

Abstract
Purpose:
Epigenetic modulators improve immune checkpoint inhibitor (ICI) efficacy and increase CD8+ effector: FoxP3+ regulatory T cell ratios in preclinical models. We conducted a multicenter phase I clinical trial combining the histone deacetylase (HDAC) inhibitor entinostat with nivolumab ± ipilimumab in advanced solid tumors.
Experimental Design:
Patients received an entinostat run-in (5 mg, weekly x 2) prior to the addition of ICIs. Dose escalation followed a modified 3+3 design (Dose level [DL]1/2: entinostat + nivolumab; DL 3/4: entinostat + nivolumab + ipilimumab). Blood and tissue samples were collected at baseline, after entinostat run-in, and after 8 weeks of combination therapy. Primary endpoints included safety and tolerability, and the recommended phase II dose (RP2D). Secondary endpoints included anti-tumor activity, change in tumor CD8/FoxP3 ratio pre- and post-therapy.
Results:
Thirty-three patients were treated across four dose levels. Treatment-related adverse events (AEs) included fatigue (65%), nausea (41%), anemia (38%), diarrhea (26%), and anorexia (26%). Grade 3/4 AEs included fatigue (n=7, 21%), anemia (n=9, 27%), and neutropenia (n=4, 12%). The RP2D was 3mg entinostat weekly, 3mg/kg q2 weeks nivolumab, and 1mg/kg q6 weeks ipilimumab (max 4 doses). The objective response rate by RECIST 1.1 was 16%, including a complete response in triple-negative breast cancer. A statistically significant increase in CD8/FoxP3 ratio was seen following the addition of ICIs to entinostat, but not post entinostat alone.
Conclusions:
The combination of entinostat, nivolumab ± ipilimumab was safe and tolerable with expected rates of irAEs. Preliminary evidence of both clinical efficacy and immune modulation supports further investigation.
Keywords: Immune checkpoint inhibitors, epigenetics, entinostat, nivolumab, ipilimumab
Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized the management of select solid tumor malignancies, but many patients still do not benefit from this approach (1). These agents have traditionally worked best in “immunogenic” cancers that naturally express adequate levels of antigens (neo-antigens), which leads to immune activation instead of immune tolerance (2). ICIs reset the tumor-specific immune response by releasing the brakes on cytotoxic T effector (Teff) cells in the tumor microenvironment (TME) (3). Investigation of the TME of immunogenic tumor types has revealed high numbers of tumor infiltrating lymphocytes (TILs), which have been shown to correlate with improved prognosis (4–7). However, tumor types with a paucity of TILs have proved to be less responsive to ICIs (8,9).
Preclinical studies have investigated therapeutic strategies to ‘prime’ the TME through an increase in TIL, activation of resident TIL, or an increase in the ratio of Teff to T regulatory (Treg) cells, with the goal of enhancing tumor cell killing. The addition of a histone deacetylase (HDAC) inhibitor (entinostat), in combination with ICIs (anti-programmed death receptor 1 [anti-PD-1] and anti-cytotoxic T-lymphocyte-associated protein 4 [anti-CTLA-4]), led to an increased Teff/Treg cell ratio that correlated with decreased tumor growth and improved survival in mice (10). Synergy between HDAC inhibitors and various immunotherapies, including ICIs, has subsequently been reported in other mouse models and in human cell lines (11–13).
We therefore hypothesized that the HDAC inhibitor entinostat in combination with ICIs would be safe and tolerable and would result in changes in the ratio of Teff/Treg, represented by the ratio of CD8/FoxP3 positive T cells in tumor biopsies. To test these hypotheses, we performed a multicenter phase I clinical trial combining entinostat, nivolumab ± ipilimumab in advanced solid tumors, incorporating blood and tissue biomarker evaluation pre- and post-therapy (NCT02453620, ETCTN-9844).
Patients and Methods
Eligibility Criteria
Patients 18 years of age or older were eligible if they had histologically or cytologically confirmed solid tumor malignancy that was metastatic or unresectable; and for whom either standard curative or palliative measures did not exist, were no longer effective, or for whom ICI was an appropriate treatment strategy. In addition, Eastern Cooperative Oncology Group (ECOG) performance status 0–1, adequate organ and pulmonary function (oxygen saturation ≥ 90% when ambulating and not requiring supplemental oxygen), less than 30% liver tumor involvement as determined locally, and stable brain metastases (4 weeks) were required. Measurable or evaluable/non-measurable disease per Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) was required, as well as an accessible non-bone tumor lesion from which serial core biopsy specimens could be obtained.
Exclusion criteria included prior immunomodulatory or ICI therapy, active autoimmune disease or history of autoimmune disease that might recur and affect vital organ function or require immune suppressive treatment, and corticosteroid use (>10 mg daily prednisone equivalent) within 14 days of enrollment. The study was registered at clinicaltrials.gov, approved by the Institutional Review Boards (IRB) of participating institutions, and participants signed a written informed consent prior to enrolment. The trial was conducted according to the principles of Good Clinical Practice and the Declaration of Helsinki.
Clinical Trial Design
This multi-site, open-label, phase I clinical trial was conducted in patients with advanced solid tumors. The dose-escalation cohort is reported in this manuscript, and an ongoing dose expansion cohort in advanced HER2-negative breast cancer will be reported at a later date. A two-week run-in of 5mg per week of oral entinostat was incorporated to help determine the effect of entinostat on immune-related parameters in tumor biopsies, prior to addition of ICIs (Table 1, Figure 1). Entinostat was taken on an empty stomach, at least 1 hour before and 2 hours after food, and a 5-HT3 receptor antagonist was administered as premedication to prevent nausea.
Table 1:
Dose Escalation
| Dose Level | Dose | ||
|---|---|---|---|
| Entinostat | Nivolumab | Ipilimumab | |
| ALL DOSE LEVELS | Entinostat 5mg/week x 2, run-in/ single agent dosing prior to proceeding to Dose Level assignment below. | ||
| Level - 1 | 3mg, weekly | 1mg/kg, q2 weeks | None |
| Level 1 | 3mg, weekly | 3mg/kg, q2 weeks | None |
| Level 2 | 5mg, weekly | 3mg/kg, q2 weeks | None |
| Level 3 | 3mg, weekly | 3mg/kg, q2 weeks | 1mg/kg, q6 weeks (max 4 doses) |
| Level 4 | 5mg, weekly | 3mg/kg, q2 weeks | 1mg/kg, q6 weeks (max 4 doses) |
Figure 1:

Study Schema
Tumor biopsy (blue arrow) and blood (red arrow) collection timepoints; Baseline, Timepoint 1 (after entinostat run-in), Timepoint 2 (after 8 weeks combination therapy) therapy.
Abbreviations: ECOG, Eastern Cooperative Oncology Group Performance Status; ICI, immune checkpoint inhibitor.
The study followed a modified 3+3 dose escalation design with four dose levels (DLs); DL1/2: entinostat 3–5mg weekly plus nivolumab 3mg/kg every 2 weeks, DL3/4: entinostat 3–5mg weekly plus nivolumab 3mg/kg every 2 weeks plus ipilimumab 1mg/kg every 6 weeks) (Table 1). The 3mg starting dose of entinostat was chosen for DL1 given that the combination of entinostat plus dual checkpoint inhibition had not previously been tested; thus providing a cautious approach in case of unexpected toxicity. At the request of the sponsor, the design required safety data from at least twelve subjects from DL2 in order to determine the safety of entinostat plus nivolumab prior to the addition of ipilimumab (DL3/4). Determination of the RP2D required that at least 6 patients were treated at the RP2D. Dose-limiting toxicity (DLT) was evaluated in the first 6 weeks of combination treatment for DL1 and 2. Once escalated from DL2, and in the first 4 weeks of therapy for DL3 and 4. Treatment continued until progressive disease or unacceptable toxicity. Treatment beyond initial RECIST 1.1-defined progression was permitted if protocol specific criteria were met.
