

Abstract
Background:
Among men with metastatic prostate cancer, about 10% have germline alterations in DNA damage response genes. Most studies have examined BRCA2 alone or an aggregate of BRCA1/2 and ATM. Emerging data suggest that ATM mutations may have distinct biology and warrant individual evaluation. The objective of this study is to determine whether response to prostate cancer systemic therapies differs between men with germline mutations in ATM (gATM) and BRCA2 (gBRCA2).
Methods:
This is an international multicenter retrospective matched cohort study of men with prostate cancer harboring gATM or gBRCA2. PSA50 response (≥50% decline in prostate-specific antigen) was compared using Fisher’s exact test.
Results and Limitations:
The study included 45 gATM and 45 gBRCA2 patients, matched on stage and year of germline testing. Patients with gATM and gBRCA2 had similar age, Gleason grade, and PSA at diagnosis. We did not observe differences in PSA50 responses to abiraterone, enzalutamide, or docetaxel in metastatic castration resistant prostate cancer between the two groups; however, 0/7 with gATM and 12/14 with gBRCA2 achieved PSA50 response to PARPi (p<0.001). Median (95% CI) overall survival from diagnosis to death was 10.9 years (9.5-not reached) vs. 9.9 years (7.1-not reached, p=0.07) for the gATM and gBRCA2 cohorts, respectively. Limitations include the retrospective design and lack of mutation zygosity data.
Conclusions:
Conventional therapies can be effective in gATM carriers and should be considered before PARPi, which shows limited efficacy in this group. Men with gATM mutations warrant prioritization for novel treatment strategies.
Keywords: ATM, BRCA2, germline, homologous recombination deficiency, PARPi, platinum, abiraterone, enzalutamide, docetaxel
Introduction
Approximately 10% of men with metastatic prostate cancer have germline (inherited) DNA damage response (gDDR) gene alterations. BRCA2 is a homologous recombination (HR) gene and is the most frequent pathogenic germline alteration in advanced prostate cancer (3–5%), followed by ATM (1.6–2%) and BRCA1 (0.8–1.3%).1–3 Several studies have shown that germline BRCA2 mutations (gBRCA2) are associated with poor prognosis and worse prostate cancer outcomes and/or increased genomic instability.3–8
Castro, et al., reported that at diagnosis, patients with prostate cancer and gBRCA1/2 mutations are more likely to have Gleason Grade Group ≥4 disease, T3/4 stage, nodal involvement, metastases, and shorter cancer-specific survival compared to non-carriers.6 The IMPACT study showed that gBRCA2 mutation carriers have a higher incidence of prostate cancer and are more likely to be diagnosed at a younger age and have clinically significant disease compared to non-carriers, whereas no difference in age or tumor characteristics was detected between gBRCA1- and non-carriers.5 Na, et al., reported that the combined gBRCA1/2 and germline ATM (gATM) mutation rate was higher in lethal prostate cancer compared to localized disease.9 However, features of tumors and treatment responses linked to gATM mutations as a separate cohort are not characterized.
gATM mutation carriers have not been well-characterized despite ATM being the second most frequently observed DNA damage response gene alteration in metastatic prostate cancer. Several retrospective and prospective studies have reported that ATM-deficient prostate tumors may have attenuated response to poly-ADP-ribose polymerase inhibitors (PARPi) and platinum chemotherapy.7,10–15 Preliminary results of the phase II TRITON2 study demonstrated radiographic response to PARPi rucaparib in 51% (50/98) of men with BRCA1/2 and only 4% (2/49) of men with ATM mutations.11,16 The U.S. Food and Drug Administration (FDA) granted rucaparib an accelerated approval for men with metastatic castration resistant prostate cancer (mCRPC) and BRCA1/2 mutations who were previously treated with docetaxel. In the phase III randomized PROfound study of the PARPi olaparib vs AR targeting agent, the primary endpoint of radiographic progression-free survival (rPFS) in men with mCRPC harboring mutations in BRCA1/2 and/or ATM (cohort A) was met, and olaparib also received FDA approval. While the primary endpoint was met for cohort A, in a post-hoc subgroup analysis of men whose prostate cancer harbored ATM alterations, olaparib did not significantly improve rPFS (median 5.4 months vs. 4.7 months for controls).12 One potential explanation for the observed differences in clinical activity of PARPi in men with BRCA2 vs. ATM mutations may relate to the distinctive roles these proteins play in HR repair, with ATM acting as a sensor of DNA double strand break and BRCA2 being a core effector of HR DNA repair.
Conventional systemic prostate cancer therapies, such as androgen receptor (AR) targeted or taxane agents, are not currently selected by biomarkers. These therapies have been reported to be effective in gBRCA1/2 carriers with prostate cancer.3,17 PROREPAIR-B, a prospective cohort study, compared response outcomes for mCRPC treatments among gBRCA2 carriers and non-carriers and showed similar response rates.3 Efficacy in patients with gATM, as a distinct cohort, has not been evaluated. Given the uncertain response to HR-deficiency targeted treatments in these men, we sought to investigate whether these patients respond to conventional biomarker-agnostic therapies. We hypothesized that, compared to men carrying gBRCA2, those carrying gATM would have a similar response to AR-targeted agents and docetaxel yet attenuated responses to platinum and PARPi therapies.
