

Abstract
Stem cell–derived β-like cells (sBC) carry the promise of providing an abundant source of insulin-producing cells for use in cell replacement therapy for patients with diabetes, potentially allowing widespread implementation of a practical cure. To achieve their clinical promise, sBC need to function comparably with mature adult β-cells, but as yet they display varying degrees of maturity. Indeed, detailed knowledge of the events resulting in human β-cell maturation remains obscure. Here we show that sBC spontaneously self-enrich into discreet islet-like cap structures within in vitro cultures, independent of exogenous maturation conditions. Multiple complementary assays demonstrate that this process is accompanied by functional maturation of the self-enriched sBC (seBC); however, the seBC still contain distinct subpopulations displaying different maturation levels. Interestingly, the surface protein ENTPD3 (also known as nucleoside triphosphate diphosphohydrolase-3 [NDPTase3]) is a specific marker of the most mature seBC population and can be used for mature seBC identification and sorting. Our results illuminate critical aspects of in vitro sBC maturation and provide important insights toward developing functionally mature sBC for diabetes cell replacement therapy.
Introduction
Cell replacement therapy using cadaveric human islets can achieve long-lasting insulin independence in patients suffering from diabetes, tremendously improving the quality of life and reducing life-threatening complications associated with exogenous insulin (1). However, the absence of an abundant source of functional β-cells has prevented the widespread application of this practical cure for diabetes.
Human pluripotent stem cells divide rapidly and can differentiate into every cell type in the human body; as such, they represent a potentially unlimited source from which to derive β-like cells in vitro (2). Indeed, glucose-responsive stem cell–derived β-like cells (sBC) have been generated by several groups including ours (3–5). Thus far, sBC tend to present with an immature, fetal-like phenotype and exhibit distinct differences when compared with cadaveric adult β-cells. For generation of more mature sBC in vitro, several approaches, including sorting of putative progenitor cells during direct differentiation (6), optimization of culture conditions (7), modeling the nutrient-sensing transition occurring at birth (8), entrainment of circadian cycles (9), and manipulation of transcription factors (10,11), have been described with varying degrees of success. Recently, methodologies that enrich for endocrine or insulin-producing cells have been used by several groups to generate a more mature sBC phenotype. We previously fluorescence-associated cell (FAC) sorted sBC using a fluorescent transgenic reporter to generate enhanced sBC (eBC) clusters that exhibit functional properties closely resembling those of adult mature β-cells (12). Others have used antibodies against the endocrine cell surface protein ITGA1 to reaggregate sBC—a strategy that resulted in improved functionality (13). Lastly, several sBC differentiation protocols now include a simple dissociation/reaggregation step that preferentially ablates progenitor cells within heterogenous pancreatic cell populations, resulting in enrichment of sBC (7,13). Collectively, these studies point toward a critical role for sBC enrichment in a three-dimensional organization as a prerequisite for functional maturation. However, the mechanism by which this enrichment influences sBC maturation and whether it occurs spontaneously in vitro remains to be investigated.
The advent of single-cell RNA-sequencing (scRNA-seq) technology has allowed in-depth analysis of human β-cell heterogeneity, suggesting the existence of distinct β-cell subtypes (14–16). More defined evidence for β-cell heterogeneity was provided by a study where antibodies were used for recognition of surface markers CD9 and ST8SIA1, showing the existence of four functionally distinct β-cell subtypes (17). The degree of heterogeneity within sBC populations is a key question in the field, and the development of a strategy to isolate different populations of sBC in culture would be valuable for furthering our understanding of sBC maturation.
Research Design and Methods
Cell Culture
Generation of sBC From Human Embryonic Stem Cells and From Induced Pluripotent Stem Cells
MEL1 human embryonic stem cells (hESC) containing the INSGFP/W reporter (18) (referred to as pINSGFP throughout) and subclones thereof (19,20), induced pluripotent stem cells (iPSC) of a patient with type 1 diabetes (T1D-iPSC) (21) (Supplementary Fig. 8), and iPSC from a healthy donor (22) (Supplementary Fig. 9), as well as pNKX6.1 GFP reporter iPSC (23), were used in this study. All pluripotent stem cell lines were maintained on hESC-qualified Matrigel (no. 354277; Corning) in mTeSR Plus media (05826; STEMCELL Technologies). MEL1 subclones were only used for bulk RNA-seq analysis experiments. Differentiation to sBC was carried out in suspension-based, low attachment suspension culture plates as previously described (19) or in a bioreactor magnetic stirring system (ABBWVS0 3A-6, ABBWVDW-1013, ABBWBP03N0S-6; REPROCELL) as described in Supplementary Materials.
Human Islet Culture
Human islets were used in this study as described in Supplementary Materials.
FACS
sBC clusters and human islets were dissociated, stained, and sorted for downstream analysis as outlined in Supplementary Materials.
Reaggregation of Sorted ENTPD3 sBC
Human umbilical vein endothelial cells (HUVEC) (C2519A; Lonza) and human mesenchymal stem cells (PT-2501; Lonza PT-2501) were grown as per the manufacture’s instruction. For reaggregation experiments a total of 1,000 sBC were sorted and reaggregated with 100 human mesenchymal stem cells and 400 HUVEC for 2 days in round bottom plates in a 50:50 mixture of maturation and HUVEC culture media as previously described (35).
Cell Characterization
Flow Cytometry
hESC and iPSC clusters were collected, dissociated, stained, and analyzed on a CYTEK Aurora as outlined in Supplementary Materials.
Content Analysis
Total insulin and proinsulin content analyses were carried out on aliquots of 1,000 sorted pINSGFP+ cells lysed in acid ethanol with commercially available ELISA kits (insulin, Alpco 80-INSHU-E01.1, and proinsulin, 80-PINHUT-CH01).
Global 5-Hydroxymethylcytosine Analysis
A total of 500 cells were sorted into Eppendorf tubes and lysed by flash freezing of pellets at −80°C. DNA was extracted with the PicoPure DNA Extraction Kit (KIT0103; Thermo Fisher Scientific) and global 5-hydroxymethylcytosine (5-hmc) percentage was determined with Quest 5-hmC DNA ELISA kit (D5425; Zymo Research) as per the manufacturer’s instructions.
Immunofluorescence
sBC and human islet clusters were fixed, prepared, and imaged as outlined in Supplementary Materials.
mtDNA Copy Number
A total of 500 cells were sorted into Eppendorf tubes and lysed by flash freezing of pellets at −80°C. DNA was extracted with PicoPure DNA Extraction Kit, and Human Mitochondrial DNA (mtDNA) Monitoring Primer Set (7246; Takara) was used to quantify the relative number of copies of human mtDNA by real-time PCR, with genomic DNA (gDNA) as the standard for normalization.