The primary endpoints were the safety and tolerability of entinostat and nivolumab with or without ipilimumab in advanced solid tumors. The primary objective was to determine the RP2D. Secondary endpoints included the changes in CD8/FoxP3 ratio in mandatory tumor biopsy samples pre- and post-therapy, and antitumor activity including overall response rate (ORR) per RECIST 1.1 and immune-related RECIST (irRECIST) (14). Exploratory objectives included evaluations of other immune-related biomarkers including tumor infiltrating lymphocytes (TILs), programmed death-ligand 1 (PD-L1) and indoleamine 2,3 dioxygenase (IDO). Common Terminology Criteria for Adverse Events (CTCAE, version 5.0) was used to grade treatment-related toxicity.
Assessments
Baseline evaluations included routine history and physical examination, measurement of oxygen saturation, complete blood counts, serum chemistries, thyroid function, hepatitis serology, pregnancy test, and radiologic evaluations. Clinical evaluations and laboratory tests were repeated every 2 weeks for the first two cycles, with clinical evaluation every four weeks thereafter. Mandatory blood and tissue samples were collected from accessible sites at baseline, after entinostat run-in (timepoint 1), and after 8 weeks of combination therapy (timepoint 2) or at the time of progressive disease (if prior to 8 weeks) (Figure 1). Treatment response was evaluated using RECIST 1.1 and irRECIST criteria every 8 weeks for the first 6 months, and every 12 weeks thereafter (15).
Upon discontinuation of treatment, patients were followed every 3 months until disease progression, then every 6 months until either 5 years post-registration or death, whichever was earlier. Patients removed from study for unacceptable adverse events (AEs) were followed until resolution or stabilization of the AE. All patients were followed for toxicity assessment for 30 days after going off-study.
Pharmacokinetic Analysis
Concentrations of entinostat were determined at serial timepoints, matching those of tumor biopsies as above, using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method (16). The steady-state minimum plasma concentrations (Cmin, ss) were calculated as an average from both timepoints provided this was a true trough sample.
Correlative Analysis
The pipeline for slide processing is described in Supplementary Methods. Briefly, following dewaxing and rehydration, epitope retrieval was performed using Ventana Ultra CC1 buffer (#6414575001, Roche Diagnostics) at 96oC for 64 minutes. Primary antibody was applied at 36oC for 60 minutes and detected using an anti-mouse HQ detection system (#7017936001 and 7017782001, Roche Diagnostics) followed by Chromomap DAB IHC detection kit (#5266645001, Roche Diagnostics), when appropriate, counterstaining was done with Mayer’s hematoxylin, followed by dehydration, and mounting. For specific immunohistochemical (IHC) staining of formalin-fixed paraffin-embedded (FFPE) slides, we used monoclonal antibodies against CD8 (#m7103, lot 20024879, Dako, 1:100) and FoxP3 (#12653, lot 6 Cell Signaling Technology, 1:75). Indoleamine 2 3-dioxygenase (IDO) staining was performed using ab228468 (clone SP260, abcam 1.19 ug/mL) and was performed on the Leica Bond RX (17). Staining for PD-L1 was performed manually using ab228462 (clone SP142, abcam 0.08 ug/mL) (18–20). Following completion of stains, each slide was digitally scanned at 20X magnification using a Hamamatsu digital slide scanner (Hamamatsu, Japan). Regions of interest of viable tissue, excluding necrosis, were annotated by a pathologist (R.A.A.) using Aperio software. Using the HALO Digital Image Analysis program (Indica Labs, Corrales, NM), automatic edge detection was manually tuned to a minimum threshold that tightly applied to each sample in batch. Areas with apparent interferences such as bubbles, dark smudges, and tissue folds were manually annotated and excluded from the analysis.
CD8 and FoxP3 staining were obtained to determine the ratio of CD8/FoxP3 as an integrated correlative secondary objective, as it depicts Teff/Treg infiltration. Positive staining for CD8 and FoxP3 was assessed as a density (number of cells/ mm2), obtained by the total number of CD8 positive cells within the total tissue area (using the Immune cell module in HALO). Regions of interest (ROIs) designated as ‘cancer’ and ‘tumor’ were also annotated (RAA); ‘cancer’ where greater than 50% of the cells being large malignant tumor cells, and ‘tumor’ where a mix of large malignant tumor cells, stroma (i.e. cancer associated fibroblasts) and tumor infiltrating lymphocytes. TILs were scored within the stroma on a hematoxylin and eosin (H&E) stained slide by a pathologist (A.C-M.) according to international consensus guidelines (21). Of note, TIL counts were not included in cases where the samples lacked stroma or if the tissue sample was largely necrotic (4,22). PD-L1 IHC was manually assessed by a pathologist (R.A.A.). The percentage of PD-L1 stained cells was estimated on tumor cells (membranous staining pattern only), and lymphocytes (all the inflammatory cells in between malignant cells, cytoplasm and membranous pattern) by the study pathologist (RAA). RAA then documented the percentage of tumor cells or lymphocytes that were found to have ‘positive’ staining. IDO staining was determined as a ratio of stained area/ total viable tissue area (using the Area quantification module in HALO). Thresholds were used to bin data and were selected based on individual biomarker distribution by settings in HALO (Version 3.0.311), the nuclear stain detection program by IndicaLabs, and existing standards (17,20,23).
Statistical Considerations
The study followed a modified 3+3 design for the combination of entinostat and nivolumab (DL1/2), and then for entinostat, nivolumab, and ipilimumab (DL3/4). Compared to the typical 3+3 design, additional patients may be treated at each DL in order to obtain additional safety information as pre-specified in the study protocol. The RP2D was determined based on the totality of information from all DLs including AE and DLT data.
Safety and tolerability were analyzed through the incidence of AEs, serious AEs, and specific laboratory abnormalities (worst grade) in each arm. Toxicities were tabulated by type and grade for all doses and presented using frequencies and percentages based on the CTCAE v5.0. All subjects who receive at least 1 dose of study drug were evaluable for toxicity.
Preliminary antitumor activity of entinostat and nivolumab with or without ipilimumab was assessed as a secondary objective based on RECIST criteria 1.1 and irRECIST. Point estimate and exact 95% CIs were to be provided based on the observed data for ORR and clinical benefit rate (proportion of patients with complete [CR], partial response [PR], or stable disease [SD]). Kaplan-Meier survival curves for progression free survival (PFS) and overall survival (OS) were to be provided.
The ratio of CD8/FoxP3 in tumor biopsies at baseline, after 2 weeks of entinostat therapy (timepoint 1), and after the combination therapy (timepoint 2) was treated as a continuous variable and summarized with descriptive statistics. Changes in the ratio of CD8/FoxP3 were graphically depicted. Wilcoxon signed-rank test was used to determine whether the observed data showed evidence of changes in the ratio of CD8/FoxP3 at different timepoints. Descriptive statistics were used to summarize TILs, PD-L1 and IDO data observed on the study.
Pharmacokinetic parameters were summarized using descriptive statistics. Entinostat Cmin, ss was correlated with safety using Wilcoxon signed rank test or Wilcoxon rank-sum test.
Multiplicity adjustment is not considered because of the exploratory nature of the correlative analyses. Statistical tests with p-value < 0.05 are considered significant. The analyses of AEs were carried out using SAS software (v9.3, SAS Institute, Cary, NC), JMP Statistical Discovery software (version 7.0.1; SAS Institute, Cary, NC) and the R statistical software suite and programming environment (version 3.6.0 www.r-project.org).
