

Abstract
Background:
The integrated stress response (ISR) is an evolutionarily conserved process to cope with intracellular and extracellular disturbances. Myocardial infarction is a leading cause of death worldwide. Coronary artery reperfusion is the most effective means to mitigate cardiac damage of myocardial infarction, which however causes additional reperfusion injury. This study aimed to investigate the role of the ISR in myocardial ischemia/reperfusion (I/R).
Methods:
Cardiac-specific gain- and loss-of-function approaches for the ISR were employed in vivo. Myocardial I/R was achieved by the ligation of the cardiac left anterior descending artery for 45 minutes, followed by reperfusion for different times. Cardiac function was assessed by echocardiography. Additionally, cultured H9c2 cells, primary rat cardiomyocytes, and mouse embryonic fibroblasts were used to dissect underlying molecular mechanisms. Moreover, tandem mass tag (TMT) labeling and mass spectrometry was conducted to identify protein targets of the ISR. Pharmacological means were tested to manipulate the ISR for therapeutic exploration.
Results:
We show that the PERK/eIF2α axis of the ISR is strongly induced by I/R in cardiomyocytes in vitro and in vivo. We further reveal a physiological role of PERK/eIF2α signaling by showing that acute activation of PERK in the heart confers robust cardioprotection against reperfusion injury. In contrast, cardiac-specific deletion of PERK aggravates cardiac responses to reperfusion. Mechanistically, the ISR directly targets mitochondrial complexes via translational suppression. We identify NDUFAF2, an assembly factor of mitochondrial complex I, as a selective target of PERK. Overexpression of PERK suppresses the protein expression of NDUFAF2 while PERK inhibition causes an increase of NDUFAF2. Silencing of NDUFAF2 significantly rescues cardiac cell survival from PERK knockdown under I/R. Further, we show that activation of PERK/eIF2α signaling reduces mitochondrial complex-derived reactive oxygen species and improves cardiac cell survival in response to I/R. Moreover, pharmacological stimulation of the ISR protects the heart against reperfusion damage, even after the restoration of occluded coronary artery, highlighting a clinical relevance for myocardial infarction treatment.
Conclusions:
These studies suggest that the ISR improves cell survival and mitigate reperfusion damage by selectively suppressing mitochondrial protein synthesis and reducing oxidative stress in the heart.
Keywords: Integrated stress response, PERK, eIF2α, mitochondrial complex, unfolded protein response, reactive oxygen species, ischemic heart disease, ischemia/reperfusion
Introduction
The integrated stress response (ISR) is an adaptive process helping organisms accommodate various disturbances.1 A number of upstream stimuli that trigger the ISR share the same hallmark: phosphorylation at serine 51 of the α subunit of eukaryotic initiation factor 2 (eIF2α). This process relies on four distinct serine/threonine kinases: heme-regulated inhibitor kinase (HRI), induced by heme deficiency;2 interferon-induced double-stranded RNA-dependent eIF2α kinase (PKR), sensing viral infection;3 PKR-like endoplasmic reticulum resident kinase (PERK), activated by endoplasmic reticulum stress;4 general control non-derepressible 2 (GCN2), stimulated by amino acid deprivation.5 During translation initiation, eIF2 forms an active ternary complex with GTP and initiator methionyl transfer RNA (eIF2·GTP·tRNAiMet), which binds 40S ribosomal subunit to assemble 43S preinitiation complex. Upon joining of 60S ribosomal subunit, GTP is hydrolyzed to GDP and eIF2 is recycled and recharged for another round of initiation. During the ISR, eIF2α phosphorylation causes tight binding between eIF2B and eIF2, preventing the latter from reuse.6 As a consequence, general translation initiation is stalled. Although ISR rewiring provides acute survival advantage to resolve stress, long-term inhibition of protein synthesis may lead to detrimental consequences.7 Despite ample understanding of the upstream regulation of the ISR, downstream signaling remains incompletely defined.
Mitochondria are the powerhouse of living cells and also the major place for free radical production, with complex I and complex III as the main sites.8, 9 In the period of ischemia, low oxygen pressure and limited ADP availability cause mitochondrial damage by disrupting electron transport chain (ETC) function. Upon reoxygenation, restoration of oxygen leads to an increase in electron leakage from the ETC, generating superoxide and oxidants.10 Previous studies have shown that metformin, an anti-diabetic drug and an inhibitor of complex I, protects cardiac myocytes against reoxygenation-associated oxidative damage.11 Moreover, genetic deficiency of complex I through deletion of an assembly subunit NDUFS4 significantly blocks reactive oxygen species (ROS) generation and improves cell survival.12
Myocardial infraction is a common cause of heart failure and mortality.13 Sudden blockage of the coronary artery results in instantaneous limitation of blood flow to myocardium. Deficient oxygen and nutrient supply may eventually cause transmural cardiomyocyte death. Timely restoration of blood flow can reduce infarct size and markedly improve long-term clinical outcomes.14 However, reperfusion of the occluded artery per se leads to additional damage, which may account for up to 40% of the final infarction.15 Despite utmost clinical needs and extensive interest, our understanding of the so-called ischemia/reperfusion (I/R) injury remains incomplete and effective therapies are missing. A number of hypotheses have been proposed to explain reperfusion damage, including oxidative stress, calcium overload, metabolic dysfunction, and inflammation. Among them, over-production of ROS, particularly from mitochondria, may play a dominant role.16 On one hand, I/R is accompanied by abrupt changes in oxygen and nutrients, which are potent inducers of the ISR. On the other hand, mitochondria-derived ROS are a major culprit of I/R damage. These connections prompted us to investigate the role of the ISR in cardiac reperfusion injury, oxidative stress, and mitochondrial homeostasis.
Here we report that cardiac I/R suppresses protein synthesis, which is mediated by the PERK/eIF2α axis of the ISR. Moreover, stimulation of PERK is necessary and sufficient to protect the heart against reperfusion injury by selectively targeting mitochondrial complexes and suppressing mitochondria-derived ROS. Further, pharmacological activation of the ISR confers robust cardioprotection at the time of reperfusion. Taken together, our studies provide the first evidence to mechanistically couple the ISR and mitochondrial homeostasis, as well as uncovering a previously unrecognized role of PERK in ischemic heart disease.
Methods
All animal experiments were approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern Medical Center. Data, methods, and study materials are available from the corresponding author upon reasonable request. Additional details of experimental procedures are included in the Data Supplement.
Statistical analyses
Data are represented as mean±SEM. Two-tailed Student’s t test was performed to compare differences between two groups. For multiple group comparisons with one variable, one-way ANOVA was conducted, followed by either Tukey’s or Dunnett’s multiple comparisons test. For multiple group comparisons with more than two variables, two-way ANOVA was conducted, followed by either Tukey’s or Sidak’s multiple comparisons test. A P value of <0.05 was considered statistically significant. Statistical analyses were performed using Graphpad Prism software 8.3.0.
Results
Ischemia/reperfusion is associated with decreased protein synthesis.
To explore global changes under I/R, we conducted an unbiased assay at the protein level using H9c2 cardiomyocytes. We cultured H9c2 cells under either normoxia, simulated ischemia (sI, 95% N2/ 5% CO2) for 4 hours, or simulated ischemia for 4 hours followed by reperfusion for 30 minutes (sI/R). We harvested total proteins for tandem mass tag (TMT) labeling and mass spectrometry (MS) (Figure 1A).
Figure 1. Ischemia/reperfusion is associated with a decrease in protein synthesis.
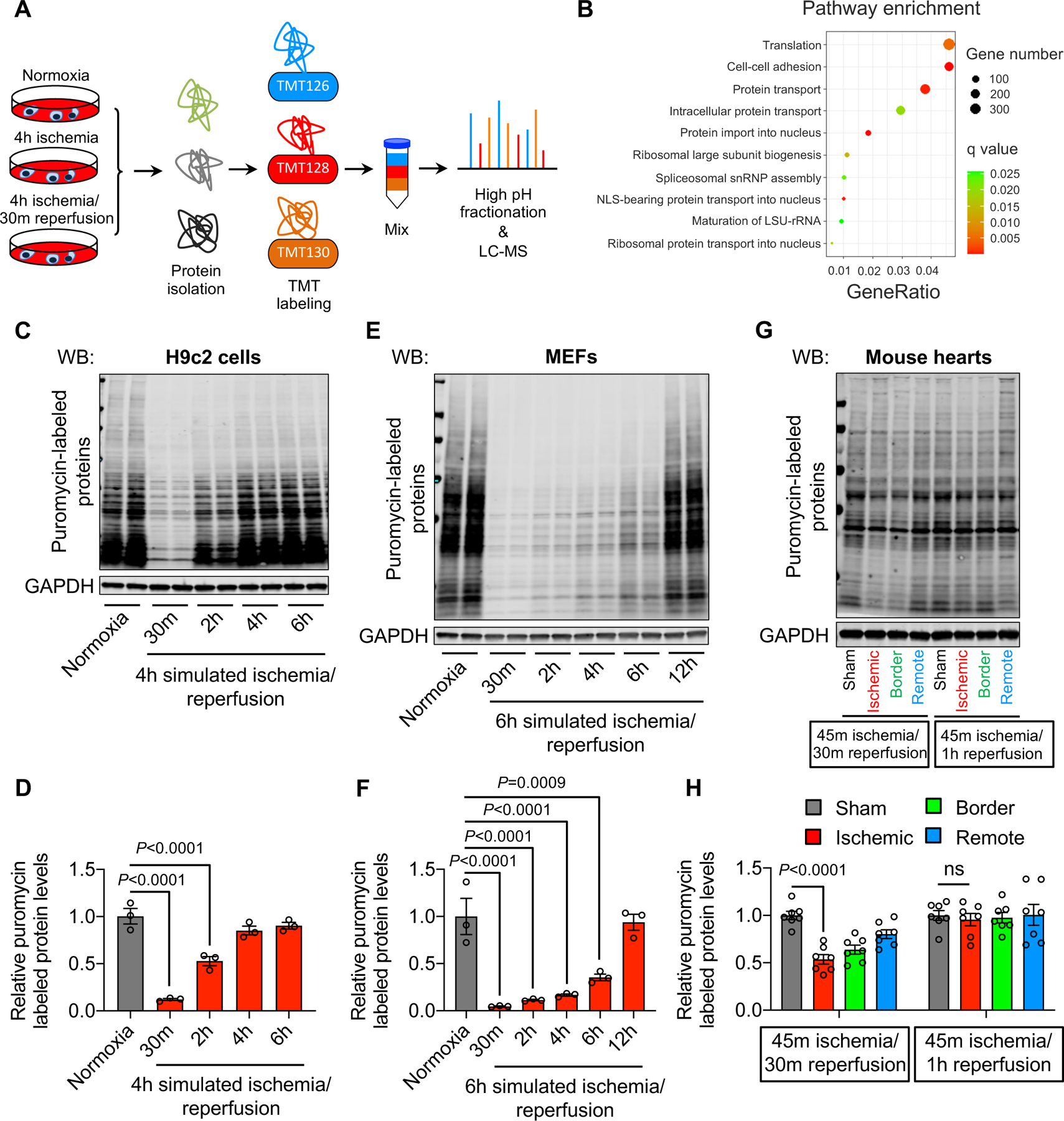
A. H9c2 cardiac myocytes were subjected to simulated ischemia (sI) for 4 hours or simulated ischemia for 4 hours, followed by reperfusion for 30 minutes (sI/R). Total proteins were isolated for tandem mass tag (TMT) labeling and mass spectrometry (MS).
