

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, with a minimal difference between its incidence rate and mortality rate. Advances in oncology over the past several decades have dramatically improved the overall survival of patients with multiple cancers due to the implementation of new techniques in early diagnosis, therapeutic drugs, and personalized therapy. However, pancreatic cancers remain recalcitrant, with a 5-year relative survival rate of <9%. The lack of measures for early diagnosis, strong resistance to chemotherapy, ineffective adjuvant chemotherapy and the unavailability of molecularly targeted therapy are responsible for the high mortality rate of this notorious disease. Genetically, PDAC progresses as a complex result of the activation of oncogenes and inactivation of tumor suppressors. Although next-generation sequencing has identified numerous new genetic alterations, their clinical implications remain unknown. Classically, oncogenic mutations in genes such as KRAS and loss-of-function mutations in tumor suppressors, such as TP53, CDNK2A, DPC4/SMAD4, and BRCA2, are frequently observed in PDAC. Currently, research on these key driver genes is still the main focus. Therefore, studies assessing the functions of these genes and their potential clinical implications are of paramount importance. In this review, we summarize the biological function of key driver genes and pharmaceutical targets in PDAC. In addition, we conclude the results of molecularly targeted therapies in clinical trials and discuss how to utilize these genetic alterations in further clinical practice.
Keywords: pancreatic cancer, KRAS, CDKN2A, TP53, SMAD4, clinical implication
Introduction
Pancreatic cancer is listed as one of the most lethal cancers, with a 5-year overall survival rate of <9%. In 2019, in the US, 56,770 new pancreatic cancer cases were confirmed, and its estimated deaths accounted for 7%–8% of all cancer-related deaths [1]. In addition, pancreatic cancer is predicted to become the second leading cause of cancer-related death in the next decade [2]. While advances in the treatment of other cancer types have dramatically improved the overall outcomes of patients, the incidence and mortality rates of pancreatic cancer have only decreased slightly over the past 30 years [1, 3–5]. The high mortality rate might be a consequence of the combination of a late diagnosis, resistance to therapies and insufficiency of the effective treatment modality.
To date, surgical resection remains the only potential curative treatment. However, only 15% of patients have resectable tumors. The majority of patients are diagnosed at an advanced stage and are mainly be treated with chemotherapy regimens. Although novel chemotherapy regimens have been established recently, such as FOLFIRINOX and gemcitabine plus nab-paclitaxel, their overall efficacy remains limited. The overall survival duration of patients with metastatic pancreatic cancer is still <1 year, regardless of treatment with FOLFIRINOX or nab-paclitaxel plus gemcitabine [6, 7]. In addition, long-lasting debates regarding the survival benefits of chemoradiation therapy exist. With research progress in molecularly targeted therapy, tumor therapy has undergone revolutionary changes. Some cancers, such as lung cancer, have entered the era of molecularly targeted therapy. However, in patients with PDAC, only the combination of gemcitabine plus erlotinib is associated with a statistically significant increase in survival compared to gemcitabine alone. However, the actual benefit is small, suggesting that only a small subset of patients might intrinsically benefit from this treatment. Nonetheless, molecularly targeted therapy remains the only hope for patients with PDAC.
Accordingly, many published studies have been conducted using the latest next-generation high-throughput sequencing technology, and it is widely accepted that pancreatic cancer is a disease of genetic alterations. The most commonly mutated genes are generated from The Cancer Genome Atlas (TCGA) data and are presented in Fig. 1. However, the number of druggable targets in individual patients is low. Thus, the classic progression model of PDAC must be re-examined, and possible solutions should be identified. PDAC originates from a series of precursor lesions, such as pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN) [8]. The dysregulation of several core pathways and a myriad of genomic alterations drive pancreatic tumorigenesis [9]. Whole-genome sequencing has revealed the main driver genes in pancreatic cancers, including KRAS, CDKN2A, TP53, and SMAD4/DPC4 [10–13]. These genes are mutated in different stages of precursor lesions, and their dysregulation promotes the differentiation and proliferation of pancreatic cancers (Fig. 2) [8]. In general, KRAS mutations emerge from stage 1 lesions (PanIN-1) to promote the initiation process, while CDKN2A mutations occur in PanIN-2 to facilitate further progression. In addition, mutations in TP53 and SMAD4 are frequently detected in PanIN-3 and invasive tumors, driving the proliferation and expansion of pancreatic cancers [14, 15]. Therefore, studies investigating the roles of these driver genes in pancreatic cancers are of paramount importance to provide additional strategies for use in clinical practice. Here, we review the biological functions of these driver genes and summarize their clinical implications reported to date.
Fig. 1. Mutation profile of pancreatic cancer in the TCGA dataset.
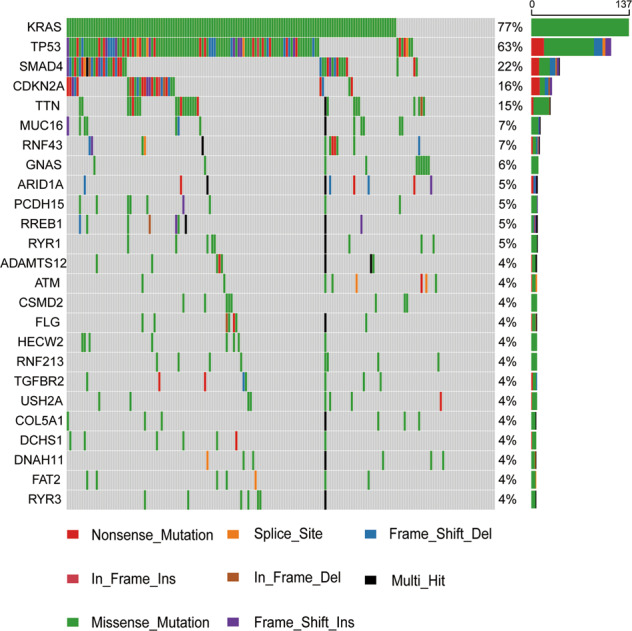
Mutation information on 178 pancreatic cancers in the TCGA dataset was analyzed. KRAS/CDKN2A/TP53/SMAD4 are the most commonly mutated genes in pancreatic cancer, with mutation rates of 77%, 63%, 22%, and 16%, respectively. In addition, missense mutations and nonsense mutations are the main alteration types.
Fig. 2. Classical progression model of pancreatic cancer.
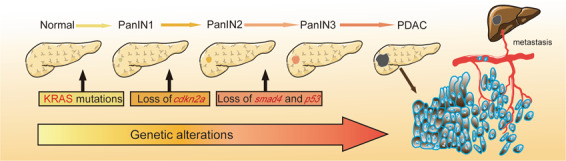
Pancreatic cancer is considered a disease of multiple genetic alterations, and mutations in KRAS/CDKN2A/TP53/SMAD4 promote the initiation and progression of precursor lesions. KRAS mutations occur in the early stage of PanIN-1; the loss of cdkn2a occurs in PanIN-2; and the loss of p53 and smad4 occurs in the later stage of precursor lesions. A series of other mutations cooperate to promote the tumorigenesis and metastasis of PDAC.
KRAS
General introduction
As the most common genetic driver in pancreatic cancers, the KRAS gene is mutated in ~93% of pancreatic cancers [16–19]. The KRAS protein is a small GTPase responsible for interacting with cell membrane growth factor receptors and controlling the switch of multiple signaling pathways and cellular processes. The proteins in the human RAS family usually consist of two functional domains: the G domain and the membrane-targeting domain. Different isoforms of the RAS family have a similar G domain but vary in the membrane-targeting domain. The G domain spans residues 1-164 and functions as a molecular switch for the downstream signaling pathway by binding to GTP/GDP. GTP-bound RAS prompts the membrane-targeting domain to interact with effector proteins and activate downstream signaling pathways, while GDP-bound RAS inactivates the process, and GTP/GDP conversion is catalyzed by the SOS1 protein. The most frequent mutation of KRAS in pancreatic cancer occurs in codon G12 of exon 2(e.g., G12D (40%) and G12C (33%)). Approximately 10% of mutations occur on codons 13, 61, 117, and 146 [20]. Point mutations in codon 12 of the KRAS oncogene prevent the conversion from GTP to GDP, resulting in the constitutive activation of downstream signaling pathways and markedly promoting carcinogenesis in various cancer types [20, 21].
KRAS mutations in pancreatic cancer
Similar to other cancers, pancreatic cancers are derived from a series of precursor lesions, among which pancreatic intraepithelial neoplasia (PanIN) is the most common lesion [22, 23]. Oncogenic KRAS mutations have been identified in 95% of PDAC tissues [9, 16, 17]. According to many studies, oncogenic KRAS mutations drive the initiation and progression of different stages of PanINs, and this process involves changes in the gene mutation rate, from 50% in PanINs to 95% in PDAC [17, 19]. In addition, factors that promote and activate KRAS, such as inflammation, oxidants and TGF-β, all contribute to the initiation and development of pancreatic tumors. In contrast, NSAIDs and antioxidants decrease KRAS activity and prevent tumor progression [19]. Nonetheless, KRAS also functions as a fundamental factor in the progression from IPMNs to PDAC [23].
Decades of research have discovered and clarified the complex picture of KRAS-regulated biological processes, including cell metabolism, tumor cell signaling, the tumor microenvironment, micropinocytosis, apoptosis, and redox homeostasis [24–28]. According to Sunil et al., the endogenous expression of KRASG12D induced all three stages of PanINs in all genetically engineered mouse models, and a small percentage of these animals developed metastatic PDAC [29]. In addition, KRAS mutations play an important role in the maintenance and proliferation of PDAC. Collins et al. constructed models with the reversible expression of oncogenic KRAS, and a reduction in KRAS expression led to rapid tumor relapse [30]. However, a KRAS mutation alone is insufficient to drive carcinogenesis, and hundreds of changes in gene expression, especially in tumor suppressor genes, cooperate to drive the formation of the final invasive PDAC [19, 31]. Moreover, due to the consecutive activation of oncogenic KRAS, the RAF/MEK/ERK pathway, PI3K/AKT/mTOR pathway, RalA/B pathway and NF-κB pathway are all activated to promote the proliferation of PDAC (Fig. 3) [32]. Our previous studies also revealed a novel KRAS/ERK/FBW7/cMyc pathway in PDAC cell lines, and all these effectors and pathways represent potential drug targets for further study [33–35].
Fig. 3. Pathways of key driver genes and therapeutic targets in pancreatic cancer.

Oncogenic mutations in KRAS activate downstream signaling pathways, such as the PI3K/Akt/mTOR, KRAS/Ral, and KRAS/Raf/MEK pathways. Therapeutic methods include directly targeting KRAS, targeting upstream EGFR, or targeting downstream effectors such as PI3K, Akt, mTOR, Raf and MEK. Loss-of-function mutations in CDKN2A/TP53/SMAD4 attenuate the tumor suppressive functions of downstream signaling pathways. Therapeutic targets for tumor suppressor genes include restoring the function of wild-type p53, HSP90 inhibitors, vaccine therapy targeting mut-p53, Wee-1 kinase inhibitors (not shown), CDK4/6 inhibitors and TGF-β inhibitors.
KRAS mutations for early diagnosis
The high mortality rate of pancreatic cancer is closely related to the fact that only a small percentage of patients are diagnosed at the early stage [36]. Considering the crucial role of oncogenic KRAS in pancreatic cancer, scientists have tried to determine its potential efficacy in diagnosis and medical treatment since its discovery. Over 30 years ago, oncogenic KRAS mutations were also detected in duodenal fluid [37]. Examinations of KRAS mutations involve endoscopic ultrasonography-guided fine needle aspiration (EUS) and circulating tumor DNA (ctDNA) analyses. EUS-FNA is the main well-established tool for collecting cytological and histological samples from locally advanced PDAC [38]. The combination of a KRAS mutation assay and cytopathology dramatically increases the sensitivity, accuracy, and negative predictive value of pancreatic cancers compared to cytopathology alone [39]. Notably, the KRAS mutation assay not only facilitates a differential diagnosis of PDAC and pseudotumorous chronic pancreatitis but also helps distinguish these diseases from autoimmune pancreatitis [40].
Liquid biopsy has rapidly emerged in recent years as a promising tool for early diagnosis, monitoring the effect of treatment and predicting prognosis. In the study performed by Cohen et al., tumor-specific KRAS mutations were detected in 30% of plasma samples from 221 patients with pancreatic cancer, and the combination of KRAS mutations and elevated CA19-9 levels was more sensitive in detecting pancreatic cancer than CA19-9 levels alone [41]. In addition, the KRAS mutation detection rates in ctDNA are notably connected with the stages of tumor progression, namely, 53% in metastatic disease and 34% in localized disease [42]. Negative detection of KRAS mutations before treatment is closely associated with a good prognosis and therapeutic response, regardless of tumor resection. In addition, the emergence of KRAS-mutated ctDNA within 1 year after surgery may predict poor overall survival [43, 44]. Moreover, KRAS mutation in exosome-derived DNA (exoDNA) is also an important factor for predicting tumor resectability and the overall survival of patients with pancreatic cancer [45, 46]. However, the detection of KRAS mutations alone in both ctRNA and exoDNA inevitably leads to false-negative results, and thus, multiple biomarkers and mutations must be evaluated simultaneously to improve the accuracy, sensitivity, and specificity of pancreatic cancer detection. In addition, liquid biopsy is not very sensitive for the early detection of tumors <10 mm in size [47].
