

Abstract
Liver fibrosis is one of the most severe pathologic consequences of chronic liver diseases, and effective therapeutic strategies are urgently needed. Proton pump inhibitors (PPIs) are H+/K+-ATPase inhibitors and currently used to treat acid-related diseases such as gastric ulcers, which have shown other therapeutic effects in addition to inhibiting acid secretion. However, few studies have focused on PPIs from the perspective of inhibiting hepatic fibrosis. In the present study, we investigated the effects of pantoprazole (PPZ), a PPI, against liver fibrosis in a bile duct ligation (BDL) rat model, human hepatic stellate cell (HSC) line LX-2 and mouse primary HSCs (pHSCs), and explored the potential mechanisms underlying the effects of PPZ in vitro and in vivo. In BDL rats, administration of PPZ (150 mg· kg−1· d−1, i.p. for 14 d) significantly attenuated liver histopathological injury, collagen accumulation, and inflammatory responses, and suppressed fibrogenesis-associated gene expression including Col1a1, Acta2, Tgfβ1, and Mmp-2. In LX-2 cells and mouse pHSCs, PPZ (100–300 μM) dose-dependently suppressed the levels of fibrogenic markers. We conducted transcriptome analysis and subsequent validation in PPZ-treated LX-2 cells, and revealed that PPZ inhibited the expression of Yes-associated protein (YAP) and its downstream targets such as CTGF, ID1, survivin, CYR61, and GLI2. Using YAP overexpression and silencing, we demonstrated that PPZ downregulated hepatic fibrogenic gene expression via YAP. Furthermore, we showed that PPZ promoted the proteasome-dependent degradation and ubiquitination of YAP, thus inhibiting HSC activation. Additionally, we showed that PPZ destabilized YAP by disrupting the interaction between a deubiquitinating enzyme OTUB2 and YAP, and subsequently blocked the progression of hepatic fibrosis.
Keywords: liver fibrosis, pantoprazole, bile duct ligation, hepatic stellate cells, YAP, OTUB2
Introduction
As one of the most common pathological consequences of chronic liver diseases, liver fibrosis has emerged as a major health problem worldwide. Liver fibrosis is a wound-healing response to various hepatic injuries, including alcohol abuse, hepatitis virus infection, cholestasis, and non-alcoholic steatohepatitis (NASH), and is characterized by the excessive accumulation of extracellular matrix (ECM) components following the activation and proliferation of hepatic stellate cells (HSCs). In the normal liver, quiescent HSCs reside in the space of Disse and are mainly responsible for the storage of vitamin A. Upon liver injury, HSCs are activated, become myofibroblast-like cells, and accumulate at the sites of tissue repair, secreting massive amounts of ECM, including fibrillar collagen [1, 2]. HSC activation is regulated by various cytokines and signalling pathways, among which transforming growth factor β (TGFβ) appears to be a key mediator. TGFβ favours HSC conversion to myofibroblast-like cells, induces ECM synthesis, and inhibits ECM degradation [3]. Thus, strategies aimed at suppressing TGFβ synthesis or signalling pathways can prevent or ameliorate liver fibrosis.
Yes-associated protein (YAP), the major effector of the Hippo pathway, plays an essential role in controlling liver cell fate and HSC activation [4, 5]. Upon activation, YAP translocates into the nucleus and primarily binds to TEA domain-containing protein (TEAD) 1–4 to promote the transcription of downstream target genes, most of which are associated with liver fibrogenesis, such as connective tissue growth factor (CTGF) and TGFβ [6]. CTGF induces the synthesis and secretion of ECM components, especially fibrillar collagens [7–9]. Interestingly, it has been reported that nuclear YAP also interacts with TGFβ-induced Smad2/3 to promote TGFβ-dependent transcriptional changes, suggesting that YAP co-ordinately regulates TGFβ signalling [10]. Moreover, experimental evidence suggests that YAP directly regulates the transcription of Notch signalling, including Notch1/2, Jag1 and Notch targets Hes1 and Sox9, whose activities are important in the progression of fibrosis [5]. Previous studies have shown that YAP stimulates profibrotic gene expression at the early stages of HSC activation [4]. In addition, elevated YAP expression strongly correlates with bile duct proliferation and fibrosis [11]. Therefore, YAP inhibition is a potential therapeutic approach in hepatic fibrosis.
Although the reversibility of liver fibrosis has been documented, few therapeutic strategies are available clinically. Thus, there is an urgent need to develop effective and safe antifibrotic strategies, especially those targeting activated HSCs. Pantoprazole (PPZ), a proton pump inhibitor (PPI), is an essential H+-K+-ATPase inhibitor that is commonly used to treat acid-related diseases such as gastric ulcers. Interestingly, accumulating evidence has demonstrated that PPIs possess other therapeutic effects in addition to inhibiting acid secretion, such as anti-inflammatory [12, 13], antioxidative [14], antifungal [15, 16], proapoptotic [14, 17, 18], autophagy-modulating [19, 20], and chemosensitizing effects [21]. However, few studies have focused on PPIs from the perspective of inhibiting hepatic fibrosis. Esomeprazole has been shown to have antifibrotic effects on CCl4-induced rats by modulating oxidative stress and inflammatory and apoptotic markers [22]. It has also been demonstrated that lansoprazole administration inhibits the progression of liver fibrosis in choline-deficient amino acid-defined (CDAA) diet-induced NASH model rats by downregulating the expression of TGF-β and interleukin (IL)-6 [23]. However, there are no data available on the effects of PPZ on cholestatic fibrosis. In this study, we investigated whether PPZ could exert antifibrotic effects on a bile duct ligation (BDL) rat model, the human HSC line LX-2, and mouse primary HSCs (pHSCs) and explored the potential mechanisms underlying the function of PPZ in vitro and in vivo.
