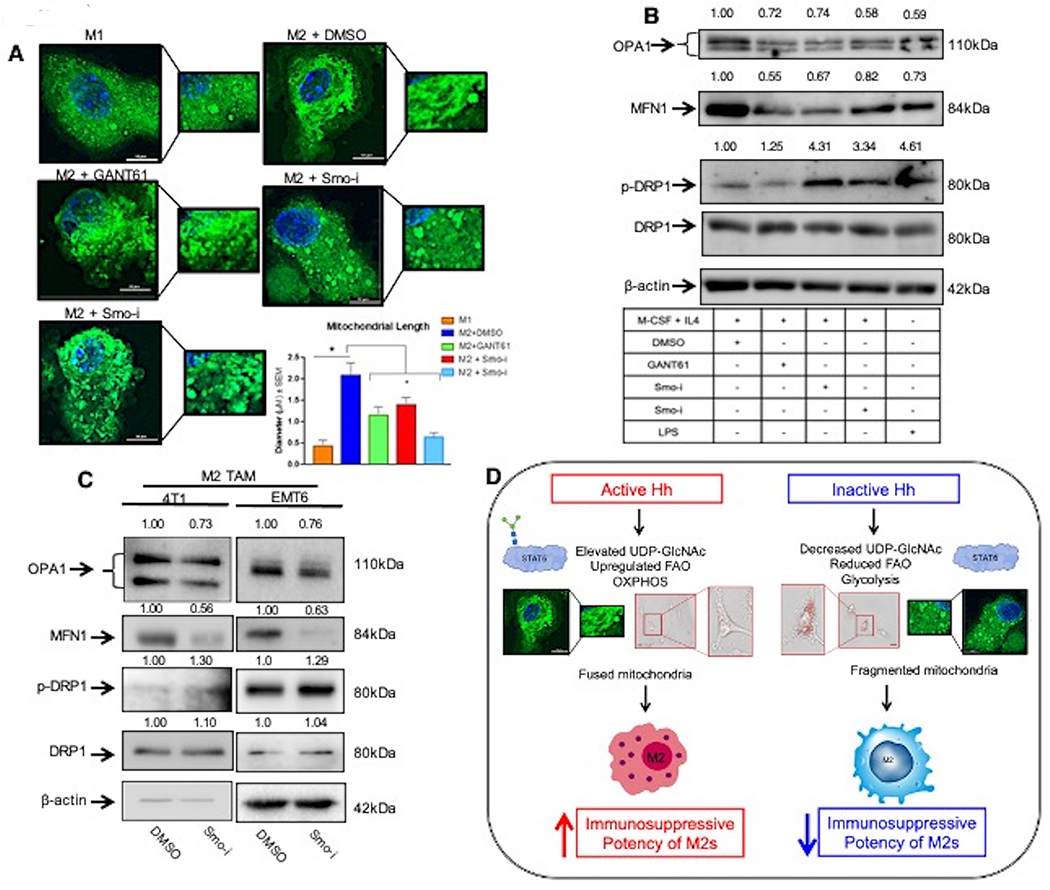
A. Macrophages were stained with Mitotracker Green and visualized via confocal imaging. Scale bar represents 10 μm. Quantitation was performed using NIS Elements. B. Western Blot for fusion (OPA1, MFN1) and fission (p-DRP1, DRP1) related proteins in BMDMs skewed to M1 or M2 macrophages +/− Hhi. C. F4/80hi Cd206hi tumor infiltrating (M2) macrophages were sorted from 4T1 and EMT6 mammary tumors from mice treated with Smo-i or vehicle control and immunoblotted for mitochondrial fusion and fission proteins. These lysates were also used for data depicted in Figure 3F. As such the same image for β-actin loading control is used. D. Schematic highlighting the role of Hh signaling in altering macrophage metabolism. Parts of this schematic were made using BioRender *P<0.05
In order to explore pharmacological relevance of these leads, we employed the 4T1-Balb/c and EMT6-Balb/c mammary tumor models (Figure 1A) and sorted tumor-infiltrating F4/80hi Cd206hi (M2) macrophages (Supplementary Figure 3H). M2 macrophages enriched from Smo-i-treated primary tumors demonstrate decreased expression of OPA1 and MFN1 and upregulated expression of p-DRP1 (Figure 8C, Supplementary Figure 8E, 8F). Inhibiting Hh activity did not significantly impact mitochondrial DNA (mtDNA) abundance (Supplementary Figure 8G). Taken together, our data indicate that Hh signaling directs major alterations in metabolic and bioenergetic programs that collectively impact alternative polarization of macrophages (Figure 8D).