B. Gene Ontology (GO) pathway analysis showed an enrichment of protein translation.
C. sI/R decreased protein synthesis. H9c2 cells were used for normoxia or sI/R. Puromycin was added 15 minutes before cell harvest. Total proteins were extracted to detect puromycin incorporation as a measure of protein synthesis.
D. Quantification of (C). n=3.
E. sI/R in mouse embryonic fibroblasts (MEFs) reduced protein synthesis. MEFs were subjected to either normoxia or sI/R. Puromycin was added 15 minutes before cell collection. Puromycin incorporation was detected by Western blotting.
F. Quantification of (E). n=3.
G. Ischemia/reperfusion (I/R) in vivo decreased protein synthesis in mouse hearts. Wild-type mice were subjected to cardiac ischemia for 45 minutes, followed by reperfusion for either 30 minutes or 1 hour. Puromycin was injected 30 minutes before heart collection. Cardiac tissues from different regions (ischemic, red; border, green; remote, blue) were separated and used for immunoblotting.
H. Quantification of (G). n=4.
One-way ANOVA was conducted, followed by Dunnett’s multiple comparisons test (D, F, H). Data are represented as mean±SEM. WB, Western blot and ns, not significant.
Out of 4,507 proteins detected, we found 991 upregulated proteins (sI and sI/R groups vs. normoxia, fold change >2) and 97 downregulated proteins (sI and sI/R groups vs. normoxia, fold change <0.5). Gene Ontology (GO) analysis showed that translation represented the top enriched pathway (Figure 1B, Figure IA in the Supplement), consistent with Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure IB in the Supplement). A comparison between sI and sI/R groups showed the same finding (Figure IC in the Supplement). These data combined suggest that the translation process is strongly affected by I/R in cardiomyocytes.
We went on to determine whether protein translation is increased or suppressed by I/R. We subjected H9c2 cells to sI for 4 hours, followed by reperfusion for various times. Approximately 15 minutes before cell harvesting, puromycin was added to culture media. Puromycin (0.5 μg/mL) was used to label newly synthesized proteins without affecting cell survival.17 Protein synthesis was then evaluated by immunoblotting for puromycin. We found that sI/R caused a significant decrease of protein synthesis in H9c2 cells (Figure 1C–D). A similar finding was discovered in mouse embryonic fibroblasts (MEFs) (Figure 1E–F). It is important to note that sI/R caused a more profound decrease of protein synthesis compared to sI only (Figure ID in the Supplement). To validate these results in vivo, we conducted I/R in mice. We ligated left anterior descending artery (LAD) for 45 minutes, followed by releasing the ligature for varying lengths of time (Figure IE in the Supplement). Puromycin was administrated 30 minutes prior to heart collection. The left ventricle was then split to three regions: ischemic, below the ligature; border, surrounding the ischemic area; remote, non-affected area. Consistent with the in vitro findings, we found that protein synthesis was significantly reduced in the ischemic area of the heart (Figures 1G–H). Importantly, this effect was transient, disappearing 1 hour after reperfusion. Taken together, these findings support that cardiac reperfusion is associated with reduced protein synthesis.
PERK/eIF2α signaling is activated by I/R.
Protein translation is regulated at multiple levels with translation initiation as the most important step. The integrated stress response (ISR), when activated by various stimuli, converges on the inhibition of translation initiation with an aim to restore cellular homeostasis. The key regulatory nexus is eIF2α, of which phosphorylation on serine 51 causes suppression of eIF2B and inhibition of translation initiation.
To evaluate whether the ISR may be involved in I/R-induced translation suppression, we first examined eIF2α phosphorylation. H9c2 cells were subjected to sI for 4 hours, followed by reperfusion for different times. We found that eIF2α phosphorylation was significantly and acutely increased (Figure 2A–B). These findings were later confirmed in primary neonatal rat ventricular myocytes (NRVMs) (Figure IIA in the Supplement). Importantly, cardiac reperfusion in vivo led to augmentation of eIF2α phosphorylation in the heart (Figure 2C–D), and this increase was transient, quickly returning to the basal level as early as 4 hours after reperfusion (Figure 2C–D, Figure IIB in the Supplement). This early and transient nature of eIF2α phosphorylation is consistent with the acute suppression of protein synthesis (Figure 1G–H), suggesting that eIF2α phosphorylation may be involved in the I/R-induced translation inhibition.
Figure 2. PERK-eIF2α signaling is activated by I/R.
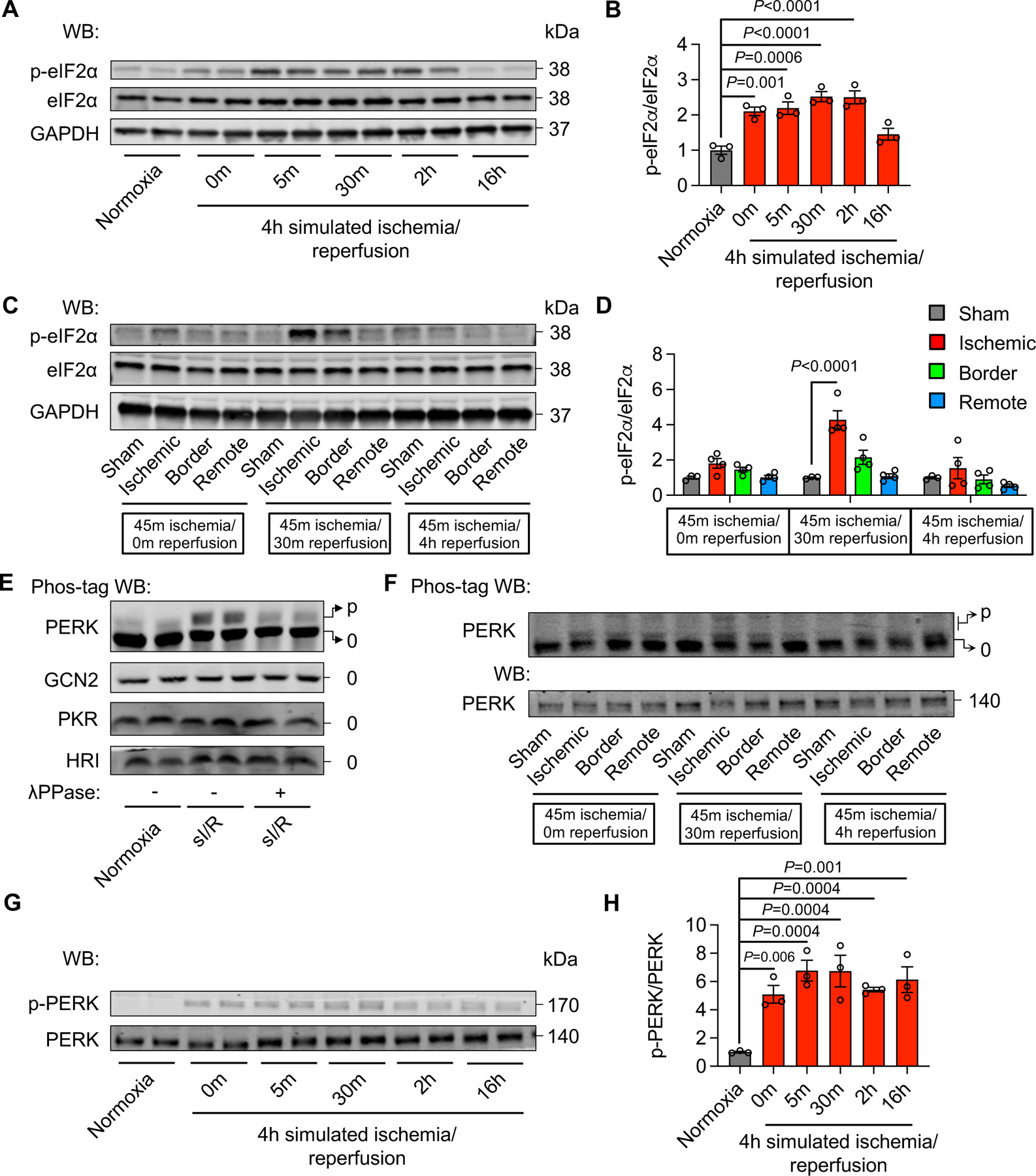
A. sI/R stimulated eIF2α phosphorylation in H9c2 cells.
B. Quantification of (A). n=3.
C. Cardiac I/R increased eIF2α phosphorylation.
D. Quantification of (C). n=3 to 4.
E. Phos-tag electrophoresis and Western blotting showed that PERK phosphorylation was elevated by sI/R in H9c2 cells, in contrast to other three eIF2α upstream kinases. p indicates phosphorylated protein; 0 indicate non-phosphorylated protein.
