

Summary
Lafora disease (LD) is a fatal childhood dementia characterized by progressive myoclonic epilepsy manifesting in the teenage years, rapid neurological decline, and death typically within ten years of onset. Mutations in either EPM2A, encoding the glycogen phosphatase laforin, or EPM2B, encoding the E3 ligase malin, cause LD. Whole exome sequencing has revealed many EPM2A variants associated with late-onset or slower disease progression. We established an empirical pipeline for characterizing the functional consequences of laforin missense mutations in vitro using complementary biochemical approaches. Analysis of 26 mutations revealed distinct functional classes associated with different outcomes that were supported by clinical cases. For example, F321C and G279C mutations have attenuated functional defects and are associated with slow progression. This pipeline enabled rapid characterization and classification of newly identified EPM2A mutations, providing clinicians and researchers genetic information to guide treatment of LD patients.
Subject areas: Disease, Biochemistry, Structural biology, Biophysics
Graphical abstract
Highlights
-
•
Lafora disease (LD) patients present with varying clinical progression
-
•
LD missense mutations differentially affect laforin function
-
•
An empirical in vitro pipeline is used to classify laforin missense mutations
-
•
Patient progression can be predicted based on mutation class
Disease; Biochemistry; Structural biology; Biophysics
Introduction
Lafora disease (LD) is a fatal progressive myoclonic epilepsy. LD patients develop normally until their adolescent years, when generalized seizures and myoclonic jerks begin. Over time, patients experience increasingly severe and frequent epileptic episodes, cognitive decline, ataxia and aphasia, leading to childhood dementia and a vegetative state (Verhalen et al., 2018). Antiseizure drugs are only palliative, and most patients do not live beyond age 30. A hallmark of LD is the presence of polyglucosan bodies, known as Lafora bodies (LBs), in most tissues (Brewer et al., 2020). Multiple studies have demonstrated that LBs are the etiological agent driving LD (Duran et al., 2014; Pederson et al., 2013; Turnbull et al., 2011, 2014). Thus, LD is classified as a member of the larger glycogen storage disease (GSD) family, which affects 1 in 20,000–43,000 newborns (Ozen, 2007).
Patients with LD carry mutations in either the epilepsy progressive myoclonus 2A (EPM2A) gene encoding laforin or the EPM2B gene encoding malin. Laforin is the founding member of the glucan phosphatase family and dephosphorylates glycogen, a soluble glucose-storage molecule synthesized by eukaryotic cells (Gentry et al., 2007; Tagliabracci et al., 2007; Worby et al., 2006). We previously defined the structural basis for binding and dephosphorylation of glycogen by laforin and characterized its quaternary structure and conformational dynamics in solution (Raththagala et al., 2015). Laforin also interacts with and is ubiquitinated by malin, an E3 ubiquitin ligase (Gentry et al., 2005; Lohi et al., 2005). It has been suggested that laforin also serves as a central glycogen-associated scaffolding protein, interacting with multiple partner proteins important for glycogen metabolism (Gentry et al., 2013) and that a possible cause of LD is the lack of a functional laforin-malin complex (Sullivan et al., 2017).
A large amount of genetic information has emerged from whole exome sequencing of rare diseases (Boycott et al., 2013). Over one hundred distinct LD-causing mutations in EPM2A have been identified, including missense and nonsense mutations and insertions and deletions (indels) (http://projects.tcag.ca/lafora/) (Ianzano et al., 2005; Singh and Ganesh, 2009). Although the number of reported mutations grows yearly, many mutations have only been identified in a single patient, often with compound heterozygosity, and published clinical details can be sparse (Aslam et al., 2017; Lesca et al., 2010). This lack of data makes genotype–phenotype correlations difficult. Computational algorithms can potentially predict pathogenicity of a variant; however, these algorithms range in performance, and results from different programs often do not correlate (Hicks et al., 2011; Potapov et al., 2009; Thusberg et al., 2011). In many cases, experimental strategies can define patient-specific pathogenesis, explain differences in disease severity, and enable personalized medicine (Kroncke et al., 2015). This approach has been successful in the cystic fibrosis (CF) field. For CF, effects of disease-causing genetic variants were difficult to predict using in silico tools; instead the functional consequences of the variants were defined through many basic biological and biochemical studies (Amaral, 2015; Kerem, 2005; Masica et al., 2012; Sheppard and Welsh, 1999; Welsh and Smith, 1993). Now, CF mutations are classified based on their functional effect(s), and mutation-specific therapies are used to treat patients (Amaral, 2015).
LD was initially considered to have a fairly homogeneous clinical course with initial seizures appearing in adolescence and death within 10 years of onset (Minassian, 2001). Before the genetic loci were identified, if patients presented with progressive myoclonic epilepsy and lived beyond the age of 30, then a diagnosis of LD was ruled out (Van Heycop Ten Ham, 1975). Even now, adult patients with milder progressive myoclonus epilepsy typically do not undergo LD screening (Ferlazzo et al., 2014). However, late-onset and slowly progressing LD have been confirmed by genetic testing in older patients (Minassian, 2001; Van Heycop Ten Ham, 1975). With the increasing use of genetic testing to confirm LD diagnosis, the neurology community has recently begun to explore the more heterogeneous progression of LD patients (Brewer et al., 2019a; Ferlazzo et al., 2014).
In this report, we describe four LD patients with EPM2A missense mutations who displayed this newly recognized spectrum of disease severity. Three patients presented with a classic aggressive LD course and one displayed a protracted course. To define the molecular basis for these differences and others previously reported, we established a pipeline to rapidly profile laforin functions and analyzed 26 laforin missense mutations, including the four in this clinical report. The mutations are segregated into five classes based on their biochemical effects. The milder effects of some laforin mutations provided an explanation for classic and slowly progressing forms of LD. Moving forward, new mutations can be quickly classified using our empirical pipeline and distinguished from benign polymorphisms. Our results showed that this system can be used to guide patient-specific clinical care and the deployment of therapeutics, which is needed to maximize the benefit from emerging LD therapeutics (Austin et al., 2019; Brewer and Gentry, 2018; Brewer et al., 2019a, 2019b; Gentry et al., 2020; Zhou et al., 2019).
Results
Clinical features of four LD patients
We describe 4 patients from four families. The patients have EPM2A mutations and displayed varying clinical courses that highlight the heterogeneous progression observed in LD patients (Table 1). Patient 1 (E28K/W165X) developed visual and generalized tonic-clonic seizures at the age of 10 years, with absence seizures manifesting at 11 years and myoclonic seizures at 13 years. He developed cognitive problems at 15 years and 1 year later started with speech difficulties and ataxia. He was wheelchair-bound at 17 years. He needs assistance for all activities of daily living and has no social interaction. Patient 2 (W32G/R241X) suffered his first visual seizure followed by a generalized tonic-clonic seizure at 10 years and absence seizures at 13 years. At age 14 years he developed myoclonic jerks, began suffering from cognitive impairment, and dropped out of school. When he was 16 years old, he presented gait unsteadiness and became wheelchair-bound. Presently, he is bedridden and mute with continuous myoclonus. Patient 3 (W32G/W165X) experienced cognitive problems at 14 years old and epilepsy with myoclonic and generalized tonic-clonic seizures at 16 years. When he was 17 years old, he became wheelchair-bound and speechless. Patient 4 (G279C/Q55X) suffered a first generalized tonic-clonic seizure at age 25, and after 7 years of evolution has no cognitive or motor impairment.
Table 1.
Clinical features, genetic findings, and classification of selected EPM2A patients
| Patient | Age at onset | Nucleic acid change | Protein mutation | 5-year status post initial seizure |
|---|---|---|---|---|
| 1 | 10 | c.82G > A c.495G > A | E28K W165X |
Epilepsy, cognitive impairment, bedridden |
| 2 | 10 | c.94T > G c.721C > T | W32G R241X |
Epilepsy, cognitive impairment, bedridden and mute |
| 3 | 14 | c.94T > G c.495G > A |
W32G W165X |
Epilepsy, cognitive impairment, bedridden and mute |
| 4 | 25 | c.835G > A c.163C > T |
G279C Q55X |
Epilepsy with no cognitive or motor impairment |
All 4 patients were compound heterozygous for a missense mutation and a nonsense mutation. Nevertheless, patients 1 and 2 presented a classic progression, patient 3 an ultra-rapid developing subtype, and patient 4 displayed a slower progression. Late-onset LD was also reported in a compound heterozygous patient with EPM2A mutations G279C/R241X (Jara-Prado et al., 2014). Therefore, it is possible that the G279C mutation is less detrimental to the function of laforin than the missense mutations, E28K and W32G, carried by patients 1, 2 and 3.
Pathogenicity predictions of EPM2A missense mutations
To define the dysfunctional basis for EPM2A missense mutations, 26 variants spanning the entire laforin protein were initially selected for analysis (Figure 1A). The variants correspond to the range of disease progression with G279C and P211L associated with a slower disease course and F321C with a very late-onset phenotype (Table 1). The 26 mutations were initially analyzed using three different in silico tools to predict mutation pathogenicity. PolyPhen-2 is a broadly used program that predicts the effect of a mutation based on sequence homology and local structural features using only amino acid sequence as input (Adzhubei et al., 2010). Site Directed Mutator (SDM) (Pandurangan et al., 2017) and Cologne University Protein Stability Analysis Tool (CUPSAT) (Parthiban et al., 2006) predict the effect of mutations on protein stability using a known protein structure. SDM predicts changes in free energy of unfolding (ΔΔG) induced by the mutation using estimated environment-specific potentials, whereas CUPSAT makes ΔΔG predictions based on atom potentials and torsion angle potentials.
Figure 1.

Pathogenicity predictions and a biochemical pipeline for EPM2A missense mutations
(A) LD missense mutations selected for study are shown mapped to the primary sequence of laforin.
(B) Correlation analysis between PolyPhen-2 pathogenicity score and ΔΔG (kcal/mol) predictions by CUPSAT and SDM for the 26 mutations selected for analysis (see Table 2).
(C) Empirical pipeline for characterizing missense mutations in vitro. The mutations were introduced into bacterial and yeast expression plasmids. Bacterially purified recombinant protein was used for biochemical assays and hydrogen deuterium exchange (HDX) studies. Yeast two-hybrid assays were performed to study laforin interactions with binding partners. The biochemical profile of the mutant was then compiled to determine the severity of the mutation, which is then linked to the clinical course of the patient.
