

Abstract
The accurate annotation of protein localization is crucial in understanding protein function in tandem with a broad range of applications such as pathological analysis and drug design. Since most proteins do not have experimentally-determined localization information, the computational prediction of protein localization has been an active research area for more than two decades. In particular, recent machine-learning advancements have fueled the development of new methods in protein localization prediction. In this review paper, we first categorize the main features and algorithms used for protein localization prediction. Then, we summarize a list of protein localization prediction tools in terms of their coverage, characteristics, and accessibility to help users find suitable tools based on their needs. Next, we evaluate some of these tools on a benchmark dataset. Finally, we provide an outlook on the future exploration of protein localization methods.
Keywords: Protein localization prediction, Computational methods, Review
1. Introduction
Cells contain well-organized compartments with different protein constituents. Although most proteins are synthesized in the cytosol, about half of them are transported into or across at least one cellular membrane to reach their functional destination [1], [2], [3]. The aberrant localization of proteins usually has harmful effects, including diseases in humans and animals and poor traits in plants [4], [5], [6], [7]. Hence, studying the mechanism of protein localization is essential in a broad range of applications, such as plant breeding, pathological analysis, and the therapeutic modification of disease-related protein mislocalization [5], [8]. Protein localization is a complicated biological process controlled by many factors, such as signal peptides, protein trafficking, protein–protein interactions, folding, and alternative splicing [5], [9]. Among these, protein localization guided by targeting peptides is the most common mechanism [10] and includes pre-sequences and internal signals [11], [12]. Pre-sequences are found at the N- or C-terminus of protein sequences with enrichment of charged or hydrophobic amino acids, while internal signals are located in the middle of a sequence. How precursor proteins are directed to their target organelles is only partially understood [11], and only a small number of targeting peptides (particularly internal signals) have been experimentally identified. According to UniProt annotation (release 2020_05), out of the reviewed 20,394 human proteins, 7348 (36.0%) proteins have localization annotation with experimental verification, while only 3608 (17.7%) proteins have known targeting peptides. Furthermore, limited sub-organelle compartment localization data are available. According to a recent search that we conducted on 16,213 human proteins in ten human organelles, 5882 (36.3%) proteins had experimentally verified organellar localization annotation, while only 3518 (21.7%) proteins had experimentally verified sub-organellar localization annotation. Targeting peptide and sub-organelle data for non-human species are even sparser.
Several experimental methods can be used for protein localization analysis. Quantitative mass spectrometric readouts allow for the identification of proteins across fractions [13], [14], [15], [16]. Spatially and temporally resolved proteomic maps in living cells can be obtained by targetable peroxidase [17], [18], [19]. Techniques such as immunofluorescence and high-resolution confocal microscopy have enabled the visual estimation of protein localization within a single cell [20], [21], [22], [23], [24]. One problem with experimental methods is that their throughput is relatively low. In addition, experimental protein localization identification requires a great deal of time and resources. Importantly, experimental and computational protein localization identification approaches are complementary to each other. Experimental annotations are typically used as true labels for computational methods. Computational models are trained using these ground truth data to predict the localization of other proteins. Due to their cost-effective, automated, and high-throughput nature, computational methods are helpful for the large-scale characterization of protein subcellular locations.
Several papers have reviewed protein localization prediction methods. The review of [25] focuses on methods for bacterial protein localization prediction. Other reviews [26], [27] mainly cover protein sequence features (such as targeting peptides) in localization prediction. The methods reviewed in [28] predict protein function taxonomies, such as the Functional Catalogue, Enzyme Commission, or Gene Ontology, rather than specific cellular components. Another review mainly discusses web-based prediction tools for human protein subcellular localization [29]. General methods and tools for protein localization prediction are introduced in the reviews of [30], [31], [32], [33], which have a scope similar to ours. However, the most recent review in the literature [30], [31], [32] was published in 2014. Many new methods have been proposed since then that have greatly improved prediction accuracy, especially deep-learning methods. This review focuses on these new methods and tools in addition to previous representative methods. A less detailed review [33] was recently published. Compared to [33], this review separates the introduction of features, algorithms, and tools in greater detail so readers can better understand their relationships. Additionally, the applicability of the tools is considered, and only actively maintained tools are listed. Users can select the tools they need based on the information summarized and access them through the links provided. All the aforementioned features make this review unique and valuable. This review is organized as follows. In 2, 3, we analyze the features and classifiers that are often associated with different methods, respectively. Many of these methods provide standalone tools and/or web services that we summarize in Section 4. For each tool, information of target compartments, used algorithm, accessibility, etc. is given. In Section 5, a summary is provided together with promising directions for future protein localization prediction methods. The relationship of the data, features, and models used in computational protein localization prediction, as well as their outputs, are shown in Fig. 1. The features and main contributions of this review are summarized as follows:
-
•
A systematic introduction of features, algorithms, methods, and tools, as well as their relationships related to protein localization.
-
•
A comprehensive list of available protein localization prediction tools, many of which became available in recent years.
-
•
Extensive evaluations of localization prediction tools/methods, providing insights on why some methods have better prediction performance than others.
-
•
Significant discussion on the future direction of protein localization studies.
Fig. 1.
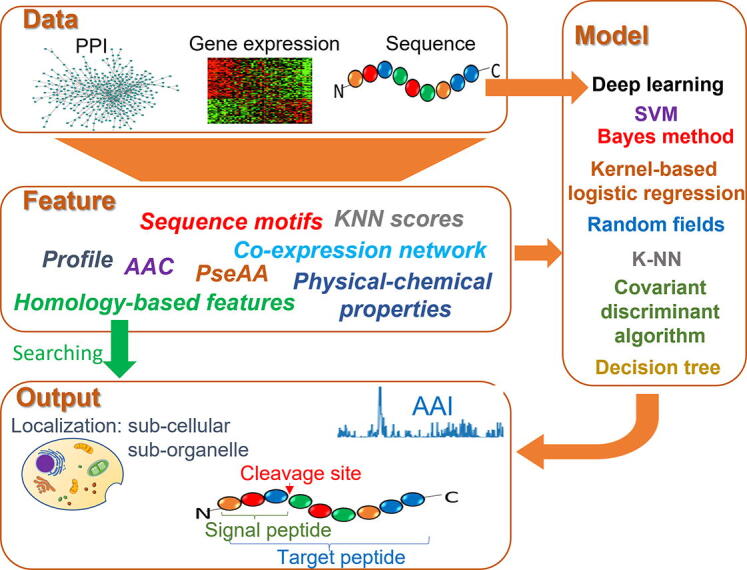
Relationships among the data, features, models, and prediction outputs in the computational prediction of protein localization. Sequence data can be converted into different features before feeding the data to a classifier model. Some classification models take raw data (e.g., one-hot-encoding of protein sequences for deep learning) as input, while others use engineered features. Localization prediction (at the sub-cellular and/or suborganellar level) is the most common output. Some methods also provide side product predictions such as target peptides, signal peptide cleavage sites, and mechanism interpretability at amino-acid-level resolution (AAI). Homology-based methods are special in the sense that they can make predictions directly based on homology-based features, such as the GO terms of homologous proteins.
2. Data and features
2.1. Sequence-based features
Protein sequences are considered the most essential source of information for protein localization prediction, particularly terminal region sequences where targeting signals are likely to be found. Protein sequence information can be obtained from databases such as UniProt [34]. In addition, many types of features have been proposed based on protein sequences.
2.1.1. Amino acid composition
The simplest feature representing a protein sequence is likely amino acid (AA) composition [35]. Given a protein sequence , the AA composition of can be expressed by
| (1) |
where are the normalized occurrence frequencies of the 20 native amino acids in protein .
2.1.2. PseAA composition
The main shortcoming of using AA composition as a feature is its lack of protein sequence order information [31]. The concept of pseudo amino acid composition (PseAA) was proposed to address this problem [36] by representing a protein as a vector :
| (2) |
where the components are given by
| (3) |
where w is a weight factor set to 0.05 in the original paper [36], and is the k-th tier correlation factor, which reflects the sequence order correlation between all of the k-th most contiguous residues as formulated by
| (4) |
As in Eq. (2), the first 20 components are associated with the conventional amino acid composition of P, whereas the remaining components are the correlation factors that reflect the first tier, second tier, and so on up to the -th tier sequence order correlation patterns. These factors incorporate sequence order effects, and is a chosen hyperparameter (integer). The calculation of integrates the hydrophobicity values (), hydrophilicity values (), and side-chain masses () for amino acids i and i + k as
| (5) |
Note that Eq. (5) is just one form for deriving the correlation factors. Other information, such as physicochemical distance and amphiphilic patterns, can also derive different types of PseAA composition.