F. Cardiac I/R increased PERK phosphorylation.
G. sI/R in H9c2 cells stimulated PERK phosphorylation.
H. Quantification of (G). n=3.
One-way ANOVA was conducted, followed by Dunnett’s multiple comparisons test (B, D, H). Data are represented as mean±SEM. WB, Western blot and sI/R, simulated ischemia/reperfusion.
There are four different upstream kinases of eIF2α—HRI, PKR, PERK, and GCN2. We went on to investigate which one is responsible for eIF2α phosphorylation by I/R. We conducted Phos-tag labeling and λ phosphatase treatment to confirm phosphorylation status. We first validated this approach by using positive stimuli for individual kinases (Figure IIC in the supplement). Next, we found that sI/R in H9c2 cells only caused the phosphorylation of PERK (Figure 2E), which was later confirmed in the heart in vivo (Figure 2F, Figure IID in the supplement). Moreover, a time course study in H9c2 cells showed an acute increase of PERK phosphorylation (Figure 2G–H), which was in accordance with eIF2α phosphorylation (Figure 2A–B) and translation suppression (Figure 1C–D). Collectively, these data suggest that the PERK/eIF2α axis of the ISR is activated by I/R to inhibit protein translation.
PERK protects the heart from reperfusion injury in vivo.
To address the role of PERK in vivo, we generated a cardiac-specific knockout mouse model to selectively eliminate PERK in the heart (cKO) (Figure 3A, Figure IIIA-B in the Supplement). We subjected these mice, along with single transgenic αMHC-Cre and PERKfl/fl controls, to cardiac ischemia for 45 minutes, followed by reperfusion for 24 hours. We then harvested the heart for triphenyltetrazolium chloride (TTC) staining to visualize cardiac infarction (Figure 3B). No difference in area at risk/left ventricle (AAR/LV) was found, suggesting that I/R surgery was similarly conducted across all genotypes (Figure 3C). Importantly, PERK cKO exacerbated cardiac injury (Figure 3C, Figure IIIC in the Supplement) and caused a more severe decline in cardiac function after I/R (Figure 3D, Figure IIID-H in the Supplement). Taken together, these data suggest that PERK is required for the heart to mount an adaptive response during I/R.
Figure 3. PERK protects the heart from reperfusion injury in vivo.
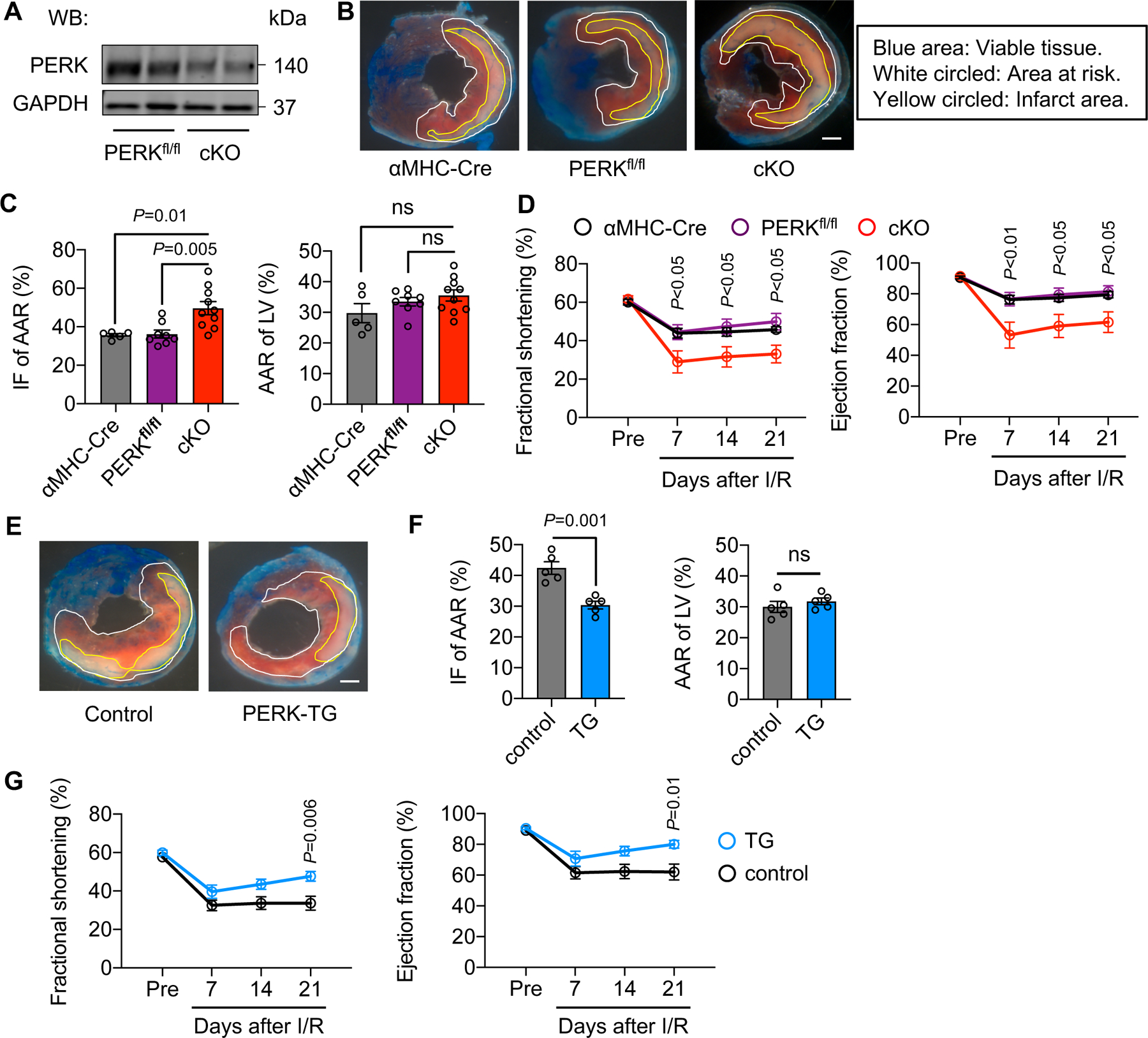
A. Validation of PERK knockout efficiency. Adult cardiomyocytes were isolated from PERKfl/fl and PERK cardiac-specific conditional knockout (cKO) mice, respectively, and used for immunoblotting.
B. PERK cKO mice and littermate controls were subjected to cardiac ischemia for 45 minutes, followed by reperfusion for 24 hours. The heart was then collected for triphenyltetrazolium chloride (TTC) staining to visualize viable tissue, area at risk, and infarct region. Different areas are circled. Scale bar: 1 mm.
C. PERK cKO led to an increase of infarct area/area at risk (IF/AAR). No difference in area at risk/left ventricle (AAR/LV) was found, suggesting that I/R surgeries were similarly conducted. n=5 to 10.
D. PERK cKO caused more severe reduction of cardiac function, as shown by decreases in fractional shortening and ejection fraction. n=5 to 7.
E. PERK transgenic (TG) mice and littermate controls were used for cardiac I/R. Dimer was administrated at the time of reperfusion. TTC staining was conducted. Control mice included αMHC-Cre only, CAG-FKBP-PERK only, and wild-type animals. Scale bar: 1 mm.
F. PERK activation conferred cardioprotection against I/R, as shown by a decrease of infarct size. Surgery was similarly conducted. n=5.
G. Cardiac function was significantly improved by PERK activation in the heart. n=6 to 9.
One-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (C). Two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (D) and Sidak’s multiple comparisons test (G). Unpaired Student’s t test was conducted (F). Data are represented as mean±SEM. WB, Western blot and ns, not significant.
We next asked whether PERK activation may protect the heart from reperfusion injury. To address this question, we generated a transgenic mouse model with PERK under the control of a universal CAG promoter.18 We placed a transcriptional/translational stop cassette between the CAG promoter and PERK, which was flanked by two loxP sites (Figure IVA in the Supplement). PERK, as a kinase, may not be activated by overexpression. Here, we engineered a fusion protein harboring the kinase domain of PERK and two FK506-binding domains (FKBP-PERK).19 We crossed FKBP-PERK transgenic mice with αMHC-Cre transgenic mice. In double transgenic mice (TG), FKBP-PERK fusion protein was only expressed in cardiomyocytes in the heart without baseline PERK activation (Figure IVB in the Supplement). We administrated a chemical dimerizer (dimer, AP20187) to trigger forced proximity of two FKBP-PERK fusion proteins and autophosphorylation of PERK ensued (Figure IVB in the Supplement). This approach allowed us to selectively activate PERK and downstream events only in cardiomyocytes, without confounding issues from other signaling branches of the ISR. We received multiple FKBP-PERK transgenic lines and confirmed that there was no basal leaky activation of PERK in the absence of dimer. In addition, dimer injection led to a significant increase of PERK phosphorylation and downstream eIF2α phosphorylation (Figure IVC in the Supplement). The FKBP-PERK transgenic line D was chosen for further analysis since eIF2α phosphorylation was significant and within the physiological level.
