

Genome-wide network modeling of individual glioblastomas identifies dysregulation of PD1 signaling in patients with poor prognosis, indicating this approach can be used to understand how gene regulation influences cancer progression.
Abstract
Glioblastoma is an aggressive cancer of the brain and spine. While analysis of glioblastoma ‘omics data has somewhat improved our understanding of the disease, it has not led to direct improvement in patient survival. Cancer survival is often characterized by differences in gene expression, but the mechanisms that drive these differences are generally unknown. We therefore set out to model the regulatory mechanisms associated with glioblastoma survival. We inferred individual patient gene regulatory networks using data from two different expression platforms from The Cancer Genome Atlas. We performed comparative network analysis between patients with long- and short-term survival. Seven pathways were identified as associated with survival, all of them involved in immune signaling; differential regulation of PD1 signaling was validated to correspond with outcome in an independent dataset from the German Glioma Network. In this pathway, transcriptional repression of genes for which treatment options are available was lost in short-term survivors; this was independent of mutational burden and only weakly associated with T-cell infiltration. Collectively, these results provide a new way to stratify patients with glioblastoma that uses network features as biomarkers to predict survival. They also identify new potential therapeutic interventions, underscoring the value of analyzing gene regulatory networks in individual patients with cancer.
Significance:
Genome-wide network modeling of individual glioblastomas identifies dysregulation of PD1 signaling in patients with poor prognosis, indicating this approach can be used to understand how gene regulation influences cancer progression.
Introduction
Microarrays and next-generation sequencing technologies have been broadly applied to the study of cancer. Large collaborative projects, such as The Cancer Genome Atlas (TCGA), have explored the ‘omics landscape for many different cancer types. Although this has somewhat improved our understanding of the biology underlying the development and progression of cancer (1), it has only led to direct improvement of patient survival for a limited subset of cancer types. For most cancers, genomic signatures associated with survival are very complex, making it difficult to point to direct targets for treatment.
An example is glioblastoma multiforme, an aggressive cancer of the brain and spine. With a median overall survival of only 15–17 months, glioblastoma has a particularly poor prognosis. Multiple gene signatures have been identified that correlate with glioblastoma survival (see, e.g., refs. 2–6), but comparison of these signatures finds very few shared genes or pathways, making interpretation of the results difficult at best. Although unsupervised clustering of gene expression analysis has identified glioblastoma subtypes that differ somewhat in patient survival (7), and integrative analysis with mutational profiles has shed some light on what could drive these subtypes (8), the causative regulatory mechanisms that distinguish these subtypes are not fully understood. We believe that, by modeling the regulatory mechanisms that mediate gene expression patterns associated with patient subtypes and survival, we will be better able to explain what influences disease progression, and may identify therapeutic interventions that advance precision medicine treatments for patients with glioblastoma.
Gene regulatory mechanisms can be modeled using gene regulatory network reconstruction approaches. An example of such an approach is PANDA (Passing Attributes between Networks for Data Assimilation; refs. 9, 10). PANDA relies on message passing (11) to infer regulatory processes by seeking consistency between transcription factor (TF) protein–protein interaction (PPI), DNA motif binding, and gene expression data. PANDA has been key to understand tissue-specific gene regulation (12), identify regulatory differences between cell lines and their tissues of origin (13), and to identify regulatory changes that may drive cancer subtypes (14). One drawback, however, of estimating regulatory networks using PANDA or other methods is that network reconstruction relies upon combining information from large, typically diverse study populations to estimate one “aggregate” network representing that dataset.
To reconstruct regulatory networks for each sample in a dataset, we developed LIONESS, or Linear Interpolation to Obtain Network Estimates for Single Samples (15). LIONESS assumes that the edges estimated in an “aggregate” network model are a linear combination of edges specific to each of the input samples. This allows estimation of individual sample edge scores using a linear equation. What this means is that, instead of only reconstructing one single network representing glioblastoma, we can “extract” distinct, reproducible networks for each individual patient from this aggregate network model. This allows us to associate individual networks and network properties with clinical endpoints, such as patient survival and response to therapy, and is therefore an exciting advance in network inference with potential applications in precision medicine. We have used this method in a number of applications including modeling 8,279 sample-specific gene regulatory networks to discover sex-biased gene regulation across 29 tissues (16) and modeling regulatory networks for more than 1,000 patients with colon cancer to find sex-linked regulatory mechanisms associated with response to chemotherapy (17).
In the study presented here, we applied PANDA and LIONESS to model gene regulatory networks for individual patients with primary glioblastoma from TCGA. We performed a comparative network analysis to identify regulatory differences between patients with long- and short-term survival (Fig. 1). We validated our results in an independent dataset of primary glioblastomas from the German Glioma Network. This integrative gene regulatory network approach allowed us to identify disrupted regulation of PD1 pathway genes as associated with glioblastoma survival. Specifically, we found that transcriptional repression of genes involved in PD1 signaling is lower in short-term survivors. This is independent of PD1 gene methylation status, and mutation burden, and weakly associated with immune infiltrate, and therefore suggests a new mechanism of heterogeneity in glioblastoma prognosis.
Figure 1.

Schematic overview of the study. Left box, overview of the approach used to reconstruct individual patient networks with PANDA and LIONESS by integrating information on PPIs between TFs, prior information on TF-DNA motif binding, and gene expression data from two platforms using data from TCGA (n = 522 and 431 patients). Right box, overview of the differential regulation analysis.
Materials and Methods
Gene expression data preprocessing
We downloaded primary glioblastoma gene expression data from TCGA using RTCGAToolbox (18) as described previously (19). This included Affymetrix HG-U133 microarray data for 525 patients and Affymetrix Human Exon 1.0 ST microarray data for 431 patients. We refer to these datasets as “discovery dataset 1” and “discovery dataset 2” in the rest of this article.
We performed batch correction on each dataset independently, using ComBat (20). Some TCGA batch numbers only contained one glioblastoma sample (two batch numbers in discovery dataset 1 and one in discovery dataset 2) and thus for those samples, batch correction was not possible. We grouped these samples together with those batches that were most similar in their expression profiles, based on Pearson similarity. We visualized the distribution of expression levels in each sample in density plots and removed three outlier samples from discovery dataset 1, resulting in a dataset that included 522 patients. We then used the quantile normalization in Bioconductor package “preprocessCore” to normalize each dataset independently (21).
We used data from the German Glioma Network (22) to for independent validation of our findings from TCGA; we refer to the German Glioma Network dataset as the “validation dataset.” These data consist of untreated, primary glioblastomas profiled on Affymetrix HG-U133 Plus 2.0 microarrays and are available in the Gene Expression Omnibus (GEO) with identifier GSE53733 (download date: April 15, 2017; ref. 22). We downloaded the normalized expression data from the “Series Matrix File.” Follow-up information was available in the form of three groups—long-term (survival >36 months, n = 23), intermediate (between 12–36 months, n = 31), and short-term (<12 months, n = 16) survival.
Finally, we used data from Kwon and colleagues (23) to compare primary (n = 25) with recurrent (n = 18) glioblastomas. These data were profiled on Illumina HumanHT-12 V4.0 expression beadchips and are available in GEO with identifier GSE62153. We used Bioconductor package “GEOquery” to download the normalized expression data (download date: June 19, 2021).
Curation of clinical data and selection of patient groups
For detailed information on how we curated the clinical data from TCGA, we refer to our previous publication (24). In short, we used RTCGAToolbox (18) to download the data, and curated these by combining data from all available Firehose versions consecutively as to retain all clinical information, with the most up-to-date information for data present across different Firehose versions.