Discussion
Hedgehog signaling is essential for effective metabolic reprogramming in immunosuppressive macrophages
Utilizing two immunocompetent orthotopic models of mammary tumors treated with the pharmacological Hh inhibitor, Vismodegib, we registered a remarkable shift in the portfolio of tumor-infiltrating macrophages. Untargeted metabolomics identified that Hh inhibition significantly reduced flux through the UDP-GlcNAc biosynthesis pathway in M2 macrophages. Relative to M1 macrophages, M2 macrophages have elevated UDP-GlcNAc, which serves as a substrate for N- or O-linked glycosylation (19)(18). While the relevance of N-Glycosylation in the M2 macrophage state is appreciated, the role of O-linked Glycosylation in M2 macrophages remains largely associative and enigmatic. Elevated O-GlcNAcylation in the TME contributes to increased tumor growth, potentially through decreased inflammatory cytokine production by TAMs (31). O-GlcNAcylation of STAT6 augmented its activity and expression of its transcriptional targets, Arg1 and Cd206, critically contributing to the immune-suppressive state of macrophages. STAT6 functions as a co-factor for PGC-1β to bind the promoters of genes responsible for upregulating lipid uptake and FAO. PGC-1β, in concert with PPARγ and STAT6, promotes lipid uptake and enhances FAO as a means for generating energy for the metabolically demanding M2 macrophages. Blocking Hh reduced the transcript levels of both Pparg and Ppargc1b, and attenuated FAO. In medulloblastoma, Hh signaling activates PPARγ and enhances the proliferative capacity and progression of tumor cells (32). As such, our work establishes a novel role for Hh signaling in orchestrating O-linked glycosylation that impinges upon STAT6-regulated FAO and consequently governing the M2 state of macrophages.
Hedgehog signaling regulates bioenergetics in immunosuppressive macrophages
Metabolic adaptations that drive the bioenergetic profiles associated with M1 and M2 macrophages are fundamental to macrophage activation and effector function (2). While M2 macrophages require an abundance of ‘fuel’, they employ programs driving OXPHOS and lipid oxidation; M1 macrophages principally employ glycolysis, yielding less ATP. Uncoupling Hh activity in M2 macrophages prompted a decrease in mitochondrial function reflected in compromised basal and maximal respiration. In contrast with our findings in macrophages, paracrine Hh signaling in hepatocellular carcinoma stimulates glycolysis in surrounding myofibroblasts resulting in increased lactate production providing an additional energy source to fuel tumor cell proliferation (33). Moreover, in brown fat and muscle cells, Hh signaling drives glycolysis by reducing OCR and increasing ECAR (34). Interestingly, Hh signaling regulates bioenergetics in a cell type specific manner that ultimately supports tumor growth. By upregulating glycolysis in tumor cells, Hh signaling helps contribute to the availability of growth-promoting molecular intermediates through the Warburg Effect. Whereas, in macrophages, Hh signaling supports OXPHOS to promote their M2 phenotype. Collectively, such findings cement the integral role of Hh signaling in regulating cellular metabolism, in both immune cells and tumor cells, to collectively foster tumor survival, thus providing a multi-faceted role for targeting Hh signaling within the TME.
These changes in cellular metabolism and bioenergetics are enabled by mitochondrial functional adaptations (29). A continuous mitochondrial network formed by mitochondrial fusion is required for FAO and OXPHOS. Consequently, cellular supplementation with lipids results in reduced mitochondrial fusion (35), leading to an accumulation of triacylglycerols and the export of free fatty acids out of the cell (36). When cells utilize glycolysis predominately for energy production, which is indicative of a state of nutrient abundance, their mitochondria rapidly undergo fission diminishing their cellular fitness (30). Zheng et al have established that hypoxia provokes mitochondrial fragmentation and diminishes the function of natural killer cells within the TME (37). Studies such as this demonstrate a close connection between hypoxia and changes in cellular metabolic profiles, suggesting that many complex aspects of TME can contribute to these alterations. Our findings indicate that Hh inhibition elicits a markedly altered bioenergetic program that mitigates the health and functionality of M2 macrophages in the TME.
Hh signaling is uniquely positioned at the nexus of several transcriptional networks. In the context of macrophages, this work shows that Hh establishes a circuit with STAT6 via its O-GlcNAcylation, resulting in a metabolic and bioenergetic portfolio that is reminiscent of inflammatory macrophages. As such, interference with the homeostasis of macrophage metabolism readily primes macrophages for phenotype reversal. This is the first time that Hh activity has been shown to regulate metabolic and bioenergetic programming of tumor-associated macrophages. Further, our work provides evidence for modulating Hh activity, and consequently macrophage metabolism, to sculpt the plasticity of macrophages.