Methods
This is an international, retrospective, matched cohort study of Consecutive patients with prostate cancer who underwent clinical germline genetic testing between 2014 and 2019 at the University of Washington (UW), Johns Hopkins (JH) Hospital, CNIO-IBIMA Genitourinary Cancer Unit, or Tulane University Cancer Center. We selected patients who had gATM or gBRCA2 mutations identified with germline genetic testing panels (Ambry Color, Invitae, Myriad, or in-house germline genetic testing at CNIO, JH and UW). Only alterations designated as pathogenic or likely pathogenic by the American College of Medical Genetics were included.18 The gBRCA2 cohort was chosen as a comparison group because it has the most characterized HR-deficient prostate cancer phenotype and established management guidelines. To facilitate comparisons, the gBRCA2 cohort was individually matched (1:1) to the gATM group by stage at diagnosis (metastatic vs. non-metastatic), year of germline testing and by center at which patients were treated.
A total of 45 patients with gATM and 45 matched gBRCA2 cases were included. Two patients included in the current study were also reported in the analysis by Marshall et al.: one gATM and one gBRCA2 mutation carrier.10 Medical records review was performed after local institutional review board approvals at participating centers.
Statistical Analysis
Baseline characteristics for gATM and gBRCA2 cohorts were compared using the Mann-Whitney test for continuous variables and Fisher’s exact test for categorical variables. The primary efficacy endpoint was the percentage of men achieving at least one prostate-specific antigen value that was ≥50% below baseline (PSA50 response). Treatment-specific PSA50 responses were compared using Fisher’s exact tests. Follow-up was calculated using reverse Kaplan-Meier estimation. Metastasis-free survival (MFS) was defined as time from diagnosis to death, last clinical evaluation, or evidence of metastasis on conventional imaging, determined at the local radiologists’ discretion and broadly consistent with the Prostate Cancer Clinical Trials Working Group 3 guidelines.19 Overall survival (OS) was defined as time from prostate cancer diagnosis to death or last clinical evaluation. Time on therapy was defined as time from initiation to termination of therapy or last clinical evaluation, and time to next treatment was defined as time from the start of treatment to the initiation of the next regimen or last clinical evaluation. OS, MFS, median time on therapy, and median time to next treatment were estimated using Kaplan-Meier methods. Differences between gATM and gBRCA2 cohorts were estimated using the log-rank test. All tests were two-sided and p <0.05 was considered statistically significant. R, version 3.6.3, was used for statistical analysis.
Results
Cohort Characteristics
The study included 90 men with prostate cancer: 45 with gATM mutations and 45 with gBRCA2 mutations. Specific mutations in gATM and gBRCA2 genes are documented in Figure 1. Baseline characteristics, including age, PSA, Gleason Grade Group, were similar in the gATM and gBRCA2 cohorts (Table 1). A similar number of patients had a family history of cancer, meeting Prostate Cancer NCCN Guidelines20 for germline testing. Distribution of pathology patterns (e.g., cribriform, neuroendocrine), definitive treatment, and anatomical sites of metastases were also similar between the two cohorts. The median follow-up time since diagnosis was 11.8 years in the gATM cohort and 8.0 years in the gBRCA2 cohort. Metastases developed in 23/28 gATM and 20/28 gBRCA2 patients after a median follow-up of 15.7 and 15.0 years, respectively, for the subgroup of men diagnosed with localized prostate cancer. Of the 12 men in the gATM cohort and 14 men in the gBRCA2 cohort for whom tumor sequencing results were available, none were reported to have somatic alterations in other HR genes.
Figure 1.