2.1.3. Homology information
As subcellular localization tends to be evolutionarily conserved [37], homology to a protein of known localization is often a good indicator of actual protein localization [38]. Such information can be derived via BLAST [39] or a more sensitive search method such as HHblits [40] against a database of proteins with known localization. One important source of known localization is the cellular component of Gene Ontology (GO) [41], which has been used to improve protein localization prediction performance [42], [43], [44], [45]. Homology information can also be obtained through protein structure similarity, as did in C-I-Tasser [46], a template-based method for protein structure and function prediction. In C-I-Tasser, the function prediction of a query protein is obtained by matching its structural model with proteins in the BioLiP function library via structure and sequence profile comparisons. Each entry in BioLiP contains GO terms so that the GO cellular localization of the query protein can be inferred.
2.1.4. Evolutionary profiles
Evolutionary profiles, represented by Position-Specific Scoring Matrices (PSSMs), etc., provide informative input for protein localization prediction. PSSMs indicate the amino acid occurrence for each position in a protein multiple sequence alignment. PSSM scores are generally given as positive or negative values. A positive score means that the given amino acid substitution occurs more frequently in the alignment than expected by chance, while a negative score indicates that the substitution occurs less frequently than expected by chance. PSSMs can be created using PSI-BLAST, which finds similar protein sequences to a query sequence and then constructs a PSSM from the resulting alignment.
The BLOSUM (BLOcks SUbstitution Matrix) matrix [47] is a substitution matrix used for scoring alignments between evolutionarily divergent protein sequences. Several BLOSUM matrices exist using different alignment databases, which are named with sequence identity thresholds in the alignments. For example, BLOSUM62 is a matrix built using sequences with less than 62% similarity (sequences with ≥62% identity were clustered). BLOSUM62 is the default matrix for protein BLAST and is among the best for detecting weak protein similarities. Encoding with BLOSUM matrices is fast and provides a viable alternative if acquiring a PSSM is slow or unsuccessful [48], [49].
One particular usage of a sequence profile is as the profile kernel of an SVM. A key feature of the SVM optimization problem is that it depends only on the inner products of the feature vectors representing the input data. Several kernel functions have been proposed to avoid the explicit transformation of input data to feature vectors, explained as follows. Let represent a mapping from the input space of protein sequences into a (possibly high-dimensional) vector space called the feature space. A string kernel is defined by , where x and y are sequences, e.g., from the alphabet of amino acids (, and the length depends on the sequence). Let represent a profile for sequence , with denoting the emission probability of amino acid in position and for each position ; a profile kernel is defined as . The Fisher-SVM method [50] is a profile-kernel method that represents each protein sequence as a vector of Fisher scores extracted from a profile Hidden Markov Model (HMM) for a protein family. Kuang et al. proposed profile-based string kernels that use probabilistic profiles, such as those produced by the PSI-BLAST algorithm, to define position-dependent mutation neighborhoods along with protein sequences for inexact matching of k-length subsequences (“k-mers”) [51]. Such profile kernels are used in LocTree2 [52], an SVM-based method for protein localization prediction.
2.1.5. Motifs
Certain sequence patterns may correlate with a specific subcellular localization due to localization signals or functional relationships [53]. This motif information can be retrieved from databases such as PROSITE [54] or by data mining. One special type of motifs represents targeting peptides, i.e., short sequences mainly present at protein termini that function like a postal code to specify an intracellular or extracellular destination [55]. Some methods predict the presence of targeting peptides as a side product in tandem with protein localization prediction [56], [57], while other methods use targeting peptides as input features to predict protein localization [53].
A sequence pattern can also be extracted through a sliding window of a k-mer sequence. The motif length k is often set based on specific needs or prior biological knowledge. For example, TetraMito [58] uses over-represented tetrapeptides (four continuous amino acids believed to encode a particular structure) as features to predict submitochondrial protein localization. A similar idea is used for sub-Golgi protein localization prediction by SubGolgi 2.0 [59], which uses an SVM classifier trained with g-gap dipeptide compositions (two amino acids with g residues between them). LOCALIZER [60] is another k-mer-based method for predicting plant and effector protein localization to chloroplasts, mitochondria, and nuclei. The motif length k varies in LOCALIZER to capture the target signals on protein sequences.
2.1.6. Physical–chemical properties
As the name suggests, this feature uses AAs' physical and chemical properties to represent protein sequences. These previously calculated properties are stored in public databases. According to Venkatarajan and Braun [61], a comprehensive list of 237 physical–chemical properties of each amino acid was compiled from the SWISS-PROT [34] and dbGET [62] databases. They showed that the number of properties could be reduced while retaining approximately the same distribution of amino acids in the feature space. Notably, the correlation coefficient between the original and regenerated distances was more than 99% using the first five eigenvectors.
2.1.7. Pre-train sequence embedding
Evolutionary information significantly benefits model prediction performance; however, as the number of proteins in databases increases, retrieving such information is often time-consuming. Additionally, evolutionary information is less powerful for small protein families, e.g., for proteins from the Dark Proteome [63]. One promising sequence embedding method uses the pre-train model adopted from Natural Language Processing (NLP). The pre-train model utilizes large, unlabeled text-corpora such as Wikipedia to conceptualize syntax and semantics. Pre-train methods such as Transformer [64], ELMo [65], Word2Vec [66], and Bert [67] employ self-learning and predict either the next word in a sentence given all previous words, the current word from a window of surrounding context words (or using the current word to predict the surrounding window of context words), or masked-out words given all unmasked words. Once trained, language models can extract features, referred to as embeddings, to use as input for subsequent supervised learning (transfer-learning). A similar strategy has been used for protein sequence embedding. SeqVec [68] uses ELMo on UniRef50 for pre-train embedding and transfer-learning for subcellular localization prediction. ProtTrans [69] employs different pre-training embedding models on UniRef and BFD data containing 2.1 billion protein sequences, which can also be used for protein localization prediction. In addition, a recent study showed that the pre-training embedding from language models followed by an attention-based deep-learning architecture could yield excellent performance in protein localization prediction even without using evolutionary information [70].
2.2. Protein interactions
If two proteins interact, they are neighbors of each other in a protein–protein-interaction (PPI) network. The localizations of the neighbors in a PPI network carry information about the localization of un-annotated proteins. For example, if the majority of a protein’s neighbors share the same localization, the protein is likely localized to the same location. The definition of protein interaction varies and can be based on physical connections or genetic regulations. Protein interaction data can be retrieved from databases such as MINT [71], DIP [72], BioGRID [73], and STRING [74].
2.3. Gene/protein expression
The rationale for using gene/protein expression as a feature is that genes/proteins in the same compartment at the organelle or suborganelle level tend to be co-expressed to perform related functions. Gene/protein expression information can be used in network form like the aforementioned protein interaction feature [75]. For example, an interaction is established if the expression correlation between two genes/proteins exceeds a predefined threshold. Gene/protein expression information can also be used to create features such as the k-nearest-neighbor (k-NN) scores in the MU-LOC method [76] or used as standalone features in the SLocX method [77]. Gene/protein expression data are widely available and can be downloaded from databases like the Gene Expression Omnibus (GEO) [78] and The Cancer Genome Atlas (TCGA) [79].
3. Classification algorithms
3.1. Support vector machine
Support vector machines (SVMs) [80] use kernel functions to map input vectors into high dimensional feature space and construct a hyperplane that maximizes the margin between different classes. SVMs can handle large feature spaces and effectively avoid overfitting.
The method proposed in [81] is an early SVM-based protein localization prediction approach. To deal with a multi-class classification problem, it uses AA composition as a feature to train SVM classifiers in a one-versus-rest fashion. pSLIP [82] employs the SVM method in conjunction with multiple physicochemical properties of amino acids to predict protein subcellular localization in eukaryotes across six different localizations. The Density-induced Support Vector Data Description (D-SVDD) is an extension of Conventional Support Vector Data Description (C-SVDD) that was introduced for a one-class classification task inspired by SVMs [83]. PLPD [84] uses AA-based and motif features to modify the D-SVDD for multi-class multi-label protein localization prediction, mainly from imbalanced training datasets. A two-level SVM system to predict protein localization is described in [85]. The first level consists of multiple SVMs using distinct AA-based features (AA composition and physical–chemical properties), and the SVM at the second level makes the final prediction. SLocX [77] uses an SVM to predict the subcellular localization of Arabidopsis proteins using gene expression and AA composition as features.