Targeting KRAS for treatment
Multiple methods targeting KRAS mutations have been proposed in the past three decades. Phase II/III studies of strategies targeting KRAS or KRAS-related pathways in pancreatic cancer are summarized in Table 1. Drugs that directly target KRAS are the most obvious option, but the lack of a potential binding pocket apparently renders this mutation “undruggable” [9, 48]. Recently, in parallel with a better understanding of KRAS biochemistry and new methods to identify potential chemicals that target KRAS, several strategies have been pursued for the direct targeting of KRAS, such as targeting nucleotide exchange and RAS-effector interactions [49]. A group of small molecules has been identified to be able to bind to KRAS and inhibit SOS-mediated nucleotide exchange, thus preventing the activation of KRAS [50, 51]. Ongoing clinical trials investigating treatments that directly target KRAS are listed in Table 2. Strategies targeting downstream effectors also represent a promising method, but the administration of single MEK, RAF, or PI3K inhibitors has been unsatisfactory in KRAS-mutated cancer [21, 52]. Indirect targeting of KRAS by inhibiting membrane localization is also a theoretically plausible option, but its clinical efficacy was extremely disappointing [53, 54]. In addition, because reovirus can induce oncolysis in the context of activated RAS signaling, clinical trials have examined the role of reovirus in PDAC [55, 56]. Moreover, the use of RNA interference (RNAi) to suppress the KRAS protein has been reported to inhibit pancreatic cancer growth in vivo, and its role is being explored in a clinical trial (NCT01676259) [57, 58]. However, despite 35 years of research on KRAS-mutated pancreatic cancer, very few effective drugs have been produced for clinical use. Moreover, most drugs that enter phase III clinical trials are closely related to KRAS pathways, indicating that strategies targeting KRAS still hold promise to conquer this disease in the future.
Table 1.
Summary of phase II or III clinical trials investigating KRAS-related therapy in patients with pancreatic cancer.
| Target gene/pathway | Tested drug | n (subjects) | Treatment setting | Therapeutic scheme | Phase | Median OS | Median PFS | Ref |
|---|---|---|---|---|---|---|---|---|
| KRAS | Tipifarnib | 688 | Untreated advanced PDAC | Gem + tipifarnib vs gem + placebo | III | 193 vs 182 days, P = 0.75 | 112 days vs 109 days, P = 0.72 | [53] |
| Pelareorep | 73 | Untreated metastatic PA | Paclitaxel/carboplatin+ pelareorep vs paclitaxel/carboplatin | II | 7.3 months vs 8.8 months, P = 0.68 | 4.9 months vs 5.2 months, P = 0.6 | [56] | |
| Ras oncogene peptide vaccines | 23 | Resectable PDAC with surgery | Single or seven peptide vaccination | II | 27.5 months | – | [201] | |
| KRAS/RAF/MEK | Pimasertib | 88 | Metastatic PC | Gem + pimasertib vs gem + placebo | II | 7.26 months vs 7.59 months, P = 0.674 | 3.75 months vs 2.83 months, P = 0.681 | [202] |
| Selumetinib, erlotinib | 46 | Previously treated advanced PDAC | Combination of selumetinib and erlotinib | II | 7.3 months (95% CI, 5.2–8.0 months) | 1.9 months (95% CI, 1.4–3.3 months) | [203] | |
| Trametinib | 160 | Metastatic PDAC | Gem + trametinib vs gem + placebo | II | 8.4 months vs 6.7 months, P = 0.453 | 16.1 weeks vs 15.1 weeks, P = 0.349 | [204] | |
| Selumetinib | 70 | Metastatic PC with failed first-line gemcitabine therapy | Selumetinib vs capecitabine | II | 5.4 months vs 5.0 months, P = 0.92 | 2.1 months vs 2.2 months, P = 0.41 | [205] | |
| Refametinib | 80 | Advanced PDAC | Refametinib + gem | II | 8.4 months (95% CI, 6.4–11.6 months) | 5.4 months (95% CI, 4.0–7.1 months) | [206] | |
| CI1040 | 15 | Advanced PC | CI1040 | II | – | – | [207] | |
| Selumetinib, MK-2206 | 120 | Metastatic PC | Selumetinib + mk-2206 vs mFOLFOX | II | 3.9 months vs 6.7 months, P = 0.15 | 1.9 months vs 2.0 months, P = 0.02 | [199] | |
| KRAS/PI3K/AKT/mTOR | Temsirolimus | 55 | Metastatic PC | Gem + temsirolimus | II | 4.95 months (95% CI 3.54–6.85) | 2.69 months (95% CI 1.74–4.95) | [208] |
| Everolimus | 31 | Advanced PC | Capecitabine + everolimus | II | 8.9 months (95% CI 4.6–13.1 months) | 3.6 months (95% CI 1.9–5.3) | [209] | |
| Everolimus, cetuximab | 31 | Locally advanced or metastatic PDAC | Everolimus, cetuximab and capecitabine | II | 5.0 months (CI 3.1–6.8 months) | – | [210] | |
| Everolimus | 33 | Metastatic PDAC with a failure of the 1st gem-based chemotherapy | Everolimus | II | 4.5 months | 1.8 months | [211] | |
| Temsirolimus, everolimus, cetuximab |
A: 5 B: 16 |
Metastatic PDAC |
A: temsirolimus B: everolimus + erlotinib |
II |
A: 44 days B: 87 days |
A: 19 days B: 49 days |
[212] | |
| Rigosertib | 160 | Untreated metastatic PDAC | Rigosertib + gem vs gem | II/III | 6.1 months vs 6.4 months, HR = 1.24 (95% CI 0.85–1.81) | 3.4 months vs 3.4 months, HR = 0.96, 95% CI 0.68–1.36 | [213] | |
| KRAS/RAF/MEK/ERK | Sorafenib | 24 | Untreated locally advanced or metastatic PDAC | Oxaliplatin, capecitabine and sorafenib | II | 8.1 months (95% CI 3.5–10.9) | 6.0 months (95% CI 2.5–9.6) | [214] |
| Sorafenib | 102 | Untreated locally advanced or metastatic PDAC | Gem + sorafenib vs gem + placebo | III | 8 months and 9.2 months, P = 0.231 | 3.8 months and 5.7 months, P = 0.902 | [215] | |
| Sorafenib |
A: 15 B: 37 |
Metastatic PC |
A: sorafenib B: gem + sorafenib |
II |
A: 4.3 months (95% CI: 3.3–8.3) B: 6.5 months (95% CI: 5.5–8) |
A: 2.3 months (95% CI: 1.2–5.7) B: 2.9 months (95% CI: 2.1–4.3) |
[216] | |
| Sorafenib, erlotinib | 38 | Advanced PDAC | sorafenib + erlotinib | II | 99.5 days (95% CI: 71–188) | Eight-week PFS rate of 46% (95% CI: 0.32–0.67) | [217] | |
| EGFR | Erlotinib | 569 | Locally advanced or metastatic PDAC | Gem + erlotinib vs gem + placebo | III | 6.24 months v 5.91 months, P = 0.038 | 3.75 months v 3.55 months, P = 0.004 | [218] |
OS overall survival, PFS progression-free survival, gem gemcitabine, tipifarnib farnesyltransferase inhibitor, pelareorep oncolytic virus, pimasertib MEK1/2 inhibitor, selumetinib MEK1/2 inhibitor, erlotinib EGFR inhibitor, refametinib MEK1/2 inhibitor, trametinib MEK1/2 inhibitor, CI1040 MEK1/2 inhibitor, MK-2206 AKT inhibitor, temsirolimus mTOR inhibitor, everolimus mTOR inhibitor, cetuximab EGFR inhibitor, rigosertib PI3K/PLK1 inhibitor, sorafenib BARF inhibitor.
Table 2.
Ongoing clinical trials targeting mutations in KRAS.
| Trial ID | Tested drugs | Treatment setting | Phase | Status | Primary outcomes |
|---|---|---|---|---|---|
| NCT04117087 | KRAS peptide vaccine, nivolumab, ipilimumab | resected PDAC with KRAS mutations | I | Recruiting | Drug-related toxicities and fold change in interferon levels |
| NCT04146298 | Mutant KRAS G12V-specific TCR transduced autologous T cells, anti-PD-1 monoclonal antibody, fludarabine, cyclophosphamide | advanced PC with the KRAS G12V mutation and HLA-A*11:01 allele | I/II | Recruiting | AEs and ORR |
| NCT04132505 | Binimetinib, hydroxychloroquine | metastatic PC with KRAS mutation | I | Recruiting | MTD |
| NCT03040986 | Selumetinib sulfate | Locally advanced or metastatic PC with KRAS G12R mutation | II | Active, not recruiting | Clinical response |
| NCT03592888 | mDC3/8-KRAS vaccine | resected PDAC with KRAS mutations | I | Recruiting | Safety and side effects |
| NCT03608631 | iExosomes with KRAS G12D siRNA | metastatic PC with KRAS G12D mutation | I | Not yet recruiting | MTD and DLTs |
| NCT03948763 | mRNA-5671/V941, pembrolizumab | KRAS mutant advanced or metastatic NSCLC, CRC or PDAC | I | Recruiting | DLTs, AEs and discontinuations |
| NCT03190941 | anti-KRAS G12V mTCR, cyclophosphamide, fludarabine, aldesleukin | Metastatic unresectable malignancy with KRAS G12V mutation. | I/II | Recruiting | Response rate and AEs |
| NCT03637491 | Avelumab, binimetinib, talazoparib | metastatic PDAC or other locally advanced or metastatic solid tumors with KRAS/NRAS mutations | Ib/II | Recruiting | DLTs and objective response |
| NCT03745326 | anti-KRAS G12D mTCR PBL, cyclophosphamide, fludarabine, aldesleukin | Metastatic unresectable malignancy with KRAS G12D mutation. | I/II | Recruiting | Response rate and AEs |
| NCT04330664 | MRTX849, TNO155 | Advanced solid tumor with KRAS G12C mutation | I/II | Recruiting | AEs and pharmacokinetics |
| NCT03146962 | Vitamin C | Cohort A: resectable CRC, PC, LC; cohort B: CRC, PC, LC with KRAS/NRAS/BRAF mutations; cohort C: CRC amenable to locoregional therapy | II | Recruiting | Change in antitumor activity in cohort A; 3-month DCR in cohort B; maximal tolerated dose in cohort C |
| NCT03329248 | GI-4000 and other agents | PC with fail in standard-of –care therapy | Ib/II | Active, not recruiting | AEs and ORR |
| NCT03387098 | GI-4000 and other agents | PC with fail in standard-of –care therapy | Ib/II | Active, not recruiting | AEs and ORR |
| NCT03136406 | GI-4000 and other agents | PC with fail in standard-of –care therapy | Ib/II | Active, not recruiting | AEs and ORR |
| NCT00389610 | Allogenic GM-CSF plasmid-transfected pancreatic tumor cell vaccine | Stage I-III PDAC with surgery | II | Active, not recruiting | Local and systemic toxicities |
| NCT04121286 | JAB-3312 | Advanced solid tumors | I | Recruiting | DLTs and recommended phase II dose |
| NCT04045496 | JAB-3312 | Advanced solid tumors | I | Recruiting | DLTs and recommended phase II dose |
The data originated from https://clinicaltrials.gov (accessed July 29, 2020).
PC pancreatic cancer, AEs adverse events, ORR objective response rate, MTD maximum tolerated dose, NSCLC non-small cell lung cancer, CRC colorectal cancer, LC lung cancer, DLTs dose-limiting toxicities, DCR disease control rate.
Compared with wild-type KRAS, mutated KRAS is highly relevant to the poor survival rate of patients with PDAC [59–61]. According to the National Comprehensive Cancer Network (NCCN) guidelines for pancreatic cancer (version 1.2020), erlotinib is the only molecular therapy that has produced significant outcomes in combination with gemcitabine. Interestingly, a survival advantage was observed in patients with tumors expressing wild-type KRAS when erlotinib was prescribed with gemcitabine/capecitabine (median survival rates of 7.9 months and 5.7 months for patients with wild-type KRAS and mutated KRAS, respectively, P = 0.005) [62]. In addition, in a phase IIb study of gemcitabine/nimotuzumab, patients with wild-type KRAS had a better overall survival rate than patients with mutated KRAS [63]. However, the role of the KRAS status in predicting the efficacy of treatment with erlotinib remains elusive, and the minimal benefit of erlotinib has prevented its use as a clinical treatment [64–66].
