

SUMMARY
The function of enhancer RNAs (eRNAs) in transcriptional regulation remains obscure. By analyzing the genome-wide nascent transcript profiles in breast cancer cells, we identify a special group of eRNAs that are essential for estrogen-induced transcriptional repression. Using eRNAs of TM4SF1 and EFEMP1 as the paradigms, we find that these RNA molecules not only stabilize promoter-enhancer interactions but also recruit liganded estrogen receptor α (ERα) to particular enhancer regions, facilitate the formation of a functional transcriptional complex, and cause gene silencing. Interestingly, ERα is shown to directly bind with eRNAs by its DNA-binding domain. These eRNAs help with the formation of a specific ERα-centered transcriptional complex and promote the association of the histone demethylase KDM2A, which dismisses RNA polymerase II from designated enhancers and suppresses the transcription of target genes. Our work demonstrates a complete mechanism underlying the action of eRNAs in modulating and refining the locus-specific transcriptional program.
In Brief
Yang et al. identified a group of eRNAs that are essential for estrogen-induced transcriptional repression by assisting with the chromatin recruitment of ERα through binding to its DNA-binding domain and facilitating the interaction of ERα with its cofactors, which leads to the dismissal of RNA polymerase II.
Graphical Abstract
INTRODUCTION
For about 30 years, enhancers were mainly considered to be DNA fragments on chromatin that control transcription in cis, until the widespread identification of transcripts derived from the active enhancers, called enhancer RNAs (eRNAs) (Andersson et al., 2014; Arner et al., 2015; Hsieh et al., 2014; Kim et al., 2010; Li et al., 2013). The discovery of eRNAs opens up new avenues for the mechanisms of action of enhancers and adds another layer of complexity to transcriptional regulation. So far, most studies focus on the roles of these non-coding RNAs (ncRNAs) in gene activation. On one hand, eRNAs serve to link the enhancers that produce them and cognate promoters, and therefore functional chromosome architecture can be formed (Li et al., 2013). On the other hand, they act to release paused RNA polymerase II (Pol II) so that transcriptional activation is induced (Schaukowitch et al., 2014; Zhao et al., 2016). Recently, more profound biological functions of eRNAs were revealed, as they are reported to directly modulate the functionality of transcription factors/cofactors. For example, eRNAs were demonstrated to stimulate the enzymatic activity of the acetyltransferase CBP or to enhance the capacity of the bromodomain-containing protein BRD4 to bind with the acetylated histones (Bose et al., 2017; Rahnamoun et al., 2018). These studies suggest that more mechanisms of eRNA-mediated transcriptional regulation await to be disclosed.
The estrogen-induced transcriptional program has been a perfect exemplar for exploring the roles of enhancers in dictating cellular responses to environmental stimuli. This is partly because the global chromatin-binding pattern revealed that the master transcription factor estrogen receptor α (ERα) is predominantly located at enhancer regions after being liganded with the estradiol (E2) (Carroll, 2016; Carroll et al., 2006). Interestingly, the strong binding intensity of functional Pol II is observed at these enhancers, and nascent transcripts are robustly transcribed from the associated DNA sequences upon E2 stimulation, suggesting that these ERα-bound enhancers can serve as active transcription units (Hah et al., 2011, 2013; Li et al., 2013). Although there is a strong association between the expression of the eRNAs and the nearby mRNAs, it is still quite controversial regarding the roles of these eRNAs in regulating E2-stimulated enhancer activity. Some evidence showed that the abundance of these transcripts was required for the enhancer-promoter looping and cohesin-mediated gene activation, indicating a structural role of these eRNAs (Li et al., 2013). However, another report failed to detect any changes in the dimensional chromatin looping or the signals of the histone marks at the active enhancers when the levels of eRNAs were drastically reduced (Hah et al., 2013). Therefore, there are many unanswered questions about the functions of E2-induced eRNAs in modulating the activities of their associated enhancers.
With an external stimulus, diverse transcriptional consequences are typically induced from different groups of enhancers. It is still debatable why the same stimulation leads to divergent effects of enhancers on transcription of cognate genes. For example, some are transactivated, whereas some are silenced. A few models have been put forward to explain this enigma, such as the association of transcriptional coactivators or corepressors with separate enhancers and chromatin accessibility remodeling (Stadhouders et al., 2012). Currently, functions of eRNAs have been mostly centered on the gene activation program (Andersson et al., 2014; Danko et al., 2015). However, it is unclear whether eRNAs help with transcriptional repression and, if yes, how. Considering the context-specific functions of eRNAs, it is conceivable that these RNA molecules may determine not only gene activation but also gene repression, depending on which subsets of enhancers they affect.
