

Abstract
Lung cancer is the leading cause of cancer‐related death worldwide. Late diagnosis and metastatic dissemination contribute to its low survival rate. Since microRNA (miRNA) deregulation triggers lung carcinogenesis, miRNAs might represent an interesting therapeutic tool for lung cancer management. We identified seven miRNAs, including miR‐126‐3p and miR‐221‐3p, that are deregulated in tumours compared with normal tissues in a series of 38 non‐small‐cell lung cancer patients. A negative correlation between these two miRNAs was associated with poor patient survival. Concomitant miR‐126‐3p replacement and miR‐221‐3p inhibition, but not modulation of either miRNA alone, reduced lung cancer cell viability by inhibiting AKT signalling. PIK3R2 and PTEN were validated as direct targets of miR‐126‐3p and miR‐221‐3p, respectively. Simultaneous miRNA modulation reduced metastatic dissemination of lung cancer cells both in vitro and in vivo through CXCR4 inhibition. Systemic delivery of a combination of miR‐126‐3p mimic and miR‐221‐3p inhibitor encapsulated in lipid nanoparticles reduced lung cancer patient‐derived xenograft growth through blockade of the PIK3R2–AKT pathway. Our findings reveal that cotargeting miR‐126‐3p and miR‐221‐3p to hamper both tumour growth and metastasis could be a new therapeutic approach for lung cancer.
Keywords: lipid nanoparticles, lung cancer, microRNA
Lung cancer is the first cause of cancer‐related deaths in the world. MicroRNAs are deregulated during lung carcinogenesis, and their modulation could represent an interesting therapeutic approach. We demonstrated that concomitant miR‐126 replacement and miR‐221 inhibition reduced lung cancer growth and metastasis both in vitro and in vivo. This study provides evidence for the use of miRNAs as therapy in lung cancer.
Abbreviations
- CCL
coated cationic lipid nanoparticles
- FACS
fluorescence‐activated cell sorter
- HUVECs
human umbilical vein endothelial cells
- ISH
in situ hybridization
- LNA
locked nucleic acid
- MILD
multicentric Italian lung detection
- Mim
microRNA‐mimic
- miRNA
microRNA
- NHBE
normal human bronchial epithelial cells
- NSCLC
non‐small‐cell lung cancer
- OS
overall survival
- PDX
patient-derived xenograft
- S.E.M.
standard errors of the mean
- SAECs
small airway epithelial cells
- SCID
severe combined immunodeficient mice
- siRNA
small interfering RNA
- TRAIL
tumour necrosis factor-related apoptosis-inducing ligand
- WB
western blot
1. Introduction
Lung cancer has the highest mortality rate among cancers, with a 5‐year survival rate of less than 18% [1]. Treatment options for lung cancer are influenced by the subtype and stage of disease. While surgery remains the gold standard in the treatment of lung cancer in patients with early‐stage disease [2], the combination of platinum‐based drugs is the standard of care for patients with advanced lung cancer [3]. Targeted therapies against epidermal growth factor and/or anaplastic lymphoma kinase have improved response rates only in a small group of patients with actionable mutations or rearrangements [4]. Moreover, over the last decade, the clinical management of lung cancer has been improved by the introduction of immune checkpoint inhibitors targeting cytotoxic T‐lymphocyte‐associated protein 4 and the programmed death 1/programmed death ligand 1 axis [5]. Unfortunately, only a small subset of patients successfully responds to this therapeutic approach as well [6], suggesting that finding alternative therapeutic strategies for lung cancer is an important clinical need.
MicroRNAs (miRNAs) are noncoding RNAs that are deregulated in different diseases, such as cancer, hepatitis and metabolic disease [7]. Based on their function, miRNAs may be considered ‘oncomirs’ when their overexpression in malignant cells inhibits tumour suppressor genes, for instance, the miR‐17‐92 cluster in lung cancer [8]. On the other hand, ‘tumour suppressor miRNAs’, such as let‐7a, are generally downregulated in cancer cells, and their loss results in aberrant expression of an oncogene [9]. miRNAs do not require perfect sequence complementarity with their target mRNAs to recognize them, implying that a single miRNA can regulate multiple genes [10] and several cellular mechanisms. This observation suggests a role for miRNAs as therapeutic tools in clinical cancer management [11].
Several studies have reported aberrant expression of miRNAs in lung tumours when compared to normal lung tissues [12], supporting the critical contribution of miRNAs to lung cancer development [13]. In our previous work, the miRNA expression profiles of 28 lung tumours and 24 paired normal lung tissues were analysed and the expression levels of the 34 identified miRNAs were revealed to be significantly different between normal lung and early‐stage lung cancer tissues [14]. Among the differentially expressed miRNAs, miR‐126‐3p was identified as an endothelial miRNA expressed at low levels in non‐small‐cell lung cancer (NSCLC) patients [15], and its replacement inhibited tumour growth by targeting EGFL7 [16]. Additionally, miR‐221 has been reported to be overexpressed in aggressive lung cancers, promoting tumour growth and invasion of lung cancer cells [17].
In the present study, miRNA expression was analysed in an independent clinical series of 38 lung cancer patients to identify the miRNAs whose expression modulation may have the greatest clinical utility. Here, we demonstrated that combined miR‐126‐3p replacement and miR‐221‐3p inhibition reduced tumour growth both in vitro and in vivo by inducing tumour necrosis factor‐related apoptosis‐inducing ligand (TRAIL)‐mediated apoptosis. Interestingly, miR‐126‐3p replacement reduced lung cancer metastatic dissemination through CXCR4 blockade both in vitro and in vivo. Importantly, the antitumour activity of these miRNAs encapsulated in lipid nanoparticles was also observed in patient‐derived xenograft (PDX) models of lung cancer.
2. Materials and methods
2.1. Population study
Tissue samples were collected from 38 lung cancer patients enrolled in the MILD trial (Table 1) [18]. Tissue specimens were obtained according to the Internal Review and the Ethics Boards of the Fondazione IRCCS Istituto Nazionale Tumori of Milan (INT 53/05), and the study was accomplished in accordance with the Declaration of Helsinki. All patients provided informed consent.
Table 1.
Clinicopathological characteristics of patients for analysis on lung tissue.
| Trial MILD (n = 38 tissues) | |
|---|---|
| Gender | |
| Male | 30 (75%) |
| Female | 10 (25%) |
| Age (years) | 62.08 + 6 (S.D.) |
| Smoking habit (Pack‐Year index) | 55.5 + 26.6 (S.D.) |
| Histotype | |
| Adenocarcinoma | 27 (67.5%) |
| Squamous cell carcinoma | 10 (25%) |
| Other | 3 (7.5%) |
| Stage | |
| Ia–Ib | 28 (70%) |
| II–III–IV | 12 (30%) |
| Prognosis | |
| Alive | 26 (65%) |
| Alive with disease | 4 (10%) |
| Dead | 8 (20%) |
| Not available | 2 (5%) |
2.2. Reagents and chemicals
Lipids are as follows: hydrogenated soy phosphatidylcholine (HSPC), cholesterol (CHE), 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[methoxy(polyethylene glycol)‐2000] (DSPE‐PEG2000) and 1,2‐dioleoyl‐3‐trimethylammonium propane (DOTAP) (Avanti Polar Lipids, Inc., Alabaster, AL, USA). miRVana™ miRNA Mimic Custom miR‐126‐3p (m126) (ID# MC12841; Ambion, Thermo Fisher Scientific, Waltham, MA, USA) and miRCURY LNA™ miRNA Custom Inhibitor were used in vivo on a large scale [negative control A (SCR), ID# 339203YC10201876‐FZA; I‐HAS‐MIR221‐3p (i221), ID# 339203YC10202385‐FZA] (QIAGEN, Hilden, Germany). All other reagents were of analytical‐grade purity or the highest available purity (Sigma‐Aldrich, St. Louis, MO, USA).
2.3. Cell lines and siRNA, plasmid and miRNA transfection
The human lung cancer cell lines, Calu1, A549 and H460, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI 1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) medium supplemented with 10% heat‐inactivated FBS and 1% penicillin–streptomycin (Sigma‐Aldrich). Immortalized bronchial–epithelial cells and their genetically modified variants (HBEC1: hTERT + Cdk4; HBEC6: hTERT + Cdk4 + sh‐p53 + KRASV12; HBEC‐KRAS: hTERT + Cdk4 + sh‐p53 + KRASV12high) were obtained from J. Minna (UT Southwestern, TX) and were described previously [19]. Human primary normal human bronchial epithelial (NHBE) cells, small airway epithelial cells (SAECs) and human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Basel, Switzerland).
Cells were transfected with constructs at the following concentrations: negative control A mirVana miRNA mimic, negative control mim‐SCR, 50 nm (Ambion, Thermo Fisher Scientific) or miRCURY locked nucleic acid (LNA)‐SCR, 50 nm, (Exiqon, QIAGEN); mirVana miRNA mimic‐126‐3p (ID: MC12841; Ambion, Thermo Fisher Scientific), 50 nm; miRCURY miRNA inhibitor LNA‐221 (ID: YI04100607‐ADA, Exiqon, QIAGEN), 50 nm; and a combination of mimic‐126+LNA‐221 25 nm + 25 nm. Lipofectamine 2000 (Thermo Fisher Scientific) was used for transfections according to the manufacturer's instructions. PTEN transfection was performed using a PTEN expression plasmid (OriGene, Rockville, MD, USA) at 72 h following the company's protocol. PIK3R2 silencing was accomplished using 50 nm PI3K p85β small interfering RNA (siRNA; Santa Cruz, Dallas, TX, USA) according to the manufacturer's instructions after 72 h of transfection. CXCR4 silencing was performed using 100 nm MISSION® esiRNA targeting human CXCR4 (EHU022821‐20UG; Sigma‐Aldrich) according to the manufacturer's instructions (72 h of transfection).
2.4. miRNA and gene expression analysis
RNA was extracted from cells at 72 h post‐transfection and from tissues using a mirVana Paris kit (Thermo Fisher Scientific). Reverse transcription and real‐time PCR were performed as previously described [20].
For gene expression analysis, cDNA synthesis was performed using 250 ng of total RNA. Relative quantification of the analysed genes was performed using a TaqMan assay (Thermo Fisher Scientific), and GAPDH was used as the endogenous control.