CDKN2A
General introduction
CDKN2A, cyclin-dependent kinase inhibitor 2A, which is located on chromosome 9p21, is one of the most important tumor suppressor genes, with a crucial role in the regulation of the cell cycle by directly or indirectly targeting CDK4/6-cyclins. Proteins encoded by CDKN2A, p14ARF and p16INK4A share exons 2 and 3 but differ in exon 1, resulting in the translation of two unrelated proteins that function through different pathways [67]. Notably, p16INK4A, one of the INK family inhibitors, binds to CDK4/6 and inhibits the activation of D-cyclins, further preventing the phosphorylation of retinoblastoma (Rb) and limiting cell cycle entry [68]. The p14ARFprotein also promotes cell cycle arrest by binding to and inactivating mouse double minute 2 homologue (MDM2), an E3 ubiquitin ligase that mediates the degradation of p53 (Fig. 3).
Biological and oncogenic functions
Numerous studies have confirmed important roles for CDKN2A in cancer development, aging and type 2 diabetes. Importantly, p16INK4A is expressed at high levels during islet regeneration, and the overexpression of p16INK4A inhibits beta-cell proliferation [69]. The expression of p16INK4Aalso increases with aging in both rodent and human islets, indicating that p16INK4A might be responsible for impaired beta-cell proliferation in aging mice [69, 70]. Increased expression of p16INK4A has also been observed in almost all rodents with aging, and this change occurs in parallel with decreased proliferative and regenerative capacity [71, 72]. In addition to its role in aging, CDKN2A is also one of the most frequently mutated tumor suppressor genes, and genetic alterations in CDKN2A have been detected in many types of cancers (30%–50% of pancreatic cancer cases) [73–76]. The inactivation of CDKN2A cooperates with KRAS mutations and drives the malignant transformation of the pancreas [77]. Loss-of-function p16INK4A mutations stimulate pancreatic neoplastic development in the intermediate or late stages, and the dysregulation of p14ARF advances tumor development and metastasis [73, 78]. Regarding PDAC, the loss of either p14ARF or p16INK4A facilitates malignant progression and differentiation [74]. In families with melanoma, genetic alterations in both p14ARF and p16INK4A dramatically increase the risk of pancreatic cancers [75]. Moreover, the deletion of CDKN2A in a mouse model promoted the tumorigenesis of pancreatic neuroendocrine tumors (PanNETs), the second most common pancreatic malignancy, and reduced survival [79]. Genetic alterations in CDKN2A in pancreatic cancer mainly include deletions, mutations, and promoter hypermethylation [80]. Approximately 40% of PanNETs harbor aberrant hypermethylation of CDKN2A, and a high level of methylation may predict the malignant behavior of PanNETs [81].
Prognostic role of CDKN2A mutations
In contrast to KRAS mutations, the prognostic role of CDKN2A mutations remains controversial among different studies of PDAC (Table 3). In 88 patients with pancreatic cancer treated with preoperative chemoradiation, the polymorphic genotypes of p16INK4A were significantly associated with a shorter median time to tumor progression, namely, 10.8 months for patients with polymorphic genotypes and 16.2 months for patients with the wild-type genotype [82]. The deletion of CDKN2A also results in poor outcomes for patients who have undergone partial pancreatoduodenectomy and radical lymphadenectomy [83]. Oshima et al. investigated the genetic status of 106 patients with PDAC undergoing radical surgery and discovered that the loss of CDKN2A was significantly correlated with lymphatic invasion and postoperative metastases [84].
Table 3.
Prognostic and predictive roles of mutant driver genes in clinical trials.
| Target gene | n (subjects) | Genetic test | Setting | Type of study | Prognostic role (mutant type compared to wild type) | Predictive role | Ref |
|---|---|---|---|---|---|---|---|
| KRAS | 136 | Surgical specimen or biopsy | Recurrent or advanced PDAC | Retrospective | A shorter OS (P = 0.001) | – | [65] |
| KRAS | 114 | Surgical specimen or biopsy | Recurrent or advanced PDAC | Retrospective | – | Wild-type KRAS for the efficacy of gem plus erlotinib (P = 0.002) | [65] |
| KRAS | 186 | Not mentioned | Advanced PC | Prospective | – | Wild-type KRAS for the efficacy of gem plus nimotuzumab (P = 0.026) | [63] |
| KRAS | 14 | ctDNA before chemotherapy | Advanced PC | Prospective | A shorter OS (P = 0.01) | – | [219] |
| KRAS | 60 | ctDNA | Advanced PDAC | Prospective | A shorter OS (P = 0.001) | KRAS mutations for the efficacy of refametinib plus gem | [206] |
| KRAS | 45 | Postoperative cfDNA | PDAC with surgery | Retrospective | A shorter OS (P = 0.044) | Early recurrence (P < 0.0001) | [220] |
| CDKN2A | 160 | Surgical specimen | PC with surgery | Retrospective | Not related | – | [99] |
| CDKN2A | 32 | Surgical specimen | PDAC with surgery | Retrospective | A shorter OS (P = 0.002) | – | [83] |
| CDKN2A | 125 | Surgical specimen or biopsy | PDAC | Retrospective | Not related | – | [132] |
| CDKN2A | 89 | Surgical specimen | PDAC with Whipple surgery | Retrospective | Not related | – | [133] |
| CDKN2A | 106 | Surgical specimen | PDAC with surgery | Retrospective | A shorter OS (P = 0.029) | lymphatic invasion (P = 0.012), postoperative widespread metastases (P < 0.001) | [84] |
| TP53 | 162 | Surgical specimen | PC with surgery | Retrospective | A shorter OS (P = 0.033) | – | [99] |
| TP53 | 106 | Surgical specimen | PDAC with surgery | Retrospective | Not related | Tumor dedifferentiation (P = 0.022), locoregional recurrence (P = 0.020) | [84] |
| TP53 | 61 | Surgical specimen or biopsy | Stage IV PDAC | Retrospective | Not related | – | [221] |
| TP53 | 32 | Surgical specimen | PDAC with surgery | Retrospective | Not related | – | [83] |
| TP53 | 89 | Surgical specimen | PDAC with Whipple surgery | Retrospective | Not related | – | [133] |
| TP53 | 125 | Surgical specimen or biopsy | PDAC | Retrospective | Not related | – | [132] |
| SMAD4 | 119 | Surgical specimen or biopsy | PDAC | Retrospective | A better OS (P = 0.0257) | SMAD4 mutation for resectability (P < 0.0001) | [132] |
| SMAD4 | 89 | Surgical specimen | PDAC with Whipple surgery | Retrospective | A shorter OS (P = 0.006) | – | [133] |
| SMAD4 | 106 | Surgical specimen | PDAC with surgery | Retrospective | A shorter OS (P < 0.001) | Tumor size (P = 0.006), lymphatic invasion (P = 0.033), and lymph node metastasis (P = 0.006) | [84] |
| SMAD4 | 41 | Cytology specimens | Locally advanced PC | Prospective | – | A distant dominant pattern of disease progression (P = 0.016) | [137] |
| SMAD4 | 76 | Carcinoma tissues (autopsy) | PC | Prospective | – | Widespread metastasis(P = 0.007) | [136] |
| SMAD4 | 249 | Surgical specimen | PDAC with Whipple surgery | Retrospective | A shorter OS (P = 0.03) | – | [222] |
| SMAD4 | 274 | Surgical specimen | PDAC with surgery | Retrospective | – | SMAD4 mutations for the efficacy of gem-based chemotherapy (P = 0.002), but not related to recurrence pattern | [135] |
| SMAD4 | 127 | Surgical specimen | PDAC with surgery | Retrospective | Not related | Not related to the recurrence pattern | [138] |
Targeting CDKN2A function for treatment
CDKN2A mainly functions by inhibiting CDK4/6. Specific drugs targeting CDKN2A have not yet been reported, but a series of therapies targeting CDK4/6 has been implicated in patients with a loss of CDKN2A. Palbociclib is an oral, small-molecule inhibitor of CDK4/6 that has been approved by the FDA as a treatment for metastatic breast cancer [85]. By inducing apoptosis and cell cycle arrest, palbociclib enhances the therapeutic efficacy of gemcitabine and inhibits the invasiveness of PDAC cells [86]. Notably, these functions are mainly effective in Rb-positive PDAC cells, suggesting that the Rb protein, which is downstream of CDKN2A, might also serve as a predictive biomarker for palbociclib. The clinical efficacy of palbociclib in PDAC is still under investigation. Ongoing clinical trials targeting CDKN2A/TP53/SMAD4 for the treatment or diagnosis of pancreatic cancers are presented in Table 4. A combination of therapeutic and predictive biomarkers might be needed in future clinical trials.
Table 4.
Ongoing clinical trials targeting P53/SMAD4/CDKN2A pathways for the treatment and diagnosis of pancreatic cancer.
| Trial ID | Target gene | Tested drugs | Treatment setting | Phase | Study type | Status | Primary outcomes |
|---|---|---|---|---|---|---|---|
| NCT02340117 | TP53 | SGT-53, nab-paclitaxel, gemcitabine | Stage IV metastatic PDAC | II | Interventional | Recruiting | PFS at 5.5 months |
| NCT02432963 | TP53 | p53MVA vaccine and pembrolizumab | Uncontrolled solid tumors with p53 overexpression or mutations | I | Interventional | Active, not recruiting | Tolerability |
| NCT02433626 | TP53 | COTI-2 and cisplatin | Advanced or recurrent malignancies | I | Interventional | Recruiting | Number of DLTs and Tmax in 6 months |
| NCT03095781 | HSP90 | XL888 and pembrolizumab | Advanced gastrointestinal malignancies | I | Interventional | Recruiting | Recommended phase II dose |
| NCT03221400 | HSP90 | PEN-866 sodium | PDAC and advanced solid malignancies | I/IIa | Interventional | Recruiting | DLTs, AEs and efficacy |
| NCT02194829 | Wee-1 | MK-1775 | Untreated PC | I/II | Interventional | Active, not recruiting | MTD and PFS |
| NCT03666832 | TGF-β | TEW-7197, FOLFOX | Metastatic PDAC who have failed in first-line chemotherapy | I/II | Interventional | Recruiting | PFS |
| NCT03454035 | CDK4/6 | Ulixertinib, palbociclib | Previously treated metastatic PC and other solid tumors | I | Interventional | Recruiting | MTD and OS |
| NCT03065062 | CDK4/6 | Palbociclib, gedatolisib | Pancreatic, and other solid tumors | I | Interventional | Recruiting | MTD, recommended phase II dose and AEs |
| NCT03891784 | CDK4/6 | Abemaciclib | Metastatic neuroendocrine tumors in the digestive system | II | Interventional | Recruiting | ORR |
| NCT03524677 | KRAS, CDKN2A, P53, SMAD4 | KRAS, CDKN2A, SMAD4 and TP53 mutation on circulating cfDNA | Non-metastatic PC | – | Observational | Recruiting | Vascular invasion and early recurrence |
The data originated from https://clinicaltrials.gov (accessed July 29, 2020).
TP53
General introduction
As the most frequently mutated tumor suppressor gene in all cancers, the TP53 gene is estimated to be mutated in 60%-70% of pancreatic cancers [17]. The TP53 gene encodes the p53 protein, which binds to specific sequences through its DNA binding domain and regulates the transcription of downstream molecules to exert its functions in various biological processes, including the cell cycle, mitochondrial respiration, cell metabolism, autophagy and stem cell maintenance and development [87]. TP53 is commonly activated by oncogenic mutations or cellular stress, such as DNA damage and oxidative stress, preventing p53 from interacting with MDM2/4 and therefore stabilizing p53. As its level increases, p53 increases the transcription of downstream genes, such as P21 and Bcl-2, thus driving cell cycle arrest and repairing or eliminating damaged cells to inhibit the accumulation of oncogenic mutations [88]. Interestingly, p53 both represses and induces the expression of different genes in a context-dependent manner [89]. Consistent with its function, mutations in TP53 usually occur in the DNA binding domain, and most of the mutations are missense mutations, providing a great opportunity for cancer cells to proliferate and survive in a mild stress environment [90]. However, mut-p53 does not simply lose its original function but rather gains other abilities to promote cancer development through different mechanisms, including remodeling the tumor microenvironment and enhancing cell metabolism [91, 92]. Once mutated, mut-p53 binds specifically to the Hsp90 chaperone machinery, a system that senses cellular stress, such as protein misfolding and oncogenic signaling. Hsp90 specifically blocks the activity of MDM2 and CHIP to prevent degradation of the p53 protein, resulting in the accumulation of dysfunctional p53 proteins in cells [91].