RESULTS
A Large Set of ERα-Bound Enhancers Produces eRNAs That Are Transcriptionally Downregulated upon E2 Treatment
To systematically characterize the activity and specificity of functional enhancers in response to E2, we integrated GRO-seq (Global Run-on Sequencing) that captures nascent transcripts (Li et al., 2013) with chromatin immunoprecipitation sequencing (ChIP-seq) that characterizes ERα-bound enhancers (Schmidt et al., 2010) in breast cancer cell line MCF-7 (Figure S1A). Out of 71,861 ERα peaks, 12,544 were located within the intergenic areas and demarcated by the active histone mark H3K27 acetylation (H3K27ac). Some of these chromatin regions led to a robust production of RNA transcripts, which indicates that these ERα-bound enhancers are active in terms of transcriptional competency. Interestingly, among all the 1,408 de novo enhancer-generated transcripts that are differentially expressed upon E2 stimulation, 908 of them were significantly upregulated, whereas 500 were actually downregulated (Figure 1A). These transcripts are generally 1- to 2-kb long and bidirectional, displaying the typical characteristics of eRNAs (Kim et al., 2010; Figure 1B). This finding identifies two groups of functional enhancers as well as eRNAs whose reactions to E2 are contradictory. In separate datasets, we confirmed the presence of two categories of enhancers, both of which showed strong ERα binding (Tsai et al., 2010b; Figure S1B) and produced eRNAs either activated or repressed following E2 treatment (Franco et al., 2015; Figure S1C). Several studies have scrutinized the functions of E2-upregulated eRNAs in gene activation (Hah et al., 2011; Li et al., 2013). However, the roles of E2-downregulated enhancers in gene regulation have just been explored (Tan et al., 2018), and it is largely unknown whether the E2-repressed eRNAs are functionally involved in the regulation of the enhancers that transcribe them.
Figure 1. Two Groups of ERα-Bound, eRNA-Producing Enhancers Were Identified, which Respond Differentially to E2 Treatment.

(A) MA plot of ERα-localized enhancers that generate differentially expressed (false discover rate [FDR] ≤ 0.05 and fold change ≥ 1.5) eRNAs upon E2 treatment.
(B) Heatmaps of GRO-seq signals for sense- and antisense-strand eRNAs under vehicle (Veh.) or estrogen treatment (E2) condition.
(C) E2-induced changes in the Pol-II-binding intensities around transcription start sites (TSSs) of coding genes whose promoters showed detectable ChIA-PETsignals with E2-upregulated (Up) or E2-downregulated (Down) enhancers.
(D) Aggregate plots showing the binding intensities of indicated factors at E2-upregulated (Up) and E2-downregulated (Down) enhancers in the absence (Veh.) orpresence (E2) of estradiol.
(E) Cumulative distribution of E2-stimulated expression changes of coding genes nearest to E2-activated or E2-repressed eRNAs. The differential expression between vehicle (Veh.) and E2 treatment conditions was presented as log2(fold change) (log2FC).
Also see Figure S1.
In an effort to get some clues about the functions of these E2-repressed eRNAs, we first investigated whether the E2-downregulated enhancers that we identified are associated with any transcriptional program. By analyzing the published Pol II chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) data in MCF-7 cells (Li et al., 2012), we found that for those ERα bound, E2-downregulated enhancers showing physical connections with the promoters of annotated coding genes, only 2.68% of the genes that they are associated with are E2 activated, and 53.57% of them are significantly suppressed by the hormone. The rest (43.75%) of these genes were barely changed compared to the vehicle condition. In line with this observation, Pol II levels at the promoters of the coding genes, which were found to interact with E2-downregulated enhancers in Pol II ChIA-PET data (Li et al., 2012), are notably decreased upon E2 treatment, whereas its levels at the promoters that interact with E2-upregulated enhancers are significantly increased (Figure 1C). This result supports the idea that E2-downregulated enhancers control the transcription of E2-repressed coding genes. We then compared the chromatin features of the E2-downregulated enhancers with E2-upregulated ones (Figure 1D). Engagement of Pol II is significantly elevated at E2-upregulated enhancers but diminished at E2-downregulated regions after adding the hormone. Similar patterns are observed for DNase I hypersensitivity sites and H3K27ac mark. Interestingly, H3K4me1, an enhancer-specific histone mark, was unaffected by E2 stimulation. Both H3K27ac and H3K4me1 indicate the transcriptionally active status of associated cis-regulatory elements, but their responses to E2 stimulation at these two types of enhancers are quite distinct. Binding intensities of master transcription factor ERα and pioneer factor FOXA1 are significantly elevated upon E2 addition, regardless of how the enhancers react to the hormone. Therefore, landscapes of histone marks and cistromes of transcription factors are inadequate to explain the contrasting functions of the two sets of enhancers in mediating E2-induced transcriptional programs. There are plausibly some unrevealed factors that determine the differential responses of these enhancers to E2. Interestingly, when we examined the levels of the cognate mRNAs, almost all of them showed the same changing trend with the eRNAs in response to E2, i.e., more than 90% of the genes that are closest to E2-repressed eRNAs are also downregulated by the hormone, and it is the same case for the genes that are nearest to E2-activated eRNAs (Figure 1E). Based on these findings, we hypothesized that eRNAs being transcribed from E2-downregulated enhancers are involved in E2-induced transcriptional repression.