2.5. Viability assay
For the viability assay, cells were seeded into a 96‐well plate (5 × 103 cells/well), and luminescence was measured after 72 h using a RealTime‐Glo MT Cell Viability Assay (Promega, Milan, Italy). Each experiment was performed in quintuplicate.
2.6. Apoptosis evaluation
Apoptosis was measured by quantifying the percentage of Annexin V/propidium iodide‐positive cells by flow cytometry (Miltenyi Biotech, Bergisch Gladbach, Germany) after 72 h of transfection, as previously described [20].
2.7. miRNA ISH
miRNA in situ hybridization (ISH) was performed on FFPE tissue sections as previously described [15]. Briefly, a combination of double DIG‐conjugated mirCURY LNA probes (Exiqon, Vedbæk, Denmark) and an automatic DAB chromogenic detection system was used for detection of miRNAs. Probes selected for ISH analysis are listed in Table S1. A scramble probe was used as negative control.
Samples were hybridized with probe mixture for 2 h, in the Dako Hybridizer, at specific probe hybridization temperature (RNA Tm −30 °C) (Table S1). After washing steps, miRNA expression was automated detected with the Ventana BenchMark ULTRA instrument using the OptiviewDAB Detection Kit (Ventana Medical Systems Arizona, Oro Valley, AZ, USA). For image analysis, stained sections were scanned with Aperio ScanScope XT (Leica Biosystems, Nussloch, Germany) and miRNAs signals were quantified as number of positive cells/total cells by counting three random field for each slide.
2.8. Proteomic array and western blot analyses
Proteins were extracted by incubation with RIPA buffer and quantified by the Bradford method. Forty‐five micrograms of protein was added to a human apoptosis array (R&D Systems, Minneapolis, MN, USA) and analysed following the manufacturer's instructions. Western blot (WB) analysis was performed as previously described [20] using the following antibodies: mouse anti‐cleaved caspase 8 (clone: 1C12; #9746), rabbit anti‐phospho‐AKT (Ser473) (clone: D9E; #4060), rabbit anti‐AKT (clone: 11E7; #4685), rabbit anti‐PIK3R2 (clone: 19H8; #4257), rabbit anti‐PTEN (#9552), rabbit anti‐TRAILR 1 (clone: D4E9; #8074), rabbit anti‐TRAILR 2 (clone: D9S1R; #42533), rabbit anti‐FADD (Human Specific; #2782), mouse anti‐FAS (clone: 4C3; #8023), rabbit anti‐p21( Waf1/Cip1; clone: 12D1 #2947), rabbit anticleaved caspase 3 (clone: Asp175; #9661) (all from Cell Signaling, Danvers, MA, USA, diluted 1 : 1000), rabbit anti‐CXCR4 (clone: UMB2; ab124824, Abcam, Cambridge, UK) mouse monoclonal anti‐β‐actin antibody (clone: AC‐74) (Sigma‐Aldrich A2228, 1 : 2000) horseradish peroxidase‐conjugated goat anti‐rabbit or goat anti‐mouse secondary antibodies (GE Healthcare, Chicago, IL, USA, 1 : 5000). Signal detection was performed via chemiluminescence (ECL, GE Healthcare) using a MINI HD9 Western Blot Imaging System (Cleaver Scientific Ltd, Warwickshire, UK). Band densities were quantified in imagej software (National Institutes of Health, Bethesda, MD, USA).
2.9. Flow cytometry
To analyse CXCR4 modulation, cells (A549, Calu1 and H460) were incubated with an anti‐human CD184 (CXCR4) monoclonal antibody, APC conjugated (clone: 12G5; # 17‐9999‐41 eBioscience, San Diego, CA, USA) diluted 1 : 100 in PBS for 15 min at room temperature and subsequently washed two times with PBS. Total events were analysed using a BD FACSCanto II with flowjo software (BD Biosciences, San Jose, CA, USA).
2.10. Migration, invasion and transendothelial migration assays
The migratory and invasive abilities of lung cancer cells (A549, Calu1 and H460) were assessed as previously described [20]. For the transendothelial migration assay, 1 × 105 HUVECs were plated into the upper chambers of 24‐well cell culture inserts coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). One day later, 2 × 104 PKH‐26‐labelled (A549, Calu1 and H460) cells were plated in the upper chambers of the inserts. For invasion, migration and transendothelial evaluation of four random fields per condition at 20× magnification were acquired after 48 h. The numbers of migrated cells (DAPI+ cells for invasion and migration assays and PKH‐26+ cells for transmigration assay) were counted using the imagej software.
2.11. Coated cationic lipid nanoparticles
The cationic lipid (DOTAP) amount is modified according to the number on negative charges of synthetic miRNA and miR‐LNA molecules [21, 22, 23]. Small, stable neutral coated cationic lipid nanoparticles (CCLs) encapsulating miRNA‐126 mimic (CCL‐126), miR‐221 LNA‐modified inhibitor (CCL‐221) or negative sequence (CCL‐SCR) molecules were prepared and purified as previously described [21, 22, 24]. The amounts and percentages of miRNAs encapsulated in the CCLs were evaluated by solubilizing lipid nanoparticle preparations with 40 mmol·L−1 sodium deoxycholate for 1.5 h at room temperature prior to spectrophotometric measurement at 260 nm. The good stability in media approximating the physiological conditions of the lipid nanoparticle preparations was evaluated by measuring the particle size, hydrophobic diameter, polydispersity index and zeta potential using a Malvern Nano ZS90 light scattering apparatus (Malvern Instruments Ltd, Worcestershire, UK).
2.12. IHC analysis
H&E staining was performed, and the levels of the Ki‐67, PTEN, PIK3R2 and cleaved caspase 3, cytokeratins and p‐AKT proteins were investigated using IHC staining. In brief, sections 2.5/3 μm thick were cut from paraffin blocks, dried, dewaxed, rehydrated and unmasked (with Dako PT‐link, EnVision™ FLEX Target Retrieval Solution, High/Low pH). Anti‐MIB1 (monoclonal; clone: MIB 1 Dako, Santa Clara, CA, USA, diluted 1 : 400), anti‐human cytokeratin (clones: AE1/AE3 – Dako – dilution 1 : 100; #M3515), anti‐PTEN (clone: A2B1: sc‐7974, diluted 1 : 50), (clone: 19H8; #4257; diluted 1 : 100), anti‐cleaved caspase 3 (clone: Asp175; #9661; diluted 1 : 100) and anti‐phospho‐AKT (Ser473) (clone: D9E; #4060; diluted 1 : 100), anti‐CXCR4 (clone: D4Z7W; #97680; diluted 1 : 800) (all from Cell Signaling) incubated with a commercially available detection kit (EnVision™ FLEX+, Dako) in an automated immunostainer (Dako Autostainer link 48). Images of sections were acquired with an Aperio ScanScope XT (Leica Biosystems, Aperio, Wetzlar, Germany) at 400× magnification, and four random fields were acquired for each condition. The percentage of positive cells was then evaluated by counting the positive cells out of the total number of cells. The Ki‐67 proliferation index was evaluated by the percentage of Ki‐67‐expressing cells among at least 500 cells counted in the areas of the strongest nuclear labelling (hot spots). For the necrosis evaluation, samples were stained with haematoxylin and eosin and the images were acquired with Aperio ScanScopeXT® (Leica Biosystems, Aperio) at 20× and 200× magnifications.
The necrosis was quantified with the imagescope software (Aperio) using a positive pixel count algorithm. The staining intensity was classified as strongly positive (red), positive (orange), weakly positive (yellow) or negative (blue). The algorithm identifies yellow, orange and red as varying degrees of pink‐positive pixel of the eosin staining while blue as negative nuclei of deep blue‐purple colour‐pixel of haematoxylin staining. The percentage of necrosis in the tumour tissue samples, defined as increased eosinophilia in haematoxylin and eosin stains, was calculated as a total‐positive pixel staining divided by the total amount of pixels.
2.13. Cytokine quantification
The plasma levels of 12 cytokines (IL‐1α, IL‐1β, IL‐2, IL‐4, IL‐5, IL‐6, IL‐10, IL‐12, IL‐13, IL‐17a, G‐CSF and GM‐CSF) in mice treated with lipid nanoparticles for 4 weeks were analysed using Mouse Common Cytokines Multi‐Analyte ELISArray Kits following the manufacturer's instructions (Qiagen). The absorbance at 450 nm was measured in an Infinite M1000 plate reader (Tecan, GmbH, Grodig/Salzburg, Austria).
Interferon‐α and interferon‐β were analysed using Mouse IFN‐alpha and beta ELISA Kit (Abcam).
2.14. Luciferase assay
To investigate whether PIK3R2, PTEN and CXCR4 are a direct target of mir‐126 and miR‐221, the 3′UTR of PIK3R2 (ID: S813973), PTEN (ID: S809030) and CXCR4 (ID: S803938) were purchased from Switchgear Genomics and analysed as previously described [20]. Predicted target sites for miR‐126 and miR‐221 were mutated by direct mutagenesis of the pLightSwitch 3′UTR vectors encoding the 3′UTR of CXCR4, PTEN and PIK3R2, using the PCR‐based QuikChange II site‐directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions and the following primers:
CXCR4Mut‐FW 5′‐GTATGTCTCGTGTCCAGACTGTAGAAAAG‐3′
CXCR4Mut‐Rev 5′‐CTTTTCTACAGTCTGGACACGAGACATAC‐3′
PTENMut‐FW 5′‐AGGTTGTTCTACTGTGTCATGTATATAC‐3′
PTENMut‐Rev 5′‐GTATATACATGACACAGTAGAACAACCT‐3′
PIK3R2Mut‐FW 5′‐GCAGGTTTTGTATTTCACGTTGTTATTG‐3′
PIK3R2Mut‐Rev 5′‐CAATAACAACGTGAAATACAAAACCTGC‐3′
The presence of the mutations was confirmed by sequencing (Eurofins Genomics, Ebersberg, Germany).