TP53 mutations in pancreatic cancer
Genetic alterations in p53 without loss of heterozygosity have been detected in early PanIN, and homozygous mutations in p53 have been observed in PanIN-3, indicating the potential for p53 to drive the carcinogenesis of PDAC [93]. Morton et al. constructed a mouse model with mut-p53 (Trp53R172H) and found that mut-p53 facilitated malignant transformation from premalignant lesions to PDAC [94]. Furthermore, the same study also showed that mut-p53 enhanced the invasion of pancreatic tumor cells and promoted lymph node metastasis. In contrast, supplementary expression from a retroviral p53 vector substantially inhibited the growth of primary pancreatic tumors [95]. In addition, when TP53 was mutated together with KRAS in the mouse pancreas, metastatic PDAC was widely distributed, and the tumor tissue exhibited a high degree of genomic instability [96].
Prognostic role of TP53 mutations
Despite the importance of TP53 mutations in tumorigenesis and progression, many studies have investigated the predictive and prognostic roles of p53 expression and reported seemingly controversial results (Table 3). Although mutations in TP53 increase the stability and accumulation of the p53 protein, immunohistochemical staining for p53 is not related to the overall survival of patients with PDAC who undergo complete pancreatic resection [59, 60, 97]. However, in patients with metastatic pancreatic cancer treated with FOLFIRINOX, high levels of p53 expression in the tumor are significantly correlated with a poor overall survival rate but not a poor PFS or response rate [98]. In patients receiving adjuvant gemcitabine treatment, p53 expression is inversely related to disease-free survival and overall survival, consistent with the high tolerance to gemcitabine cytotoxicity exhibited by a mut-p53 cell line [99, 100]. While mutated and wild-type p53 do not result in a difference in patient survival, an analysis of cancer genomic data showed that specific mutation types, such as mutations at Arg248 and Arg282, result in a notably poor outcome in several tumors [101]. Moreover, the abnormal expression of p53 predicts a high risk of locoregional recurrence (P = 0.020) [84]. In summary, genetic alterations in TP53 are capable of predicting advanced tumor progression, but their prognostic status requires further study.
Treatments targeting TP53 mutations
Approximately half of all human cancers harbor mutations in TP53, and multiple strategies targeting TP53 have been proposed. Gain-of-function mutations in TP53 markedly increase the proliferation and metastasis of tumor cells, consistent with the decreased function of wild-type p53. Therefore, the identification of compounds or therapies that restore wild-type p53 activity or delete the mut-p53 protein shows promise for treating TP53-mutated cancers [102]. Several compounds have been reported to restore the transcriptional activity of the mut-p53 protein, such as PRIMA-1 and APR-246, which bind to thiols in the core domain of mut-p53 [103]. PRIMA-1 rescues the function of mut-p53 and induces apoptosis and cell cycle arrest in pancreatic cancer cells [104]. In addition, PRIMA-1 increases the sensitivity of p53-mutated PDAC cells to gemcitabine and erlotinib. An inhibitor of histone deacetylase 6 (HDAC6), a positive regulator of HSP90, also blocks the oncogenic activity of mut-p53 by increasing the degradation of mut-p53 without altering wild-type p53 [105]. Furthermore, HSP90 inhibitors exhibited significant efficacy in reducing the growth and angiogenesis of pancreatic cancer both in vitro and in vivo but failed in a phase II study of patients with advanced pancreatic cancer [106–108].
Furthermore, the restoration of wild-type p53 by delivering nanoparticles carrying plasmid DNA induces apoptosis in pancreatic cancer cells [109]. The combination of wt-p53-expressing plasmid DNA and gemcitabine significantly inhibited tumor proliferation compared with gemcitabine alone, with 77.3% and 61.7% reductions in tumor growth, respectively [109]. A phase II study is ongoing to evaluate the efficacy of the combination of targeted p53 gene therapy plus gemcitabine/nab-paclitaxel in patients with metastatic pancreatic cancer (NCT02340117). Moreover, Chung et al. immunized patients with solid tumors with a p53-expressing modified vaccinia Ankara virus (p53MVA), which activates peripheral T cells to kill cells overexpressing mut-p53, and a clinical response was observed in 3/11 patients [110].
Because cancer cells with mut-p53 lose control over the G1 checkpoint and rely heavily on the G2 checkpoint to repair cellular DNA damage, treatments that inhibit the key regulator of the G2 checkpoint, Wee1 kinase, are predicted to abrogate the DNA repair process and induce synthetic lethality in TP53-mutated cancer cells [111]. A phase II clinical trial evaluated the efficacy of adavosertib (AZD1775), a Wee1 kinase inhibitor, in combination with gemcitabine and radiotherapy in patients with locally advanced pancreatic cancer and reported an extremely encouraging result: a median OS duration of 21.7 months and a median PFS duration of 9.7 months [112]. Although the median OS duration was 15.2 months for patients receiving chemoradiotherapy in LAP07 trials, Wee1 inhibitors dramatically improved clinical outcomes and hence provided opportunities for future treatment [113]. Last but not least, downstream molecules of mut-p53 and the ability to block the formation of mut-p53 complexes and other proteins represent targets for anticancer drugs [102, 114].
SMAD4
General introduction
SMAD4 (Sma (Caenorhabditis elegans) mothers against decapentaplegia homologue 4), also known as DPC4 (deleted in pancreatic cancer, locus 4), is a tumor suppressor gene that is mutated in a wide range of diseases and cancers, particularly in pancreatic cancers, with a mutation rate of ~20%–50% [11, 115]. The SMAD family consists of 8 proteins and plays a crucial role in mediating TGF-β signaling. Although SMAD4 is not obligatory for the activation of TGF-β signaling pathways, it is indispensable for producing a strong signaling response [116]. SMAD4 shuttles between the nucleus and cytoplasm and forms a heterodimeric complex with SMAD2/SMAD3, which is phosphorylated by activated TGF-β receptors. The SMAD complex subsequently enters the nucleus and interacts with downstream proteins to regulate the transcription of target genes. The E3 ubiquitin ligase ectodermin targets SMAD4 for degradation in the nucleus and antagonizes TGF-β signaling, thus blocking downstream pathways regulating cell differentiation and proliferation [117]. In contrast, the deubiquitinase USP9x reverses the ubiquitination caused by ectodermin and restores the function of SMAD complexes [118].
SMAD4 mutations in pancreatic cancer
The TGF-β/SMAD4 signaling pathway mediates the growth of cancer cells by promoting cell cycle arrest, apoptosis and DNA damage repair, while genetic alterations in SMAD4 attenuate the tumor suppressor function of the TGF-β pathway [119, 120]. In contrast, enhancement of the epithelial-mesenchymal transition (EMT) process in a SMAD4-dependent manner is commonly presumed to increase the invasion and metastasis of cancer cells [121]. As one of the driver genes, SMAD4 is mutated in half of PDAC cases, with homozygous deletions occurring in 30% of cases and chromosome allelic loss existing in 20% of cases, and the mutations remarkably decrease immunohistochemical staining for the SMAD4 protein [122, 123]. In addition, loss of SMAD4 expression has been detected in high-grade precursor lesions rather than in low-grade lesions, suggesting that the inactivation of SMAD4 promotes progression to a later stage of tumorigenesis [124]. Similarly, SMAD4 deletion alone in a transgenic mouse model was insufficient to initiate the development of PDAC, while SMAD4 inactivation substantially enhanced the progression of KRASG12D-initiated neoplasms [125]. In addition, in mouse models of PDAC carrying mutations in both KRAS and TRP53, the inactivation of SMAD4 increased metastasis, but the expression of wild-type SMAD4 decreased metastasis and increased proliferation [126]. Our previous studies have also demonstrated a disparity in progression and migration that might result from TGF-β-induced autophagy and PGK1-mediated metabolic reprogramming, depending on the SMAD4 status [127, 128]. Alterations in SMAD4 also regulate the differentiation of PDAC: SMAD4 insufficiency is beneficial to retain epithelial features, while wild-type SMAD4 promotes the EMT process [125]. Surprisingly, the EMT process is dispensable for pancreatic cancer metastasis but promotes the proapoptotic function of the TGF-β signaling pathway [129, 130]. SMAD4 deletion remarkably increases the resistance of both PDAC cell lines and mouse models to radiotherapy, and this decrease in radiosensitivity is correlated with the induction of ROS production and autophagy [131].
Prognostic and predictive roles of SMAD4 mutations
The relationship between SMAD4 mutations and clinical outcomes has been extensively investigated in numerous studies but remains elusive [132–134]. Andrew et al. investigated the relationship between SMAD4 expression and overall survival in 119 patients with PDAC and showed that the loss of SMAD4 expression was remarkably associated with an improved median survival in patients who underwent pancreatic resection in the univariate analysis (13.6 vs 6.4 months, P = 0.0257) [132]. However, the majority of other studies reported shorter overall survival in patients with SMAD4-inactivated PDAC (Table 4). The loss of SMAD4 also predicts a significant benefit from postoperative adjuvant chemotherapy (P = 0.002) [135]. The loss of SMAD4 is also significantly associated with an increased metastatic burden, and wild-type SMAD4 is related to local recurrence, indicating that systematic chemotherapy might achieve satisfactory outcomes in patients with SMAD4 mutations [136, 137]. However, different studies have reported contradictory findings regarding the role of the SMAD4 status in predicting the recurrence pattern of patients with resected PDAC [135, 138].
Treatments targeting SMAD4
Although several anticancer agents targeting SMAD4-deficient cells have been discovered, no results from animal models or human trials have been published to date [139, 140]. Since the TGF-β signaling pathway promotes the progression and metastasis of PDAC in the absence of SMAD4, strategies targeting TGF-β might provide new methods for clinical treatments. Vactosertib (TEW-7197), an inhibitor of TGF-β signaling, combined with nanoliposomal irinotecan and 5-FU dramatically improved the survival outcome of an animal model of pancreatic cancer and suppressed the migration and invasion of pancreatic cancer cells [141]. Furthermore, a phase Ib clinical trial is ongoing to evaluate the efficacy of vactosertib with FOLFOX in patients with metastatic pancreatic cancers (NCT03666832). Synthetic lethality is another potential option available for targeting SMAD4 mutations, as the loss of SMAD4 is commonly accompanied by a passenger deletion of mitochondrial malic enzymes 2 (ME2), a housekeeping gene that functions with ME3 to sustain NADPH synthesis in mitochondria. Inhibition of ME3 substantially slows the growth and proliferation of ME2-null pancreatic cells, suggesting that compounds targeting ME3 are promising treatments for patients with SMAD4 mutations [142]. Moreover, due to the potential ability of wild-type SMAD4 to predict locally advanced pancreatic cancer, patients with intact SMAD4 expression might benefit more from intense local therapy than systematic chemotherapy. However, no ongoing registered clinical trial is evaluating the role of the SMAD4 status in radiotherapy. Further clinical trials examining the SMAD4 status might be able to improve the efficacy of radiotherapy in the treatment of local pancreatic cancers.
Other genes
BRCA1/2
BRCA1/2 are the most common genes mutated in familial pancreatic cancers, and mutations in BRCA1/2 increase the risk of pancreatic cancer susceptibility [143]. Germline BRCA1/2 mutations occur in 4%–7% of all pancreatic cancers [144]. As the key factors involved in DNA damage repair, BRCA1/2 cooperate to mediate recombination between homologous DNA sequences to repair double-strand DNA breaks (DSBs), while mutations in BRCA1/2 lead to inappropriate DSB repair during the cell cycle and gross chromosomal rearrangements [145]. In addition, as one of the genes involved in Fanconi’s anemia (FA) pathways, BRCA2 and other FA proteins, together with BRCA1, are required for the repair of DNA interstrand cross-links [146]. In BRCA1/2-deficient cells, the accumulation of DSBs and genomic instability drive malignant transformation and progression [147]. Likewise, poly (ADP-ribose) polymerase 1 (PARP1) is responsible for the repair of single-strand DNA breaks (SSBs), and the simultaneous dysregulation of PARP1 and BRCA1/2 results in genomic instability and cell death (Fig. 4). This synthetic lethality provides evidence for the ability of PARP1 to serve as a potential target for the treatment of BRCA1/2-mutated cancers.
Fig. 4. DNA damage repair pathway and the mechanism of PARP inhibitors.