Recent SVM-based methods include SubMitoPred [86], which uses Pfam domain information to predict mitochondrial proteins and their sub-mitochondrial localization. ERPred [87] predicts ER-resident proteins by training an SVM with a combination of amino acid compositions from different parts of proteins. SubNucPred [88] predicts protein localization for 10 sub-nuclear locations sequentially by combining the presence or absence of a unique Pfam domain and an amino acid composition-based SVM model. CELLO2GO [89] combines an SVM-based localization prediction method with BLAST homology search. When homologous proteins with known localizations are available, their GO terms are used as possible functional annotations for a queried protein. Otherwise, the SVM classifier provides localization prediction. MultiP-SChlo [90] is another SVM-based method that predicts subchloroplast protein localization with multiple labels based on features such as PseAAC and AA properties. MKLoc [91] is an SVM-based method for multi-label protein localization prediction where protein sequences are represented by a 30-dimensional feature vector consisting of PseAAC, physical–chemical properties, motifs from PROSITE, and GO annotations. LocTree3 [42] improves upon LocTree2 [52] by including information about homologs, if available, through a PSI-BLAST search. MitoFates [92] is a prediction method for cleavable N-terminal mitochondrial targeting signals and their cleavage sites. Besides classical features such as AA composition, sequence profiles, and physical–chemical properties, MitoFates introduces novel sequence features, including positively charged amphiphilicity and presequence motifs, and trains an SVM classifier using these features. SChloro [93] converts a protein sequence into a PSSM profile and Kyte-Doolittle scale (average hydrophobicity). Two layers of SVMs are designed to predict targeting signal and membrane protein information. The final output predicts six sub-chloroplastic localizations by integrating the predictions from previous layers.
3.2. Probabilistic methods
3.2.1. Bayes method
Probabilistic models, specifically Bayesian methods such as the Bayes Optimal Classifier or Bayesian Networks, make the most probable prediction for a new example. Bayesian methods use the Bayes Theorem [94] for calculating a conditional probability. They are also closely related to the Maximum a Posteriori (MAP), a probabilistic framework that finds the most probable hypothesis for a training dataset. In large real-world applications, the Bayes method usually assumes that different features are independent of each other, known as Naïve Bayes.
PSORT-B [53] and subsequent versions of it [95], [96] (with higher prediction coverage and refined subcategories), construct six analytical modules based on features including homology, motifs, and signal peptides. A query protein undergoes each of the six analyses and the results are combined using a Bayesian Network to generate a final probability value for each localization site.
3.2.2. Kernel-based logistic regression
When determining the probability of a protein to be localized at a specific location given a PPI network, kernel-based logistic regression (KLR) considers the localization information of all the proteins in the network. The KLR model can be formulated as follows [97]. Given a protein–protein interaction network with N proteins , some of which have unknown localization, let
| (6) |
represent the protein set excluding protein i. Let
| (7) |
represent the summed distances of protein i to proteins targeting localization L, where is the kernel function for calculating the distances between two proteins in the network. Then, the KLR model is given by
| (8) |
which means that the logit of , and the probability of protein i targeting location L is linear based on the summed distances of proteins targeting L or another location. Then, we have
| (9) |
Note that the probability of being in each localization is calculated separately as a binary classification problem.
NetLoc [75] applies KLR to protein networks based on different relationships, including physical PPI, genetic PPI, and coexpression. In NetLoc, networks with high connectivity and a high percentage of interacting protein pairs targeting the same location lead to better prediction performance.
3.2.3. Random Fields
Given a probability space, a random field T(x) defined in is a function such that for every fixed , T(x) is a random variable on the probability space [98]. Markov Random Fields (MRFs) and Conditional Random Fields (CRFs) have been used for protein localization prediction [56], [99]. An MRF of a graph G is a set of random variables corresponding to the nodes in G (random field) with a joint distribution that is Markov-constrained for G. In other words, the joint probability distribution associated with the MRF is subject to the Markov constraint given by G: for any two variables, and , the value of is conditionally independent of given its neighbors . In this case, the joint probability distribution factorizes according to G. In contrast, we can describe a CRF for a graph G as a set of random variables corresponding to the nodes in G, a subset of which are assumed always to be observed, and remaining variables with a conditional distribution that is Markov-constrained for G. Both MRFs and CRFs typically fit a model that can be used for conditional inference in diverse settings. The main difference is that an MRF has no consistently designated “observed variables” and requires a joint distribution over all variables that adhere to the Markov constraints of G.
CRFs are used for signal peptide cleavage site prediction in DeepSig [99] and specific signal peptide prediction in SignalP 5.0 [56]. A tissue-specific subcellular localization prediction method is proposed in [100] using multi-label MRF. A tissue-specific network was constructed from generic physical PPI networks and tissue-specific functional associations, and tissue-specific localization annotations were obtained from HPA [101].
3.3. Distance-based methods
3.3.1. k-nearest Neighbors (k-NN) classification
The k-NN algorithm is a nonparametric method used for classification and regression [102]. In both cases, the input consists of the k closest training examples in the data set. The output depends on whether the k-NN model is used for classification or regression. In k-NN classification, the output is class membership. An object is classified by a plurality vote of its neighbors, and assigned to the most common class of its k nearest neighbors (k is typically a small positive integer). If k = 1, then the object is simply assigned to the class of the single nearest neighbor.
WoLF PSORT [103] converts protein amino acid sequences into numerical localization features such as targeting signals, amino acid composition, and functional motifs. After conversion, a k-NN classifier is used for prediction. An idea similar to k-NN is used in [104], where a physical interaction network was obtained from BioGRID [73], and GO Cellular Component annotation was mapped onto the network, if available, for the corresponding protein (node). For a query protein, the percentage of its interactors associated with each target localization is calculated. The top two localizations are then reported as the prediction.
3.3.2. Covariant discriminant algorithm based on Mahalanobis distance
The Mahalanobis distance [105] is a measure of the distance between a point P and a distribution D. It is essentially a multidimensional generalization to measure how many standard deviations away P is from the mean of D. This distance is zero if P is at the mean of D and grows as P moves away from the mean along each principal component axis. If each of these axes is re-scaled to have unit variance, then the Mahalanobis distance corresponds to the standard Euclidean distance in the transformed space. The Mahalanobis distance is thus unitless and scale-invariant and takes the correlations in a data set into account.
The Mahalanobis distance of an observation from a set of observations with mean and covariance matrix S is defined as:
| (10) |
The similarity between standard vector (normalized occurrence frequencies of the 20 AA from class ) and protein is characterized by the covariant discriminant, as defined by Liu and Chou in [106]:
| (11) |
where the first term is the squared Mahalanobis distance, and is the i-th eigenvalue of covariance matrix S.
The covariant discriminant algorithm is used in general protein localization prediction in [106], as well as in apoptosis protein localization prediction [107] and Golgi protein subtype prediction [108]. The features used in these methods are AA composition or Pseudo AAC.
3.4. Neural network/deep learning
An artificial neural network (ANN) is based on a collection of connected units or nodes called artificial neurons that loosely model the neurons in a biological brain. Each connection, like the synapses in a biological brain, can transmit a signal to other neurons. Each artificial neuron receives a signal and processes it, and the output of each neuron is computed by a non-linear function of the sum of its inputs. Increased GPU computing power and distributed computing allow the use of larger networks, which is known as “deep learning” [109]. Deep learning has become the hottest field in machine learning, and different architectures have been proposed, such as deep neural networks (DNNs) [110], convolutional neural networks (CNNs) [109], recurrent neural networks (RNNs) [111], [112], and attention mechanisms [113]. These deep learning methods, as well as traditional ANNs, have been applied in protein localization prediction. Due to the abstract feature extraction capability of deep learning models, artificial feature engineering is sometimes not required. Raw protein sequences can be given as inputs for many deep learning localization prediction methods [114], [115]. Among different deep learning architectures, RNNs are inherently suitable for processing protein sequences. Notably, a widely-adopted implementation of RNN, Long Short-Term Memory (LSTM), captures long-distance dependencies well [116]. LSTMs have been successfully applied in machine translation [117], [118], [119] and speech recognition [120], [121]. The methods used for these tasks can be applied to protein localization prediction by considering protein sequences as sentences and amino acids as words. CNNs are most commonly applied to analyze visual imagery [122]. A CNN uses shared-weight convolution kernels to slide along input features and provide feature maps for downstream calculations. The pooling operation reduces data dimension by combining the outputs of neuron clusters at one layer into a single neuron in the next layer. It is often desirable to apply CNNs to long protein sequences at the cost of losing single residue resolution for improved computational efficiency [48], [49], [123]. Moreover, CNN filters can be used to build position-weight matrices (PWMs) of sequence motifs, which can improve model interpretability [123]. The attention mechanism technique mimics cognitive attention [113] as it enhances the essential parts of input data and fades out the rest. This increases the signal-to-noise ratio and elucidates the contribution of features to the final prediction [48], [124], e.g., determines which amino acids are responsible for protein localization.