PolyPhen-2 produced predictions for the mutants ranging from “benign” to “probably damaging” (Table 2). Although PolyPhen-2 predicted that the mutations F5S, V7A, F84L, N148Y, and E210K were benign, all of these mutations are associated with typical LD (Gomez-Garre et al., 2000; Salar et al., 2012; Singh et al., 2008), with the exception of F5S for which no clinical information has been published (http://projects.tcag.ca/lafora/) (Ianzano et al., 2005). Both SDM and CUPSAT predicted that most mutations would be destabilizing (Table 2). However, a positive ΔΔG (increased stability) for W32G, Y294N, and P301L was predicted by at least one program. These predictions conflict with our experimental data showing that each of these mutations is destabilizing (Raththagala et al., 2015). Strikingly, there was no obvious clustering of mutations or consensus predictions among the 3 in silico tools (Figure 1B).
Table 2.
Predicted pathogenicity of selected EPM2A missense mutations
| PolyPhen-2 |
CUPSAT |
SDM |
|||||
|---|---|---|---|---|---|---|---|
| Probability score | Prediction | ΔΔG (kcal/mol) | Predicted overall stability | ΔΔG (kcal/mol) | Outcome | ||
| F5S | Lafora Epilepsy Mutation Database(Ianzano et al., 2005) | 0.198 | Benign | −4.045 | Destabilizing | −2.96 | Reduced stability |
| V7A | Salar et al. (2012) | 0.092 | Benign | −6.71 | Destabilizing | −2.71 | Reduced stability |
| S25P | Ganesh et al. (2002) | 1 | Probably damaging | 0.56 | Stabilizing | −0.78 | Reduced stability |
| E28K | (Minassian et al., 2000) Current report |
1 | Probably damaging | −0.76 | Destabilizing | −1.09 | Reduced stability |
| W32G | (Minassian et al., 2000) Current report |
1 | Probably damaging | −0.595 | Destabilizing | 2.76 | Increased stability |
| E56K | Lesca et al. (2010) | 1 | probably damaging | 2.605 | Stabilizing | −0.87 | Reduced stability |
| F84L | Gomez-Garre et al. (2000) | 0.003 | Benign | −2.705 | Destabilizing | −1.89 | Reduced stability |
| Y86D | Jara-Prado et al. (2014) | 0.863 | Possibly damaging | −1.68 | Destabilizing | −2.77 | Reduced stability |
| K87T | Lesca et al. (2010) | 1 | Probably damaging | −0.26 | Destabilizing | −0.63 | Reduced stability |
| R91P | Ianzano et al. (2004) | 0.988 | Probably damaging | −0.145 | Destabilizing | −1.26 | Reduced stability |
| R108C | Ganesh et al. (2002) and Minassian et al. (2000) | 1 | Probably damaging | 1.31 | Stabilizing | −0.28 | Reduced stability |
| K140N | Singh et al. (2008) | 0.915 | Possibly damaging | −1.975 | Destabilizing | −0.86 | Reduced stability |
| N148Y | Singh et al. (2008) | 0.085 | Benign | 0 | No effect | 1.21 | Increased stability |
| T187A | Ki et al. (2003) | 0.999 | Probably damaging | −1.06 | Destabilizing | 0.06 | Increased stability |
| A188G | Singh and Ganesh (2009) | 1 | Probably damaging | −5.965 | Destabilizing | −0.92 | Reduced stability |
| E210K | Singh et al. (2008) | 0.381 | Benign | 0.28 | Stabilizing | −0.04 | Reduced stability |
| P211L∗ | Lafora Epilepsy Mutation Database (Ianzano et al., 2005) | 0.867 | Possibly damaging | −1.74 | Destabilizing | −0.58 | Reduced stability |
| E224I | Ganesh et al. (2002) | 0.812 | Possibly damaging | 0.585 | Stabilizing | −0.57 | Reduced stability |
| G240S | Gomez-Garre et al. (2000) | 0.815 | Possibly damaging | 1.11 | Stabilizing | −0.01 | Reduced stability |
| P246A | Singh and Ganesh (2009) | 0.985 | Probably damaging | −2.71 | Destabilizing | 2.19 | Increased stability |
| G279C∗ | (Jara-Prado et al., 2014) Current report |
1 | Probably damaging | −1.76 | Destabilizing | −0.01 | Reduced stability |
| G279S∗ | Franceschetti et al. (2006), Gomez-Garre et al. (2000), Minassian et al. (2000) and Serratosa et al. (1999) | 1 | Probably damaging | −1.73 | Destabilizing | −2.32 | Reduced stability |
| Y294N | Gomez-Garre et al. (2000) and Serratosa et al. (1999) | 0.999 | Probably damaging | 1.44 | Stabilizing | −2.3 | Reduced stability |
| P301L | Gomez-Garre et al. (2000) | 1 | Probably damaging | −4.335 | Destabilizing | 1.26 | Increased stability |
| L310W | Singh et al. (2008) | 1 | Probably damaging | −2.275 | Destabilizing | −1.24 | Reduced stability |
| F321C∗∗ | Lynch et al. (2016) | 1 | Probably damaging | −2.5 | Destabilizing | −0.19 | Reduced stability |
Mutations explicitly associated with a slow or late-onset phenotype in the compound heterozygous state are indicated by (∗) or in the homozygous state by (∗∗), according to the Lafora Epilepsy Mutation Database (http://projects.tcag.ca/lafora/), the indicated reference, or the current study. For CUPSAT results, ΔΔG was predicted for residues in both subunits of the laforin structure (chains A and C from PDB:4rkk); values shown represent an average. Green indicates benign or stabilizing (≥0 in CUPSAT or SDM); orange indicates possibly damaging or destabilizing (between −2 and 0 in CUPSAT or SDM); red indicates probably damaging or destabilizing (<−2 in CUPSAT or SDM).
These analyses demonstrated the need for an alternative strategy to understand laforin mutants in LD patients. Therefore, we designed an experimental pipeline to empirically characterize the range of effects of laforin missense mutations in vitro (Figure 1C). Protein stability, carbohydrate binding, and conformational dynamics were measured using purified proteins, and functional interactions with partner proteins were determined by co-expression in yeast.
Effect of laforin missense mutations on stability and substrate binding
Laforin is a constitutive dimer with each subunit containing a carbohydrate binding module (CBM) and a dual specificity phosphatase (DSP) domain (Figure 2A) (Raththagala et al., 2015). Disease-associated missense mutations fall into all 4 regions of the laforin structure: the CBM, the DSP domain, the intra-subunit interface between the CBM and the DSP domain, and the inter-subunit interface between the dimers (Figure 2B). Of the selected mutants, 22/26 could be purified and were subsequently analyzed. The exceptions were F5S, Y86D, and R108C, affecting core residues of the CBM, and T187A of the DSP domain, none of which could be expressed as soluble proteins. The stability of each purified mutant was determined by differential scanning fluorimetry (DSF). Since short (7 glucosyl units, DP7) or long (24 glucosyl units, DP24) oligosaccharides stabilize laforin, binding of glucans to laforin can also be assessed by DSF (Raththagala et al., 2015). We performed DSF on WT laforin and mutants first in the absence of glucan to establish a baseline thermal stability, and then in the presence of either DP7 or DP24 to measure glucan binding.
Figure 2.

Stability, carbohydrate binding and phosphatase activity of LD mutants
(A) Surface structure of laforin bound to maltohexaose, i.e. DP6 (PDB:4rkk) (Raththagala et al., 2015). One homodimer is shown with the CBM and DSP domain of one subunit shown in light and dark shades of blue, respectively. Maltohexaose molecules, shown in green, were bound to the CBM and DSP domain in each subunit of the dimer.
(B) Ribbon diagram of one subunit of the laforin dimer. Residues affected by missense mutations are shown in orange. Phosphate bound to the active site is shown in purple, and glucans bound to the CBM are shown in green. Mutations in the CBM are boxed in pink, those in the CBM-DSP domain interface in yellow, those in the DSP domain in green, and those in the dimer interface in purple.
(C) Mutant stability measured by melting temperature (Tm). For mutants displaying biphasic melting, the first (primary) peak is represented by the filled gray bar; the empty bar indicates the approximate Tm corresponding to the second peak.
(D) The difference in ΔTm displayed for each mutant in the presence of 10 mM DP7 compared to 10 mM DP24, indicated as ΔΔTm.
(E) Specific activity of laforin mutants with the indicated substrates. Activity of each mutant was normalized to WT activity. In (C), (D) and (E), all assays were performed in triplicate, graphs represent the average ± SD, and WT levels are indicated with a dashed line.
(F–J) Correlation scatterplots of Tm versus glycogen specific activity (r = −0.005929, p = 0.9786); C3-P and C6-P starch specific activity (r = 0.2141, p = 0.3266 for C3-P; r = 0.09289, p = 0.6734 for C6-P); PolyPhen-2 score (r = −0.4068, p = 0.0603), and CUPSAT (r = 0.2033, p = 0.3641) and SDM (r = 0.3972, p = 0.0672) predictions for ΔΔG (kcal/mol). Also see Figure S1.
WT laforin exhibited a single melting transition and a melting temperature (Tm) of 49.6°C (Figures 2C and S1A). DP7 and DP24 stabilized WT laforin (ΔTm) by 4.6 and 7.8°C, respectively (Figure S1B). The 3.2°C difference between DP7 and DP24 binding (ΔΔTm) reflected the established preference of laforin for long glucan chains (Figure 2D) (Chan et al., 2004; Raththagala et al., 2015). Laforin W32G and K87T were slightly destabilized with Tm values of 44–45°C (Figure 2C), but these mutants showed minimal shifts of <1°C in the presence of glucans, indicating that these were stable but had virtually no glucan binding (Figures 2D and S1B). These results are consistent with direct glucan engagement by W32 and K87 in the laforin structure (Raththagala et al., 2015).
Mutants with alterations of CBM core residues (V7A, E28K, F84L, or R91P) and at the CBM-DSP interface (E56K, Y294N, or P301L) were highly destabilized with Tm values of 40°C or less (Figure 2C). Notably, some of these mutants (E28K, R91P, Y294N, and P301L) displayed a biphasic melting profile with a second Tm appearing between 45 and 53°C (Figures 2C and S1A). Furthermore, the mutants with biphasic transitions had an initial Tm of less than 37°C (Figure 2C), indicating they are at a high risk for unfolding at physiological temperatures. Compared to WT laforin, CBM core mutants or CBM-DSP interface mutants exhibited a greater change in Tm upon addition of glucan, suggesting enhanced substrate-induced stabilization (Figures 2D and S1B). However, the absolute Tm values achieved by these mutants in the presence of DP7 and DP24 was not higher than WT laforin (Figure S1B), indicating the observed increase in ΔΔTm is likely to be compensatory due to the inherent instability of these mutants. With the exception of W32G and K87T, in which glucan-binding residues were affected, all of the CBM and CBM-DSP mutants displayed a preference for DP24 like WT laforin (Figure 2D). S25P was the only CBM mutant with no change in stability or binding compared to WT laforin (Figures 2C, 2D, S1A, and S1B).