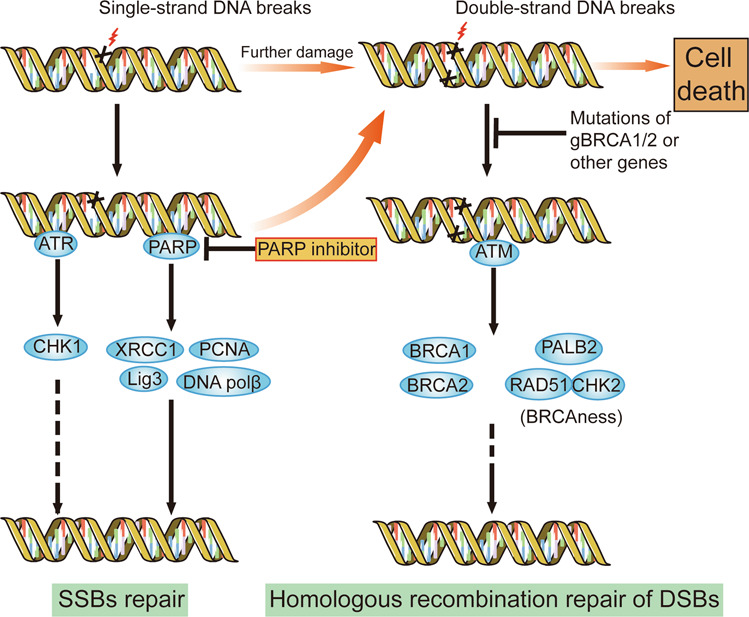
DNA damage repair mainly includes the repair of DSBs and SSBs. PARP and ATR/CHK1 are responsible for SSB repair, while ATM, BRCA1/2 and other BRCAness-related genes are necessary for the homologous recombination repair of DSBs. PARP inhibitors block the repair of SSBs and increase DSBs. Mutations in germline BRCA1/2 or other BRCAness-related genes impair the homologous recombination repair of DSBs, leading to the accumulation of DSBs. The dysfunction of two pathways causes synthetic lethality, genomic instability and cell death.
In parallel with the roles of BRCA1/2 in DNA damage repair, advanced pancreatic cancers with BRCA 1/2 mutations also respond well to chemotherapies containing DNA-interacting regimens, such as platinum compounds [148, 149]. PARP inhibitors are effective in patients carrying germline BRCA mutations with several types of advanced cancers, including breast cancer, prostate cancer and ovarian cancer [150–152]. In a phase III clinical trial of 154 patients with germline BRCA-mutated metastatic pancreatic cancers, the PARP inhibitor olaparib dramatically prolonged progression-free survival (PFS) compared with the placebo (7.4 months vs 3.8 months, respectively), while no statistically significant difference in overall survival was observed between the olaparib and placebo groups. According to the NCCN guidelines version 1.2020, olaparib has been highlighted as a maintenance therapy for patients with metastatic PDAC carrying germline BRCA1/2 mutations who have not experienced disease progression. Furthermore, ongoing clinical trials are evaluating the efficacy of PARP inhibitors against both germline and somatic mutations in BRCA1/2 (NCT03601923). In addition, genetic alterations in the PALB2, CHK2, ATM and RAD51 genes result in defects in homologous recombination and DNA repair in the absence of BRCA1/2 mutations, and this phenocopy of BRCA1/2 mutations is defined as BRCAness [153]. Concerning mutations in other genes involved in BRCAness, their response to PARP inhibitors is also being investigated in patients with advanced PDAC (NCT03601923 and NCT04171700).
ATM
ATM, ataxia telangiectasia mutated, is an indispensable gene that senses and repairs DNA damage [154]. As a serine/threonine kinase, ATM reacts to cellular DSBs by phosphorylating downstream proteins to activate multiple cellular processes, including cell cycle arrest, apoptosis and DNA repair [154]. Hu et al. conducted a large case-control study of 3030 patients with pancreatic cancer and found that germline mutations in ATM occurred in 2.3% of patients compared with 0.37% in the normal population (OR, 5.71; 95% CI, 4.38–7.33) [155]. In addition, next-generation sequencing of two pedigrees of familial pancreatic cancers revealed that loss-of-function mutations are the main type of ATM mutation and are highly correlated with the predisposition to familial pancreatic cancer [156]. Based on these studies, ATM mutations promote the tumorigenesis and malignancy of pancreatic cancers.
In contrast to BRCA1/2 mutations, the loss of ATM is an independent prognostic factor for poor overall survival in patients with resectable pancreatic cancers [157–159]. Lukas et al. constructed a transgenic mouse model and cell lines with ATM deletion and showed that the loss of ATM enhanced the malignant features of pancreatic cancer cells, such as genomic instability and migratory properties, and promoted proliferation under metabolic stress [160]. In addition, in the same study, both in vitro and in vivo experiments confirmed that ATM deficiency increases radiosensitivity and induces synthetic lethality in combination with PARP inhibitors. A phase II proof-of-concept trial is being conducted to investigate the role of the PARP inhibitor niraparib in treating advanced PDAC patients carrying mutations in ATM and other BRCAness-related genes (NCT03601923).
In addition to PARP1, the ATR (Ataxia telangiectasia and Rad3-related) protein also mediates SSB repair, and its inhibitors are capable of inducing synthetic lethality in the context of ATM deletion [161]. The PARP inhibitor olaparib or the ATR inhibitor VE-822 dramatically inhibited the growth of ATM-deficient pancreatic cancer in vitro and in vivo [160]. Moreover, the combination of olaparib, an ATR inhibitor, and cisplatin has been tested in a clinical trial for refractory solid tumors (NCT02723864). Two distinct kinase signaling cascades account for the response to cellular DNA damage, as checkpoint kinase 1 (CHK1) participates in ATR-mediated SSB repair, while checkpoint kinase 2 (CHK2) is involved in ATM-mediated DSB repair [162]. Inhibitors of CHK1 or CHK2 are capable of eliciting the cytotoxicity associated with chemotherapy in pancreatic cancer cells, and multiple studies have been performed to determine their efficacy in clinical practice [163–165]. Homologous recombination-related gene mutations are estimated to occur in 15.4% of PDACs, indicating that a considerable percentage of patients with pancreatic cancer might benefit from inhibitors targeting the DNA repair process [166]. Moreover, although numerous potential targets in the DNA repair process have been identified, PARP inhibitors are the only drugs available for personalized therapy in patients with pancreatic cancers.
PALB2
PALB2, partner and localizer of breast cancer 2 (BRCA2), interacts with BRCA1/2 and modulates the localization of BRCA2 to facilitate the homologous recombination process during DNA damage repair [167]. Similar to BRCA2, PALB2 is also one of the genes involved in Fanconi’s anemia pathways, and mutations in BRCA2 and PALB2 have been confirmed to increase the susceptibility to breast cancer [146]. In addition, PALB2 is mutated in 0.6%–3% of hereditary pancreatic cancers, while the mutation rate varies in different populations [168–170].
While PALB2 mutations are estimated to increase the risk of breast cancers 5–9-fold in different age groups, the relation between PALB2 mutations and the predisposition to pancreatic cancer remains controversial [155, 166, 171, 172]. In a single case report of metastatic and gemcitabine-resistant pancreatic cancer, Maria et al. found that mitomycin C and cisplatin significantly prolonged symptom-free survival to at least 3 years, and exome sequencing revealed the connection of PALB2 mutations with high sensitivity to DNA damaging agents [173]. Moreover, interstrand crosslinking agents dramatically inhibited pancreatic cancer growth in mouse models with deletions of PALB2 and BRCA1/2 [174]. In a phase I study of the PARP inhibitor talazoparib, a clinical benefit was observed in a patient with a PALB2 mutation, suggesting an expanded range of potential targets of the PARP inhibitor [175]. Due to its close relationship with BRCA1/2, several ongoing clinical trials are investigating the role of the PARP inhibitor in treating PALB2-mutated pancreatic cancers (NCT04300114 and NCT03337087).
BRAF
The BRAF protein is a member of the RAF family of serine/threonine protein kinases. With the wide prevalence of KRAS mutations in pancreatic cancer, downstream signaling pathways, such as RAF/MEK/ERK, PI3K/AKT/mTOR, and RalA/B, all enhance cancer progression, proliferation and differentiation [176]. As one of the most common mutations in melanoma, BRAF mutations, mainly the BRAF V600E point mutation, have been detected in 1.4%-3% of pancreatic cancers and are mutually exclusive with KRAS mutations [177–179]. According to Eric et al.,the BRAF V600E mutation alone was sufficient to drive PanIN lesions in the mouse pancreas, while the combination of BRAF and TP53 mutations led to the formation of PDAC [180]. In the same study, MEK1/2 inhibitors induced a profound survival benefit in mice with PDAC. Therefore, BRAF mutations and RAF/MEK/ERK signaling play a pivotal role in the initiation and progression of PDAC expressing wild-type KRAS.
BRAF inhibitors, including vemurafenib and dabrafenib, have been used in clinical practice and dramatically changed the treatment of melanoma expressing the BRAF V600 mutant[181]. In addition, the combination of dabrafenib and the MEK inhibitor trametinib significantly improved the PFS outcome of patients with metastatic melanoma carrying the BRAF V600 mutation compared with dabrafenib alone [182]. The efficacy of dabrafenib and trametinib has also been verified during adjuvant therapy for patients with stage III melanoma carrying BRAF V600 mutations [183]. Kazimierz et al. described a patient with advanced PDAC carrying BRAF mutations, and dabrafenib dramatically improved the patient’s clinical condition for 6 months [184]. However, no clinical trials have been conducted to examine the efficacy of BRAF inhibitors in patients with metastatic pancreatic cancers.
Challenges and perspectives
What factors might be responsible for the failure of various molecularly targeted therapies: from the laboratory to the clinic
For the past several decades, a milieu of genetic targets have been tested for potential efficacy, with many of them succeeding in the preclinical stage but failing in clinical trials. Many mechanisms have been identified to explain this frustrating condition, including the complex biological features and microenvironment of PDAC [185, 186]. The high density of stromal cells and interaction between tumor cells and the microenvironment might lead to an unsatisfactory drug response and chemotherapy resistance [187]. As a result, reliable preclinical tools that imitate real tumor biology in the human body are difficult to establish. Different cell lines and genetically engineered mouse models have been widely used to examine the functions of genetic drivers. However, the cell lines only recapitulate a small group of patients with PDAC, and the function of the extracellular matrix is easily neglected in cell-based experiments [188]. Genetically engineered mouse models represent state-of-the-art methods to investigate the functions of genetic alterations and responses to new drugs, but species disparity and other underlying mechanisms, such as the effect of microbes, limit the ability of this model to assess new chemotherapies and detailed pancreatic tumor biology [176]. As only a small percentage of targeted drugs have entered phase III trials or clinical practice in the past several decades, we must reflect on our research strategies and obtain additional insights into the biology of PDAC.
Recently, organoids have emerged as new techniques and reliable models of human organs and diseases in vitro. Compared to the low cellularity of the primary tumor tissue, organoids are derived from cancer cells and possess high neoplastic purity, which may assist researchers in identifying more actionable genetic alterations. In addition, organoids avoid the differences between human tumors and mouse models because they are directly constructed from the tumor tissue rather than from injecting tumor cells into mice [189]. Tiriac and colleagues successfully generated pancreatic cancer organoids from human samples and found that the organoids exhibit high similarity to the primary tumor specimens in terms of the genetic hallmarks. They also analyzed the therapeutic profile of organoids in response to different treatments, and this pharmacotranscriptomic signature showed high concordance with chemotherapy sensitivities [190]. Moreover, alterations in the KRAS or TP53 gene in organoids promote the initiation and progression of PDAC [191]. Without the highly dense stroma, the organoid potentially represents a more efficient preclinical tool to test the effects of molecularly targeted therapies on pancreatic cancer cells. Notably, coculture of cancer cells, the stroma and other peritumoral components in an organoid model can provide an environment that is similar to that of pancreatic cancer in humans. Therefore, new tools and therapeutic methods to identify and examine more molecular medicines are needed in future studies.
What is the role of genetic testing in the treatment of pancreatic cancer?
A precise therapy based on genomic data might represent a new era in cancer treatment. A mixture of genetic mutations endow pancreatic cancer with different properties in different patients, and personalized therapy based on the mutation types might provide unprecedented clinical benefits. An analysis of molecular profiles revealed that pancreatic cancer is not a single disease, and this heterogeneous disease has been divided into several subgroups with various responses to chemotherapy [192]. In particular, patients with a defect in homologous recombination exhibit a satisfactory clinical response to platinum-based chemotherapy. Olaparib, the only orphan drug used to treat pancreatic cancer, has potential therapeutic efficacy in patients not only with germline BRCA1/2 mutations but also with other BRCAness-related gene mutations. The anti-PD-1 receptor antibody pembrolizumab dramatically reduced tumor progression in patients with solid tumors presenting with high microsatellite instability or mismatch repair deficiency, which accounts for <2% of patients with PDAC [193]. Moreover, the FDA approved the TRK inhibitors larotrectinib and entrectinib as treatments for solid tumors with NTRK gene infusions, and TRK inhibitors were tolerated and promoted relatively prolonged survival [194]. Therefore, the identification of genetic alterations provides opportunities to administer precise chemotherapy, particularly in patients in whom first-line therapy has failed. According to the NCCN guidelines version 1.2020, actionable targets include fusions of ALK, NRG1, NTRK and ROS1, mutations in BRAF, BRCA1/2, HER2, KRAS and PALB2, and mismatch repair deficiency. In addition, an examination of the response of xenograft tumors can predict the chemotherapy scheme resulting in the greatest sensitivity to increase the overall survival rate [173].