2.15. PDXs and in vivo assays
Patient‐derived xenografts were established as previously described [25]. The PDX used for the experiment, derived from a lung cancer patient with a squamous cell carcinoma, stage IIIa. Mice were maintained in the Animal Facility of Fondazione IRCCS Istituto Nazionale dei Tumori. Animal experiments were authorized by the Institutional Animal Welfare Body and Italian Ministry of Health and were performed in accordance with national laws (D.lgs 26/2014). All experiments were carried out with female severe combined immunodeficient (SCID) mice aged 7–10 weeks (Charles River Laboratories, Calco, Italy). All animals were kept in IVC cages (max 5 animals/cage) in the Animal Facility of the ‘Fondazione IRCCS Istituto nazionale dei Tumori’. (SPF animal facility with 20 Hepa‐filtered air change/hour). Cages were maintained in rooms with controlled temperature and humidity, and 12/12‐light/dark cycles. Before starting experimentation, animals underwent at least 7 days of adaptation period. Cages were enriched with nesting material.
Lung cancer cells transfected (A549 and Calu1) with the miR‐126‐3p mimic, miR‐221‐3p inhibitor or SCR control were harvested 24 h post‐transfection and resuspended in Matrigel/RPMI (1 : 1). A total of 5 × 105 cells were injected subcutaneously into both flanks of the mice (n = 5 tumours/group).
For the lung colonization assay, 5 × 105 A549 or H460 cells transiently transfected with the miR‐126‐3p mimic or miR‐221‐3p inhibitor were injected intravenously into immunodeficient mice (n = 4 for each cell line). For the experimental metastasis assay, 3 weeks after injection, mice were sacrificed, and the lungs were harvested and analysed by both IHC staining and flow cytometry. Detection of tumour cells within dissociated murine lung tissue was performed by FACS analysis to exclude dead cells and MHC‐I‐positive murine cells, as previously reported [24].
Aerosol inhalation was performed by nebulizing 1.5 mg·kg−1 miR‐126‐3p in 200 µL of PBS in a 2‐chamber BIAS flow generator (model AIR421; EMMS Supporting Science, Edinburgh, UK). In brief, beginning 2 days after injection of H460 cells, mice were treated twice weekly for two weeks, and cell dissemination was analysed as described above.
For CXCR4 inhibition experiment, H460 cells (1 × 105) in Matrigel (Corning, New York, NY, USA) + culture medium (1 : 1) were injected subcutaneously into the flanks of SCID mice. Mice were treated i.p. with 2 mg·kg−1 anti‐CXCR4 peptide R twice daily starting on the day after cell injection, and tumour growth was monitored by calliper measurement. At the end of the observation period (5 weeks), mice were sacrificed, and lungs were harvested for FACS and IHC analyses.
Mice bearing a PDX tumour in either flank were treated i.p. with CCL‐SCR, CCL‐126, CCL‐221 or an equimolar combination of CCL‐126 and CCL‐221 (CCL‐combo; total amount: 1.5 mg·kg−1) twice weekly for four weeks. Each time, mice received a total volume of 200 µL of a freshly diluted solution of lipid nanoparticles in HEPES‐buffered saline. The dose and schedule of treatment were selected based on our previous study [24]. Tumour growth was measured weekly using a calliper, and the results were analysed using graphpad prism 5 software (GraphPad Software, San Diego, CA, USA).
2.16. Statistical analysis
For the overall survival (OS) endpoint, the time‐to‐event occurrence was computed from the date of the surgery to the date when the event was recorded or the patient was censored at the date of the last follow‐up assessment (for event‐free patients). Survival curves were estimated using the Kaplan–Meier method and were compared by the log‐rank test. The Spearman correlation coefficient was also calculated. Statistical analyses were performed using graphpad prism 5 software. The results are presented as the mean ± standard errors of the mean (S.E.M.) values for quantitative data. Statistically significant differences were determined with Student's t‐test when comparing two groups or ANOVA test for multiple comparisons. For in vivo tumour growth, two‐way ANOVA test was used. P‐values < 0.05 were considered statistically significant.
3. Results
3.1. Analysis of miRNA levels in lung tumour tissues
In our previous work [14], a microarray analysis of 28 tumours and 24 paired normal lung tissues from patients in a low‐dose computed tomography screening trial cohort was performed. In that study, 56 miRNAs were identified as differentially expressed between tumour and normal tissues. Among those miRNAs, 30 miRNAs with a potential role in lung carcinogenesis were identified by correlation analysis with the clinical characteristics of the patients. The association of miRNAs with disease aggressiveness was evaluated by RT‐PCR analysis of the expression of the previously identified miRNAs (Table S2) in 38 lung tumours and paired normal tissues from patients in the Multicentric Italian Lung Detection (MILD) screening trial [14]. Among the 30 analysed miRNAs, miR‐210, miR‐21 and miR‐221‐3p were overexpressed in lung tumours, whereas miR‐30a, miR‐126‐3p, miR‐451 and miR‐486 were downregulated in lung tumours compared with normal tissues (Fig. 1A). Furthermore, the deregulation of these miRNAs in tumour cells was confirmed and quantified by miRNA ISH in tissues from lung cancer patients (Fig. 1B and Fig. S1A).
Fig. 1.
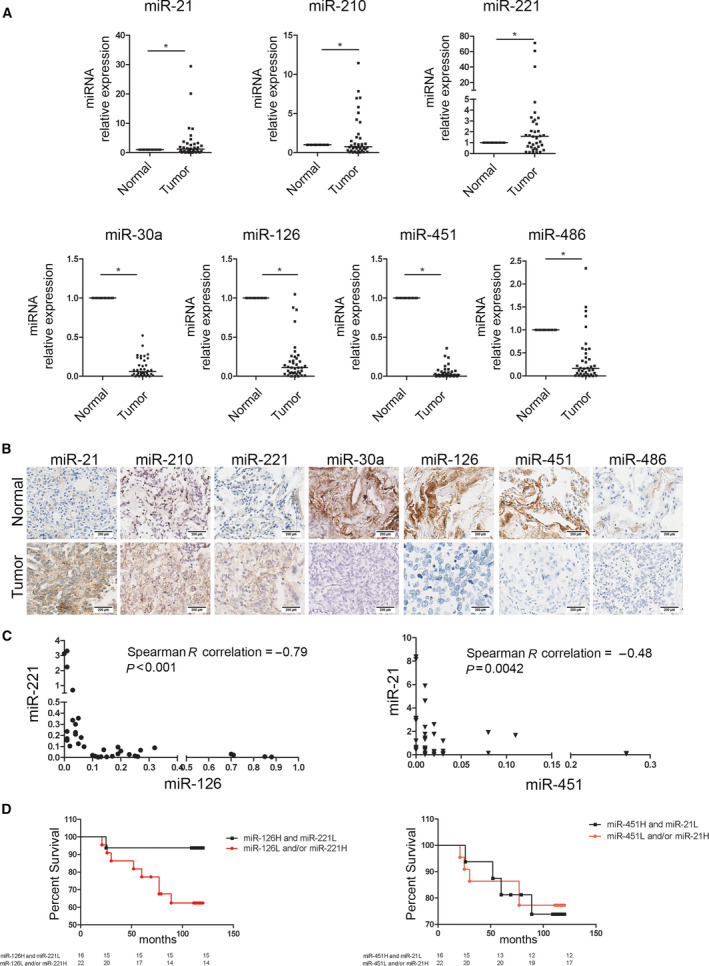
miRNAs deregulated in lung cancer tissues. (A) Dot plots showing miRNA median levels in lung cancers compared with distant normal tissues (n = 38 paired samples). *P < 0.05 versus controls. Data are expressed as mean ± standard error of the mean (S.E.M.). Statistically significant differences were determined with Student's t‐test. (B) miRNA levels in lung cancer tissues compared with normal tissues evaluated by miRNA ISH (n = 3). Scale bar: 200 µm. (C) Correlation graphs according to miR‐126/miR‐221 or miR‐451/miR‐21 expression levels in tumour tissues. (D) Kaplan–Meier curves illustrate the association with OS considering the presence of one or two unfavourable markers according to miRNA expression.
Potential additive or synergistic effects of miRNAs deregulated in lung cancer tissues were assessed by correlation analysis between all the significantly modulated miRNAs to identify a more efficient anticancer strategy (Table S3).
Among all the differentially modulated miRNAs in lung cancer, we observed a significant negative correlation between miR‐126‐3p and miR‐221‐3p levels and between miR‐21 and miR‐451 levels, suggesting that these miRNAs could synergistically affect lung carcinogenesis (Fig. 1C). These findings led us to investigate the clinical utility of these miRNAs. Patient survival was evaluated according to the miRNA levels in tumour cells, with the median values of the investigated miRNAs used as the cut‐off values. As illustrated in Fig. 1D, 94% of patients with high miR‐126‐3p and low miR‐221‐3p levels in cancer tissues survived at 10 years, whereas the survival rate of patients with one or both unfavourable markers (miR‐126‐3p low and/or miR‐221‐3p high) was significantly lower 63% (Hazard Ratio 4.080, P = 0.0369) (Table S4). No differences were observed in the analysis of miR‐21 and miR‐451 levels according to patient survival (Fig. 1D and Table S5).
The expression of the identified miRNAs was confirmed in normal (NHBE, SAEC), nontumorigenic (HBEC1, HBEC6, HBEC‐KRASV12high) and tumorigenic (H460, Calu1, A549) lung cell lines (Fig. S1B).
3.2. Concomitant modulation of miR‐126‐3p and miR‐221‐3p exhibits antitumour activity in vitro
To identify a potential therapeutic approach for lung cancer management, we developed a cotargeting strategy by simultaneously modulating miR‐126‐3p and miR‐221‐3p using commercially available synthetic oligonucleotides: a miR‐126‐3p mimic (m126) and a miR‐221‐3p inhibitor (i221). In vitro experiments were performed in A549 and Calu1 cells, which were selected because of the opposite miRNA levels detected in these cell lines (Fig. S1C,D).