We subjected PERK TG mice and littermate controls to cardiac ischemia for 45 minutes. Dimer was administrated upon reperfusion. TTC staining was conducted 24 hours later (Figure 3E). Control mice included αMHC-Cre and FKBP-PERK single transgenic mice. I/R was similarly conducted for both groups. TG mice showed an improved response to I/R (Figure 3F, Figure IVD in the Supplement). Moreover, PERK overexpression and activation in the heart led to better recovery in cardiac function after I/R (Figure 3G, Figure IVE-I in the Supplement). These findings together suggest that PERK is necessary and sufficient to confer cardioprotection against reperfusion injury in vivo.
PERK/eIF2α signaling protects cardiomyocytes from reperfusion injury in vitro.
We next asked whether the protective effect of PERK/eIF2α is cardiomyocyte-autonomous. We silenced PERK (Figure 4A). Consistent with the protective role of PERK in vivo, knockdown of PERK led to more severe cell death under sI/R in both H9c2 cells and NRVMs (Figure 4B, Figure VA in the Supplement). The exacerbated cell death was further confirmed by SYTOX Green staining (Figure VB in the Supplement). We then used an independent approach to validate these findings. We treated H9c2 cells with a PERK inhibitor (PERKi) which suppressed PERK phosphorylation and eIF2α activation (Figure 4C). Importantly, PERKi treatment exacerbated sI/R-induced cell death (Figure 4D). Along this line, PERK−/− MEFs showed less cell survival compared to PERK+/+ MEFs under sI/R (Figure 4E, Figure VC in the Supplement).
Figure 4. PERK/eIF2α signaling protects cardiomyocytes from reperfusion injury in vitro.
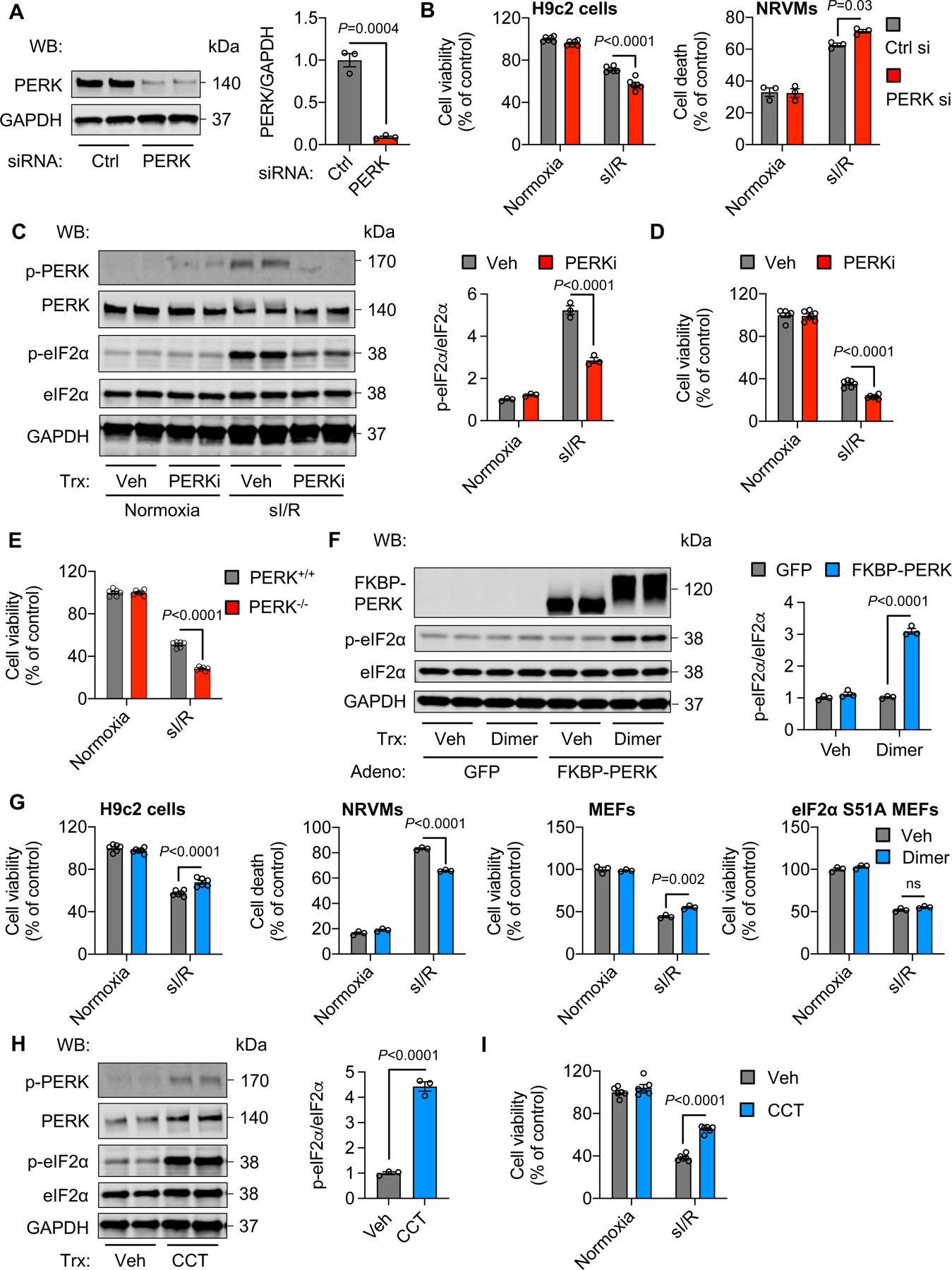
A. siRNA-mediated knockdown of PERK in H9c2 cells. n=3.
B. PERK silencing exacerbated sI/R-induced cell death in H9c2 cells and NRVMs, respectively. Note that cell viability was determined in H9c2 cells, whereas cell death was assessed in NRVMs. n=3 to 6.
C. PERK inhibitor (PERKi) suppressed PERK phosphorylation and downstream eIF2α activation in H9c2 cells. n=3.
D. PERK inhibition caused more severe cell death after sI/R. n=6.
E. PERK−/− MEFs showed a higher degree of cell death compared to PERK+/+ cells in response to sI/R. n=6.
F. Adenovirus-mediated PERK overexpression and activation stimulated PERK phosphorylation and downstream eIF2α signaling. n=3.
G. PERK activation improved cell survival after sI/R for H9c2 cells, NRVMs, and MEFs, respectively. No protective effect was found in MEFs with eIF2α serine 51 mutation (S51A). Note that the sI/R condition for wild-type MEFs was sI for 4 hours, followed by reperfusion for 2 hours, while the sI/R condition for S51A mutant MEFs was sI for 2 hours, followed by reperfusion for 1 hour since S51A mutant MEFs were more susceptible to reperfusion damage. n=3 to 6. ns, not significant.
H. CCT, a PERK activator, stimulated PERK and increased eIF2α phosphorylation in H9c2 cells. n=3.
I. Cell viability was improved by CCT treatment after sI/R. n=6.
Unpaired Student’s t test was conducted (A, H). Two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (B-G, I). Data are represented as mean±SEM. WB, Western blot; NRVMs, neonatal rat ventricular myocytes; sI/R, simulated ischemia/reperfusion, Ctrl, control; si, small interfering; Veh, vehicle; and MEFs, mouse embryonic fibroblasts.
We next took gain-of-function approaches to further examine the role of PERK in response to reperfusion. We cloned the FKBP-PERK fragment into a mammalian expression plasmid and transfected to HEK293 cells to validate the functionality of the FKBP-PERK fusion protein in vitro. We treated the cells with dimer and detected PERK and eIF2α phosphorylation (Figure VD in the Supplement). We then generated adenoviruses expressing the fusion protein FKBP-PERK and used dimer to activate PERK (Figure 4F, Figure VE in the Supplement). PERK activation was protective against sI/R-induced cell death in H9c2 cells, NRVMs, and MEFs, respectively (Figure 4G). SYTOX Green staining further confirmed the protective role of PERK (Figure VB in the Supplement). Importantly, MEFs with eIF2α phosphorylation site serine 51 mutation did not show a beneficial effect under PERK activation (Figure 4G). In addition, treatment with a PERK kinase activator (CCT) conferred protection against reperfusion-induced cell death (Figure 4H–I). Taken together, these data support that the PERK/eIF2α axis of the ISR protects cells from reperfusion injury in vitro.
Activation of PERK/eIF2α signaling suppresses protein synthesis.
We have shown that cardiac I/R is associated with inhibition of protein synthesis, which is caused by the PERK/eIF2α axis of the ISR. Moreover, PERK is necessary and sufficient to protect the heart from reperfusion injury. We next asked whether suppression of protein synthesis is the underlying mechanism of PERK-mediated cardioprotection.
In PERK TG mice, we administrated dimer to activated PERK and its downstream target eIF2α (Figure VIA-B in the Supplement). We found that activation of PERK caused a decrease in protein synthesis in vivo (Figure 5A). Further, PERK activation in H9c2 cells led to translation inhibition (Figure 5B, Figure VIC-D in the Supplement). Additionally, dimer-induced activation of PERK in NRVMs was also associated with a decrease of protein synthesis (Figure VIE-H in the Supplement). Accordingly, transgenic overexpression and activation of PERK in the heart further reduced translation in comparison to control mice after reperfusion, whereas PERK cKO prevented the reperfusion-induced decrease of protein synthesis (Figure 5C). To further evaluate the role of translation inhibition in cardiomyocyte response under reperfusion, we activated PERK in H9c2 cells and suppressed general protein synthesis by cycloheximide. We found that PERK activation and cycloheximide treatment protected the cells from sI/R injury, respectively. However, no additive effect was observed (Figure VII in the Supplement). We next activated PERK/eIF2α signaling in H9c2 cells and suppressed eIF2α phosphorylation with ISRIB, a small-molecule ISR inhibitor.20, 21 Importantly, ISRIB treatment diminished the PERK-induced increase of eIF2α phosphorylation and protection against reperfusion injury (Figure 5D–E, Figure VIJ in the Supplement). Similarly, PERK cKO hearts showed a decrease in eIF2α phosphorylation after I/R (Figure VIK in the Supplement). These findings collectively suggest that the activation of PERK/eIF2α signaling protects cardiomyocytes from I/R, which is mediated by translation inhibition.