However, many limitations in genetic testing still exist, since only a small percentage of patients with genomic results ultimately receive genome-guided therapy [195, 196]. First, at least 20 days are needed to determine the genomic profile, which is a relatively long period to wait in some cases. Second, even if the genomic profile is available, only 10% of patients have actionable genetic alterations that have been verified in clinical trials, and the clinical benefit of precision medicine in a single individual remains unclear. Moreover, the high cost of genetic sequencing and the related processes may also limit its clinical applications.
Personalized therapy and combinatorial therapy in pancreatic cancer
The increasing prevalence of next-generation sequencing has made it possible to identify druggable genetic alterations. As discussed before, patients with germline BRCA mutations, ALK fusions, mismatch repair deficiency and other aberrations have benefitted from targeted therapies. However, only a small percentage of patients harbor these aberrations. Moreover, the feasibility of identifying and utilizing actionable aberrations as a routine clinical practice still needs to be confirmed and normalized in additional trials [197]. Notably, the aforementioned targeted therapies are related more to cancer treatment than the characteristics of pancreatic cancer. Transcriptional profile analysis has uncovered subtypes of PDAC and their different responses to chemotherapies. Our previous research also demonstrated that patients with a high strain ratio in EUS respond well to gemcitabine plus nab-paclitaxel, which indicates that a high strain ratio could provide guidance for the utility of stroma-disrupting agents [198]. Therefore, integration of the clinical and molecular information of pancreatic cancers is of significance to identify subgroups to offer more strategies for personalized therapies.
Due to the high malignancy of pancreatic cancer, only a few drugs can be applied to systemic therapy (Table 5). Since the end of the last century, gemcitabine-based therapies have been widely tested in clinical trials, but only a few combinations have been proven to significantly extend overall survival with systematic therapy. The combined use of targeted drugs has also been reported in several clinical trials, but most failed to provide better efficacy. In the clinical trial SWOG S1115, the combination of AKT and MEK inhibitors did not result in improved overall survival [199]. In addition, the high toxicity of combinatorial therapy remains an important issue, and some studies reported that only patients with a good performance benefitted from gemcitabine-based combinatorial therapy [200]. However, the success of FOLFIRINOX and nab-paclitaxel plus gemcitabine in the past decade suggests that combinatorial therapies of different chemotherapeutic agents could still be inevitable in further trials. Appropriate combinations and subgroup analyses might also be necessary in further trials.
Table 5.
Summary of drugs showing favorable result in phase III clinical trials of systematic therapy.
| Tested drug | N (subjects) | Treatment setting | Therapeutic scheme | Median OS | Median PFS | Year | Ref |
|---|---|---|---|---|---|---|---|
| Gem | 126 | Advanced pancreatic cancer | Gem vs 5-FU | 5.56 months vs 4.41 months, P = 0.0025 | – | 1997 | [223] |
| Erlotinib | 569 | Locally advanced or metastatic PDAC | Gem + erlotinib vs gem + placebo | 6.24 months v 5.91 months, P = 0.038 | 3.75 months v 3.55 months, P = 0.004 | 2007 | [218] |
| Capecitabine | 533 | Advanced pancreatic cancer | Capecitabine + gem vs gem | 7.1 months vs 6.2 months, P = 0.08 | 5.3 months vs 3.8 months, P = 0.004 | 2009 | [224] |
| Nab-paclitaxel + gem | 861 | Advanced pancreatic cancer | Nab-paclitaxel + gem vs gem | 8.5 months vs 6.7 months, P < 0.0001 | 5.5 months vs 3.7 months, P < 0.0001 | 2013 | [7] |
| FOLFIRINOX | 342 | Metastatic pancreatic cancer | FOLFIRINOX vs gem | 11.1 months vs 6.8 months, P < 0.0001 | 6.4 months vs 3.3 months, P < 0.0001 | 2011 | [6] |
| S-1 | 377 | Resected pancreatic cancer | S-1 vs gem | 46.5 months vs 25.5 months, P < 0.0001 | 22.9 months vs 11.9 months, P < 0.0001a | 2016 | [225] |
| mFOLFIRINOX | 493 | Resected pancreatic cancer | mFOLFIRINOX vs gem | 54.4 months vs 35.0 months, P = 0.003 | 21.6 months vs 12.8 months, P < 0.001 | 2018 | [226] |
| Olaparib | 154 | Germline BRCA-mutated metastatic pancreatic cancer. | Olaparib vs placebo | 18.9 months vs 18.1 months, P = 0.68 | 7.4 months vs. 3.8 months, P = 0.004 | 2019 | [144] |
aThe relapse-free survival of S-1/gem in adjuvant therapy.
Conclusions
KRAS, CDKN2A, TP53, and SMAD4 have been confirmed to be mutated in a wide range of pancreatic cancers and play a crucial role in driving tumorigenesis and metastasis through different mechanisms. Here, we conducted a general review of the biological functions and clinical implications of these four driver genes in pancreatic cancer. Despite tremendous efforts to investigate the functions of driver gene mutations, with the majority of studies focused on KRAS, the detailed mechanism remains elusive, and clinical trials have seldom reported significant benefits on overall outcomes. Considering the sophisticated crosstalk among these altered genes, monotherapy inhibiting a single target is unlikely to produce a remarkable clinical benefit, and a combination of multiple targeted drugs might provide further opportunities for treatment.
Acknowledgements
This work was supported by grants from Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), National Science Foundation for Distinguished Young Scholars of China [81625016], National Natural Science Foundation of China (No. 81871950 and 81972250), Shanghai Municipal Commission of Health and Family Planning (No. 2018YQ06).
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential competing interests.
Footnotes
These authors contributed equally: Hai-feng Hu, Zeng Ye
Contributor Information
Qi-feng Zhuo, Email: zhuoqifeng@fudanpci.org.
Shun-rong Ji, Email: jishunrong@fudanpci.org.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21.. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–48.. doi: 10.1038/s41575-018-0005-x. [DOI] [PubMed] [Google Scholar]
- 4.Maisonneuve P. Epidemiology and burden of pancreatic cancer. Presse Med. 2019;48:e113–e23.. doi: 10.1016/j.lpm.2019.02.030. [DOI] [PubMed] [Google Scholar]
- 5.Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10–27. doi: 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25.. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 7.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703.. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Adv Anat Pathol. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 9.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 11.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88.. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553–65.. doi: 10.1038/nrc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–72. [PubMed] [Google Scholar]
- 16.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54.. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 17.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140–1. doi: 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- 18.Haigis KM. KRAS alleles: the devil is in the detail. Trends Cancer. 2017;3:686–97. doi: 10.1016/j.trecan.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–9. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–68.. doi: 10.1038/s41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- 21.Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers (Basel) 2016;8:45. doi: 10.3390/cancers8040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–53.. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omori Y, Ono Y, Tanino M, Karasaki H, Yamaguchi H, Furukawa T, et al. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology. 2019;156:647–61 e2. doi: 10.1053/j.gastro.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165:1818. doi: 10.1016/j.cell.2016.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70.. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W, et al. Metabolic plasticity in heterogeneous pancreatic ductal adenocarcinoma. Biochim Biophys Acta. 2016;1866:177–88.. doi: 10.1016/j.bbcan.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50.. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 30.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7:e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–20.. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. doi: 10.1016/j.pharmthera.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H, et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res. 2015;25:561–73.. doi: 10.1038/cr.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji SR, Qin Y, Liang C, Huang R, Shi S, Liu J, et al. FBW7 (F-box andWDRepeat Domain-Containing 7) negatively regulates glucose metabolism by targeting the c-Myc/TXNIP (thioredoxin-binding protein) axis in pancreatic cancer. Clin Cancer Res. 2016;22:3950–60.. doi: 10.1158/1078-0432.CCR-15-2380. [DOI] [PubMed] [Google Scholar]
- 35.Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W, et al. Oncogenic KRAS targets MUC16/CA125 in pancreatic ductal adenocarcinoma. Mol Cancer Res. 2017;15:201–12.. doi: 10.1158/1541-7786.MCR-16-0296. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Sanagapalli S, Stoita A. Challenges in diagnosis of pancreatic cancer. World J Gastroenterol. 2018;24:2047–60.. doi: 10.3748/wjg.v24.i19.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilentz RE, Chung CH, Sturm PD, Musler A, Sohn TA, Offerhaus GJ, et al. K-ras mutations in the duodenal fluid of patients with pancreatic carcinoma. Cancer-Am Cancer Soc. 1998;82:96–103. doi: 10.1002/(sici)1097-0142(19980101)82:1<96::aid-cncr11>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Lisotti A, Frazzoni L, Fuccio L, Serrani M, Cominardi A, Bazzoli F, et al. Repeat EUS-FNA of pancreatic masses after nondiagnostic or inconclusive results: systematic review and meta-analysis. Gastrointest Endosc. 2020;91:1234–41.e4. doi: 10.1016/j.gie.2020.01.034. [DOI] [PubMed] [Google Scholar]
- 39.Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur J Cancer. 2016;54:75–83. doi: 10.1016/j.ejca.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Khalid A, Dewitt J, Ohori NP, Chen JH, Fasanella KE, Sanders M, et al. EUS-FNA mutational analysis in differentiating autoimmune pancreatitis and pancreatic cancer. Pancreatology. 2011;11:482–6. doi: 10.1159/000331505. [DOI] [PubMed] [Google Scholar]
- 41.Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA. 2017;114:10202–7. doi: 10.1073/pnas.1704961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard V, Kim DU, San Lucas FA, Castillo J, Allenson K, Mulu FC, et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic cancer. Gastroenterology. 2019;156:108–18 e4. doi: 10.1053/j.gastro.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe F, Suzuki K, Tamaki S, Abe I, Endo Y, Takayama Y, et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS One. 2019;14:e0227366. doi: 10.1371/journal.pone.0227366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perets R, Greenberg O, Shentzer T, Semenisty V, Epelbaum R, Bick T, et al. Mutant KRAS circulating tumor DNA is an accurate tool for pancreatic cancer monitoring. Oncologist. 2018;23:566–72.. doi: 10.1634/theoncologist.2017-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28:741–7. doi: 10.1093/annonc/mdx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang S, Che SP, Kurywchak P, Tavormina JL, Gansmo LB, Correa de Sampaio P, et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol Ther. 2017;18:158–65.. doi: 10.1080/15384047.2017.1281499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiala C, Diamandis EP. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018;16:166. doi: 10.1186/s12916-018-1157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–81.. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 49.Winter JJ, Anderson M, Blades K, Brassington C, Breeze AL, Chresta C, et al. Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. J Med Chem. 2015;58:2265–74. doi: 10.1021/jm501660t. [DOI] [PubMed] [Google Scholar]
- 50.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci USA. 2012;109:5299–304.. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu S, Jang H, Zhang J, Nussinov R. Inhibitors of Ras-SOS interactions. ChemMedChem. 2016;11:814–21. doi: 10.1002/cmdc.201500481. [DOI] [PubMed] [Google Scholar]
- 52.Holderfield M. Efforts to develop KRAS inhibitors. Cold Spring Harb Perspect Med. 2018;8:a031864. doi: 10.1101/cshperspect.a031864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 54.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21:1819–27.. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong J, Mita MM. Activated ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol. 2014;4:167. doi: 10.3389/fonc.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noonan AM, Farren MR, Geyer SM, Huang Y, Tahiri S, Ahn D, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther. 2016;24:1150–8. doi: 10.1038/mt.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20723–8. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strand MS, Krasnick BA, Pan H, Zhang X, Bi Y, Brooks C, et al. Precision delivery of RAS-inhibiting siRNA to KRAS driven cancer via peptide-based nanoparticles. Oncotarget. 2019;10:4761–75.. doi: 10.18632/oncotarget.27109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin SH, Kim SC, Hong SM, Kim YH, Song KB, Park KM, et al. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013;42:216–22.. doi: 10.1097/MPA.0b013e31825b6ab0. [DOI] [PubMed] [Google Scholar]
- 60.Sinn BV, Striefler JK, Rudl MA, Lehmann A, Bahra M, Denkert C, et al. KRAS mutations in codon 12 or 13 are associated with worse prognosis in pancreatic ductal adenocarcinoma. Pancreas. 2014;43:578–83.. doi: 10.1097/MPA.0000000000000077. [DOI] [PubMed] [Google Scholar]
- 61.Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48:640–6. doi: 10.1007/s00535-012-0664-2. [DOI] [PubMed] [Google Scholar]