Mutations of buried DSP domain residues (G279C, G279S, and L310W) resulted in destabilized proteins with Tm values of 36–41°C (Figure 2C). In contrast, other DSP domain mutations (K140N, N148Y, A188G, G240S, E210K, P211L, E224I, or G240S) had little effect on protein stability (Figures 2C and S1A). All of the DSP mutants were still bound to DP7 and DP24 and displayed a preference for long glucans (Figures 2D and S1B). We previously demonstrated that WT laforin is a dimer in solution, that F321C is monomeric, and that dimerization is linked to the preferential binding of laforin for long glucans (Raththagala et al., 2015; Sharma et al., 2018). Here, we found that F321C, affecting the dimer interface, had a moderately destabilizing effect (Figures 2C and S1B), and this mutant displayed no preferential binding to DP24, as indicated by ΔΔTm values of <1°C (Figures 2D and S1B).
Activity of laforin missense mutants
We tested the phosphatase activity of the mutants using multiple substrates. Although the natural biological substrate is glycogen, laforin preferentially binds to glucans with long chains in vitro and in vivo (Chan et al., 2004; Raththagala et al., 2015; Wang and Roach, 2004). Furthermore, LBs resemble plant starch in that they are insoluble and contain more phosphate and longer chains than are present in soluble glycogen (Brewer et al., 2019b, 2020). Glycogen, LBs, and starch are phosphorylated at the C3 and C6 hydroxyls of glucosyl units (DePaoli-Roach et al., 2014; Nitschke et al., 2013; Ritte et al., 2006). Site-specificity assays indicate that laforin dephosphorylates both positions with a slight preference for C6-phosphate (C6-P) (Meekins et al., 2016). Thus, we evaluated the activity of WT laforin and the mutants against glycogen, C3-P-labeled starch, and C6-labeled starch (Figures 2E, S1C, and S1D).
Most of the mutants had similar activity or within 50% of WT laforin against all of the substrates (Figures 2E, S1C, and S1D). Y294N had slightly increased activity toward glycogen compared to WT laforin. Despite binding glucans similarly to WT laforin (Figure 2D), S25P had reduced activity toward all substrates tested (Figures 2E, S1C, and S1D). Consistent with their impaired glucan binding (Figure 2D), W32G and K87T had significantly impaired phosphatase activity toward all substrates (Figures 2E, S1C, and S1D). P301L and F321C were notable for their profoundly imbalanced activity toward the different substrates (Figures 2E, S1C, and S1D). P301L had almost no activity toward C6-P, but retained partial activity toward C3-P and glycogen. F321C only exhibited activity toward glycogen. The glucan specificity data (Figure 2D) and activity data (Figure 2E) indicated that F321C had reduced interactions with and activity toward LB-like substrates.
Thermal stability and phosphatase activities were not correlated (Figures 2F and 2G). Furthermore, when we compared the Tm of the purified mutants with the in silico predictions, no correlation between Tm and PolyPhen-2 score or the ΔΔG predicted with CUPSAT or SDM was detected (Figures 2H–2J), further emphasizing the need for experimental approaches to define this system.
Mutation-induced decoupling of the CBM and DSP domain
WT laforin melts with a single sharp peak (Figure S1A). Most laforin mutants displayed a melting profile of similar shape, even when the curve shifted left due to reduced stability. The biphasic transitions observed in E28K, R91P, Y294N, and P301L suggested decoupling between the CBM and DSP domain in these mutants. To test this hypothesis, we purified these regions of the protein separately. The Tm value of the CBM (38.2°C) and of the DSP domain (51.7°C) (Figure 3A), were similar to the Tm values of the prominent P301L transitions (31.8 and 52.5°C, respectively) (Figures 2C and S1A), supporting our hypothesis. When the CBM and DSP domain were incubated with increasing concentrations of DP7, the CBM exhibited a similar binding curve to that of full-length laforin, whereas the DSP domain did not exhibit binding even at high concentrations of DP7 (Figure 3B). These results confirmed that the CBM is necessary and sufficient for carbohydrate binding.
Figure 3.

Stability and glucan binding of WT laforin, the purified CBM and DSP domain, and mutants with biphasic melting profiles
(A) Thermal stability of the individual CBM or DSP domain compared to that of full-length laforin (FL).
(B) Binding of full-length laforin, the CBM, and the DSP domain to DP7 as measured by the change in thermal stability. In (A) and (B) bar graphs represent the average of triplicate reactions ± SD.
(C) Melting curves of WT laforin and Y294N in the presence of increasing concentrations of glycogen. Curves are representative of triplicate reactions.
(D) Residues in the CBM core and CBM-DSP interface affected by missense mutations are shown in orange. Glucan is shown in green.
Glycogen did not affect the shape of the melting curve of WT laforin (Figure 3C). However, the melting curve of the decoupled mutant Y294N gradually shifted to a single transition with increasing concentrations of glycogen (Figure 3C). This phenomenon was also observed with DP7 for the other decoupled mutants (Figure S1A), suggesting that glucan binding restores the interaction between the CBM and DSP domain in these mutants. The importance of such intramolecular interactions is supported by the finding that the CBM core and CBM-DSP interface are “hotspots” for laforin missense mutations and that many affect solvent-inaccessible residues (Figure 3D).
Solution dynamics of LD mutants determined by hydrogen-deuterium exchange (HDX)
The biochemical effects underlying slow or late-onset cases of LD could not be understood from stability, binding, or phosphatase activity measurements of the associated mutants. To further understand the structural and functional perturbations that may alter laforin behavior, we performed hydrogen deuterium exchange (HDX) experiments on a subset of LD mutants. HDX reveals the dynamics of protein conformational states in solution by quantifying exposure to a deuterated solvent over time. We selected key mutants associated with a range of biochemical activities and clinical outcomes: R91P (associated with typical LD; displayed biphasic melting, severe destabilization, and no change in activity), G279C (associated with a slower disease progression; destabilized with little change in glucan binding or activity), P211L (associated with a slower disease progression; no change in stability or glucan binding, and little change in specific activity), and F321C (associated with very late-onset LD; destabilized with altered specificity for and activity toward long glucan substrates). For WT laforin and all 4 mutants, we achieved 100% peptide coverage with slight differences in the pattern of pepsin digestion among them (Figure S2). Residues with an average deuteration change of 10% or more were mapped onto the laforin structure (Figure 4).
Figure 4.

HDX of select LD mutants
(A–D) (Left) Deuteration incorporation for each residue of the indicated mutant was determined from deuteration of overlapping peptides and compared with that of WT laforin. The optimized value for change in deuteration compared to WT laforin is plotted. Positive changes indicate an increase in solvent accessibility in the mutant; negative changes indicate a decrease in solvent accessibility in the mutant. The significance thresholds that were used for mapping significantly changed peptides onto the laforin crystal structure are marked by orange (10%) and red (20%) lines. The CBM and major motifs of the DSP domain, the recognition domain, V-loop, D-loop, PTP-loop, and R-motif, are indicated. (Right) Deuteration changes induced by the mutations are mapped onto one subunit of the laforin structure. Gray indicates a maximum change ≤10% from WT laforin, orange a change between 11 and 20% and red a change >20%. The side chain of the mutated residue is shown in stick representation and the residue is labeled. Structural features are labeled in black. Also see Figure S2, Datas S1, and S2.
R91P, a mutant with CBM-DSP decoupling, yielded increases in deuteration exceeding 10% throughout the CBM (Figure 4A). P211L caused small changes in deuteration with only residues 200–216 in the V-loop of the DSP domain, including P211, exceeding 10% (Figure 4B). G279C caused deuteration increases throughout the DSP domain (Figure 4C): Increased solvent accessibility was observed in the D loop, R-motif, and helices α8 and α11 of the DSP domain, and α10 of the CBM-DSP interface. F321C affected similar regions of the DSP domain as did G279C but to a greater extent, both in terms of the number of residues and the difference compared to WT laforin (Figure 4D). Unlike G279C, F321C had increased deuteration in the catalytic PTP-loop and the adjacent α9 helix also exhibited significantly increased deuteration. The greater solvent accessibility at the DSP–DSP dimer interface is consistent with the loss of dimerization in F321C (Sharma et al., 2018).
Effects of LD mutations on protein-protein interactions
Our results indicated that some LD mutants are pathogenic because they impair glucan binding (W32G and K87T) or are unstable (E28K, R91P, Y294N, and P301L). F321C only impairs preferential binding and activity toward long glucans and is associated with very late onset (Lynch et al., 2016). However, many LD mutants maintained phosphatase activity and displayed little to no destabilization, indicating there are additional disease-relevant functions of laforin. Laforin interacts with multiple proteins involved in glycogen metabolism. Therefore, to understand the effects of patient mutations on laforin interactions in a cellular context, we assessed the impact of these mutations on the interaction with other laforin-binding partners.
Malin and PTG (protein targeting to glycogen) are well-characterized laforin-binding partners (Fernandez-Sanchez et al., 2003; Gentry et al., 2005; Lohi et al., 2005; Roma-Mateo et al., 2011; Sanchez-Martin et al., 2015; Worby et al., 2008). To measure their interaction with WT and mutant laforin, we used a directed yeast two-hybrid assay (Gentry et al., 2005; Lohi et al., 2005; Roma-Mateo et al., 2011; Solaz-Fuster et al., 2008; Vernia et al., 2009). WT laforin and mutants were expressed as bait proteins fused with the DNA-binding protein LexA. WT malin and PTG were expressed as prey fused with the Gal4 activation domain (GAD). β-galactosidase (β-gal) activity was used to quantify interaction between bait and prey.
Five laforin mutations (S25P, W32G, R91P, Y294N, and P301L) abolished the interaction with either PTG, malin, or both and another 8 resulted in a reduction of 50% or greater (Figure 5A), highlighting the importance of laforin as a glycogen-associated protein scaffold. Mutations W32G or K87T at the CBM glucan-binding site reduced the PTG interaction without impairing the interaction with malin (Figure 5A). In contrast, F321C specifically impaired the malin interaction (Figure 5A). For S25P, which retained some activity toward glycogen and starch and had stability and glucan-interaction preference comparable to WT laforin (Figures 2C–2E), the interaction with both malin and PTG was abolished (Figure 5A).
Figure 5.

Effects of LD mutations on the interaction of laforin with malin and PTG
(A) Interaction of WT laforin and mutants with GAD, GAD-PTG, and GAD-malin fusion proteins. The dotted line indicates WT level of malin interaction, and the dashed line indicates WT level of PTG interaction.