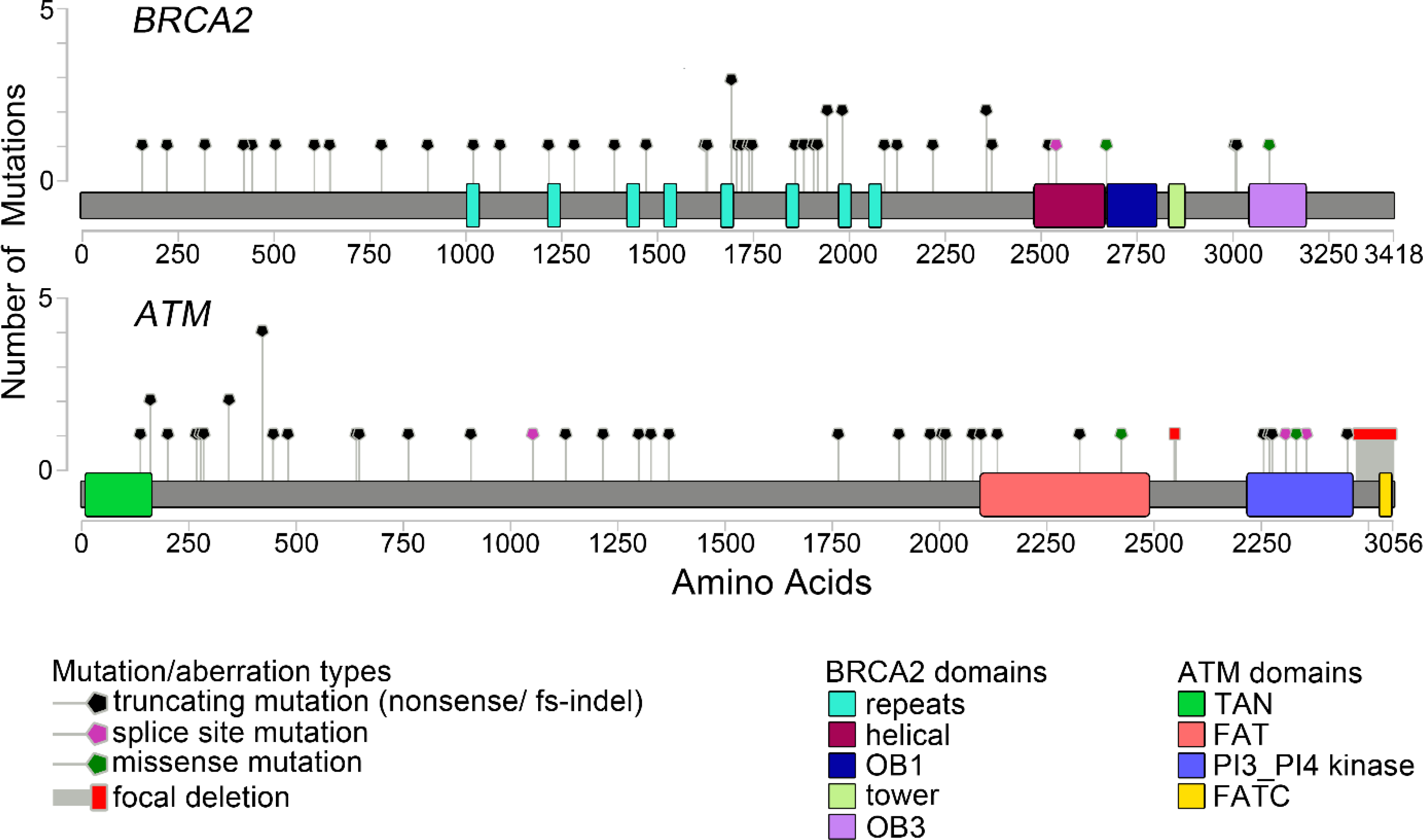
Distribution of ATM and BRCA2 Mutations
Table 1.
Patient Characteristics
| Characteristics | gATM | gBRCA2 | P |
|---|---|---|---|
| Number of patients | 45 | 45 | |
| Stage M1 at diagnosis (%) | 17 (38) | 17 (38) | |
| Age (median [IQR]) | 58 [54, 66] | 62 [55, 67] | 0.2 |
| PSA (median [IQR]) | 24 [9, 76] | 11 [6, 46] | 0.13 |
| Grade (%) | |||
| 2 | 6 (17) | 4 (11) | |
| 3 | 7 (20) | 5 (14) | |
| 4 | 5 (14) | 8 (22) | |
| 5 | 17 (49) | 20 (54) | |
| Family history of cancer meeting Prostate Cancer NCCN Guidelines for germline testing20 (%) | 25 (60) | 29 (71) | 0.4 |
| Known other primary cancers (%) | 5 (11) | 4 (9) | >0.9 |
| Pathology (%) | |||
| acinar | 24 (80) | 22 (76) | |
| ductal | 3 (10) | 3 (10) | |
| intraductal | 0 ( 0) | 1 ( 3) | |
| cribriform | 1 ( 3) | 1 ( 3) | |
| neuroendocrine | 2 ( 7) | 2 ( 7) | |
| Prostatectomy (%) | 20 (44) | 22 (50) | 0.7 |
| Radiotherapy (%) | 22 (51) | 24 (56) | 0.8 |
| Bone metastasis at the time of diagnosis (%) | 14 (31) | 15 (33) | >0.9 |
| Nodal metastasis at the time of diagnosis (%) | 13 (29) | 11 (24) | 0.8 |
| Visceral metastasis at the time of diagnosis (%) | 1 ( 2) | 3 ( 7) | 0.6 |
PSA50 Response Rates
Responses to systemic therapies in the mCRPC setting, as measured by PSA50, are summarized in Table 2. Comparing patients with gATM versus gBRCA2 mutations, there was no evident difference in PSA50 response to abiraterone: 9/16 (56%) vs. 11/19 (58%); to enzalutamide: 9/16 (56%) vs. 8/12 (67%); or to docetaxel: 9/13 (69%) vs. 9/16 (60%). Only 1 of 3 patients with gATM vs. 5 of 7 patients with gBRCA2 responded to platinum, numbers are too small to draw conclusions. In contrast, there appeared to be a difference in responses to PARPi—0/7 (0%) patients with gATM mutations responded vs. 12/14 (86%) patients with gBRCA2 mutations (p<0.001).