Several neural network/deep learning-based methods have been proposed for protein localization prediction. SCLpred [114] is an N-to-1 neural network for protein localization prediction capable of mapping a whole sequence into fixed-length properties so that no predefined feature is needed. A similar method was later used in SCLpred-EMS [125] to predict proteins in the endomembrane system and secretory pathway. DeepLoc [49] applies the CNN method, bidirectional LSTM [112], and the attention mechanism for predicting localization and detecting the regions in a protein sequence that contribute to localization prediction. The length of the embedding is the same as the input sequence, while the attention weight of each amino acid is a combination of several CNN filters of different receptive fields. This reduces the interpretation resolution of the model. The researchers also apply different embedding methods and illustrate that PSSM achieves significantly better performance than BLOSUM62 at the cost of increased computing time. MU-LOC [76] provides two models (SVM and DNN) to predict mitochondrial protein in plants. The features used include AA composition, PSSM, and gene expression. MULocDeep [48], developed from the same group that developed MU-LOC, is a recently developed deep learning method that extends target localization coverage to 10 main subcellular compartments and their suborganellar compartments with 44 localization classes in total. Its deep learning model consists of a bidirectional LSTM and a multi-head self-attention mechanism [124]. In addition to protein localization prediction, it sheds light on the mechanism of localization by highlighting regions on protein sequences as likely targeting peptides. DeepMito [126] is another deep learning method for sub-mitochondrial localization prediction using CNNs. Its features include physical–chemical properties and PSSM in addition to the one-hot encoding of raw sequences.
Some methods do not predict localization directly; rather, they predict the presence and location of targeting peptides from which the localization of corresponding proteins can be roughly inferred. For example, DeepSig [99] and SignalP 5.0 [56] predict signal peptides and their cleavage sites using deep-learning methods. DeepSig uses a CNN, while SignalP 5.0 applies a CNN, bidirectional LSTM, and a CRF for specific signal peptide prediction. TargetP 2.0 [57] is a deep learning model constructed by bidirectional LSTM and a multi-attention mechanism to predict N-terminal targeting signals that direct proteins to the secretory pathways, mitochondria, and chloroplasts, or other plastids. One attention head was assigned to each target class and trained as the second loss function to focus on the peptide cleavage site.
3.5. Decision tree-based methods
For prediction problems involving large-scale labeled data, neural networks tend to outperform other algorithms or frameworks. However, when it comes to small- to medium-sized data, decision tree-based algorithms are often considered optimal. A decision tree is a flowchart-like structure in which each internal node represents a “test” on an attribute, where each branch represents the outcome of the test, and each leaf node represents a class label. Decision tree-based methods have evolved over the years. For example, bagging (bootstrap aggregating) combines the predictions of multiple decision trees through a majority voting mechanism, random forests select only a subset of features at random to build a forest of decision trees, and boosting is achieved by sequentially minimizing the errors of previous models. Gradient boosting employs the gradient descent algorithm to minimize errors in sequential models. XGBoost [127] optimizes gradient boosting through parallel processing, tree-pruning, handling missing values, and regularization to avoid overfitting.
Decision-tree-based methods have also been applied to protein localization problems. Pang et al. developed a CNN-XGBoost model [128] to predict protein subcellular localization. A CNN acts as a feature extractor to automatically obtain features from a protein sequence, and an XGBoost classifier functions as a recognizer based on the output of the CNN. SubMito-XGBoost [129] extracts protein sequence-based features including g-gap dipeptide composition, PseAAC, and PSSM as feature vectors for boosting to predict protein submitochondrial localization. A similar study [130] extracts feature vectors of protein sequences using PSSM for a random forest model. Both [129], [130] apply the synthetic minority oversampling technique (SMOTE) to balance samples [131].
4. Tools
Many of the aforementioned methods mention web servers or standalone tools, but some of these are inaccessible due to lack of maintenance. We summarize a list of available protein localization prediction tools regarding their coverage, algorithms, accessibility, and other characteristics. These localization prediction tools (at the subcellular or suborganellar level) are shown in Table 1. Note that the BUSCA [132] and SubCons [133] tools are web servers that integrate different computational tools for protein subcellular localization prediction. The localization coverage of some tools, e.g., DeepSig and SignalP 2.0, is marked as SP (secretory pathway) in Table 1 because they are signal peptide prediction tools. Signal peptides direct proteins toward the secretory pathway, where the proteins are either located inside certain organelles (the endoplasmic reticulum, Golgi, or endosomes), secreted from the cell, or inserted into cellular membranes. Thus, the specific localization of these proteins is not unique. Some tools consider the secretory pathway as a low-resolution localization. For example, TargetP 2.0 predicts the presence of signal peptides and also predicts the targeting peptide for mitochondrial proteins and plastid proteins where unique protein localization can be inferred.
Table 1.
Summary of protein localization prediction tools.
| Tool | Cov_lv1 | Cov_lv2 | Species kingdom | Algorithm | Metrics | Year | Web server | Standalone |
|---|---|---|---|---|---|---|---|---|
| BUSCA [132] | 1–4,7,11–14 | Eu,Pro | Integrated method | F1, MCC | 2018 | http://busca.biocomp.unibo.it/ | ||
| CELLO2GO [89] | 1–6,8–11,15 | Eu,Pro,V | SVM and homology search | Acc | 2014 | http://cello.life.nctu.edu.tw/cello2go/ | ||
| MULocDeep [48] | 1–10 | 1–10 | Eu | LSTM + attention | Acc, MCC, Rec, Prec, ROC_AUC, P&R_AUC | 2021 | http://mu-loc.org/ | √ |
| DeepLoc [49] | 1–10 | Eu | CNN + LSTM + attention | Acc, MCC, Gorodkin measure | 2017 | https://services.healthtech.dtu.dk/service.php?DeepLoc-1.0 | ||
| TargetP 2.0 [57] | SP,4,7 | Eu,Pro | LSTM + attention | Prec, Rec, F1, MCC | 2019 | https://services.healthtech.dtu.dk/service.php?TargetP-2.0 | ||
| MU-LOC [76] | 4 | P | SVM and neural network | Acc, Prec, F1, MCC | 2018 | http://136.32.161.178/ | √ | |
| LocTree3 [42] | 1–4,6–11 | Eu,Pro | SVM and homology search | Acc, Std | 2014 | https://rostlab.org/services/loctree3/ | ||
| MitoFates [92] | 4 | Eu | SVM | Prec, Rec, MCC, ROC_AUC | 2015 | http://mitf.cbrc.jp/MitoFates/cgi-bin/top.cgi | √ | |
| LOCALIZER [60] | 1,4,7 | P | SVM | SN, SP, PPV, MCC, Acc | 2017 | http://localizer.csiro.au/ | √ | |
| SignalP 5.0 [56] | SP | Eu,Pro | CNN, bidirectional LSTM, and CRF | MCC, Rec, Prec | 2019 | http://www.cbs.dtu.dk/services/SignalP/ | √ | |
| DeepSig [99] | SP | Eu,Bac | CNN and CRF | MCC, FPR, F1 | 2018 | https://deepsig.biocomp.unibo.it/welcome/default/index | √ | |
| PSORTb 3.0 [96] | 2,3,14–16 | Bac | SVM and homology search | Prec, Rec, Acc, MCC | 2010 | https://www.psort.org/psortb/ | √ | |
| WoLF PSORT [103] | 1–4,7,11 | Eu | k-NN classifier | Acc | 2007 | https://wolfpsort.hgc.jp/ | ||
| SubCons [133] | 1–4,6,8–11 | Hum | Integrated method | F1, MCC | 2017 | https://subcons.bioinfo.se/ | ||
| TPpred 3.0 [136] | 4,7 | Eu | Integrated method | MCC, Prec, Rec | 2015 | https://tppred3.biocomp.unibo.it/tppred3 | √ | |
| MultiLoc2 [44] | 1–4,6–11 | Eu | SVM | SN, SP, MCC | 2009 | https://abi-services.informatik.uni-tuebingen.de/multiloc2/webloc.cgi | √ | |
| YLoc [45] | 1–4,6–11 | Eu | Naïve Bayes and entropy-based discretization | F1, Acc | 2010 | https://abi-services.informatik.uni-tuebingen.de/yloc/webloc.cgi | √ | |
| SCLpred-EMS | SP | Eu | Neural network | SP, SN, FPR, MCC | 2020 | http://distilldeep.ucd.ie/SCLpred2/ | ||
| ERPred [87] | 6 | Eu | SVM | Acc, SN, SP, MCC | 2017 | http://proteininformatics.org/mkumar/erpred/index.html | √ | |
| SeqVec[68] | 1–10 | Eu | Language Model + FNN | Acc, MCC, FPR | 2019 | https://embed.protein.properties/ | √ | |
| ProtTrans [69] | 1–10 | Eu | Language Model + FNN | Acc | 2020 | https://embed.protein.properties/ | √ | |
| LA [70] | 1–10 | Eu | Language Model + attention | Acc | 2021 | https://embed.protein.properties/ | √ | |
| DeepMito [126] | 4 | Eu | CNN | MCC, GCC | 2019 | http://busca.biocomp.unibo.it/deepmito/ | √ | |
| SubGolgi v2 [59] | 8 | 8 | Eu | SVM | SN, Acc, MCC | 2013 | http://lin-group.cn/server/subGolgi2 | |
| TetraMito [58] | 4 | Eu | SVM | SN, Acc, MCC | 2013 | http://lin-group.cn/server/TetraMito | ||
| Schloro [93] | 7 | P | SVM | Acc, Rec, Prec, F1, ROC_AUC, MCC | 2017 | https://schloro.biocomp.unibo.it/welcome/default/index | √ | |
| SubMitoPred [86] | 4 | 4 | Eu | SVM | Acc | 2017 | http://proteininformatics.org/mkumar/submitopred/ | √ |
| SubNucPred [88] | 1 | Eu | SVM | Acc, SN, SP, MCC | 2014 | http://proteininformatics.org/mkumar/subnucpred/index.html | √ |
The localization coverage codes are: 1. nucleus; 2. cytoplasm; 3. extracellular; 4. mitochondrion; 5. cell membrane; 6. endoplasmic reticulum; 7. plastid/chloroplast; 8. Golgi apparatus; 9. lysosome/vacuole; 10. peroxisome; 11. plasma membrane; 12. organelle membrane; 13. endomembrane system; 14. outer membrane; 15. periplasmic; 16. cell wall; SP. secretory pathway.