Quantitative RT-PCR
Total RNA was isolated with RNeasy Micro kit (74104; QIAGEN) and reverse transcribed with the iScript cDNA kit (1708891; Bio-Rad Laboratories) as per the manufacturer’s instructions. Quantitative PCR analysis was performed on the Bio-Rad CFX96 Real Time System using TaqMan probes (Insulin, Hs00355773_m1 and ENTPD3, Hs00154325_m1 (both from Thermo Fisher Scientific) and GAPDH, 10031285 (Bio-Rad Laboratories)).
scRNA-seq and Bulk RNA-seq
scRNA-seq libraries were generated with use of the 10x Genomics 3′ end platform and analyzed as outlined in Supplementary Materials. Total RNA was isolated from cell cultures with RNeasy kits from QIAGEN. Sequencing libraries were generated with the NEBNext Ultra II Directional RNA Library kit with NEBNext rRNA depletion. Paired-end sequencing reads were trimmed with use of cutadapt (v1.16 [24]), aligned with STAR (v 2.5.2a [25]), and exonic read counts were quantified with featureCounts from the subread package (v1.6.2 [26]). Differentially expressed genes were identified with DESeq2 (v1.24.0 [27]). Heat maps were generated using ComplexHeatmap and ordered using hierarchical clustering of Euclidean distances with the complete method (28).
Data and Code Availability
All sequencing data were deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) database (GSE142290). Analysis scripts and interactive UCSC cellbrowser are provided at a GitHub repository (https://github.com/rnabioco/sebeta).
Functional Characterization
Calcium imaging, image, activity and coordinated area analysis was performed as described in Supplementary Materials. Dynamic insulin secretion was measured with a Biorep Technologies perifusion machine (PERI4-02-0230-FA-ORB) as described in Supplementary Materials.
Results
Immature sBC Spontaneously Self-Organize to Form Caps Within Cell Clusters That Contain Matured Self-Enriched β-Like Cells
We used a previously described hESC line that contains a green fluorescent protein (GFP) reporter gene under the control of the endogenous insulin promoter (herein referred to as pINSGFP) (18). These cells underwent a suspension culture-based direct differentiation protocol to generate glucose responsive, but largely immature sBC after ∼23 days (imBC) (Fig. 1A) (4,12,19). imBC exhibit readily detectable insulin secretion in static glucose-stimulated insulin secretion (GSIS) assays while failing to display a response to elevated glucose concentrations in dynamic GSIS (dGSIS) assays (12,19). Use of GFP expression to visualize individual imBC revealed a heterogeneous distribution of insulin expression throughout individual clusters (Fig. 1B). Intriguingly, extending the culture period of sBC clusters by 1 week resulted in spontaneous self-aggregation of imBC into discrete self-enriched β-like cell (seBC) caps (Fig. 1B). seBC cap formation was not dependent on TGFβ inhibition or the presence of T3 thyroid hormone, as imBC rearrangement was also observed in a minimal culture media without factors Alk5i and T3 that could potentially exhibit confounding effects. However, sBC cultured in minimal media showed reduced levels of insulin expression, indicating that optimal insulin expression is dependent on addition of factors at this culture stage (Supplementary Fig. 1A–C). The percentage of pINSGFP+ cells remained constant during the self-aggregation process (Fig. 1C), suggesting that cap formation is not due to de novo production of sBC but, rather, is a result of active rearrangement of existing cells within each cluster. The intensity of pINSGFP fluorescence, which correlates with insulin expression, was significantly higher in seBC when compared with imBC (Fig. 1D). Analysis of common endocrine and β-cell markers and hormones by immunofluorescence staining showed no obvious differences in expression intensity or pattern in imBC and seBC clusters (Fig. 1E and F). Quantitative analysis of single and polyhormonal cells similarly showed no significant changes at the imBC and seBC stages (Supplementary Fig. 1D). Since mitochondrial number is known to increase with β-cell maturation (12), we stained sBC mitochondria for mtFA and quantified the staining intensity in imBC and seBC clusters. This analysis demonstrated significantly stronger mtFA staining intensity in seBC compared with the dispersed imBC cells, indicating an increased number of mitochondria (Fig. 1G and H). With use of the pINSGFP reporter line, imBC and seBC were FAC sorted at day 23 and day 30, respectively (Fig. 1I), for global transcriptomic analysis. Overall, we found 158 and 53 genes significantly up- or downregulated, respectively, in seBC compared with imBC (adjusted P value <0.05) (Fig. 1J). However, in accordance with our immunofluorescence analysis, seBC exhibited no differences in common markers of β-cell identity. Gene Ontology (GO) analysis of differentially expressed genes indicated significant enrichment of genes associated with cell morphogenesis and differentiation in seBC (Fig. 1K). Analysis of mtDNA in sorted imBC and seBC showed a significant increase in seBC (Fig. 1L), further supporting the observed increase in mitochondrial staining. Global levels of 5-hmc have recently been suggested to increase with β-cell maturation (29); quantification of global 5-hmc in DNA isolated from sorted imBC and seBC by ELISA demonstrated a threefold increase in the percentage of 5-hmc levels in seBC (Fig. 1M). Finally, we FAC sorted aliquots of 1,000 pINSGFP+ cells from imBC and seBC to quantify total insulin and proinsulin. seBC were found to contain twice as much insulin as imBC (Fig. 1N), and the proinsulin-to-insulin molar ratio was found to be significantly lower in seBC than imBC (Fig. 1O), indicating a profile of more mature insulin processing and storage in seBC. Taken together, these data demonstrate a more mature phenotype for seBC at the protein, RNA, DNA, and mitochondrial level compared with imBC.
Figure 1.

