

Abstract
Purpose:
The development of allogeneic chimeric antigen receptor (CAR) T-cell therapies for off-the-shelf use is a major goal that faces two main immunologic challenges, namely the risk of graft-versus-host disease (GvHD) induction by the transferred cells and the rejection by the host immune system limiting their persistence. In this work we assessed the direct and indirect antitumor effect of allogeneic CAR-engineered invariant natural killer T (iNKT) cells, a cell population without GvHD-induction potential that displays immunomodulatory properties.
Experimental Design:
After assessing murine CAR iNKT cells direct antitumor effects in vitro and in vivo, we employed an immunocompetent mouse model of B-cell lymphoma to assess the interaction between allogeneic CAR iNKT cells and endogenous immune cells.
Results:
We demonstrate that allogeneic CAR iNKT cells exerted potent direct and indirect antitumor activity when administered across major MHC barriers by inducing tumor-specific antitumor immunity through host CD8 T-cell cross-priming.
Conclusions:
In addition to their known direct cytotoxic effect, allogeneic CAR iNKT cells induce host CD8 T-cell antitumor responses, resulting in a potent antitumor effect lasting longer than the physical persistence of the allogeneic cells. The utilization of off-the-shelf allogeneic CAR iNKT cells could meet significant unmet needs in the clinic.
Translational Relevance.
The use of autologous cells for the generation of chimeric antigen receptor (CAR) T cells represents a significant limitation to the widespread use of this therapeutic approach. The development of allogeneic CAR T cells for off-the-shelf use still faces two major obstacles, namely the risk of graft-versus-host disease (GvHD) induction and the rejection by the host immune system. In this work, we show that allogeneic CAR-engineered invariant natural killer T (iNKT) cells, a cell population without GvHD-induction potential and with strong immunomodulatory properties, exerted potent antitumor activity by inducing tumor-specific antitumor immunity through host CD8 T-cell cross-priming. The induction of host antitumor immunity by allogeneic CAR iNKT cells resulted in a long-lasting antitumor effect going beyond their physical persistence. We believe that allogeneic CAR iNKT cells are promising candidates for the development of off-the-shelf CAR therapies, an important unmet need in the clinic.
Introduction
Chimeric antigen receptor (CAR) T cells have resulted in dramatic and effective therapy for a range of relapsed and refractory malignancies. The use of autologous cells for the generation of CAR T cells represents a significant limitation to their widespread use for a number of significant reasons, including the impact of disease and treatment on the T-cell product, costs of individual production, and the time required to produce the cellular product for patients with often rapidly progressive disease. The development of universal allogeneic CAR T cells could address these challenges yet faces two major limitations, namely the risk of graft-versus-host disease (GvHD) induction by the allogeneic cells that recognize host tissues and the rejection of the CAR modified cells by the host immune system. Ablation of the T-cell receptor (TCR; refs. 1–4) or use of non–MHC-restricted innate lymphocytes have been attempted to prevent GvHD (reviewed in ref. 5). Similarly, ablation of MHC class-I molecules to limit rejection by the host immune system (6) has been employed in preclinical models.
Invariant Natural Killer T (iNKT) cells are a rare subset of innate lymphocytes representing less than 1% of the total lymphocyte population both in humans and mice. iNKT cells express a semi-invariant TCR recognizing glycolipids presented in the context of the monomorphic, MHC-like molecule CD1d. Because of their peculiar TCR constitution and antigen recognition modality, iNKT cells do not display any GvHD induction potential and can even prevent GvHD (reviewed in ref. 7). iNKT cells display potent direct antitumor activity through production of cytotoxic molecules (8). Several groups successfully generated human CAR iNKT cells provided with antitumor potential as assessed in vitro and in xenogeneic murine models (9–13). These studies revealed several advantages of using CAR iNKT cells over conventional CAR T cells, including their lack of induction of xeno-GvHD (9), their preferential migration to tumor sites (9), and their capacity of CAR iNKT to target both the natural ligand CD1d and the CAR-targeted antigen (12). In addition to their direct cytotoxic effect, iNKT cells are known for their strong immunomodulatory effect. In particular, iNKT cells induce CD8 T-cell cross-priming (14, 15) through the licensing of CD103+ CD8alpha dendritic cells (16–18), allowing the establishment of long-lasting antitumor CD8 T-cell responses in murine models (19–22).
In this study, we tested the hypothesis that induction of host CD8 T-cell cross-priming by allogeneic CAR iNKT cells would allow the establishment of an antitumor immunity lasting beyond the physical persistence of the transferred cells. Taking advantage of the immunoadjuvant role of iNKT cells and their lack of GvHD-inducing potential, we demonstrate that allogeneic CAR iNKT cells exert, in addition to their previously reported direct antitumor effect (9–13), an indirect effect through the induction of host CD8 T-cell cross-priming.
Materials and Methods
Mice
BALB/cJ (H-2Kd) and FVB/NJ (H-2Kq) mice were purchased from The Jackson Laboratory. Firefly Luciferase (Luc+) transgenic FVB/N mice have been reported previously (23) and were bred in our animal facility at Stanford University. BALB/c Rag1−/−gamma-chain−/− and BALB/c BATF3−/− mouse strains were kind gifts of Dr. Irving Weissman and Dr. Samuel Strober, respectively, and were bred in our animal facility at Stanford University. All procedures performed on animals were approved by Stanford University's Institutional Animal Care and Use Committee and were in compliance with the guidelines of humane care of laboratory animals.