Table 2.
PSA50 Response
| Therapy | Prior | gATM | gBRCA2 | P |
|---|---|---|---|---|
| Abiraterone | Overall | 9/16 (56%) | 11/19 (58%) | >0.9 |
| Pre-enza | 9/14 (64%) | 10/17 (59%) | ||
| Post-enza | 0/2 ( 0%) | 1/2 (50%) | ||
| Enzalutamide | Overall | 9/16 (56%) | 8/12 (67%) | 0.7 |
| Pre-abi | 7/10 (70%) | 5/7 (71%) | ||
| Post-abi | 2/6 (33%) | 3/5 (60%) | ||
| Docetaxel | Overall | 9/13 (69%) | 9/16 (56%) | 0.7 |
| Pre-abi/enza | 7/9 (78%) | 4/7 (57%) | ||
| Post-abi/enza | 2/4 (50%) | 5/9 (56%) | ||
| PARPi | Overall | 0/7 ( 0%) | 12/14 (86%) | <0.001 |
| Pre-plat | 0/3 ( 0%) | 10/11 (91%) | ||
| Post-plat | 0/4 ( 0%) | 2/3 (67%) |
Time on Treatment
Median time on mCRPC treatment for the gATM and gBRCA2 cohorts is shown in Table 3. Overall, for abiraterone, enzalutamide, and docetaxel, there was no evidence of different duration from the start to the end of treatment between the cohorts. In the mCRPC setting, median (95% CI) time on AR-targeted therapies in gATM compared to gBRCA2 cohort was 9.7 (6.5–23) vs. 6.4 (5.4–15.5) months for abiraterone (p=0.5); 6.5 (4.6-not reached) vs 9 (4.9-not reached) months for enzalutamide (p>0.9); and 5.1 (3.7-not reached) vs. 4 (3–6) months for docetaxel-based chemotherapy (p=0.06). Median time on platinum-based chemotherapy in the mCRPC setting was 3 (1-not reached) months in the gATM cohort compared to 6 (4-not reached) months in the gBRCA2 cohort (p=0.11). We observed a difference in treatment duration on PARPi: 3 (2-not reached) months in the gATM cohort compared to 12 (6.9-not reached) months in the gBRCA2 cohort (p=0.004). Time on treatment for each therapy is shown in Supplemental Figures 5 A–E.
Table 3.
Time on Treatment
| Therapy | Setting | gATM | gBRCA2 | P | ||
|---|---|---|---|---|---|---|
| Number of pts | Median time on therapy (95% CI) | Number of pts | Median time on therapy (95% CI) | |||
| Abiraterone | Overall | 19 | 9.71 (6.5–23) | 24 | 6.44 (5–15.5) | 0.6 |
| HSPC | 2 | 3 (3–N/A) | 5 | 6 (5–N/A) | >0.9 | |
| CRPC | 17 | 9.71 (6.5–23) | 19 | 6.44 (5.38–15.5) | 0.5 | |
| Enzalutamide | CRPC | 16 | 6.5 (4.62–N/A) | 12 | 9 (4.92–N/A) | >0.9 |
| PARPi | CRPC | 7 | 3 (2–N/A) | 15 | 12 (6.9–N/A) | 0.004 |
| Platinum | CRPC | 3 | 3 (1–N/A) | 7 | 6 (4–N/A) | 0.11 |
| Docetaxel | Overall | 18 | 4.13 (4–7) | 21 | 4 (3–6) | 0.12 |
| HSPC | 5 | 4 (N/A–N/A) | 4 | 4.5 (3–N/A) | 0.4 | |
| CRPC | 13 | 5.12 (3.7–N/A) | 17 | 4 (3–6) | 0.06 | |
| Median time to next therapy (CI 95%) | Median time to next therapy (CI 95%) | |||||
| CRPC | 13 | 10.47 (6.47–N/A) | 15 | 7 (4.16–12.82) | 0.15 | |
Pts - patients; HSPC – hormone sensitive prostate cancer; CRPC – castration resistant prostate cancer.
Overall Survival
During the study follow-up period, 15/45 (33.3%) gATM and 18/45 (40%) gBRCA2 patients died. Median (95% CI) OS from diagnosis to death was 10.9 years (9.5-not reached) vs. 9.9 years (7.1-not reached, p=0.07) for the gATM and gBRCA2 cohorts, respectively (Figure 2). There was no evidence of OS difference between gATM and gBRCA2 cohorts when analyzing subgroups of patients initially diagnosed with localized (not reached vs 9.9 years, respectively, p=0.07) or metastatic disease (8.7 vs 3.6 years, respectively, p=0.4; Supplemental Figure 3).