sBC self-enrich to form insulin+ enriched islet-like caps. A: Schematic representation of stepwise differentiation of human pluripotent stem cell clusters toward β-like cells in suspension. B: Representative live image of green fluorescent protein driven by the endogenous insulin promoter (pINSGFP) of clusters during imBC (day ∼23) and seBC (day ∼30) differentiation stages (scale bars indicate 200 μm). C: Flow cytometric quantification of GFP expression in imBC and seBC clusters (n = 7 independent differentiation experiments). D: Quantification of GFP intensity in imBC and seBC clusters (n = 6 independent differentiation experiments). E and F: Immunofluorescence analysis of sections from imBC and seBC clusters, respectively, for endocrine and β-cell markers. G: Immunofluorescence analysis of sections from imBC clusters, seBC clusters, and human islets for mitochondria-specific mtFA protein. H: Quantification of mtFA fluorescence intensity in imBC and seBC cells (n = 5 independent differentiation experiments with 10 clusters analyzed per experiment). I: Schematic representation of pINSGFP+ cell sorting from imBC and seBC clusters. J: Bulk RNA-seq analysis of pINSGFP+ sorted imBC vs. seBC. Top 30 genes significantly up- and downregulated with adjusted P value <0.05 are shown (n = 3 independent differentiation experiments). K: GO of differentially regulated genes. L: Quantitative PCR analysis of mtDNA normalized to gDNA in pINSGFP+ sorted cells (n = 3 independent differentiation experiments with 3 × 500 cells collected for analysis from each). M: Global levels of 5-hmc in pINSGFP+ sorted cells (n = 3 independent differentiation experiments). N and O: Total insulin content (N) and proinsulin-to-insulin content ratios (O) per 1,000 pINSGFP+ sorted cells (n = 3 independent differentiation experiments with 3 × 1,000 cells collected per experiment). *P < 0.05; **P < 0.01; ***P < 0.001. Error bars are representative of the mean ± SD. Scale bars represent 20 μm unless otherwise indicated. AU, arbitrary units; ns, not significant.
To more directly investigate the functional maturation state of seBC, we conducted dGSIS assays via islet perifusion. Clusters (20–30) of imBC, seBC, or human islets were subjected to a sequence of different glucose concentrations (0.5 mmol/L, 16.7 mmol/L) and 10 nmol/L exendin-4 and 30 mmol/L KCl challenges (Fig. 2A). As expected, human islets exhibited a characteristic first- and second-phase insulin secretion in response to a 16.7 mmol/L glucose challenge that was efficiently diminished by subsequent exposure to 0.5 mmol/L glucose. Membrane depolarization with 30 mmol/L KCl resulted in a maximal secretion that was similar to the observed peak at first-phase secretion in response to 16.7 mmol/L glucose alone. imBC clusters exhibited minimal elevated insulin secretion in response to increased glucose levels and showed exaggerated insulin secretion in response to KCl membrane depolarization. In contrast, seBC displayed low insulin secretion at 0.5 mmol/L glucose and a significant increase in secretion in response to stimulation with 16.7 mmol/L glucose, with a typical first- and second-phase profile. seBC clusters efficiently and rapidly reduced insulin secretion upon return to 0.5 mmol/L glucose levels, and membrane depolarization resulted in insulin secretion comparable with that of the first-phase peak, thus exhibiting a dGSIS profile similar to that of human islets. As with the sorted seBC analyzed in Fig. 1, seBC clusters recovered after dGSIS had higher insulin content than imBC clusters, while human islets exhibited levels comparable with seBC (Fig. 2B). The fold change in insulin secretion from additional perifusion experiments was calculated (Fig. 2C–E); seBC and human islets showed a significant increase in insulin secretion in response to high glucose that was comparable with membrane depolarization with KCl, while imBC showed a significant increase in insulin secretion upon KCL exposure but not in response to high glucose.
Figure 2.

seBC demonstrate functional maturity. A: Representative perifusion analysis of imBC, seBC, and hIslets; 20–25 clusters were analyzed per sample, and data are presented as % of total insulin in cluster pellet recovered. Total insulin content of pellet recovered after perifusion (B) and relative insulin secretion during perifusion of imBC (n = 3 independent differentiation experiments), seBC (n = 5 independent differentiation experiments), and hIslet (n = 3 independent human islets prep) (C, D, and E, respectively) (data normalized to basal [0.5 mmol/L glucose] secretion). F: Representative images of imBC (left) and seBC clusters (right) displaying pINSGFP, Ca2+ indicator (Rhod2 AM) labeling, and overlay; scale bar is 10 µm. G: Color map (top) of islets displayed in F showing magnitude and extent of Ca2+ elevations at 2 mmol/L and 11 mmol/L glucose; scale bar is 10 µm, and white arrows point to cells chosen for time courses (bottom). Representative time courses (bottom) of individual cells from the imBC and seBC clusters displayed in F at 2 mmol/L and 11 mmol/L glucose; scale bar is 20% change from mean. H: Fraction of area within intact cluster showing elevations in Ca2+ activity at 2 mmol/L and 11 mmol/L glucose (n = 3 independent differentiation experiments with >10 clusters measured per condition). Human islet data quantified in the same manner is included for reference (21). I: Fold change in Ca2+ activity when glucose is elevated from 2 mmol/L to 11 mmol/L glucose (n = 3 independent differentiation experiments with >10 clusters measured per condition). *P < 0.05; **P < 0.01; ***P < 0.001. B–E: Error bars are representative of the mean ± SD. H and I: Error bars are representative of the mean ± SEM. AU, arbitrary units; Gluc, glucose; ns, not significant; sec, seconds.
Highly sensitive Ca2+ imaging has previously been used to accurately assay β-cell function of both mice and humans (30). Intact imBC and seBC clusters were incubated with Rhod2 AM calcium binding dye and then exposed to 2 mmol/L and 11 mmol/L glucose concentrations; uptake of Ca2+ into individual cells was recorded by fluorescence imaging (Fig. 2F) and oscillations in Ca2+ uptake quantified over time (Fig. 2G). Both imBC and seBC clusters were found to exhibit robust β-cell function, evidenced by a significant increase in the Ca2+ active area upon exposure to elevated glucose (Fig. 2H and I and Supplementary Fig. 2A). However, seBC displayed a significantly larger response compared with imBC. Interestingly, seBC clusters also present with significantly lower basal Ca2+ active areas than imBC clusters, indicating reduced insulin secretion, a feature specific to mature β-cells (31) (Fig. 2H). Coordination of Ca2+ dynamics of whole clusters was not changed between imBC and seBC but was within the range of what has previously been reported for human islets (30) (Supplementary Fig. 2B).