CAR iNKT and conventional CAR T generation
Murine CD19.28z CAR iNKT and conventional CAR T cells specifically recognizing the murine CD19 molecule were generated using an adaptation of previously reported protocols (24). Murine CD19 (mCD19) CAR stable producer cell line (25) was kindly provided by Dr. Terry J. Fry. iNKT cells were negatively enriched from FVB/N mouse spleen single-cell suspensions and using a mixture of biotinylated mAbs (GR-1, clone: RB6–8C5; CD8a, clone: 53–6.7; CD19, clone: 6D5; TCRγδ, clone: GL3; TER119/erythroid cell, clone: TER-119; CD62L, clone: MEL-14; BioLegend) and negative selection by anti-biotin microbeads (BD IMag Streptavidin Particles Plus DM, BD Biosciences). The enriched fraction (typically 10–30% enrichment) was then stimulated for 5 days with a synthetic analog of α-galactosylceramide (KRN7000, 100 ng/mL, REGiMMUNE) in the presence of human IL2 (100 UI/mL; NCI Repository) and human IL15 (100 ng/mL; NCI Repository). Cells were grown in DMEM media supplemented with 10% heat-inactivated FBS, 1 mmol/L sodium pyruvate, 2 mmol/L glutamine, 0.1 mmol/L nonessential amino acids, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C with 5% CO2. Conventional T cells were enriched from FVB/N mouse spleen single-cell suspensions using the mouse Pan T Cell Isolation Kit II (Miltenyi Biotec) according to the manufacturer's protocol. T cells were activated for 24 hours with Dynabeads Mouse T-Activator CD3/CD28 (Life Technologies) in the presence of human IL2 (30 U/ml) and murine IL-7 (10 ng/mL; PeproTech) in RPMI1640 media supplemented with 10% heat-inactivated FBS, 1 mmol/L sodium pyruvate, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C with 5% CO2. Activated cells were then transduced by culturing them for 48 hours in retronectin-coated plates loaded with supernatant harvested from the stable producer line 48 hours after culture. Invariant NKT cell purity was evaluated by flow cytometry using PE-conjugated PBS-57–loaded mCD1d tetramer (NIH Tetramer Facility) and TCR-β (clone H57–597; BioLegend). Transduction efficacy was measured by flow cytometry after protein L staining (26). CAR iNKT cell numbers were adjusted based on transduction efficacy before in vitro or in vivo use and the same number of untransduced iNKT cells was used as control. In experiments comparing CAR iNKT and conventional CAR T cells, percentages of transduced cells were adjusted to the same transduction efficacy (50%).
In vitro cytotoxic assay
In vitro cytotoxic assays were performed as described previously (27). Briefly, murine CD19.28z CAR iNKT or conventional CAR T cells were co-cultured for 24 hours with luciferase-transduced A20 cells (A20yfp+/luc+; ref. 28). A20yfp+/luc+ cells tested negative for Mycoplasma by PCR in October 2017, were cryopreserved in liquid nitrogen, thawed, and cultured for maximum 1 week before use.
In vivo bioluminescence imaging
In vivo bioluminescence imaging (BLI) was performed as previously described (27), using an IVIS Spectrum imaging system (Perkin Elmer) and Living Image Software 4.1 (Perkin Elmer) or using an Ami LED-illumination based imaging system (Spectral Instruments Imaging) with Aura Software (Spectral Instruments Imaging).
In vivo murine tumor models
We employed two systemic B-cell lymphoma mouse models reported previously (28). Briefly, CD19-expressing BCL1luc+ (5 × 104) or A20luc+ cells (2 × 104) resuspended in PBS were injected intravenously by tail vein into alymphoid BALB/c (H-2Kd) Rag1−/− gamma-chain−/− mice. For tumor induction in immunocompetent mice, tumor cells were injected intravenously into sublethally (4.4 Gy) irradiated BALB/c mice. For syngeneic bone marrow transplantation, BALB/c mice were lethally irradiated (8.8 Gy in 2 doses administered 4 hours apart) and transplanted with syngeneic BALB/c bone marrow cells (5 × 106) after T-cell depletion using CD4 and CD8 MicroBeads (Miltenyi Biotec). For retransfer experiments, bone marrow cells from alymphoid BALB/c (H-2Kd) Rag1−/− gamma-chain−/− mice were used to exclude any potential contribution from bone marrow–derived T or NK cells after reconstitution.
Flow cytometry analysis
In vitro cultured cells or ex vivo isolated cells were resuspended in phosphate-buffered saline (PBS) containing 2% FBS. Extracellular staining was preceded by incubation with purified FC blocking reagent (Miltenyi Biotech). Cells were stained with: TIM3 (clone: RMT3–23) APC, CD62 L (clone: MEL-14) AF700, CD19 (clone: 6D5) APCFire750, CD44 (clone: IM7) PerCpCy5.5, PD-1 (clone: 29F.1A12) BV605, CD8a (clone: 53–6.7) BV650, NK1.1 (clone: PK136) BV711, ICOS (clone: C398.4A) BV785, CD25 (clone: PC61.5) PE, TCRβ (clone: H57–597) PE/Dazzle594, and Thy1.1 (clone: HIS51) PeCy7. All antibodies were purchased from BioLegend. Dead cells were excluded using Fixable Viability Dye eFluor 506 (eBioscience). Samples were acquired on a BD LSR II flow cytometer (BD Biosciences), and analysis was performed with FlowJo 10.5.0 software (Tree Star).
RNA and TCR sequencing analysis
Host CD4 and CD8 T cells were FACS-sorted from pooled spleens from 3 mice treated with allogeneic CAR iNKT or untreated control, frozen in TRizol, and conserved at −80°C. RNA was extracted using the TRizol RNA isolation method (Thermo Fisher Scientific) combined with the RNeasy MinElute Cleanup (Qiagen). Full-length cDNA was generated using the Clontech SMARTer v4 Kit (Takara Bio USA, Inc.) prior to library generation with the Nextera XT DNA Library Prep Kit (Illumina, Inc.). Libraries were pooled for sequencing on the Illumina HiSeq 4000 platform (75 bp, paired-end). Sequencing reads were checked using FastQC v.0.11.7. Estimated transcript counts and transcripts per million (TPM) for the mouse genome assembly GRCm38 (mm10) were obtained using the pseudo-aligner Kallisto. Transcript-level abundance was quantified and summarized into gene level using the tximport R package. Differential gene expression was performed using the DESeq2 R package version 1.22.221, using FDR < 0.05. Gene-set enrichment analysis conducted using the fgsea R package. For TCR sequencing, libraries were prepared from the synthesized full-length cDNA using the nested PCR method reported previously (29, 30). Sequencing was performed by using the Illumina MiSeq platform after Illumina paired-end adapters incorporation. TCRβ sequence analysis was performed with VDJFasta. After total count normalization, downstream analysis was performed on the 1,000 most represented clonotypes across the samples using the FactoMineR and factoextra R packages.