First, we verified that the transfection with negative sequence for mimic (mim‐SCR) and for inhibitor (LNA‐SCR) did not alter proliferation of lung cancer cells as shown in Fig. S2A. Interestingly, compared with the negative sequence (SCR) control, only combined replacement of miR‐126‐3p with inhibition of miR‐221‐3p decreased the viability of all lung tumour cell lines after 72 h (Fig. 2A). Since single miRNA modulation did not affect proliferation, our results suggested a cooperative antitumour effect exerted by combined replacement of miR‐126‐3p and inhibition of miR‐221‐3p in lung cancer cells. Moreover, the antiproliferative effect of the combined treatment was also confirmed after 120 h post‐transfection (Fig. S2B). The proliferation rate of nontumorigenic human bronchial epithelial cells (HBEC‐KRASV12high) was analysed to demonstrate whether miRNA modulation affects normal epithelial cells. As shown in Fig. 2B, we did not observe any significant changes in cell viability at 72 h after combined miR‐126‐3p replacement and miR‐221‐3p inhibition.
Fig. 2.
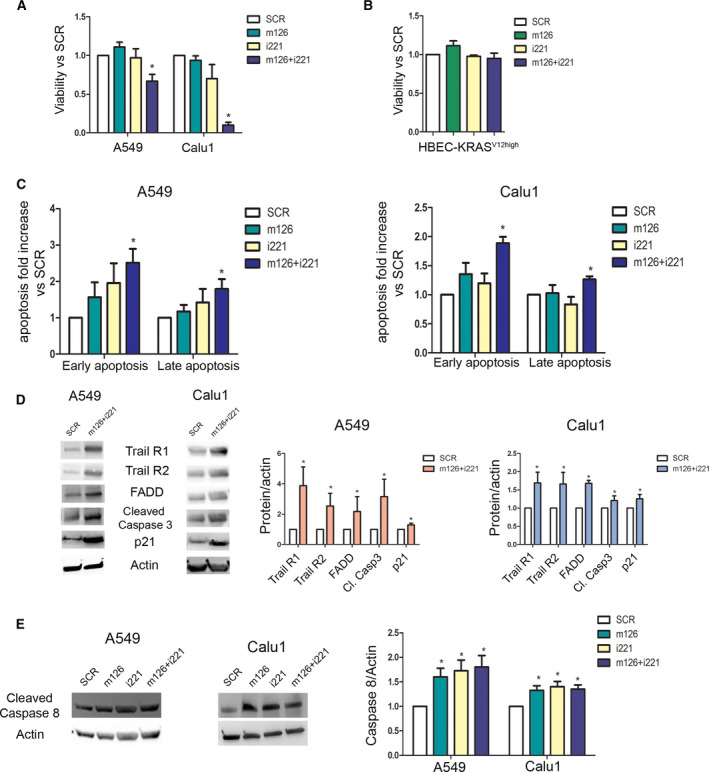
miR‐126‐3p and miR‐221‐3p modulation exhibited anti‐tumour activity in vitro. (A) Bar plots illustrate viability rate after miR‐126‐3p replacement and miR‐221‐3p inhibition compared with scramble (SCR) control (n = 5 for each cell). (B) Bar plots illustrate viability rate after miR‐126‐3p replacement and miR‐221‐3p inhibition compared with SCR control (n = 5 for each cell) in HBEC‐KRASV12high cells. (C) Flow cytometry analysis showed an increase in the number of early apoptotic (Annexin V+/PI−) and late apoptotic (Annexin V+/PI+) cells (n = 5 for each cell). (D) Representative images (left) and quantification (right) of western blot indicating the activation of apoptosis after miR‐126‐3p and miR‐221‐3p modulation (n = 3 for each cell). (E) Western blot band images (left) and quantification (right) demonstrated induction of caspase 8 after miRNA transfection (n = 5). Statistically significant differences were determined with Student's t‐test when comparing two groups or ANOVA test for multiple comparisons.*P < 0.05 versus SCR. Data are expressed as mean ± S.E.M.
Moreover, the effects on apoptosis and the cell cycle were examined to better comprehend the means by which miRNA modulation caused the reduction in cancer cell viability. Only simultaneous modulation of both miRNAs resulted in increased numbers of early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cells at 72 h (Fig. 2C and Fig. S2C) and 120 h post‐transfection (Fig. S2E). However, we did not observe any significant changes in the cell cycle (Fig. S3A). We evaluated apoptosis in transfected HBEC‐KRASV12high cells and observed no changes in the numbers of apoptotic cells after 72 h compared with those in SCR control‐transfected cells (Fig. S3B). The activation of the apoptotic cascade was studied by analysis of a multiple protein array that detects 35 apoptosis‐related proteins. In all cell lines, simultaneous blockade of miR‐221‐3p and supplementation of miR‐126‐3p (m126+i221) increased the expression of TRAIL receptors 1 and 2 with concomitant upregulation of FADD and Fas, implying activation of the TRAIL‐dependent apoptotic pathway at 72 h post‐transfection (Fig. S3C). The activation of the apoptotic process was confirmed by the significant increase in the cleaved caspase 3 level in all m126+i221‐treated cells (Fig. S3D). Interestingly, the p21 level increased after modulation of miR‐126‐3p and miR‐221‐3p (Fig. S3D). The activation of the apoptotic cascade evidenced in Fig. S3C,D was also confirmed and quantified by WB analysis (Fig. 2D). An increase in the cleaved caspase 8 protein level after miRNA deregulation was observed in both cell lines (Fig. 2E), indicating the induction of TRAIL‐mediated apoptosis.
3.3. PIK3R2 and PTEN are direct targets of miR‐126‐3p and miR‐221‐3p
Based on the observed in vitro results, putative targets of miR‐126‐3p and miR‐221‐3p, primarily those encoding proteins involved in the AKT pathway, were investigated by an in silico approach. This analysis revealed that the 3′UTRs of PIK3R2 and PTEN contain complementary binding sequences for miR‐126‐3p and miR‐221‐3p, respectively (Fig. 3A). The direct binding of these miRNAs with their targets was further demonstrated by a luciferase assay in A549 cells. Interestingly, we observed downmodulation of the luciferase activity of the PIK3R2 reporter after cotransfection with the miR‐126‐3p mimic and an increase in the PTEN reporter luciferase activity after cotransfection with miR‐221‐3p inhibitor (Fig. 3B). No modulation of luciferase activity was observed after cotransfection with an empty vector containing no 3′UTR or by site‐directed mutagenesis in the putative miRNA‐binding sites (Fig. 3B).
Fig. 3.
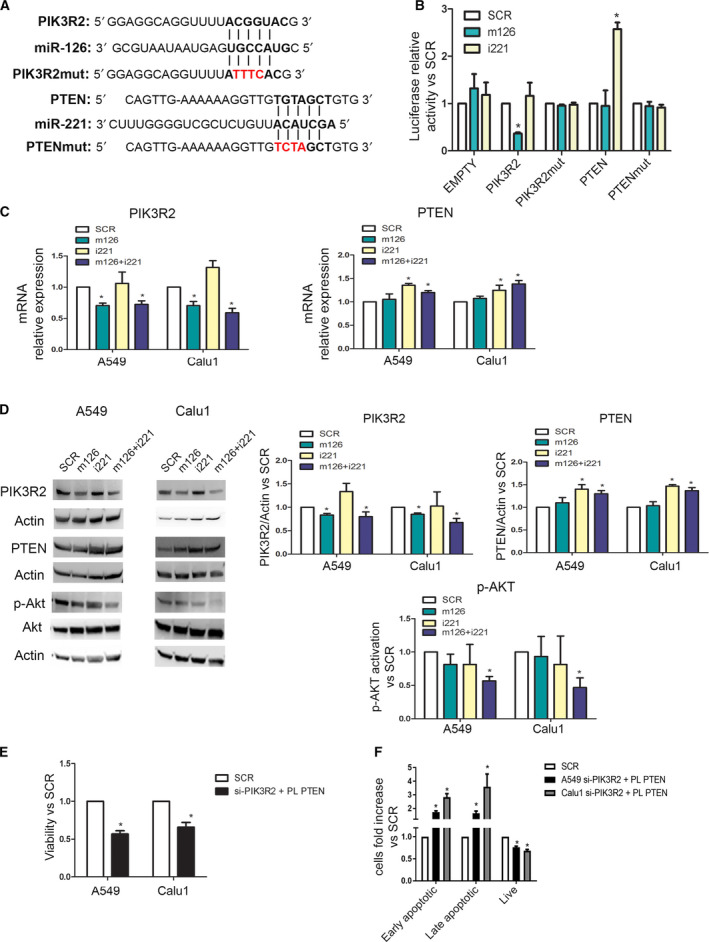
PIK3R2 and PTEN are direct targets of miR‐126‐3p and miR‐221‐3p. (A) miR‐126‐3p and miR‐221‐3p seed sequence alignments with the 3′UTRs of PIK3R2 and PTEN, respectively. (B) Bar graphs showing average luciferase activity of A549 transfected with PIK3R2, PTEN and EMPTY 3′UTR wild‐type or mutated in combination with miR‐126‐3p mimics or miR‐221‐3p inhibitors or control (n = 3). (C) Real‐time analysis of PIK3R2 and PTEN mRNA after miR‐126‐3p upregulation and miR‐221‐3p inhibition (n = 3 for each cell line). (D) Western blot bands and histogram revealed PIK3R2 downmodulation after miR‐126‐3p replacement. Inhibition of miR‐221‐3p increases PTEN expression as illustrated by western blot and bands' quantification (n = 3 for each cell). Phospo‐Akt (left) and histogram quantification (right) were observed after concomitant miRNA modulation (n = 3). (E) Silencing of PIK3R2 and overexpression of PTEN reduced viability of cancer cells (n = 5 for each cell). (F) The number of apoptotic cells was higher after silencing of PIK3R2 and PTEN upregulation (n = 5 for each cell). *P < 0.05 versus controls. Data are expressed as mean ± S.E.M.
A significant reduction in PIK3R2 expression at either the mRNA (Fig. 3C) or protein (Fig. 3D) level was detected in all cell lines 72 h after miR‐126‐3p replacement alone or in combination with miR‐221‐3p inhibition. Furthermore, downmodulation of miR‐221‐3p expression in lung cancer cells increased the intracellular level of PTEN, as detected by qPCR (Fig. 3C) and western blotting (Fig. 3D). Importantly, only simultaneous miR‐126‐3p replacement and miR‐221‐3p inhibition resulted in blockade of AKT phosphorylation, as evaluated and quantified by WB (Fig. 3D) that may subtend the observed reduction in cancer cell viability (Fig. 2A).