Figure 2.
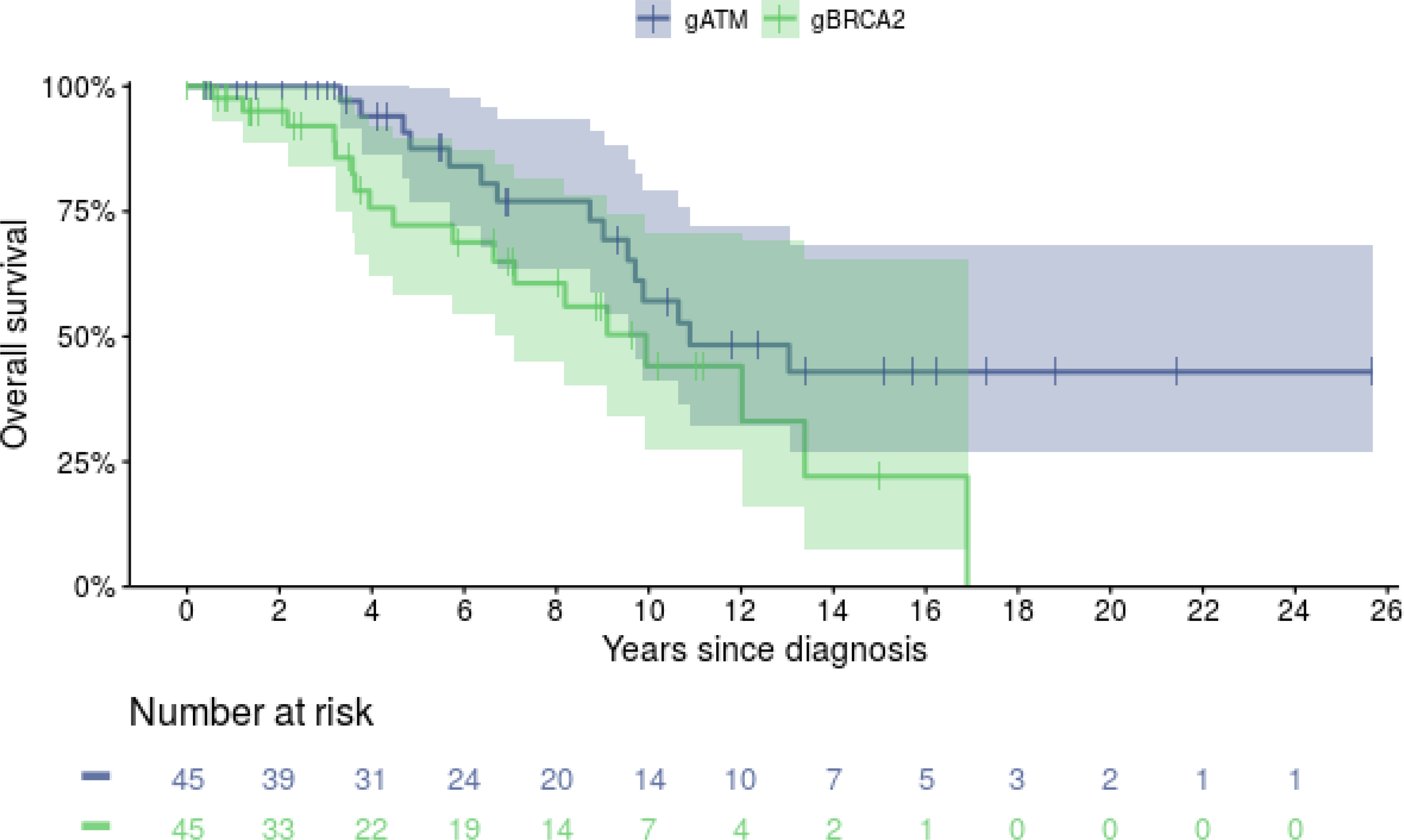
Overall Survival
Among the 28 patients in each cohort diagnosed with localized prostate cancer, median (95% CI) MFS was 5.7 years (5.1–11.1) vs 5.0 years (4.1–7.0, p=0.13) for the gATM and gBRCA2 cohorts, respectively (Supplemental Figure 4).
Discussion
Prostate tumors with alterations in DDR genes, particularly those in the HR repair pathway, represent a group of interest particularly in light of recent FDA approvals of the PARP inhibitors rucaparib and olaparib. While broadly grouped with gBRCA1/2 carriers, patients with prostate cancer in the setting of gATM mutations have not been characterized as an independent cohort. This study focuses on patients with prostate cancer and gATM mutations and describes responses to conventional and emerging systemic therapies with the aim of improving our understanding of therapeutic approaches for these patients.