- 62.Heinemann V, Vehling-Kaiser U, Waldschmidt D, Kettner E, Marten A, Winkelmann C, et al. Gemcitabine plus erlotinib followed by capecitabine versus capecitabine plus erlotinib followed by gemcitabine in advanced pancreatic cancer: final results of a randomised phase 3 trial of the ‘Arbeitsgemeinschaft Internistische Onkologie’ (AIO-PK0104) Gut. 2013;62:751–9. doi: 10.1136/gutjnl-2012-302759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schultheis B, Reuter D, Ebert MP, Siveke J, Kerkhoff A, Berdel WE, et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: a multicenter, randomized phase IIb study. Ann Oncol. 2017;28:2429–35.. doi: 10.1093/annonc/mdx343. [DOI] [PubMed] [Google Scholar]
- 64.Lee JW, Lee JH, Shim BY, Kim SH, Chung MJ, Kye BH, et al. KRAS mutation status is not a predictor for tumor response and survival in rectal cancer patients who received preoperative radiotherapy with 5-fluoropyrimidine followed by curative surgery. Med (Baltim) 2015;94:e1284. doi: 10.1097/MD.0000000000001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim ST, Lim DH, Jang KT, Lim T, Lee J, Choi YL, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther. 2011;10:1993–9. doi: 10.1158/1535-7163.MCT-11-0269. [DOI] [PubMed] [Google Scholar]
- 66.Propper D, Davidenko I, Bridgewater J, Kupcinskas L, Fittipaldo A, Hillenbach C, et al. Phase II, randomized, biomarker identification trial (MARK) for erlotinib in patients with advanced pancreatic carcinoma. Ann Oncol. 2014;25:1384–90.. doi: 10.1093/annonc/mdu176. [DOI] [PubMed] [Google Scholar]
- 67.Kong Y, Sharma RB, Nwosu BU, Alonso LC. Islet biology, the CDKN2A/B locus and type 2 diabetes risk. Diabetologia. 2016;59:1579–93.. doi: 10.1007/s00125-016-3967-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75.. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–7. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 70.Taneera J, Fadista J, Ahlqvist E, Zhang M, Wierup N, Renstrom E, et al. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol Cell Endocrinol. 2013;375:35–42. doi: 10.1016/j.mce.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–52.. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–307.. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ozenne P, Eymin B, Brambilla E, Gazzeri S. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. 2010;127:2239–47.. doi: 10.1002/ijc.25511. [DOI] [PubMed] [Google Scholar]
- 74.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–52.. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghiorzo P, Pastorino L, Bonelli L, Cusano R, Nicora A, Zupo S, et al. INK4/ARF germline alterations in pancreatic cancer patients. Ann Oncol. 2004;15:70–8. doi: 10.1093/annonc/mdg498. [DOI] [PubMed] [Google Scholar]
- 76.Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, et al. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30:2087–97.. doi: 10.1038/onc.2010.614. [DOI] [PubMed] [Google Scholar]
- 77.Singh SK, Ellenrieder V. Senescence in pancreatic carcinogenesis: from signalling to chromatin remodelling and epigenetics. Gut. 2013;62:1364–72.. doi: 10.1136/gutjnl-2012-302793. [DOI] [PubMed] [Google Scholar]
- 78.Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160:1573–81.. doi: 10.1016/S0002-9440(10)61104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azzopardi S, Pang S, Klimstra DS, Du YN. p53 and p16(Ink4a)/p19(Arf) loss promotes different pancreatic tumor types from PyMT-expressing progenitor cells. Neoplasia. 2016;18:610–7. doi: 10.1016/j.neo.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 81.House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, et al. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–31. doi: 10.1097/01.sla.0000086659.49569.9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen J, Li D, Killary AM, Sen S, Amos CI, Evans DB, et al. Polymorphisms of p16, p27, p73, and MDM2 modulate response and survival of pancreatic cancer patients treated with preoperative chemoradiation. Ann Surg Oncol. 2009;16:431–9. doi: 10.1245/s10434-008-0220-8. [DOI] [PubMed] [Google Scholar]
- 83.Luo Y, Tian L, Feng Y, Yi M, Chen X, Huang Q. The predictive role of p16 deletion, p53 deletion, and polysomy 9 and 17 in pancreatic ductal adenocarcinoma. Pathol Oncol Res. 2013;19:35–40. doi: 10.1007/s12253-012-9555-3. [DOI] [PubMed] [Google Scholar]
- 84.Oshima M, Okano K, Muraki S, Haba R, Maeba T, Suzuki Y, et al. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg. 2013;258:336–46.. doi: 10.1097/SLA.0b013e3182827a65. [DOI] [PubMed] [Google Scholar]
- 85.Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21:4760–6. doi: 10.1158/1078-0432.CCR-15-1185. [DOI] [PubMed] [Google Scholar]
- 86.Chou A, Froio D, Nagrial AM, Parkin A, Murphy KJ, Chin VT, et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut. 2018;67:2142–55.. doi: 10.1136/gutjnl-2017-315144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Junttila MR, Evan GI. p53-a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–9. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 88.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170:1062–78.. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37.. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 90.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mantovani F, Collavin L, Del, Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212. doi: 10.1038/s41418-018-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baumgart M, Werther M, Bockholt A, Scheurer M, Ruschoff J, Dietmaier W, et al. Genomic instability at both the base pair level and the chromosomal level is detectable in earliest PanIN lesions in tissues of chronic pancreatitis. Pancreas. 2010;39:1093–103. doi: 10.1097/MPA.0b013e3181dc62f6. [DOI] [PubMed] [Google Scholar]
- 94.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–51. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lillemoe KD, Hwang, Thompson JC, Townsend CM, Vickers SM, Beauchamp RD, et al. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene - Discussion. Surgery. 1998;124:150–1. [PubMed] [Google Scholar]
- 96.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 97.Smith RA, Tang J, Tudur-Smith C, Neoptolemos JP, Ghaneh P. Meta-analysis of immunohistochemical prognostic markers in resected pancreatic cancer. Br J Cancer. 2011;104:1440–51. doi: 10.1038/bjc.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vitellius C, Eymerit-Morin C, Luet D, Fizanne L, Foubert F, Bertrais S, et al. Relationship between the expression of O(6)-methylguanine-DNA methyltransferase (MGMT) and p53, and the clinical response in metastatic pancreatic adenocarcinoma treated with FOLFIRINOX. Clin Drug Investig. 2017;37:669–77. doi: 10.1007/s40261-017-0522-3. [DOI] [PubMed] [Google Scholar]
- 99.Striefler JK, Sinn M, Pelzer U, Juhling A, Wislocka L, Bahra M, et al. P53 overexpression and Ki67-index are associated with outcome in ductal pancreatic adenocarcinoma with adjuvant gemcitabine treatment. Pathol Res Pr. 2016;212:726–34. doi: 10.1016/j.prp.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 100.Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR, Dumontet C. Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int J Cancer. 2002;97:439–45. doi: 10.1002/ijc.1628. [DOI] [PubMed] [Google Scholar]
- 101.Xu J, Wang J, Hu Y, Qian J, Xu B, Chen H, et al. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014;5:e1108. doi: 10.1038/cddis.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parrales A, Iwakuma T. Targeting oncogenic mutant p53 for cancer therapy. Front Oncol. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–88. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 104.Izetti P, Hautefeuille A, Abujamra AL, de Farias CB, Giacomazzi J, Alemar B, et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs. 2014;32:783–94. doi: 10.1007/s10637-014-0090-9. [DOI] [PubMed] [Google Scholar]
- 105.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–13. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moser C, Lang SA, Hackl C, Wagner C, Scheiffert E, Schlitt HJ, et al. Targeting HSP90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 2012;32:2551–61. [PubMed] [Google Scholar]
- 107.Nagaraju GP, Mezina A, Shaib WL, Landry J, El-Rayes BF. Targeting the Janus-activated kinase-2-STAT3 signalling pathway in pancreatic cancer using the HSP90 inhibitor ganetespib. Eur J Cancer. 2016;52:109–19. doi: 10.1016/j.ejca.2015.10.057. [DOI] [PubMed] [Google Scholar]
- 108.Renouf DJ, Hedley D, Krzyzanowska MK, Schmuck M, Wang L, Moore MJ. A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pancreatic cancer. Cancer Chemother Pharmacol. 2016;78:541–5. doi: 10.1007/s00280-016-3102-y. [DOI] [PubMed] [Google Scholar]
- 109.Xu J, Singh A, Amiji MM. Redox-responsive targeted gelatin nanoparticles for delivery of combination wt-p53 expressing plasmid DNA and gemcitabine in the treatment of pancreatic cancer. BMC Cancer. 2014;14:75. doi: 10.1186/1471-2407-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chung V, Kos FJ, Hardwick N, Yuan Y, Chao J, Li D, et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin Transl Oncol. 2019;21:363–72. doi: 10.1007/s12094-018-1932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G2 checkpoint abrogator. Cancer Res. 2001;61:8211–7. [PubMed] [Google Scholar]
- 112.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37:2643–50. doi: 10.1200/JCO.19.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA. 2016;315:1844–53. doi: 10.1001/jama.2016.4324. [DOI] [PubMed] [Google Scholar]
- 114.Mantovani F, Walerych D, Sal GD. Targeting mutant p53 in cancer: a long road to precision therapy. FEBS J. 2017;284:837–50. doi: 10.1111/febs.13948. [DOI] [PubMed] [Google Scholar]
- 115.McCarthy AJ, Chetty R. Smad4/DPC4. J Clin Pathol. 2018;71:661–4. doi: 10.1136/jclinpath-2018-205095. [DOI] [PubMed] [Google Scholar]
- 116.Wrana JL. The secret life of Smad4. Cell. 2009;136:13–4. doi: 10.1016/j.cell.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 117.Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, et al. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell. 2005;121:87–99. doi: 10.1016/j.cell.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 118.Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–35. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 119.Zhao M, Mishra L, Deng CX. The role of TGF-beta/SMAD4 signaling in cancer. Int J Biol Sci. 2018;14:111–23. doi: 10.7150/ijbs.23230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grau AM, Zhang L, Wang W, Ruan S, Evans DB, Abbruzzese JL, et al. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res. 1997;57:3929–34. [PubMed] [Google Scholar]
- 121.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586:1959–70. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 122.Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006;97:1–7. doi: 10.1111/j.1349-7006.2005.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156:37–43. doi: 10.1016/S0002-9440(10)64703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]
- 125.Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–46. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell. 2015;161:1345–60. doi: 10.1016/j.cell.2015.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liang C, Shi S, Qin Y, Meng Q, Hua J, Hu Q, et al. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer. Gut. 2020;69:888–900. doi: 10.1136/gutjnl-2018-317163. [DOI] [PubMed] [Google Scholar]
- 128.Liang C, Xu J, Meng Q, Zhang B, Liu J, Hua J, et al. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy. 2020;16:486–500. doi: 10.1080/15548627.2019.1628540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-beta tumor suppression through a lethal EMT. Cell. 2016;164:1015–30. doi: 10.1016/j.cell.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang F, Xia X, Yang C, Shen J, Mai J, Kim HC, et al. SMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin Cancer Res. 2018;24:3176–85. doi: 10.1158/1078-0432.CCR-17-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Biankin AV, Morey AL, Lee CS, Kench JG, Biankin SA, Hook HC, et al. DPC4/Smad4 expression and outcome in pancreatic ductal adenocarcinoma. J Clin Oncol. 2002;20:4531–42. doi: 10.1200/JCO.2002.12.063. [DOI] [PubMed] [Google Scholar]
- 133.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674–9. doi: 10.1158/1078-0432.CCR-09-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Singh P, Srinivasan R, Wig JD. SMAD4 genetic alterations predict a worse prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2012;41:541–6. doi: 10.1097/MPA.0b013e318247d6af. [DOI] [PubMed] [Google Scholar]
- 135.Bachet JB, Marechal R, Demetter P, Bonnetain F, Couvelard A, Svrcek M, et al. Contribution of CXCR4 and SMAD4 in predicting disease progression pattern and benefit from adjuvant chemotherapy in resected pancreatic adenocarcinoma. Ann Oncol. 2012;23:2327–35. doi: 10.1093/annonc/mdr617. [DOI] [PubMed] [Google Scholar]
- 136.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–13. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Crane CH, Varadhachary GR, Yordy JS, Staerkel GA, Javle MM, Safran H, et al. Phase II trial of cetuximab, gemcitabine, and oxaliplatin followed by chemoradiation with cetuximab for locally advanced (T4) pancreatic adenocarcinoma: correlation of Smad4(Dpc4) immunostaining with pattern of disease progression. J Clin Oncol. 2011;29:3037–43. doi: 10.1200/JCO.2010.33.8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Winter JM, Tang LH, Klimstra DS, Liu W, Linkov I, Brennan MF, et al. Failure patterns in resected pancreas adenocarcinoma: lack of predicted benefit to SMAD4 expression. Ann Surg. 2013;258:331–5. doi: 10.1097/SLA.0b013e31827fe9ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang H, Stephens B, Von Hoff DD, Han H. Identification and characterization of a novel anticancer agent with selectivity against deleted in pancreatic cancer locus 4 (DPC4)-deficient pancreatic and colon cancer cells. Pancreas. 2009;38:551–7. doi: 10.1097/MPA.0b013e31819d7415. [DOI] [PubMed] [Google Scholar]
- 140.Wang H, Han H, Von, Hoff DD. Identification of an agent selectively targeting DPC4 (deleted in pancreatic cancer locus 4)-deficient pancreatic cancer cells. Cancer Res. 2006;66:9722–30. doi: 10.1158/0008-5472.CAN-05-4602. [DOI] [PubMed] [Google Scholar]