To demonstrate that the antitumour effects of miR‐126‐3p and miR‐221‐3p were caused by the modulation of PIK3R2 and PTEN, PIK3R2 was silenced using a commercially available siRNA, and PTEN was overexpressed by transfection of a specific plasmid (Fig. S4A). After PIK3R2 silencing and PTEN overexpression, a reduction in cell viability compared with that in the control group was observed at 72 h post‐transfection (Fig. 3E). Moreover, an increase in the number of apoptotic cells (Fig. 3F) and concomitant blockade of AKT phosphorylation were observed in these cells after 72 h (Fig. S3A).
3.4. Concomitant modulation of miR‐126‐3p and miR‐221‐3p reduces lung metastatic dissemination
The potential role of the observed miRNAs in blockade of the lung cancer dissemination process was evaluated by analysing both the migratory and invasive abilities of three lung cancer cell lines (A549, Calu1 and H460). Based on our previous work [26], we analysed the anti‐metastatic activity of miR‐126 and miR‐221 also in the metastatic H460 lung cancer cells.
Interestingly, marked reductions in the cell migratory (Fig. 4A and Fig. S5A) and invasive (Fig. 4B and Fig. S5B) abilities after modulation of miR‐126‐3p alone or in combination with miR‐221‐3p were seen compared with those observed after transfection with the SCR control. Simultaneous miRNA modulation was more effective compared with miR‐126 alone, except for H460 cells likely due to the absence of miR‐221 (Fig. S1C). The in vitro transendothelial migration assay demonstrated that combination of miR‐126‐3p mimics and miR‐221 inhibitors reduced the number of cancer disseminating cells with respect to that of the corresponding control cells (Fig. 4C and Fig. S5C). To investigate whether the observed effect on migration is proliferation‐independent, we evaluated proliferation and demonstrated that it was not modulated by miRNA modulation after 24 h (Fig. S6A). Overall, these in vitro data indicate that combined miR‐126 replacement and miR‐221 inhibition are a candidate target that can inhibit the lung cancer dissemination process.
Fig. 4.
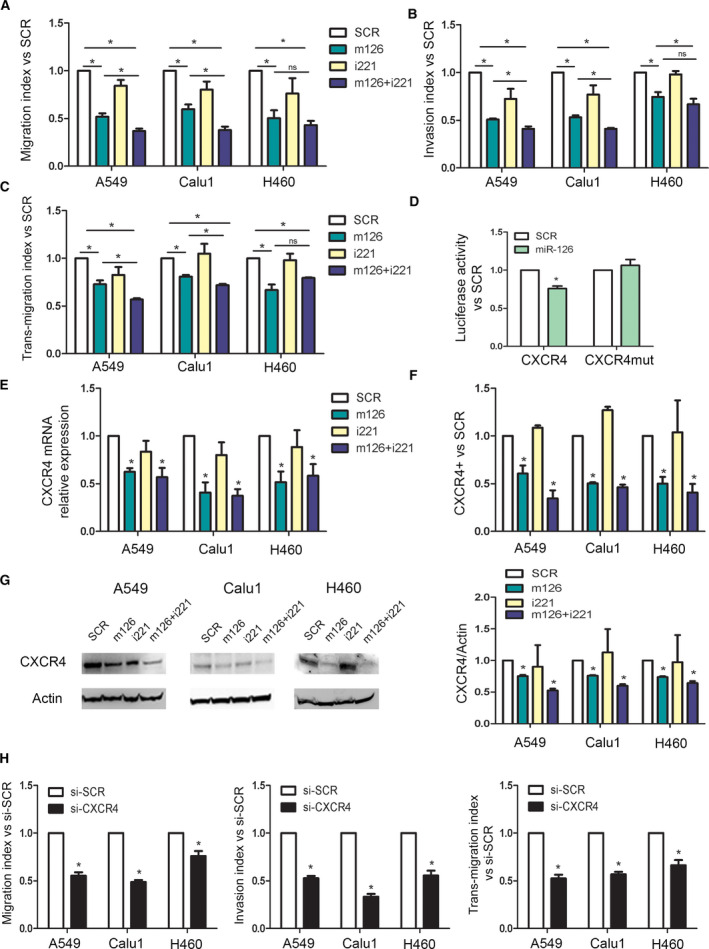
miR‐126‐3p replacement and miR‐221 inhibition reduced lung dissemination by inhibiting CXCR4 levels. (A) Migration index after miR‐126‐3p and miR‐221‐3p modulation of three lung cancer cells (n = 5). (B) Invasive capacity of lung cancer cells transfected with miR‐126/221 (n = 4). (C) Number of transmigrated lung cancer cells through endothelial monolayer after miRNA modulation (n = 3). (D) Luciferase activity for 3′UTR of CXCR4 wild‐type and mutated sequence after miR‐126 and SCR transfection in A549 cells (n = 4). CXCR4 levels after miR‐126‐3p replenishment and miR‐221‐3p inhibition evaluated by qPCR (n = 3) (E) and flow cytometry (n = 3) (F). (G) Representative images and quantification of CXCR4 in all the three cell lines after miRNA modulation (n = 3). (H) Migration, invasion and transendothelial index of lung cancer cells transient transfected with CXCR4 siRNA and controls (n = 3 for each cell line). Statistically significant differences were determined with Student's t‐test when comparing two groups or ANOVA test for multiple comparisons. *P < 0.05 versus controls. Data are expressed as mean ± S.E.M.
In silico analysis also revealed miR‐126‐3p binding sites in the 3′UTR of CXCR4, a well‐known receptor involved in metastatic dissemination of lung cancer cells [26]. We confirmed that miR‐126‐3p transfection in A549 caused significant reductions in luciferase activity in CXCR4 3′UTR whereas this inhibitory effect was absent after mutation in the binding site (Fig. 4D and Fig. S7A). miR‐126‐3p reduced CXCR4 mRNA (Fig. 4E) and protein expression using flow cytometry (Fig. 4F) and WB analysis (Fig. 4G). Inhibition of the CXCR4 pathway in transfected cells was also confirmed by the reduction in the phosphorylation of AKT, a downstream signalling molecule in the CXCR4 axis, compared with that in control cells (Fig. S7B).
Moreover, to better comprehend the key role of CXCR4 modulation in lung cancer dissemination, CXCR4 expression was silenced with a commercially available siRNA (si‐CXCR4) (Fig. S7C), and alterations in the migratory and invasive capacities were examined. Interestingly, si‐CXCR4 cells showed reduced migration, invasion and transendothelial migration compared with si‐SCR cells (Fig. 4H) that may be driven by reduction in AKT phosphorylation (Fig. S7C).
3.5. Combined miR‐126‐3p and miR‐221‐3p modulation reduces xenograft tumour growth in immunodeficient mice
Subcutaneous (s.c.) injection of A549 cells transiently transfected with both the miR‐126‐3p mimic and miR‐221 inhibitor into SCID mice resulted in a 30% reduction in tumour growth (Fig. 5A and Fig. S8A) compared to that in mice injected with SCR and nontransfected (CTR) control cells after 32 days, adding important in vivo functional validation. The antitumour efficacy of combined miR‐126‐3p and miR‐221‐3p modulation was supported by the decrease in the number of Ki‐67‐expressing proliferating cells in tumours generated from miR‐modulated cells compared with those generated from SCR‐treated cells and the increase in the necrotic area (Fig. S8A). miR‐126‐3p and miR‐221‐3p modulation exerted its antitumoral effect through the induction of apoptotic mechanisms in cancer cells, as shown by the increase in cleaved caspase 3‐positive cells and by blockade of AKT phosphorylation in treated tumours (Fig. 5A and Fig. S8A). A similar effect was also observed after subcutaneous injection of Calu1 cells transiently transfected with the combination of the miR‐126‐3p mimic and miR‐221‐3p inhibitor (Fig. 5B and Fig. S8B). Despite the similar size of tumours from Calu1 cells was observed, immunohistochemical (IHC) staining for Ki‐67 revealed a decrease in the mitotic index in m126+i221 Calu1 tumours compared with SCR Calu1 tumours and an increase in the necrotic area, as evaluated by IHC staining (Fig. S8B). Compared to SCR Calu1 tumours, m126+i221 Calu1 tumours also displayed an increase in the number of cleaved caspase 3‐positive cells and a downmodulation of phosphor‐AKT (Fig. 5B and Fig. S8B), overall suggesting that mir126+i221 treatment was able to induce apoptosis and antitumour effect also in this NSCLC cell line.
Fig. 5.