Current approaches to target TAMs involve depleting M2 macrophages or re-polarizing them to M1-like macrophages. The allure of targeting Hh signaling stems from the availability of several FDA-approved inhibitors already in use in the clinic, making it applicable to different tumor types. Our work supports the possibility that Hh blockade is multi-dimensional in its effect on recalibrating the metabolism and consequently the functions of immunosuppressive macrophages. Therefore, inhibition of Hh activity could present a dual-pronged approach that reduces the abundance of M2 TAMs and perhaps, promotes M2 to M1 trans-differentiation by inherently changing their metabolic status, thus promoting a robust immunogenic response to cancer.
Supplementary Material
Statement of Significance.
These findings illustrate that Hh activity regulates a metabolic and bioenergetic regulatory program in tumor-associated macrophages that promotes their immune suppressive polarization.
Acknowledgements
The authors acknowledge funding from the following sources: Department of Defense (W81XWH-14-1-0516, W81XWH-18-1-0036, W81XWH-10-1-0755), NCI R01CA169202, O’Neal Invests, and The Breast Cancer Research Foundation of Alabama (BCRFA), all awarded to L. A. Shevde. The work is supported in part by BXAl3374 (VA) and CA194048 (NCI/NIH) to R. S. Samant. The authors would like to thank Melissa J. Sammy for her technical assistance with experimental procedures; the UAB Bio-Analytical Redox Biology (BARB) Core, the UAB High Resolution Imaging Facility, the UAB Comprehensive Flow Cytometry Core supported by NIH Grants P30 AR048311 and P30 AI027667, and UAB Comprehensive Cancer Center’s Preclinical Imaging Shared Facility supported by NIH grant P30 CA013148. The authors thank the Juan Liu and the Jason Locasale laboratory at Duke University for their assistance with the metabolomics analysis.
Abbreviations
- ARG1
Arginase 1
- ATP
Adenosine triphosphate
- BMDMs
Bone Marrow-Derived Macrophages
- CCL2
C-C Motif Chemokine Ligand 2
- CCR2
C-C Motif Chemokine Receptor 2
- Cd206
Cluster of Differentiation 206
- DHH
Desert Hedgehog
- DRP1
Dynamin-related protein 1
- ECAR
Extracellular Acidification Rate
- FAO
Fatty Acid Oxidation
- FCCP
Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- Fizz1
Resistin like molecule alpha 1
- GLI
Glioma-Associated Oncogene
- HBP
Hexosamine Biosynthetic Pathway
- Hh
Hedgehog
- Hhi
Hedgehog inhibitor
- IL-4
Interleukin 4
- IL-10
Interleukin 10
- IL-12
Interleukin 12
- IL-13
Interleukin 13
- IFN-γ
Interferon Gamma
- IHH
Indian Hedgehog
- Kif7
Kinesin Family Member 7
- M1
Classically Activated Macrophages
- M2
Alternatively Activated Macrophages
- MFN1
Mitofusin-1
- mtDNA
Mitochondrial DNA
- OCR
Oxygen Consumption Rate
- OGA
O-GlcNAcase
- OGT
O-GlcNAc Transferase
- OPA1
Optic Atrophy Protein 1
- OXPHOS
Oxidative Phosphorylation
- p-DRP1
Phosphorylated Dynamin-related protein 1
- PD-L1
Programmed Death Ligand-1
- PGC-1β
Peroxisome proliferator-activated receptor gamma coactivator 1-beta
- PPARγ
Peroxisome proliferator-activated receptor gamma
- Ptch1
Patched 1
- SHH
Sonic Hedgehog
- SMO
Smoothened
- Smo-i
Smoothened Inhibitor
- STAT6
Signal Transducer and Activator of Transcription 6
- SUFU
Suppressor of fused protein
- TAMs
Tumor-Associated Macrophages
- TCA
Tricarboxylic Acid cycle
- TGF-β
Transforming Growth Factor Beta
- TME
Tumor microenvironment
- TNF-α
Tumor Necrosis Factor alpha
- UDP-GlcNAc
Uridine diphosphate N-acetylglucosamine
Footnotes
Declaration of Interests
The authors declare no competing conflicts of interest for this work.