These data demonstrate that sBC generated after ∼3 weeks in vitro are immature but self-enrich and mature during extended culture into seBC that are both phenotypically and functionally akin to cadaveric human islets.
seBC Are Heterogeneous and Comprise Subpopulations of Cells With Varying Maturity Expression Profiles
For molecular characterization of this novel population of in vitro differentiated cells, pINSGFP+ seBC were FAC sorted and profiled via scRNA-seq using the 10x Genomics platform (Fig. 3A). A total of 4,143 cells were assigned to seven distinct subpopulations based on marker gene expression (Fig. 3B); seBC subpopulations were distinguished by INS and FEV expression, among other genes, into mature and immature subpopulations, respectively. Two polyhormonal subpopulations expressing transcripts for SST or GCG along with INS were identified. With expression of IGF2 or CD9 we identified two additional subpopulations of seBC. Finally, a small proliferative (Ki67+) subpopulation was also found (Fig. 3C and Supplementary Table 1). RNA velocity analysis identified a differentiation trajectory from the immature subpopulations toward the most mature subpopulation of seBC (Fig. 3D), while Markov diffusion modeling of the RNA velocity allowed estimation of the probable differentiation start point (Fig. 3E) as the polyhormonal and proliferative seBC subpopulations and end point (Fig. 3F) as the mature seBC subpopulation. Inferred trajectory of differentiation through the various subpopulations shows a drift from polyhormonal seBC toward mature seBC with two key branch points along the predicted trajectory (Fig. 3G). While it has been shown that polyhormonal cells primarily give rise to α-like cells (4), induction of NKX6.1 expression in such cells can result in an sBC phenotype (32). In-depth analysis of the branch points and their gene expression demonstrates that the first branch point is primarily composed of the proliferative cell subpopulation (Supplementary Fig. 3A). However, the second branch is enriched for cells from the immature seBC FEV+ and CD9+ β-cell subpopulations, suggesting that the CD9+ β-cell subpopulation may be generated through a trajectory distinct from that of the dominant mature seBC population (Supplementary Fig. 3B). Analysis of gene expression dynamics across pseudotime demonstrated increasing expression of INS, IAPP, and LMO1 along the differentiation axis, concomitant with decreasing expression of SST, GCG, and APOA1/C3, known markers of the less differentiated polyhormonal subpopulations (Fig. 3H). Finally, GO analysis of the mature seBC population revealed significant enrichment of genes associated with insulin processing, β-cell development, hormone activity, and K+ channel activity, further strengthening the identity of the subpopulation.
Figure 3.

scRNA-seq profiling of β-cell differentiation defines temporal dynamics of β-cell maturation. A: Schematic representation of seBC production and sorting. B: tSNE projection of 4,143 seBC. Cells are colored by inferred cell type based on marker gene expression. C: Heat map showing scaled abundance of the top 10 marker genes for each cell type identified by scRNA-seq analysis. D: tSNE projection with RNA velocity vector estimates overlaid. E and F: Differentiation start point (E) and end point (F) modeled using a Markov diffusion process on RNA velocity transmission probabilities. Start and end points were sampled from a uniform 100 × 100 grid and then imputed for all cells using K = 10 K-nearest neighbor pooling. Values range from 0 (yellow) to 1 (dark blue). G: Trajectory inference (monocle 2) analysis with cells colored by cell type. H: Pseudotime estimates overlaid on original tSNE projection. I: Heat map of scaled gene expression of genes with varying expression across pseudotime. Genes were clustered into two clusters with k-means clustering, and expression values were smoothed using cubic-spline interpolation. J: GO term enrichment analysis of marker genes of most mature seBC population identified by RNA velocity end point analysis (>0.8 end point density). hPSC, human pluripotent stem cell; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Recently, we reported that artificial reaggregation of quasi-pure, FAC-sorted imBC into eBC clusters results in improved maturation (12). For testing of whether eBC generation from seBC results in further maturation, seBC sorted and reaggregated for 4 days were profiled by scRNA-seq (Supplementary Fig. 4A). A total of 4,178 cells were assigned to seven different subpopulations based on marker gene expression (Supplementary Fig. 4B and C and Supplementary Table 2). RNA velocity analysis identified a trajectory from immature polyhormonal subpopulations to the most mature β-cell populations, similar to the trajectory observed in seBC (Supplementary Fig. 4D–G). Alignment of the seBC and eBC scRNA-seq data sets into the same t-distributed stochastic neighbor embedding (tSNE) projection revealed that similar subpopulations are present in both samples, with the exception of a minor unknown cell population found in the eBC data set (Supplementary Fig. 4H and I). Taken together, these data indicate that phenotypically and functionally mature seBC present as distinct subpopulations with different maturation levels. Our analysis further suggests that under the culture conditions used seBC exhibit a trajectory toward the most mature subpopulation.
Ectonucleoside Triphosphate Diphosphohydrolase 3 Marks Most Mature β-Like Cells
Detailed analysis of the most mature seBC subpopulation revealed significant enrichment of the cell-surface marker ectonucleoside triphosphate diphosphohydrolase 3 (ENTPD3) (also known as nucleoside triphosphate diphosphohydrolase-3 [NTPDase3]), recently described as a marker of mature human β-cells in vivo (33) (Fig. 4A). Ectonucleotidases are a family of membrane-bound nucleotide metabolizing enzymes that regulate extracellular ATP levels by degrading ATP and related nucleotides (34). ENTPD3 transcripts are significantly increased in seBC compared with imBC and while undetectable at the protein level in imBC, ENTPD3 is readily expressed in CPEP+ cells within seBC clusters, strongly marking CPEP+ caps (Fig. 4B and C). While pINSGFP-based cell sorting allows for collection of all seBC, addition of an ENTPD3-specific antibody directly conjugated to Alexa Fluor 555 allows the specific isolation of a presumptive, most mature INS+ENTPD3+ seBC subpopulation, equaling ∼30% of the total pINSGFP+ seBC population, as compared with unstained controls (Fig. 4D–F and Supplementary Fig. 4A and B). For in-depth analysis, seBC were sorted into “mature” INS+ENTPD3+ and “immature” INS+ENTPD3− seBC subpopulations (Fig. 4G). Differential analysis of RNA collected for bulk RNA sequencing allowed compilation of a novel list of maturation-associated genes, with 314 and 356 genes up- and downregulated, respectively (Fig. 4H–J and Supplementary Table 3). GO analysis of differentially expressed genes revealed significant enrichment for genes encoding cell membrane proteins, in particular those involved in ion channel activity in mature INS+ENTPD3+ seBC (Fig. 4I). For further characterization of INS+ENTPD3+ and INS+ENTPD3− seBC, 1,000 cells from each subpopulation were FAC sorted and analyzed by ELISA for insulin and proinsulin content. To allow direct comparison with human β-cells, we sorted ENTPD3+ cells from human islet preps to an average purity of 90% insulin-expressing cells (Supplementary Fig. 6). INS+ENTPD3+ seBC have significantly higher insulin content than INS+ENTPD3− cells (Fig. 4K); however, levels are lower in comparison with FAC sorted ENTPD3+ cadaveric β-cells. The proinsulinto-insulin molar ratio of INS+ENTPD3− cells is significantly higher compared with INS+ENTPD3+, suggesting more efficient insulin bioprocessing in the INS+ENTPD3+ seBC subpopulation (Fig. 4L). The observed proinsulinto-insulin ratio of INS+ENTPD3+ seBC is comparable with that of ENTPD3+ cadaveric β-cells, further strengthening the idea that INS+ENTPD3+ seBC represent a mature β-cell subpopulation. mtDNA copy number is also significantly increased in INS+ENTPD3+ seBC compared with immature seBC and within the range of primary human islet (hIslet) ENTPD3+ cells (Fig. 4M). No significant difference was detected in global 5-hmc levels across the three cell types.