Statistical analysis
The Mann–Whitney U test was used in cross-sectional analyses to determine statistical significance. Survival curves were represented with the Kaplan–Meier method and compared by log-rank test. Statistical analyses were performed using Prism 8 (GraphPad Software) and R version 3.5.1 Comprehensive R Archive Network (CRAN) project (http://cran.us.r-project.org) with R studio version 1.1.453.
Results
Allogeneic CAR iNKT cell antitumor effect is significantly enhanced in the presence of host lymphocytes
To study the interaction of allogeneic CD19-specific CAR iNKT cells with the host immune system, we utilized a fully murine experimental system and transduced murine iNKT cells expanded ex vivo from FVB/N mice with a previously reported CAR construct (24) composed of the variable region cloned from the 1D3 hybridoma recognizing murine CD19 linked to a portion of the murine CD28 molecule and to the cytoplasmic region of the murine CD3-ζ molecule (CD19.28z CAR; Fig. 1A). The cytotoxic potential of CD19.28z-CAR iNKT was confirmed by in vitro cytotoxic assays against the CD19-expressing A20 lymphoma cell line, revealing dose-dependent cytotoxicity of the CD19.28z-CAR iNKT cells (Fig. 1B). As predicted, untransduced iNKT did not display any significant cytotoxic effect against A20 cells (Fig. 1B) according to their lack of expression of CD1d. We next evaluated in vivo the direct antitumor effect of allogeneic CAR iNKT cells using BALB/c (H-2Kd) Rag1−/− gamma-chain−/− mice as recipients (Fig. 1C and F). FVB/N (H-2Kq) derived allogeneic CAR iNKT cells significantly controlled tumor growth (Fig. 1D) and improved animal survival (Fig. 1E) compared with both untreated mice and mice receiving untransduced iNKT cells after administration to MHC-mismatched immunodeficient mice receiving CD19-expressing BCL1 B-cell lymphoma cells. In a second, more aggressive model of B-cell lymphoma using A20 cells (Fig. 1F), allogeneic CAR iNKT minimally affected tumor growth as revealed by BLI (Fig. 1G) and slightly but significantly improved survival (Fig. 1H) compared with untreated mice and mice treated with untransduced iNKT cells.
Figure 1.

In vitro and in vivo antitumor activity of murine CAR iNKT cells. A, Representative FACS-plot of untransduced (left) and mCD19.28z-CAR–transduced (right) murine iNKT cells. iNKT were identified as PBS-57 CD1d tetramer–positive cells and CAR transduction was quantified by Protein L staining. B, Mean and SD of cytotoxicity relative to the untreated control at different Effector:Target (E:T) ratios. Results are representative of two independent experiments performed in triplicate. C and F, Schematic representation of the BCL1luc+ (C) and A20luc+ (F) into Rag1−/− gamma-chain−/− recipient experiments. D and G, Representative in vivo bioluminescence (BLI) images of BCL1luc+ (D) and A20luc+ (G) tumor cell progression in Rag1−/− gamma-chain−/− treated with untransduced iNKT cells (blue boxes and dots), CAR iNKT cells (red boxes and dots), or untreated (gray box and dots). E and H, Survival of mice receiving BCL1luc+ (E) or A20luc+ (H) and treated with untransduced iNKT cells (blue lines), CAR iNKT cells (red lines), or left untreated (NT, gray lines). Results are pooled from two independent experiments with a total of 6 to 9 mice per group. BLI results were compared using a nonparametric Mann–Whitney U test and P values are shown when significant. Survival curves were plotted using the Kaplan–Meier method and compared by log-rank test. P values are indicated when significant.
To assess the interplay between the transferred CAR iNKT cells and the host immune cells, we employed the A20 tumor model to test the antitumor activity mediated by allogeneic CAR iNKT cells in an immunocompetent model (Fig. 2A) using as recipients wild-type BALB/c mice receiving sublethal irradiation (4.4 Gy) leading to a partial and transient lymphopenia. The antitumor effect of 1 × 106 allogeneic untransduced iNKT and CAR iNKT cells was greatly enhanced in this partially lymphopenic model, leading to long-term survival of all treated mice (Fig. 2B). Interestingly, a dose as low as 5 × 104 untransduced iNKT cells (Fig. 2C) was sufficient to significantly extend animal survival (Fig. 2D) and the addition of the CAR further improved the effect of iNKT leading to long-term survival of all CAR iNKT-treated mice (Fig. 2D). To further stress the model, we tested the antitumor effect of untransduced iNKT and CAR iNKT cells in a high-burden, pre-established tumor model in which high numbers (2.5 × 105) of A20 cells were injected 7 days before the adoptive transfer of the effector cells (Fig. 2F). In this model, untransduced iNKT displayed a minimal although statistically significant effect (Fig. 2F), whereas the administration of CAR iNKT cells significantly improved animal survival compared with both untreated mice and mice receiving untransduced iNKT cells (Fig. 2F).
Figure 2.