References
- 1.Alessandrini F, Menotti L, Avitabile E, Appolloni I, Ceresa D, Marubbi D, et al. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019;38:4467–79 [DOI] [PubMed] [Google Scholar]
- 2.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol 2017;38:395–406 [DOI] [PubMed] [Google Scholar]
- 4.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol 2016;212:379–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front Immunol 2017;8:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 2017;18:452–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013;14:416–29 [DOI] [PubMed] [Google Scholar]
- 8.Choudhry Z, Rikani AA, Choudhry AM, Tariq S, Zakaria F, Asghar MW, et al. Sonic hedgehog signalling pathway: a complex network. Ann Neurosci 2014;21:28–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caro I, Low JA. The role of the hedgehog signaling pathway in the development of basal cell carcinoma and opportunities for treatment. Clin Cancer Res 2010;16:3335–9 [DOI] [PubMed] [Google Scholar]
- 10.Hanna A, Metge BJ, Bailey SK, Chen D, Chandrashekar DS, Varambally S, et al. Inhibition of Hedgehog signaling reprograms the dysfunctional immune microenvironment in breast cancer. Oncoimmunology 2019;8:1548241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petty AJ, Dai R, Lapalombella R, Baiocchi RA, Benson DM, Li Z, et al. Hedgehog-induced PD-L1 on tumor-associated macrophages is critical for suppression of tumor-infiltrating CD8+ T cell function. JCI Insight 2021;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D, et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest 2019;129:5151–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JM, Recht L, Strober S. The Promise of Targeting Macrophages in Cancer Therapy. Clin Cancer Res 2017;23:3241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017;32:654–68 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Sadhukhan S, Sun S, Wagner GR, Hirschey MD, Qi L, et al. High-Resolution Metabolomics with Acyl-CoA Profiling Reveals Widespread Remodeling in Response to Diet. Mol Cell Proteomics 2015;14:1489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem 2014;86:2175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015;42:419–30 [DOI] [PubMed] [Google Scholar]
- 19.Langston PK, Shibata M, Horng T. Metabolism Supports Macrophage Activation. Front Immunol 2017;8:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ezekowitz RA, Sastry K, Bailly P, Warner A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J Exp Med 1990;172:1785–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paveley RA, Aynsley SA, Turner JD, Bourke CD, Jenkins SJ, Cook PC, et al. The Mannose Receptor (CD206) is an important pattern recognition receptor (PRR) in the detection of the infective stage of the helminth Schistosoma mansoni and modulates IFNgamma production. Int J Parasitol 2011;41:1335–45 [DOI] [PubMed] [Google Scholar]
- 22.Gewinner C, Hart G, Zachara N, Cole R, Beisenherz-Huss C, Groner B. The coactivator of transcription CREB-binding protein interacts preferentially with the glycosylated form of Stat5. J Biol Chem 2004;279:3563–72 [DOI] [PubMed] [Google Scholar]
- 23.Zhao M, Xiong X, Ren K, Xu B, Cheng M, Sahu C, et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chawla A Control of macrophage activation and function by PPARs. Circ Res 2010;106:1559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varga T, Mounier R, Horvath A, Cuvellier S, Dumont F, Poliska S, et al. Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J Immunol 2016;196:4771–82 [DOI] [PubMed] [Google Scholar]
- 26.Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol 2015;33:49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016;16:553–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol 2014;5:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab 2016;27:105–17 [DOI] [PubMed] [Google Scholar]
- 30.Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 2012;32:309–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriwaki K, Asahi M. Augmented TME O-GlcNAcylation Promotes Tumor Proliferation through the Inhibition of p38 MAPK. Mol Cancer Res 2017;15:1287–98 [DOI] [PubMed] [Google Scholar]
- 32.Bhatia B, Potts CR, Guldal C, Choi S, Korshunov A, Pfister S, et al. Hedgehog-mediated regulation of PPARgamma controls metabolic patterns in neural precursors and shh-driven medulloblastoma. Acta Neuropathol 2012;123:587–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan IS, Guy CD, Chen Y, Lu J, Swiderska-Syn M, Michelotti GA, et al. Paracrine Hedgehog signaling drives metabolic changes in hepatocellular carcinoma. Cancer Res 2012;72:6344–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 2012;151:414–26 [DOI] [PubMed] [Google Scholar]
- 35.Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, et al. Serine Availability Influences Mitochondrial Dynamics and Function through Lipid Metabolism. Cell Rep 2018;22:3507–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walch L, Copic A, Jackson CL. Fatty acid metabolism meets organelle dynamics. Dev Cell 2015;32:657–8 [DOI] [PubMed] [Google Scholar]
- 37.Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol 2019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.