Figure 5. Activation of PERK/eIF2α signaling inhibits protein synthesis.
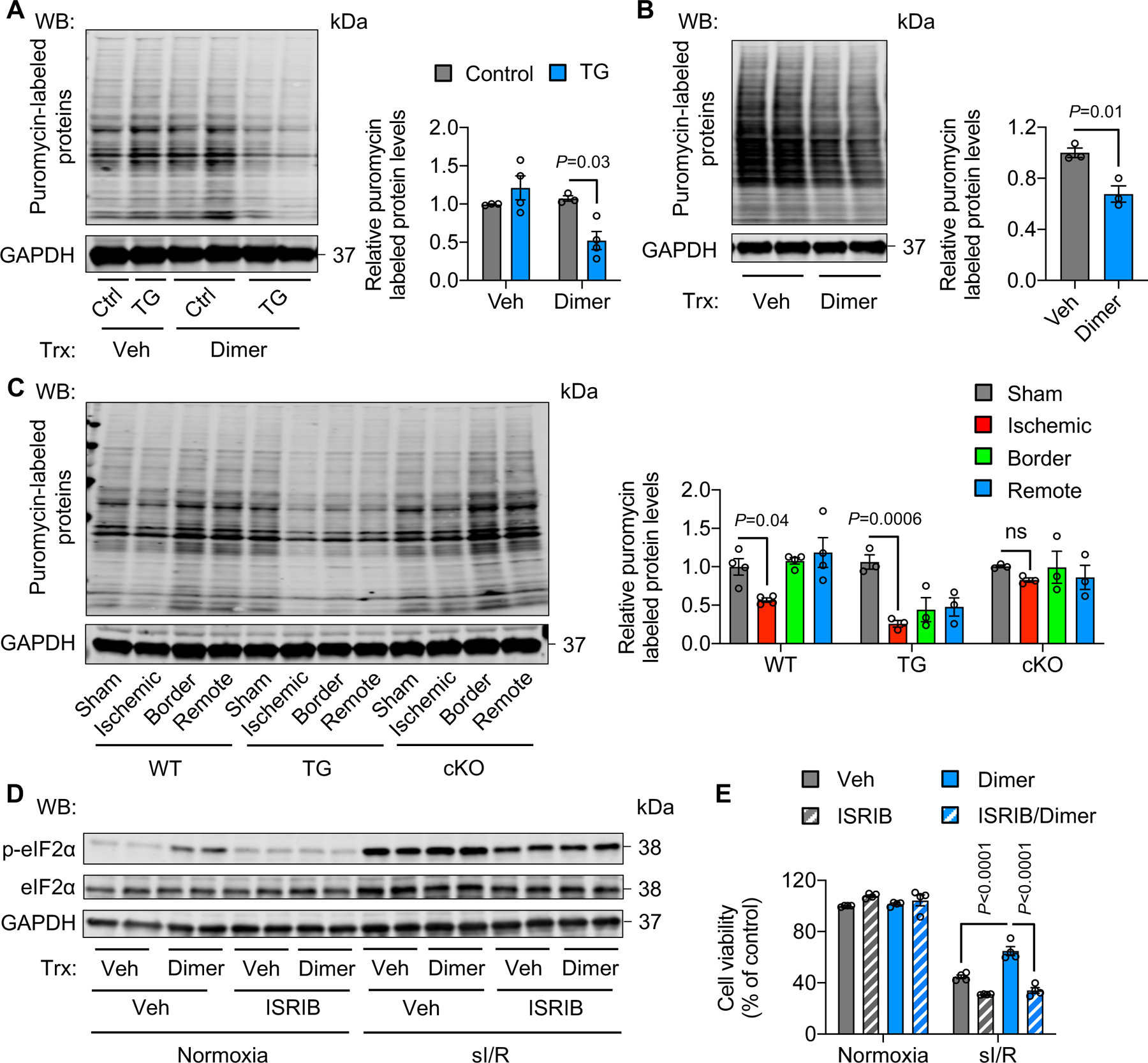
A. PERK activation by dimer in PERK TG mice decreased protein synthesis. Dimer was injected to TG mice for 2 hours. Approximately 30 minutes before heart collection, puromycin was administrated to label newly synthesized proteins. n=3.
B. Activation of PERK in H9c2 cells reduced protein synthesis. Adenoviruses expressing FKBP-PERK were used to infect H9c2 cells, which were then treated with dimer for 30 minutes. Puromycin was added to label newly synthesized proteins 15 minutes before cell harvest. n=3.
C. PERK TG mice showed a lower level of protein synthesis after I/R in comparison to wild-type controls. In contrast, PERK cKO prevented the decrease of protein synthesis. n=3 to 4.
D. ISRIB, an eIF2α phosphorylation inhibitor, suppressed eIF2α phosphorylation in PERK-activated H9c2 cells.
E. ISRIB treatment abolished the protective effect of PERK under sI/R. n=3.
Two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (A, E). Unpaired Student’s t test was conducted (B). One-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (C). Data are represented as mean±SEM. WB, Western blot; WT, wild-type; TG, transgenic; cKO, cardiac-specific knockout; not significant; Veh, vehicle; and sI/R, simulated ischemia/reperfusion.
PERK suppresses protein synthesis of mitochondrial complexes.
A conventional view about ISR-mediated translation regulation is global initiation inhibition. Here, we asked whether the activation of PERK/eIF2α signaling by I/R may show selectivity of targeting. We overexpressed and activated PERK in H9c2 cells. We subjected the cells to sI for 4 hours, followed by different times of reperfusion, i.e. 30 minutes and 8 hours, respectively. We isolated total proteins and labeled them with TMT for MS (Figure VIIA in the Supplement). We confirmed that PERK was activated and protein synthesis was suppressed (Figure VIIB-C in the Supplement). Importantly, GO analysis showed an enrichment of mitochondrial proteins between normoxia and sI/R (Figure 6A). Further analysis highlighted mitochondrial complex components being selectively targeted (Figure VIID in the Supplement). In contrast, RNA-seq analysis did not identify mitochondria as a significantly affected cellular component (Figure VIIE-F in the Supplement), highlighting the importance of translation regulation by PERK. Consistently, mitochondrial complex I activity was significantly decreased in PERK transgenic hearts after I/R (Figure VIIIA-B in the Supplement). In H9c2 cells, PERK activation reduced key components of individual mitochondrial complexes (Figure VIIIC in the Supplement). Mitochondria are one of the major sites of reactive oxygen species (ROS) production, which is a central player of I/R injury in the heart.22 Previous studies have shown that inhibition of mitochondrial complex I reduces ROS and protects cardiomyocytes from reperfusion injury.12, 23 Next, we asked whether the suppression of mitochondrial complex protein translation may be an underlying mechanism of PERK/eIF2α signaling-mediated cardioprotection against reperfusion damage.
Figure 6. PERK activation suppresses the synthesis of mitochondrial complex proteins.
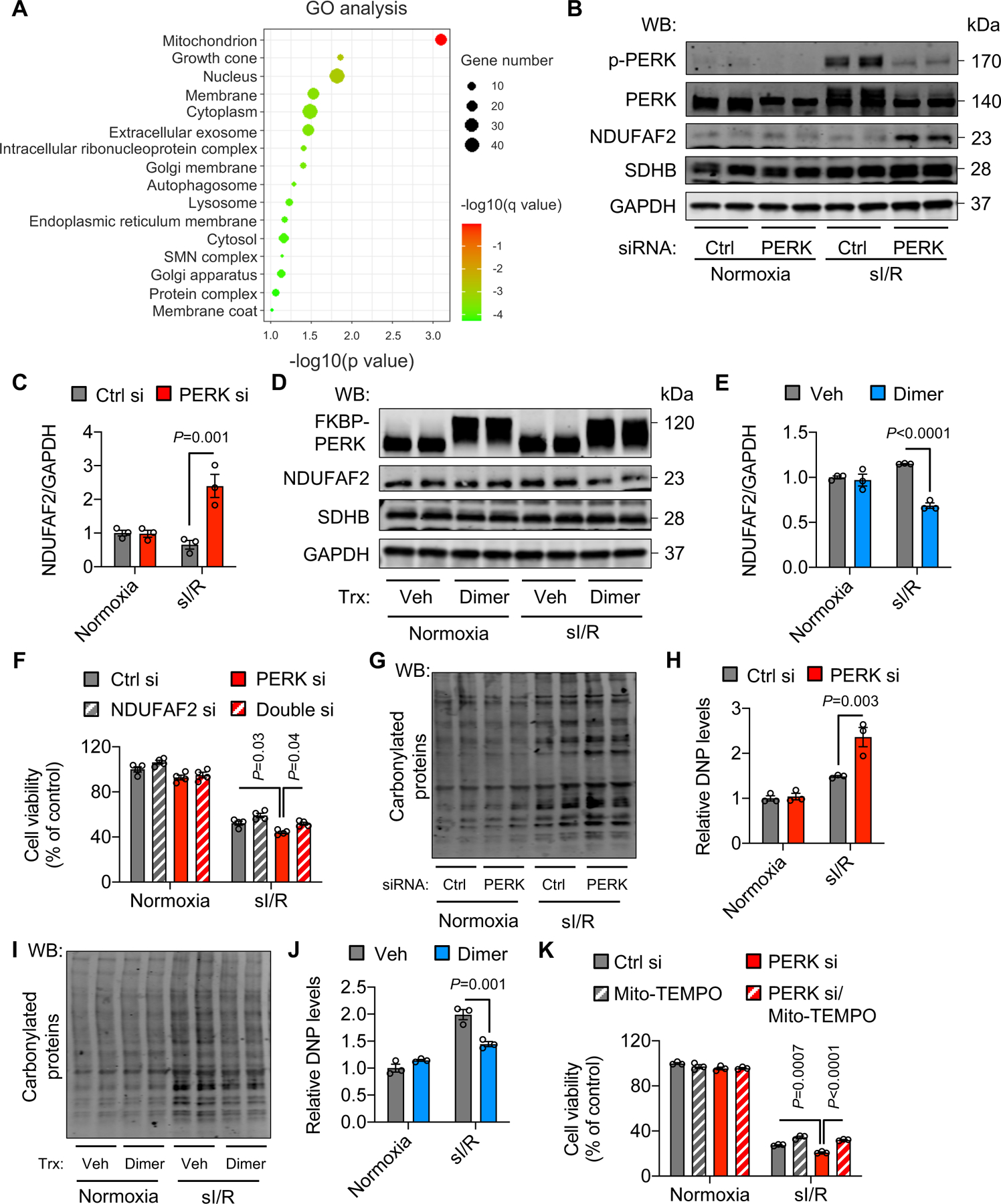
A. PERK activation in H9c2 cells was stimulated by dimer treatment. Thirty minutes after sI/R, total proteins were extracted for TMT-labeled MS. GO analysis identified mitochondrion as the most enriched cellular component.