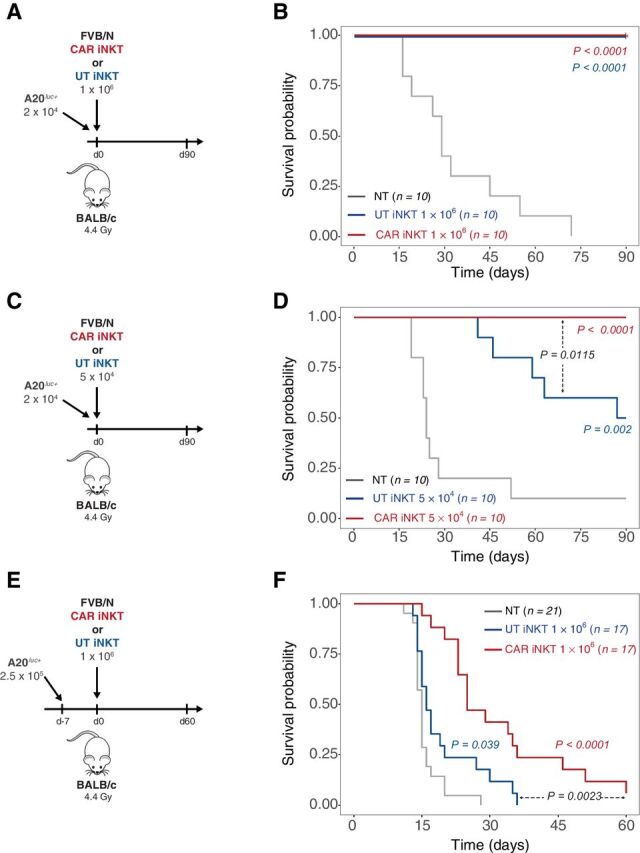
Allogeneic CAR iNKT-cell antitumor effect is greatly enhanced by the presence of host lymphocytes. A, C, and E, Schematic representation of the experiments employing A20luc+ cells into sublethally (4.4 Gy) irradiated WT BALB/c mice. B, D, and F, Survival of mice receiving A20luc+ cells and treated with untransduced iNKT cells (blue lines), CAR iNKT cells (red lines), or left untreated (NT, gray lines). Results are pooled from two independent experiments with a total of 10 to 21 mice per group. Survival curves were plotted using the Kaplan–Meier method and compared by log-rank test. P values are indicated when significant.
Collectively, these in vitro and in vivo data confirm the direct antitumor effect of murine CAR iNKT cells and revealed an improved effect of untransduced iNKT and, even more, of CAR iNKT cells in the presence of host lymphocytes.
Host CD8 T-cell cross-priming contributes to the indirect antitumor effect of allogeneic CAR iNKT cells
The striking difference in allogeneic CAR iNKT effect observed in mice with partial lymphopenia (Fig. 2B and D) compared with genetically alymphoid mice (Fig. 1H) suggested a role for host-derived lymphocytes in the antitumor effect. To test the hypothesis that host CD8 T-cell cross-priming mediates the indirect antitumor effect of allogeneic CAR iNKT cells, we employed as recipients BALB/c BATF3−/− mice, in which CD8 T-cell cross-priming is impaired as a result of the absence of BATF3-dependent CD103+ CD8alpha+ dendritic cells (31).
The effect of allogeneic CAR iNKT cells was partially abrogated in A20-receiving BATF3−/− mice as compared to WT mice (Fig. 3A and B), supporting the hypothesis that the impact of allogeneic CAR iNKT cells is mediated, at least partially, by the activation of host CD8 T cells via their cross-priming. To further assess the synergistic effect of allogeneic CAR iNKT cells and host-derived CD8 T cells, we employed an autologous bone marrow transplantation model, co-administering allogeneic FVB/N CAR iNKT with syngeneic BALB/c CD8 T cells at the time of transplantation with T-cell–depleted syngeneic BALB/c bone marrow cells and transfer of A20 lymphoma cells into lethally irradiated (8.8 Gy) BALB/c recipients. Co-administration of allogeneic CAR iNKT and autologous CD8 T cells resulted in a synergistic effect, significantly improving tumor control (Fig. 3C) and animal survival (Fig. 3D) compared to mice receiving no treatment, as well as to mice receiving either allogeneic CAR iNKT or autologous CD8 T cells alone. Collectively these data indicate that CD8 T-cell cross-priming is necessary for allogeneic CAR iNKT cells to exert their full antitumor effect and suggest a synergy between these two cytotoxic T-cell compartments.
Figure 3.

Indirect antitumor effect of allogeneic CAR iNKT cells is dependent on host CD8 T-cell cross-priming. Representative in vivo BLI images of A20luc+ cell progression (A) and survival (B) of sublethally (4.4 Gy) irradiated WT or BATF3−/− BALB/c mice treated or not with 106 CAR iNKT cells. Representative in vivo BLI images of A20luc+ cell progression (C) and survival (D) of lethally (8.8 Gy) irradiated WT BALB/c mice transplanted with syngeneic BALB/c TCD-BM and treated with syngeneic CD8 T cells (4 × 106; green symbols and line), CAR iNKT cells (106; blue symbols and line), or both (red symbols and line). Untreated controls are depicted in gray. BLI results were compared using a nonparametric Mann–Whitney U test and P values are shown. Survival curves were plotted using the Kaplan–Meier method and compared by log-rank test. P values are indicated when significant.
Allogeneic CAR iNKT-cell treatment modulates host CD8 T-cell phenotype, transcriptome, and TCR repertoire
To gain further insights into the impact of CAR iNKT cells on host T cells, we performed phenotypic analysis of host T cells recovered at day 7 and 14 after treatment with allogeneic CAR iNKT cells. At these timepoints, allogeneic CAR iNKT cells were already undetectable as revealed by in vivo tracking by bioluminescence (Supplementary Fig. S1A), flow cytometry (data not shown), and as suggested by the progressive increase of B-cell numbers (Supplementary Fig. S1B). We observed a significant increase in the number of CD8 T cells recovered at day 7 and day 14 from the spleen of mice treated with CAR iNKT cells compared with untreated mice (Fig. 4A). Immunophenotypic analysis revealed higher proportions of cells with a central memory (CD62L+ CD44+) and reduced proportions of cells with an effector (CD62L– CD44–) or effector memory (CD62L− CD44+) phenotype in CD8 T cells recovered at day 7 after allogeneic CAR iNKT treatment compared with untreated mice (Fig. 4B). CD4 T-cell numbers were increased at day 7 but not at day 14 after allogeneic CAR iNKT treatment (Supplementary Fig. S2A), and CD4 T-cell phenotype was only minimally affected by CAR iNKT treatment (Supplementary Fig. S2B). A transcriptomic analysis performed on CD8 T cells FACS-sorted at day 14 revealed the upregulation of genes associated with cytotoxic antitumor activity (Lyz2, Gzma, Gzmm, Fasl) and the downregulation of genes involved with responses to type I interferon (Irf7, Ifitm1, Ifi27l2a; Fig. 4C). Gene Set Enrichment Analysis (GSEA) for Gene Ontology (GO) Biological Processes confirmed the upregulation of antitumor gene sets (Fig. 4D) and the downregulation of the type I IFN signature. In agreement with our phenotypic results, a GSEA performed using two well-established memory CD8 T-cell gene signatures revealed enrichment in CD8 T-cell memory genes (Fig. 4E and F). Transcriptomic analysis of CD4 T cells showed a similar downregulation of genes involved in responses to type I interferon (Irf7, Ifit1, Ifit3; Supplementary Fig. S2C) but did not reveal any consistent pattern of expression of genes involved in antitumor activity or cellular differentiation (Supplementary Fig. S2C). To assess the impact of CAR iNKT treatment on the TCR repertoire of CD8 T cells, we performed paired TCR-β sequencing. Hierarchical clustering based on the 1,000 most represented TCR clonotypes revealed a closer relationship between the TCR repertoire of CD8 T cells recovered from mice receiving CAR iNKT cells compared with CD8 T cells from untreated mice (Fig. 4G). Accordingly, principal component analysis (PCA) showed close similarity in the TCR repertoire of CD8 T cells from allogeneic CAR iNKT-treated mice, whereas cells from untreated mice displayed high heterogeneity (Fig. 4H). Analysis of the TCR repertoire of CD4 T cells did not reveal any impact of allogeneic CAR iNKT treatment (Supplementary Fig. S2D). Collectively, these results indicate that allogeneic CAR iNKT-cell treatment shaped the host CD8 T-cell compartment phenotypically, transcriptomically, and in terms of clonal repertoire.