Results
Patient Characteristics
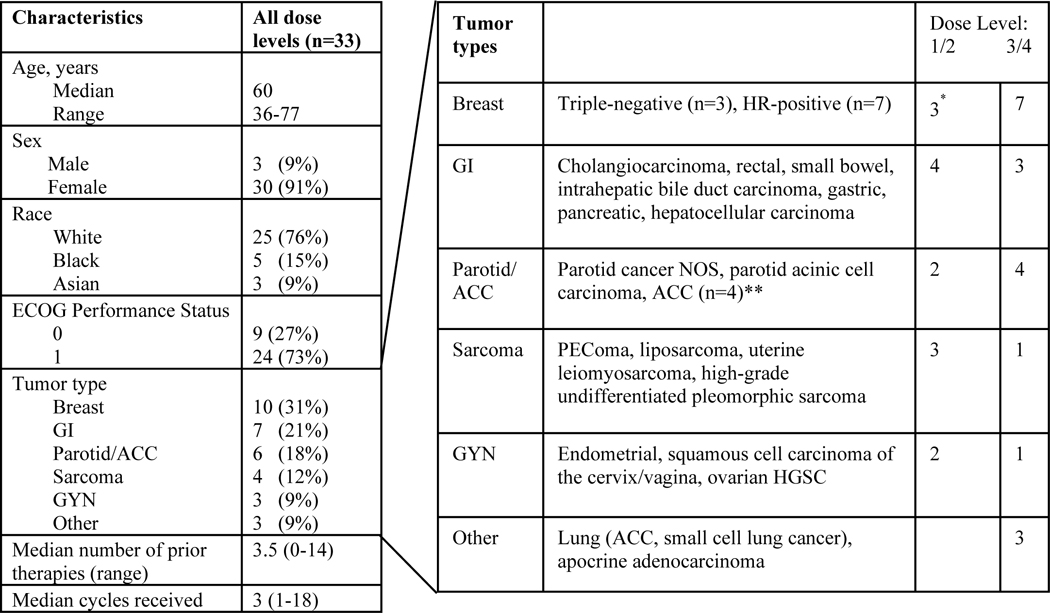
Thirty-five patients were consented across four National Cancer Institute (NCI) sites from February 2016 to May 2018. Thirty-three patients with a variety of solid tumors were eligible and treated in the dose escalation cohort (Table 2), with no patients remaining on therapy at time of data cut (March 2, 2020). Median age was 60 years (range 36–77), the majority enrolled were female (91%), and the median number of prior regimens for advanced disease was 3.5 (range 0–14). Off-study reasons included progressive disease (58%), AE/dose omission per protocol (33%), or withdrawal of consent (9%).
Table 2:
Patient Characteristics
|
Abbreviations: ACC, adenoid cystic carcinoma; ECOG, Eastern Cooperative Oncology Group; GI, gastrointestinal; GYN, gynecologic; HGSC, high grade serous carcinoma; HR, hormone-receptor; NOS, not otherwise specified
Included 2 patients with HR-positive breast and 1 with triple-negative breast cancer.
2 patients with ACC received DL 1/2 and 2 received DL 3/4
Treatment and Treatment Safety
All patients received the pre-defined starting doses of entinostat, nivolumab and ipilimumab per assigned DL. Median cycles of treatment received were 3 (range 1–18). Hematological and non-hematological toxicities are shown in Table 3, with the most common AEs including fatigue (65%), nausea (41%), anemia (38%), diarrhea (26%), pain (26%), and anorexia (26%). Grade 3 and 4 drug-related toxicities were infrequent. DLT occurred in one patient in DL2 (grade 3 pneumonitis) and one in DL4 (grade 3 allergic reaction). Possible immune-related (ir) AEs included hypothyroidism, hyperthyroidism (n=1, [DL3]), colitis (n=1 [DL2], n=1 [DL3]), pneumonitis (n=2 [DL2], n=1 [DL4]), and meningoencephalitis and myasthenia gravis (n=1, [DL3]) (Table 3). DL3 was determined to be the RP2D based on review of AEs and DLTs experienced across DL3 and DL4.
Table 3:
Adverse Events Related to Treatment in ≥ 4 patients
| Dose Level 1 (n=3) | Dose Level 2 (n=14) | Dose Level 3 (n=7) | Dose Level 4 (n=9) | (n=33) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adverse Event (n, %) | ≤G2 | G3 | G4 | Total | ≤G2 | G3 | G4 | Total | ≤G2 | G3 | G4 | Total | ≤G2 | G3 | G4 | Total | Total |
| Fatigue | 1(33) | 0 | 1(33) | 4(29) | 4(29) | 0 | 8(57) | 4(57) | 1(14) | 5(71) | 6(67) | 2(22) | 8(89) | 22(65) | |||
| Nausea | 2(67) | 2(67) | 6(43) | 6(43) | 2(29) | 2(29) | 4(44) | 4(44) | 14(41) | ||||||||
| Anemia | 1(33) | 1(33) | 1(7) | 6(43) | 7(50) | 2(29) | 2(29) | 1(11) | 2(22) | 3(33) | 13(38) | ||||||
| Diarrhea | 1(33) | 1(33) | 1(7) | 1(7) | 2(29) | 1(14) | 3(43) | 4(44) | 4(44) | 9(26) | |||||||
| Pain | 3(21) | 3(21) | 3(43) | 1(14) | 4(57) | 2(22) | 2(22) | 9(26) | |||||||||
| Anorexia | 3(21) | 3(21) | 4(57) | 4(57) | 2(22) | 2(22) | 9(26) | ||||||||||
| AST/ALT increased | 2(14) | 2(14) | 4(29) | 1(14) | 1(14) | 2(29) | 1(11) | 1(11) | 2(22) | 8(24) | |||||||
| Rash | 1(7) | 1(7) | 4(57) | 4(57) | 2(22) | 2(22) | 4(44) | 8(24) | |||||||||
| ALP increased | 4(29) | 1 (7) | 5(36) | 1(14) | 1(14) | 2(22) | 2(22) | 8(24) | |||||||||
| Thrombocytopenia | 4(29) | 4(29) | 2(29) | 2(29) | 2(22) | 2(22) | 8(24) | ||||||||||
| Vomiting | 5(36) | 5(36) | 1(14) | 1(14) | 1(11) | 1(11) | 7(21) | ||||||||||
| Neutropenia | 1(7) | 4(29) | 5(36) | 2(29) | 2(29) | 7(21) | |||||||||||
| Weight loss | 2(67) | 2(67) | 3(21) | 3(21) | 2(29) | 2(29) | 7(21) | ||||||||||
| Hypokalemia | 1(33) | 1(33) | 2(14) | 2(14) | 1(14) | 1(14) | 2(22) | 2(22) | 6(18) | ||||||||
| Dry mouth | 4(29) | 4(29) | 1(14) | 1(14) | 1(11) | 1(11) | 6(18) | ||||||||||
| Hypoalbuminemia | 2(14) | 1(7) | 3(21) | 1(14) | 1(14) | 2(22) | 2(22) | 6(18) | |||||||||
| Hyponatremia | 2(14) | 2(14) | 1(7) | 5(36) | 1(14) | 1(14) | 6(18) | ||||||||||
| Lipase increased | 1(7) | 1(7) | 2(14) | 1(14) | 1(14) | 2(29) | 1(11) | 1(11) | 2(22) | 6(18) | |||||||
| Leukopenia | 3(21) | 3(21) | 2(29) | 2(29) | 1(11) | 1(11) | 6(18) | ||||||||||
| Lymphopenia | 1(33) | 1(33) | 2(14) | 1(7) | 3(21) | 2(29) | 2(29) | 6(18) | |||||||||
| Pruritus | 2(14) | 2(14) | 2(29) | 2(29) | 2(22) | 2(22) | 6(18) | ||||||||||
| Dysgeusia | 1(33) | 1(33) | 3(21) | 3(21) | 2(22) | 2(22) | 6(18) | ||||||||||
| Hypophosphatemia | 3(21) | 1(7) | 4(29) | 1(14) | 1(14) | 5(15) | |||||||||||
| Hypothyroidism | 3(21) | 3(21) | 1(14) | 1(14) | 1(11) | 1(11) | 5(15) | ||||||||||
| Constipation | 1(33) | 1(33) | 1(7) | 1(7) | 1(14) | 1(14) | 1(11) | 1(11) | 4(12) | ||||||||
| Dizziness | 1(7) | 1(7) | 2(29) | 2(29) | 1 (11) | 1(11) | 4(12) | ||||||||||
| Dyspnea | 1(33) | 1(33) | 2(14) | 2(14) | 1(14) | 1(14) | 4(12) | ||||||||||
| Hypocalcemia | 3(21) | 3(21) | 1(14) | 1(14) | 4(12) | ||||||||||||
| Peripheral neuropathy | 1(7) | 1(7) | 2(29) | 2(29) | 1(11) | 1(11) | 4(12) | ||||||||||
| Amylase increased | 3(21) | 3(21) | 1(14) | 1(14) | 4(12) | ||||||||||||
Adverse events (AEs) related to treatment in ≥ 4 patients. Note: Number of worst grade adverse events (AEs) possibly, probably, or definitely attributed to study drug administration. Toxicities graded per the NCI Common Terminology Criteria for Adverse Events (CTCAE) Version 5 criteria. Note: Possible immune-related AEs not included in Table 3 due to frequency < 4 include hyperthyroidism, colitis, pneumonitis, meningoencephalitis and myasthenia gravis. Abbreviations: ≤G2=Grade 1 and 2, G3=Grade 3, G4=Grade 4. AST/ALT, aspartate/alanine aminotransferase; ALP, alkaline phosphatase.