Figure 4.
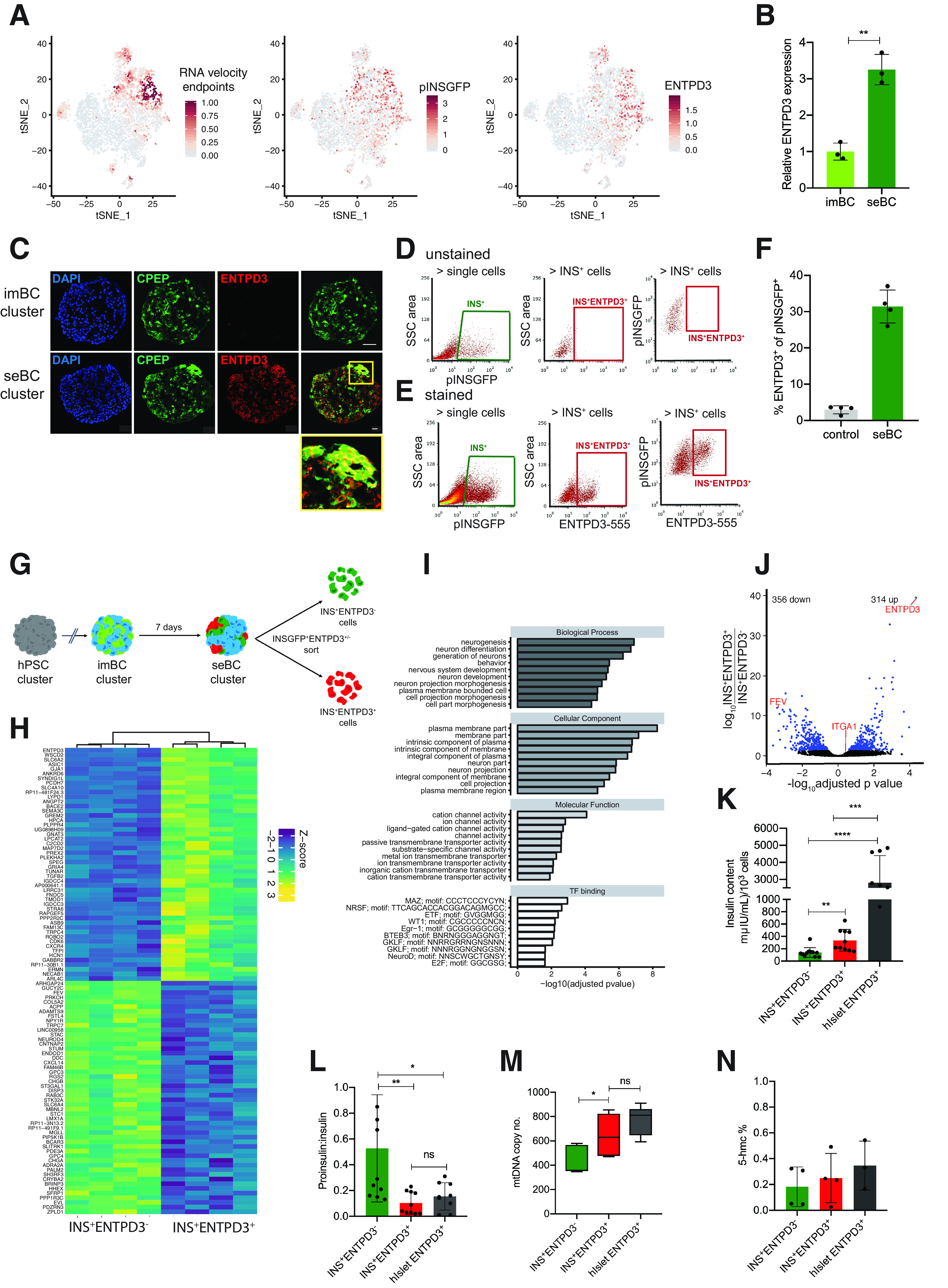
Ectonucleoside triphosphate diphosphohydrolase 3 marks mature β-like cells. A: tSNE projection of RNA velocity end points and pINSGFP transgene and ENTPD3 expression in 4,143 seBC. B: Relative ENTPD3 gene expression in pINSGFP+ imBC and seBC (n = 3 independent differentiation experiments). C: Immunofluorescence staining for C-peptide (CPEP) and ENTPD3 in sections of imBC and seBC clusters (scale bar represents 20 μm). D and E: Representative sorting gates for pINSGFP+ENTPD3+/− cells in unstained negative control cells (D) and with direct conjugated ENTPD3 antibody (E). F: Quantification of the percentage of ENTPD3+ cells within total pINSGFP+ population by FACS (n = 4 independent differentiation experiments). G: Schematic representation of pINSGFP+ENTPD3+/− sorting from seBC. H: Bulk RNA-seq analysis of INS+ENTPD3+ vs INS+ENTPD3− cells sorted from seBC clusters (uncurated list of top 30 genes significantly up- and down regulated as per adjusted P value <0.05; n = 4 independent differentiation experiments). I: GO term enrichment analysis of differentially expressed genes identified in bulk RNA seq. J: Volcano plot of differential expression analysis of INS+ENTPD3+ vs. INS+ENTPD3−. K: Insulin content per 1,000 INS+ENTPD3+/− sorted cells from seBC and human islets (n = 3 independent differentiation experiments or human islets preps, with 3 × 1,000 cells analyzed per experiment). L: Proinsulin-to-insulin content ratio of INS+ENTPD3+/− sorted cells from seBC and human islets (n = 3 independent differentiation experiments or human islets preps, with 3 × 1,000 cells analyzed per experiment). M: Quantitative PCR analysis of mtDNA normalized to gDNA in INS+ENTPD3+ vs. INS+ENTPD3− sorted cells from seBC and human islets; n = 3 independent differentiation experiments or human islets preps, with 3 × 500 cells analyzed per experiment). N: Global levels of 5-hmc in INS+ENTPD3+ vs. INS+ENTPD3− sorted cells from seBC day 30 and human islets (n = 4 independent differentiation experiments, with 1 × 500 cells analyzed per experiment). *P < 0.05; **P < 0.01; ***P < 0.001. Error bars are representative of the mean ± SD. ns, not significant.