Figure 4.
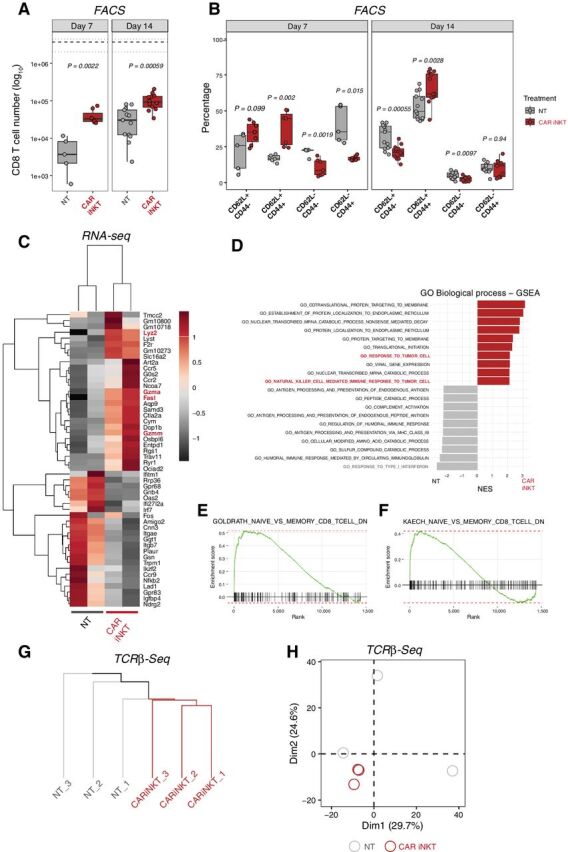
Allogeneic CAR iNKT-cell treatment modulates host CD8 T-cell number, phenotype, transcriptome, and TCR repertoire. Number (A) and immunophenotype (B) of host CD8 T cells recovered from spleen 7 and 14 days after tumor induction in mice treated with allogeneic CAR iNKT cells (red boxes and symbols) or untreated (gray boxes and symbols). Median (black dashed line) and upper/lower range (gray dotted lines) of CD8 T-cell counts in naive mice are represented. Results are pooled from two independent experiments with a total of 5 to 13 mice per group. Groups were compared using a nonparametric Mann–Whitney U test and P values are shown. C, Heatmap representing differentially expressed genes in host CD8 T cells FACS-sorted from recipients treated or not with allogeneic CAR iNKT cells. Expression for each gene is scaled (z-scored) across single rows. Each column represents independent experiments with one to two biological replicates per experiment. D, Top 10 enriched terms/pathways in CD8 T cells from untreated (gray bars) and CAR iNKT-cell–treated (red bars) animals revealed by GO Biological Process analysis using GSEA. E and F, Enrichment plots displaying the distribution of the enrichment scores for the genes downregulated during transition from naive CD8 T cells versus memory CD8 T cells according to the Luckey and colleagues (E; ref. 66) or Kaech and colleagues (F; ref. 67) signatures. Gene signatures were obtained from Molecular Signatures Database (MSigDB; C7: immunologic signatures). G and H, Hierarchical clustering (G) and PCA (H) of the top 1,000 clonotypes based on TCRβ sequencing of CD8 T cells from hosts treated with allogeneic CAR iNKT cells (red) or left untreated (gray).
Allogeneic CAR iNKT-cell treatment induces long-lasting host CD8 T-cell tumor-specific responses
To formally prove that allogeneic CAR iNKT cells induce tumor-specific host immune responses, at day 60 after treatment, we recovered splenocytes from mice receiving A20 lymphoma cells and treated with allogeneic CAR iNKT cells. Recovered splenocytes were transferred into new lethally irradiated BALB/c recipients together with bone marrow from Rag1−/− gamma-chain−/− BALB/c mice and A20 cells (Fig. 5A). Unprimed splenocytes from mice receiving only sublethal irradiation were used as control. Splenocytes primed in the presence of allogeneic CAR iNKT significantly extended the survival of mice compared with both untreated mice and mice receiving unprimed splenocytes (Fig. 5B, left). To assess the contribution of CD8 T cells to this protective effect, we performed the same experiment retransferring only allogeneic CAR iNKT-primed or unprimed CD8 T cells. As shown in Fig. 5B (right), host CD8 T cells from allogeneic CAR iNKT-treated mice significantly extended animal survival compared with both mice left untreated or receiving unprimed CD8 T cells. Collectively, these experiments formally demonstrate that allogeneic CAR iNKT treatment induced a long-lasting tumor-specific host CD8-dependent antitumor immunity in allogeneic recipients.