The reasons for modification of entinostat dose included fatigue, decreased white blood cell (WBC)/absolute neutrophil count (ANC), transaminase elevation, rash, and metabolic AEs. The reasons for modification of nivolumab dose included fatigue, transaminase/ lipase/ amylase/ creatinine elevations, and arthralgia. Lastly, ipilimumab modifications were made due to fatigue, ALP elevation and arthralgias.
Treatment Efficacy
The ORR by RECIST 1.1 was 16% (4/25 patients evaluable for objective response, 95% CI [6.4%, 35%]); including one complete response (CR) in a patient with triple-negative breast cancer (TNBC) who discontinued therapy after 3 cycles due to AEs. Three partial responses (PRs) occurred in one patient with hormone receptor (HR)-positive breast cancer, one with cholangiocarcinoma, and one with high grade undifferentiated pleomorphic sarcoma. ORR by irRECIST was 20% (5/25 evaluable patients, 95% CI [8.9%, 39%]), which included in addition to responses noted above, a PR in an additional patient with TNBC (DL 2). The median PFS was 6.1 months (95% CI [2.7, 23.8]) for the entire cohort; 4.3 months (95% CI [2.7, 13.5]) for DL1/2, and 23.8 months (95% CI = [2.4, NA]) for DL 3/4 (Supplementary Figure S1A). The median overall survival (OS) was 10.6 months (95% CI [4.5, 22.7]) for the entire cohort; 7.6 months (95% CI [4.3, 12.8]) for those in DL1/2, and 20.3 months (95% CI [3.3, NA]) for DL 3/4 (Supplementary Figure S1B). The difference in the survival outcomes between DL1/2 and DL3/4 may reflect the tumor types enrolled in those DLs; for instance, the majority (75%) of patients with sarcoma (n=4) received DL1/2, whereas all of the patients with ACC (n=4) received DL3/4 (Table 2). Stable disease as the best response was observed in 44% (11/25 patients, 95% CI [27%, 63%]): HR-positive breast cancer (n=3), adenoid cystic carcinoma (ACC, n=3), and one each of parotid, rectal, small bowel, apocrine adenocarcinoma, and lung cancer. Clinical benefit rate was 60% (15/25 patients with measurable disease (95% CI [41%, 77%]), and did not differ between DLs (Supplemental Table S1). Three patients were treated beyond progression with none having a subsequent treatment response.
Pharmacokinetics
Pharmacokinetic data were obtained from 30 patients. Entinostat trough concentrations were 0.79 ± 0.52 ng/mL. Higher entinostat trough concentrations correlated with first cycle anemia (p=0.02) and increased amylase (p=0.04). There was no other statistically significant correlation between the worst grade of toxicity and entinostat exposure.
Evaluation of CD8/FoxP3 Ratio
Research biopsies were collected in all 33 patients at baseline, in 85% of patients (28/33) at timepoint 1, and in 42% of patients (14/33) at timepoint 2. Supplementary Table S2 outlines the number of correlative analyses that were performed at each timepoint. A statistically significant increase in the CD8/FoxP3 ratio was observed across DLs between timepoints 1 and 2 (Wilcoxon signed-rank test, p = 0.002) (Figure 2A), which was not observed between baseline and later timepoints. The median ratio was 4.11, 3.56, and 9.03 for baseline, timepoint 1, and timepoint 2, respectively (Figure 2B). Of note, the two patients with the most dramatic increase in CD8/FoxP3 ratios between these timepoints had HR-positive breast cancer (Figure 2A). These patients both experienced treatment-related toxicity; one each of grade 3 colitis and grade 3 lipase elevation. Lastly, one of these patients had a PR while the other experienced stable disease. There was an observed increase in CD8/FoxP3 ratio in those patients who experienced a treatment response (n=4) (Supplementary Figure S2A) however, this was not statistically significant. Of note, an increase was observed after addition of entinostat alone (baseline vs. timepoint 1) but was more pronounced following addition of ICIs. Analyses of CD8/FoxP3 ratio by timepoint includes all dose levels combined. Of note, in an exploratory analysis, patients who received entinostat + nivolumab + ipilimumab [DL 3/4] had higher observed median ratios of CD8/FoxP3 at baseline and timepoint 1 when compared to those who received entinostat + nivolumab alone [DL 1/2] (Supplementary Figure S2B & S2C). The densities of CD8 and FoxP3 are presented in Supplementary Figures S3–S4.
Figure 2:

Ratio of CD8/FoxP3
A. Log-transformed ratios of CD8 density to FoxP3 density at different timepoints. Box plots show the median, upper and lower quartiles, and range for the corresponding ratios. P-value is from Wilcoxon signed-rank test. B. Table showing summary statistics for CD8/FoxP3. Median and mean CD8/FoxP3 ratios at each timepoint. *Two patients with largest increase in ratio with HR-positive breast cancer.
An exploratory analysis was performed to evaluate entinostat as an immunologic primer for ICI therapy. We examined CD8/FoxP3 ratios by tumor type and demonstrated that following entinostat run-in, (baseline vs. timepoint 1), the CD8/FoxP3 ratio in ROI designated as ‘cancer’, appears to increase in breast and GI tumors, largely driven by decreased FoxP3 infiltration; whereas the trend is less consistent in parotid/ACC and all other tumor types (Supplementary Figure S5). We did not, however, observe a clear trend in CD8/FoxP3 ratios in ROIs designated as ‘tumor’ following entinostat run-in, regardless of tumor type (Supplementary Figure S6). Lastly, an additional exploratory analysis of CD8 T cell density in ROIs designated as ‘cancer’ revealed that the median density was higher in patients with tumor response vs. those without response, following entinostat run-in (Supplementary Figure S7). In ROIs designated as ‘tumor’, CD8 T cell density demonstrated a trend towards being decreased in patients without response vs. those with response, following entinostat run-in (Supplementary Figure S7).
Evaluation of TIL
The median percent stromal TIL observed across all timepoints was 3% (range 3–25%) (Supplementary Table 5). Evaluation of change in TIL before and after addition of ICI was only possible in 9/33 (27%) patients; with 6/33 (18%) having paired baseline to timepoint 2 samples, and 3/33 (9%) paired timepoint 1 to timepoint 2 samples. These results indicate the difficulties in obtaining serial TIL in patients with advanced solid tumors. No significant changes in TILs between timepoints, nor significant correlation of TIL with response, was detected in this small patient cohort.
Evaluation of PD-L1 and indoleamine 2,3-dioxygenase (IDO)
At baseline, 34.5% (10/29) of patients evaluated for PD-L1 had ≥ 1% PD-L1 positive tumor cells in tumor biopsies as assessed by membranous staining (Supplementary Table S4, Figure 3A). Of those patients who experienced a treatment response (n=4), only the patient with TNBC and a CR had baseline expression of PD-L1 > 1% (Figure 3B). At baseline, 25% (7/28) of evaluated patients had ≥ 1% PD-L1 positive lymphocytes in tumor biopsies (Figure 3A). There was no significant correlation between PD-L1 expression in tumor cells or lymphocytes and response to treatment (Supplementary Figure S8A &S8B).