Among men diagnosed with prostate cancer, those carrying gBRCA2 mutations are recognized to have a more aggressive phenotype (Supplemental Table 3).6 Another retrospective study, albeit with limited numbers of gATM carriers, found that gBRCA1/2 and gATM are associated with earlier age of death and shorter cancer-specific survival.9 Dedicated attention is warranted for gATM mutation carriers to further define specific prostate cancer risks and response to treatment.
Our data support the concept that while ATM-deficient prostate cancer may share features with BRCA2-deficient tumors, such as enrichment in the metastatic setting and response to non-targeted agents, they have distinct clinical characteristics. For example, we observed an attenuated response to PARPi in the gATM cohort compared to the gBRCA2 cohort, consistent with a retrospective study by Marshall et al., in which 0/8 patients with germline or somatic ATM mutations responded to PARPi.10 This difference in sensitivity to PARPi may partially be explained by different roles for ATM and BRCA2 in the HR repair pathway. ATM’s primary role is to recognize double-strand break and to activate downstream HR repair proteins, such as Chk2.21–23 Once activated, Chk2 has an overlapping function with ATM and phosphorylates the core HR repair pathway effectors, e.g., BRCA1, BRCA2.21 Chk2 can be activated by proteins other than ATM, such as DNA-dependent protein kinase, suggesting that HR repair pathway can be activated even in cells with loss of ATM function.22 These mechanistic differences in ATM and BRCA2 may account for observed differences in sensitivity to HR-targeted therapies between the two cohorts of our study. In addition, Neeb, et al., have recently reported that ATM protein expression as measured by ATM IHC is not perfectly overlapping with ATM mutations identified by NGS and suggest that protein expression may be another factor for treatment selection, potentially more predictive than DNA sequencing.7
Abiraterone, enzalutamide, and docetaxel have mechanisms of action largely independent of BRCA2 and ATM. A previous study reported that these therapies are similarly effective in gBRCA2 mutation carriers compared to non-carriers and gBRCA2 mutation carriers might benefit from upfront androgen-directed therapy rather than taxanes.3 We observed comparable PSA50 response rates in the two cohorts in our study. Thus, our data suggest that abiraterone, enzalutamide, and docetaxel should be offered to patients with mCRPC who carry gATM mutations.
Recent data suggest that platinum chemotherapy is effective in patients with BRCA2 mutations.24–26 In our study, patients with gATM mutations appeared to have a reduced response to platinum chemotherapy compared to the gBRCA2 cohort, but this comparison was not statistically significant owing to the small numbers. However, our observations are consistent with other studies reporting disappointing responses to platinum chemotherapy among ATM mutation carriers with prostate cancer.15,26 To date, reported numbers of patients with mCRPC and ATM alterations treated with platinum chemotherapy remain small and further studies are needed.
Our data highlight the need to explore new targeted therapies in patients with mCRPC and ATM alterations. Preclinical data suggest that ATM-deficient prostate tumors may be sensitive to ATR inhibitors, which, when combined with PARPi, result in apoptosis in PARPi-resistant prostate cancer cell lines.7,28 Several ongoing clinical trials are evaluating ATR inhibitors in prostate cancer (e.g., NCT04267939, NCT03787680).
We did not observe a significant difference in OS between the two cohorts, although this could be attributable to the limited numbers of patients and deaths and to different proportions of men receiving PARPi in the two groups. More men in gBRCA2 cohort received PARPi, which has a proven OS benefit for these patients.12,29
There are a number of important limitations to our study. First, this is a non-randomized retrospective study with a relatively small sample size. Second, the indications for germline testing in prostate cancer have been and remain evolving, so there are likely differences in practice from 2014 to 2019, as well as ascertainment biases. We attempted to minimize confounding effect by matching cases by year of testing; we acknowledge that men undergoing germline testing 2014–2019 will have been largely those with a strong family history of cancer and/or aggressive phenotype, although both gATM and gBRCA2 cohorts are likely to have been similarly affected. Third, the two cohorts are matched only for the year of testing, stage at diagnosis and treatment center; other patient characteristics were not matched. Fourth, the study does not include a control group of men without gATM and gBRCA2 mutations, which limits broader implications for treatment response. Fifth, the study does not include radiographic response assessment or confirmed PSA50 responses, limiting treatment response assessments. Clinical practices at different institutions may vary. For example, imaging was performed at clinician discretion without predefined standard intervals, which may have affected the time on treatment and MFS assessments. Finally, somatic alterations in other genes, mutation zygosity and protein expression were not fully addressed, but interference from clonal hematopoiesis of indeterminant potential would be less of an issue.30 Nevertheless, given the greater prevalence of gATM mutations31,32 in general population, compared to gBRCA2 mutations,33,34 we believe that specific examination of gATM remains important to this patient population.