Figure 5.

Allogeneic CAR iNKT-primed host CD8 T cells display long-lasting antitumor immunity. A, Schematic representation of the sequential adoptive transfer experiment. Host splenocytes or CD8 T cells were recovered after 60 days from sublethally irradiated BALB/c mice, injected with A20luc+ cells, and treated with CAR iNKT cells (primed cells). Splenocytes or CD8 T cells recovered after 60 days from sublethally irradiated BALB/c mice were used as controls (unprimed cells). Primed or unprimed host splenocytes (5 × 106 cells) were transferred, after lethal irradiation, to a new set of BALB/c mice receiving A20luc+ cells together with bone marrow cells from syngeneic Rag1−/− gamma-chain−/− BALB/c mice. Alternatively, primed or unprimed host CD8 T cells (1 × 106 cells) were transferred. B, Survival of transplanted mice receiving primed (red line) or unprimed (blue line) splenocytes (left) or CD8 T cells (right). Untreated controls are depicted in gray. Results are pooled from two independent experiments with a total of 10 to 14 mice per group. Survival curves were plotted using the Kaplan–Meier method and compared by log-rank test. P values are indicated when significant.
Allogeneic CAR iNKT cells outperform conventional CAR T cells in the presence of host lymphocytes
To assess the advantage that this indirect antitumor effect could confer to allogeneic CAR iNKT cells over allogeneic conventional CAR T cells, we compared these two populations. Given the potent direct antitumor activity of conventional CAR T cells (Supplementary Fig. S3A), a dose of as little as 2.5 × 105 conventional CAR T cells was sufficient to significantly extend mouse survival when administered into alymphoid animals (Supplementary Fig. S3B), and this dose was selected for comparison to CAR iNKT cells. As shown in Fig. 6A and B, during partial lymphopenia conventional CAR T cells significantly extended animal survival, whereas CAR iNKT cells dramatically outperformed conventional CAR T cells leading to tumor control and survival of all treated mice. Collectively, these results demonstrate that allogeneic CAR iNKT cells were significantly more effective than allogeneic conventional CAR T cells in inducing extended tumor control in immunocompetent hosts.
Figure 6.

Allogeneic CAR iNKT cells are more effective than allogeneic conventional CAR T cells. Representative in vivo BLI images of A20luc+ cell progression (A) and survival (B) of sublethally (4.4 Gy) irradiated BALB/c mice treated with 2.5 × 105 allogeneic CAR iNKT cells (red curve and symbols), 2.5 × 105 allogeneic conventional CAR T cells (green curve and symbols), or untreated (gray curve and symbols). Results are pooled from two independent experiments with a total of 10 mice per group. BLI results were compared using a nonparametric Mann–Whitney U test and P values are shown. Survival curves were plotted using the Kaplan–Meier method and compared by log-rank test. P values are indicated when significant.
Discussion
In this study, we demonstrated in a murine model of CD19+ lymphoma that allogeneic CAR iNKT cells exert, in addition to their previously reported direct antitumor effect (9, 10, 12), an even stronger indirect antitumor effect mediated by the induction of host immunity.
The potential contribution of the host immune system in the effect of CAR T cells has been shown in preclinical (32–42) and clinical (43–45) studies. Recent studies indicate that immunogenic cell death eliciting endogenous cell responses contribute to the effects of adoptively transferred immune effector cells, including TCR-transgenic T cells (46, 47) and NK cells (46). We hypothesized that induction of bystander host antitumor responses might be a particularly interesting approach in the allogeneic setting, as CAR cells administered across major MHC barriers will be invariably rejected by the host immune system. After confirming in a fully murine model the previously reported direct cytotoxic effect of CAR iNKT cells (9–13), we demonstrate that allogeneic CAR iNKT cells efficiently induce antitumor immune responses in the recipient. Using BATF3−/− mice, a classically employed model of conventional type 1 dendritic cell (cDC1) deficiency (38, 46, 31, 48), we show that host CD8 T-cell cross-priming is necessary for the full action of CAR iNKT cells. However, BATF3 deficiency affects several lineages, including CD8 T cells (49, 50) and Treg (51). To circumvent this limitation, we completed our analysis with an adoptive transfer model using wild-type recipients and wild-type CD8 T cells, indicating a synergy between allogeneic CAR iNKT cells and host-type CD8 T cells. Importantly, we show that retransfer of CAR iNKT-primed CD8 T cells allow for the transfer of protective antitumor immunity. In our study, we focused our attention on the induction of host CD8 T-cell responses that, lasting longer than the physical persistence of the administered allogeneic cells, would provide long-term antitumor protection. However, we cannot exclude that other mechanisms, including host NK-cell activation (52) or killing of tumor-associated macrophages (53), can contribute to the early effects of CAR iNKT cells in our model.
iNKT cells are an ideal platform for off-the-shelf immunotherapies given their lack of GvHD-induction potential (7) without need for deletion of their endogenous TCR, a manipulation that has been recently shown to alter the CAR T-cell homeostasis and persistence (54). Moreover, despite being a rare lymphocyte population, iNKT cells can be easily expanded ex vivo to numbers needed for clinical uses (55–58) and several clinical trials using ex vivo expanded autologous iNKT cells have been already successfully conducted (59–61). However, previous reports indicate that the ability of iNKT cells to expand in vitro may vary widely among individuals (62), a potential limitation for generation of autologous or allogeneic MHC-matched products. Use of allogeneic, off-the-shelf iNKT cells to be administered across MHC barriers will circumvent this potential limitation as universal donors whose iNKT cells display optimal expansion potential can be selected. Moreover, our results indicate that extremely low numbers of CAR iNKT cells persisting for a very limited time are able to induce a potent, long-lasting antitumor effect through their immunomodulatory role. Such an effect is in accordance with what we previously reported in the GvHD settings, where similarly low numbers (5 × 10e4) of CD4+ iNKT cells were able to efficiently prevent GvHD induced by conventional T cells in a major MHC-mismatch mouse model of bone marrow transplantation (63, 64), even when rapidly rejected third-party cells were employed (65).