For testing of the functionality of INS+ENTPD3+ seBC directly, immature INS+ENTPD3− and mature INS+ ENTPD3+ cells were sorted and reaggregated in the presence of endothelial and mesenchymal support cells for 48 h followed by dGSIS assay (Fig. 5A). We added supporting cells to aid in the formation of robust cell clusters after β-cell sorting as previously described (17,35), to allow cluster handling for downstream perifusion assays. We found that with reaggregation of sorted INS+ENTPD3− or INS+ENTPD3+ cells without the addition of supporting cells more time was required for forming of robust clusters. However, INS+ENTPD3− clusters started to coexpress the maturation marker ENTPD3 after longer culture periods, indicating that seBC maturation is a dynamic and potentially continuous process within differentiation cultures (Supplementary Fig. 7). Due to this observation, functional analysis of clusters cultured for longer periods of time was prevented and the use of support cells to stabilize clusters was needed. Reaggregated immature INS+ENTPD3− clusters were found to be non–glucose responsive but responded to membrane depolarization with KCl (Fig. 5B and D). In contrast, mature INS+ENTPD3+ clusters readily responded to 16.7 mmol/L glucose, exendin-4, and KCl and regulated insulin secretion dynamically (Fig. 5B and D). Clusters from each condition were recovered after dGSIS and tested for total insulin content; mature INS+ENTPD3+ clusters were found to contain more insulin than the immature clusters (Fig. 5C), consistent with previous data (Fig. 4).
Figure 5.

INS+ENTPD3+ cells show improved function. A: Schematic representation of pINS+ENTPD3+/− cells sorted from seBC clusters reaggregated (reagg) in the presence of support cells, HUVEC, and mesenchymal stem cells for 48 h. B: Perifusion analysis of INS+ENTPD3− and INS+ENTPD3+ clusters; 20–25 clusters were analyzed per condition, and data are presented as % of total insulin in cluster pellet recovered (n = 2 independent differentiation experiments). C: Total insulin content of clusters recovered following perifusion analysis (n = 2 independent differentiation experiments). D: Relative insulin secretion during perifusion of INS+ENTPD3− and INS+ENTPD3+ clusters (n = 2 independent experiments) (data normalized to basal [0.5 mmol/L glucose] secretion). Gluc, glucose; hPSC, human pluripotent stem cell.
While the pINSGFP reporter line is an excellent research tool, the clinical applications of this transgenic line are limited. Thus, we established iPSC from a donor with type 1 diabetes (T1D-iPSC) through episomal reprogramming of peripheral blood mononuclear cells as previously reported (21) (Supplementary Fig. 8) and iPSC from a healthy control donor as reported (22, referred to as CTRL) (Supplementary Fig. 9). In addition, we used a previously described iPSC line that contains a GFP reporter under the control of the NKX6.1 promoter (referred to as pNKX6.1-GFP) (23). All three iPSC were differentiated for 30 days with use of an iPSC differentiation protocol, and protein expression of specific lineage markers at key differentiation stages was quantified by flow cytometry (Fig. 6A and B). Dependent on the iPSC line, we obtained between 20 and 50% CPEP+NKX6.1+ cells at day 23, indicating efficient production of sBC. After an additional 7 days in culture, ∼30% CPEP+ENTPD3+ cells could be readily identified (Fig. 6B). Immunofluorescence staining of CTRL-, T1D-, and pNKX6.1-GFP–derived seBC revealed formation of INS+ENTPD3+ caps within clusters (Fig. 6C). Using the pan endocrine marker HPi1, previously described (17), we sorted ENTPD3− and ENTPD3+ from CTRL-iPSC–derived seBC clusters and reaggregated them in the presence of supporting cells for 48 h before assaying function via perifusion (Fig. 6D). Reaggregated immature HPi1+ENTPD3− clusters did not respond to glucose but secreted insulin readily after membrane depolarization with KCl (Fig. 6E). In contrast, mature HPi1+ENTPD3+ clusters readily responded to 16.7 mmol/L glucose and regulated insulin secretion dynamically (Fig. 6E), similar to what was observed with hESC-derived INS+ENTPD3+ clusters. Taken together, these data show that the surface protein ENTPD3 can be used as a marker to identify and FAC sort the most mature β-cell subpopulation of hESC- and iPSC-derived sBC, as characterized by gene expression, insulin storage, insulin bioprocessing, mtDNA copy number, and β-cell function. In fact, ENTPD3+ seBC are comparable with ENTPD3+ cadaveric β-cells sorted from human islets by a number of different assay parameters.
Figure 6.

INS+ENTPD3+ cells are present in patient-derived seBC. A: Schematic representation of stepwise differentiation of iPSC clusters toward β-like cells in suspension. B: Representative flow cytometry analysis of iPSC differentiation at definitive endoderm (DE), pancreatic endoderm (PE), imBC, and seBC for specific lineage markers (n = 2 independent differentiation experiments). C: Immunofluorescence staining of seBC clusters derived from CTRL-iPSC, T1D-iPSC, and iPSC containing a GFP reporter under the contorl of the NKX6.1 promoter (scale bar represents 20 μm) (n = 5 independent differentiation experiments). D: Schematic representation of pINS+ENTPD3+/− cells sorted from iPSC-derived seBC clusters reaggregated in the presence of support cells, HUVEC, and mesenchymal stem cells (MSC) for 48 h. E: Representative perifusion analysis of INS+ENTPD3− and INS+ENTPD3+ clusters; 35–45 clusters were analyzed per condition, and data are presented as % of total insulin in cluster pellet recovered. EE, early endocrine.