Conclusions
Our data provide evidence that standard therapies may be similarly effective in gATM- and gBRCA2-associated prostate cancer, whereas PARPi appear less effective in gATM-associated prostate cancer. We did not find that abiraterone, enzalutamide, and docetaxel were less effective in patients with prostate cancer with gATM mutations and thus these agents should remain standard of care options for patients. This important subgroup of patients should continue to be studied and incorporated into clinical trials—especially those incorporating novel agents and combination strategies, e.g., ATR inhibitors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Agnes Gawne for data collection assistance. We gratefully acknowledge funding support from the Institute for Prostate Cancer Research, Prostate Cancer Foundation, Washington State Medical Oncology Society, Advancing Cancer Treatment, Patrick Walsh Prostate Cancer Research Fund, and NCI award numbers CA097186 (Pacific Northwest Prostate Cancer SPORE), P30 CA015704, P30 CA006973–52, R01 CA238384, R50 CA221836, and T32CA009515; PCF YI VALor award, CDMRP Awards W81XWH-17–2-0043, W81XWH-15–1-0430, PC170503, PC171001, DOD EIRA award W81XWH-17–1-0380 and the DOD Clinical Consortium award W81XWH-16-PCRP-CCRSA.
Funding statement: gratefully acknowledge funding support from the Institute for Prostate Cancer Research, the Prostate Cancer Foundation, the Washington State Medical Oncology Society, Patrick Walsh Prostate Cancer Research Fund, and NCI award numbers CA097186 (Pacific Northwest Prostate Cancer SPORE), P30 CA015704, R50 CA221836, and T32CA009515, CDMRP Awards PC170503, PC171001, 5P30 CA006973–52, R01 CA238384, and the DOD Clinical Consortium award W81XWH-16-PCRP-CCRSA.
Conflict of Interest Statement:
Alexandra O. Sokolova: nothing to disclose
Catherine H. Marshall: Consulting from Dendreon, Bayer, McGraw-Hill Publishing Company. Travel from Dava Oncology.
Rebeca Lozano: speaker fees from Roche, Janssen, Sanofi and Bayer. Travel support from Roche, Janssen, Sanofi and Astellas Pharma.
Roman Gulati: nothing to disclose
Elisa M. Ledet: nothing to disclose
Navonil De Sarkar: nothing to disclose
Petros Grivas: received grants from Bavarian Nordic, Bristol Myers Squibb, Clovis Oncology, Debiopharm, EMD Serono, GlaxoSmithKline, Immunomedics, Kure It Cancer Research, Merck & Co., Mirati Therapeutics, Pfizer, QED Therapeutics. Dr. Grivas has received consulting fees from AstraZeneca, Astellas Pharma, Bayer, Bristol Myers Squibb, Clovis Oncology, Dyania Health, Driver, EMD Serono, Exelixis, Foundation Medicine, Genentech/Roche, Genzyme, GlaxoSmithKline, Guardant Health, Heron Therapeutics, Immunomedics/Gilead, Infinity Pharmaceuticals, Janssen, Merck & Co., Mirati Therapeutics, Pfizer, QED Therapeutics, Regeneron Pharmaceuticals, Seattle Genetics, 4D Pharma PLC.Celestia S. Higano: Institutional research funding: Aptevo, Aragon, Astellas, AstraZeneca, Clovis, Dendreon, eFFECTOR Therapeutics, Emergent, Ferring, Genentech, Hoffman-Laroche, Medivation, Pfizer; Consulting, scientific advisory boards: Astellas, Bayer, Blue Earth Diagnositics, Clovis, Dendreon, Ferring, Hinova, Janssen, Merck, Orion, Pfizer, Tolmar, Carrick Therapeutics, Novartis, Genentech; Other: spouse holds stock and former officer of CTI Biopharma
Bruce Montgomery: Institutional research funding: Astellas, AstraZeneca, Beigene, Clovis, Janssen.
Peter S. Nelson: Consultant for Bristol Myers Squibb, Astellas, and Janssen Pharmaceuticals and received fees from UpToDate.
David Olmos: grant research support (to the institution) from AstraZeneca, Astellas, Bayer, Jansen. Advisory board and speaker fees from AstraZeneca, Astellas, Bayer, BioOncotech (Uncompensated), Clovis, Daiichi-Sankyo, Jansen, MSD, Pfizer. Travel support from AstraZeneca, Astellas, Bayer, F. Hoffman La Roche, Genetech, Ipsen, Jansen
Vadim Sokolov: nothing to disclose
Michael T. Schweizer: Paid consultant for Janssen and Resverlogix. Research funding from Bristol Myers Squibb, Merck, Immunomedics, Janssen, AstraZeneca, Pfizer, Madison Vaccines, Tmunity and Hoffman-La Roche
Todd A. Yezefski: consultant for Dendreon
Evan Y. Yu: Consulting – Advanced Accelerator Applications, Bayer, Clovis, Janssen, Merck. Research to institution – Bayer, Blue Earth, Dendreon, Merck, Taiho
Channing J. Paller: nothing to disclose
Oliver Sartor: Grant support to institution for AAA Pharma, AstraZeneca, Bayer, Endocyte, Progenics, Novartis, and Janssen, consulting fees from Astellas, Blue Earth Diagnostics, EMD Serono, Pfizer, Constellation, Dendreon, Bristol-Myers Squibb, Invitae, Merck, Innocrin, and Sotio, Consulting fees from AAA Pharma, AstraZeneca, Bayer, Endocyte, Progenics, Novartis, Janssen, Astellas, Blue Earth Diagnostics, EMD Serono, Pfizer, Constellation, Noria Therapeutics, Clovis, Myriad, Noxopharm, Point Biopharm, Tenebio, Theragnostics, Telix, Clarity Pharmaceuticals, and Fusion
Elena Castro: honoraria and consulting CLOVIS, Astra Zeneca, Astellas, Jansen, MSD, Roche, Pfizer; research funding Astra Zeneca, Bayer, Janssen, travel accommodations Astra Zeneca, Bayer, Janssen
Emmanuel S. Antonarakis: has served as a paid consultant/advisor for Invitae, Janssen, Pfizer, Sanofi, Dendreon, Merck, Bristol-Myers Squibb, AstraZeneca, Clovis, Bayer, Constellation, Eli Lilly and Amgen; and has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Bayer, Merck, Bristol-Myers Squibb, AstraZeneca, ESSA and Constellation.