A phase I clinical trial employing CD19-specific allogeneic CAR iNKT cells for patients with relapsed or refractory B-cell malignancies is currently ongoing (ANCHOR; NCT03774654). In analogy to what performed with conventional CAR T cells, this clinical trial involves the administration of a lymphodepleting regimen containing fludarabine and cyclophosphamide before CAR iNKT-cell infusion. Our results indicate that a major component of allogeneic CAR iNKT cells' effect derives from their interplay with the host immune system, an interaction that can significantly be impaired by the lymphodepleting conditioning. Future studies will determine whether the conventional fludarabine/cyclophosphamide lymphodepletion interferes with CAR iNKT-cell effect and will test alternative regimens to optimize both the homeostasis and the immunoadjuvant effect of the administered product.
In conclusion, our results represent the first demonstration of an immunoadjuvant effect exerted by an allogeneic CAR cell product toward the host immune system, resulting in long-lasting antitumor effects that go beyond the physical persistence of the allogeneic cells.
Data and Materials Availability
Sequencing data can be found under accession number PRJNA742379 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA742379).
Authors' Disclosures
F. Simonetta reports grants from Swiss Cancer League and American Society for Transplantation and Cellular Therapy during the conduct of the study. C.L. Mackall reports grants from NIH, California Institute for Regenerative Medicine, and Parker Institute for Cancer Immunotherapy during the conduct of the study. C.L. Mackall also reports grants and personal fees from Lyell Immunopharma, as well as personal fees from Syncopation Life Sciences, Neoimmune Tech, Immatics, GlaxoSmithKline, Bristol Myers Squibb, Apricity Health, Allogene, Vor, Bryologyx, and Red Tree Capital outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Acknowledgments
This work was supported by funding from R01 CA23158201 (to R.S. Negrin), P01 CA49605 (to R.S. Negrin), the Parker Institute for Cancer Immunotherapy (to R.S. Negrin), an American Society for Blood and Marrow Transplantation New Investigator Award 2018 (to F. Simonetta), the Geneva University Hospitals Fellowship (to F. Simonetta), the Swiss Cancer League BIL KLS 3806-02-2016 (to F. Simonetta), the Fondation de Bienfaisance Valeria Rossi di Montelera Eugenio Litta Fellowship (to F. Simonetta), the Dubois-Ferrière-Dinu-Lipatti Foundation (to F. Simonetta), the Virginia and D.K. Ludwig Fund for Cancer Research (to C.L. Mackall), and a St Baldrick's/Stand Up to Cancer Pediatric Dream Team Translational Cancer Research Grant (to C.L. Mackall). Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.L. Mackall is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. Flow cytometry analysis and sorting were performed on instruments in the Stanford Shared FACS Facility purchased using a NIH S10 Shared Instrumentation Grant (S10RR027431–01). Sequencing was performed on instruments in the Stanford Functional Genomics Facility, including the Illumina HiSeq 4000 purchased using a NIH S10 Shared Instrumentation Grant (S10OD018220).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
F. Simonetta: Conceptualization, data curation, formal analysis, funding acquisition, investigation, visualization, methodology, writing–original draft, project administration. J.K. Lohmeyer: Investigation, writing–review and editing. T. Hirai: Conceptualization, investigation, methodology, writing–review and editing. K. Maas-Bauer: Investigation, writing–review and editing. M. Alvarez: Investigation, methodology, writing–review and editing. A.S. Wenokur: Investigation, writing–review and editing. J. Baker: Investigation, methodology, project administration, writing–review and editing. A. Aalipour: Resources, investigation, methodology, writing–review and editing. X. Ji: Methodology, writing–review and editing. S. Haile: Methodology, writing–review and editing. C.L. Mackall: Resources, methodology, writing–review and editing. R.S. Negrin: Conceptualization, resources, supervision, funding acquisition, validation, writing–review and editing.
References
- 1. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res 2015;75:3853–64. [DOI] [PubMed] [Google Scholar]
- 2. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543:113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther 2017;25:949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiebking V, Lee CM, Mostrel N, Lahiri P, Bak R, Bao G, et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica 2021;106:847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: What lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov 2021;11:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013;122:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mavers M, Maas-Bauer K, Negrin RS. Invariant natural killer T cells as suppressors of graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Front Immunol 2017;8:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wingender G, Krebs P, Beutler B, Kronenberg M. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J Immunol 2010;185:2721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014;124:2824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest 2016;126:2341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ngai H, Tian G, Courtney AN, Ravari SB, Guo L, Liu B, et al. IL-21 selectively protects CD62L+ NKT cells and enhances their effector functions for adoptive immunotherapy. J Immunol 2018;201:2141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell 2018;34:596–610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res 2019;25:7126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med 2003;198:267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol 2003;171:5140–7. [DOI] [PubMed] [Google Scholar]
- 16. Farrand KJ, Dickgreber N, Stoitzner P, Ronchese F, Petersen TR, Hermans IF. Langerin+CD8α+ dendritic cells are critical for cross-priming and IL-12 production in response to systemic antigens. J Immunol 2009;183:7732–42. [DOI] [PubMed] [Google Scholar]
- 17. Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell–licensed DCs. Nat Immunol 2010;11:313–20. [DOI] [PubMed] [Google Scholar]
- 18. Valente M, Dölen Y, van Dinther E, Vimeux L, Fallet M, Feuillet V, et al. Cross-talk between iNKT cells and CD8 T cells in the spleen requires the IL-4/CCL17 axis for the generation of short-lived effector cells. Proc Natl Acad Sci U S A 2019;116:25816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, et al. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol 2000;12:987–94. [DOI] [PubMed] [Google Scholar]