Cov_lv1 represents subcellular localization coverage, and Cov_lv2 indicates that suborganellar localization predictions are provided for the organelle.
The species kingdom codes are: Eu (Eukaryota, including animal, plant, and fungi); Pro (Prokaryota, including Bacteria and Archaea); V (Virus); P (Plant); Bac (Bacteria); Hum (Human).
The metrics codes are: MCC (Matthews correlation coefficient), Acc (accuracy), SN (sensitivity), SP (specificity), Prec (precision), Rec (recall), ROC_AUC (area under receiver operating characteristic curve), P&R_AUC (area under precision & recall curve), GCC (Generalized Correlation Coefficient), PPV (positive predictive value), FPR (false positive rate).
To assess prediction tools, competitions can provide large-scale blind tests for objective evaluation. A well-known example is the CASP [134] in the protein structure prediction field. For protein localization prediction, the Critical Assessment of protein Function Annotation algorithms (CAFA) [135] is a good platform for such a purpose. CAFA requires a method to provide prediction in the form of cellular component ontology (CCO) terms. However, most methods reviewed in this paper predict UniProt's localization annotations rather than the CCO terms, and hence may not be assessed at CAFA directly. DeepLoc is a state-of-the-art method, and their dataset is often used by new methods for training and testing, as well as method comparison. Here, we used the DeepLoc dataset as a benchmark to evaluate some of the tools. The DeepLoc dataset was extracted from the UniProt database, release 2016_04. The protein dataset was filtered using the following criteria: eukaryotic, complete protein, encoded in the nucleus, longer than 40 amino acids, and experimentally verified (ECO:0000269) single localization annotation. Similar locations or subclasses of the same location were mapped to 10 main locations to increase the number of proteins per compartment (refer to Table 1 in [49] for details regarding the class distribution). A total of 13,858 proteins were obtained after the filtering process. PSI-CD-HIT [137] was used to cluster proteins with 30% identity or a 10−6 E-value cutoff, and the alignment was required to cover 80% of the shorter sequences, resulting in 8410 clusters for the whole dataset. The five-fold datasets generated had approximately the same number of proteins at each location. Four of the datasets were used for training and validation, and one was held out for testing. In this way, the redundancy between the training and testing datasets was reduced.
The DeepLoc, MULocDeep, SeqVec, ProtVec, and ProtTrans methods were stringently trained and tested using the training and testing samples in the DeepLoc dataset. LocTree2, MultiLoc2, CELLO, WoLF PSORT, YLoc, SherLoc2, and iLoc-Euk were run on the testing samples in the DeepLoc dataset. Thus, their performance is potentially overestimated because redundancy control was not performed. All the evaluated methods could be applied to proteins in eukaryotic cells. In the cases where a method predicted more than ten locations, the predicted locations were mapped onto the ten locations in the DeepLoc dataset. Overall accuracy is used as the evaluation criterion. The evaluation performance is directly cited from [48], [49], [68], [69], [70]. As shown in Fig. 2, the deep learning-based methods (DeepLoc, MULocDeep, ProtTrans, and SeqVec) have overall better performance than the other methods, except for ProtVec [138], which uses Word2Vec, a context-independent embedding method. DeepLoc_PSSM achieves better performance than DeepLoc_BLOSUM, indicating that evolutionary information enhances localization prediction. By comparing the performance of pre-trained methods (ProtTrans and SeqVec) with other deep learning methods (DeepLoc and MULocDeep), we find that a simple deep learning architecture with pre-train embedding can achieve competitive or even better performance than delicately designed deep-learning models using evolutionary profile features.
Fig. 2.

Evaluations of protein localization methods/tools. The criterion is the overall prediction accuracy for 10 main localizations. DeepLoc_PSSM and DeepLoc_BLOSUM are DeepLoc methods with PSSM and BLOSUM62 embedding, respectively. ProtT5_MLP and ProtBert_MLP are simple feed-forward neural networks in the ProtTrans method but using pre-train embeddings by T5 and Bert, respectively. ProtT5_LA and ProtBert_LA use the same two pre-trained models as above but are followed by an attention-based neural network.
5. Discussion and outlook
The computational prediction of protein localization has significantly improved prediction accuracy and localization mechanism studies over past two decades, especially with deep learning. However, the current methods still have limitations. For example, an 80% overall prediction accuracy shown in Fig. 2 does not mean that the localization prediction problem is 80% solved. In particular, many suborganellar localizations do not have sufficient data to build reliable prediction models. In this section, we discuss several areas for future exploration of localization analysis methods.
Protein localization problems have several biological characteristics. Many proteins can localize to more than one compartment. Some proteins are tissue-/cell type-specific, meaning their localization varies between different tissues or cell types. Proteins expressed at the correct location but with altered efficiency or concentration can also lead to illness. Thus, quantitively measuring or predicting protein localization in different tissues or cell types are in great demand. Additionally, proteins may be mislocalized due to mutations, which may have disease consequences [5]. Predicting mislocalization due to mutations is also challenging because it requires more sensitive methods with individual residue resolution.
Researchers could also pay more attention to biological interpretability when designing future localization analysis models. The mechanism of protein localization is complicated. In addition to targeting peptides, which are considered in some existing methods, other phenomena can affect/control protein localization. The trafficking machinery in cells controls the transport of molecules across membranes of organelles. Dysregulation of the protein trafficking machinery can have dramatic effects on general protein transport processes [139]. For example, the homozygous mutation R391H in the nucleoporin NUP155 has been shown to reduce nuclear envelope permeability and affect the export of Hsp70 mRNA and import of HSP70 protein [140]. Another fairly common method that affects protein localization involves binding partners that carry bound proteins between compartments. This mechanism allows for indirect control of protein localization by regulating the localization and concentration levels of binding partners, similar to the role of import receptors [9]. However, the prediction of protein localization changes affected by other proteins has not been explored. Furthermore, some localization signals are not contained within the linear peptide sequence of a cargo protein but are formed by the arrangement of amino acid residues on its surface. One advantage of such an arrangement is that conformational changes induced by allosteric events can disrupt or reform the localization signal transiently in response to the state of the protein [9]. Making protein localization analysis methods interpretable would allow us to answer “how” besides “where” a protein localizes, which has implications in pathology and drug design. The corresponding training data for such methods is currently lacking but may become available in the near future.
CRediT authorship contribution statement
Yuexu Jiang: Conceptualization, Investigation, Visualization, Validation, Writing - original draft. Duolin Wang: Visualization, Writing - review & editing. Weiwei Wang: Validation. Dong Xu: Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the US National Institutes of Health grants R35-GM126985 and R21-LM012790. We like to thank Dr. Ian Max Møller for the helpful discussions.