Discussion
Here we demonstrate that with extended culture imBC organize into seBC that exhibit a more mature β-cell phenotype. seBC were found to be heterogenous and comprise populations of cells with varying degrees of β-cell maturity. The most mature seBC can be identified and FAC sorted by the surface protein ENTPD3 (also known as NTPDase3), recently described as a marker of mature human primary β-cells (33). Taken together, our work illuminates critical aspects of in vitro sBC maturation and provides important insight into developing functionally mature sBC for diabetes cell replacement therapy.
Until now, detailed knowledge of the underlying processes of human β-cell maturation has been largely absent. In this study, we have uncovered self-organization of insulin-expressing cells into discrete islet-like caps as a critical element of sBC maturation in vitro. We used multiple assays to demonstrate that imBC functionally mature into seBC by extending the culture period. This observation is in line with data from several independent studies that show prolonging of the last culture stage (often referred to as maturation stage) results in gradual functional improvement of sBC (7,9,13). Indeed, our data further show that self-enrichment occurs in minimal and maturation media, suggesting self-enrichment as a common mechanism of sBC maturation across different differentiation protocols.
Unexpectedly, we discovered that the functionally mature seBC population exhibits distinct subpopulations. While a significant subpopulation could be classified as most mature sBC based on β-cell marker gene expression, other subpopulations with interesting expression profiles were also identified. We find a small but distinct CD9 sBC subpopulation; CD9 has recently been shown to mark less functional β-cells in cadaveric islet preparations, and their frequency is increased in patients with diabetes (17). Additionally, a distinct IGF2+ population exists in sBC; IGF2 has been shown to be involved in the development of β-cells and is associated with the development of diabetes (36). Furthermore, we confirm the existence of previously described fetal-like sBC and sBC with cohormone transcript expression (37) and a small subpopulation of proliferating sBC (38). How these different sBC subpopulations interact and potentially (inter)change, for example, upon transplantation, needs to be determined in the future. Our own RNA velocity and pseudotime analysis suggest that the less differentiated sBC subpopulations develop into the more mature sBC phenotype over time, at least with the culture conditions used.
Markers for mature β-cells have previously been identified, but currently only surface proteins that can enrich for stem cell derived–endocrine cells, but not sBC, are available (13). We used our scRNA-seq data to search for surface markers specifically expressed on mature seBC and successfully identified ENTPD3 as a marker of such cells. Comparing ENTPD3 expression levels with those of the recently described endocrine marker ITGA1 using our data sets suggested greater specificity of ENTPD3 for mature sBC (Supplementary Fig. 10A–C). Flow-based quantification verified the use of ITGA1 to enrich for sBC but also showed significantly higher enrichment of single positive insulin-expressing sBC with use of ENTPD3 (Supplementary Fig. 10D and E). Although ENTPD3 has been show to label mature human β-cells in vivo after the age of 5 years (33), it is important to note that ENTPD3 marks some somatostatin-expressing cells in addition to β-cells. Global transcriptomic analysis from ENTPD3+ and ENTPD3− seBC provided us with a novel list of genes that are up- and downregulated upon sBC maturation in vitro. Surprisingly, this list did not include genes, especially transcription factors, commonly associated with β-cell generation and function. In line with recent work that suggests inhibition of TGFb signaling interferes with sBC maturation in vitro (7), we find TGFb2 significantly upregulated in mature ENTPD3+ seBC, despite an ALK inhibitor present in the media of these cultures. This apparent contradiction further highlights the need for optimized culture conditions during sBC maturation that will now be feasible with use of our novel surface marker ENTPD3.
Recently, great progress has been made in generating sBC at high efficiency using direct differentiation approaches, but our current understanding of sBC maturation is still incomplete. There is increasing recognition that mature sBC provide distinct advantages for cell replacement therapy strategies; most notably, mature sBC with improved function will reduce the total dose of cells required and reduce the potential risk from uncontrolled proliferation of less differentiated cells within grafts. Thus, a better understanding of sBC maturation is of critical importance. We present compelling evidence for several important observations, including molecular mechanisms and markers pertaining to sBC maturation in vitro. We anticipate that our findings will further accelerate current research efforts to generate clinically relevant sBC for cell replacement therapy in patients with diabetes.
Article Information
Acknowledgments. The authors thank the Alberta Islet Distribution Program (39,40), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)-funded Integrated Islet Distribution Program (IIDP) (Research Resource Identifier [RRID]: SCR _014387) at City of Hope (National Institutes of Health [NIH] grant 2UC4DK098085), and the JDRF-funded IIDP Islet Award Initiative. The authors also thank the Scharp-Lacy Research Institute, Southern California Islet Cell Resources Center, University of Alberta, University of Wisconsin, University of Pennsylvania, University of Miami, and University of Illinois Chicago for supplying human islet samples. The authors thank the organ donors and their families. Finally, the authors thank Dr. Lori Sussel and Dr. Howard Davidson, Barbara Davis Center for Diabetes, University of Colorado, Medical Campus, Aurora, CO, and Dr. Kevin Docherty, The Institute of Medical Sciences, University of Aberdeen, Scotland, for helpful comments and Scott Beard, Barbara Davis Center for Diabetes, University of Colorado, Medical Campus, Aurora, CO, for expert technical support.
Funding. This work was supported by NIDDK and National Institute of Allergy and Infectious Diseases, NIH, grants R01DK120444 (to H.A.R.), R21AI140044 (H.A.R.), K12DK094712 (K.A.R.), UC4 DK104194 (C.E.M.), R01 DK127497 (C.E.M.), UG3 DK122638 (C.E.M.), P01 AI042288 (C.E.M.), and P30-DK116073 (Colorado Diabetes Research Center) and a pilot grant from the RNA Bioscience Initiative (H.A.R.), funding from the Children’s Diabetes Foundation (H.A.R.), a new investigator award from the NIDDK-supported Human Islets Research Network (RRID: SCR_014393, grant UC24 DK1041162), the Culshaw Junior Investigator Award in Diabetes, a University of Colorado Grubstake award, and a grant from the JDRF (2-SRA-2019-781-S-B).
Duality of Interest. H.A.R. is a consultant and scientific advisory board member to Sigilon therapeutics, consultant to Eli Lilly, and scientific advisory board member at Prellis Biologics. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. F.M.D. and H.A.R. contributed to the study conception and study design. F.M.D., K.A.R., R.C.-G., J.M.D., A.H.S., M.S.H., S.P.M.W., L.H.A., K.E.S., M.A.W., C.E.M., T.M.T., and H.A.R. contributed to execution of experiments. F.M.D., K.A.R., R.C.-G., J.M.D., A.H.S., M.S.H., R.K.P.B., and H.A.R. contributed to data analysis and interpretation. F.M.D., K.A.R., and H.A.R. contributed to manuscript writing. F.M.D., K.A.R., R.C.-G., J.M.D., A.H.S., M.S.H., S.P.M.W., L.H.A., K.E.S., M.A.W., C.E.M., T.M.T., R.K.P.B., and H.A.R. gave final approval of the manuscript. H.A.R. serves as guarantor, taking responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.15124980.