- 20. Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al. NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol 2013;4:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dashtsoodol N, Shigeura T, Tashiro T, Aihara M, Chikanishi T, Okada H, et al. Natural killer T cell-targeted immunotherapy mediating long-term memory responses and strong antitumor activity. Front Immunol 2017;8:1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghinnagow R, Meester JD, Cruz LJ, Aspord C, Corgnac S, Macho-Fernandez E, et al. Co-delivery of the NKT agonist α-galactosylceramide and tumor antigens to cross-priming dendritic cells breaks tolerance to self-antigens and promotes antitumor responses. OncoImmunology 2017;6:e1339855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, et al. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood 2005;106:1113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 2010;116:3875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qin H, Ishii K, Nguyen S, Su PP, Burk CR, Kim B-H, et al. Murine pre-B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood 2018;132:1899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med 2012;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simonetta F, Alam IS, Lohmeyer JK, Sahaf B, Good Z, Chen W, et al. Molecular imaging of chimeric antigen receptor T cells by ICOS-ImmunoPET. Clin Cancer Res 2021;27:1058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edinger M, Cao Y-A, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 2003;101:640–8. [DOI] [PubMed] [Google Scholar]
- 29. Han A, Glanville J, Hansmann L, Davis MM. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat Biotechnol 2014;32:684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 2019;572:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322:1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res 2014;2:154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 2014;20:972–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics 2018;8:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brossart P. The role of antigen-spreading in the efficacy of immunotherapies. Clin Cancer Res 2020;26:4442–7. [DOI] [PubMed] [Google Scholar]
- 36. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol 2020;21:914–26. [DOI] [PubMed] [Google Scholar]
- 37. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell 2019;35:473–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuhn NF, Lopez AV, Li X, Cai W, Daniyan AF, Brentjens RJ. CD103+ cDC1 and endogenous CD8+ T cells are necessary for improved CD40L-overexpressing CAR T cell antitumor function. Nat Commun 2020;11:6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alizadeh D, Wong RA, Gholamin S, Maker M, Aftabizadeh M, Yang X, et al. IFNg is critical for CAR T cell mediated myeloid activation and induction of endogenous immunity. Cancer Discov 2021;11:2248–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boulch M, Cazaux M, Loe-Mie Y, Thibaut R, Corre B, Lemaître F, et al. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci Immunol 2021;6:eabd4344. [DOI] [PubMed] [Google Scholar]
- 41. Murad JP, Tilakawardane D, Park AK, Lopez LS, Young CA, Gibson J, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther 2021;29:2335–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O'Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun 2021;12:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014;2:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X, Hu Y, Liu X, Yu J, Xu P, Wei G, et al. Quantitative characterization of T-cell repertoire alteration in Chinese patients with B-cell acute lymphocyte leukemia after CAR-T therapy. Bone Marrow Transplant 2019;54:2072–80. [DOI] [PubMed] [Google Scholar]
- 45. Chen P-H, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K, et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 2020;5:e134612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Minute L, Teijeira A, Sanchez-Paulete AR, Ochoa MC, Alvarez M, Otano I, et al. Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer 2020;8:e000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jaime-Sanchez P, Uranga-Murillo I, Aguilo N, Khouili SC, Arias MA, Sancho D, et al. Cell death induced by cytotoxic CD8+ T cells is immunogenic and primes caspase-3–dependent spread immunity against endogenous tumor antigens. J Immunother Cancer 2020;8:e000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017;31:711–23.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ataide MA, Komander K, Knöpper K, Peters AE, Wu H, Eickhoff S, et al. BATF3 programs CD8+ T cell memory. Nat Immunol 2020;21:1397–407. [DOI] [PubMed] [Google Scholar]
- 50. Qiu Z, Khairallah C, Romanov G, Sheridan BS. Cutting edge: Batf3 expression by CD8 T cells critically regulates the development of memory populations. J Immunol 2020;205:901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 costimulation inhibits Foxp3 expression and Treg induction via BATF3-dependent and independent mechanisms. Cell Rep 2018;24:607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol 2001;167:3114–22. [DOI] [PubMed] [Google Scholar]
- 53. Song L, Asgharzadeh S, Salo J, Engell K, Wu H, Sposto R, et al. Vα24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest 2009;119:1524–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stenger D, Stief TA, Käuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020;136:1407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Exley MA, Hou R, Shaulov A, Tonti E, Dellabona P, Casorati G, et al. Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR α-chain CDR3 loop. Eur J Immunol 2008;38:1756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chiba A, Cohen N, Brigl M, Brennan PJ, Besra GS, Brenner MB. Rapid and reliable generation of invariant natural killer T-cell lines in vitro. Immunology 2009;128:324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. East JE, Sun W, Webb TJ. Artificial antigen presenting cell (aAPC) mediated activation and expansion of natural killer T cells. J Vis Exp 2012;29:4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mavers M, Simonetta F, Lee AW, Hirai T, Maas-Bauer K, Alvarez M, et al. IL-2 plus IL-15 leads to enhanced ex vivo expansion of human invariant natural killer T cells. Biol Blood Marrow Transplant 2018;24:S208–9. [Google Scholar]
- 59. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non–small cell lung cancer. Clin Cancer Res 2006;12:6079–86. [DOI] [PubMed] [Google Scholar]
- 60. Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol 2011;138:255–65. [DOI] [PubMed] [Google Scholar]
- 61. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: A phase I clinical trial. Clin Cancer Res 2017;23:3510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rubio M-T, Bouillié M, Bouazza N, Coman T, Trebeden-Nègre H, Gomez A, et al. Pre-transplant donor CD4- invariant NKT cell expansion capacity predicts the occurrence of acute graft-versus-host disease. Leukemia 2017;31:903–12. [DOI] [PubMed] [Google Scholar]
- 63. Schneidawind D, Pierini A, Alvarez M, Pan Y, Baker J, Buechele C, et al. CD4+ invariant natural killer T cells protect from murine GVHD lethality through expansion of donor CD4+CD25+FoxP3+ regulatory T cells. Blood 2014;124:3320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maas-Bauer K, Lohmeyer JK, Hirai T, Lopes Ramos T, Fazal FM, Litzenburger UM, et al. Invariant natural killer T cell subsets have diverse graft-versus-host-disease-preventing and anti-tumor effects. Blood 2021;138:858–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schneidawind D, Baker J, Pierini A, Buechele C, Luong RH, Meyer EH, et al. Third-party CD4+ invariant natural killer T cells protect from murine GVHD lethality. Blood 2015;125:3491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, Mathis D. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci USA 2006;103:3304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 2002;111:837–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3