E2-Repressed eRNAs Control E2-Induced Transcriptional Repression of Nearby Coding Genes
To prove our hypothesis, we clustered all the E2-regulated eRNAs according to their expression changes in response to E2 and found a clear-cut arrangement of the nearby mRNAs into E2-activated and E2-repressed groups (Figure 2A). We chose two representative genes, TM4SF1 and EFEMP1, for further investigation. Both genes were ranked at the top of the list, whose expression was drastically reduced after E2 addition. IGV (Integrative Genomics Viewer) view illustrated that three distal chromatin elements on the TM4SF1 gene are characterized with all the active enhancer features and produce bidirectional, E2-repressed eRNAs. They are henceforth referred to as Enh1–3 (Figure S2A). The same chromatin characteristics are found at the EFEMP1 enhancer region. We first examined the response kinetics of TM4SF1 eRNAs, pre-mRNA, and mature mRNA to E2 stimulation over various time points (Figure 2B). We found that in both ERα-positive breast cancer cell lines MCF-7 and ZR-75–1, all TM4SF1 RNA molecules were transiently upregulated within 5–10 min after E2 addition and then constantly decreased over the rest of the treatment duration. Interestingly, the rates of reduction for TM4SF1 eRNAs were faster than those of pre-mRNA and mature mRNA transcripts. A similar result was obtained in the case of EFEMP1 (Figure S2B). These findings implied a potential function of eRNAs in triggering E2-mediated suppression of the coding genes. So, we designed the small interfering RNAs (siRNAs) that specifically target either strand of the eRNAs of TM4SF1 and EFEMP1. Interestingly, although both strands were efficiently knocked down, only depletion of the sense-strand transcripts (Figure 2C), but not the antisense-strand ones (Figure S2C), significantly decreased the levels of linked mRNAs, suggesting a strand-specific function of these eRNAs. In addition, after knocking down eRNAs of TM4SF1 or EFEMP1, we did not observe any changes in the transcript level of the control gene COMMD2, which is located distal to the selective enhancers and is irresponsive to E2 (Figure S2D).
Figure 2. E2-Repressed eRNAs Play Essential Roles in the Regulation of E2-Mediated, ERα-Dependent Transcriptional Repression.

(A) Heatmaps showing expression levels of eRNAs and their nearest genes upon E2 treatment. eRNAs expression in two biological replicates (rep1 and rep2) was Z score transformed and normalized to rep1 under vehicle condition. The ranking of TM4SF1 and EFEMP1 was specified, and the scales of eRNA or mRNA expression (exp.) were indicated.
(B) Expression of mRNA, pre-mRNA, and eRNAs of TM4SF1 in breast cancer cells at different time points after 100-nM E2 stimulation. The forward and reverse primers for detecting TM4SF1 mRNA were designed based on sequences in exon 4 and 3, respectively, whereas the target sequences of the primers for detecting pre-mRNA are located in intron 3.
(C) Expression of sense (S)-strand eRNA and mRNA of TM4SF1 and EFEMP1 upon knock down of indicated eRNAs. The strand information was defined according to the transcription direction of the coding genes.
(D) Expression of specified RNA species in MCF-7 cells upon treatment with ethanol (Veh.), 1 nM estradiol (E2) for 3 h, or 1 nM estradiol and 1 μM fulvestrant for 3 h (E2 + Ful.).
(E) E2-induced expression changes in indicated mRNAs after knocking down the specified eRNAs in MCF-7 cells treated with 100 nM E2 for 3 h.
Data in (B)–(E) are presented as mean ± SD with three biological and technical replicates. Expression was all normalized to the GAPDH mRNA level. Statistical significance was calculated using the t test. *p < 0.05; **p < 0.005; ***p < 0.0005; NS, not significant.
Also see Figures S2 and S3.