B. PERK silencing increased NDUFAF2 expression. In contrast, SDHB, a component of complex II, was not affected.
C. Quantification of (B). n=3.
D. Activation of PERK decreased NDUFAF2 protein expression.
E. Quantification of (D). n=3.
F. NDUFAF2 silencing rescued cell death from PERK knockdown after sI/R. n=4.
G. PERK silencing led to an increase of reactive oxygen species (ROS) after sI/R. Protein carbonylation assay was conducted to determine relative ROS modifications.
H. Quantification of (G). n=3.
I. PERK activation decreased protein carbonylation.
J. Quantification of (I). n=3.
K. Mito-TEMPO, a mitochondrial superoxide scavenger, rescued cell death from PERK knockdown. n=3.
Two-way ANOVA was conducted for, followed by Tukey’s multiple comparisons test (C, E, F, H, J, K). Data are represented as mean±SEM. WB, Western blot; sI/R, simulated ischemia/reperfusion; Trx, treatment; Veh, vehicle; si, small interfering; Ctrl, control; and DNP, dinitrophenyl hydrazone.
We first validated the inhibition of mitochondrial complex protein expression by PERK. We chose NDUFAF2, which displayed the highest level of suppression by PERK (Figure VIID in the Supplement). Although sI alone did not increase the turnover rate of NDUFAF2, sI/R strongly enhanced NDUFAF2 turnover (Figure IXA-B in the Supplement), highlighting reperfusion-specific regulation of NDUFAF2. NDUFAF2 is a complex I assembly factor. Mutation of NDUFAF2 causes deficiency of complex I activity.24 We silenced PERK in H9c2 cells and then conducted sI/R. We found that knockdown of PERK led to an increase of NDUFAF2 expression after sI/R (Figure 6B–C), consistent with the notion that PERK inhibits NDUFAF2 expression. In contrast, PERK activation caused a significant decrease of NDUFAF2 protein level (Figure 6D–E). More importantly, we found that NDUFAF2 knockdown rescued the deterioration of cell survival from PERK silencing (Figure 6F, Figure XA in the Supplement). In addition, inhibition of mitochondrial complex I activity by metformin improved cell survival under sI/R (Figure XB in the Supplement), lending further support for a key role of mitochondrial complex I in the pathophysiology of reperfusion injury.
Consistent with an essential contribution of mitochondria to ROS production and suppression of mitochondrial activity by PERK, silencing of PERK caused an increase of ROS, as shown by the elevation of protein carbonylation (Figure 6G–H), whereas overexpression and activation of PERK reduced ROS levels (Figure 6I–J). These findings were further confirmed by ROS fluorescent staining under the conditions of PERK silencing and PERK activation, respectively (Figure XC in the Supplement). Importantly, treatment of Mito-TEMPO, a mitochondrial superoxide scavenger, significantly alleviated PERK silencing-induced cell death (Figure 6K). Furthermore, mitochondrial activity, as measured by oxygen consumption rate (OCR), was increased in PERK knockdown cells and reduced by PERK activation after sI/R (Figure XD in the Supplement). In aggregate, these findings strongly suggest that the PERK/eIF2α axis of the ISR selectively targets mitochondrial complexes to decelerate electron transfer, reduce ROS production, and protect cardiomyocytes from reperfusion damage.
ISR activation protects the heart from reperfusion injury in vivo.
We have shown that genetic activation of PERK protects the heart from I/R. Next, we asked whether this pathway may be manipulated pharmacologically to confer cardioprotection in vivo. Salubrinal (Salu) is a selective inhibitor of eIF2α phosphatase complexes.25 We first tested whether Salu can prolong the phosphorylation status of eIF2α under I/R in vivo.
We injected Salu right after reperfusion and harvested the heart at different times, including 30 minutes, 2 hours, and 4 hours post reperfusion. We also tested a condition of one injection 6 hours after reperfusion and collected the heart 30 minutes later. Immunoblotting showed that Salu administration upon reperfusion maintained a higher level of eIF2α phosphorylation up to 4 hours (Figure 7A). In contrast, Salu injection at 6 hours post reperfusion did not affect eIF2α phosphorylation, likely due to complete diminishing of eIF2α phosphorylation at this late time point.
Figure 7. ISR activation protects the heart from reperfusion injury in vivo.
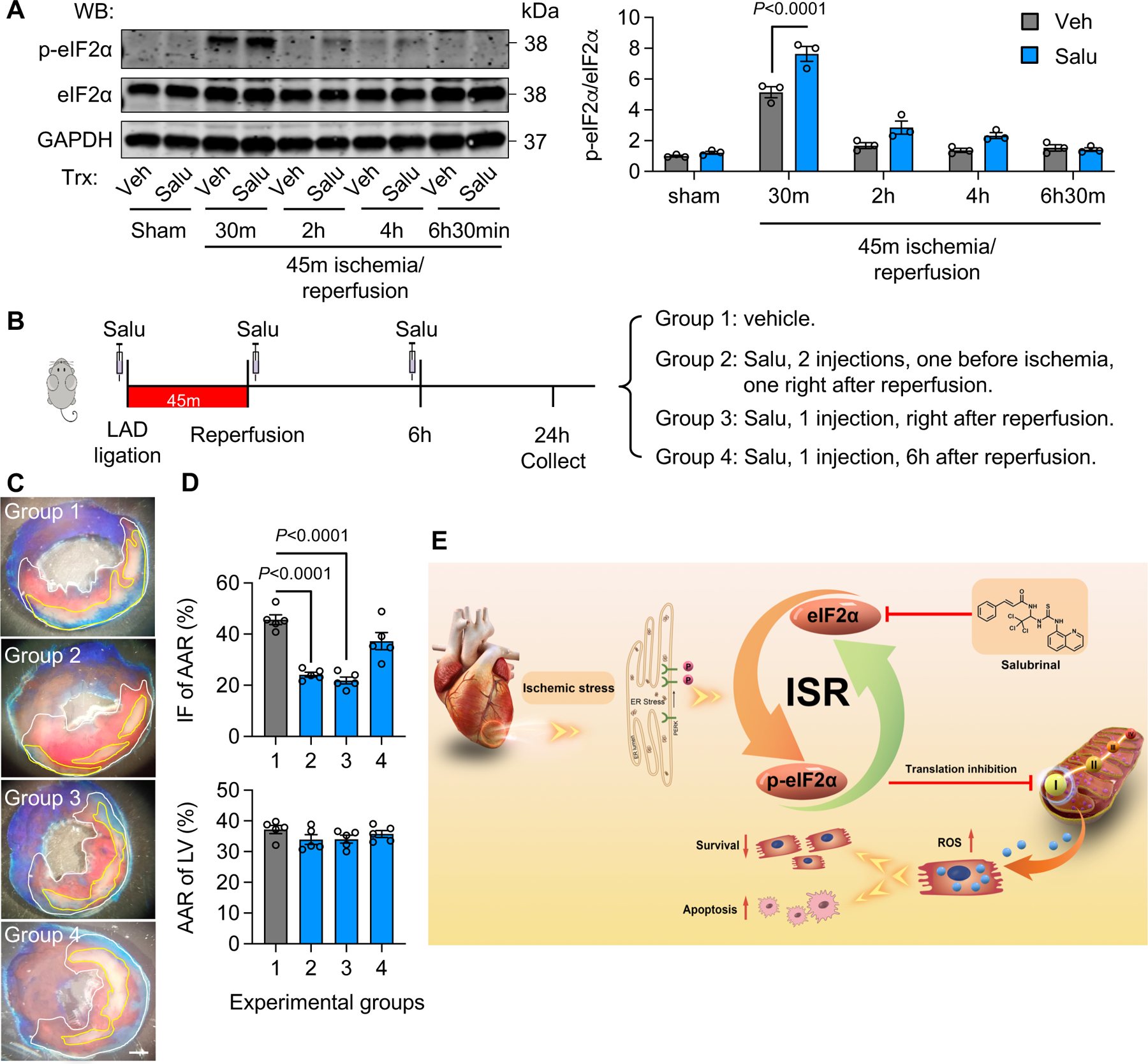
A. Salu (salubrinal) injection enhanced eIF2α phosphorylation in vivo. While the injection right after reperfusion augmented eIF2α phosphorylation, Salu administration 6 hours post reperfusion had no effect. n=3.