Materials and methods
Reagents
Pantoprazole sodium was purchased from Yangzi River Pharmaceutical Group LTD (Jiangsu, China). TGFβ1 was acquired from R&D System (Minneapolis, MN, USA). The pcDNA3.1/Flag-YAP and pcDNA3.1/HA-Ubiquitin plasmids were purchased from YouBio (Changsha, China). The siRNA-YAP, Protein A/G Plus-agarose, and anti-TEAD2 antibody (sc-67115) were purchased from Santa Cruz Biotechnology Inc (CA, USA). The anti-α-smooth muscle actin antibody (α-SMA, A5228) and cycloheximide (CHX) were obtained from Sigma-Aldrich (St Louis, MO, USA), and MG132 was acquired from Selleck Chemicals (Shanghai, China). Anti-collagen type I α 1 (COL1Α1, PAB17205) and anti-TGFβ1 (ab179695) antibodies were purchased from Abnova (Colorado, USA) and Abcam (Cambridge, UK), respectively. Anti-matrix metalloproteinase 2 (MMP2, 10373-2-AP), anti-inhibitor of DNA binding 1 (ID1, 18475-1-AP), anti-Flag-tag (20543‐1‐AP), anti-HA-tag (51064‐2‐AP), and anti-Lamin B1 (66095-1-Ig) antibodies were purchased from Proteintech (Wuhan, China). Antibodies against CTGF (#86641), cysteine-rich protein 61 (CYR61, #14479), IL-6 (#12153), IL-1β (#12242), phospho-Smad2/3 (#8828), Smad2/3 (#8685), tumour necrosis factor-α (TNFα, #6945), survivin (#2808), YAP (#14074), and GAPDH (#5174) were purchased from Cell Signalling Technology (Danvers, USA). The anti-OTUB2 (ER64479) antibody was acquired from HuaAn Biotechnology (Hangzhou, China).
Animal experiments
Male Sprague-Dawley rats (8 weeks old, 180–200 g, SPF class) were acquired from the Institute of Laboratory Animal Science (Chinese Academy of Medical Sciences, Beijing, China). The research protocols were performed in accordance with the Good Laboratory Practice regulations for non-clinical laboratory studies of drugs issued by the National Scientific and Technological Committee of the People’s Republic of China. The rats were housed in an environmentally controlled room (pathogen-free, 20–24 °C, 40%–60% relative humidity, and 12 h alternating light/dark cycle) and provided standard rat feedstuff and water ad libitum. Rats were randomly divided into three groups (n = 6): the sham, BDL-normal saline (NS), and BDL-PPZ groups. Twelve rats underwent BDL. These rats were anaesthetized with isoflurane. We opened the abdomen, isolated the choledochal duct, and then ligated the bile duct using surgical sutures. Twenty-four hours after surgery, BDL rats received daily i.p. injections of NS or 150 mg· kg−1 PPZ resuspended in NS for 14 d. A sham operation served as a healthy control. Body weight was measured daily. After 14 d, the animals were sacrificed in a random order after an overnight fast. Rat liver maximum lobule samples were collected for further analyses.
Serum and liver tissue biochemistry
Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), gamma glutamyl transpeptidase (GGT), cholesterol (CHO), low-density lipoprotein cholesterol (LDL), high-density lipoprotein cholesterol (HDL), triglyceride (TG), total bile acid (TBA), and total bilirubin (TBiL) concentrations were analyzed with a Hitachi 7100 Analyzer using kits purchased from Zhongsheng Beikong Biotechnology (Beijing, China). Liver tissue TBA level was determined using a kit obtained from Nanjing Jiancheng Company (Nanjing, China) according to the manufacturer’s instructions.
Histological analysis of liver tissue samples
The liver tissue was fixed with formalin, embedded in paraffin, and cut into sections. Liver tissue sections were stained with hematoxylin and eosin (H&E) and Sirius red. Liver necrosis, inflammation, and bile duct proliferation were quantified on a 1−5 scale in a blinded manner using a Leica DM1000 microscope. The liver hydroxyproline concentration was determined utilizing commercially available kits (Nanjing Jiancheng, Nanjing, China) according to the manufacturer’s instructions.
Immunohistochemistry
Liver sections were deparaffinized, hydrated, and incubated in citrate buffer at 95 °C for 40 min for antigen retrieval. After the removal of endogenous catalase with 3% H2O2 for 30 min, the sections were blocked with 5% bovine serum albumin for 1–2 h followed by incubation overnight at 4 °C with primary antibody against α-SMA (1:250 dilution). After being washed three times with phosphate-buffered saline, tissue sections were incubated with secondary antibody for 20 min at 37 °C and then washed three times. Colour was developed by incubation with 3,3'-diaminobenzidine tetrahydrochloride for 2–5 min, and specific staining was visualized by light microscopy.
Cell culture, isolation, and treatment
The human HSC line LX-2 was cultured in DMEM/GlutaMAX I medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S, Invitrogen, Carlsbad, CA, USA). After the cells reached 70%−80% confluence, the cells were starved in a serum-free medium for 24 h and treated with TGF-β1 (2 ng· mL−1) and different concentrations of PPZ (dissolved in NS) for an additional 24 h. Mouse pHSCs were isolated according to previously reported procedures [24]. These cells were cultured in DMEM (high glucose) with 10% FBS and 1% antibiotics, plated in untreated flasks, and presented a fibroblast-like morphology after 7 d. Therefore, these cells did not need starvation or TGFβ1-induced activation. HEK293T cells (RRID: CVCL_0063) were cultured in basic DMEM with 10% FBS and 1% antibiotics. All cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. LX-2 and HEK293T cells were transfected with plasmids or siRNAs in the presence of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and serum-free Opti-MEM (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Quantitative real-time PCR
Total RNA from cells or liver tissue samples was extracted and purified using an ultrapure RNA Kit (CWBIO, Beijing, China), and reverse-transcribed into cDNAs using a Transcriptor First Strand cDNA Synthesis Kit (Roche). The relative expression of specific genes was determined using TaqMan primers/probes (Applied Biosystems, Foster City, CA, USA) with an ABI QuantStudio 3 real-time PCR system. The data were calculated using the equation 2−ΔΔCt and normalized to GAPDH.
Immunoprecipitation and Western blot analysis
Cells or liver tissues were lysed with RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) containing 1% PMSF. After the samples were centrifuged at 12,000 ×g for 15 min, the total protein concentrations were determined using a BCA protein assay kit (Thermo Scientific, USA). Equal amounts of lysate were immunoprecipitated with appropriate antibodies overnight at 4 °C and then incubated with Protein A/G Plus-agarose for 4 h at 4 °C. Thereafter, the precipitants were washed five times with washing buffer and eluted with 2× SDS loading buffer for 10 min at 95 °C. The immunoprecipitated proteins were then separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA). The membranes were blocked in 5% (w/v) non-fat milk before being incubated with primary antibodies (1:1000 in primary antibody diluent) at 4 °C overnight. Immunoblots were developed using horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000) and visualized by enhanced chemiluminescent (ECL) reagents (Millipore, USA) with a Tanon 5200 imaging system (Shanghai, China). The relative protein level of each band was normalized to GAPDH.