We used a survival threshold of 1.7 years (620 days) to define long-term and short-term survival. We estimated this threshold by taking the third quartile of the last day to follow-up available in discovery dataset 1 to ensure a meaningful (e.g., not too short) threshold while maintaining relatively balanced sample groups. This threshold also falls in between thresholds previously used to define survival groups (Supplementary Table S1). It divided the patient population of discovery dataset 1 into a group of 127 relatively long-term survivors who survived for at least 1.7 years, 336 short-term survivors who deceased within 1.7 years, and 59 patients for whom not enough follow-up data were available (“censored” patients). We excluded the latter group from our analyses. Similarly, the group of patients in discovery dataset 2 was divided into 116 long-term, 275 short-term, and 40 censored patients. We note that, for the significant pathways identified in the comparison of networks for these survival groups (see below), we repeated the analysis for a wide range of survival thresholds—all thresholds that resulted in a minimum group size of 10 patients—to ensure the robustness of our results.
Modeling single-sample networks
We used the MATLAB version of the PANDA network reconstruction algorithm (available in the netZoo repository https://github.com/netZoo/netZooM) to estimate aggregate gene regulatory networks. PANDA incorporates regulatory information from three types of data: gene expression data (see above), PPI data, and a “prior” network based on a TF motif scan to their putative target genes, which is used to initialize the algorithm.
To build the motif prior, we used a set of 695 TF motifs from the Catalogue of Inferred Sequence Binding Preferences (CIS-BP; ref. 25), which we had selected previously (12). We scanned these motifs to promoters as described previously (26). After intersecting the prior to only include genes and TFs with expression data (see above) and at least one significant promoter hit, this process resulted in an initial map of potential regulatory interactions involving 650 TFs targeting 10,701 genes.
We estimated an initial PPI network between all TFs in our motif prior using interaction scores from StringDb v10 (27), as described in Sonawane and colleagues (12). PPI interaction scores were divided by 1,000 to have them range [0,1] and self-interactions were set equal to one.
For each dataset, we built an aggregate network using PANDA and then used the LIONESS equation in MATLAB to extract networks for individual samples.
Community structure comparison
To perform a comparative analysis of regulatory network community structures between the two patient groups, we averaged the single-sample networks in each of the groups. We filtered these networks for canonical edges (prior edges representing putative TF–DNA binding) and then performed community structure analysis using the bipartite community structure detection algorithm in CONDOR (28). As CONDOR requires positive edge weights, we transformed the network edges as in Sonawane and colleagues (12) using the following equation, before applying CONDOR:
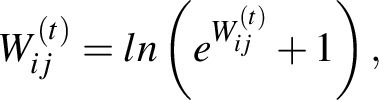 |
where 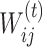 is the edge weight calculated by PANDA between a TF (i) and gene (j) in a particular network (t).
is the edge weight calculated by PANDA between a TF (i) and gene (j) in a particular network (t).
We used ALPACA (29)—a method that uses message passing and modularity optimization to identify structural changes in complex networks—to compare the community structure of the long-term survival network with that of the short-term network set as a baseline, and vice versa. We reported differential communities that included a minimum of 10 nodes. To identify enrichment in Gene Ontology (GO) terms, we used communities that included at least 50 genes. We compared the top 50 genes based on their ALPACA scores with the background of all genes in the network model, using the “classical” Fisher exact test from R package “topGO.” We corrected P values for multiple testing using the Benjamini and Hochberg approach and reported those GO terms with FDR < 0.25 and gene set size <50 as significantly enriched.
LIMMA analysis
For each tumor sample, we calculated gene targeting scores by taking the sum of all edge weights pointing to a gene (this is the same as a weighted gene “degree”). This measure is representative of the amount of regulation a gene receives from the entire set of TFs available in our network models (17). We performed a Bayesian statistical analysis using LIMMA (30) comparing gene targeting scores in long-term survivors with those in short-term survivors, correcting for the patient's age at diagnosis of the primary tumor, their sex, and whether they were treated with neoadjuvant therapy or not. We repeated this analysis on network edge weights, so that we could visualize edges connecting to genes in the PD1 signaling pathway.
We note that we also evaluated other relevant clinical features in glioblastoma, including 1p19q co-deletion, IDH1/2 [isocitrate dehydrogenase (IDH)] status, and MGMT promoter methylation status. 1p19q co-deletion status (available for 96% of patients) was, as expected in this primary cohort, only deleted in a small fraction (0.4%) of patients. We ran linear models correcting for MGMT and IDH status, and reported the results when correcting for IDH status (available for 79% of patients) as well. As MGMT status was only available for 53% of patients, we did not correct for this variable.
For the analysis in the validation dataset of primary tumors, we compared the long-term group with the short-term group as defined in Reifenberger and colleagues (22). We used the dataset from Kwon and colleagues (23) to perform an additional validation, comparing recurrences with primary tumors.
Preranked gene set enrichment analysis
We performed preranked gene set enrichment analysis (GSEA; ref. 31) on the t-statistics from the LIMMA analysis to identify Reactome gene sets enriched for differentially targeted genes. We defined gene sets with FDR < 0.05 and absolute GSEA enrichment score (ES) > 0.5 to be significantly differentially targeted. We evaluated gene sets that had less than 50 genes, so as to exclude some of the more general pathways.
Cellular composition analysis
We used xCell (32) to estimate cell compositions in each tumor sample in each of the three datasets. xCell is a novel gene signature-based method that integrates the advantages of gene set enrichment with deconvolution approaches to infer 64 immune and stromal cell types. The scores calculated by xCell approximate cell type fractions, and adjust for overlap between closely related cell types. Finally, the method calculates P values for the null hypothesis that a cell type is not in the mixture and thereby allows for filtering out cell types that are likely not present in the sample. We applied the xCell pipeline to each of the datasets and used the default threshold of 0.2 to filter out cell types not present in the datasets.
We used CIBERSORTx (33) as a second deconvolution approach to validate the findings obtained with xCell. We used the median values of expression for the single-cell RNA sequencing (RNA-seq) data provided by the Allen Institute for Brain Science, available at https://portal.brain-map.org/atlases-and-data/rnaseq/human-m1-10x as reference for the human brain (M1–10X, 127 cell types; ref. 34). We used the CIBERSORTx "impute cell fractions" module with default parameters (relative mode, no batch correction, no quantile normalization, and no permutations for significance analysis; ref. 33). We then compared the identified cell fraction compositions between the two survival groups using the Wilcoxon rank-sum test.
P-values from xCell and CIBERSORTx were corrected for multiple testing using the Benjamini and Hochberg method (35).
Association with mutation load
We downloaded and preprocessed glioblastoma mutation data as previously described in Kuijjer and colleagues (24). We selected data corresponding to patients that we used in our network analysis (n = 232). We calculated patient-specific tumor mutation scores based on all available genes (n = 26,076) using SAMBAR [https://github.com/kuijjerlab/SAMBAR and Kuijjer and colleagues (24)], which corrects the number of somatic mutations per gene based on the gene length and then sums these scores to obtain a patient-specific tumor mutation burden score. We calculated PD1 pathway targeting scores by dividing the sum of the PD1 gene targeting scores in each sample by the number of PD1 pathway genes available in our dataset (n = 13). We then correlated the patient mutation scores with these PD1 pathway targeting scores using Spearman correlation. In addition, we assessed the association between PD1 pathway targeting scores and mutation scores in individual Reactome pathways, across patients. As for most pathways only a subset of patients had non-zero mutation scores, we used Pearson correlation (which can handle ties) to calculate these pathway-specific scores.