References
- 1. Shapiro AMJ, Ricordi C, Hering BJ, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med 2006;355:1318–1330 [DOI] [PubMed] [Google Scholar]
- 2. Thomson JA, Kalishman J, Golos TG, et al. Isolation of a primate embryonic stem cell line. Proc Natl Acad Sci U S A 1995;92:7844–7848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rezania A, Bruin JE, Arora P, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014;32:1121–1133 [DOI] [PubMed] [Google Scholar]
- 4. Russ HA, Parent AV, Ringler JJ, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J 2015;34:1759–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pagliuca FW, Millman JR, Gürtler M, et al. Generation of functional human pancreatic β cells in vitro. Cell 2014;159:428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mahaddalkar PU, Scheibner K, Pfluger S, et al. Generation of pancreatic β cells from CD177+ anterior definitive endoderm. Nat Biotechnol 2020;38: 1061–1072 [DOI] [PubMed] [Google Scholar]
- 7. Velazco-Cruz L, Song J, Maxwell KG, et al. Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Reports 2019;12:351–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Helman A, Cangelosi AL, Davis JC, et al. A nutrient-sensing transition at birth triggers glucose-responsive insulin secretion. Cell Metab 2020;31: 1004–1016.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvarez-Dominguez JR, Donaghey J, Rasouli N, et al. Circadian entrainment triggers maturation of human in vitro islets. Cell Stem Cell 2020;26:108–122.e10 [DOI] [PubMed] [Google Scholar]
- 10. Velazco-Cruz L, Goedegebuure MM, Maxwell KG, Augsornworawat P, Hogrebe NJ, Millman JR. SIX2 regulates human β cell differentiation from stem cells and functional maturation in vitro. Cell Rep 2020;31:107687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu S, Russ HA, Wang X, et al. Human pancreatic beta-like cells converted from fibroblasts. Nat Commun 2016;7:10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nair GG, Liu JS, Russ HA, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol 2019;21:263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Veres A, Faust AL, Bushnell HL, et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 2019;569:368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Segerstolpe Å, Palasantza A, Eliasson P, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 2016;24:593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baron M, Veres A, Wolock SL, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 2016;3:346–360.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muraro MJ, Dharmadhikari G, Grün D, et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst 2016;3:385–394.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dorrell C, Schug J, Canaday PS, et al. Human islets contain four distinct subtypes of β cells. Nat Commun 2016;7:11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Micallef SJ, Li X, Schiesser JV, et al. INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia 2012;55:694–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faleo G, Russ HA, Wisel S, et al. Mitigating ischemic injury of stem cell-derived insulin-producing cells after transplant. Stem Cell Reports 2017;9:807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou X, Nair GG, Russ HA, et al. LIN28B impairs the transition of hESC-derived β cells from the juvenile to adult state. Stem Cell Reports 2020;14:9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hudish LI, Bubak A, Triolo TM, et al. Modeling hypoxia-induced neuropathies using a fast and scalable human motor neuron differentiation system. Stem Cell Reports 2020;14:1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor JP, Cash MN, Santostefano KE, Nakanishi M, Terada N, Wallet MA. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J Leukoc Biol 2018;103:1225–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gupta SK, Wesolowska-Andersen A, Ringgaard AK, et al. NKX6.1 induced pluripotent stem cell reporter lines for isolation and analysis of functionally relevant neuronal and pancreas populations. Stem Cell Res (Amst) 2018;29:220–231 [DOI] [PubMed] [Google Scholar]
- 24. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17:10 [Google Scholar]
- 25. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–930 [DOI] [PubMed] [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847–2849 [DOI] [PubMed] [Google Scholar]
- 29. Gurlo T, Georgia S, Dhawan S. DNA hydroxymethylation regulates beta-cell maturation and expansion (Abstract). Diabetes 2018;67(Suppl. 1). DOI: 10.2337/db18-50-OR [Google Scholar]
- 30. Westacott MJ, Farnsworth NL, St Clair JR, et al. Age-dependent decline in the coordinated [Ca2+] and insulin secretory dynamics in human pancreatic islets. Diabetes 2017;66:2436–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 2012;30:261–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petersen MBK, Azad A, Ingvorsen C, et al. Single-cell gene expression analysis of a human ESC model of pancreatic endocrine development reveals different paths to β-cell differentiation. Stem Cell Reports 2017;9:1246–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saunders DC, Brissova M, Phillips N, et al. Ectonucleoside triphosphate diphosphohydrolase-3 antibody targets adult human pancreatic β cells for in vitro and in vivo analysis. Cell Metab 2019;29:745–754.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Syed SK, Kauffman AL, Beavers LS, et al. Ectonucleotidase NTPDase3 is abundant in pancreatic β-cells and regulates glucose-induced insulin secretion. Am J Physiol Endocrinol Metab 2013;305:E1319–E1326 [DOI] [PubMed] [Google Scholar]
- 35. Furuyama K, Chera S, van Gurp L, et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 2019;567:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Casellas A, Mallol C, Salavert A, et al. Insulin-like growth factor 2 overexpression induces β-cell dysfunction and increases beta-cell susceptibility to damage. J Biol Chem 2015;290:16772–16785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hrvatin S, O’Donnell CW, Deng F, et al. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci U S A 2014;111:3038–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosado-Olivieri EA, Aigha II, Kenty JH, Melton DA. Identification of a LIF-responsive, replication-competent subpopulation of human β cells. Cell Metab 2020;31:327–338.e6 [DOI] [PubMed] [Google Scholar]
- 39. Lyon J, Manning Fox JE, Spigelman AF, et al. Research-focused isolation of human islets from donors with and without diabetes at the Alberta Diabetes Institute IsletCore. Endocrinology 2016;157:560–569 [DOI] [PubMed] [Google Scholar]
- 40. Kin T, O’Gorman D, Schroeder A, et al. Human islet distribution program for basic research at a single center. Transplant Proc 2011;43:3195–3197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequencing data were deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) database (GSE142290). Analysis scripts and interactive UCSC cellbrowser are provided at a GitHub repository (https://github.com/rnabioco/sebeta).