B. Schematic of experimental design.
C. Salu injection protected the heart from reperfusion injury. Salu was injected for either two times (one before ligation and one right after reperfusion, group 2), one time right after reperfusion (group 3), or one time 6 hours post reperfusion (group 4). TTC staining was conducted 24 hours later to determine infarct area. Scale bar: 1 mm.
D. Both two injections (group 2) and one injection right after reperfusion (group 3) protected the heart against I/R damage. In contrast, one injection 6 hours post reperfusion (group 4) was not significantly effective. n=5.
E. The working model. Cardiac I/R stimulates the PERK/eIF2α axis of the ISR and in turn attenuates mitochondrial complex protein translation. As a result, oxidative stress from mitochondria is decreased and cardiac cell survival is improved. Salubrinal, an activator of the ISR, stimulates the ISR and protects the heart against reperfusion injury.
One-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (A, D). Data are represented as mean±SEM. WB, Western blot and Veh, vehicle.
We next asked whether Salu administration may protect the heart from reperfusion injury. Here, we tested 4 different treatment schemes (Figure 7B). Group 1 received vehicle treatment. Group 2 received two Salu injections, one before cardiac ischemia and one right after reperfusion. Group 3 only received one injection upon reperfusion, while group 4 was injected once 6 hours post reperfusion. We collected the heart 24 hours after reperfusion and conducted TTC staining to determine cardiac damage (Figure 7C). The two-injection group 2 showed significant protection against reperfusion damage (Figure 7D). Importantly, even one injection at reperfusion (group 3) elicited a similar beneficial effect (Figure 7D). Consistent with the inability to maintain eIF2α phosphorylation, one injection 6 hours post reperfusion (group 4) did not show cardioprotection. Taken together, these data suggest that pharmacological stimulation of the ISR protects the heart from reperfusion damage upon restoration of coronary blood flow (Figure 7E), highlighting a relevance in clinical use to tackle ischemic heart disease.
Discussion
Prompt restoration of coronary blood flow is the most effective means to mitigate cardiac injury and salvage viable myocardium under myocardial infarction. Reperfusion of occluded coronary arteries however causes additional damage, which contributes significantly to the final infarction. Currently, no effective therapeutics are available to curb reperfusion injury. We show that protein synthesis is suppressed by reperfusion in the heart. The PERK/eIF2α axis of the ISR is induced to inhibit translation initiation. Both gain- and loss-of-function experiments highlight a requisite role of PERK in protecting the heart against reperfusion damage in vivo. Mechanistically, PERK/eIF2α signaling selectively targets mitochondrial complexes and thereby reduces ROS production from mitochondria. More importantly, pharmacological activation of the PERK/eIF2α axis of the ISR, even at the moment of restoration of coronary blood flow, protects the heart from reperfusion injury, providing a new approach for therapeutic exploration to counteract ischemic heart disease.
The ISR in the heart
In response to extrinsic and intrinsic disturbances, the ISR is activated to fine-tune protein synthesis.7 Four upstream kinases converge on the phosphorylation of eIF2α and as a result, global protein translation is attenuated. On the other hand, a number of targets of the ISR are selectively induced to enhance protein-folding capacity, detoxify ROS, etc. Depending on intensity and duration, persistent ISR may cause cell death to eliminate terminally injured cells. Previous studies have shown that the ISR exerts critical roles in cardiac homeostasis.26 Wang et al. found that PKR is significantly increased in failing hearts.27 PKR deficiency in vivo protects the heart from hemodynamic stress. Likewise, GCN2 knockout leads to improvements in cardiac function after pressure overload.28 In contrast, cardiac specific deletion of PERK exacerbates cardiac response under pressure overload.29 Despite these discrepancies, pharmacological stimulation of the ISR by salubrinal protects pressure overload-induced hypertrophy and pathological cardiac remodeling.30 These findings collectively suggest that the ISR is activated in the heart by hemodynamic stress and highlight that the PERK/eIF2α axis of the ISR may exert distinct actions in comparison to other three kinase cascades.
Myocardial I/R is a different stressor compared to chronic pressure overload. Abrupt changes in oxygen pressure and nutrient availability in ischemic heart disease lead to sudden alterations in ROS levels, calcium homeostasis, amino acid concentrations, etc. Most, if not all, of these processes are potent inducers of the ISR. However, our understanding of the ISR in the heart in response to I/R remains elusive. We found that eIF2α phosphorylation is initiated during ischemia only by PERK and that this event is maintained at a high level throughout the early reperfusion period. Interesting, Kumar et al. reported that brain I/R also causes an increase of eIF2α phosphorylation, which is mostly mediated by PERK, not HRI or GCN2.31 The activation of PERK/eIF2α signaling attenuates protein translation at ischemia, a process further enhanced upon reperfusion. These findings not only reveal an essential role of the PERK/eIF2α axis of the ISR in cardiac proteostasis, but also provide a promising target to curb reperfusion injury.
Mitochondrial ROS production in cardiac I/R
Mitochondria are the major organelle for ROS generation. Previous studies have identified a number of distinct sites in mitochondria from which ROS are formed. Among them, complex I contributes the most. NDUFS4 is a complex I assembly factor. Cardiac specific knockout of NDUFS4 accelerates heart failure development under chronic pressure overload, likely due to the imbalance of NAD+/NADH and diminished SIRT3 activity.23 However, cardiomyocyte-restricted deletion of NDUFS4 suppresses ROS production from complex I and improves cardiomyocyte survival under I/R.12 These findings suggest a critical role of mitochondrial complex I in ROS generation in I/R and highlight it as a promising target for intervention.
Accumulating evidence indicates that selective inhibition of complex I ROS production leads to beneficial effects in the heart under I/R. Early studies by Lesnefsky et al. showed that rotenone, an irreversible inhibitor of complex I, attenuates cardiac injury from ischemia.32 Further work strengthened the role of complex I in ischemic injury in the heart by using a reversible complex I inhibitor (amobarbital).33 Recent studies showed that rotenone treatment during I/R is sufficient to protect cardiomyocytes, highlighting a critical role of complex I-derived ROS in reperfusion injury.12 Consistent with these findings, metformin, a complex I inhibitor, provides significant cardioprotection even when administrated at the time of reperfusion.34 Recently, Chouchani et al. found that succinate accumulated during ischemia stimulates extensive ROS production by reverse electron transport at complex I.35 Moreover, Ainscow and colleagues screened and identified specific inhibitors against reverse electron transport at site IQ of complex I. Treatment with these inhibitors reduces reperfusion injury without affecting oxidative phosphorylation.36 These data suggest reverse electron transport in complex I may be a major source of ROS production. However, other studies indicate that electron transport at both directions may contribute. Zhang et al. found that dimethyl succinate, a cell-permeable derivative of succinate, when used solely, causes less cell death compared with glucose.12 Furthermore, rotenone, a chemical conferring cardioprotection, inhibits both forward and reverse electron transport. Collectively, mitochondrial complex I represents a druggable target to alleviate I/R injury likely with contributions from both forward and reverse electron transport.
Crosstalk between the ISR and mitochondrial homeostasis
Previous studies have shown that inhibition of protein synthesis protects the heart from I/R injury. In vivo treatment with rapamycin protects the heart from I/R.37 This finding has been recapitulated in vitro using cultured adult cardiomyocytes.37 In addition, in an ex vivo study, treatment with cycloheximide, a protein synthesis inhibitor, improves cardiac recovery from I/R, and this effect is more prominent when cycloheximide is administrated during reperfusion.38
The central event of the ISR is suppression of protein translation. Our conclusion regarding the cardioprotective role of the PERK/eIF2α axis of the ISR is consistent with previous findings. Importantly, we showed that the PERK/eIF2α pathway selectively targets components of mitochondrial complexes. Interestingly, mitochondrial unfolded protein response is not affected by PERK/eIF2α signaling (data not shown). As a result, ROS production from mitochondrial complexes is decreased. In addition, inhibition of mitochondrial complex protein synthesis may confer other protective benefits. By decreasing mitochondrial complex protein synthesis, more ATP may be preserved for other vital processes. Taken together, PERK/eIF2α-mediated protein translation inhibition may protect the heart from reperfusion injury by reducing ROS production and decreasing ATP demand on mitochondria.
The ISR and the unfolded protein response in cardiac I/R
The PERK/eIF2α axis of the ISR is also one of the three signaling transducers of the unfolded protein response (UPR).39 Previous studies have shown that the UPR plays an essential role in protecting the heart against I/R.40, 41 Jin et al. found that the deletion of ATF6, a branch of the UPR, exacerbates cardiac response under I/R.42 On the other hand, pharmacological activation of ATF6 protects the heart against reperfusion injury.43 Importantly, this protection is not limited to the heart, but also includes the brain, liver, and kidney under I/R conditions. Likewise, we found that the XBP1s signaling of the UPR is necessary and sufficient to protect the heart from reperfusion injury.44 Consistently, pre-conditioning with an ER stress inducer reduces cardiac I/R damage in diabetic rats.45 Our findings about the cardioprotection by PERK supports a common theme that acute activation of the ISR and the UPR exerts a beneficial effect under I/R conditions.