Transcriptome analysis
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and a NucleoSpin RNA clean-up kit (Macherey-Nagel, Germany) were used to isolate and purify the total RNA, respectively. RNA samples from four independent experiments were mixed equally in each group. Oligo(dT)-attached magnetic beads were used to purify mRNA. Then first-strand cDNA was generated using random hexamer-primed reverse transcription, followed by a second-strand cDNA synthesis, and A-tailing mix and RNA index adapters were added by incubating to carry out end repair. After amplification and purification, the cDNA products were validated on the Agilent Technologies 2100 bioanalyzer for quality control, heated denatured, and circularized by the splint oligo sequence to get the final library. The final library was amplified to make DNA nanoballs (DNBs) which were loaded into the patterned nanoarray, and pair-end 100 bases reads were generated on BGIseq500 platform (BGI-Shenzhen, China). The gene expression was considered significant if the fold change value was >1.5 or <0.67, and P < 0.05.
Nuclear and cytoplasmic protein extraction
Nuclear and cytoplasmic proteins were extracted from LX-2 cells with a Nuclear-Cytosol Extraction Kit (Applygen Technologies Inc., Beijing, China) according to the manufacturer’s instructions. The extracts were mixed with loading buffer and evaluated by Western blotting as described above.
Statistical analysis
All in vitro experiments were repeated three times. For all animal experiments, six randomized animals were included in each group. The data are reported as the mean ± SD and were analyzed by one-way analysis of variance (ANOVA). All statistical analyses were performed using SPSS/Win 13.0 software. A P value < 0.05 was considered statistically significant.
Results
PPZ attenuated BDL‐induced liver injury in rats
BDL generally results in severe hepatocellular injury and liver fibrosis. The serum and liver tissue levels of biochemical markers were significantly increased in BDL rats compared with sham rats. Significant reductions in levels of the liver enzymes ALT and AST were observed when the BDL rats were treated with PPZ (Table 1). Notably, our results showed that PPZ decreased the serum TBA and TBiL and hepatic TBA levels in BDL rats, suggesting that PPZ affected bile acid metabolism (Table 1). Figure 1a shows the gross appearance of representative rat livers from the sham, BDL‐NS, and BDL‐PPZ groups. BDL resulted in hepatomegaly, irregularity of the liver surface, and loss of the normal ratio of hepatic lobes, and these effects were significantly improved after PPZ administration. H&E staining demonstrated that the histological structure of the liver in the BDL group was severely damaged, paralleled by concomitant extensive parenchyma necrosis and newly formed bile ducts. PPZ administration substantially improved these pathological changes compared with those of the BDL group (Fig. 1b). In a double-blinded assessment, the PPZ-treated group had significantly lower bile duct proliferation and inflammation scores than the BDL group (Fig. 1c). The notable upregulation of hepatic IL-6, IL-1β, and TNFα mRNA and protein levels in BDL rats compared with sham rats suggests that BDL could induce potent inflammatory reactions in rats. After PPZ treatment, the expression levels of these inflammatory cytokines were obviously reduced (Fig. 1d, e), which was consistent with the reduction in inflammation observed by H&E staining. Next, we evaluated the antifibrotic effect of PPZ by Sirius red staining and hydroxyproline concentration assays. In BDL rats, Sirius red-stained collagen fibrils extended to not only the portal areas but also the hepatic parenchyma, and this effect was markedly attenuated by PPZ treatment (Fig. 2a). Additionally, PPZ dramatically reduced the BDL-induced increase in liver hydroxyproline concentration (Fig. 2b). These results indicated that PPZ could ameliorate BDL-induced liver injury and exert a therapeutic effect on hepatic fibrosis.
Table 1.
Serum and liver tissue biochemical analysis of BDL rats.
| Sham (n = 6) | BDL-NS (n = 6) | BDL-PPZ (n = 6) | |
|---|---|---|---|
| Serum AST (U· L−1) | 167.71 ± 41.43 | 711.67 ± 119.18## | 349.33 ± 204.71** |
| Serum ALT (U· L−1) | 27.25 ± 6.34 | 100.33 ± 28.06## | 50.00 ± 24.75** |
| Serum ALP (U· L−1) | 188.25 ± 62.37 | 391.33 ± 74.07## | 268.17 ± 125.01 |
| Serum GGT (U· L−1) | / | 52.50 ± 16.02## | 30.17 ± 36.69 |
| Serum CHO (mmol· L−1) | 1.50 ± 0.33 | 3.57 ± 0.09## | 3.28 ± 0.67 |
| Serum LDL (mmol· L−1) | 0.23 ± 0.07 | 2.23 ± 0.18## | 1.57 ± 0.93 |
| Serum HDL (mmol· L−1) | 0.65 ± 0.09 | 0.61 ± 0.04 | 0.71 ± 0.13 |
| Serum TG (mmol· L−1) | 0.46 ± 0.02 | 0.94 ± 0.13## | 0.89 ± 0.55 |
| Serum TBA (μmol· L−1) | 6.33 ± 1.43 | 255.23 ± 50.81## | 95.45 ± 89.95** |
| Serum TBiL (μmol· L−1) | 0.55 ± 0.52 | 168.60 ± 22.63## | 69.35 ± 70.75* |
| Liver tissue TBA (nmol· mg−1) | 67.8 ± 45.3 | 184.25 ± 75.50## | 90.41 ± 50.60* |
Values are presented as the mean ± SD (n = 6 per group).
AST aspartate aminotransferase, ALT alanine aminotransferase, ALP alkaline phosphatase, GGT gamma-glutamyl transpeptidase, CHO cholesterol, LDL low-density lipoprotein cholesterol, HDL high-density lipoprotein cholesterol, TG triglyceride, TBA total bile acid, TBiL total bilirubin.
##P < 0.01 vs. the sham group; *P < 0.05, **P < 0.01, vs. the BDL-NS group.