(B) Triple hybrid experiment demonstrating noncompetitive binding of malin and PTG to laforin. LexA-laforin was expressed with GAD-malin and pWS-PTG or with GAD-PTG and pWS-malin. In (A) and (B) bar graphs represent the average of triplicate reactions ±SD.
(C–G) Correlation between Tm and GAD-malin interaction (r = 0.6087, p = 0.0021), Tm and GAD-PTG interaction (r = 0.499, p = 0.0154), GAD-malin and GAD-PTG interactions (r = 0.6462, p = 0.0009), glycogen activity and GAD-malin interaction (r = 0.01087, p = 0.9607), and glycogen activity and GAD-PTG interaction (r = 0.3646, p = 0.0872). In e, inset corresponds to the boxed region. Dot color corresponds to mutation location. See Figure S3 for the full 2D correlation analysis and Table S1 for all correlation coefficients and p values.
We noticed a general pattern to these interaction data: mutations at the CBM glucan binding site specifically affected the PTG interaction, mutations at the dimer interface specifically affected the malin interaction, and centrally located mutations affected both interactions. Therefore, we hypothesized that the binding sites for PTG and malin are spatially distinct. This arrangement is logical because PTG is a laforin-dependent substrate for the E3 ubiquitin ligase activity of malin (Worby et al., 2008). Therefore, both PTG and malin likely interact with laforin at the same time (Worby et al., 2008). We tested this hypothesis by performing yeast triple hybrid experiments in which laforin was expressed in the presence of both malin and PTG. One partner was expressed as prey fused to GAD, and the other was expressed with only a hemagglutinin A (HA) tag. If the two binding partners had overlapping binding sites, the HA-tagged partner would compete with the other, resulting in reduced β-gal activity. No difference in β-gal activity was observed when HA-tagged PTG was expressed in addition to LexA-laforin and GAD-malin or when HA-tagged malin was expressed with LexA-laforin and GAD-PTG (Figure 5B). These data indicated that PTG and malin have independent binding sites on laforin. Collectively, the results indicated that these binding sites are differentially affected by LD missense mutations because of local structural perturbations.
Correlation of defects in laforin
To determine whether the measured effects of the mutations on laforin biochemistry or function were correlated, we performed pairwise correlation analyses between Tm, ΔΔTm, specific activities, PTG interaction, and malin interaction measurements (Figure S3; Table S1). Stability (Tm) was significantly positively correlated with the malin interaction (Figure 5C, p = 0.0021) and with the PTG interaction (Figure 5D, p = 0.0154). A strong positive correlation was identified between PTG and malin interaction (Figure 5E, p = 0.0009). However, no significant correlation was found between glycogen specific activity and the malin or PTG interaction (Figures 5F and 5G). As expected, strong correlations were found between glycogen and starch activities, and between C6-P starch activity and ΔΔTm, because starch dephosphorylation requires substrate specificity for which ΔΔTm is a proxy (Figure S3; Table S1).
In conclusion, these data illustrated that LD mutants primarily affect the ability of laforin to bind to glucan substrates or protein-binding partners. LD mutations induce unique structural perturbations that affect the ability of laforin to bind to interacting partners, such as malin and PTG. In addition, mutants appeared to group into particular patterns of functional effects based on the location of the mutation within the structure.
Discussion
Here, we presented four LD clinical cases with different outcomes and defined the molecular defects of EPM2A missense mutations affecting laforin function. Genotype–phenotype correlations in the LD population are extremely difficult due to the small number of patients, high allelic heterogeneity, frequent under-diagnosis, and limited clinical data. We employed an integrated experimental pipeline and defined mutation pathogenicity for 26 patient mutations using biochemical methods.
Functional classes of EPM2A missense mutations
Using the biochemical and functional properties of the laforin mutants that we studied, we propose a classification of laforin mutants to predict their pathogenecity (Table 3). Class I mutations directly affect CBM carbohydrate binding. The CBM is essential for carbohydrate binding and W32 and K87 are primary residues involved in glucan interaction. Class I mutations are only slightly destabilizing to the laforin structure, and their phosphatase activity is greatly reduced because of impaired substrate affinity. These mutations also compromise the laforin interaction with PTG. Although both W32G and K87T still interact with malin, they cannot bind glycogen and thus cannot target malin to glycogen-associated substrates. Based on our biochemical results, these mutations are highly detrimental to all aspects of laforin function; consequently, they lead to a severe phenotype with rapid progression, as we observed in patients 2 and 3 (Table 1).
Table 3.
Classification of LD-causing EPM2A missense mutations based on biochemical and clinical data
| Functional class | Structural group | Mutations | General effects | Predicted pathogenicity |
|---|---|---|---|---|
| Class I | CBM | W32G K87T |
Severely impaired carbohydrate binding and phosphatase activity; decreased PTG interaction | Severe |
| Class IIa | CBM | (F5S) V7A E28K F84L (Y86D) F88L R91P (R108C) |
Destabilized and decoupled CBM and DSP domain; mild or moderate effects on phosphatase activity; severely impaired PTG and malin interaction. | Severe |
| Class IIb | CBM-DSP interface | E56K Y294N P301L |
||
| Class III | DSP domain | N148Y N163D∗ (T187A) A188G L310W G279S∗ G279C∗ |
Destabilized; variable effects on phosphatase activity; impaired malin and PTG interaction. | Variable |
| Class IV | DSP domain (V-loop) | E210K P211L∗ E224I |
Slightly destabilized; slight decrease in activity; slight or no decrease in malin and PTG interaction. | Moderate |
| Class V | Dimer interface | F321C∗∗ | Destabilized; mild effects on phosphatase activity; impaired malin interaction; loss of dimerization and preferential binding to long oligosaccharides | Mild |
| Unknown | Various | S25P K140N G240S P246A |
Mild or no effect on stability or activity. S25P displays no interaction with malin or PTG. |
Unknown |
Asterisk(s) indicates this mutation has been associated with a milder phenotype, either in the compound heterozygous (∗) or homozygous (∗∗) state. Mutants in parentheses are likely classifications but were not fully characterized due to their insolubility in E. coli. N163D was previously characterized (Garcia-Gimeno et al., 2018).
Class IIa mutations affect the CBM core and Class IIb mutations affect the CBM-DSP interface (Table 3). Although structurally distinct, Class IIa and IIb mutations produce similar biochemical defects: they destabilize laforin and impair interactions with both PTG and malin. Most of these mutations have little or no effect on phosphatase activity, with the exception of P301L, which may have compromised catalytic activity due to its severe instability. Patients either homozygous or compound heterozygous for Class II mutations have been reported to exhibit a classic clinical course, such as patient 1 in the present report, who was compound heterozygous for the E28K mutation. Class II mutations may also be associated with early-onset LD because of their severe impairment of laforin stability and binding to other proteins. A family of three children all homozygous for R108C experienced seizure onset at ages 5, 8, and 10 with early onset learning disability that manifested at 4 years old (Ganesh et al., 2002). Another patient homozygous for Y86D displayed early onset learning problems (Jara-Prado et al., 2014). Y86D and R108C were two of the four mutations that could not be expressed and purified from E. coli as soluble proteins, strongly suggesting that Y86D and R108C are severely destabilizing mutations. Based on their location within the CBM, we predict that these mutants are likely to have similar biochemical effects and clinical outcomes as other Class IIa mutants.
Class III mutations affect the DSP domain (Table 3) and have variable effects on laforin stability and on PTG and malin interactions. All Class III mutations have mild or moderate effects on phosphatase activity, do not affect carbohydrate binding, and impair the interaction between laforin and PTG and malin. For LD patients, Class III mutations are associated with variable disease progression (Ki et al., 2003; Singh et al., 2008). We report that G279C is associated with a slower clinical course (patient 4, Table 1), which is consistent with previous reports of patients carrying the G279C or G279S mutation. One patient (G279C/R241X) did not present with seizures until the age of 21 years old, and cognitive impairment did not develop until the patient was 24 years old (Jara-Prado et al., 2014). Another patient (G279S/R241X) had seizure onset at age 17 but lived beyond the age of 40 (Ferlazzo et al., 2014). Because Class III mutants retained some interaction with malin and PTG and show no major impairment in glucan interaction, they may delay LD onset. We published a study on a compound heterozygous patient carrying the missense mutation N163D and the nonsense mutation Y112X (Garcia-Gimeno et al., 2018). This patient had her first seizure at 16 and at 28 years old she was still very cognitively engaged and able to walk. In that study, we reported that N163D had no effect on laforin stability, carbohydrate binding, or phosphatase activity but impaired the interaction of laforin with its binding partners. N163 is in the DSP domain and the biochemical profile of N163D (Garcia-Gimeno et al., 2018) is consistent with other mutants in Class III (Table 3). Therefore, mutations in class III can produce both atypically moderate and typically severe phenotypes.
Class IV mutations affect a surface-exposed region of the V-loop adjacent to the dimer interface (Table 3). These mutations display no major defects in laforin stability, carbohydrate binding, phosphatase activity, or interaction with PTG or malin. However, laforin interacts with additional proteins, including AMP-activated protein kinase (AMPK), glycogen synthase, and R6, another regulatory subunit of protein phosphatase 1 (PP1) (Gentry et al., 2009; Moreno et al., 2010; Rubio-Villena et al., 2013; Vilchez et al., 2007). This region is likely important for binding to one or more of these additional proteins. Because class IV mutations have a moderate effect on laforin function related to glycogen, they may be generally associated with a less severe disease progression.
Class V mutations affect laforin dimerization and therefore substrate specificity. F321C renders laforin monomeric, and impaired dimerization leads to a loss of specific binding to substrates with long glucans like LBs (Raththagala et al., 2015; Sharma et al., 2018). Although we only characterized a single class V mutant, we predict the following: the reduced stability of F321C is likely a result of dimerization loss, and these mutations do not affect the interaction of laforin with PTG, but they reduce the malin interaction. F321C is associated with a homozygous case of extremely mild LD in which the patient had a history of seizures that were relatively controlled with sodium valproate and lived to the age of 56 (Lynch et al., 2016). The biochemical effects of F321C and the extremely unusual clinical outcome are so unique that a separate class is merited, but the class could easily expand to include other mutants with similar effects.