Association with methylation data
We downloaded TCGA methylation beta value data for glioblastoma samples from the GDC Data Portal on June 9, 2019 (https://portal.gdc.cancer.gov/repository). We selected only primary tumor samples used in our network analysis, which included 246 samples obtained from the Illumina Infinium HumanMethylation27 (27K) BeadChip array and 93 samples from the Illumina Infinium HumanMethylation450 (450K) BeadChip array. We subsetted to probes for genes in the PD1 pathway, which included 19 probes on the 27K platform, and 274 probes on the 450K platform. We performed quantile normalization on the methylation beta values, followed by a Bayesian statistical analysis using LIMMA as described above. For each platform, we compared the normalized beta values between short-term and long-term survivors, adjusting for age, sex, and whether the patient was treated with neoadjuvant therapy or not. We adjusted the P values using the Benjamini and Hochberg method (35).
Validation in protein abundance data
We downloaded protein abundance data for glioblastoma samples from TCGA using Bioconductor package “RTCGA.RPPA” (accessed April 13, 2018; ref. 36). We subsetted these data to primary tumors only and to samples corresponding to patients that were available in our network analysis (n = 233). We selected PD1 pathway proteins in the RPPA data based on whether they corresponded to PD1 pathway genes from the Reactome signature (18 genes in total). We identified 3 of 208 proteins as components of the PD1 pathway: Lck, p62-LCK-ligand, and PDCD4. We compared protein abundance of these three proteins between the two survival groups using a t test, and corrected for the FDR using the Benjamini and Hochberg method (35).
Data and code availability
The input data, clinical information, code to reproduce all individual patient networks, and the entire collection of reconstructed networks used in this study are available on https://grand.networkmedicine.org/cancers/GBM_cancer/. Code to reproduce the results and figures is available at http://netbooks.networkmedicine.org/ under “Case studies—Gene regulatory network analysis in Glioblastoma.”
Results
Data features
We investigated the regulatory processes that drive survival differences in glioblastoma by performing network analysis in two discovery datasets followed by a validation in two independent datasets (Fig. 1). For the discovery datasets, we analyzed microarray data of primary glioblastoma tumor samples from TCGA, which were profiled on two different platforms—Affymetrix HG-U133 arrays (discovery dataset 1) and Affymetrix Human Exon 1.0 ST arrays (discovery dataset 2). We removed potential outlier samples based on gene expression densities and retained 522 and 431 samples, respectively, for the primary analysis. These included 424 samples obtained from the same patients. Thus, by assessing reproducibility of our network models in these two discovery datasets, we could identify whether the network models are robust across platforms.
For the independent validation dataset of primary tumors, we downloaded normalized microarray data from the German Glioma Network (22) from GEO, which were profiled on Affymetrix HG-U133 Plus 2.0 arrays. This validation dataset included 70 primary, untreated glioblastoma samples. Patient clinical features for the discovery dataset are summarized in Table 1. For this validation dataset, no clinicopathologic features except for survival information were available. In addition, we performed a comparison of primary and recurrent glioblastomas profiled on Illumina HumanHT-12 V4.0 expression beadchips. This second validation dataset included a total of 43 samples (23).
Table 1.
Clinical features for patients available in the two discovery datasets from TCGA.
| Discovery dataset 1 | Discovery dataset 2 | |||||
|---|---|---|---|---|---|---|
| Long-term surv. | Short-term surv. | P | Long-term surv. | Short-term surv. | P | |
| Number of patients | 127 | 336 | – | 116 | 275 | – |
| Sex | ||||||
| Female | 55 | 122 | 0.198 | 49 | 96 | 0.172 |
| Male | 72 | 214 | 67 | 179 | ||
| Age | 49.2 ± 14.7 | 60.7 ± 13.2 | 4.34e-13 | 49.7 ± 14.5 | 60.0 ± 13.7 | 4.55e-10 |
| Year of diagnosis | 2003 ± 4.4 | 2004 ± 5.1 | 0.162 | 2003 ± 4.2 | 2003 ± 4.9 | 0.797 |
| Race | ||||||
| Asian | 5 | 4 | 0.178 | 5 | 4 | 0.200 |
| Black | 8 | 17 | 8 | 14 | ||
| White | 113 | 298 | 102 | 241 | ||
| Not reported | 1 | 17 | 1 | 16 | ||
| Ethnicity | ||||||
| Hispanic or latino | 2 | 8 | 0.731 | 1 | 7 | 0.443 |
| Not hispanic or latino | 111 | 276 | 105 | 234 | ||
| Not reported | 14 | 52 | 10 | 34 | ||
| Neoadjuvant therapy | ||||||
| Yes | 68 | 160 | 0.250 | 64 | 137 | 0.375 |
| No | 57 | 172 | 51 | 134 | ||
| Not reported | 2 | 4 | 1 | 4 | ||
Note: t test was performed for age and year of diagnosis. For other clinical features, Fisher exact test was performed, excluding unreported values.
Abbreviation: Surv., survival.
To identify regulatory gene network differences associated with survival, we divided the patient population into two groups (see Materials and Methods). The first group had overall survival of more than 1.7 years (127 and 116 patients for discovery datasets 1 and 2, respectively) while the second group died within 1.7 years of initial diagnosis (336 and 275 patients, respectively); patients who were alive, but for whom we did not have follow-up data >1.7 years were excluded from the analysis (59 and 40 patients, respectively). The number of long-term and short-term survivors in each dataset were well matched, and we controlled the downstream analysis of the discovery data for potential differences between the two outcome groups for age, sex, and neoadjuvant treatment status. We note that we also evaluated correction for IDH status, as well as multiple survival thresholds (see Materials and Methods and below).
Patient-specific glioblastoma networks
In previous studies, we found gene regulatory network analysis identified altered phenotype-relevant biological processes that were not observed using other analytic methods. Specifically, we found that TFs often demonstrate altered regulatory roles through changes in their targeting patterns that are independent of their own mRNA expression levels (12, 13, 17), highlighting the importance of integrative frameworks that include information on putative TF-DNA binding sites to model regulatory mechanisms. Therefore, we performed a gene regulatory network analysis of glioblastoma using the PANDA and LIONESS integrative network modeling framework (Fig. 1), reconstructing regulatory networks for each individual in our discovery populations and independently for the validation sets.
PANDA uses a message passing algorithm to seek consistency between evidence provided by multiple datasets. PANDA starts with a TF to target gene prior network that is based on a motif scan mapping TF binding sites to the promoter of their putative target genes. The method then integrates this prior network of putative interactions with PPIs between TFs and with target gene expression data. It does this through identifying overlaps in targeting patterns in the three input networks, rather than through estimating correlations between TFs or between TFs and their target genes [as we previously showed in Sonawane and colleagues (12), expression levels of important regulatory factors are not always correlated with their targets]. The resulting gene regulatory network consists of weighted edges between each TF-target gene pair. These edge weights reflect the strength of the inferred regulatory relationship.
We initiated PANDA with TF binding sites from CIS-BP (25), PPI data from StringDb (27), and expression data from either the discovery or the validation datasets to model aggregate gene regulatory networks based on all samples. We then used LIONESS to estimate each individual patient-specific regulatory network. These individual patient networks allow us to associate network properties with clinical information.