More surprisingly, the eRNA that was produced from TM4SF1 Enh2 had a much more limited effect on TM4SF1 mRNA expression than the other two eRNAs (Figure 2C), indicating that only a specific group of eRNAs, but not all, participate in transcriptional regulation. We inspected the organization of the chromatin that spans the entire TM4SF1 gene (Figure S2E). Interestingly, in both duplicates of Pol II ChIA-PET data, physical contacts between the promoter and Enh1 or Enh1 and Enh3 are consistently observed. However, Enh2 may be connected to Enh3, but with neither Enh1 nor the promoter. Instead, we found a strong CTCF (CCCTC-binding factor) ChIA-PET signal right on the border of Enh2, which joins to another chromatin site located downstream of the TM4SF1 gene. Both ends of the looping structure are marked with the co-localization of CTCF and two key structural components of the cohesin complex, RAD21 (Hakimi et al., 2002) and STAG1 (Losada et al., 2000). Considering the well-established role of CTCF and cohesin complex in chromatin insulation (Zuin et al., 2014), it is plausible that Enh2 is insulated from the transcriptionally active unit that encompasses the promoter and Enh1 of TM4SF1. As for Enh3, it is located relatively far from this insulation site and therefore may get away with the effect of CTCF and the cohesin complex. The unique chromatin topology surrounding the TM4SF1 gene may explain the contrasting functions of distinct enhancers as well as the transcribed eRNAs, and all these speculations definitely need further proof. Altogether, we identified a group of eRNAs whose function in gene regulation is highly specific, as they orchestrate the transcription of associated mRNAs in a strand-, location-, and gene-dependent manner.
Then, we asked whether these eRNAs could indeed modulate the repressive transcriptional program that is elicited by the estrogen-ERα axis. First, E2-induced downregulation of mRNAs and eRNAs was rescued by ERα degrader fulvestrant in MCF-7 (Figure 2D), indicating that intact ERα signaling is required for E2-mediated suppression of both eRNAs and cognate mRNAs. This was confirmed in ZR-75–1 cells (Figure S3A). A similar expression pattern in response to the treatment was observed for mRNAs and eRNAs of four extra genes in both MCF-7 (Figure S3B) and ZR-75–1 (Figure S3C). Second, a E2-induced reduction of mRNAs was completely abolished when eRNAs were knocked down using either siRNAs (Figure 2E) or locked nucleic acid (LNA) probes (Figure S3D). As a control, degradation of the nonfunctional eRNA2 of TM4SF1 had no effect on the response of mRNA to E2 stimulation (Figure S3E). Taken together, all these results indicated that the set of eRNAs we identified played an important role in dictating E2-mediated transcriptional repression.
E2-Repressed eRNAs Recruit ERα to E2-Downregulated Enhancers
Now, we have demonstrated that E2-repressed eRNAs help govern the specific transcriptional profile from E2-downregulated, ERα-bound enhancers. To explore the underlying mechanism, we first tested whether E2-repressed eRNAs are also involved in the promoter-enhancer connection because E2-activated eRNAs were shown to facilitate the formation of functioning chromatin topology in order to activate target genes (Lietal.,2013). Quantitative PCR following the 3C assay showed that fragments B and C on Enh3 interact with its promoter, which was diminished upon E2 treatment. When we knocked down the local eRNA transcript, the dimensional chromatin structure was disrupted and no longer responsive to the ligand (Figure 3A). 3C products were visualized on the gel with expected sizes (Figure S4A) and were further confirmed by traditional Sanger sequencing (Figure S4B). The result suggested that, similar to E2-activated eRNAs, E2-repressed eRNAs also have the structural role of linking functional enhancers and promoters. However, it could not explain how they elicit disparate transcriptional responses to E2.
Figure 3. E2-Repressed eRNAs Assist in Recruiting ERα to Specific Enhancers.

(A) 3C analysis of the interaction between the TM4SF1 promoter (TM4SF1 pro.) and Enh3 upon knock down of eRNA3 in MCF-7 receiving vehicle (Veh.) or 100 nM E2 for 3 h. Bottom: illustrative diagram of the anchor region and selected docking sites (A–D).
(B) Scatterplot showing the significance of transcription factor binding motifs in E2-downregulated (y axis) and E2-upregulated (x axis), ERα-bound enhancers. Forkhead (FKH) motif and estrogen response element (ERE) were marked.
(C) ERα binding at indicated cis-regulatory elements near TM4SF1 gene (left) and expression of TM4SF1 eRNAs (right) in MCF-7 cells treated with 100 nM E2 for indicated lengths of time.
(D) FC of ERα ChIP-seq peak intensities at each indicated time point after E2 stimulation at E2-downregulated enhancers relative to those at E2-upregulated enhancers.
(E) E2-induced recruitment of ERα to Enh1 and 3 of TM4SF1 after knocking down indicated eRNAs in MCF-7 with 100-nM E2 treatment for various lengths of time. ChIP with normal IgG antibody in cells transfected with nontargeting siRNA (siCtrl) was included as a negative control.
(F) TM4SF1 native promoter (TM4SF1 pro.)-driven luciferase activity in the absence (blue bars) or presence (light pink bars) of BoxB-fused sense-strand eRNA1 of TM4SF1 (BoxB-TM4SF1e1-S) in MCF-7 cells treated with 100 nM E2 for 5 min. Top: schematic diagram of RNA-tethering luciferase assay.
Data in (A), (C), (E), and (F) are presented as mean ± SD with three biological and technical replicates. p values in (A), (E), and (F) were calculated using the t test and in (B) were obtained using the hypergeometric distribution in the HOMER program (Heinz et al., 2010). *p < 0.05; **p < 0.001. Also see Figure S4.