Although these classes explain the defect induced by most of the patient mutations analyzed, no clear defect was identified in K140N, G240S, P246A, and the reason for the impaired interaction of S25P with malin and PTG is unclear. Thus, there are additional unidentified factors causing pathogenicity, and these mutants have not yet been classified. It is possible that some pathogenic mutations alter post-translational modifications of laforin. An in vitro study showed that laforin is phosphorylated at S25 by AMPK, and this phosphorylation enhances the malin-laforin interaction in yeast (Roma-Mateo et al., 2011). This result could explain the absence of both malin and PTG interaction in our study, but the hypothesis requires further validation. There is strong in vitro evidence that laforin is ubiquitinated by malin (Gentry et al., 2005; Lohi et al., 2005; Sanchez-Martin et al., 2015). Although the specific lysine residue(s) that are ubiquitinated have not been identified, only 3 of the 11 laforin lysine residues are surface exposed: K140, K219, and K323. It is possible that K140 is a primary ubiquitination site and that impaired laforin ubiquitination leads to LD by a yet unknown mechanism. G240S and P246A are centrally located in the DSP domain between the active site and dimer interface. These mutations have little to no effect on stability, glucan binding or interaction with PTG or malin; therefore, like Class IV mutations, they may affect interactions with other laforin binding partners.
Late-onset LD and genetic effects
An obvious additional complicating factor for predicting clinical progression is that many patients are compound heterozygotes, carrying two different chromosomal aberrations that affect the same gene. Because LD is a recessive disease, it is likely that a disease threshold exists for decreased laforin function (Figure 6A). Our data indicated that the laforin function most relevant to LD progression is its ability to coordinately bind glycogen, malin, and other glycogen-associated proteins. When laforin function is above a specific threshold then individuals are completely healthy, for example in parents heterozygous for LD mutations. Below this threshold, there is likely a gradation of how rapidly patients progress down a clinical LD path. Both the type of mutation and whether the mutation is in the homozygous or heterozygous state influences disease progression. For example, the slowest onset form of LD associated with an EPM2A mutation was reported in a patient homozygous for the Class V mutation F321C (Lynch et al., 2016). In contrast, compound heterozygous patients carrying a class III or IV mutation in addition to a deleterious mutation, such as a nonsense mutation or indel, would experience a clinical course with slower progression than classic LD and yet still more rapid than patients with class V mutations (Garcia-Gimeno et al., 2018; Jara-Prado et al., 2014). The most severe and rapidly progressing LD cases are homozygous or compound heterozygous patients with class I or II mutations that lead to nonfunctional protein. Patients with these mutations are likely indistinguishable from patients with only nonsense mutations, indels, or a combination thereof.
Figure 6.

An empirical pipeline to facilitate LD personalized medicine
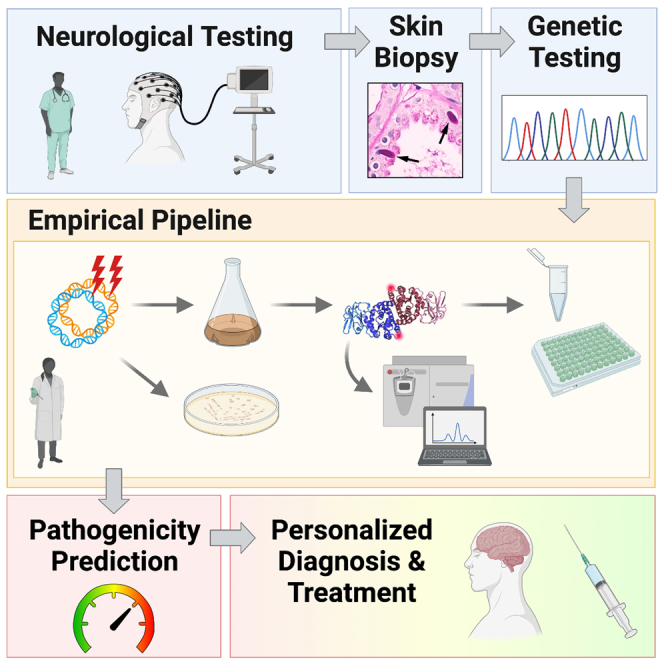
(A) LD progression is dependent on the type of mutation(s) carried by LD patients and where this places them with regard to the disease threshold of laforin function.
(B) A suspected LD diagnosis will be confirmed by genetic testing and classified for prognosis. Skin biopsies should also be performed to facilitate comparisons of LB load between patients. Nonsense, frameshift, or indel mutations in either EPM2A or EPM2B would be complete loss-of-function mutations and the most pathogenic. If known loss-of-function or EPM2A missense mutation(s) are identified, patient progression could be predicted immediately. If new EPM2A missense mutations are identified, mutant proteins could be characterized and classified within a matter of weeks to predict patient-specific clinical progression.
In addition to the class of mutation, modifier effects are certain to play a role in clinical manifestation. These effects could be either genetic or environmental. Patients carrying the same mutations in ethnic isolates show some phenotypic variability (Gomez-Abad et al., 2007), and siblings carrying identical mutations sometimes display differences in disease progression, suggesting a role for other genetic or epigenetic factors (Ganesh et al., 2002; Gomez-Garre et al., 2007; Jara-Prado et al., 2014; Lohi et al., 2007). For example, a PTG variant has been reported to contribute to a slower disease course (Guerrero et al., 2011). Differences in medical care can also influence clinical progression (Minassian et al., 2000). However, a clinically homogeneous patient progression is often reported within families and among genetic isolates (Baykan et al., 2005; Turnbull et al., 2008).
An empirical pipeline for personalized medicine
We are currently expanding our analysis to include all known EPM2A missense mutations. In the future, when genetic testing confirms an LD diagnosis and reveals a previously unidentified EPM2A missense mutation, the effects of the mutation could be quickly biochemically assayed and the mutant classified (Figure 6B). If a known EPM2A missense mutation is detected, no assays would be required because the biochemical profiles will already be known. Skin biopsies should also be performed to supplement the diagnosis and determine whether LB enrichment correlates with disease severity or progression. Although biochemical studies have not yet been performed for EPM2B mutations, case studies indicate specific EPM2B mutations can also cause late-onset or slow LD (Ferlazzo et al., 2014; Lanoiselee et al., 2014). This empirical pipeline would permit clinicians to make a more accurate prognosis for LD patients based on the mutations they carry.
Genetic screening during pregnancy or at birth is already widely employed to identify genetic diseases and chromosomal abnormalities (Green et al., 2004)(). Life-threatening disorders, even those as rare as LD, may soon be added to these early genetic screens and more cases of mild or late-onset LD will be identified. We anticipate that as therapies become available, pre-symptomatic detection of LD would be valuable because early treatment will have a greater chance of ameliorating symptoms or even preventing development of LD. However, benign polymorphisms also need to be correctly differentiated from mild, moderate, and severely pathogenic mutations to prevent a false diagnosis and unnecessarily aggressive treatment. Our data revealed distinct characteristics of pathogenic mutations, differentiated mild from severe mutations, and may facilitate differentiation of benign polymorphisms from pathogenic mutations to prevent false diagnosis.
Preclinical studies of LD therapeutics are currently underway with multiple lines of treatment in development (Brewer and Gentry, 2018; Brewer et al., 2019a; Gentry et al., 2020). The first published therapeutic strategy uses an antibody-enzyme fusion that degrades LBs, the toxic carbohydrate aggregates that cause LD (Austin et al., 2019; Brewer et al., 2019b; Zhou et al., 2019). This drug is administered directly into the central nervous system, eliminating brain LBs and correcting the cerebral metabolic phenotype in LD mouse models. Another promising treatment in development involves antisense oligonucleotides to reduce glycogen synthase, which halted LB formation in a preliminary mouse study (Brewer and Gentry, 2018). A third strategy uses small molecules to inhibit glycogen synthase activity (Tang et al., 2020). A final strategy consists of repurposing approved drugs, such as metformin, that have shown improved outcomes in mouse models (Sanchez-Elexpuru et al., 2017). With the large number of new mutations arising in new LD patients, our biochemical pipeline will be extremely useful for providing patients with a personalized diagnosis and treatment strategy once therapies become clinically available.
Limitations of the study
Although the present study includes some clinical detail from four LD patients, a natural history study of a larger group of patients (NCT03876522) is currently underway that includes an analysis of electroencephalographic (EEG) features, neurological testing, skin biopsy, and other phenotypic factors. Additional clinical information from patients with EPM2A mutations will increase the accuracy with which predictions of disease progression can be made from our empirical pipeline. This pipeline is an in vitro system utilizing recombinant proteins purified from E. coli and expressed in yeast. Future studies in mammalian cell culture and patient-derived cells will be used to further validate the effects of laforin missense mutations in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| OneShot BL21(DE3) Chemically Completent E. coli | ThermoFisher | C600003 |
| Biological samples | ||
| Maltoheptaose (DP7) | Elicityl | GLU317 |
| Maltodextrin (DP24) | Elicityl | GLU310 |
| Glycogen from rabbit muscle | Gift from Anna-DePaoli-Roach (Tagliabracci et al., 2007) | |
| Glycogen from rabbit liver | Sigma | G8876 |
| phosphate-free Arabidopsis (sex1-3) starch | Gift from Diana Santelia (Meekins et al., 2015) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Invitrogen SYPRO Orange Protein Gel Stain | ThermoFisher | S6650 |
| Critical commercial assays | ||
| QuikChange Lightning Site-Directed Mutagenesis Kit |
Agilent | 210519 |
| Q5 Site-Directed Mutagenesis | New England BioLabs | E0554 |
| Pi ColorLock Gold Phosphate Detection system | Novus Biologicals | 30-301-25 |
| Recombinant DNA | ||
| pEG202 | Pascual Sanz Lab (Garcia-Gimeno et al., 2018) | NovoPro Labs #V010323 |
| pET28b+ | Gift from Jack Dixon Lab (Raththagala et al., 2015) | Sigma cat #69865 |
| pACT2 | Pascual Sanz Lab (Garcia-Gimeno et al., 2018) | ClonTech (discontinued) |
| Software and algorithms | ||
| Proteome Discoverer software v1.3 | Thermo Scientific | N/A |
| HDXaminer | Sierra Analytics | N/A |
| PyMol 2.0 | Schrödinger | N/A |
| Prism 8 | GraphPad | N/A |
| Other | ||
| [β 33P]-ATP | Hartman Analytic | SCF-327 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew Gentry (matthew.gentry@uky.edu).
Materials availability
Plasmids and purified proteins generated for this study are available from the lead contact upon request.
Method details
Cloning, protein expression, and protein purification
All pET28b and pEG202 laforin mutants were generated by site-directed mutagenesis (QuickChange Lightning, Agilent; Q5 Site-Directed Mutagenesis, New England BioLabs; GENEWIZ custom Site-Directed Mutagenesis). All pET28b mutants were expressed in OneShot BL21(DE3) Chemically Competent E. coli cells (ThermoFisher) and purified using immobilized metal affinity chromatography and a Profinia Purification System (BioRad) and size exclusion chromatography via an ÄKTA fast protein liquid chromatography system (GE Healthcare). Purity of proteins was determined by SDS-polyacrylamide gel electrophoresis (PAGE) with Coomassie staining.