Fig. 1. PPZ attenuated BDL-induced liver injury in rats.

a Gross morphology of representative rat livers from the sham, BDL‐NS, and BDL‐PPZ groups. b Liver pathological changes were detected by H&E staining (magnification × 400). c Blinded quantitative assessment of liver bile duct proliferation, inflammation, and necrosis. d Hepatic mRNA expression levels of Il-6, Il-1β, and Tnfα in rats from the sham, BDL‐NS, and BDL‐PPZ groups. e Western blot analysis and semi‐quantitation of IL-6, IL-1β, and TNFα levels in each group of rats. The data are presented as the mean ± SD, n = 6. #P < 0.05, ##P < 0.01 vs. the sham group; *P < 0.05, **P < 0.01 vs. the BDL‐NS group.
Fig. 2. PPZ significantly ameliorated liver fibrosis in BDL rats.

a The degree of liver collagen accumulation was determined by Sirius red staining (magnification × 400). b Liver hydroxyproline levels were determined by a hydroxyproline assay kit. c The mRNA expression levels of Col1a1, Acta2, Tgfβ1, and Mmp-2 were analyzed by real-time PCR. d Western blot analysis and semi‐quantitation of COL1A1, α‐SMA, MMP-2, TGFβ1, P-Smad2/3, and Smad2/3 expression in the sham, BDL‐NS, and BDL‐PPZ groups. e Immunohistochemical analysis of α‐SMA expression in liver samples. For b−e, the data are presented as the mean ± SD, n = 6. #P < 0.05, ##P < 0.01 vs. the sham group; *P < 0.05, **P < 0.01 vs. the BDL‐NS group.
PPZ significantly reduced hepatic fibrogenic gene expression in BDL rats
To further verify the antifibrotic activity of PPZ in vivo, we measured the expression of fibrogenic markers, including COL1A1, α-SMA, TGFβ1, and MMP-2. COL1A1 is the most abundant component of the ECM and is markedly increased in liver fibrosis. α-SMA (encoded by ACTA2) is a typical biomarker of activated HSCs. MMP-2 is an endogenous protease that remodels the ECM, and its rapid accumulation promotes liver fibrogenesis [25]. Our results demonstrated that PPZ significantly lowered the BDL-induced increases in the mRNA expression of fibrogenic markers (Fig. 2c). As shown in Fig. 2d, the protein levels of COL1A1, α-SMA, TGFβ1, and MMP-2 were also markedly reduced by PPZ administration. In addition, the phosphorylation of Smad2/3 was downregulated by PPZ, indicating an inhibitory effect on the TGFβ1/Smad pathway, whose activation promotes the transcription of most fibrosis-related genes. Immunohistochemical analysis substantiated the PPZ-mediated repression of α-SMA expression (Fig. 2e). These findings suggested that PPZ treatment could alleviate BDL-induced liver fibrosis in rats.
PPZ markedly suppressed hepatic fibrogenic gene expression in HSCs
The next step was to evaluate the antifibrotic activity of PPZ in the liver in vitro. The human HSC line LX-2 was starved with serum-free medium for 24 h before TGFβ1 stimulation with or without PPZ treatment. Mouse pHSCs can self-activate during culture in a normal medium. As shown in Fig. 3a, PPZ dose-dependently downregulated the mRNA expression of various fibrogenic markers, including COL1A1, α-SMA, MMP-2 and TGFβ1, in LX-2 cells. The protein expression of these fibrogenic markers was also attenuated in a dose-dependent manner after treatment with PPZ in both LX-2 cells and mouse pHSCs. Furthermore, consistent with the in vivo results, the phosphorylation of Smad2/3 was also downregulated in HSCs by PPZ (Fig. 3b, c). These results indicated that PPZ could serve as a potent antifibrogenic agent for human/mouse HSCs.
Fig. 3. PPZ markedly suppressed the expression of hepatic fibrosis-related genes in HSCs.

LX-2 cells were starved for 24 h and then treated with TGF-β1 (2 ng· mL−1) and different concentrations of PPZ for an additional 24 h. a The mRNA expression levels of COL1A1, ACTA2, MMP-2, and TGFβ1 in LX-2 cells were measured by real-time PCR. b Western blot analysis and semi‐quantitation of COL1A1, MMP-2, α‐SMA, TGFβ1, P-Smad2/3, and Smad2/3 protein levels in LX-2 cells. The data are presented as the mean ± SD of three independent experiments. #P < 0.05, ##P < 0.01 vs. the control group; *P < 0.05, **P < 0.01 vs. the TGFβ1-treated group. c Mouse pHSCs were treated with 100 and 200 µM PPZ for 24 h after 7 d of isolation. The protein levels of COL1A1, MMP-2, α‐SMA, TGFβ1, P-Smad2/3, and Smad2/3 were measured using Western blot analysis. The data are presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. the control group of mouse pHSCs.
To investigate the genome-wide alterations in PPZ-treated LX-2 cells, we conducted transcriptome sequencing experiments and analyzed the gene expression profiles. As shown in Fig. 4a, 269 upregulated genes and 401 downregulated genes were identified in LX-2 cells treated with 2 ng· mL−1 TGFβ1 compared with untreated cells. Furthermore, 723 genes were upregulated and 559 genes were downregulated in PPZ-treated LX-2 cells compared with TGFβ1-treated cells. We found that a total of 212 genes were included in the intersection between two sets of significantly altered genes. These genes are involved in a variety of biological processes and signalling pathways, some of which are closely associated with liver fibrosis (Fig. 4b, c). Among these genes, 87 have been documented to be involved in hepatic fibrosis (Fig. 4d). CTGF and ID1, two well-known target genes of the Hippo pathway effector YAP, were downregulated 0.32-fold and 0.48-fold, respectively, suggesting that PPZ could inhibit YAP signaling.
Fig. 4. Transcriptome analysis of LX-2 cells.

LX-2 cells were starved for 24 h and then treated with 2 ng· mL−1 TGFβ1 (2 T) and 200 µM PPZ for an additional 24 h. The total RNA was extracted for transcriptome analysis. a Number of genes significantly altered in the transcriptome analysis. b Gene ontology (GO) analysis (biological process) based on the classification of gene numbers, expression, and significance probability. c KEGG pathways were classified by DAVID functional annotation bioinformatics of differential genes in transcriptome analysis. d Expression profiling and heat map of the abovementioned grouping of 60 fibrogenesis-related genes.