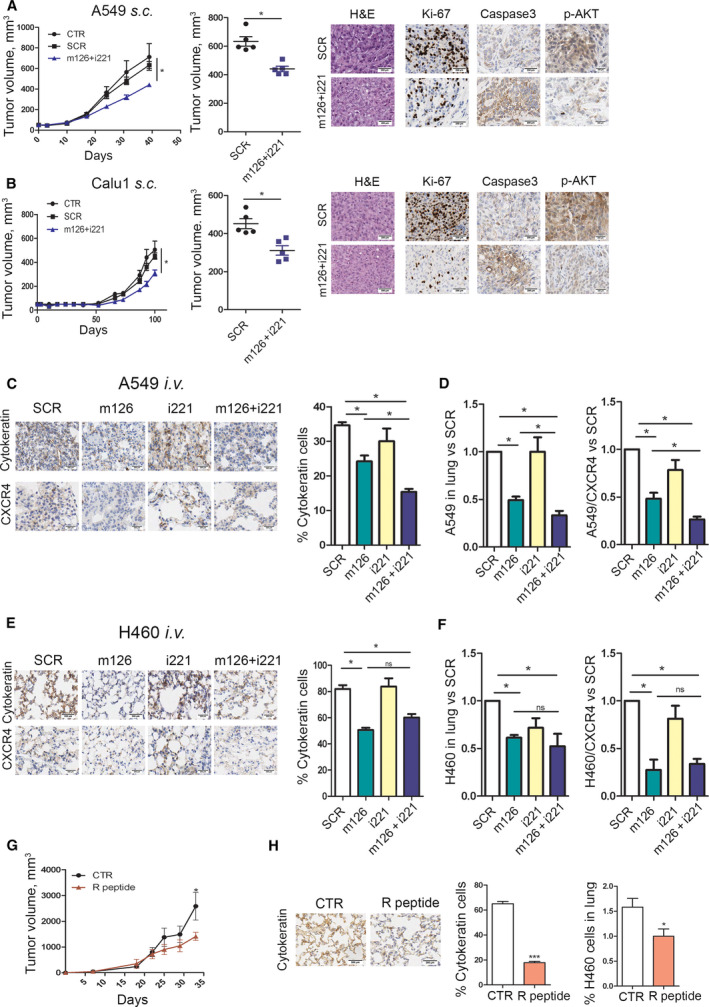
Combined replenishment of miR‐126‐3p and inhibition of miR‐221‐3p reduced xenograft growth in vivo. (A) A549 transfected with both miR‐126‐3p mimic (m126) and miR‐221‐3p inhibitor (i221) showed a reduction in tumour growth in immunodeficient mice compared with SCR and CTR controls. Ki‐67, cleaved caspase 3 and phosphor‐AKT quantification and representative images in A549 m126+i221 cells compared with controls. Necrosis was evaluated on haematoxylin & eosin (H&E) staining in subcutaneously implanted xenograft after miRNA transfection (n = 5 for each group) Scale bar: 200 µm (B) miR‐126‐3p replacement and miR‐221‐3p inhibition reduced tumour growth in subcutaneous Calu1 xenografts in immunodeficient mice. Ki‐67, cleaved caspase 3 and phospho‐AKT count and staining revealed a reduction in proliferation rate and increase in apoptosis of these cells. Necrosis was evaluated on H&E staining (n = 5 for each group). Scale bar: 200 µm. (C) Pan‐cytokeratin and CXCR4 staining and quantification in the lungs of mice treated with A549 transfected miR‐126 and/or miR‐221 alone or in combination (n = 4 for each group). Scale bar: 200 µm. (D) Quantification of A549 and A549/CXCR4+ cells in the lungs of mice by flow cytometry (n = 4). (E) Representative images and quantification of pan‐cytokeratin and CXCR4 staining of the lungs from H460 transfected with miR‐126‐3p, miR‐221 inhibitors alone or in combination and control (n = 4 mice for each group). Scale bar: 200 µm. (F) Flow cytometric quantification of H460‐disseminating cells and H460‐CXCR4 positive in the lung of treated mice (n = 4 mice for each group). (G) Tumour growth curves of mice treated with R peptide and CTR (n = 4 for each group). Scale bar: 200 µm. (H) Pan‐cytokeratin staining and flow cytometric quantification of the lungs from H460 treated with R peptide (n = 4 for each group). Data are expressed as mean ± S.E.M. Statistically significant differences were determined with Student's t‐test when comparing two groups or ANOVA test for multiple comparisons *P < 0.05, ***P < 0.0001.
3.6. Combined miR‐126‐3p and miR‐221‐3p administration reduced lung cancer cell dissemination in mice
Since subcutaneous lung cancer xenografts do not disseminate efficiently to the lungs to form overt metastases [26], we performed an experimental metastasis assay in immunocompromised mice to show the efficacy of miRNA modulation in inhibiting A549 and H460 metastasis in two orthotopic models of lung cancer. Specifically, 5 × 105 lung cancer cells transiently transfected with the miR‐126‐3p mimic and/or miR‐221 inhibitor or a control SCR sequence were injected into SCID mice via the tail vein, and lung nodule growth was monitored after cellular injection. First, we observed that combination of miR‐126 replacement and miR‐221 inhibition was able to reduce the growth of A549 cells in the lungs as shown by H&E and pan‐cytokeratin staining (Fig. 5C). Moreover, we observed a reduction in CXCR4‐positive cells compared with controls (Fig. 5C). Interestingly, also miR‐126 transfection alone was able to significantly reduce metastatic growth (Fig. 5C). Furthermore, by using an already validated gating strategy (Fig. S8C) [26], we observed a reduction in the human cells in the lungs of m126+i221 A549 cells compared with controls (Fig. 5D and Fig. S8D). H&E and pan‐cytokeratin staining demonstrated a reduction in the number of metastatic H460 cells in m126+i221 compared with control tumours (Fig. 5E and Fig. S8E). These data were also confirmed by flow cytometric analysis, which revealed a 40% reduction in the number of H460 cells in the lungs of mice injected with m126+i221 cells compared with that in the lungs of mice injected with control cells (Fig. 5F). Furthermore, replacement of miR‐126‐3p and miR‐221‐3p inhibition induced a reduction in the CXCR4 level in H460 cells in the lung 3 weeks postintravenous (i.v.) injection, as evaluated by determining CXCR4‐positive cells (Fig. 5F). No statistical difference was observed between m126+i221 H460 and m126 H460 in terms of metastasis reduction likely due to the absence of miR‐221 in this cell line.
To further investigate a new method for miRNA administration that could be potential used for clinical applications, we performed a preliminary experiment by treating the lungs via nebulized aerosol inhalation. So to further confirm the importance of miR‐126‐3p in inhibiting metastatic dissemination of lung cancer cells, we injected H460 cells into SCID mice via the tail vein, and after 1 week, mice were treated twice with naked miR‐126‐3p mimics (n = 4 for each group). After confirmation of miR‐126‐3p replacement in the lungs (Fig. S9A), we observed a reduction in H460 cells compared with that in mice treated with the SCR control, as evaluated by IHC staining (Fig. S9B) and flow cytometry (Fig. S9C). Interestingly, miR‐126‐3p mimic treatment reduced the number of CXCR4‐positive H460 cells in the lungs compared with that in mice treated with the SCR control (Fig. S9D).
A cyclic peptide inhibitor of CXCR4 (peptide R), designed as an SDF‐1 mimetic peptide, and already validated to inhibit CXCR4 downstream pathways and to exert anti‐metastatic effects, was utilized to better explain the role of CXCR4 inhibition in the modulation of lung cancer metastatic potential [27, 28]. In detail, H460 cells were inoculated subcutaneously into SCID mice, and the mice were treated daily with peptide R (2 mg·kg−1). We observed a slight reduction in subcutaneous tumour growth after 32 days (Fig. 5G) but a consistent reduction in the dissemination of H460 cells to the lungs (a reduction of 60% compared with CTR), as evaluated by both IHC staining and flow cytometry (Fig. 5H).
3.7. CCL‐combo administration reduces tumour growth in PDX models
The efficacy of combined modulation of miR‐126‐3p and miR‐221‐3p as a novel therapeutic approach for lung cancer management was evaluated by using neutral CCLs [24, 29] encapsulating miR‐126‐3p mimic and miR‐221‐3p inhibitor molecules administered alone (CCL‐126 and CCL‐221, respectively) or in combination (CCL‐combo). In the PDX model, mice were treated intraperitoneally (i.p.) twice weekly for four weeks with CCLs. Figure 6A shows the clear reduction (33%) in the volume of CCL‐combo‐treated PDXs compared to PDXs treated with CCLs containing a negative sequence (CCL‐SCR). PDXs treated with CCL‐126 or CCL‐221 alone did not show a significant reduction in tumour growth compared to those treated with CCL‐SCR. Effective CCL delivery was confirmed by ISH (Fig. 6B,C). The miR‐126/miR‐221 combination also decreased the number of Ki‐67‐positive proliferating cells and also AKT phosphorylation in PDXs (Fig. 6B,C). The reduction in tumour proliferation was confirmed by the 20% increase in the necrotic area observed only in CCL‐combo‐treated tumours and not in tumours treated with CCL‐SCR or CCL‐126/221 alone (Fig. 6B,C). Interestingly, CCL‐combo treatment reduced tumour growth by activating the apoptotic cascade, as demonstrated by the increase in caspase 3‐positive cells (Fig. 6B,C). IHC staining demonstrated the increase in PTEN and downmodulation of PIK3R2 in CCL‐combo treated PDXs (Fig. 6B,C). The modulation of miRNAs and their direct targets PIK3R2 and PTEN was confirmed by real‐time PCR (Fig. S10B,C). Interestingly, cell cycle arrest was also demonstrated by the increase in p21 mRNA expression in CCL‐combo‐treated tumours (Fig. S10C). The safety of our lipid nanoparticles was previously demonstrated in vivo [24, 29]. To exclude nonspecific interferon induction after lipid nanoparticle treatment, we evaluated the levels of interferon alpha (IFN‐α) and beta (IFN‐β) at the end of CCL treatment. Compared to CCL‐SCR, CCL‐combo reduced the IFN‐α expression whereas and CCL‐126 and CCL‐221 alone did not modulate the plasma levels of IFN‐α and β in treated mice (Fig. 6D). Additionally, a panel of 12 pro‐inflammatory cytokines was evaluated to exclude potential chronic toxic effects of the lipid nanoparticles. No appreciable changes in the plasma levels of these cytokines, except for a small increase in IL‐12, were observed in mice treated with CCL‐combo or CCL‐SCR. Therefore, immunological responses to the carrier can be excluded (Fig. 6D). Collectively, these data confirmed the importance of combined treatment with a miR‐126‐3p mimic and a miR‐221‐3p inhibitor to reduce lung cancer growth in vivo.
Fig. 6.