References
- 1.Schnell D.J., Hebert D.N. Protein translocons: multifunctional mediators of protein translocation across membranes. Cell. 2003;112(4):491–505. doi: 10.1016/s0092-8674(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 2.Wickner W., Schekman R. Protein translocation across biological membranes. Science. 2005;310(5753):1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 3.Neupert W., Herrmann J.M. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 4.Davis J.R., Kakar M., Lim C.S. Controlling protein compartmentalization to overcome disease. Pharm Res. 2007;24(1):17–27. doi: 10.1007/s11095-006-9133-z. [DOI] [PubMed] [Google Scholar]
- 5.Hung M.C., Link W. Protein localization in disease and therapy. J Cell Sci. 2011;124(Pt 20):3381–3392. doi: 10.1242/jcs.089110. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez J.A., Schüchner S., Au W.W., Fabbro M., Henderson B.R. Nuclear–cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene. 2004;23(10):1809–1820. doi: 10.1038/sj.onc.1207302. [DOI] [PubMed] [Google Scholar]
- 7.Marques-Bueno M.M., Moreno-Romero J., Abas L., De Michele R., Martinez M.C. A dominant negative mutant of protein kinase CK2 exhibits altered auxin responses in Arabidopsis. Plant J. 2011;67(1):169–180. doi: 10.1111/j.1365-313X.2011.04585.x. [DOI] [PubMed] [Google Scholar]
- 8.Thevissen K., de Mello T.P., Xu D., Blankenship J., Vandenbosch D., Idkowiak-Baldys J. The plant defensin RsAFP2 induces cell wall stress, septin mislocalization and accumulation of ceramides in Candida albicans. Mol Microbiol. 2012;84(1):166–180. doi: 10.1111/j.1365-2958.2012.08017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauer N.C., Doetsch P.W., Corbett A.H. Mechanisms regulating protein localization. Traffic. 2015;16(10):1039–1061. doi: 10.1111/tra.12310. [DOI] [PubMed] [Google Scholar]
- 10.Hagmann M. Protein zip codes make Nobel journey. Science. 1999;286(5440):666. doi: 10.1126/science.286.5440.666. [DOI] [PubMed] [Google Scholar]
- 11.Chacinska A., Koehler C.M., Milenkovic D., Lithgow T., Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138(4):628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt O., Pfanner N., Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11(9):655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 13.Jakobsen L., Vanselow K., Skogs M., Toyoda Y., Lundberg E., Poser I. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011;30(8):1520–1535. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christoforou A., Mulvey C.M., Breckels L.M., Geladaki A., Hurrell T., Hayward P.C. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun. 2016;7:8992. doi: 10.1038/ncomms9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itzhak D.N., Tyanova S., Cox J., Borner G.H. Global, quantitative and dynamic mapping of protein subcellular localization. Elife. 2016;5 doi: 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orre L.M., Vesterlund M., Pan Y., Arslan T., Zhu Y., Fernandez Woodbridge A. Proteome-wide mapping of protein localization and relocalization. Mol Cell. 2019;73(1):166–182 e167. doi: 10.1016/j.molcel.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 17.Rhee H.W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung V., Zou P., Rhee H.W., Udeshi N.D., Cracan V., Svinkina T. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell. 2014;55(2):332–341. doi: 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S.Y., Kang M.G., Park J.S., Lee G., Ting A.Y., Rhee H.W. APEX fingerprinting reveals the subcellular localization of proteins of interest. Cell Rep. 2016;15(8):1837–1847. doi: 10.1016/j.celrep.2016.04.064. [DOI] [PubMed] [Google Scholar]
- 20.Chong Y.T., Koh J.L., Friesen H., Duffy S.K., Cox M.J., Moses A. Yeast proteome dynamics from single cell imaging and automated analysis. Cell. 2015;161(6):1413–1424. doi: 10.1016/j.cell.2015.04.051. [DOI] [PubMed] [Google Scholar]
- 21.Barbe L., Lundberg E., Oksvold P., Stenius A., Lewin E., Bjorling E. Toward a confocal subcellular atlas of the human proteome. Mol Cell Proteomics. 2008;7(3):499–508. doi: 10.1074/mcp.M700325-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Stadler C., Skogs M., Brismar H., Uhlen M., Lundberg E. A single fixation protocol for proteome-wide immunofluorescence localization studies. J Proteomics. 2010;73(6):1067–1078. doi: 10.1016/j.jprot.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Thul P.J., Akesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H. A subcellular map of the human proteome. Science. 2017;356(6340) doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 24.Burns T.J., Frei A.P., Gherardini P.F., Bava F.A., Batchelder J.E., Yoshiyasu Y. High-throughput precision measurement of subcellular localization in single cells. Cytometry A. 2017;91(2):180–189. doi: 10.1002/cyto.a.23054. [DOI] [PubMed] [Google Scholar]
- 25.Gardy J.L., Brinkman F.S. Methods for predicting bacterial protein subcellular localization. Nat Rev Microbiol. 2006;4(10):741–751. doi: 10.1038/nrmicro1494. [DOI] [PubMed] [Google Scholar]
- 26.Nakai K. Protein sorting signals and prediction of subcellular localization. Adv Protein Chem. 2000;54:277–344. doi: 10.1016/s0065-3233(00)54009-1. [DOI] [PubMed] [Google Scholar]
- 27.Imai K., Nakai K. Tools for the recognition of sorting signals and the prediction of subcellular localization of proteins from their amino acid sequences. Front Genet. 2020;11 doi: 10.3389/fgene.2020.607812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonetta R., Valentino G. Machine learning techniques for protein function prediction. Proteins. 2020;88(3):397–413. doi: 10.1002/prot.25832. [DOI] [PubMed] [Google Scholar]
- 29.Shen Y., Ding Y., Tang J., Zou Q., Guo F. Critical evaluation of web-based prediction tools for human protein subcellular localization. Brief Bioinform. 2020;21(5):1628–1640. doi: 10.1093/bib/bbz106. [DOI] [PubMed] [Google Scholar]
- 30.Donnes P., Hoglund A. Predicting protein subcellular localization: past, present, and future. Genomics Proteomics Bioinformatics. 2004;2(4):209–215. doi: 10.1016/S1672-0229(04)02027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou K.C., Shen H.B. Recent progress in protein subcellular location prediction. Anal Biochem. 2007;370(1):1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z., Zou Q., Jiang Y., Ju Y., Zeng X. Review of protein subcellular localization prediction. Curr Bioinform. 2014;9(3):331–342. [Google Scholar]
- 33.Kumar R., Dhanda S.K. Bird eye view of protein subcellular localization prediction. Life (Basel) 2020;10(12) doi: 10.3390/life10120347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou K.C. A novel approach to predicting protein structural classes in a (20–1)-D amino acid composition space. Proteins. 1995;21(4):319–344. doi: 10.1002/prot.340210406. [DOI] [PubMed] [Google Scholar]
- 36.Chou K. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins: Struct Funct Bioinf. 2001;43(3):246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 37.Nair R., Rost B. Sequence conserved for subcellular localization. Protein Sci. 2002;11(12):2836–2847. doi: 10.1110/ps.0207402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joshi T., Xu D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genomics. 2007;8(1):1–10. doi: 10.1186/1471-2164-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 40.Remmert M., Biegert A., Hauser A., Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 41.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldberg T., Hecht M., Hamp T., Karl T., Yachdav G., Ahmed N. LocTree3 prediction of localization. Nucleic Acids Res. 2014;42(Web Server issue):W350–W355. doi: 10.1093/nar/gku396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briesemeister S., Blum T., Brady S., Lam Y., Kohlbacher O., Shatkay H. SherLoc2: a high-accuracy hybrid method for predicting subcellular localization of proteins. J Proteome Res. 2009;8(11):5363–5366. doi: 10.1021/pr900665y. [DOI] [PubMed] [Google Scholar]
- 44.Blum T., Briesemeister S., Kohlbacher O. MultiLoc2: integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinf. 2009;10(1):274. doi: 10.1186/1471-2105-10-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Briesemeister S, Rahnenfuhrer J, Kohlbacher O: YLoc--an interpretable web server for predicting subcellular localization. Nucleic Acids Res 2010, 38(Web Server issue):W497-502. [DOI] [PMC free article] [PubMed]
- 46.Zheng W, Zhang C, Li Y, Pearce R, Bell EW, Zhang Y: Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Reports Methods 2021:100014. [DOI] [PMC free article] [PubMed]
- 47.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci. 1992;89(22):10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang Y., Wang D., Yao Y., Eubel H., Künzler P., Møller I.M. MULocDeep: A deep-learning framework for protein subcellular and suborganellar localization prediction with residue-level interpretation. Comput Struct Biotechnol J. 2021;19:4825–4839. doi: 10.1016/j.csbj.2021.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Almagro Armenteros J.J., Sonderby C.K., Sonderby S.K., Nielsen H., Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33(21):3387–3395. doi: 10.1093/bioinformatics/btx431. [DOI] [PubMed] [Google Scholar]