PPZ inhibited YAP signalling
To verify the blockade of YAP signalling by PPZ, we further measured the expression of YAP and its target genes in vitro and in vivo. PPZ administration dramatically downregulated the protein level of YAP and the most highly characterized target gene, CTGF, in BDL rat liver samples, LX-2 cells, and mouse pHSCs (Fig. 5a, c, e). Both the mRNA and protein levels of survivin, a well-known YAP-inducible gene, were significantly diminished in PPZ-treated BDL rats compared to untreated rats (Fig. 5a, b). Given the documented roles of survivin in enhancing proliferation and suppressing apoptosis [26, 27], our findings suggested that the downregulation of survivin could contribute to the inhibitory effect of PPZ on hepatomegaly and bile duct proliferation in BDL rats. The activation of transcription factor ID1 in HSCs represents a critical mediator of transdifferentiation that might be involved in hepatic fibrogenesis [28]. CYR61 has been identified as an important constituent of the fibrotic liver by proteomic analysis of ECM from the HSCs [29]. Glioblastoma protein 2 (GLI2) is an important transcription factor in regulating HSC activation and liver fibrosis [30]. In LX-2 cells, PPZ treatment dose‐dependently suppressed the protein expression of ID1 and CYR61 and the mRNA expression of GLI2, which was due to the blockade of YAP and was conducive to the antifibrotic effects of PPZ (Fig. 5c, d). These results confirmed that YAP, as a key transcription factor associated with liver fibrosis, was inhibited by PPZ treatment in vitro and in vivo.
Fig. 5. PPZ inhibited YAP and the expression of its downstream target genes.

a The protein levels of YAP, CTGF, and survivin in rat liver samples. b The mRNA levels of Ctgf and survivin in rat liver samples. For a, b, the data are presented as the mean ± SD, n = 6. #P < 0.05, ##P < 0.01 vs. the sham group; *P < 0.05, **P < 0.01 vs. the BDL‐NS group. c The protein levels of YAP, CTGF, ID1, and CYR61 in LX-2 cells. d The mRNA levels of CTGF, survivin, and GLI2 in LX-2 cells. For c, d, the data are presented as the mean ± SD of three independent experiments. #P < 0.05, ##P < 0.01 vs. the control group; *P < 0.05, **P < 0.01 vs. the TGFβ1-treated group. e The protein levels of YAP and CTGF in mouse pHSCs. The data are presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. the control group of mouse pHSCs. f The nuclear and cytoplasmic proteins were isolated from LX-2 cells after PPZ treatment and analyzed by Western blotting. g The interaction between YAP and TEAD2 was investigated using immunoprecipitation assays in LX-2 cells.
YAP activation is associated with its translocation from the cytoplasm into the nucleus. Therefore, we assayed whether PPZ affected YAP localization. Indeed, our results showed that the translocation of YAP from the cytoplasm to the nucleus was suppressed by PPZ in a dose-dependent manner. Notably, however, cytoplasmic YAP levels were also attenuated after PPZ treatment, suggesting that the stability of YAP might be damaged (Fig. 5f). The interaction between nuclear YAP and TEAD2 is an important premise in controlling a complex transcriptional programme. Coimmunoprecipitation analyses showed that PPZ strongly inhibited YAP/TEAD2 complex formation (Fig. 5g). These results demonstrated that PPZ inhibited the translocation of YAP from the cytoplasm to the nucleus, interfering with the formation of YAP/TEAD2 conjugates and consequently decreasing the expression of downstream target genes.
PPZ downregulated hepatic fibrogenic gene expression via YAP
Next, we sought to determine whether YAP plays a role in PPZ-mediated downregulation of fibrogenic gene expression. LX-2 cells were transfected with pcDNA3.1/Flag-YAP plasmids before TGFβ1 stimulation and PPZ treatment, and YAP protein was effectively overexpressed, resulting in higher expression of fibrogenic markers, including COL1A1, α-SMA, and TGFβ1, than that in vector-transfected cells. As expected, the inhibitory effect of PPZ on fibrogenic marker expression was weakened by YAP overexpression (Fig. 6a). In contrast, the suppressive effect of PPZ was potentiated when the expression of YAP was knocked down by RNA interference (Fig. 6b). As evidenced by the knockdown and overexpression experiments, YAP could be a target of PPZ in downregulating the expression of hepatic fibrogenic genes in LX-2 cells.
Fig. 6. PPZ downregulated the expression of hepatic fibrogenic genes via YAP.

LX-2 cells were transfected with pcDNA3.1/Flag-YAP plasmids (2.5 µg) (a) or siRNA-YAP (50 nM) (b) for 24 h, and followed by 2 ng· mL−1 TGFβ1 (2T) and 200 µM PPZ treatment for an additional 24 h. The protein expression levels of YAP and fibrogenic markers, including COL1A1, α-SMA, and TGFβ1, were measured by Western blotting. The data are presented as the mean ± SD of three independent experiments. #P < 0.05, ##P < 0.01 vs. the pcDNA3.1/si-Control + 2T + PPZ group; *P < 0.05, **P < 0.01 vs. the pcDNA3.1/Flag-YAP/si-YAP + 2T group; △P < 0.05 vs. the pcDNA3.1/Flag-YAP/si-YAP + 2T + PPZ group.
PPZ promoted the proteasome‐dependent degradation and ubiquitination of YAP
To further determine whether the inhibitory effect of PPZ on the YAP protein was due to the blockade of protein synthesis, LX-2 cells were treated with the protein translation inhibitor CHX. In the absence of protein translation, the YAP protein level decreased in a time-dependent manner (Fig. 7a). However, a more rapid reduction in YAP protein levels was observed in LX-2 cells treated with PPZ in the presence of CHX, indicating that PPZ-induced attenuation of YAP was not due to the blockade of protein translation but to posttranslational modification. We next explored whether PPZ affected the stability of the YAP protein. As shown in Fig. 7b, after treatment with the proteasome inhibitor MG132, the PPZ-induced reduction in YAP was significantly reversed, suggesting that PPZ decreased the cellular level of the YAP protein through a proteasome-dependent degradation pathway. A series of bands representing the ubiquitinated YAP protein was observed in lysates from HEK293T cells transfected with Flag-YAP and HA-Ubiquitin plasmids (Fig. 7c). Indeed, YAP ubiquitination was markedly augmented, and degradation was increased in the presence of PPZ. Taken together, these results indicated that PPZ reduced the YAP protein level via proteasome‐dependent degradation and ubiquitination pathways.