One striking difference we noticed between these two groups of ERα-bound enhancers is that the estrogen response element (ERE) is only significantly enriched at E2-upregulated enhancers (p = 1E-198), not E2-downregulated ones (Figure 3B). This indicates that ERα binds to chromatin DNA directly at E2-upregulated enhancers but indirectly at E2-downregulated ones. Even though this distinction has been reported previously (Guertin et al., 2014; Tan et al., 2018), the factors that help to recruit ERα were never pinned down. Interestingly, we found that ERα was loaded onto the promoters (Enh1 and Enh3) of TM4SF1 at rates highly coincident with the E2-induced expression of eRNAs (Figure 3C). However, it does not bind to the Enh2 region, which generates the nonfunctional eRNA2. This bi-phase DNA-binding pattern of ERα was also detected at enhancer regions of other E2-repressed genes (Figure S4C). It prompted us to examine the genome-wide chromatin recruitment of ERα within short periods of E2 treatment. Strikingly, rapid and abundant accumulation of ERα right after E2 stimulation was universally seen at all the E2-downregulated enhancers we identified, which was confirmed in two independent ERα ChIP-seq data sets at various time points of hormone treatment (Figures 3D and S4D) (Honkela et al., 2015). In contrast, ERα binding at E2-upregulated enhancers peaked at about 40 min of E2 treatment, which is consistent with the classical DNA-binding pattern of ERα (Shang et al., 2000).
All these observations made us speculate that E2-repressed eRNAs might be one of the unknown factors that facilitate the indirect recruitment of ERα to E2-downregulated enhancers. Therefore, we knocked down the functional eRNA1 and 3 of TM4SF1 and detected a significant reduction of ERα binding intensities at the corresponding enhancers by ChIP-qPCR (Figure 3E). eRNAs seem to act in cis and locally, as knockdown of eRNA1 of TM4SF1 had no effect on ERα recruitment to enhancer of MYC, a typical E2-activated gene (Bourdeau et al., 2008; Figure S4E). To further prove that the eRNAs per se contribute to chromatin loading and functionality of ERα, we carried out a GAL4-BoxB tethering luciferase reporter assay in MCF-7 cells (Figure 3F). We fused the sense-strand eRNA1 of TM4SF1 to BoxB viral RNA so that the chimeric eRNA (BoxB-TM4SF1e1-S) would be bound by the RNA-binding domain of the λN protein that was linked to the GAL4 DNA-binding domain (DBD) (λN-GAL4). Meanwhile, we subcloned 1.2-kb TM4SF1 Enh1 harboring an ERα-binding site at the center into the reporter plasmid, where the basal expression of Luc gene was driven by the TM4SF1 promoter. The alignment direction and order of TM4SF1 Enh1 and promoter relative to the luciferase gene are the same as how they are manifested on human genome relative to TM4SF1. We put 4 times upstream activating sequence (UAS) motifs right downstream of TM4SF1 Enh1 so that the aforementioned chimeric eRNA could be artificially tethered to the plasmid. After acute treatment with E2 for only 5 min, we observed a more than 2.5-fold increase in the luciferase activity when full-length TM4SF1 Enh1 was present instead of random DNA. However, the elevation was notably mitigated once the DNA sequences that transcribe the sense-strand eRNA1 of TM4SF1 were deleted. To the contrary, when the chimeric eRNA containing the sense-strand eRNA1 was exogenously introduced, the Luc gene was activated in the presence of both full-length and deleted forms of TM4SF1 Enh1. This result indicated that the transcribed eRNA itself is important for ERα competency, likely by assisting with recruitment of ERα to target loci.
ERα Specifically and Directly Binds with E2-Repressed eRNAs
Now, we sought to examine whether eRNA and ERα do interact with each other. To this end, we performed an in vitro RNA pull-down assay (Figure 4A). In-vitro-transcribed sense-strand TM4SF1 eRNA1 was labeled with biotin and incubated with the nuclear extract from MCF-7. Just like in the case of sense-strand HOTAIR RNA that was previously shown to directly interact with ERα and modulate its transcriptional activity (Xue et al., 2016), we detected the robust presence of ERα in the eRNA1 precipitates by using streptavidin beads. This observation was reproducible under both mild and stringent washing conditions. Furthermore, RNA immunoprecipitation (RIP) showed that the ERα-specific antibody pulled down noticeably more eRNA1 and 3 of TM4SF1 than the control immunoglobulin G (IgG) after MCF-7 cells were treated with E2 for only 5 min (Figure 4B). Although a comparable amount of ERα protein was immunoprecipitated down, such eRNA-ERα interaction was undetectable at later time points of E2 treatment, which was in agreement with the kinetics of ERα chromatin binding. Even more appealing, the ERα antibody could only pull down the sense-strand eRNAs, but not the antisense-strand ones. Taken together, our data suggested that ERα associates with the functional E2-repressed eRNAs.