- 141.Hong E, Park S, Ooshima A, Hong CP, Park J, Heo JS, et al. Inhibition of TGF-beta signalling in combination with nal-IRI plus 5-Fluorouracil/Leucovorin suppresses invasion and prolongs survival in pancreatic tumour mouse models. Sci Rep. 2020;10:2935. doi: 10.1038/s41598-020-59893-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dey P, Baddour J, Muller F, Wu CC, Wang H, Liao WT, et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature. 2017;542:119–23. doi: 10.1038/nature21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33:3124–9. doi: 10.1200/JCO.2014.59.7401. [DOI] [PubMed] [Google Scholar]
- 144.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–27. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–82. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 146.D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909–19. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kowalewski A, Szylberg L, Saganek M, Napiontek W, Antosik P, Grzanka D. Emerging strategies in BRCA-positive pancreatic cancer. J Cancer Res Clin Oncol. 2018;144:1503–7. doi: 10.1007/s00432-018-2666-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111:1132–8. doi: 10.1038/bjc.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Palacio S, McMurry HS, Ali R, Donenberg T, Silva-Smith R, Wideroff G, et al. DNA damage repair deficiency as a predictive biomarker for FOLFIRINOX efficacy in metastatic pancreatic cancer. J Gastrointest Oncol. 2019;10:1133–9. doi: 10.21037/jgo.2019.09.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–63. doi: 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379:2495–505. doi: 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 152.Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74. doi: 10.1016/S1470-2045(19)30684-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 154.Nanda N, Roberts NJ. ATM serine/threonine kinase and its role in pancreatic risk. Genes (Basel). 2020;11:108. [DOI] [PMC free article] [PubMed]
- 155.Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319:2401–9. doi: 10.1001/jama.2018.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–6. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim H, Saka B, Knight S, Borges M, Childs E, Klein A, et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin Cancer Res. 2014;20:1865–72. doi: 10.1158/1078-0432.CCR-13-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kamphues C, Bova R, Bahra M, Klauschen F, Muckenhuber A, Sinn BV, et al. Ataxia-telangiectasia-mutated protein kinase levels stratify patients with pancreatic adenocarcinoma into prognostic subgroups with loss being a strong indicator of poor survival. Pancreas. 2015;44:296–301. doi: 10.1097/MPA.0000000000000248. [DOI] [PubMed] [Google Scholar]
- 159.Golan T, Sella T, O’Reilly EM, Katz MHG, Epelbaum R, Kelsen DP, et al. Overall survival and clinical characteristics of BRCA mutation carriers with stage I/II pancreatic cancer. Br J Cancer. 2017;116:697–702. doi: 10.1038/bjc.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 2017;77:5576–90. doi: 10.1158/0008-5472.CAN-17-0634. [DOI] [PubMed] [Google Scholar]
- 161.Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15:1781–91. doi: 10.1158/1535-7163.MCT-15-0945. [DOI] [PubMed] [Google Scholar]
- 162.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 163.Armstrong SA, Schultz CW, Azimi-Sadjadi A, Brody JR, Pishvaian MJ. ATM dysfunction in pancreatic adenocarcinoma and associated therapeutic implications. Mol Cancer Ther. 2019;18:1899–908. doi: 10.1158/1535-7163.MCT-19-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kawamura D, Takemoto Y, Nishimoto A, Ueno K, Hosoyama T, Shirasawa B, et al. Enhancement of cytotoxic effects of gemcitabine by Dclk1 inhibition through suppression of Chk1 phosphorylation in human pancreatic cancer cells. Oncol Rep. 2017;38:3238–44. doi: 10.3892/or.2017.5974. [DOI] [PubMed] [Google Scholar]
- 165.Duong HQ, Hong YB, Kim JS, Lee HS, Yi YW, Kim YJ, et al. Inhibition of checkpoint kinase 2 (CHK2) enhances sensitivity of pancreatic adenocarcinoma cells to gemcitabine. J Cell Mol Med. 2013;17:1261–70. doi: 10.1111/jcmm.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen WJ, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol. 2018;2018:1–13. doi: 10.1200/PO.17.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nepomuceno TC, De Gregoriis G, de Oliveira FMB, Suarez-Kurtz G, Monteiro AN, Carvalho MA, et al. The role of PALB2 in the DNA damage response and cancer predisposition. Int J Mol Sci. 2017;18:1886. doi: 10.3390/ijms18091886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17:569–77. doi: 10.1038/gim.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Harinck F, Kluijt I, van Mil SE, Waisfisz Q, van Os TA, Aalfs CM, et al. Routine testing for PALB2 mutations in familial pancreatic cancer families and breast cancer families with pancreatic cancer is not indicated. Eur J Hum Genet. 2012;20:577–9. doi: 10.1038/ejhg.2011.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hu C, LaDuca H, Shimelis H, Polley EC, Lilyquist J, Hart SN, et al. Multigene hereditary cancer panels reveal high-risk pancreatic cancer susceptibility genes. JCO Precis Oncol. 2018;2:PO.17.00291. doi: 10.1200/PO.17.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10:3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Park D, Shakya R, Koivisto C, Pitarresi JR, Szabolcs M, Kladney R, et al. Murine models for familial pancreatic cancer: histopathology, latency and drug sensitivity among cancers of Palb2, Brca1 and Brca2 mutant mouse strains. PLoS One. 2019;14:e0226714. doi: 10.1371/journal.pone.0226714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7:620–9. doi: 10.1158/2159-8290.CD-16-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Perez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142:1079–92. doi: 10.1053/j.gastro.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 177.Davis EJ, Johnson DB, Sosman JA, Chandra S. Melanoma: what do all the mutations mean? Cancer-Am Cancer Soc. 2018;124:3490–9. doi: 10.1002/cncr.31345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, et al. A central role for RAF->MEK->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–93. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Foster SA, Whalen DM, Ozen A, Wongchenko MJ, Yin J, Yen I, et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29:477–93. doi: 10.1016/j.ccell.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 181.Zhang W. BRAF inhibitors: the current and the future. Curr Opin Pharmacol. 2015;23:68–73. doi: 10.1016/j.coph.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 182.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 183.Long GV, Hauschild A, Santinami M, Atkinson V, Mandala M, Chiarion-Sileni V, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1813–23. doi: 10.1056/NEJMoa1708539. [DOI] [PubMed] [Google Scholar]
- 184.Wrzeszczynski KO, Rahman S, Frank MO, Arora K, Shah M, Geiger H, et al. Identification of targetable BRAF DeltaN486_P490 variant by whole-genome sequencing leading to dabrafenib-induced remission of a BRAF-mutant pancreatic adenocarcinoma. Cold Spring Harb Mol Case Stud. 2019;5. [DOI] [PMC free article] [PubMed]
- 185.DuFort CC, DelGiorno KE, Hingorani SR. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology. 2016;150:1545–57 e2. doi: 10.1053/j.gastro.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–97. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 187.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends Cancer. 2016;2:176–90. doi: 10.1016/j.trecan.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21:571–84. [DOI] [PMC free article] [PubMed]
- 190.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018;8:1112–29. doi: 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–38. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu SD, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–U140. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.O’Reilly EM, Hechtman JF. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann Oncol. 2019;30:Viii36–i40. doi: 10.1093/annonc/mdz385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hayashi H, Tanishima S, Fujii K, Mori R, Okamura Y, Yanagita E, et al. Genomic testing for pancreatic cancer in clinical practice as real-world evidence. Pancreatology. 2018;18:647–54. doi: 10.1016/j.pan.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 196.Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res. 2017;23:6094–100. doi: 10.1158/1078-0432.CCR-17-0899. [DOI] [PubMed] [Google Scholar]
- 197.Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res. 2015;21:2029–37. doi: 10.1158/1078-0432.CCR-15-0426. [DOI] [PubMed] [Google Scholar]
- 198.Shi S, Liang C, Xu J, Meng Q, Hua J, Yang X, et al. The strain ratio as obtained by endoscopic ultrasonography elastography correlates with the stroma proportion and the prognosis of local pancreatic cancer. Ann Surg. 2020;271:559–65. doi: 10.1097/SLA.0000000000002998. [DOI] [PubMed] [Google Scholar]
- 199.Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3:516–22. doi: 10.1001/jamaoncol.2016.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C. Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer. 2008;8:82. doi: 10.1186/1471-2407-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Weden S, Klemp M, Gladhaug IP, Moller M, Eriksen JA, Gaudernack G, et al. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer. 2011;128:1120–8. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 202.Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer. 2018;143:2053–64. doi: 10.1002/ijc.31603. [DOI] [PubMed] [Google Scholar]
- 203.Ko AH, Bekaii-Saab T, Van Ziffle J, Mirzoeva OM, Joseph NM, Talasaz A, et al. A multicenter, open-label phase II clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin Cancer Res. 2016;22:61–8. doi: 10.1158/1078-0432.CCR-15-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50:2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 205.Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30:1216–23. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- 206.Van Laethem JL, Riess H, Jassem J, Haas M, Martens UM, Weekes C, et al. Phase I/II study of refametinib (BAY 86-9766) in combination with gemcitabine in advanced pancreatic cancer. Target Oncol. 2017;12:97–109. doi: 10.1007/s11523-016-0469-y. [DOI] [PubMed] [Google Scholar]
- 207.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 208.Karavasilis V, Samantas E, Koliou GA, Kalogera-Fountzila A, Pentheroudakis G, Varthalitis I, et al. Gemcitabine combined with the mTOR inhibitor temsirolimus in patients with locally advanced or metastatic pancreatic cancer. a hellenic cooperative oncology group phase I/II study. Target Oncol. 2018;13:715–24. doi: 10.1007/s11523-018-0605-y. [DOI] [PubMed] [Google Scholar]
- 209.Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol. 2015;75:1135–41. doi: 10.1007/s00280-015-2730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kordes S, Richel DJ, Klumpen HJ, Weterman MJ, Stevens AJ, Wilmink JW. A phase I/II, non-randomized, feasibility/safety and efficacy study of the combination of everolimus, cetuximab and capecitabine in patients with advanced pancreatic cancer. Invest New Drugs. 2013;31:85–91. doi: 10.1007/s10637-012-9802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27:193–8. doi: 10.1200/JCO.2008.18.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.O’Neil BH, Scott AJ, Ma WW, Cohen SJ, Aisner DL, Menter AR, et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann Oncol. 2016;27:1180. doi: 10.1093/annonc/mdw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Makielski RJ, Lubner SJ, Mulkerin DL, Traynor AM, Groteluschen D, Eickhoff J, et al. A phase II study of sorafenib, oxaliplatin, and 2 days of high-dose capecitabine in advanced pancreas cancer. Cancer Chemother Pharmacol. 2015;76:317–23. doi: 10.1007/s00280-015-2783-y. [DOI] [PubMed] [Google Scholar]
- 215.Goncalves A, Gilabert M, Francois E, Dahan L, Perrier H, Lamy R, et al. BAYPAN study: a double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol. 2012;23:2799–805. doi: 10.1093/annonc/mds135. [DOI] [PubMed] [Google Scholar]
- 216.El-Khoueiry AB, Ramanathan RK, Yang DY, Zhang W, Shibata S, Wright JJ, et al. A randomized phase II of gemcitabine and sorafenib versus sorafenib alone in patients with metastatic pancreatic cancer. Invest New Drugs. 2012;30:1175–83. doi: 10.1007/s10637-011-9658-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cardin DB, Goff L, Li CI, Shyr Y, Winkler C, DeVore R, et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014;3:572–9. doi: 10.1002/cam4.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 219.Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016;10:635–43. doi: 10.1016/j.molonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nakano Y, Kitago M, Matsuda S, Nakamura Y, Fujita Y, Imai S, et al. KRAS mutations in cell-free DNA from preoperative and postoperative sera as a pancreatic cancer marker: a retrospective study. Br J Cancer. 2018;118:662–9. doi: 10.1038/bjc.2017.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Temraz S, Shamseddine A, Mukherji D, Charafeddine M, Tfayli A, Assi H, et al. Ki67 and P53 in relation to disease progression in metastatic pancreatic cancer: a single institution analysis. Pathol Oncol Res. 2019;25:1059–66. doi: 10.1007/s12253-018-0464-y. [DOI] [PubMed] [Google Scholar]
- 222.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115–21. [PubMed] [Google Scholar]
- 223.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 224.Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27:5513–8. doi: 10.1200/JCO.2009.24.2446. [DOI] [PubMed] [Google Scholar]
- 225.Uesaka K, Boku N, Fukutomi A, Okamura Y, Konishi M, Matsumoto I, et al. Adjuvant chemotherapy of S-1 versus gemcitabine for resected pancreatic cancer: a phase 3, open-label, randomised, non-inferiority trial (JASPAC 01) Lancet. 2016;388:248–57. doi: 10.1016/S0140-6736(16)30583-9. [DOI] [PubMed] [Google Scholar]
- 226.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379:2395–406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]