Figure 3:

Summary of Biomarkers of Immunogenicity
A. Average percentage of tumor cells or lymphocytes demonstrating PD-L1 staining. PD-L1 staining, and evaluation was done manually from biopsies obtained from N= 28 patients at baseline. B. Representative images of immunostains for CD8+ T cells, FoxP3+ T cells, PDL1 in immune cells and IDO in immune cells, at each timepoint. C. Average IDO staining in all samples by timepoint did not reveal significant differences. N=28 patients for baseline, 28 for timepoint 1 and 14 for timepoint 2.
The median IDO staining was 0.124 (range 0.003–8.025) at baseline, 0.078 (range 0.008–5.244) at timepoint 1, and 0.1325 (range 0.008–1.444) at timepoint 2 (Figure 3C). Expression of IDO did not correlate with response (Supplementary Figure S8C). Examples of these IHC stains are shown in Figure 3B.
Discussion
In this multicenter phase I clinical trial, we have demonstrated that the combination of entinostat, nivolumab ± ipilimumab is safe and tolerable, with expected rates of irAEs (24). We have also identified 3mg weekly entinostat, 3mg/kg every 2 weeks nivolumab, and 1mg/kg every 6 weeks ipilimumab (max 4 doses) as the RP2D. Our trial was designed prior to flat dosing recommendations for ICI therapies, and future investigations will take into account these recommendations (25). We report an ORR of 16% and a clinical benefit rate of 60% with this combination in a heterogeneous cohort of patients. Interestingly, of ten patients with breast cancer enrolled, 2/10 experienced a treatment response (PR in HR-positive, CR in TNBC), and 4/10 experienced stable disease. The most robust increases in CD8/FoxP3 ratio were also observed in two patients with HR-positive breast cancer who experienced a PR and stable disease (Figure 2). Three additional patients who experienced stable disease had ACC and remained on treatment for 6, 13, and 17 cycles; which may be consistent with disease biology observed in ACC (26).
To our knowledge, this is the first published study that investigates treatment with an HDAC inhibitor in combination with dual ICI therapy. Trials combining dual anti-PD-1 and anti-CTLA-4 blockade have shown variable ORR; ranging from 12–18% in metaplastic breast cancer (27) to 69% in MSI-H/dMMR colorectal cancer as examples (28). The ORR we have observed in this trial is thus comparable with reported studies performed with dual ICI in breast cancer. The combination of entinostat with single agent PD-L1 inhibitor has also been reported in a number of tumor types. These studies have reported promising clinical activity with the combination of entinostat and anti-PD-L1 therapy when compared to PD-L1 alone; as noted by a PFS benefit in patients with microsatellite stable colorectal cancer (29), melanoma (30), and non-small cell lung cancer (31). However, patients with ovarian cancer (32) and TNBC (33) have not benefited from a similar strategy. Future investigation of combination approaches with HDAC inhibition and dual ICIs may yield improved responses.
Based on results from clinical and preclinical studies, we had hypothesized that the addition of entinostat would prime the TME and make tumors more susceptible to immune infiltration following treatment with ICI. To optimally examine this hypothesis, we included a two-week window of entinostat treatment alone, with tumor biopsies pre- and post-entinostat. The goal for patient tissue collection at three timepoints was to dissect the biologic changes of the TME in response to entinostat alone, and following the addition of ICIs. We chose a short window (i.e. 2-week run-in) due to the nature of a phase I trial; i.e the need to start combination therapy sooner than later in patients with pretreated cancers. Future trials may facilitate further evaluation of the role of entinostat as a priming agent.
Our integrated correlative analysis investigated ratios of CD8/FoxP3 immunostains, which has been shown to correlate with outcome in multiple tumor types (34). We expected to see significant changes in these T cell populations following treatment with ICIs but were unsure if T cell populations specifically would be affected by entinostat priming versus other immune cell types. Our results confirm that after a 2-week run-in with entinostat, there were no significant changes in CD8/FoxP3 ratios overall, however, there was a trend toward an increase in this ratio in patients responding to treatment. In addition, our exploratory analysis of more homogenous tumor types within this cohort suggest that the CD8/FoxP3 ratio increases in breast and GI tumors following entinostat run-in. These data lead us to hypothesize that the priming effects of entinostat may be more easily measured in homogenous tumor types and are potentially tumor type specific. This also suggests that the shift in T cell infiltration may have begun following sensitization with entinostat alone in these patients. Finally, the exploratory observation of increased CD8 T cell density in ‘cancer’ in patients with irRECIST response suggests that priming with entinostat could affect tumor cell immunogenicity leading to increased interaction with CD8 T cells. Evaluation of different ROIs within the tumor may thus be helpful in suggesting potential mechanisms of response. Future analysis of a larger homogenous cohort of patients i.e. our planned expansion cohort in breast cancer, are likely to improve our understanding of entinostat’s priming capacity.
Upon treatment with ICI therapy, we also evaluated the change in both CD8 and FoxP3 levels in biopsy samples. Our findings support our pre-clinical observations (10,13) that there are higher percentages of CD8+ T cells in the TME following addition of ICIs. The increased ratios of CD8/FoxP3 observed in this study appear to be driven by a higher increase in CD8+ T cell density from baseline to timepoint 2 and from timepoint 1 to timepoint 2 (Supplementary Figure 4). This observation supports the hypothesis that ICIs promote cytotoxic T cell infiltration and allow activation of resident T cells when administered with this treatment schedule (10,35). In addition, we investigated immunohistochemical expression of PD-L1 and IDO as surrogate markers of T cell activation (36–38) as a first step toward understanding mechanisms of response. Our evaluation of TIL did not show an association between TIL infiltration and response, although the significance of this is limited due to the small sample size and heterogenous cohort. Also, the median average TIL of 3% is relatively low in this cohort as compared to previously reported percentages (7,39). These findings are likely reflective of the heterogeneity of tumor types represented (17 different tumor types), the fact that no prior ICI therapy was permitted prior to study entry (tumor types for whom ICI therapy not approved), and that the majority of the biopsies were taken from metastatic sites (40). TIL infiltration is reportedly lower in metastatic disease sites (40).
Strengths of this study include the robust preclinical rationale, the incorporation of a two-week run-in of entinostat to facilitate evaluation of important biological questions as well as pharmacokinetic endpoints, and the multicenter design of the clinical trial. In addition, close collaboration between the basic science, pathology, and clinical trial investigators facilitated the study design, optimal tissue handling and storage, identification of the most relevant tissue sections for slide preparation, and division of samples to enable availability for more complex future studies such as imaging mass cytometry and sequencing (Supplementary Methods). Select biospecimens representative of a single tumor type collected from this study will be used to investigate the infiltration of other important immune cells types to the TME such as myeloid derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) which have become increasingly important with regard to biomarkers of response to ICIs in various tumor types (42–44) and improved efficacy of ICIs in preclinical models (10,13,45,46).
Limitations of the study include the single arm design, which was required for dose escalation purposes, and the heterogenous patient population enrolled. Pooled clinical and correlative analyses from a more homogenous population of patients with breast cancer treated in the dose escalation and expansion cohorts may provide further insight into the role of these agents in breast cancer. Another potential limitation may relate to performance of the SP142 clone used for PD-L1 staining however, we have optimized our assay with use of this antibody, and it shows comparable performance to the other antibodies. Finally, research tumor biopsies at the 8-week timepoint were not always obtained, which may limit investigation of mechanisms of response. The majority of missing biopsies for timepoint 2 were due to either disease progression or technical difficulties related to biopsy collection; both realities in early phase drug and biomarker development. In addition, 3 of the 4 patients experiencing treatment response did not have biopsies at timepoint 2; likely due to the treatment response itself. Thes limitations, however, highlight the importance of alternative strategies such as liquid biopsies and imaging-based methods to develop predictive biomarkers of response to anti-cancer therapies.