Figure 4. E2-Repressed eRNAs Directly Bind to ERα and Facilitate ERα Binding at E2-Downregulated Enhancers.

(A) In vitro RNA pull-down assay by incubating the in-vitro-transcribed, biotin-labeled TM4SF1 eRNA1 with the nuclear extract from MCF-7 cells treated with 100 nM E2 for 3 h. Empty Ctrl, streptavidin beads with no RNAs; TM4SF1e1-S, sense-strand eRNA1 of TM4SF1; HOTAIR-AS, antisense-strand HOTAIR as negative control; HOTAIR-S, sense-strand HOTAIR as positive control.
(B) RIP of ERα in MCF-7 cells treated with 100 nM E2 for indicated duration of time. Normal IgG antibody was included as a negative control, and data were presented as the FC to GAPDH mRNA levels. S, sense-strand; AS, antisense-strand. Bottom: western blot showing immunoprecipitation (IP) efficiency. Arrowhead, IgG heavy chain.
(C) Violin plot comparing the log2FC of ERα peak intensities before (−) and after (+) RNase addition under vehicle (Veh.) or E2 (100 nM for 30 min) treatment condition at E2-upregulated (E2-Up Enh) and E2-downregulated (E2-Down Enh) enhancers.
(D) Rank of ERα-bound, E2-upregulated (red dots), and E2-downregulated (blue dots) enhancers based on the ratios of E2-induced ERα chromatin binding intensities with RNase treatment (+RNase) to those without RNase treatment (−RNase). The E2-induced ERα chromatin binding was evaluated by comparing the ERα ChIP-seq peak signals between E2- and vehicle-treatment conditions (E2/Veh.). Insert: percentages of E2-downregulated (blue bars) and E2-upregulated enhancers (red bars) in each group of ERα ChIP-seq peak signals (E2/Veh.): increased, log2[ERα peak intensities (E2/Veh.) + RNase/ERα peak intensities (E2/Veh.) − RNase ≥ log2(1.2); no change (NC), −log2(1.2) ≤ log2[ratios] ≤ log2(1.2); or decreased, log2[ratios] ≤ −log2(1.2) after adding RNase compared to no RNase treatment. Numbers in white, the proportions of E2-downregulated enhancers in the aforementioned groups of ERα peaks.
(E) Heatmap showing eRNA enrichment in the ERα PAR-CLIP immunoprecipitates from MCF-7 cells that were treated with vehicle (Veh.) or 100 nM E2 for 5 min (E2.5min) or 3 h (E2.3hr). Data from three biological replicates (1, 2, and 3) were presented. FC, levels of specified eRNAs in the ERα immunoprecipitates upon E2 treatment compared to the vehicle condition; log2(fold enrichment) (FE), FE of indicated eRNAs in the ERα immunoprecipitates over the input RNAs. The final results in (B) and (E) were obtained from three biological and technical replicates; the data in (B) are presented as mean ± SD; P values in (B), (C), and (E) were determined using t test. *p < 0.05; **p < 0.001. Also see Figure S5.
To find out whether an interaction with eRNAs affects ERα chromatin binding globally and specifically, we treated the crosslinked chromatin with RNase A/T and then mapped the ERα cistrome by ChIP-seq. It is intriguing to see contrasting effects of RNase on E2-stimulated ERα binding at two different types of enhancers (Figures 4C and S5A). At E2-upregulated enhancers, ERα chromatin association after the addition of the hormone was still significantly augmented in the presence of RNase. Conversely, E2-prompted ERα binding at E2-downregulated enhancers was much less noticeable when RNase was present. We further categorized ERα-bound enhancers into three groups based on the E2-induced ERα peak intensity ratios between conditions with and without RNase treatment: increased after the addition of RNase, no change, or decreased. These categories of enhancers represent the cis-regulatory elements where E2-stimulated ERα chromatin association is hindered, barely affected, or dependent on RNA integrity. Interestingly, for enhancers that are susceptible to RNA degradation, they are enriched for E2-downregulated ones (55% for decreased, 40% for no change, and 25% for increased groups) (Figure 4D). These data indicated that disruption of ERα DNA occupancy due to decomposition of RNA molecules is more prominent at E2-downregulated enhancers than at E2-upregulated ones. To further signify the biological consequence of RNA decay at these two sets of ERα-bound enhancers, we examined chromatin binding of cohesin component RAD21 under the same RNase and hormone treatment conditions. RAD21 is well demonstrated with a strong role in chromosomal interactions (Hadjur et al., 2009; Kagey et al., 2010). Consistent with a prior report (Li et al., 2013) and our above finding that both E2-activated and E2-repressed eRNAs were involved in enhancer-promoter looping, RNase-mediated degradation of eRNAs had a severe impact on the DNA-binding capability of RAD21, with similar intensity at both E2-upregulated and E2-downregulated enhancers (Figure S5B). It is intriguing to note that binding of RAD21 at E2-upregulated enhancers was severely affected by RNase but still sensitive to E2. This implies that RNAs and ERα may contribute to RAD21 chromatin association independently. However, recruitment of RAD21 to E2-downregulated enhancers and its response to the ligand at these sites were totally impaired after RNA digestion. This, from another perspective, confirmed the dominant role of RNA molecules in constructing the function of these particular ERα-bound enhancers.