Differential scanning fluorimetry (DSF)
Experiments were performed using a CFX96 Real-Time PCR system (BioRad). Individual reactions contained 2 μM protein and 5X SYPRO Orange Protein Gel Stain (Invitrogen). DP7/maltoheptaose (Elicityl), DP24 maltodextrins (Elicityl), or rabbit liver glycogen (Sigma) were used as substrates in DSF reactions. Melting was monitored from 20 to 90°C at a ramp rate of 1°C/50 sec. Melting temperature (Tm) was calculated from a Gaussian fit of the first derivative of the melting curve. Data analyses and binding fits were determined using the Prism software (Graphpad).
Glycogen dephosphorylation assays
Glycogen purified from rabbit muscle was a gift from Anna De-Paoli Roach (DePaoli-Roach et al., 2014; Tagliabracci et al., 2007). Phosphate release was quantified using the Pi ColorLock Gold Phosphate Detection system (Novus Biologicals), a commercial reagent based on the malachite green assay for detecting inorganic phosphate (Sherwood et al., 2013; Tagliabracci et al., 2007). Assays were performed in 100 μL reactions containing 2.5 μg enzyme, 2 mM DTT, and phosphatase buffer (100 mM sodium acetate, 50 mM bis-Tris, 50 mM Tris-HCl, pH 6.5) at 25°C. Mutants were assayed in the linear range with respect to time, enzyme amount, and substrate concentration. A time course of phosphate release was performed with 5 and 10 mg/ml glycogen to determine the assay length within the linear phase of dephosphorylation with sufficient signal to detect phosphate release, which was consistent with a previous report (Tagliabracci et al., 2007). For specific activity determinations, reactions were performed for 30 minutes with 10 mg/ml glycogen.
Site-specific dephosphorylation assays
Phosphate-free Arabidopsis (sex1-3) starch was a gift from Diana Santelia (ETH Zurich). Radiolabeled starch was prepared in two steps by incubating sex1-3 starch first with purified glucan water dikinase (GWD), which phosphorylates C6 hydroxyls, and phospho-glucan water dikinase (PWD), which phosphorylates C3 hydroxyls (Meekins et al., 2014, 2015; Santelia et al., 2011). Starch was divided into 2 aliquots of 300 mg each: C6-labeled starch was prepared with [β 33P]-ATP (0.5 μCi/mg starch) added during the GWD incubation and only unlabeled ATP in the PWD incubation, and C3-labeled starch was prepared with only unlabeled ATP during the GWD incubation and [β 33P]-ATP (0.5 μCi/mg starch) added during the PWD incubation. [β 33P]-ATP was obtained from Hartman Analytic.
First, both aliquots of starch were incubated with GWD (0.5 μg/mg starch) on a rotator in a with phosphorylation buffer (10 μM ATP, 50 mM HEPES pH 7.5, 1 mM EDTA, 6 mM MgCl2, 0.5% Triton X-100) for 12–16 hours at 25°C; [β 33P]-ATP was added only to the tube for C6-labeling. Reactions were quenched by microcentrifugation (5 minutes at 13,000 rpm), removal of the supernatant, and resuspension of the starch in 10% SDS. Then starch was washed in 2% SDS with 2 mM ATP and then in 0.05% Triton X-100 to remove unbound phosphate. 33P incorporation was counted in triplicate by adding 4 μL of the suspension (0.2 mg starch) to 3 mL of scintillation liquid and measured using a 1900 TR liquid scintillation counter (Packard). In the second step, the starch was incubated with PWD (1 μg/mg starch) in phosphorylation buffer for 12–16 hours at 25°C; [β 33P]-ATP was added only to the tube for C3-labeling. After reactions were quenched in 10% SDS, starch was washed in 2% SDS with 2 mM ATP and then in 0.05% Triton X-100, followed by counting in the scintillation counter. C6- and C3-phosphate incorporation was determined by counts per million (cpm) per mg starch detected after each phosphorylation step.
Dephosphorylation reactions were performed in a volume of 150 μL with 50 ng of enzyme in dephosphorylation buffer (100 mM sodium acetate, 50 mM bis-Tris, 50 mM Tris-HCl, pH 6.5, 0.05% Triton X-100, 1 μg/μL BSA, and 2 mM DTT) and 3 mg/ml of either C6- or C3-labeled starch. After 2.5 minutes on a rotating wheel at 25°C, reactions were quenched by adding 50 μL of 10% SDS, and then microcentrifuged for 5 minutes at 13,000 rpm to pellet the starch. 150 μL of the supernatant was added to 3 mL scintillation liquid, and 33P release was quantified using the scintillation counter.
Yeast two-hybrid assays
For yeast two-hybrid assays, pEG202 laforin encoding a LexA-laforin fusion protein and pACT2, pACT2-malin and pACT2-PTG encoding Gal4 activation domain (GAD) and GAD fusions have been previously described (Garcia-Gimeno et al., 2018; Guerrero et al., 2011; Roma-Mateo et al., 2011; Solaz-Fuster et al., 2008). pWS-malin and pWS-PTG encoding HA-tagged proteins were used for the triple hybrid assay. Saccharomyces cerevisiae were transformed with the indicated plasmids, and transformants were grown in selective SC medium. Extracts were prepared as described previously (Roma-Mateo et al., 2011). Yeast two-hybrid assays were performed by screening transformants for β-galactosidase activity using a filter lift assay (Garcia-Gimeno et al., 2018). The strength of the interaction was determined by measuring β-galactosidase activity in permeabilized yeast cells and expressed in Miller units.
Hydrogen deuterium exchange (HDX)
Quenching conditions for optimal sequence coverage of laforin was previously established as 0.08M GuHCl, 0.1M Glycine, 16.6% Glycerol, pH 2.4 (Raththagala et al., 2015). Functional HDX experiments were initiated by dilution of 3 μl of stock solution (WT laforin or laforin mutants at 1 mg/ml) into 9 μl of D2O buffer (8.3 mM Tris, 150 mM NaCl, pDREAD 7.2) and the mixture was incubated at 0°C. The exchange reactions were quenched at various times (10, 100, 1000, 10,000 and 100,000 sec) by the addition 18 μl of the optimal quench solution, and the quenched samples were flash frozen with dry ice. Un-deuterated and equilibrium-deuterated control samples were also prepared as previously described (Raththagala et al., 2015). All frozen samples were passed over an immobilized pepsin column (16 μl) at a flow rate of 25 μl/min, and digested peptides were collected on a C18 trap column (Optimize Tech, Opti-Trap, 0.2 × 2 mm) for desalting. Peptide separation was performed on a C18 reverse phase column (Agilent, Poroshell 120, 0.3 × 35 mm, 2.7 μl) with a linear gradient of 8–48% B over 30 min (A: 0.05% TFA in H2O; B: 80% acetonitrile, 0.01% TFA, and 20% H2O). Mass spectrometry analysis was performed on the Orbitrap Elite mass spectrometer (Thermo Fisher Sci), adjusted for HDX experiments (Stefely et al., 2015). The resolution of the instrument was set at 120,000 at m/z 400.
Proteome Discoverer software (v1.3, Thermo Scientific) was used to identify the sequence of the digested peptide ions from their MS/MS data. HDXaminer (Sierra Analytics, Modesto, CA) was utilized to confirm the peptide identification and calculate the centroids of isotopic envelopes of all the peptides. The level of deuterium incorporation of each peptide was calculated by applying back-exchange correction (Zhang and Smith, 1993). The ribbon maps (Data S1) were generated from deuteration level of overlapping peptides to improve the resolution of the HDX data, and difference maps (Data S2) show changes in mutants compared to WT laforin.
Structural and statistical analysis
PyMol 2.0 was used for structural analysis and generating molecular graphics (Schrodinger, 2010). All statistical tests were performed using Prism Software (GraphPad). Correlation coefficients were determined using the nonparametric Spearman correlation.
Acknowledgments
The authors thank Nancy R. Gough (BioSerendipity, LLC) for editing support as well as members of the Gentry and Vander Kooi labs for vigorous discussions regarding the data. This work was supported by the National Institutes of Health (P01 NS097197 to M.S.G., J.M.S. and P.S., R35 NS116824 to M.S.G., and F31 NS093892 to M.K.B), National Science Foundation (DBI2018007 and MCB1817414 to M.S.G.), and Epilepsy Foundation New Therapy Commercialization Grant to M.S.G. M.K.B. received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (No. 754510M). This project also received funding from the Spanish Ministry of Science and Innovation (SAF2017-83151-R to P.S. and RTI2018-095784b-100SAF to J.M.S).
Author contributions
M.S.G. and C.W.V.K. conceived the study. M.M.C. and J.S. collected and analyzed clinical data. M.K.B. and J.W. generated mutants. M.K.B., M.S.G., C.W.V.K., Z.S., S.S., and J.W. purified proteins, performed assays, and analyzed data. R.V., M.A.G.G., and P.S. performed Y2H experiments. S.L. performed HDX experiments and analyzed data. M.K.B., C.W.V.K. and M.S.G. analyzed data and wrote the paper.