Heather H. Cheng; Research funding to institution from Clovis, Janssen, Sanofi, Medivation/Astellas, Color Foundation; Consultancy to AstraZeneca; Royalties from UpToDate
Footnotes
Ethics approval statement: This study was approved by IRB board at each participating site.
Data availability statement:
Data available on request due to privacy/ethical restrictions
References
- 1.Pritchard CC et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 375, 443–453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicolosi P et al. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 5, 523–528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castro E et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 37, 490–503 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Cheng HH, Sokolova AO, Schaeffer EM, Small EJ & Higano CS Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. JNCCN 17, 515–521 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Page EC et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 76, 831–842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro E et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 31, 1748–1757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neeb A et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 0, (2020). [DOI] [PubMed] [Google Scholar]
- 8.Carter HB et al. Germline Mutations in ATM and BRCA1/2 Are Associated with Grade Reclassification in Men on Active Surveillance for Prostate Cancer. Eur. Urol. (2018) doi: 10.1016/j.eururo.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Na R et al. Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and are Associated with Early Age at Death. Eur. Urol. 71, 740–747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall CH et al. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 76, 452–458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abida W et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. JCO.20.01035 (2020) doi: 10.1200/JCO.20.01035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Bono J et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 382, 2091–2102 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Sokolova AO, Yu EY & Cheng HH Honing in on PARPi Response in Prostate Cancer: from HR Pathway to Gene-by-Gene Granularity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 26, 2439–2440 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Athie A et al. Targeting DNA Repair Defects for Precision Medicine in Prostate Cancer. Curr. Oncol. Rep. 21, 42 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Mota JM et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 355–366 (2020) doi: 10.1200/PO.19.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abida W et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 26, 2487–2496 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson AS et al. Impact of mutations in homologous recombination repair genes on treatment outcomes for metastatic castration resistant prostate cancer. PLoS ONE 15, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karbassi I et al. A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders. Hum. Mutat. 37, 127–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scher HI et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J. Clin. Oncol. 34, 1402–1418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Comprehensive Cancer Network. Prostate Cancer (Version 2.2020). https://www.nccn.org/professionals/physician_gls/pdf/prostate_blocks.pdf. Accessed November 10, 2020.
- 21.Zannini L, Delia D & Buscemi G CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 6, 442–457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J & Stern DF Regulation of CHK2 by DNA-dependent protein kinase. J. Biol. Chem. 280, 12041–12050 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Lavin MF, Delia D & Chessa L ATM and the DNA damage response. Workshop on ataxia-telangiectasia and related syndromes. EMBO Rep. 7, 154–160 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pomerantz MM et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 123, 3532–3539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng HH, Pritchard CC, Boyd T, Nelson PS & Montgomery B Biallelic inactivation of BRCA2 in platinum sensitive, metastatic castration resistant prostate cancer. Eur. Urol. 69, 992–995 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmid S et al. Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA Netw. Open 3, e2021692 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mota JM et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 355–366 (2020) doi: 10.1200/PO.19.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rafiei S et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 80, 2094–2100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hussain M et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 383, 2345–2357 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Sokolova AO et al. Complexities of Next-Generation Sequencing in Solid Tumors: Case Studies. J. Natl. Compr. Canc. Netw. 18, 1150–1155 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Broeks A et al. ATM-Heterozygous Germline Mutations Contribute to Breast Cancer–Susceptibility. Am. J. Hum. Genet. 66, 494–500 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall MJ et al. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. (Phila. Pa.) 14, 433–440 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxwell KN, Domchek SM, Nathanson KL & Robson ME Population Frequency of Germline BRCA1/2 Mutations. J. Clin. Oncol. 34, 4183–4185 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br. J. Cancer 83, 1301–1308 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request due to privacy/ethical restrictions