- 50.Jaakkola T., Diekhans M., Haussler D. A discriminative framework for detecting remote protein homologies. J Comput Biol. 2000;7(1–2):95–114. doi: 10.1089/10665270050081405. [DOI] [PubMed] [Google Scholar]
- 51.Kuang R., Ie E., Wang K., Wang K., Siddiqi M., Freund Y. Profile-based string kernels for remote homology detection and motif extraction. J Bioinf Comput Biol. 2005;3(03):527–550. doi: 10.1142/s021972000500120x. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg T., Hamp T., Rost B. LocTree2 predicts localization for all domains of life. Bioinformatics. 2012;28(18):i458–i465. doi: 10.1093/bioinformatics/bts390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardy J.L., Spencer C., Wang K., Ester M., Tusnady G.E., Simon I. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 2003;31(13):3613–3617. doi: 10.1093/nar/gkg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sigrist C.J., Cerutti L., Hulo N., Gattiker A., Falquet L., Pagni M. PROSITE: a documented database using patterns and profiles as motif descriptors. Briefings Bioinf. 2002;3(3):265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 55.Blobel G., Dobberstein B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol. 1975;67(3):835–851. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Almagro Armenteros J.J., Tsirigos K.D., Sonderby C.K., Petersen T.N., Winther O., Brunak S. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–423. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 57.Almagro Armenteros J.J., Salvatore M., Emanuelsson O., Winther O., von Heijne G., Elofsson A. Detecting sequence signals in targeting peptides using deep learning. Life Sci Alliance. 2019;2(5) doi: 10.26508/lsa.201900429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin H., Chen W., Yuan L., Li Z., Ding H. Using over-represented tetrapeptides to predict protein submitochondria locations. Acta Biotheor. 2013;61(2):259–268. doi: 10.1007/s10441-013-9181-9. [DOI] [PubMed] [Google Scholar]
- 59.Ding H., Guo S.-H., Deng E.-Z., Yuan L.-F., Guo F.-B., Huang J. Prediction of Golgi-resident protein types by using feature selection technique. Chemometr Intell Lab Syst. 2013;124:9–13. [Google Scholar]
- 60.Sperschneider J., Catanzariti A.-M., DeBoer K., Petre B., Gardiner D.M., Singh K.B. LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Sci Rep. 2017;7(1):1–14. doi: 10.1038/srep44598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Venkatarajan M.S., Braun W. New quantitative descriptors of amino acids based on multidimensional scaling of a large number of physical–chemical properties. Mol Model Annual. 2001;7(12):445–453. [Google Scholar]
- 62.Kawashima S., Ogata H., Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 1999;27(1):368–369. doi: 10.1093/nar/27.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perdigão N., Heinrich J., Stolte C., Sabir K.S., Buckley M.J., Tabor B. Unexpected features of the dark proteome. Proc Natl Acad Sci. 2015;112(52):15898–15903. doi: 10.1073/pnas.1508380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser L, Polosukhin I: Attention is all you need. arXiv preprint arXiv:03762 2017.
- 65.Peters ME, Neumann M, Iyyer M, Gardner M, Clark C, Lee K, Zettlemoyer L. Deep contextualized word representations. arXiv preprint arXiv:05365 2018.
- 66.Mikolov T., Sutskever I., Chen K., Corrado G.S., Dean J. Advances in neural information processing systems. 2013. Distributed representations of words and phrases and their compositionality; pp. 3111–3119. [Google Scholar]
- 67.Devlin J, Chang M-W, Lee K, Toutanova K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:04805 2018.
- 68.Heinzinger M., Elnaggar A., Wang Y., Dallago C., Nechaev D., Matthes F. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinf. 2019;20(1):1–17. doi: 10.1186/s12859-019-3220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elnaggar A, Heinzinger M, Dallago C, Rihawi G, Wang Y, Jones L, et al. ProtTrans: towards cracking the language of Life's code through self-supervised deep learning and high performance computing. arXiv preprint arXiv:06225 2020.
- 70.Stärk H, Dallago C, Heinzinger M, Rost B. Light attention predicts protein location from the language of life. 2021:2021.2004.2025.441334. [DOI] [PMC free article] [PubMed]
- 71.Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 2012;40(Database issue):D857-861. [DOI] [PMC free article] [PubMed]
- 72.Xenarios I., Salwinski L., Duan X.J., Higney P., Kim S.M., Eisenberg D. DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30(1):303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ananda MM, Hu J. NetLoc: Network based protein localization prediction using protein-protein interaction and co-expression networks. In: 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM): 2010. IEEE: 142-148.
- 76.Zhang N., Rao R.S.P., Salvato F., Havelund J.F., Moller I.M., Thelen J.J. MU-LOC: A machine-learning method for predicting mitochondrially localized proteins in plants. Front Plant Sci. 2018;9:634. doi: 10.3389/fpls.2018.00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryngajllo M., Childs L., Lohse M., Giorgi F.M., Lude A., Selbig J. SLocX: Predicting subcellular localization of Arabidopsis proteins leveraging gene expression data. Front Plant Sci. 2011;2:43. doi: 10.3389/fpls.2011.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomczak K., Czerwinska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19(1A):A68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cortes C., Vapnik V. Support-vector networks. Machine Learning. 1995;20(3):273–297. [Google Scholar]
- 81.Hua S., Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17(8):721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 82.Sarda D., Chua G.H., Li K.B., Krishnan A. pSLIP: SVM based protein subcellular localization prediction using multiple physicochemical properties. BMC Bioinf. 2005;6:152. doi: 10.1186/1471-2105-6-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tax D.M., Duin R.P. Support vector data description. Machine Learning. 2004;54(1):45–66. [Google Scholar]
- 84.Lee K., Kim D.W., Na D., Lee K.H., Lee D. PLPD: reliable protein localization prediction from imbalanced and overlapped datasets. Nucleic Acids Res. 2006;34(17):4655–4666. doi: 10.1093/nar/gkl638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu C.S., Chen Y.C., Lu C.H., Hwang J.K. Prediction of protein subcellular localization. Proteins. 2006;64(3):643–651. doi: 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- 86.Kumar R., Kumari B., Kumar M. Proteome-wide prediction and annotation of mitochondrial and sub-mitochondrial proteins by incorporating domain information. Mitochondrion. 2018;42:11–22. doi: 10.1016/j.mito.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 87.Kumar R., Kumari B., Kumar M. Prediction of endoplasmic reticulum resident proteins using fragmented amino acid composition and support vector machine. PeerJ. 2017;5 doi: 10.7717/peerj.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kumar R., Jain S., Kumari B., Kumar M. Protein sub-nuclear localization prediction using SVM and Pfam domain information. PLoS ONE. 2014;9(6) doi: 10.1371/journal.pone.0098345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu C.S., Cheng C.W., Su W.C., Chang K.C., Huang S.W., Hwang J.K. CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE. 2014;9(6) doi: 10.1371/journal.pone.0099368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X., Zhang W., Zhang Q., Li G.Z. MultiP-SChlo: multi-label protein subchloroplast localization prediction with Chou's pseudo amino acid composition and a novel multi-label classifier. Bioinformatics. 2015;31(16):2639–2645. doi: 10.1093/bioinformatics/btv212. [DOI] [PubMed] [Google Scholar]
- 91.Hasan M.A.M., Ahmad S., Molla M.K.I. Protein subcellular localization prediction using multiple kernel learning based support vector machine. Mol BioSyst. 2017;13(4):785–795. doi: 10.1039/c6mb00860g. [DOI] [PubMed] [Google Scholar]
- 92.Fukasawa Y., Tsuji J., Fu S.C., Tomii K., Horton P., Imai K. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics. 2015;14(4):1113–1126. doi: 10.1074/mcp.M114.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Savojardo C., Martelli P.L., Fariselli P., Casadio R. SChloro: directing Viridiplantae proteins to six chloroplastic sub-compartments. Bioinformatics. 2017;33(3):347–353. doi: 10.1093/bioinformatics/btw656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Joyce J. Bayes' theorem. The Stanford Encyclopedia of Philosophy 2003.
- 95.Gardy J.L., Laird M.R., Chen F., Rey S., Walsh C.J., Ester M. PSORTb vol 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21(5):617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- 96.Yu N.Y., Wagner J.R., Laird M.R., Melli G., Rey S., Lo R. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee H., Tu Z., Deng M., Sun F., Chen T. Diffusion kernel-based logistic regression models for protein function prediction. OMICS. 2006;10(1):40–55. doi: 10.1089/omi.2006.10.40. [DOI] [PubMed] [Google Scholar]
- 98.Chung MK. Introduction to random fields. arXiv preprint arXiv:09660 2020.