In conclusion, our study highlights the feasibility of conducting multicenter studies that use novel epigenetic and ICI agents, with incorporation of serial tissue biopsies for biomarker development in patients with advanced cancer. A strong collaboration between the laboratory and the clinic was present from study design through completion and will be critical for the effective design of future trials. This study paves the way for novel combination therapies that have the potential to improve response rates to ICIs in traditionally less responsive tumor types.
Supplementary Material
Supplementary Table S1: Observations by Dose Level (Efficacy and PD-L1)
ORR- overall response rate, irRECIST- immune related RECIST criteria, SD- standard deviation, P-values were derived from Fisher’s Exact test based on complete data only.
Supplementary Table S2: Tissue for Correlative Analyses
*In 2/12 cases, only tumor bed was noted (no tumor visualized) consistent with complete tumor regression; and were included in the analysis.
Supplementary Table S3: Tumor Infiltration Lymphocyte Data
For each timepoint a TIL range and average TIL percentage (Percent TIL) is listed. *In these cases, only tumor bed was noted (no tumor visualized) consistent with complete tumor regression. Abbreviations: ACC-adenoid cystic carcinoma, CC-Cholangiocarcinoma, Apocrine AdenoCa- apocrine adenocarcinoma
Supplementary Figure S1: Progression-Free and Overall Survival
A. Progression free survival (PFS) and B. Overall Survival (OS) curves for all patients (Red), Dose Levels 1 and 2 (Green), and Dose Levels 3 and 4 (Blue). Table below each curve lists number of patients at risk for death at a given timepoint used to determine probability in curve above.
Supplementary Figure S2: CD8/FoxP3 Ratio A. Log-transformed ratios of CD8 to FoxP3 for responders (defined as those patients who responded to treatment by RECIST criteria) vs. non-responders. B. Log-transformed density of CD8/FoxP3 ratio at different timepoints at Dose Levels 1/2 (left) or 3/4 (right). Box plots show the median, upper and lower quartiles, and range of the corresponding densities for each timepoint. C. Table of summary statistics of CD8/FoxP3 ratio at each time point for all patients, patients on Dose Levels 1/2 and patients on Dose Levels 3/4. *P-values are derived from Wilcoxon rank sum test between the two dose level categories
Supplementary Figure S3: CD8 Density
A. Densities of CD8 at different timepoints for all patients across Dose Levels. B. Patients on Dose Level 1/2, and patients on Dose Level 3/4. C. Table showing summary statistics of CD8 density at different timepoints for all patients, Dose Level 1/2, and Dose Level 3/4. *P-values are derived from Wilcoxon rank sum test between the two dose level categories.
Supplementary Figure S4: FoxP3 Density
A. Densities of FoxP3 at different timepoints for all patients. B. Dose Level 1/2, and Dose Level 3/4 C. Table showing summary statistics of FoxP3 density at different timepoints for all patients, Dose Level 1/2, and Dose Level 3/4.*P-values are derived from Wilcoxon rank sum test between the two dose level categories
Supplementary Figure S5: CD8, FoxP3 and ratio by tumor type in ‘cancer’ ROI
Densities of CD8 and FoxP3 at different timepoints for patients with designated tumor types (Breast, GI, Parotid/ACC, Other-which includes GYN/Sarcoma/Other) across all Dose Levels. Ratios of CD8 to FoxP3 for distinct tumor types at different timepoints. ROI ‘cancer’ is defined as area of biopsy with greater than 50% of the cells are large malignant tumor cells as identified in each specimen by the study pathologist RAA.
Supplementary Figure S6: CD8, FoxP3 and ratio by tumor type in ‘tumor’ ROI
Densities of CD8 and FoxP3 at different timepoints for patients with designated tumor types (Breast, GI, Parotid/ACC, Other-which includes GYN/Sarcoma/Other) across all Dose Levels. Ratios of CD8 to FoxP3 for distinct tumor types at different timepoints. ROI of ‘tumor’ is defined as a mix of large malignant tumor cells, stroma, i.e. cancer associated fibroblasts and tumor infiltrating lymphocytes, as identified in each specimen by the study pathologist RAA.
Supplementary Figure S7: CD8, density in ‘cancer’ vs. ‘tumor’ ROIs
Density of CD8 at different timepoints for all patients across all Dose Levels and according to irRECIST response. ROI of ‘tumor’ is defined as a mix of large malignant tumor cells, stroma, i.e. cancer associated fibroblasts and tumor infiltrating lymphocytes, ‘ROI ‘cancer’ is defined as area of biopsy with greater than 50% of the cells are large malignant tumor cells as identified in each specimen by the study pathologist RAA.
Supplementary Figure S8: PD-L1 and IDO Staining
A: PD-L1 staining in tumor cells in responders (defined as those patients who responded to treatment by RECIST criteria) and non-responders at each timepoint. B: PD-L1 staining in lymphocytes in responders and non-responders at each timepoint. C. IDO staining in responders and non-responders at each timepoint.
Translational Relevance.
This multicenter phase I clinical trial investigated the combination of the histone deacetylase inhibitor, entinostat, and nivolumab with or without ipilimumab in advanced solid tumors, based on strong preclinical rationale. Serial tumor biopsies and blood samples were collected to investigate changes in immune infiltration pre- and post-therapy, including biopsies pre- and post- entinostat treatment alone. Adverse events observed were expected for the agents under investigation; and a recommended phase II dose was determined. The objective response rate by RECIST 1.1 was 16%, including one complete response in a patient with triple-negative breast cancer. Correlative studies revealed a significant increase in the ratio of CD8/FoxP3 expression in tumor biopsy following checkpoint inhibitor treatment, but not after entinostat treatment alone. Stromal tumor infiltrating lymphocyte evaluation, PD-L1, and IDO immunostains were performed as measures of immunogenicity. This drug combination is being investigated further in an expansion cohort of patients with HER2-negative breast cancer.
Acknowledgements:
We thank the patients who volunteered to participate in this study; NCI Cancer Therapy Evaluation Program (CTEP); Bristol Myers Squibb and Syndax Pharmaceuticals for supply of study drug via NCI, and the research teams and physicians at participating sites. We thank James Leatherman from Johns Hopkins University for his assistance in finalizing correlative results. We thank Gary Rosner ScD and Michael Carducci MD for valuable input during study conduct.
Additional information:
Financial Support: National Cancer Institute (grant P30CA006973, P30CA014089, UM1CA186689, UM1CA186690, UM1CA186691, U24CA247648, and UM1CA186717) the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Bloomberg Kimmel Institute for Immunotherapy, Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973 and UL1 TR 001079), the Shared Instrument Grant (1S10RR026824-01), the Clinical Protocol and Data Management facilities (P30 CA006973 and P30CA 047904). The Cancer Therapy Evaluation Program supplied entinostat, nivolumab and ipilimumab. Support also provided by the Commonwealth Foundation Grant (2018-2020), NCCN (Young Investigator Award 2015), Mary Kay Foundation (Cancer Research Grant Program 2015), V Foundation (Translational Award 2017). Tower Cancer Research Foundation (Career Development Award -ERT 2020).
RC has received research grants to institution from Novartis, Puma Biotechnology, Merck, Genentech, Macrogenics; and an unrestricted educational grant from Pfizer. MAR has received research grants to institution from Celgene Corporation, Cullinan Apollo, and RenovoRx; MAR’s spouse is employed by GlaxoSmithKline. VS received research grants to institution from Abbvie, Biocept, Pfizer, Novartis, and Puma Biotechnology, and she is a member of the Data Safety Monitoring Board, Immunomedics, Inc. ACM has received research grants to institution from HeritX, Genentech and Bristol-Myers Squibb and serves as a consultant to Bristol-Myers Squibb. AB is a paid consultant for Lilly, Novartis, Pfizer, Astrazeneca, Eisai, Roche, Sanofi. PL reports personal fees from Abbvie, Agios, Five Prime, GenMab, Halozyme, Roche-Genentech, Genentech, Cytomx, Takeda, SOTIO, Cybrexa, Agenus, Tyme, IQVIA, TRIGR, Pfizer, ImmunoMet, Black Diamond, Glaxo-Smith Kline, QED Therapeutics, Astra Zeneca, EMD Serono, Shattuck, Astellas, Salarius, Silverback, MacroGenics, Kyowa Kirin, Kineta, Zentalis, Molecular Templates, ABL Bio, SK Life Science, STCube, these are all outside the submitted work. JE reports ongoing collaboration with Roche on new approaches to Precision Medicine Tumor Boards. VC is part of the speaker’s bureau for Ipsen and Coherus, he is a Perthera and Pfizer Consultant. RAA has received research support from Bristol-Myers Squibb, Merck, StandUp2Cancer, FLXbio and is a paid consultant for Bristol-Myers Squibb, Merck, Incyte, AstraZenca and FLXbio. EMJ is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, and Genocea. She receives funding from Lustgarten Foundation and Bristol Myer Squibb. She is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the C3 Cancer Institute.