Immune system network modules are associated with glioblastoma survival
As a baseline for our individual patient network analysis, we first compared the robustness of the condition-specific networks modeled on the two discovery datasets. We calculated network similarity using Pearson correlation and found that the networks were highly reproducible, with Pearson R = 0.94, P < 2.2e-16 (see Supplementary Fig. S1). We next evaluated whether there were global network structural differences between the long-term and short-term survivors. To do this, we merged the individual patient networks of each discovery dataset into two condition-specific networks by, for all edges, taking the average of the edge weight across the patient group. For each discovery dataset, this resulted in one condition-specific network representing individuals with long-term survival and one representing individuals with short-term survival. We then used CONDOR (28), a community detection algorithm specifically designed to detect modules in bipartite networks, and applied ALPACA (29) to extract differential network modules that distinguish the long-term from the short-term survival network and vice versa. ALPACA calculates a “differential modularity” score that compares the density of modules in the “perturbed” network to the expected density of modules in the “baseline” network.
ALPACA found 14 differential modules in the long-term versus short-term comparison in discovery dataset 1 and 17 differential modules in discovery dataset 2. We performed GO term analysis on the top 50 genes with highest differential modularity scores in these modules [as described in Padi and colleagues (29)], and identified two modules that had significant overrepresentation of GO terms in each of the discovery datasets. In total, we identified overrepresentation of 19 GO terms (see Fig. 2A). The three GO terms with the highest enrichment scores were significant in both discovery datasets. These terms included “innate immune response in mucosa,” “mucosal immune response,” and “organ or tissue specific immune response.” This result suggests that the regulatory network in glioblastoma changes its topology when the disease becomes more aggressive and that this rewiring involves genes with a role in immune response.
Figure 2.

Differential network topology between long- and short-term survivors. A, Differential network modules significantly enriched for overrepresentation of GO terms. Modules are shown for both discovery datasets (D1 and D2). Two modules were significant in D1 and three in D2 (indicated with M1–M3). The color in the heatmap indicates the enrichment of genes in the module as observed/expected value (Obs/Exp), with purple representing enriched modules in discovery dataset 1 and green representing enriched modules in discovery dataset 2. B, “Beeswarm” plot visualizing the distribution of average log differential modularity scores from ALPACA for TFs and genes (Gene). Higher scores mean higher differential modularity. TFs and genes with significant differential modularity between the two survival groups are labeled.
In addition to analyzing global differences in network module structure, we pulled out the TFs and target genes with the greatest contributions to the observed differences in network topology. We averaged the differential modularity scores from ALPACA across the networks modeled on the two discovery datasets. To extract the top differential nodes, we transformed these scores to a log scale. We then calculated the median and interquartile range (IQR) for TFs and target genes separately, as these are bipartite networks with different targeting properties—TFs are fewer in number and generally regulate a large number of genes, while there are more target genes than TFs—therefore, in- and out-degree values cannot be directly compared (see Fig. 2B for their distribution).
We identified four TFs for which differential modularity deviated more than 1.5×IQR from the median (Fig. 2B). The top TF was BRCA1, which plays an important role in DNA damage repair, and is affected in many cancer types (37). Other TFs included ZBTB33, CREB3L1, and CTCF. ZBTB33 can promote histone deacetylation and the formation of repressive chromatin and plays a role in the Wnt signaling pathway. CREB3L1 is required to protect astrocytes from endoplasmatic reticulum-induced stress (38), and is activated in response to virus infection (39). CTCF can bind to a large number of sites in the human genome and is known to regulate important cellular processes, both in neuronal development and in the immune system (40).
For 12 target genes, differential modularity scores deviated with more than 3×IQR from the median (Fig. 2B). These include cancer-associated genes such as PPP2R3C, a serine/threonine phosphatase involved in PI3K/Akt signaling and B-cell survival, and DSN1, encoding a kinetochore protein. Genes known to be expressed in the brain (e.g., TACR2, TRPV4, and HSPBAP1) were also among the top target genes, as well as genes that may act as TFs (RBPJL and TOX4).
As ALPACA is not symmetric, we also contrasted short-term with long-term community structures and found similar results, with immune signaling pathways significantly enriched in differential communities in both datasets (see Supplementary Fig. S2A for enriched GO terms; Supplementary Fig. S2B for the distribution of ALPACA scores).
Regulation of PD1 pathway genes is associated with glioblastoma survival
After having analyzed the structural differences between condition-specific networks, we set out to analyze networks for each individual patient. We hypothesized that these individual patient network models, parameterized by the “weights” assigned to TF/target gene interactions (edges) we estimate, are predictive of clinical outcome. The basis for this hypothesis is that the edge weights provide an estimate of which gene regulatory programs are active in each individual.
We estimated whether alteration of the inferred regulatory networks was predictive of patient survival. For each gene in each sample, we calculated a “gene targeting score” equal to the sum of edge weights in the regulatory model. We used Bayesian statistical analysis (LIMMA) to test for significant associations of the gene targeting scores with survival. We corrected this analysis for patient age, sex, and neoadjuvant treatment status. We used GSEA (31) using Reactome (41) signatures from MSigDb (42) on the moderated t-statistic from the LIMMA analysis to identify pathways significantly differentially targeted between good and poor survivors (FDR < 0.05). We performed this analysis on both discovery datasets.
We identified 54 and 46 pathways (FDR < 0.05, absolute ES >0.5, gene set size <50) in discovery dataset 1 and 2, respectively. Seven pathways were significant in both datasets and each of these seven pathways had a role in immune function (Fig. 3), confirming our findings from the ALPACA comparison. We note that we also repeated the analysis with correction for IDH status (available for 79% of patients) in addition to the covariates mentioned above. All seven pathways identified in the analysis without correction for IDH status remained significant after correcting for IDH status.
Figure 3.
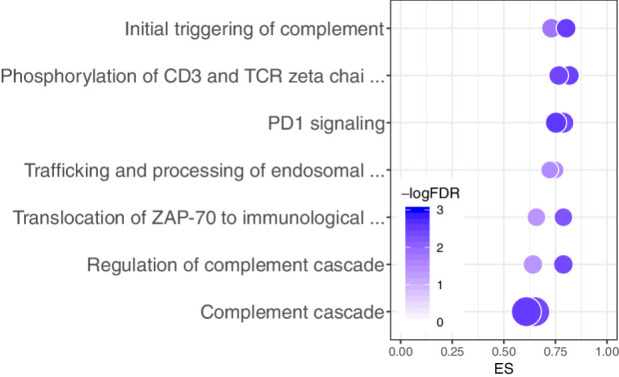
Bubble plot representation of the seven pathways that were significantly differentially regulated between long- and short-term survivors in both datasets from TCGA. For each pathway, two bubbles are shown—one for each dataset—representing the ES on the x-axis, the -logFDR (color), and the number of genes represented in the pathway (bubble size).
To validate these results, we repeated our network analysis pipeline on an independent validation dataset from the German Glioma Network. Even though the sample size of this validation dataset was considerably lower than those in the discovery datasets, we identified one of the seven pathways as significantly enriched (FDR = 0.017), with the same direction of enrichment (ES = −0.67)—the PD1 signaling pathway.
Finally, to ensure that our results were stable across multiple survival thresholds, we repeated the comparative network analysis in the two discovery datasets across multiple survival thresholds, which we ranged from zero to the maximum follow-up, with intervals of 0.1 years, keeping comparisons that had group sizes of at least 10 patients. We found that the PD1 signaling pathway was stable across a wide range of thresholds (see Supplementary Fig. S3A for discovery dataset 1 and Supplementary Fig. S3B for discovery dataset 2).