Declaration of interests
M.S.G. is a consultant for Maze Therapeutics, Enable Therapeutics, Glut1 Deficiency Syndrome Foundation, and Chelsea’s Hope. M.S.G. and C.V.K. are founders of Atterogen, LLC.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103276.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M.D. Novel personalized therapies for cystic fibrosis: treating the basic defect in all patients. J. Intern. Med. 2015;277:155–166. doi: 10.1111/joim.12314. [DOI] [PubMed] [Google Scholar]
- Aslam Z., Lee E., Badshah M., Naeem M., Kang C. Whole exome sequencing identified a novel missense mutation in EPM2A underlying Lafora disease in a Pakistani family. Seizure-Eur. J. Epilepsy. 2017;51:200–203. doi: 10.1016/j.seizure.2017.08.012. [DOI] [PubMed] [Google Scholar]
- Austin G.L., Simmons Z.R., Klier J.E., Rondon A., Hodges B.L., Shaffer R., Aziz N.M., McKnight T.R., Pauly J.R., Armstrong D.D. Central nervous system delivery and biodistribution analysis of an antibody–enzyme fusion for the treatment of Lafora disease. Mol. Pharm. 2019;16:3791–3801. doi: 10.1021/acs.molpharmaceut.9b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baykan B., Striano P., Gianotti S., Bebek N., Gennaro E., Gurses C., Zara F. Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation. Epilepsia. 2005;46:1695–1697. doi: 10.1111/j.1528-1167.2005.00272.x. [DOI] [PubMed] [Google Scholar]
- Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- Brewer M.K., Gentry M.S. The 3rd international Lafora epilepsy workshop: evidence for a cure. Epilepsy Behav. 2018;81:125–127. doi: 10.1016/j.yebeh.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer M.K., Grossman T.R., McKnight T.R., Goldberg Y.P., Landy H., Gentry M.S. The 4th international Lafora epilepsy workshop: shifting paradigms, paths to treatment, and hope for patients. Epilepsy Behav. 2019;90:284–286. doi: 10.1016/j.yebeh.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer M.K., Putaux J.-L., Rondon A., Uittenbogaard A., Sullivan M.A., Gentry M.S. Polyglucosan body structure in Lafora disease. Carbohydr. Polym. 2020;240:116260. doi: 10.1016/j.carbpol.2020.116260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer M.K., Uittenbogaard A., Austin G.L., Segvich D.M., DePaoli-Roach A., Roach P.J., McCarthy J.J., Simmons Z.R., Brandon J.A., Zhou Z. Targeting pathogenic Lafora bodies in Lafora disease using an antibody-enzyme fusion. Cell Metab. 2019;30:689–705. doi: 10.1016/j.cmet.2019.07.002. e686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.M., Ackerley C.A., Lohi H., Ianzano L., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum. Mol. Genet. 2004;13:1117–1129. doi: 10.1093/hmg/ddh130. [DOI] [PubMed] [Google Scholar]
- DePaoli-Roach A.A., Contreras C.J., Segvich D.M., Heiss C., Ishihara M., Azadi P., Roach P.J. Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J. Biol. Chem. 2014;290:841–850. doi: 10.1074/jbc.M114.607796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J., Gruart A., Garcia-Rocha M., Delgado-Garcia J.M., Guinovart J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 2014;23:3147–3156. doi: 10.1093/hmg/ddu024. [DOI] [PubMed] [Google Scholar]
- Ferlazzo E., Canafoglia L., Michelucci R., Gambardella A., Gennaro E., Pasini E., Riguzzi P., Plasmati R., Volpi L., Labate A. Mild Lafora disease: clinical, neurophysiologic, and genetic findings. Epilepsia. 2014;55 doi: 10.1111/epi.12806. e129–133. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sanchez M.E., Criado-Garcia O., Heath K.E., Garcia-Fojeda B., Medrano-Fernandez I., Gomez-Garre P., Sanz P., Serratosa J.M., Rodriguez de Cordoba S. Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum. Mol. Genet. 2003;12:3161–3171. doi: 10.1093/hmg/ddg340. [DOI] [PubMed] [Google Scholar]
- Franceschetti S., Gambardella A., Canafoglia L., Striano P., Lohi H., Gennaro E., Ianzano L., Veggiotti P., Sofia V., Biondi R.J.E. Clinical and genetic findings in 26 Italian patients with Lafora disease. Epilepsia. 2006;47:640–643. doi: 10.1111/j.1528-1167.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- Ganesh S., Delgado-Escueta A.V., Suzuki T., Francheschetti S., Riggio C., Avanzini G., Rabinowicz A., Bohlega S., Bailey J., Alonso M.E. Genotype–phenotype correlations for EPM2A mutations in Lafora's progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype. Hum. Mol. Genet. 2002;11:1263–1271. doi: 10.1093/hmg/11.11.1263. [DOI] [PubMed] [Google Scholar]
- Garcia-Gimeno M.A., Rodilla-Ramirez P.N., Viana R., Salas-Puig X., Brewer M.K., Gentry M.S., Sanz P. A novel EPM2A mutation yields a slow progression form of Lafora disease. Epilepsy Res. 2018;145:169–177. doi: 10.1016/j.eplepsyres.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Afawi Z., Armstrong D.D., Delgado-Escueta A.V., Goldberg Y.P., Grossman T.R., Guinovart J.J., Harris F., Hurley T.D., Michelucci R. The 5th international Lafora epilepsy workshop: basic science elucidating therapeutic options and preparing for therapies in the clinic. Epilepsy Behav. 2020;103:106839. doi: 10.1016/j.yebeh.2019.106839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Dixon J.E., Worby C.A. Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem. Sci. 2009;34:628–639. doi: 10.1016/j.tibs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Dowen R.H., 3rd, Worby C.A., Mattoo S., Ecker J.R., Dixon J.E. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J. Cell. Biol. 2007;178:477–488. doi: 10.1083/jcb.200704094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Roma-Mateo C., Sanz P. Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii. FEBS J. 2013;280:525–537. doi: 10.1111/j.1742-4658.2012.08549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Worby C.A., Dixon J.E. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. U S A. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Garre P., Gutierrez-Delicado E., Gomez-Abad C., Morales-Corraliza J., Villanueva V.E., Rodriguez de Cordoba S., Larrauri J., Gutierrez M., Berciano J., Serratosa J.M. Hepatic disease as the first manifestation of progressive myoclonus epilepsy of Lafora. Neurology. 2007;68:1369–1373. doi: 10.1212/01.wnl.0000260061.37559.67. [DOI] [PubMed] [Google Scholar]
- Gomez-Garre P., Sanz Y., Rodriguez De Cordoba S.R., Serratosa J.M. Mutational spectrum of the EPM2A gene in progressive myoclonus epilepsy of Lafora: high degree of allelic heterogeneity and prevalence of deletions. Eur. J. Hum. Genet. 2000;8:946–954. doi: 10.1038/sj.ejhg.5200571. [DOI] [PubMed] [Google Scholar]
- Gomez-Abad C., Afawi Z., Korczyn A.D., Misk A., Shalev S.A., Spiegel R., Lerman-Sagie T., Lev D., Kron K.L., Gómez-Garre P. Founder effect with variable age at onset in Arab families with Lafora disease and EPM2A mutation. Epilepsia. 2007;48:1011–1014. doi: 10.1111/j.1528-1167.2007.01004.x. [DOI] [PubMed] [Google Scholar]
- Green J.M., Hewison J., Bekker H.L., Bryant L.D., Cuckle H.S. Psychosocial aspects of genetic screening of pregnant women and newborns: a systematic review. Health Technol Assess. 2004;8:1–109. doi: 10.3310/hta8330. [DOI] [PubMed] [Google Scholar]
- Guerrero R., Vernia S., Sanz R., Abreu-Rodríguez I., Almaraz C., García-Hoyos M., Michelucci R., Tassinari C.A., Riguzzi P., Nobile C. A PTG variant contributes to a milder phenotype in Lafora disease. PLoS One. 2011;6:e21294. doi: 10.1371/journal.pone.0021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks S., Wheeler D.A., Plon S.E., Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianzano L., Young E.J., Zhao X.C., Chan E.M., Rodriguez M.T., Torrado M.V., Scherer S.W., Minassian B.A. Loss of function of the cytoplasmic isoform of the protein laforin (EPM2A) causes Lafora progressive myoclonus epilepsy. Hum. Mutat. 2004;23:170–176. doi: 10.1002/humu.10306. [DOI] [PubMed] [Google Scholar]
- Ianzano L., Zhang J., Chan E.M., Zhao X., Lohi H., Scherer S.W., Minassian B.A. Lafora progressive myoclonus epilepsy mutation database-EPM2A and NHLRC1 (EMP2B) genes. Hum. Mutat. 2005;26:397. doi: 10.1002/humu.9376. [DOI] [PubMed] [Google Scholar]
- Jara-Prado A., Ochoa A., Alonso M.E., Lima Villeda G.A., Fernandez-Valverde F., Ruano-Calderon L., Vargas-Canas S., Duron R.M., Delgado-Escueta A.V., Martinez-Juarez I.E. Late onset Lafora disease and novel EPM2A mutations: breaking paradigms. Epilepsy Res. 2014;108:1501–1510. doi: 10.1016/j.eplepsyres.2014.08.017. [DOI] [PubMed] [Google Scholar]
- Kerem E. Pharmacological induction of CFTR function in patients with cystic fibrosis: mutation-specific therapy. Pediatr. Pulmonol. 2005;40:183–196. doi: 10.1002/ppul.20200. [DOI] [PubMed] [Google Scholar]
- Ki C.S., Kong S.Y., Seo D.W., Hong S.B., Kim H.J., Kim J.W. Two novel mutations in the EPM2A gene in a Korean patient with Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 2003;48:51–54. doi: 10.1007/s100380300006. [DOI] [PubMed] [Google Scholar]
- Kroncke B.M., Vanoye C.G., Meiler J., George A.L., Jr., Sanders C.R. Personalized biochemistry and biophysics. Biochemistry. 2015;54:2551–2559. doi: 10.1021/acs.biochem.5b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoiselee H.M., Genton P., Lesca G., Brault F., De Toffol B. Are c.436G>A mutations less severe forms of Lafora disease? A case report. Epilepsy Behav. Case Rep. 2014;2:19–21. doi: 10.1016/j.ebcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesca G., Boutry-Kryza N., de Toffol B., Milh M., Steschenko D., Lemesle-Martin M., Maillard L., Foletti G., Rudolf G., Nielsen J.E. Novel mutations in EPM2A and NHLRC1 widen the spectrum of Lafora disease. Epilepsia. 2010;51:1691–1698. doi: 10.1111/j.1528-1167.2010.02692.x. [DOI] [PubMed] [Google Scholar]
- Lohi H., Ianzano L., Zhao X.-C., Chan E.M., Turnbull J., Scherer S.W., Ackerley C.A., Minassian B.A. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 2005;14:2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- Lohi H., Turnbull J., Zhao X.C., Pullenayegum S., Ianzano L., Yahyaoui M., Mikati M.A., Franceschetti S., Zara F., Minassian B.A. Genetic diagnosis in Lafora disease: genotype–phenotype correlations and diagnostic pitfalls. Neurology. 2007;68:996–1001. doi: 10.1212/01.wnl.0000258561.02248.2f. [DOI] [PubMed] [Google Scholar]