Footnotes
Publisher's Disclaimer: Disclaimers: All other authors have nothing to disclose.
References:
- 1.Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ B. American Society of Clinical Oncology (ASCO); 2019;147–64. [DOI] [PubMed] [Google Scholar]
- 2.Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012. page 307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan MR, Schmid P. The emerging world of breast cancer immunotherapy. Breast. Churchill Livingstone; 2018;37:200–6. [DOI] [PubMed] [Google Scholar]
- 4.Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol [Internet]. 2017. [cited 2017 Dec 18]; Available from: http://linkinghub.elsevier.com/retrieve/pii/S147020451730904X [DOI] [PubMed] [Google Scholar]
- 5.Solinas C, Gombos A, Latifyan S, Piccart-Gebhart M, Kok M, Buisseret L. Targeting immune checkpoints in breast cancer: an update of early results. ESMO Open [Internet]. 2017. [cited 2017 Dec 18];2:e000255. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29177095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tormoen GW, Crittenden MR, Gough MJ. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. Elsevier Inc; 2018. page 520–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors [Internet]. Adv. Anat. Pathol. Lippincott Williams and Wilkins; 2017. [cited 2020 Nov 16]. page 311–35. Available from: /pmc/articles/PMC5638696/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loupakis F, Depetris I, Biason P, Intini R, Prete AA, Leone F, et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes . Oncologist [Internet]. Wiley; 2020. [cited 2020 Nov 16];25:481–7. Available from: /pmc/articles/PMC7288636/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou Y, Zou X, Zheng S, Tang H, Zhang L, Liu P, et al. Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: a systematic review and meta-analysis [Internet]. Ther. Adv. Med. Oncol. SAGE Publications Inc; 2020. [cited 2020 Nov 16]. Available from: /pmc/articles/PMC7436841/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A [Internet]. 2014;111:11774–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25071169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res [Internet]. 2017;23:5187–201. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28698201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. Ling MT, editor. PLoS One [Internet]. 2012. [cited 2018 Jan 6];7:e30815. Available from: http://dx.plos.org/10.1371/journal.pone.0030815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res [Internet]. 2018. [cited 2018 Oct 30];canimm.0070.2018. Available from: http://cancerimmunolres.aacrjournals.org/lookup/doi/10.1158/2326-6066.CIR-18-0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 16.Zhao M, Rudek MA, Mnasakanyan A, Hartke C, Pili R, Baker SD. A liquid chromatography/tandem mass spectrometry assay to quantitate MS-275 in human plasma. J Pharm Biomed Anal. 2007;43:784–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim AK, Gani F, Layman AJ, Besharati S, Zhu Q, Succaria F, et al. Multiple immune-suppressive mechanisms in fibrolamellar carcinoma. Cancer Immunol Res. American Association for Cancer Research Inc; 2019;7:805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaule P, Smithy JW, Toki M, Rehman J, Patell-Socha F, Cougot D, et al. A quantitative comparison of antibodies to programmed cell death 1 ligand 1. JAMA Oncol [Internet]. American Medical Association; 2017. [cited 2021 Apr 15];3:256–9. Available from: /pmc/articles/PMC5491359/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non–small cell lung cancer. JAMA Oncol [Internet]. American Medical Association; 2017. [cited 2021 Apr 15];3:1051–8. Available from: /pmc/articles/PMC5650234/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunshine JC, Nguyen PL, Kaunitz GJ, Cottrell TR, Berry S, Esandrio J, et al. PD-L1 expression in melanoma: A quantitative immunohistochemical antibody comparison. Clin Cancer Res. American Association for Cancer Research Inc.; 2017;23:4938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. 2014. [cited 2020 May 11]; Available from: https://academic.oup.com/annonc/article-abstract/26/2/259/2800585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dieci MV, Radosevic-Robin N, Fineberg S, van den Eynden G, Ternes N, Penault-Llorca F, et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: A report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol [Internet]. 2017. [cited 2018 May 16]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/29024776 [DOI] [PubMed] [Google Scholar]
- 23.Takada K, Kashiwagi S, Goto W, Asano Y, Takahashi K, Takashima T, et al. Use of the tumor-infiltrating CD8 to FOXP3 lymphocyte ratio in predicting treatment responses to combination therapy with pertuzumab, trastuzumab, and docetaxel for advanced HER2-positive breast cancer. J Transl Med [Internet]. 2018. [cited 2020 Jan 6];16:86. Available from: https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-018-1460-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination [Internet]. Nat. Rev. Clin. Oncol. Nature Publishing Group; 2016. [cited 2020 Nov 25]. page 473–86. Available from: https://pubmed-ncbi-nlm-nih-gov.libproxy2.usc.edu/27141885/ [DOI] [PubMed] [Google Scholar]
- 25.Lebbé C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, et al. Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: Results from the phase IIIb/IV CheckMate 511 trial. J Clin Oncol. American Society of Clinical Oncology; 2019. page 867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon PM, Chakraborty S, Moskaluk CA, Joshi PJ, Thomas CY. Adenoid cystic carcinoma: A review of recent advances, molecular targets, and clinical trials [Internet]. Head Neck. John Wiley and Sons Inc; 2016. [cited 2020 Nov 25]. page 620–7. Available from: https://pubmed-ncbi-nlm-nih-gov.libproxy2.usc.edu/25487882/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams S, Othus M, Patel SP, Chae YK, Miller K, Chugh R, et al. Dual anti-CTLA-4 and anti-PD-1 blockade in metaplastic carcinoma of the breast: Dart (SWOG S1609, Cohort 36). J Clin Oncol [Internet]. 2020. [cited 2020 Jun 2];38:1073–1073. Available from: https://ascopubs.org/doi/10.1200/JCO.2020.38.15_suppl.1073 [Google Scholar]
- 28.Lenz H-J, Lonardi S, Zagonel V, Van Cutsem E, Limon ML, Wong M, et al. Nivolumab (NIVO) + low-dose ipilimumab (IPI) as first-line (1L) therapy in microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): Two-year clinical update. J Clin Oncol [Internet]. 2020. [cited 2020 Jun 2];38:4040–4040. Available from: https://ascopubs.org/doi/10.1200/JCO.2020.38.15_suppl.4040 [Google Scholar]
- 29.Azad NS, Shirai K, McRee AJ, Opyrchal M, Johnson DB, Ordentlich P, et al. ENCORE 601: A phase 2 study of entinostat in combination with pembrolizumab in patients with microsatellite stable metastatic colorectal cancer. J Clin Oncol. American Society of Clinical Oncology (ASCO); 2018;36:3557–3557. [Google Scholar]
- 30.Agarwala SS, Moschos SJ, Johnson ML, Opyrchal M, Gabrilovich D, Danaher P, et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma progressing on or after a PD-1/L1 blocking antibody. J Clin Oncol. American Society of Clinical Oncology (ASCO); 2018;36:9530–9530. [Google Scholar]
- 31.Gandhi L, Janne PA, Opyrchal M, Ramalingam SS, Rybkin II, Hafez N, et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with non-small cell lung cancer (NSCLC) previously treated with anti-PD-(L)1 therapy. J Clin Oncol. American Society of Clinical Oncology (ASCO); 2018;36:9036–9036. [Google Scholar]
- 32.Cadoo KA, Meyers ML, Burger RA, Armstrong DK, Penson RT, Gordon MS, et al. A phase II randomized study of avelumab plus entinostat versus avelumab plus placebo in patients (pts) with advanced epithelial ovarian cancer (EOC). J Clin Oncol. American Society of Clinical Oncology (ASCO); 2019;37:5511–5511. [Google Scholar]