Loss of transcriptional repression of PD1 signaling in patients with short-term survival
We next visualized the gene regulatory network around the PD1 signaling pathway and found that genes belonging to this pathway are overexpressed in the short-term survival group, but that their expression levels are repressed in the group of patients with better survival (Fig. 4). This repression of PD1 signaling genes is lost in patients with the worst survival profiles. In total, we found 105 TFs to be involved in differential targeting of the PD1 signaling pathway, indicating that the regulatory network rewiring involved in glioblastoma survival is complex. In addition, several TFs were involved in differential regulation of multiple PD1 target genes (these are indicated with a larger font size in the figure). Examples of such TFs are GSX2, ZIC2, HMGA1, GATA4, ZKSCAN1, and SMARCC1.
Figure 4.

Network representation of the PD1 signaling pathway in discovery dataset 1. Node size corresponds to the number of edges connected in the graph; line width of edges corresponds to their significance in differential regulation. Gene targeting scores (or “degrees”) are shown with a circle surrounding the node. Red, higher expression/targeting in short-term survivors; blue, higher expression/targeting in the long-term survivors.
GSX2 and ZIC2 are involved in brain development (43, 44). GSX2 is a class II Homeobox gene that is found primarily in the brain and is involved in brain development and neuronal differentiation. It was previously reported as part of a three-gene signature that can predict outcome in low-grade glioma (43). HMGA1 can promote cell growth in glioma cells (45). Overexpression of this TF was shown to correlate with proliferation, invasion, and angiogenesis in glioblastoma (46). Hypermethylation of the GATA4 promoter has been identified in glioblastoma and several other cancer types (47). ZKSCAN1 and SMARCC1 have not yet been identified in glioblastoma, but play a role in other cancer types. ZKSCAN1 was shown to be involved in cell proliferation, migration, and invasion in hepatocellular carcinoma (48), while SMARCC1 has been associated with colorectal cancer survival (49). These TFs are involved in metastasis and invasion, brain development, cell proliferation, and cancer. They could be potentially targeted to improve survival or to boost PD-1/PD-L1 inhibition in glioblastoma.
Loss of transcriptional repression of PD1 signaling in recurrent compared with primary tumors
To determine whether PD1 signaling regulation is associated with tumor aggressiveness and progression in general, we modeled networks on a second independent validation set, which included recurrent and primary glioblastomas. We repeated our analysis pipeline, comparing networks modeled on recurrent with networks modeled on primary tumors. A total of four pathways were significantly differentially regulated, of which, the PD1 signaling was the top ranked pathway (ES = −0.72, FDR = 0.046). This indicates that PD1 signaling repression is lower in recurrent tumors compared with primary tumors, and thus strengthens our results that loss of repression of the pathway is associated with tumor progression.
PD1 targeting scores weakly associate with CD8-positive naïve T-cell fractions
Because a recent study had shown that differences in coexpression, which are used as input in PANDA, may be caused by different cellular compositions in bulk tissues (50), we tested whether there were differences in immune cell composition in samples from the two patient groups. To do this, we used xCell (32) to calculate the cell type composition in each sample. We then performed a t test to determine whether our sample groups were enriched for specific cell types. We identified significant enrichment for CD8-positive naïve T cells in long-term survivors in the two discovery datasets (FDR < 0.1), as well as CD4-positive memory T cells in discovery dataset 2 (see Supplementary Fig. S4A for discovery dataset 1, Supplementary Fig. S4B for discovery dataset 2, and Supplementary Fig. S4C for the validation dataset). However, while we identified a nominal significance (P < 0.05) for CD8-positive naïve T cells in the validation dataset as well, this was not significant after correcting for multiple testing.
We did not observe significant differences in other cell types, nor in the total immune scores (Supplementary Fig. S5A). We then correlated the total immune scores (the sum of xCell scores of all immune cell types) with the PD1 pathway targeting scores to determine whether there was any other dependence between targeting scores and immune infiltrate, but did not observe any significant correlations in the discovery datasets (Supplementary Fig. S5B). We also did not observe significant differences in correlation coefficients between the two survival groups.
Deconvoluting with CIBERSORTx (33), using single-cell RNA-seq from human brain as a baseline, did not identify significant differences (FDR < 0.05) in cell types between the two survival groups in any of the cohorts, confirming the results obtained with xCell.
Together, these results indicate that while PD1 targeting scores may somewhat associate with infiltration of CD8-positive naïve T cells, they cannot solely be explained by the amount of immune infiltration in the tumor samples. The association between repression of PD1 signaling and survival is therefore not likely to be caused solely by differences in immune composition, but rather by regulatory differences in the cancer cells.
Differential regulation of PD1 signaling is independent of overall tumor mutation load
We next tested whether differential regulation of PD1 signaling was associated with tumor mutation burden. A higher mutation burden has been associated with a higher level of neoantigen presentation that may facilitate immune recognition of cancer and thus activate an antitumor immune response. It has also been associated with better response to immune checkpoint inhibitors (51).
For this and the following analyses, we focused on comparison of PD1 targeting scores from discovery dataset 1. We downloaded and preprocessed mutation data for patients with glioblastoma as described in Kuijjer and colleagues (24) and selected data for those patients we used in our network analysis (n = 235). We calculated the somatic mutation burden in these tumors using the overall mutation rate scores obtained with SAMBAR—a mathematical approach that converts and de-sparsifies somatic mutations in genes into pathway mutation scores (24). We used Spearman correlation to compare PD1 pathway targeting with the overall mutation rates. We did not identify any significant association between mutation burden and targeting of PD1 signaling genes [Fig. 5A, Spearman R = 0.016, P = 0.81 for the PD1 targeting score, range of Spearman R for individual genes [−0.14, 0.075)], indicating that regulation of PD1 signaling is independent of mutation burden in glioblastoma.
Figure 5.

Association of PD1 pathway targeting with mutation data. A, Smooth scatterplot comparing PD1 targeting scores with mutation load. B, Results from the association of PD1 pathway scores with mutation scores in individual pathways. The three most significant pathways are labeled. Purple dots, pathways with positive correlation; yellow dots, pathways with negative correlation.
PD1 pathway targeting weakly associates with mutations in cell-cycle genes
To identify whether there was an association between targeting of the PD1 pathway and mutations in specific biological pathways, we correlated PD1 pathway targeting scores with mutation scores of individual biological pathways from SAMBAR (see Materials and Methods). We identified one pathway that significantly correlated (FDR < 0.1) with PD1 targeting scores, although the correlation coefficient was rather low (Pearson R = 0.26), indicating a weak association—the Sigma-Aldrich pathway “G1- and S-phases” (Fig. 5B). This pathway consists of 15 genes, including cyclin D1, cyclin-dependent kinases and their inhibitors, E2F TFs, MDM2, and TP53. A high mutation score in this pathway associated with a high targeting of the PD1 pathway (repression). Non-canonical functions of cell-cycle genes include regulating the immune system (52), and this could be a potential mechanism for this association. The top pathways with FDR < 0.25 include two pathways associated with RAR/RXR and CTCF TF activity, respectively, indicating that mutations in these pathways may influence targeting of PD1 signaling. While both RAR/RXR and CTCF are transcriptional regulators, the genes available in these pathways are targets of these TFs, and the pathways do not include the TFs themselves. It is yet unclear why mutational patterns in targets of these TFs show a trend in correlating with PD1 pathway targeting scores.
Validation of the PD1 signaling pathway in other ‘omics data types
Finally, we analyzed differential regulation of PD1 signaling in the context of other 'omics data types, including methylation and protein abundance data.
First, we downloaded methylation data from glioblastoma samples from TCGA, and subsetted these to samples from the same patients we used in our network analysis. Data from two methylation arrays were available—Illumina Infinium Human Methylation 450K BeadChips (available for 93 patients in our cohort), which included 274 probes associated with PD1 genes, and Illumina Infinium HumanMethylation27 BeadChips (available for 246 patients in our cohort), including 19 probes associated with these genes. We performed LIMMA analyses to determine whether any of these probes were differentially methylated between the long- and short-term survival groups. We did not identify any significant differentially methylated probes, indicating that the repression of PD1 pathway genes in the long-term survival group is not driven by methylation, but directly by TF/target gene interactions.