- Lynch D.S., Wood N.W., Houlden H. Late-onset Lafora disease with prominent parkinsonism due to a rare mutation in EPM2A. Neurol. Genet. 2016;2:e101. doi: 10.1212/NXG.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masica D.L., Sosnay P.R., Cutting G.R., Karchin R. Phenotype-optimized sequence ensembles substantially improve prediction of disease-causing mutation in cystic fibrosis. Hum. Mutat. 2012;33:1267–1274. doi: 10.1002/humu.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins D.A., Raththagala M., Auger K.D., Turner B.D., Santelia D., Kotting O., Gentry M.S., Vander Kooi C.W. Mechanistic insights into glucan phosphatase activity against polyglucan substrates. J. Biol. Chem. 2015;290:23361–23370. doi: 10.1074/jbc.M115.658203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins D.A., Raththagala M., Husodo S., White C.J., Guo H.F., Kotting O. Phosphoglucan-bound structure of starch phosphatase Starch Excess4 reveals the mechanism for C6 specificity. Proc. Natl. Acad. Sci. U S A. 2014;111:7272–7277. doi: 10.1073/pnas.1400757111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins D.A., Vander Kooi C.W., Gentry M.S. Structural mechanisms of plant glucan phosphatases in starch metabolism. FEBS J. 2016;283:2427–2447. doi: 10.1111/febs.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian B.A. Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001;25:21–29. doi: 10.1016/s0887-8994(00)00276-9. [DOI] [PubMed] [Google Scholar]
- Minassian B.A., Ianzano L., Meloche M., Andermann E., Rouleau G.A., Delgado-Escueta A.V., Scherer S.W. Mutation spectrum and predicted function of laforin in Lafora's progressive myoclonus epilepsy. Neurology. 2000;55:341–346. doi: 10.1212/wnl.55.3.341. [DOI] [PubMed] [Google Scholar]
- Moreno D., Towler M.C., Hardie D.G., Knecht E., Sanz P. The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol. Biol. Cell. 2010;21:2578–2588. doi: 10.1091/mbc.E10-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke F., Wang P., Schmieder P., Girard J.M., Awrey D.E., Wang T., Israelian J., Zhao X., Turnbull J., Heydenreich M. Hyperphosphorylation of glucosyl c6 carbons and altered structure of glycogen in the neurodegenerative epilepsy lafora disease. Cell Metab. 2013;17:756–767. doi: 10.1016/j.cmet.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Ozen H. Glycogen storage diseases: new perspectives. World J. Gastroenterol. 2007;13:2541–2553. doi: 10.3748/wjg.v13.i18.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandurangan A.P., Ochoa-Montaño B., Ascher D.B., Blundell T.L. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45:W229–W235. doi: 10.1093/nar/gkx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthiban V., Gromiha M.M., Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239–W242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson B.A., Turnbull J., Epp J.R., Weaver S.A., Zhao X., Pencea N., Roach P.J., Frankland P.W., Ackerley C.A., Minassian B.A. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann. Neurol. 2013;74:297–300. doi: 10.1002/ana.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov V., Cohen M., Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng. Des. Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- Raththagala M., Brewer M.K., Parker M.W., Sherwood A.R., Wong B.K., Hsu S., Bridges T.M., Paasch B.C., Hellman L.M., Husodo S. Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Mol. Cel. 2015;57:261–272. doi: 10.1016/j.molcel.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritte G., Heydenreich M., Mahlow S., Haebel S., Kotting O., Steup M. Phosphorylation of C6- and C3-positions of glucosyl residues in starch is catalysed by distinct dikinases. FEBS Lett. 2006;580:4872–4876. doi: 10.1016/j.febslet.2006.07.085. [DOI] [PubMed] [Google Scholar]
- Roma-Mateo C., Solaz-Fuster Mdel C., Gimeno-Alcaniz J.V., Dukhande V.V., Donderis J., Worby C.A., Marina A., Criado O., Koller A., Rodriguez De Cordoba S. Laforin, a dual-specificity phosphatase involved in Lafora disease, is phosphorylated at Ser25 by AMP-activated protein kinase. Biochem. J. 2011;439:265–275. doi: 10.1042/BJ20110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Villena C., Garcia-Gimeno M.A., Sanz P. Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int. J. Biochem. Cell Biol. 2013;45:1479–1488. doi: 10.1016/j.biocel.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Salar S., Yeni N., Gunduz A., Guler A., Gokcay A., Velioglu S., Gundogdu A., Hande Caglayan S. Four novel and two recurrent NHLRC1 (EPM2B) and EPM2A gene mutations leading to Lafora disease in six Turkish families. Epilepsy Res. 2012;98:273–276. doi: 10.1016/j.eplepsyres.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Sanchez-Elexpuru G., Serratosa J.M., Sanz P., Sanchez M.P. 4-Phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of Lafora disease. Neuroreport. 2017;28:268–271. doi: 10.1097/WNR.0000000000000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin P., Roma-Mateo C., Viana R., Sanz P. Ubiquitin conjugating enzyme E2-N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin E3-ubiquitin ligase complex. Int. J. Biochem. Cell Biol. 2015;69:204–214. doi: 10.1016/j.biocel.2015.10.030. [DOI] [PubMed] [Google Scholar]
- Santelia D., Kotting O., Seung D., Schubert M., Thalmann M., Bischof S., Meekins D.A., Lutz A., Patron N., Gentry M.S. The phosphoglucan phosphatase like sex Four2 dephosphorylates starch at the C3-position in Arabidopsis. Plant Cell. 2011;23:4096–4111. doi: 10.1105/tpc.111.092155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger L.J.V. The PyMOL molecular graphics system. 2010;1 [Google Scholar]
- Serratosa J.M., Gomez-Garre P., Gallardo M.E., Anta B., de Bernabe D.B., Lindhout D., Augustijn P.B., Tassinari C.A., Malafosse R.M., Topcu M. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum. Mol. Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- Sharma S., Vander Kooi C.D., Gentry M.S., Vander Kooi C.W. Oligomerization and carbohydrate binding of glucan phosphatases. Anal. Biochem. 2018;563:51–55. doi: 10.1016/j.ab.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D.N., Welsh M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Sherwood A.R., Paasch B.C., Worby C.A., Gentry M.S. A malachite green-based assay to assess glucan phosphatase activity. Anal. Biochem. 2013;435:54–56. doi: 10.1016/j.ab.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum. Mutat. 2009;30:715–723. doi: 10.1002/humu.20954. [DOI] [PubMed] [Google Scholar]
- Singh S., Satishchandra P., Shankar S.K., Ganesh S. Lafora disease in the Indian population: EPM2A and NHLRC1 gene mutations and their impact on subcellular localization of laforin and malin. Hum. Mutat. 2008;29:E1–E12. doi: 10.1002/humu.20737. [DOI] [PubMed] [Google Scholar]
- Solaz-Fuster M.C., Gimeno-Alcaniz J.V., Ros S., Fernandez-Sanchez M.E., Garcia-Fojeda B., Criado Garcia O., Vilchez D., Dominguez J., Garcia-Rocha M., Sanchez-Piris M. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 2008;17:667–678. doi: 10.1093/hmg/ddm339. [DOI] [PubMed] [Google Scholar]
- Stefely J.A., Reidenbach A.G., Ulbrich A., Oruganty K., Floyd B.J., Jochem A., Saunders J.M., Johnson I.E., Minogue C.E., Wrobel R.L. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol. Cell. 2015;57:83–94. doi: 10.1016/j.molcel.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M.A., Nitschke S., Steup M., Minassian B.A., Nitschke F. Pathogenesis of lafora disease: transition of soluble glycogen to insoluble polyglucosan. Int. J. Mol. Sci. 2017;18:1743. doi: 10.3390/ijms18081743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci V.S., Turnbull J., Wang W., Girard J.M., Zhao X., Skurat A.V., Delgado-Escueta A.V., Minassian B.A., Depaoli-Roach A.A., Roach P.J. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. U S A. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B., Frasinyuk M.S., Chikwana V.M., Mahalingan K.K., Morgan C.A., Segvich D.M., Bondarenko S.P., Mrug G.P., Wyrebek P., Watt D.S. Discovery and development of small-molecule inhibitors of glycogen synthase. J. Med. Chem. 2020;63:3538–3551. doi: 10.1021/acs.jmedchem.9b01851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J., Olatubosun A., Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011;32:358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- Turnbull J., Depaoli-Roach A.A., Zhao X., Cortez M.A., Pencea N., Tiberia E., Piliguian M., Roach P.J., Wang P., Ackerley C.A. PTG depletion removes lafora bodies and rescues the fatal epilepsy of lafora disease. PLoS Genet. 2011;7:e1002037. doi: 10.1371/journal.pgen.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J., Epp J.R., Goldsmith D., Zhao X., Pencea N., Wang P., Frankland P.W., Ackerley C.A., Minassian B.A. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann. Neurol. 2014;75:442–446. doi: 10.1002/ana.24104. [DOI] [PubMed] [Google Scholar]
- Turnbull J., Kumar S., Ren Z.-P., Muralitharan S., Naranian T., Ackerley C.A., Minassian B.A. Lafora progressive myoclonus epilepsy: disease course homogeneity in a genetic isolate. J. Child Neurol. 2008;23:240–242. doi: 10.1177/0883073807309245. [DOI] [PubMed] [Google Scholar]
- Van Heycop Ten Ham M.W. In: Handbook of Clinical Neurology. Vinken P.J., Bruyn G.W., editors. North Holland Publishing Company; 1975. Lafora disease, a form of progressive myoclonus epilepsy; pp. 382–422. [Google Scholar]
- Verhalen B., Arnold S., Minassian B.A. Lafora disease: a review of molecular mechanisms and pathology. Neuropediatrics. 2018;49:357. doi: 10.1055/s-0038-1675238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S., Solaz-Fuster M.C., Gimeno-Alcaniz J.V., Rubio T., Garcia-Haro L., Foretz M., Rodriguez de Cordoba S., Sanz P. AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunt of the R5/PTG-PP1 holoenzyme and accelerates its downregulation by the laforin-malin complex. J. Biol. Chem. 2009;284:8247–8255. doi: 10.1074/jbc.M808492200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D., Ros S., Cifuentes D., Pujadas L., Valles J., Garcia-Fojeda B., Criado-Garcia O., Fernandez-Sanchez E., Medrano-Fernandez I., Dominguez J. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007;10:1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- Wang W., Roach P.J. Glycogen and related polysaccharides inhibit the laforin dual-specificity protein phosphatase. Biochem. Biophys. Res. Commun. 2004;325:726–730. doi: 10.1016/j.bbrc.2004.10.083. [DOI] [PubMed] [Google Scholar]
- Welsh M.J., Smith A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Worby C.A., Gentry M.S., Dixon J.E. Laforin: a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby C.A., Gentry M.S., Dixon J.E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG) J. Biol. Chem. 2008;283:4069–4076. doi: 10.1074/jbc.M708712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Smith D.L. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Austin G.L., Shaffer R., Armstrong D.D., Gentry M.S. Antibody-mediated enzyme therapeutics and applications in glycogen storage diseases. Trends Mol. Med. 2019;25:1094–1109. doi: 10.1016/j.molmed.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.