To directly demonstrate that ERα can physically associate with eRNA molecules, we performed ERα PAR-CLIP (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation) in MCF-7 cells treated with or without E2 (Figure 4E). After the cells were incubated with the photoagent 4-thiouridine (4-SU) and crosslinked using an ultraviolet light, we collected the precipitates that were pulled down by a specific ERα antibody at two time points after E2 addition, namely, 5 min and 3 h. We found a significant enrichment of E2-repressed eRNAs in the ERα immunoprecipitates upon the hormone stimulation. Interestingly, the accumulation of these eRNAs in the ERα pull-down samples was noticeably decreased at the 3-h time point. This pattern matches with the chromatin-binding kinetics of ERα at the E2-downregulated enhancers. For E2-activated eRNAs, however, we generally detected much lower levels in the ERα immunoprecipitates. We concluded that ERα directly binds to E2-repressed eRNAs immediately upon E2 stimulation, which may facilitate the chromatin recruitment of the nuclear receptor.
DBD of ERα Mediates Its Interaction with E2-Repressed eRNAs
Next, we aimed to explore the biochemical mechanism underlying the eRNA-ERα interaction. Thus, we deleted each functional domain on the ERα protein (Figure S6A) and replaced the endogenous ERα in MCF-7 cells with either the wild-type or deletion mutants. Expression levels of the endogenous and substituted ERα all looked alike (Figure S6B). We first checked which truncation form(s) compromises E2-mediated transcriptional repression and found that when either activation function 2 (AF2) or DBD was missing, eRNAs of TM4SF1 and EFEMP1 were no longer suppressed by E2 (Figure 5A) and neither were their cognate mRNAs (Figure S6C). We confirmed the loss-of-function effects of these two truncated ERα forms on the responses of four extra E2-repressed eRNAs to the hormone (Figure S6D). We then examined the association of these functionally dead mutants with the eRNAs by RIP-qPCR. Comparable amounts of the wild-type and mutant ERα were immunoprecipitated down (Figure S6E). The full-length ERα and AF2-deleted mutant pulled down significant amounts of E2-repressed eRNAs upon the hormone treatment compared to the control vector, but the DBD-truncation mutant failed to do so (Figure 5B). To further corroborate this finding, we purified recombinant ERα fragments that cover different parts of the protein from bacteria (Figure S6F) (Zhang et al., 2013) and incubated them with in-vitro-transcribed eRNA1 of TM4SF1 (Figure 5C). Although similar amounts of glutathione S-transferase (GST)-tagged fragments were applied, only fragment 2, which contains the DBD domain of ERα, could efficiently pull down eRNA1 in vitro. Again, these data firmly concluded that the DBD domain mediates the direct interaction between ERα and E2-repressed eRNAs. All our results implied distinct mechanisms of AF2- or DBD-mediated gene silencing upon E2. Because AF2 is well-known for its role in E2-dependent transcriptional programs through binding with a list of ERα cofactors (Robyr et al., 2000), it is possible that the association of ERα with some essential corepressor was lost after AF2 deletion, which led to a blockade of E2-induced gene silencing. However, a DBD-mediated interaction with eRNAs at least partially accounts for the transcriptional repression in response to E2 treatment.
Figure 5. DNA-Binding Domain of ERα Mediates Its Association with E2-Repressed eRNAs.

(A) Expression of specified eRNAs in MCF-7 cells where endogenous ERα was replaced with hemagglutinin (HA)-tagged empty vector (Vec), wild-type ERα (WT), or different truncated forms with (+) or without (−) 100-nM E2 treatment for 3 h.
(B) RIP-qPCR analysis of indicated eRNAs in the anti-HA immunoprecipitates from MCF-7. The replacement system was the same as described in (A), and cells were treated with 100 nM E2 for 5 min.
(C) SYBR-gold staining of TM4SF1 eRNA1 in the input (1/10 input) or the pull-down samples from the mixture with indicated ERα truncation proteins by glutathione beads (Pull down). Bottom: Coomassie blue staining of GST protein (GST) and GST-tagged recombinant ERα fragments (Fr-1, −2, and −3). Arrows, specific protein bands.
(D) Aggregate plots of the ChIP-seq peak signals of the WT or the P-Box mutant (P-Box) at E2-upregulated (E2-Up Enh) or E2-downregulated enhancers (E2-Down Enh).