To validate protein abundance levels of PD1 signaling genes, we downloaded reverse phase protein array (RPPA) data from glioblastoma samples from TCGA, and subsetted these to samples from the same patients we used in our network analysis (n = 233). We evaluated differential abundance of the three PD1 signaling proteins for which abundance data was available between the long-term and short-term survivors and identified one protein with significant differential abundance: p62-Lck-ligand (translated by the SQSTM gene). This protein had lower abundance in patients with long-term survival (FDR = 0.0058, Fig. 6), further strengthening the involvement of the PD1 signaling pathway in glioblastoma survival.
Figure 6.
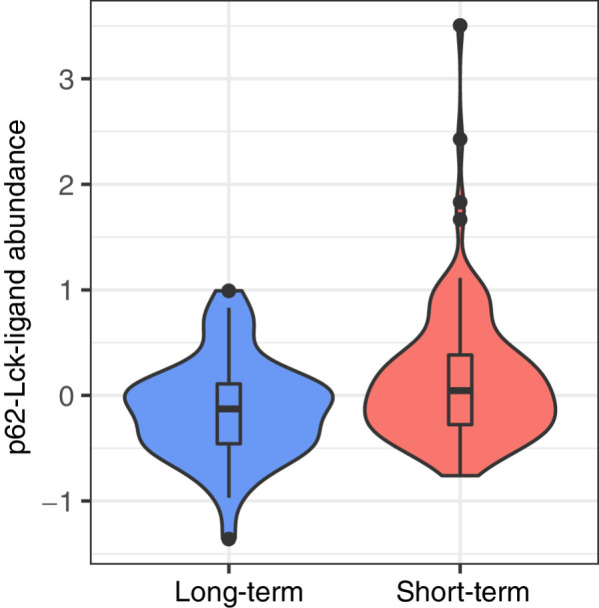
Validation analysis in RPPA data. p62-Lck-ligand protein abundance is significantly higher in short-term survivors compared to long-term survivors (FDR = 0.0058).
Discussion
Transcriptomic analysis has previously identified numerous gene sets as potentially associated with survival in glioblastoma. However, many of the published prognostic signatures are not reproducible. Therefore, we hypothesized that alternative mechanisms may explain the heterogeneity in glioblastoma survival. Most notably, previous studies have not reported alterations in PD1 pathway expression to be associated with survival. Using an integrative network approach to model gene regulation in individual tumors, we found the regulation of PD1 signaling to be repressed in primary glioblastomas of patients who exhibited relatively long-term survival, loss of this repression in patients with worse outcome, and loss of repression of the pathway in recurrent compared with primary tumors. This signature of PD1 pathway repression was independent of total tumor mutation burden and methylation, and only weakly associated with immune cell infiltrate, suggesting that repression of the PD1 signaling pathway is an independent prognostic signature in glioblastoma that could potentially be used as a biomarker to predict survival.
PD1 signaling pathway plays an inhibitory regulatory role not only during T-cell exhaustion, leading to suppression of T-cell responses, but also during naïve-to-effector CD8 T-cell differentiation (53, 54). Consistent with this biological role of the PD1 pathway and the association of immune infiltration of tumors, particularly lymphocytes, with better prognosis across cancers (55, 56), we found a nominally significant higher abundance of CD8-positive naïve T cells in long-term survivors compared with short-term survivors. However, there was no significant correlation between CD8-positive naïve T cells abundance levels and PD1 targeting scores, indicating that immune infiltration does not completely explain the association between repression of PD1 signaling and better survival. Our results thus indicate that PD1 pathway regulatory differences occur in cancer cells, or in a combination of cancer and immune cells.
Immunotherapy, and in specific immune checkpoint inhibitors and PD-1/PD-L1 inhibitors have transformed the field of cancer treatment and have been shown effective in different cancer types. While such inhibitors have already been used in a clinical setting in glioblastoma, it is still unknown whether PD1 blockade gives clinical benefit in the disease, in part because glioblastoma is a highly heterogeneous disease (57). As we have shown here, patient-specific regulatory network modeling can help determine whether the pathway is likely to be transcriptionally active in the tumor, and thus may indicate whether it can potentially be targeted. Thus, tumor-specific regulatory networks could be used as a potentially new way to stratify patients with glioblastoma.
Large-scale genome-wide network models as measured here with the PANDA and LIONESS approaches could—in addition to their potential to identify biomarkers—also highlight new avenues for treatment. For example, specific TFs that drive activation of the PD1 pathway could potentially be targeted in the short-term survival group. While TFs have historically been viewed as undruggable, recent advances have made it possible to target them and this is an emerging field in cancer research (58). Many of the TFs that we found to differentially regulate multiple PD1 pathway genes have previously been associated with metastasis, migration, and survival in other cancer types, such as GSX2, ZIC2, HMGA1, and GATA4. This makes them promising targets for treatment that may help revert the PD1 signature toward its repressive state.
Regulatory network rewiring—such as that of the PD1 pathway that we observed in this study—can be sometimes inferred from subtle changes in gene expression, including patient-specific changes in different genes that affect the same biological pathway. Importantly, regulatory network changes do not necessarily derive from differences in the expression levels of TFs themselves, as we previously found in an analysis of gene regulatory networks across 38 tissues (12). Instead, tissue-specific gene expression is regulated by TFs that change their targeting patterns to activate tissue-specific regulatory roles. Differences in these targeting patterns can be caused by a multitude of mechanisms, including the residency time of the TF on the DNA, the TF's protein abundance levels as well as the abundance of other regulatory factors in the cell (59), or epigenetic and posttranscriptional regulation (60, 61). These mechanisms are likely to drive regulatory heterogeneity in cancer in the same way as they drive tissue specificity. We would therefore like to stress the importance of system-wide network modeling in cancer to better understand drivers of heterogeneity in cancer.
In summary, our network analysis uncovered patterns of transcriptional regulation that differentiate long- and short-term glioblastoma survivors and identified differences in regulatory processes involved in immune regulation that can potentially be targeted in the clinic. These results underscore the importance of analyzing gene regulatory networks in addition to exploring differential gene expression, and illustrate how alterations of network structure may be predictive of patient survival and identify possible regulatory targets for therapeutic intervention. Most importantly, the comparative network analysis approach outlined here can be used to investigate the molecular features that drive prognosis in other cancers and complex diseases and thus has the potential to expand the use of individualized network medicine in disease study and management.
Authors' Disclosures
J. Quackenbush reports personal fees from Caris Life Sciences, Guardant Health, and Natera outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
We collected studies in glioblastoma that included survival thresholds through a PubMed search (search term: glioblastoma short-term prognostic signature). As can be seen from this table, a wide range of survival thresholds is used, with thresholds for short-term survival ranging between 7-24 months, and thresholds for long-term survival between 13-36 months after initial diagnosis. The threshold of 1.7yrs, which we chose for our comparison in the discovery datasets, corresponds to 20 months, which lies right in between these two ranges.
Smooth scatterplot visualizing the correlation between edge weights in the condition-specific network modeled on discovery dataset 1 and the condition-specific network modeled on discovery dataset 2. Pearson correlation coefficient (R) and p-value (p) are indicated in the plot.