There are several limitations in our study. First of all, the sample sizes of several experiments are small and sample homogeneity of variance may not be uniform across different groups. In addition, although acute stimulation of PERK in mice is cardioprotective, it remains to be determined whether prolonged activation is detrimental or beneficial. Recently, Molkentin and colleagues showed that constitutive overexpression and activation of PERK in the heart promotes cardiac atrophy and lethality.46 Therefore, more work is warranted to dissect and compare the acute vs. chronic effects of PERK/eIF2α signaling under different pathological conditions.
Conclusions and perspectives
Timely reperfusion is the most important approach to mitigate cardiac damage from myocardial infarction. However, reperfusion causes additional injury to the heart. Here, we uncover that the PERK/eIF2α axis of the ISR is potently activated in the heart by reperfusion. Both in vivo and in vitro studies demonstrate a protective role of this axis against reperfusion injury. Further proteomic analysis reveals that PERK/eIF2α signaling selectively targets mitochondrial complex components for translation inhibition and reduces complex-associated ROS production. Importantly, a pharmacological activator of the ISR protects the heart from reperfusion in vivo. Our findings together unveil a critical role of the PERK/eIF2α axis of the ISR in cardiac reperfusion injury, which may advance our understanding of I/R pathophysiology and provide a promising target to curb ischemic heart disease.
Supplementary Material
Clinical Perspective.
What Is New?
The integrated stress response (ISR) is activated by ischemia/reperfusion (I/R) in the heart.
The PERK branch of the ISR protects the heart from I/R injury through inhibition of protein synthesis.
Mitochondrial complex proteins are selectively suppressed and oxidative stress is reduced by the ISR.
What Are the Clinical Implications?
The ISR is cardioprotective against cardiac I/R injury.
Pharmacological stimulation of the ISR by salubrinal at reperfusion reduces heart damage and improves cardiac outcomes under I/R.
Acknowledgements
We thank the Proteomics Core (Andrew Lemoff) of University of Texas Southwestern Medical Center (UTSW) for help with proteomics and the Molecular Pathology Core (John Shelton) of UTSW for help with histology. We are grateful to the Animal Resource Center of UTSW for mouse generation and maintenance. We thank Erica Niewold for technical assistance and manuscript editing. G.Z. and Z.V.W. conceived and designed the study. G.Z. performed most experiments with the help from X.W. (transgenic mouse model generation), C.L. and Q.L. (animal analysis), Y.A.A. (Seahorse analysis), and X. L. (NRVM culture). G.Z. and Z.V.W. wrote the manuscript with help from Y.D., T.G.G., and P.E.S. All authors revised and approved the manuscript.
Sources of Funding
This work was supported by grants from the American Heart Association (17IRG33460191 and 19IPLOI34760325 to Z.V.W. and 20POST35210756 to X.W.), the American Diabetes Association (7-20-IBS-218 to Z.V.W. and 1-19-JDF-082 to Y.D.), and the National Institute of Health (R01-DK-126975 to Y.D. and R01-HL-137723 to Z.V.W.).
Non-standard Abbreviations and Acronyms
- αMHC
α-myosin heavy chain
- CAG
cytomegalovirus early enhance element; promoter, the first exon, and the first intron of chicken β-actin gene; splice acceptor of rabbit β-globin gene
- cKO
cardiac-specific conditional knockout
- EF
ejection fraction
- eIF2α
α subunit of eukaryotic initiation factor 2
- FKBP
FK506-binding protein
- FS
fractional shortening
- GCN2
general control non-derepressible 2
- GO
gene ontology
- HRI
heme-regulated inhibitor kinase
- I/R
ischemia/reperfusion
- ISR
integrated stress response
- KEGG
Kyoto encyclopedia of genes and genomes
- LAD
left anterior descending artery
- MEFs
mouse embryonic fibroblasts
- NRVMs
neonatal rat ventricular myocytes
- OCR
oxygen consumption rate
- PERK
PKR-like endoplasmic reticulum resident kinase
- PKR
interferon induced double-stranded RNA-dependent eIF2α kinase
- ROS
reactive oxygen species
- Salu
salubrinal
- sI/R
simulated ischemia/reperfusion
- TG
transgenics
- TMT
tandem mass tag
- TTC
triphenyltetrazolium chloride
Footnotes
Disclosures
The authors declare no competing interests.
References
- 1.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galabru J, Katze MG, Robert N, Hovanessian AG. The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced protein kinase. Eur J Biochem. 1989;178:581–589. [DOI] [PubMed] [Google Scholar]
- 4.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. [DOI] [PubMed] [Google Scholar]
- 5.Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, Machiah DK, Lawson B, Hakimpour P, Wang YC, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016;531:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clemens MJ, Bushell M, Jeffrey IW, Pain VM, Morley SJ. Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ. 2000;7:603–15. [DOI] [PubMed] [Google Scholar]
- 7.Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292:16804–16809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 11.Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, Thompson J, Hu Y, Li J, Lesnefsky EJ. Mitochondrial complex I inhibition by metformin limits reperfusion injury. J Pharmacol Exp Ther. 2019;369:282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Gong G, Wang P, Zhang Z, Kolwicz SC, Rabinovitch PS, Tian R, Wang W. Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J Mol Cell Cardiol. 2018;123:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics-2020 update: A report from the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 14.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 15.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Zhang Y, Ding G, May HI, Xu J, Gillette TG, Wang H, Wang ZV. Temporal dynamics of cardiac hypertrophic growth in response to pressure overload. Am J Physiol Heart Circ Physiol. 2017;313:H1119–h1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang G, Wang X, Bi X, Li C, Deng Y, Al-Hashimi AA, Luo X, Gillette TG, Austin RC, Wang Y, et al. GRP78 (glucose-regulated protein of 78 kDa) promotes cardiomyocyte growth through activation of GATA4 (GATA-binding protein 4). Hypertension. 2019;73:390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, Walter P. Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science. 2018;359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science. 2018;359:1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254–263. [DOI] [PubMed] [Google Scholar]
- 23.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr., Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest. 2005;115:2784–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. [DOI] [PubMed] [Google Scholar]
- 26.Santos-Ribeiro D, Godinas L, Pilette C, Perros F. The integrated stress response system in cardiovascular disease. Drug Discov Today. 2018;23:920–929. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Xu X, Fassett J, Kwak D, Liu X, Hu X, Falls TJ, Bell JC, Li H, Bitterman P, et al. Double-stranded RNA-dependent protein kinase deficiency protects the heart from systolic overload-induced congestive heart failure. Circulation. 2014;129:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, Wang H, Guo H, Xu D, Yan S, et al. Loss of the eukaryotic initiation factor 2α kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy. Hypertension. 2014;63:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, Wei Y, Cavener DR, Hu X, Hall J, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64:738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rani S, Sreenivasaiah PK, Cho C, Kim DH. Salubrinal alleviates pressure overload-induced cardiac hypertrophy by inhibiting rndoplasmic reticulum stress pathway. Mol Cells. 2017;40:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, et al. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–1421. [DOI] [PubMed] [Google Scholar]
- 32.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. [DOI] [PubMed] [Google Scholar]
- 33.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. [DOI] [PubMed] [Google Scholar]
- 34.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. [DOI] [PubMed] [Google Scholar]
- 35.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT, et al. Suppressors of superoxide-H(2)O(2) production at site I(Q) of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 2016;24:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–264. [DOI] [PubMed] [Google Scholar]
- 38.Musat-Marcu S, Gunter HE, Jugdutt BI, Docherty JC. Inhibition of apoptosis after ischemia-reperfusion in rat myocardium by cycloheximide. J Mol Cell Cardiol. 1999;31:1073–1082. [DOI] [PubMed] [Google Scholar]
- 39.Arrieta A, Blackwood EA, Glembotski CC. ER protein quality control and the Unfolded protein response in the heart. Curr Top Microbiol Immunol. 2018;414:193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Xu L, Gillette TG, Jiang X, Wang ZV. The unfolded protein response in ischemic heart disease. J Mol Cell Cardiol. 2018;117:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120:862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, Wiseman RL, Glembotski CC. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. 2019;10:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. 2014;156:1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan B, Liu S, Li X, Zhong Y, Tong F, Yang S. Preconditioning with endoplasmic reticulum stress alleviated heart ischemia/reperfusion injury via modulating IRE1/ATF6/RACK1/PERK and PGC-1α in diabetes mellitus. Biomed Pharmacother. 2019;118:109407. [DOI] [PubMed] [Google Scholar]
- 46.Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ, Accornero F, Sargent MA, Molkentin JD. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat Commun. 2021;12:3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, et al. Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ Res. 2018;122:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran DH, May HI, Li Q, Luo X, Huang J, Zhang G, Niewold E, Wang X, Gillette TG, Deng Y, et al. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat Commun. 2020;11:1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai C, Li Q, May HI, Li C, Zhang G, Sharma G, Sherry AD, Malloy CR, Khemtong C, Zhang Y, et al. Lactate dehydrogenase A governs cardiac hypertrophic growth in response to hemodynamic stress. Cell Rep. 2020;32:108087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Deng Y, Zhang G, Li C, Ding G, May HI, Tran DH, Luo X, Jiang DS, Li DL, et al. Spliced X-box binding protein 1 stimulates adaptive growth through activation of mTOR. Circulation. 2019;140:566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Esumi K, Nishida M, Shaw D, Smith TW, Marsh JD. NADH measurements in adult rat myocytes during simulated ischemia. Am J Physiol. 1991;260:H1743–h1752. [DOI] [PubMed] [Google Scholar]
- 52.Qi L, Yang L, Chen H. Detecting and quantitating physiological endoplasmic reticulum stress. Methods Enzymol. 2011;490:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An YA, Crewe C, Asterholm IW, Sun K, Chen S, Zhang F, Shao M, Funcke JB, Zhang Z, Straub L, et al. Dysregulation of amyloid precursor protein impairs adipose tissue mitochondrial function and promotes obesity. Nat Metab. 2019;1:1243–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.