Fig. 7. PPZ promoted the proteasome-dependent degradation and ubiquitination of YAP.

a LX-2 cells were starved for 24 h and then treated with TGF-β1 (2 ng· mL−1) and CHX (20 µg· mL−1) with or without PPZ (200 µM) for the indicated times. The protein expression of YAP was measured by Western blotting. b LX-2 cells were treated with TGF-β1 (2 ng· mL−1), CHX (20 µg ·mL−1) and MG132 (10 µM) with or without PPZ (200 µM) for the indicated times. Changes in the protein levels of YAP are shown. For a, b, the data are presented as the mean ± SD of three independent experiments. *P < 0.05 vs. the 2 ng· mL−1 TGF-β1 group. c HEK293T cells were transfected with Flag-YAP (2.5 µg) and HA-Ubiquitin (2.5 µg) plasmids and subsequently treated with PPZ. Whole-cell extracts were immunoprecipitated with anti‐Flag and blotted with an anti‐HA antibody. d The ubiquitination of endogenous YAP in LX-2 cells transfected with Flag-OTUB2 (2.5 µg) or vector. e The interaction between YAP and OTUB2 in LX-2 cells was investigated using immunoprecipitation assay.
Otubain-2 (OTUB2), a deubiquitinating enzyme, has been demonstrated to activate YAP by directly deubiquitinating and stabilizing YAP in breast cancer [31]. Consistent with this, we found that OTUB2 overexpression significantly decreased the ubiquitination of endogenous YAP in LX‐2 cells (Fig. 7d). We then examined whether OTUB2 contributed to PPZ-mediated reduction of YAP stability in LX‐2 cells. As shown in Fig. 7e, the protein level of OTUB2 was not altered, but the interaction between OTUB2 and YAP in LX‐2 cells was significantly disrupted by PPZ treatment, suggesting that PPZ attenuated the stabilizing effect of OTUB2 on YAP. Thus, PPZ might downregulate the YAP signalling pathway by disrupting the interaction between OTUB2 and YAP, subsequently inhibiting HSC activation and blocking the progression of liver fibrogenesis.
Discussion
Common BDL is a well-known experimental model to cause cholestatic injury and periportal biliary fibrosis, which etiologically and pathogenically resembles cholestatic fibrosis in humans [32]. Toxic bile acid accumulates in the liver and damages hepatocytes, leading to oxidative stress, an inflammatory response, and subsequent HSC activation and consecutive fibrosis. Advanced liver fibrosis ultimately results in cirrhosis, liver failure, and hepatocellular carcinoma and often requires liver transplantation [33]. Although liver fibrosis is reversible in both experimental models and patients, few therapeutic strategies are available in clinical practice due to limited efficacy and side effects. The present study elucidated the potent effects of PPZ, a PPI, on liver fibrosis in a BDL rat model, verified these effects in HSCs, and explored the underlying mechanisms of action.
The elevated serum levels of the liver enzymes ALT and AST induced by BDL were significantly reduced by PPZ treatment. In addition, the gross appearance of rat livers and the H&E staining results showed that PPZ markedly ameliorated BDL-induced liver injury. Notably, serum and liver tissue TBA levels were dramatically decreased in BDL rats after PPZ supplementation, suggesting that PPZ could regulate bile acid synthesis or transport. This was the first time PPZ has been shown to have an anticholestatic effect in a BDL rat model. However, the detailed regulatory mechanism needs further exploration. After BDL, persistent cholestasis occurs, which is followed rapidly by inflammatory cell infiltration and cytokine overproduction, in which Kupffer cells (liver macrophages) demonstrate critical and divergent functions. Upon activation, Kupffer cells rapidly produce many proinflammatory cytokines and chemokines, such as platelet-derived growth factor (PDGF), TGF-β, TNF-α, IL-6, IL-1β, IL-10, and IL-12, resulting in HSC activation and hepatocyte death [34, 35]. Consistent with previous studies showing that PPIs have potent anti-inflammatory effects, PPZ treatment markedly diminished the levels of inflammatory cytokines such as IL-6, TNFα, and IL-1β in BDL rats. These results indicated that PPZ could reduce BDL-induced liver injury and improve liver function.
Importantly, PPZ exerted striking effects against hepatic fibrosis in the BDL model, as supported by decreases in the positive areas stained by Sirius red, liver hydroxyproline levels, and the expression of fibrogenic markers. Next, the inhibitory effect of PPZ on fibrogenic gene expression was confirmed in vitro. It is well known that the TGFβ1/Smad2/3 signalling pathway plays a crucial role in HSC activation during liver fibrogenesis. Upon phosphorylation by activated TGFβ1, Smad2 and Smad3 translocate into the nucleus and regulate the transcription of downstream fibrogenesis-related genes, including COL1A1, α-SMA, MMP-2, and TGFβ1 [36]. In this study, the phosphorylation of Smad2 and Smad3 was stimulated in BDL rats and HSCs but was dramatically repressed by PPZ administration, suggesting that PPZ-mediated inhibition of hepatic fibrosis was associated with the downregulation of the TGFβ1/Smad2/3 pathway.