- 99.Savojardo C., Martelli P.L., Fariselli P., Casadio R. DeepSig: deep learning improves signal peptide detection in proteins. Bioinformatics. 2018;34(10):1690–1696. doi: 10.1093/bioinformatics/btx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu L., Hofestädt R., Ester M. Tissue-specific subcellular localization prediction using multi-label Markov random fields. IEEE/ACM Trans Comput Biol Bioinf. 2019;16(5):1471–1482. doi: 10.1109/TCBB.2019.2897683. [DOI] [PubMed] [Google Scholar]
- 101.Thul P.J., Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018;27(1):233–244. doi: 10.1002/pro.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Altman N.S. An introduction to kernel and nearest-neighbor nonparametric regression. Am Stat. 1992;46(3):175–185. [Google Scholar]
- 103.Horton P., Park K.J., Obayashi T., Fujita N., Harada H., Adams Collier C.J. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(Web Server issue):W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garapati H.S., Male G., Mishra K. Predicting subcellular localization of proteins using protein-protein interaction data. Genomics. 2020;112(3):2361–2368. doi: 10.1016/j.ygeno.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 105.Chandra M.P. Proceedings of the National Institute of Sciences of India. 1936. On the generalised distance in statistics; pp. 49–55. [Google Scholar]
- 106.Chou K.-C., Elrod D.W. Protein subcellular location prediction. Protein Eng. 1999;12(2):107–118. doi: 10.1093/protein/12.2.107. [DOI] [PubMed] [Google Scholar]
- 107.Zhou G.P., Doctor K. Subcellular location prediction of apoptosis proteins. Proteins. 2003;50(1):44–48. doi: 10.1002/prot.10251. [DOI] [PubMed] [Google Scholar]
- 108.Ding H., Liu L., Guo F.-B., Huang J., Lin H. Identify Golgi protein types with modified mahalanobis discriminant algorithm and pseudo amino acid composition. Protein Peptide Letters. 2011;18(1):58–63. doi: 10.2174/092986611794328708. [DOI] [PubMed] [Google Scholar]
- 109.Goodfellow I., Bengio Y., Courville A., Bengio Y. vol. 1. MIT Press; Cambridge: 2016. (Deep learning). [Google Scholar]
- 110.Bengio Y. Now Publishers Inc; 2009. Learning deep architectures for AI. [Google Scholar]
- 111.Rumelhart D.E., Hinton G.E., Williams R.J. Learning representations by back-propagating errors. Nature. 1986;323(6088):533–536. [Google Scholar]
- 112.Hochreiter S., Schmidhuber J. Long short-term memory. Neural Comput. 1997;9(8):1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 113.Bahdanau D, Cho K, Bengio Y. Neural machine translation by jointly learning to align and translate. arXiv preprint 2014.
- 114.Mooney C., Wang Y.H., Pollastri G. SCLpred: protein subcellular localization prediction by N-to-1 neural networks. Bioinformatics. 2011;27(20):2812–2819. doi: 10.1093/bioinformatics/btr494. [DOI] [PubMed] [Google Scholar]
- 115.Wang X., Jin Y., Zhang Q. DeepPred-SubMito: A novel submitochondrial localization predictor based on multi-channel convolutional neural network and dataset balancing treatment. Int J Mol Sci. 2020;21(16):5710. doi: 10.3390/ijms21165710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pascanu R, Mikolov T, Bengio Y. On the difficulty of training recurrent neural networks. In: Proceedings of the 30th International Conference on Machine Learning; Proceedings of Machine Learning Research: Edited by Sanjoy D, David M. PMLR 2013: 1310--1318.
- 117.Kalchbrenner N., Blunsom P. Proceedings of the 2013 conference on empirical methods in natural language processing. 2013. Recurrent continuous translation models; pp. 1700–1709. [Google Scholar]
- 118.Cho K, Van Merriënboer B, Gulcehre C, Bahdanau D, Bougares F, Schwenk H, et al. Learning phrase representations using RNN encoder-decoder for statistical machine translation. arXiv preprint arXiv:03762 2014.
- 119.Sutskever I., Vinyals O., Le Q.V. Advances in neural information processing systems. 2014. Sequence to sequence learning with neural networks; pp. 3104–3112. [Google Scholar]
- 120.Sak H., Senior A.W., Beaufays F. Long short-term memory recurrent neural network architectures for large scale acoustic modeling. Interspeech. 2014:338–342. [Google Scholar]
- 121.Li X, Wu X. Constructing long short-term memory based deep recurrent neural networks for large vocabulary speech recognition. In: 2015 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP): 2015. IEEE: 4520-4524.
- 122.Valueva M.V., Nagornov N., Lyakhov P.A., Valuev G.V., Chervyakov N. Application of the residue number system to reduce hardware costs of the convolutional neural network implementation. Math Comput Simul. 2020;177:232–243. [Google Scholar]
- 123.Wang D., Zhang Z., Jiang Y., Mao Z., Wang D., Lin H. DM3Loc: multi-label mRNA subcellular localization prediction and analysis based on multi-head self-attention mechanism. Nucleic Acids Res. 2021 doi: 10.1093/nar/gkab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin Z, Feng M, Santos CNd, Yu M, Xiang B, Zhou B, Bengio Y: A structured self-attentive sentence embedding. arXiv preprint 2017.
- 125.Kaleel M., Zheng Y., Chen J., Feng X., Simpson J.C., Pollastri G. SCLpred-EMS: Subcellular localization prediction of endomembrane system and secretory pathway proteins by deep N-to-1 convolutional neural networks. Bioinformatics. 2020;36(11):3343–3349. doi: 10.1093/bioinformatics/btaa156. [DOI] [PubMed] [Google Scholar]
- 126.Savojardo C., Bruciaferri N., Tartari G., Martelli P.L., Casadio R. DeepMito: accurate prediction of protein submitochondrial localization using convolutional neural networks. Bioinformatics. 2019;36(1):56–64. doi: 10.1093/bioinformatics/btz512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen T, Guestrin C: Xgboost: A scalable tree boosting system. In: Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining: 2016. 785-794.
- 128.Pang L., Wang J., Zhao L., Wang C., Zhan H. A novel protein subcellular localization method with CNN-XGBoost model for Alzheimer's disease. Front Genet. 2019;9:751. doi: 10.3389/fgene.2018.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu B., Qiu W., Chen C., Ma A., Jiang J., Zhou H. SubMito-XGBoost: predicting protein submitochondrial localization by fusing multiple feature information and eXtreme gradient boosting. Bioinformatics. 2020;36(4):1074–1081. doi: 10.1093/bioinformatics/btz734. [DOI] [PubMed] [Google Scholar]
- 130.Wu L., Huang S., Wu F., Jiang Q., Yao S., Jin X. Protein subnuclear localization based on radius-SMOTE and kernel linear discriminant analysis combined with random forest. Electronics. 2020;9(10):1566. [Google Scholar]
- 131.Chawla N.V., Bowyer K.W., Hall L.O., Kegelmeyer W.P. SMOTE: synthetic minority over-sampling technique. J Artif Intell Res. 2002;16:321–357. [Google Scholar]
- 132.Savojardo C., Martelli P.L., Fariselli P., Profiti G., Casadio R. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018;46(W1):W459–W466. doi: 10.1093/nar/gky320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Salvatore M., Warholm P., Shu N., Basile W., Elofsson A. SubCons: a new ensemble method for improved human subcellular localization predictions. Bioinformatics. 2017;33(16):2464–2470. doi: 10.1093/bioinformatics/btx219. [DOI] [PubMed] [Google Scholar]
- 134.Kryshtafovych A., Schwede T., Topf M., Fidelis K., Moult J. Critical assessment of methods of protein structure prediction (CASP)—Round XIII. Proteins: Struct Funct Bioinf. 2019;87(12):1011–1020. doi: 10.1002/prot.25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhou N., Jiang Y., Bergquist T.R., Lee A.J., Kacsoh B.Z., Crocker A.W. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019;20(1):244. doi: 10.1186/s13059-019-1835-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Savojardo C., Martelli P.L., Fariselli P., Casadio R. TPpred3 detects and discriminates mitochondrial and chloroplastic targeting peptides in eukaryotic proteins. Bioinformatics. 2015;31(20):3269–3275. doi: 10.1093/bioinformatics/btv367. [DOI] [PubMed] [Google Scholar]
- 137.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 138.Asgari E, Mofrad MRJPo. Continuous distributed representation of biological sequences for deep proteomics and genomics. 2015, 10(11):e0141287. [DOI] [PMC free article] [PubMed]
- 139.Chahine M.N., Pierce G.N. Therapeutic targeting of nuclear protein import in pathological cell conditions. Pharmacol Rev. 2009;61(3):358–372. doi: 10.1124/pr.108.000620. [DOI] [PubMed] [Google Scholar]
- 140.Zhang X., Chen S., Yoo S., Chakrabarti S., Zhang T., Ke T. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135(6):1017–1027. doi: 10.1016/j.cell.2008.10.022. [DOI] [PubMed] [Google Scholar]