Differential network topology contrasting the short-term with the long-term survival network. A) Differential network modules significantly enriched for overrepresentation of Gene Ontology terms. Modules are shown for both discovery datasets (D1 and D2). Two modules were significant in both dataset (indicated with M1 and M2). The color in the heatmap indicates the enrichment of genes in the module as observed/expected value (Obs/Exp), with purple representing enriched modules in discovery dataset 1 and green enriched modules in discovery dataset 2. B) "Beeswarm" plot visualizing the distribution of average log differential modularity scores from ALPACA, for transcription factors (TF) and genes (Gene). Higher scores mean higher differential modularity. TFs and genes with significant differential modularity between the two survival groups are labeled.
To ensure that our results were stable across multiple survival thresholds, we repeated the comparative network analysis in the two discovery datasets across multiple survival thresholds, which we ranged from zero to the maximum follow-up, with intervals of 0.1 years, keeping comparisons that had group sizes of at least ten patients. This figure reports the -log10FDR and GSEA enrichment scores (ES) for the different thresholds, for A) discovery dataset 1 and B) discovery dataset 2. Significant results (FDR<0.05) are highlighted with red circles.
xCell values for cell types with significant abundance levels (xCell scores >0.2 in at least one sample) as well as total immune and microenvironment scores. Boxplots represent the median and first and third quantiles, whisker represent 1.5Ã-IQR and are visualized for the long-term and short-term survival groups separately, in the two discovery datasets (A-B) and the validation dataset (C). Cell types with significantly different abundance between long- and short-term survival groups are indicated with asterisks (t-test, **FDR<0.05, *FDR<0.1).
A) Comparison of distributions of immune scores from xCell between the two survival groups in the discovery and validation datasets. In the boxplots, boxes represent the median and first and third quantiles, whiskers represent 1.5Ã-IQR. B) Association of the immune scores with PD1 targeting scores in the discovery and validation datasets. Regression lines with confidence intervals of 0.95 are shown for the long-term (blue) and short-term (red) groups.
Acknowledgments
The authors thank Harold Wang for initial work on the project, Megha Padi for helpful insights on ALPACA, Roel Verhaak for suggestions on covariates to correct for in glioblastoma, the Kuijjer and Quackenbush groups for helpful discussions, and Elisa Bjørgø and Ingrid Kjelsvik for administrative support. This work was supported by the Norwegian Research Council, Helse Sør-Øst, and University of Oslo through the Centre for Molecular Medicine Norway (187615 to M.L. Kuijjer, T. Belova, D. Osorio), the Norwegian Research Council (313932 to M.L. Kuijjer), the NCI/NIH (P50CA165962 to M.L. Kuijjer; R35CA220523 to C.M. Lopes-Ramos, M. Ben Guebila, J. Quackenbush; U24CA231846 to M. Ben Guebila, J. Quackenbush), the Charles A. King Trust Postdoctoral Research Fellowship Program, Sara Elizabeth O'Brien Trust, Bank of America, N.A., Co-Trustees (M.L. Kuijjer), and the Marie Skłodowska-Curie Postdoctoral Scientia Fellows program from the Faculty of Medicine at the University of Oslo, co-funded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant (801133, D. Osorio).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
C.M. Lopes-Ramos: Data curation, formal analysis, supervision, investigation, visualization, methodology, writing–original draft, writing–review and editing. T. Belova: Formal analysis, investigation, visualization, writing–review and editing. T.H. Brunner: Formal analysis, investigation, visualization, writing–review and editing. M. Ben Guebila: Data curation, writing–review and editing. D. Osorio: Data curation, formal analysis, investigation, visualization, writing–review and editing. J. Quackenbush: Conceptualization, resources, supervision, funding acquisition, writing–review and editing. M.L. Kuijjer: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2. Ovaska K, Laakso M, Haapa-Paananen S, Louhimo R, Chen P, Aittomäki V, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med 2010;2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arimappamagan A, Somasundaram K, Thennarasu K, Peddagangannagari S, Srinivasan H, Shailaja BC, et al. A fourteen gene GBM prognostic signature identifies association of immune response pathway and mesenchymal subtype with high risk group. PLoS One 2013;8:e62042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim Y-W, Koul D, Kim SH, Lucio-Eterovic AK, Freire PR, Yao J, et al. Identification of prognostic gene signatures of glioblastoma: a study based on TCGA data analysis. Neuro-oncol 2013;15:829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel VN, Gokulrangan G, Chowdhury SA, Chen Y, Sloan AE, Koyutürk M, et al. Network signatures of survival in glioblastoma multiforme. PLoS Comput Biol 2013;9:e1003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Irshad K, Mohapatra SK, Srivastava C, Garg H, Mishra S, Dikshit B, et al. A combined gene signature of hypoxia and notch pathway in human glioblastoma and its prognostic relevance. PLoS One 2015;10:e0118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glass K, Huttenhower C, Quackenbush J, Yuan G-C. Passing messages between biological networks to refine predicted interactions. PLoS One 2013;8:e64832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van IJzendoorn DG, Glass K, Quackenbush J, Kuijjer ML. PyPanda: a Python package for gene regulatory network reconstruction. Bioinformatics 2016;32:3363–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frey BJ, Dueck D. Clustering by passing messages between data points. Science 2007;315:972–6. [DOI] [PubMed] [Google Scholar]
- 12. Sonawane AR, Platig J, Fagny M, Chen C-Y, Paulson JN, Lopes-Ramos CM, et al. Understanding tissue-specific gene regulation. Cell Rep 2017;21:1077–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopes-Ramos CM, Paulson JN, Chen C-Y, Kuijjer ML, Fagny M, Platig J, et al. Regulatory network changes between cell lines and their tissues of origin. BMC Genomics 2017;18:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glass K, Quackenbush J, Spentzos D, Haibe-Kains B, Yuan G-C. A network model for angiogenesis in ovarian cancer. BMC Bioinformatics 2015;16:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuijjer ML, Tung MG, Yuan G, Quackenbush J, Glass K. Estimating sample-specific regulatory networks. iScience 2019;14:226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopes-Ramos CM, Chen C-Y, Kuijjer ML, Paulson JN, Sonawane AR, Fagny M, et al. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep 2020;31:107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lopes-Ramos CM, Kuijjer ML, Ogino S, Fuchs CS, DeMeo DL, Glass K, et al. Gene regulatory network analysis identifies sex-linked differences in colon cancer drug metabolism. Cancer Res 2018;78:5538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samur MK. RTCGAToolbox: a new tool for exporting TCGA Firehose data. PLoS One 2014;9:e106397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barnett I, Malik N, Kuijjer ML, Mucha PJ, Onnela J-P. EndNote: feature-based classification of networks. Netw Sci 2019;7:438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolstad BM, Bolstad MBM. Package ‘preprocessCore.’; 2013.
- 22. Reifenberger G, Weber RG, Riehmer V, Kaulich K, Willscher E, Wirth H, et al. Molecular characterization of long-term survivors of glioblastoma using genome-and transcriptome-wide profiling. Int J Cancer 2014;135:1822–31. [DOI] [PubMed] [Google Scholar]
- 23. Kwon SM, Kang S-H, Park C-K, Jung S, Park ES, Lee J-S, et al. Recurrent glioblastomas reveal molecular subtypes associated with mechanistic implications of drug-resistance. PLoS One 2015;10:e0140528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuijjer ML, Paulson JN, Salzman P, Ding W, Quackenbush J. Cancer subtype identification using somatic mutation data. Br J Cancer 2018;118:1492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014;158:1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill KE, Kelly AD, Kuijjer ML, Barry W, Rattani A, Garbutt CC, et al. An imprinted non-coding genomic cluster at 14q32 defines clinically relevant molecular subtypes in osteosarcoma across multiple independent datasets. J Hematol Oncol 2017;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Platig J, Castaldi PJ, DeMeo D, Quackenbush J. Bipartite community structure of eQTLs. PLoS Comput Biol 2016;12:e1005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Padi M, Quackenbush J. Detecting phenotype-driven transitions in regulatory network structure. NPJ Syst Biol Appl 2018;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miller JA, Gouwens NW, Tasic B, Collman F, van Velthoven CT, Bakken TE, et al. Common cell type nomenclature for the mammalian brain. eLife 2020;9:e59928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 1995;57:289–300. [Google Scholar]
- 36. Chodor W. RTCGA.RPPA: RPPA datasets from The Cancer Genome Atlas Project; 2015. Available from: https://CRAN.R-project.org/package=vegan.