The Hippo pathway downstream effector YAP has been demonstrated to be an important regulator of organ size and regeneration. Based on previous findings, liver-specific overexpression of Yap in mice induced an increase in liver size (hyperplasia) by promoting cell proliferation and suppressing apoptosis, which was completely reversible by disrupting Yap expression [37, 38]. Notably, there was massive induction of antiapoptotic regulators, such as survivin, a gene that has been implicated as a cell death inhibitor as well as a mitotic regulator. Survivin knockdown by RNA interference significantly reduced the ability of Yap to induce anchorage-independent cell growth [38]. In addition, patients with chronic cholestatic disorders or mice with biliary obstruction show YAP activation in bile epithelial cells as well as hepatocytes, and YAP deletion in BDL mice compromises bile epithelial cell and hepatocyte proliferation [39]. Consistent with these findings, we found that Yap and survivin were overexpressed in BDL liver tissues, along with the increased liver size and bile duct proliferation compared to those of the sham group, all of which could be reversed by PPZ treatment. CTGF, the most highly characterized YAP target gene, was induced earlier than classical markers of HSC activation, and YAP may regulate gene expression at the early stages of HSC activation [4]. We found that CTGF was strongly reduced by PPZ in vitro and in vivo, suggesting that the downregulation of profibrotic genes might be dependent on CTGF. TGFβ1 has been previously reported to be a target gene of YAP [6]. In our study, elevated YAP expression and nuclear localization were detected in LX-2 cells after TGFβ1 treatment. Additionally, previous studies showed that nuclear YAP could cooperate with TGFβ-induced Smad2/3 to promote TGFβ-dependent transcriptional changes [10]. These evidences conformed the mutual regulation and crosstalk between YAP and TGF signalling, which was implicated in the initiation and perpetuation of HSC activation in fibrogenesis. YAP exerts its effects by translocating into the nucleus and binding to TEADs. Restraining the nuclear translocation of YAP and subsequently disrupting the YAP/TEAD2 interaction might be favourable in ameliorating liver fibrosis [40], which was consistent with our findings. YAP overexpression weakened the PPZ‐induced downregulation of fibroblastic marker expression, while silencing YAP induced the opposite consequences. These results verified that PPZ reduces fibrogenesis by inhibiting YAP signalling.
In vitro, the expression of YAP was markedly reduced after PPZ treatment in LX-2 cells when the translation was blocked, suggesting that the downregulation of YAP by PPZ occurred at the posttranscriptional level. Furthermore, the treatment of LX-2 cells with MG132 in combination with PPZ revealed that PPZ decreased the protein level of YAP through the ubiquitin-proteasome-dependent pathway, a common process for regulating protein stability [41]. Recently, Zhang et al. identified a conserved SUMO interaction motif (SIM) domain in YAP that mediates the specific binding between YAP and poly-SUMOylated OTUB2, a deubiquitinating enzyme, and controls YAP polyubiquitination and protein activity [31]. Based on the documented deubiquitinating regulatory mechanism, we first confirmed the OTUB2-YAP interaction in LX-2 cells. As expected, OTUB2 overexpression significantly decreased the ubiquitination of endogenous YAP in LX‐2 cells. PPZ treatment did not change OTUB2 expression but disrupted the interaction between OTUB2 and YAP, thereby promoting YAP degradation. The increase of YAP degradation further restrained the nuclear translocation of YAP, disrupted the YAP/TEAD2 interaction, and inhibited the transcription of downstream target genes exerting a repressive effect on liver fibrosis (Fig. 8). However, whether OTUB2 has a direct regulatory effect on liver fibrosis remains unknown and requires further exploration.
Fig. 8. The anti-hepatic fibrosis mechanism of PPZ.
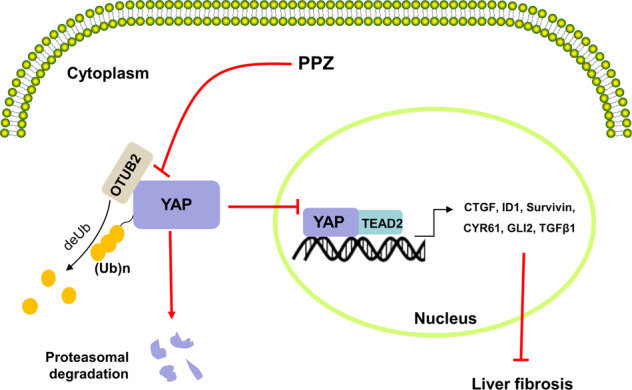
PPZ disrupted the interaction between OTUB2 and YAP, thereby promoting YAP degradation. The increase of YAP degradation further restrained the nuclear translocation of YAP, disrupted the YAP/TEAD2 interaction, and inhibited the transcription of downstream target genes exerting a repressive effect on liver fibrosis.
In conclusion, our study demonstrated that PPZ ameliorated BDL-induced liver damage and fibrosis in rats. We also evaluated the antifibrotic effects of PPZ on LX-2 cells and mouse pHSCs and revealed that the underlying mechanism of PPZ involved YAP signalling. PPZ promoted the proteasome‐dependent degradation and ubiquitination of YAP by disrupting the interaction between OTUB2 and YAP, thus inhibiting HSC activation and blocking the progression of hepatic fibrosis. Based on these findings, PPZ exerts therapeutic effects on liver fibrosis in vitro and in vivo and may be a promising strategy for hepatic fibrosis treatment in the future.
Supplementary Information
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 81673497 and 81903695), the CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-001 and 2019-I2M-1-001), and the Natural Science Foundation of Shandong Province [ZR2020MH419].
Author contributions
HWH and XLG designed the experiments. WXN and NZ performed animal experiments. ZNL and MXG performed the experiments on the cell model. YYB and YR acquired and analyzed all data. ZNL wrote and revised the paper. Every author read and approved the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Xiu-li Guo, Email: guoxl@sdu.edu.cn.