- 33.O’Shaughnessy J, Moroose RL, Babu S, Baramidze K, Chan D, Leitner SP, et al. Results of ENCORE 602 (TRIO025), a phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab with or without entinostat in patients with advanced triple-negative breast cancer (aTNBC). J Clin Oncol [Internet]. 2020. [cited 2020 Jun 2];38:1014–1014. Available from: https://ascopubs.org/doi/10.1200/JCO.2020.38.15_suppl.1014 [Google Scholar]
- 34.DeLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res 2012. page 3022–9. [DOI] [PubMed] [Google Scholar]
- 35.Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer [Internet]. J. Exp. Clin. Cancer Res. BioMed Central Ltd; 2019. [cited 2020 Nov 25]. page 1–12. Available from: 10.1186/s13046-019-1259-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance [Internet]. Trends Immunol. Elsevier Ltd; 2016. [cited 2021 Apr 12]. page 193–207. Available from: /pmc/articles/PMC4916957/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 Inhibitors: From bench to bedside [Internet]. Cancer Res. American Association for Cancer Research Inc; 2017. [cited 2021 Apr 13]. page 6795–811. Available from: www.aacrjournals.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Llosa NJ, Luber B, Tam AJ, Smith KN, Siegel N, Awan AH, et al. Intratumoral adaptive immunosuppression and type 17 immunity in mismatch repair proficient colorectal tumors. Clin Cancer Res [Internet]. American Association for Cancer Research Inc.; 2019. [cited 2021 Apr 13];25:5250–9. Available from: http://clincancerres.aacrjournals.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanton SE, Adams S, Disis ML. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review [Internet]. JAMA Oncol. JAMA Oncol; 2016. [cited 2020 Nov 25]. page 1354–60. Available from: https://pubmed-ncbi-nlm-nih-gov.libproxy2.usc.edu/27355489/ [DOI] [PubMed] [Google Scholar]
- 40.Cimino-Mathews A, Ye X, Meeker A, Argani P, Emens LA. Metastatic triple-negative breast cancers at first relapse have fewer tumor-infiltrating lymphocytes than their matched primary breast tumors: A pilot study. Hum Pathol [Internet]. Hum Pathol; 2013. [cited 2020 Nov 25];44:2055–63. Available from: https://pubmed-ncbi-nlm-nih-gov.libproxy2.usc.edu/23701942/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Tilburg CM, Witt R, Heiss M, Pajtler KW, Plass C, Poschke I, et al. INFORM2 NivEnt: The first trial of the INFORM2 biomarker driven phase I/II trial series: the combination of nivolumab and entinostat in children and adolescents with refractory high-risk malignancies. BMC Cancer [Internet]. BMC Cancer; 2020. [cited 2020 Jun 12];20:523. Available from: https://bmccancer.biomedcentral.com/articles/10.1186/s12885-020-07008-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res [Internet]. 2007;67:9518–27. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17909062 [DOI] [PubMed] [Google Scholar]
- 43.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) [Internet]. 2014;6:1670–90. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25125485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umansky V, Sevko A. Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron [Internet]. 2013;6:169–77. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23242672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-derived suppressor cells: Ductile targets in disease. Front. Immunol. Frontiers Media S.A; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med [Internet]. American Association for the Advancement of Science; 2020. [cited 2020 Feb 24];12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32051227 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Observations by Dose Level (Efficacy and PD-L1)
ORR- overall response rate, irRECIST- immune related RECIST criteria, SD- standard deviation, P-values were derived from Fisher’s Exact test based on complete data only.
Supplementary Table S2: Tissue for Correlative Analyses
*In 2/12 cases, only tumor bed was noted (no tumor visualized) consistent with complete tumor regression; and were included in the analysis.
Supplementary Table S3: Tumor Infiltration Lymphocyte Data
For each timepoint a TIL range and average TIL percentage (Percent TIL) is listed. *In these cases, only tumor bed was noted (no tumor visualized) consistent with complete tumor regression. Abbreviations: ACC-adenoid cystic carcinoma, CC-Cholangiocarcinoma, Apocrine AdenoCa- apocrine adenocarcinoma
Supplementary Figure S1: Progression-Free and Overall Survival
A. Progression free survival (PFS) and B. Overall Survival (OS) curves for all patients (Red), Dose Levels 1 and 2 (Green), and Dose Levels 3 and 4 (Blue). Table below each curve lists number of patients at risk for death at a given timepoint used to determine probability in curve above.
Supplementary Figure S2: CD8/FoxP3 Ratio A. Log-transformed ratios of CD8 to FoxP3 for responders (defined as those patients who responded to treatment by RECIST criteria) vs. non-responders. B. Log-transformed density of CD8/FoxP3 ratio at different timepoints at Dose Levels 1/2 (left) or 3/4 (right). Box plots show the median, upper and lower quartiles, and range of the corresponding densities for each timepoint. C. Table of summary statistics of CD8/FoxP3 ratio at each time point for all patients, patients on Dose Levels 1/2 and patients on Dose Levels 3/4. *P-values are derived from Wilcoxon rank sum test between the two dose level categories
Supplementary Figure S3: CD8 Density
A. Densities of CD8 at different timepoints for all patients across Dose Levels. B. Patients on Dose Level 1/2, and patients on Dose Level 3/4. C. Table showing summary statistics of CD8 density at different timepoints for all patients, Dose Level 1/2, and Dose Level 3/4. *P-values are derived from Wilcoxon rank sum test between the two dose level categories.
Supplementary Figure S4: FoxP3 Density
A. Densities of FoxP3 at different timepoints for all patients. B. Dose Level 1/2, and Dose Level 3/4 C. Table showing summary statistics of FoxP3 density at different timepoints for all patients, Dose Level 1/2, and Dose Level 3/4.*P-values are derived from Wilcoxon rank sum test between the two dose level categories
Supplementary Figure S5: CD8, FoxP3 and ratio by tumor type in ‘cancer’ ROI
Densities of CD8 and FoxP3 at different timepoints for patients with designated tumor types (Breast, GI, Parotid/ACC, Other-which includes GYN/Sarcoma/Other) across all Dose Levels. Ratios of CD8 to FoxP3 for distinct tumor types at different timepoints. ROI ‘cancer’ is defined as area of biopsy with greater than 50% of the cells are large malignant tumor cells as identified in each specimen by the study pathologist RAA.
Supplementary Figure S6: CD8, FoxP3 and ratio by tumor type in ‘tumor’ ROI
Densities of CD8 and FoxP3 at different timepoints for patients with designated tumor types (Breast, GI, Parotid/ACC, Other-which includes GYN/Sarcoma/Other) across all Dose Levels. Ratios of CD8 to FoxP3 for distinct tumor types at different timepoints. ROI of ‘tumor’ is defined as a mix of large malignant tumor cells, stroma, i.e. cancer associated fibroblasts and tumor infiltrating lymphocytes, as identified in each specimen by the study pathologist RAA.
Supplementary Figure S7: CD8, density in ‘cancer’ vs. ‘tumor’ ROIs
Density of CD8 at different timepoints for all patients across all Dose Levels and according to irRECIST response. ROI of ‘tumor’ is defined as a mix of large malignant tumor cells, stroma, i.e. cancer associated fibroblasts and tumor infiltrating lymphocytes, ‘ROI ‘cancer’ is defined as area of biopsy with greater than 50% of the cells are large malignant tumor cells as identified in each specimen by the study pathologist RAA.
Supplementary Figure S8: PD-L1 and IDO Staining
A: PD-L1 staining in tumor cells in responders (defined as those patients who responded to treatment by RECIST criteria) and non-responders at each timepoint. B: PD-L1 staining in lymphocytes in responders and non-responders at each timepoint. C. IDO staining in responders and non-responders at each timepoint.