- 37. Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature 2020;578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, et al. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol 2005;7:186–94. [DOI] [PubMed] [Google Scholar]
- 39. Denard B, Seemann J, Chen Q, Gay A, Huang H, Chen Y, et al. The membrane-bound transcription factor CREB3L1 is activated in response to virus infection to inhibit proliferation of virus-infected cells. Cell Host Microbe 2011;10:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seitan VC, Krangel MS, Merkenschlager M. Cohesin, CTCF and lymphocyte antigen receptor locus rearrangement. Trends Immunol 2012;33:153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2013;42:D472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liberzon A. A description of the molecular signatures database (MSigDB) web site. Methods Mol Biol 2014;1150:153–60. [DOI] [PubMed] [Google Scholar]
- 43. Zeng W-J, Yang Y-L, Liu Z-Z, Wen Z-P, Chen Y-H, Hu X-L, et al. Integrative analysis of DNA methylation and gene expression identify a three-gene signature for predicting prognosis in lower-grade gliomas. Cell Physiol Biochem 2018;47:428–39. [DOI] [PubMed] [Google Scholar]
- 44. Salero E, Pérez-Sen R, Aruga J, Giménez C, Zafra F. Transcription factors Zic1 and Zic2 bind and transactivate the apolipoprotein E gene promoter. J Biol Chem 2001;276:1881–8. [DOI] [PubMed] [Google Scholar]
- 45. Wang J, Xu X, Mo S, Tian Y, Wu J, Zhang J, et al. Involvement of microRNA-1297, a new regulator of HMGA1, in the regulation of glioma cell growth in vivo and in vitro. Am J Transl Res 2016;8:2149–58. [PMC free article] [PubMed] [Google Scholar]
- 46. Colamaio M, Tosti N, Puca F, Mari A, Gattordo R, Kuzay Y, et al. HMGA1 silencing reduces stemness and temozolomide resistance in glioblastoma stem cells. Expert Opin Ther Targets 2016;20:1169–79. [DOI] [PubMed] [Google Scholar]
- 47. Vaitkienė P, Skiriutė D, Skauminas K, Tamašauskas A. GATA4 and DcR1 methylation in glioblastomas. Diagn Pathol 2013;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yao Z, Luo J, Hu K, Lin J, Huang H, Wang Q, et al. ZKSCAN1 gene and its related circular RNA (circZKSCAN1) both inhibit hepatocellular carcinoma cell growth, migration, and invasion but through different signaling pathways. Mol Oncol 2017;11:422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Andersen CL, Christensen LL, Thorsen K, Schepeler T, Sørensen FB, Verspaget HW, et al. Dysregulation of the transcription factors SOX4, CBFB and SMARCC1 correlates with outcome of colorectal cancer. Br J Cancer 2009;100:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farahbod M, Pavlidis P. Differential coexpression in human tissues and the confounding effect of mean expression levels. Bioinformatics 2018;35:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol 2016;17:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A 2018;115:4749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pages F, Galon J, Dieu-Nosjean M, Tartour E, Sautes-Fridman C, Fridman W. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 2010;29:1093–102. [DOI] [PubMed] [Google Scholar]
- 56. Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic analysis of immune cell infiltrates across 11 tumor types. J Natl Cancer Inst 2016;108:djw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Khasraw M, Reardon DA, Weller M, Sampson JH. PD-1 inhibitors: do they have a future in the treatment of glioblastoma? Clin Cancer Res 2020;26:5287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bushweller JH. Targeting transcription factors in cancer—from undruggable to reality. Nat Rev Cancer 2019;19:611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell 2018;172:650–65. [DOI] [PubMed] [Google Scholar]
- 60. Kuijjer ML, Fagny M, Marin A, Quackenbush J, Glass K. PUMA: PANDA using microRNA associations. Bioinformatics 2020;36:4765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017;356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We collected studies in glioblastoma that included survival thresholds through a PubMed search (search term: glioblastoma short-term prognostic signature). As can be seen from this table, a wide range of survival thresholds is used, with thresholds for short-term survival ranging between 7-24 months, and thresholds for long-term survival between 13-36 months after initial diagnosis. The threshold of 1.7yrs, which we chose for our comparison in the discovery datasets, corresponds to 20 months, which lies right in between these two ranges.
Smooth scatterplot visualizing the correlation between edge weights in the condition-specific network modeled on discovery dataset 1 and the condition-specific network modeled on discovery dataset 2. Pearson correlation coefficient (R) and p-value (p) are indicated in the plot.
Differential network topology contrasting the short-term with the long-term survival network. A) Differential network modules significantly enriched for overrepresentation of Gene Ontology terms. Modules are shown for both discovery datasets (D1 and D2). Two modules were significant in both dataset (indicated with M1 and M2). The color in the heatmap indicates the enrichment of genes in the module as observed/expected value (Obs/Exp), with purple representing enriched modules in discovery dataset 1 and green enriched modules in discovery dataset 2. B) "Beeswarm" plot visualizing the distribution of average log differential modularity scores from ALPACA, for transcription factors (TF) and genes (Gene). Higher scores mean higher differential modularity. TFs and genes with significant differential modularity between the two survival groups are labeled.
To ensure that our results were stable across multiple survival thresholds, we repeated the comparative network analysis in the two discovery datasets across multiple survival thresholds, which we ranged from zero to the maximum follow-up, with intervals of 0.1 years, keeping comparisons that had group sizes of at least ten patients. This figure reports the -log10FDR and GSEA enrichment scores (ES) for the different thresholds, for A) discovery dataset 1 and B) discovery dataset 2. Significant results (FDR<0.05) are highlighted with red circles.
xCell values for cell types with significant abundance levels (xCell scores >0.2 in at least one sample) as well as total immune and microenvironment scores. Boxplots represent the median and first and third quantiles, whisker represent 1.5Ã-IQR and are visualized for the long-term and short-term survival groups separately, in the two discovery datasets (A-B) and the validation dataset (C). Cell types with significantly different abundance between long- and short-term survival groups are indicated with asterisks (t-test, **FDR<0.05, *FDR<0.1).
A) Comparison of distributions of immune scores from xCell between the two survival groups in the discovery and validation datasets. In the boxplots, boxes represent the median and first and third quantiles, whiskers represent 1.5Ã-IQR. B) Association of the immune scores with PD1 targeting scores in the discovery and validation datasets. Regression lines with confidence intervals of 0.95 are shown for the long-term (blue) and short-term (red) groups.
Data Availability Statement
The input data, clinical information, code to reproduce all individual patient networks, and the entire collection of reconstructed networks used in this study are available on https://grand.networkmedicine.org/cancers/GBM_cancer/. Code to reproduce the results and figures is available at http://netbooks.networkmedicine.org/ under “Case studies—Gene regulatory network analysis in Glioblastoma.”