Hong-wei He, Email: hehwei@imb.pumc.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00754-w.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830–41. doi: 10.1136/gutjnl-2014-306842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shek FW, Benyon RC. How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur J Gastroenterol Hepatol. 2004;16:123–6. doi: 10.1097/00042737-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–88. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–38. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, et al. The p130 isoform of angiomotin is required for YAP-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal. 2013;6:ra77. doi: 10.1126/scisignal.2004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams EJ, Gaca MD, Brigstock DR, Arthur MJ, Benyon RC. Increased expression of connective tissue growth factor in fibrotic human liver and in activated hepatic stellate cells. J Hepatol. 2000;32:754–61. doi: 10.1016/s0168-8278(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi H, Hayashi N, Hayashi K, Yamataka A, Lane GJ, Miyano T. Connective tissue growth factor and progressive fibrosis in biliary atresia. Pediatr Surg Int. 2005;21:12–6. doi: 10.1007/s00383-004-1254-z. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Charrier AL, Leask A, French SW, Brigstock DR. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J Hepatol. 2011;55:399–406. doi: 10.1016/j.jhep.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiemer SE, Szymaniak AD, Varelas X. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem. 2014;289:13461–74. doi: 10.1074/jbc.M113.529115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol. 2015;63:962–70. doi: 10.1016/j.jhep.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang S, Chen M, Ding X, Zhang X, Zou X. Proton pump inhibitor selectively suppresses proliferation and restores the chemosensitivity of gastric cancer cells by inhibiting STAT3 signaling pathway. Int Immunopharmacol. 2013;17:585–92. doi: 10.1016/j.intimp.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 13.Kim YJ, Lee JS, Hong KS, Chung JW, Kim JH, Hahm KB. Novel application of proton pump inhibitor for the prevention of colitis-induced colorectal carcinogenesis beyond acid suppression. Cancer Prev Res (Phila) 2010;3:963–74. doi: 10.1158/1940-6207.CAPR-10-0033. [DOI] [PubMed] [Google Scholar]
- 14.De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007;67:5408–17. doi: 10.1158/0008-5472.CAN-06-4095. [DOI] [PubMed] [Google Scholar]
- 15.Liu NN, Köhler JR. Antagonism of fluconazole and a proton pump inhibitor against Candida albicans. Antimicrob Agents Chemother. 2015;60:1145–7. doi: 10.1128/AAC.02043-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu M, Yan H, Yu C, Yuan L, Sun S. Proton pump inhibitors act synergistically with fluconazole against resistant Candida albicans. Sci Rep. 2020;10:498. doi: 10.1038/s41598-019-57174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scaringi L, Cornacchione P, Ayroldi E, Corazzi L, Capodicasa E, Rossi R, et al. Omeprazole induces apoptosis in Jurkat cells. Int J Immunopathol Pharmacol. 2004;17:331–42. doi: 10.1177/039463200401700313. [DOI] [PubMed] [Google Scholar]
- 18.Yeo M, Kim DK, Kim YB, Oh TY, Lee JE, Cho SW, et al. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin Cancer Res. 2004;10:8687–96. doi: 10.1158/1078-0432.CCR-04-1065. [DOI] [PubMed] [Google Scholar]
- 19.Udelnow A, Kreyes A, Ellinger S, Landfester K, Walther P, Klapperstueck T, et al. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE. 2011;6:e20143. doi: 10.1371/journal.pone.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1:e87. doi: 10.1038/cddis.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu ZN, Shi ZY, Dang YF, Cheng YN, Guan YH, Hao ZJ, et al. Pantoprazole pretreatment elevates sensitivity to vincristine in drug-resistant oral epidermoid carcinoma in vitro and in vivo. Biomed Pharmacother. 2019;120:109478. doi: 10.1016/j.biopha.2019.109478. [DOI] [PubMed] [Google Scholar]
- 22.Eltahir HM, Nazmy MH. Esomeprazole ameliorates CCl4 induced liver fibrosis in rats via modulating oxidative stress, inflammatory, fibrogenic, and apoptotic markers. Biomed Pharmacother. 2018;97:1356–65. doi: 10.1016/j.biopha.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 23.Nishi T, Yamamoto Y, Yamagishi N, Iguchi M, Tamai H, Ito T, et al. Lansoprazole prevents the progression of liver fibrosis in non-alcoholic steatohepatitis model rats. J Pharm Pharmacol. 2018;70:383–92. doi: 10.1111/jphp.12870. [DOI] [PubMed] [Google Scholar]
- 24.Mederacke I, Dapito DH, Affò S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305–15. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roeb E. Matrix metalloproteinases and liver fibrosis (translational aspects) Matrix Biol. 2018;68-69:463–73. doi: 10.1016/j.matbio.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 27.Altieri DC. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr Opin Cell Biol. 2006;18:609–15. doi: 10.1016/j.ceb.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM, et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology. 2006;43:1032–41. doi: 10.1002/hep.21135. [DOI] [PubMed] [Google Scholar]
- 29.Rashid ST, Humphries JD, Byron A, Dhar A, Askari JA, Selley JN, et al. Proteomic analysis of extracellular matrix from the hepatic stellate cell line LX-2 identifies CYR61 and Wnt-5a as novel constituents of fibrotic liver. J Proteome Res. 2012;11:4052–64. doi: 10.1021/pr3000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan J, Hu B, Shi W, Wang X, Shen J, Chen Y, et al. Gli2-regulated activation of hepatic stellate cells and liver fibrosis by TGF-β signaling. Am J Physiol Gastrointest Liver Physiol. 2021;320:G720–28. doi: 10.1152/ajpgi.00310.2020. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Du J, Wang S, Shao L, Jin K, Li F, et al. OTUB2 promotes cancer metastasis via hippo-independent activation of YAP and TAZ. Mol Cell. 2019;73:7–21. doi: 10.1016/j.molcel.2018.10.030. [DOI] [PubMed] [Google Scholar]
- 32.Yanguas SC, Cogliati B, Willebrords J, Maes M, Colle I, van den Bossche B, et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90:1025–48. doi: 10.1007/s00204-015-1543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman SL. Hepatic fibrosis—overview. Toxicology. 2008;254:120–9. doi: 10.1016/j.tox.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Duwaerts CC, Gehring S, Cheng CW, van Rooijen N, Gregory SH. Contrasting responses of Kupffer cells and inflammatory mononuclear phagocytes to biliary obstruction in a mouse model of cholestatic liver injury. Liver Int. 2013;33:255–65. doi: 10.1111/liv.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novo E, Cannito S, Paternostro C, Bocca C, Miglietta A, Parola M. Cellular and molecular mechanisms in liver fibrogenesis. Arch Biochem Biophys. 2014;548:20–37. doi: 10.1016/j.abb.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 36.Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 37.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–60. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 38.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–33. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai H, Zhang N, Xu Y, Chen Q, Khan M, Potter JJ, et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology. 2012;56:1097–107. doi: 10.1002/hep.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge M, Liu H, Zhang Y, Li N, Zhao S, Zhao W, et al. The anti-hepatic fibrosis effects of dihydrotanshinone I are mediated by disrupting the Yes-associated protein and transcriptional enhancer factor D2 complex and stimulating autophagy. Br J Pharmacol. 2017;174:1147–60. doi: 10.1111/bph.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.