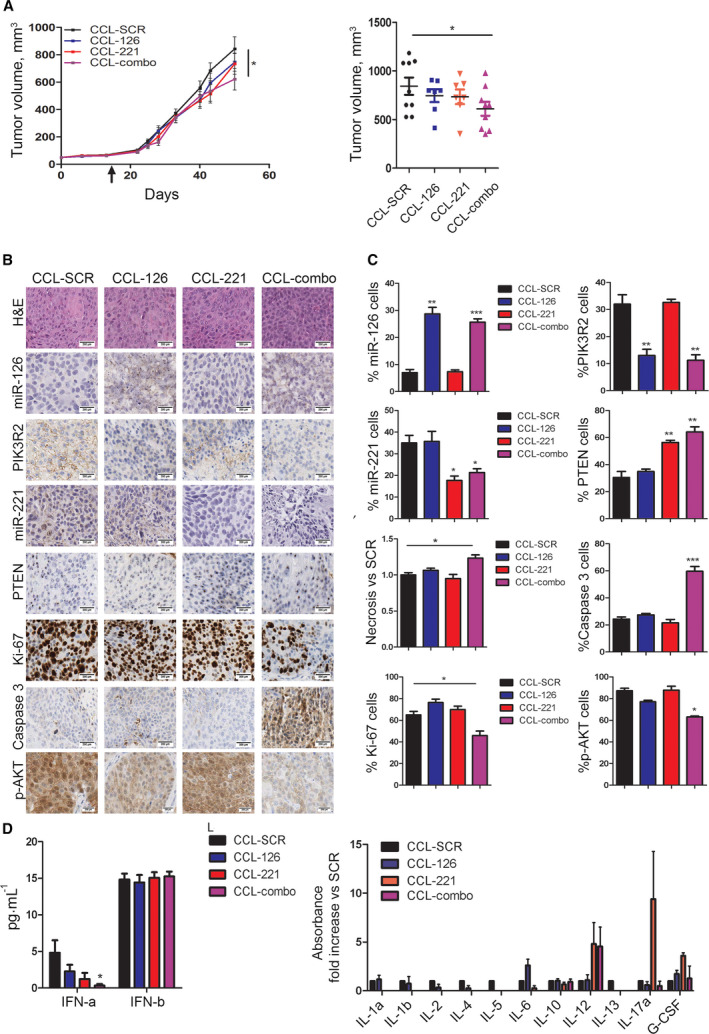
Combined replenishment of miR‐126‐3p and inhibition of miR‐221‐3p reduced tumour growth in vivo. (A) Tumour growth curves and tumour volumes of PDX bearing mice, treated with 1.5 mg·kg−1 of negative miRNA inhibitor sequence (SCR) (n = 9) or a equimolar miR‐126‐3p mimic or miR‐221‐3p inhibitor sequence alone (n = 7) or in combination (combo) (n = 9) entrapped in neutral lipid nanoparticles (CCL), two times a week for four weeks. (B) Immunohistochemistry (IHC) images and graphs (C) show the proliferation index, miR‐126‐3p, miR‐221‐3p, necrosis and PTEN, PIK3R2 and CXCR4 protein levels at the end of treatment compared with those in control mice (n = 7). Scale bar: 200 µm. (D) Bar graphs illustrated plasma levels of interferon (IFN)‐α and β in the CCL‐treated mice (left) (n = 5). Graphs illustrated 12 plasma pro‐inflammatory cytokine levels in the CCL‐combo‐treated mice twice after 4 weeks of treatment (right) (n = 3). Data are presented as means ± S.E.M. *P < 0.05 versus SCR, **P < 0.01, ***P < 0.001. Black arrow indicates the starting of the treatment.
4. Conclusion
Several works have elucidated the importance of miRNAs as crucial modulators of cancer development [10]. In particular, we and others have demonstrated that several miRNAs are deregulated in lung cancer tissues and are responsible for the abnormal phenotype of cancer cells [20, 30]. In the current study, upregulation of miR‐210, miR‐21 and miR‐221/222, together with downmodulation of miR‐126‐3p, miR‐486, miR‐451 and miR‐30a, was validated in an independent series of early‐stage (70% stage I) lung cancer patients enrolled in the MILD lung cancer screening trial.
In addition to the previously reported antitumour effects of modulating single miRNAs in cancer cells, our findings here highlighted that combined modulation of two miRNAs has to be envisaged to increase antiproliferative and anti‐metastatic dissemination effects in lung cancer both in vitro and in vivo.
Downregulation of miRNA‐126 has been described in different malignancies such as colorectal, liver, breast and renal cancers [31, 32] and in lung tissues [15, 16, 33, 34]. This downregulation was also confirmed in the current study, revealing this miRNA as a tumour suppressor miRNA. Interestingly, a study by Song and colleagues showed that miR‐126 replacement reduced the proliferative, migratory and invasive abilities of only one lung cancer cell line [34]. In our previous study, H460 lung cancer cells injected intravenously into SCID mice invaded the lung parenchyma and produced fully developed metastatic nodules [26]. In a similar lung colonization assay, miR‐126 reduced the ability of tumour cells to colonize the lung parenchyma and generate metastases, suggesting that this miRNA plays a role in metastatic processes. Indeed, simultaneous miR‐126 replacement and miR‐221 inhibition reduced the migratory and invasive capacities of lung cancer cells through blockade of the CXCR4 axis. The CXCR4 axis plays a pivotal role in the regulation of cancer cell dissemination and colonization of distant organs [35, 36]. CXCR4/SDF‐1 axis targeting has been demonstrated in several preclinical and clinical studies as a promising therapeutic strategy for metastatic disease in different cancers, including NSCLC [35, 37, 38]. The anti‐metastatic potential of combined miR‐126 and miR‐221 modulation was further demonstrated by an in vivo colonization assay using two different lung cancer cells.
An important overexpressed miRNA cluster in lung cancer is miR‐221/222, which has previously been described to be upregulated in breast cancer, colorectal cancer and glioblastoma [39]. miR‐221 has been reported to play a role in lung cancer metastasis aggressiveness [40] and to be associated with the poor prognosis of NSCLC patients [41]. A previous study demonstrated that miR‐221 overexpression induced TRAIL activation [42]. In particular, transfection with miR‐221/222 inhibitors rendered lung cancer cells sensitive to TRAIL through p27. Interestingly, our data revealed that inhibition of miR‐221‐3p induced apoptosis by activating TRAIL in the two studied lung cancer cell lines. Importantly, we showed that downmodulation of miR‐221‐3p increased PTEN protein expression, leading to a reduction in AKT phosphorylation with a negative effect on cell viability, only in combination with miR‐126‐3p replacement. The potential therapeutic use of miRNA modulators in lung cancer is based on the rationale that miRNAs can revert malignant phenotypes and aggressiveness [43]. Our study demonstrated that only combined miR‐126‐3p restoration and miR‐221‐3p inhibition reduced tumour growth by simultaneously inhibiting the PI3 kinase (PI3K)/AKT/PTEN pathway in PDX models. We observed that combined miR‐126‐3p and miR‐221‐3p modulation is well tolerated in normal cells with adequate levels of endogenously expressed miRNAs, while in tumour cells, it restores a balance that reactivates physiological mechanisms of gene/pathway regulation, leading to antitumour effects.
Here, we established a proof of concept for a new therapeutic strategy based on combined administration of lipid nanoparticles containing a miR‐126‐3p mimic and a miR‐221‐3p inhibitor. These nanoparticles showed the ability to reduce the growth of lung cancer PDXs in immunodeficient mice. The observed mild reduction in tumour growth was probably due to the low delivery efficiency of the untargeted lipid nanoparticles, as reported in our previous work [24]. To optimize the antitumour efficacy of our lipid nanoparticles, careful selection and addition of lung cancer‐specific ligands to the lipid surface could result in an improvement essential for the therapeutic use of these compounds in lung cancer. Furthermore, since miRNAs have shown slight but significant therapeutic potential in cancers, future strategies combining neutral lipid nanoparticles encapsulating synthetic miRNAs with standard chemotherapeutic agents could improve the efficacy of the antitumour activity of these novel drugs. Despite challenges such as the development of novel delivery carriers or the onset of chemoresistance, the combination of miRNAs and chemotherapeutic agents constitutes an interesting approach for lung cancer treatment.
Our study reports a potential new therapeutic approach for lung cancer management that simultaneously targets two deregulated miRNAs in lung cancer tissues. Interestingly, only when miR‐126‐3p and miR‐221‐3p were modulated simultaneously was antitumour activity observed, in contrast to when each miRNA was modulated alone, suggesting that combined modulation of miRNAs could be a new approach for anticancer therapy.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
DDP, FP, MM, OF, GB designed the study, acquired, analysed and drafted the article; FP, GC, MMe, MS, CB, PS, CBr, IP acquired and analysed data; MMi, PP, MP, UP, GS designed the study, interpreted data, revised critically the article. All authors read and approved the final manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.13036.
Supporting information
Fig. S1. miRNAs de‐regulation in lung cancer.
Fig. S2. Proliferation and apoptosis analysis.
Fig. S3. Apoptosis analysis.
Fig. S4. PIK3R2 and PTEN are miR‐126 and miR‐221 targets.
Fig. S5. Migratory and invasive capacity of lung cancer cells.
Fig. S6. Proliferation analysis after 24 h post transfection.
Fig. S7. CXCR4 is fundamental for metastatic dissemination.
Fig. S8. In vivo assays.
Fig. S9. miR‐126 replacement using nebulized aerosol inhalation.
Fig. S10. PDX was treated with lipid‐nanoparticles.
Table S1. List of detection probes.
Table S2. List of miRNAs analyzed in lung cancer tissues.
Table S3. Correlation analysis between de‐regulated miRNAs in lung cancer tissues.
Table S4. Contingency table of lung cancer patients.
Table S5. Contingency table of lung cancer patients.
Acknowledgements
We thank Laura Caleca for her technical help. The study was supported by grants from the Italian Association for Cancer Research [Investigator Grant Nos. 14318 and 18812 to GS, 14231 and 18474 to MP]; OF was supported by the Cariplo Foundation Young Investigator Grant 2015 no. 2015‐0901. DDP was partially supported by a Post‐Doctoral Fellowship of Fondazione Umberto Veronesi.
Daniela Di Paolo and Francesca Pontis contributed equally to this work
Orazio Fortunato and Gabriella Sozzi are co‐last authors
Data accessibility
The data that support the findings of this study are available from the corresponding author orazio.fortunato@istitutotumori.mi.it upon reasonable request.
References
- 1. Siegel RL, Miller KD & Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- 2. Herbst RS, Morgensztern D & Boshoff C (2018) The biology and management of non‐small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- 3. Chatwal MS & Tanvetyanon T (2018) Combination chemotherapy and immunotherapy in metastatic non‐small cell lung cancer: a setback for personalized medicine? Transl Lung Cancer Res 7, S208–S210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng L‐C, Ho K‐K, Au JS‐K, Chung L‐P, Wong MP et al. (2009) The EML4‐ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild‐type EGFR and KRAS. Cancer 115, 1723–1733. [DOI] [PubMed] [Google Scholar]
- 5. Giroux Leprieur E, Dumenil C, Julie C, Giraud V, Dumoulin J, Labrune S & Chinet T (2017) Immunotherapy revolutionises non‐small‐cell lung cancer therapy: results, perspectives and new challenges. Eur J Cancer 78, 16–23. [DOI] [PubMed] [Google Scholar]
- 6. Boeri M, Milione M, Proto C, Signorelli D, Lo Russo G, Galeone C, Verri C, Mensah M, Centonze G, Martinetti A et al. (2019) Circulating miRNAs and PD‐L1 tumor expression are associated with survival in advanced NSCLC patients treated with immunotherapy: a prospective study. Clin Cancer Res 25, 2166–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rupaimoole R, Calin GA, Lopez‐Berestein G & Sood AK (2016) miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov 6, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mogilyansky E & Rigoutsos I (2013) The miR‐17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 20, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia Y, Zhu Y, Zhou X & Chen Y (2014) Low expression of let‐7 predicts poor prognosis in patients with multiple cancers: a meta‐analysis. Tumour Biol 35, 5143–5148. [DOI] [PubMed] [Google Scholar]
- 10. Di Leva G, Garofalo M & Croce CM (2014) MicroRNAs in cancer. Annu Rev Pathol 9, 287–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barger JF & Nana‐Sinkam SP (2015) MicroRNA as tools and therapeutics in lung cancer. Respir Med 109, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seijo LM, Peled N, Ajona D, Boeri M, Field JK, Sozzi G, Pio R, Zulueta JJ, Spira A, Massion PP et al. (2019) Biomarkers in lung cancer screening: achievements, promises, and challenges. J Thorac Oncol 14, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fortunato O, Boeri M, Verri C, Moro M & Sozzi G (2014) Therapeutic use of microRNAs in lung cancer. Biomed Res Int 2014, 756975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boeri M, Verri C, Conte D, Roz L, Modena P, Facchinetti F, Calabrò E, Croce CM, Pastorino U & Sozzi G (2011) MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci USA 108, 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fortunato O, Borzi C, Milione M, Centonze G, Conte D, Boeri M, Verri C, Moro M, Facchinetti F, Andriani F et al. (2018) Circulating mir‐320a promotes immunosuppressive macrophages M2 phenotype associated with lung cancer risk. Int J Cancer 144, 2746–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun Y, Bai Y, Zhang F, Wang YU, Guo Y & Guo L (2010) miR‐126 inhibits non‐small cell lung cancer cells proliferation by targeting EGFL7. Biochem Biophys Res Commun 391, 1483–1489. [DOI] [PubMed] [Google Scholar]
- 17. Yin Z, Xu M & Li P (2017) miRNA‐221 acts as an oncogenic role by directly targeting TIMP2 in non‐small‐cell lung carcinoma. Gene 620, 46–53. [DOI] [PubMed] [Google Scholar]
- 18. Pastorino U, Silva M, Sestini S, Sabia F, Boeri M, Cantarutti A, Sverzellati N, Sozzi G, Corrao G & Marchianò A (2019) Prolonged lung cancer screening reduced 10‐year mortality in the MILD trial. Ann Oncol 30, 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato M, Larsen JE, Lee W, Sun H, Shames DS, Dalvi MP, Ramirez RD, Tang H, DiMaio JM, Gao B et al. (2013) Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res 11, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fortunato O, Boeri M, Moro M, Verri C, Mensah M, Conte D, Caleca L, Roz L, Pastorino U & Sozzi G (2014) Mir‐660 is downregulated in lung cancer patients and its replacement inhibits lung tumorigenesis by targeting MDM2‐p53 interaction. Cell Death Dis 5, e1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C, Carosio R, Loi M, Pagnan G, Emionite L, Cilli M et al. (2011) Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol Ther 19, 2201–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Paolo D, Brignole C, Pastorino F, Carosio R, Zorzoli A, Rossi M, Loi M, Pagnan G, Emionite L, Cilli M et al. (2011) Neuroblastoma‐targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol Ther 19, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perri P, DiPaolo D, Priddy L, Fiore A, Brignole C, Pastorino F, Brown D & Ponzoni M (2014) MicroRNA replacement and RNAi‐mediated silencing of ALK as combined targeted therapies for neuroblastoma. Cancer Res 74(Suppl), Meeting Abstract 1453. [Google Scholar]
- 24. Moro M, Di Paolo D, Milione M, Centonze G, Bornaghi V, Borzi C, Gandellini P, Perri P, Pastorino U, Ponzoni M et al. (2019) Coated cationic lipid‐nanoparticles entrapping miR‐660 inhibit tumor growth in patient‐derived xenografts lung cancer models. J Control Release 308, 44–56. [DOI] [PubMed] [Google Scholar]
- 25. Moro M, Bertolini G, Caserini R, Borzi C, Boeri M, Fabbri A, Leone G, Gasparini P, Galeone C, Pelosi G et al. (2017) Establishment of patient derived xenografts as functional testing of lung cancer aggressiveness. Sci Rep 7, 6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertolini G, D'Amico L, Moro M, Landoni E, Perego P, Miceli R, Gatti L, Andriani F, Wong D, Caserini R et al. (2015) Microenvironment‐modulated metastatic CD133+/CXCR4+/EpCAM‐ lung cancer‐initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res 75, 3636–3649. [DOI] [PubMed] [Google Scholar]
- 27. Portella L, Vitale R, De Luca S, D'Alterio C, Ieranò C, Napolitano M, Riccio A, Polimeno MN, Monfregola L, Barbieri A et al. (2013) Preclinical development of a novel class of CXCR4 antagonist impairing solid tumors growth and metastases. PLoS One 8, e74548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. D'Alterio C, Zannetti A, Trotta AM, Ieranò C, Napolitano M, Rea G, Greco A, Maiolino P, Albanese S, Scognamiglio G et al. (2020) New CXCR4 antagonist peptide R (Pep R) improves standard therapy in colorectal cancer. Cancers (Basel) 12, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Paolo D, Pastorino F, Brignole C, Corrias MV, Emionite L, Cilli M, Tamma R, Priddy L, Amaro A, Ferrari D et al. (2020) Combined replenishment of miR‐34a and let‐7b by targeted nanoparticles inhibits tumor growth in neuroblastoma preclinical models. Small 16, e1906426. [DOI] [PubMed] [Google Scholar]
- 30. Kohlhapp FJ, Mitra AK, Lengyel E & Peter ME (2015) MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34, 5857–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sasahira T, Kurihara M, Bhawal UK, Ueda N, Shimomoto T, Yamamoto K, Kirita T & Kuniyasu H (2012) Downregulation of miR‐126 induces angiogenesis and lymphangiogenesis by activation of VEGF‐A in oral cancer. Br J Cancer 107, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu W, Chen H, Wong N, Haynes W, Baker CM & Wang X (2017) Pseudohypoxia induced by miR‐126 deactivation promotes migration and therapeutic resistance in renal cell carcinoma. Cancer Lett 394, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu B, Peng XC, Zheng XL, Wang J & Qin YW (2009) MiR‐126 restoration down‐regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 66, 169–175. [DOI] [PubMed] [Google Scholar]
- 34. Song L, Li D, Gu Y, Wen Z‐M, Jie J, Zhao D & Peng L‐P (2016) MicroRNA‐126 targeting PIK3R2 inhibits NSCLC A549 cell proliferation, migration, and invasion by regulation of PTEN/PI3K/AKT pathway. Clin Lung Cancer 17, e65–e75. [DOI] [PubMed] [Google Scholar]
- 35. Scala S (2015) Molecular pathways: targeting the CXCR4‐CXCL12 axis‐untapped potential in the tumor microenvironment. Clin Cancer Res 21, 4278–4285. [DOI] [PubMed] [Google Scholar]
- 36. Kawaguchi N, Zhang TT & Nakanishi T (2019) Involvement of CXCR4 in normal and abnormal development. Cells 8, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eckert F, Schilbach K, Klumpp L, Bardoscia L, Sezgin EC, Schwab M, Zips D & Huber SM (2018) Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front Immunol 9, 3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang L, Wang Z, Liu X & Liu F (2014) High‐level C‐X‐C chemokine receptor type 4 expression correlates with brain‐specific metastasis following complete resection of non‐small cell lung cancer. Oncol Lett 7, 1871–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravegnini G, Cargnin S, Sammarini G, Zanotti F, Bermejo JL, Hrelia P, Terrazzino S & Angelini S (2019) Prognostic role of miR‐221 and miR‐222 expression in cancer patients. A systematic review and meta‐analysis. Cancers (Basel) 11, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin Q, Mao W, Shu Y, Lin F, Liu S, Shen H, Gao W, Li S & Shen D (2012) A cluster of specified microRNAs in peripheral blood as biomarkers for metastatic non‐small‐cell lung cancer by stem‐loop RT‐PCR. J Cancer Res Clin Oncol 138, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Acunzo M, Visone R, Romano G, Veronese A, Lovat F, Palmieri D, Bottoni A, Garofalo M, Gasparini P, Condorelli G et al. (2012) miR‐130a targets MET and induces TRAIL‐sensitivity in NSCLC by downregulating miR‐221 and 222. Oncogene 31, 634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garofalo M, Quintavalle C, Di Leva G, Zanca C, Romano G, Taccioli C, Liu CG, Croce CM & Condorelli G (2008) MicroRNA signatures of TRAIL resistance in human non‐small cell lung cancer. Oncogene 27, 3845–3855. [DOI] [PubMed] [Google Scholar]
- 43. Rupaimoole R & Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16, 203–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. miRNAs de‐regulation in lung cancer.
Fig. S2. Proliferation and apoptosis analysis.
Fig. S3. Apoptosis analysis.
Fig. S4. PIK3R2 and PTEN are miR‐126 and miR‐221 targets.
Fig. S5. Migratory and invasive capacity of lung cancer cells.
Fig. S6. Proliferation analysis after 24 h post transfection.
Fig. S7. CXCR4 is fundamental for metastatic dissemination.
Fig. S8. In vivo assays.
Fig. S9. miR‐126 replacement using nebulized aerosol inhalation.
Fig. S10. PDX was treated with lipid‐nanoparticles.
Table S1. List of detection probes.
Table S2. List of miRNAs analyzed in lung cancer tissues.
Table S3. Correlation analysis between de‐regulated miRNAs in lung cancer tissues.
Table S4. Contingency table of lung cancer patients.
Table S5. Contingency table of lung cancer patients.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author orazio.fortunato@istitutotumori.mi.it upon reasonable request.