(E) RIP-qPCR showing the relative enrichment of specified eRNAs in the anti-FLAG precipitates from MCF-7 expressing inducible FLAG-tagged WT or P-Box.Cells were treated with 100 nM E2 for 5 min. The substituted proteins were induced by adding 2 μg/mL doxycycline (DOX) overnight.
(F) ChIP of inducible biotinylated P-Box ERα using streptavidin beads at indicated enhancers after knocking down TM4SF1 eRNA1 in MCF-7 upon 100-nM E2 treatment for 30 min. Condition of DOX induction was the same as in (E). Data were normalized to qPCR signals detecting the GAPDH promoter, a negative control site.
Data in (A), (B), (E), and (F) are presented as mean ± SD with three biological and technical replicates. </p/> p values were calculated by t test, *p < 0.05; **p < 0.001. Also see Figures S6 and S7.
As our above results demonstrated that E2-repressed eRNAs played a critical role in ERα chromatin binding, we mapped cistromes of the wild-type ERα and the DBD- or AF2-deletion mutants (Figure S7A). DBD-truncated ERα lost its ability to associate with not only E2-upregulated enhancers that contains ERE but also E2-downregulated enhancers that lack ERE, although it was translocated into the nuclear compartment as efficiently as the wild-type ERα upon E2 (Figure S7B). Consequently, all the E2-responsive genes were neither activated nor repressed by the ligand (Figure S7C). On the contrary, depletion of the AF2 domain maintained DNA-binding capacity, just like the wild-type ERa. This is congruent with the observation that the AF2-deletion mutant could still bind with E2-repressed eRNAs. One may argue that expulsion of the DBD-deleted ERα from E2-downregulated enhancers is due to the fact that the entire DNA-binding structure was damaged rather than the loss of interaction with the eRNAs. To address this concern, we exploited the MCF-7 cell line that expresses proximal box (P-Box) mutant ERα (Liu et al., 2014), which harbors mutations at three conserved amino acids within the P-Box domain so that the capacity of recognizing the ERE motif is totally lost (Stender et al., 2010). Global chromatin localizations revealed that the P-Box-mutated ERα could only localize at E2-downregulated, but not -upregulated, enhancers with an intensity similar to that of the wild-type ERα, either upon E2 stimulation (Figure 5D) or under regular proliferating conditions (Figure S7D). This means that the P-Box mutation impedes the direct ERα-DNA interaction but not the indirect chromatin recruitment of the nuclear receptor. Concordantly, this mutant ERα could pull down similar amounts of E2-repressed eRNAs as the wild-type ERα does (Figure 5E) when both proteins were immunoprecipitated down efficiently (Figure S7E). Recruitment of the P-Box-mutated ERα to specific E2-downregulated enhancers is highly dependent on each individual eRNAs because when eRNA1 of TM4SF1 was knocked down, its presence at TM4SF1 Enh1 was diminished, but its binding at EFEMP1 Enh was unaffected (Figure 5F). In summary, we demonstrated that E2-repressed eRNAs directly bind with ERα at its DBD domain, which plays a critical role in the indirect recruitment of ERα to E2-downregulated enhancers.
E2-Repressed eRNAs Promote the ERα-KDM2A Association and Help Dismiss Pol II from E2-Downregulated Enhancers
Recently, the histone demethylase KDM2A was shown to orchestrate the ERα-dependent transcriptional repression program (Tan et al., 2018). It was docked at E2-downregulated enhancers by binding with the DBD domain of ERα. Subsequently, the E3 ubiquitin ligase NEDD4 was tethered and degraded Pol II, resulting in gene silencing (Anindya et al., 2007). Indeed, after E2 was added into MCF-7 cells, the association between NEDD4 and Pol II became more robust, which was accompanied by a noticeable reduction in the total level of Pol II protein (Figure S8A). Considering our above findings of the ERα-eRNA interaction, we surmised that E2-repressed eRNAs regulated the chromatin binding of KDM2A as well as Pol II. We first confirmed that eRNAs of TM4SF1 and EFEMP1 as well as their associated mRNAs were no longer suppressed by E2 when KDM2A was knocked down (Figure 6A). Second, KDM2A bound at E2-downregulated enhancers with kinetics similar to that of ERα (Figure 6B). Coincidentally, total levels of Pol II at these regulatory elements were reduced gradually, reaching equilibrium when KDM2A was completely released from these loci. We further knocked down TM4SF1 eRNA1 and found that E2-induced recruitment of KDM2A to TM4SF1 Enh1 was totally blocked, whereas KDM2A binding at the EFEMP1 enhancer was not affected (Figure 6C). This indicated that eRNAs controlled the chromatin localization of KDM2A in a site-specific way. We further examined Pol II levels at these enhancer areas (Figure 6D). The reduction of Pol II at TM4SF1 Enh1 following E2 treatment was mitigated when eRNA1 was depleted, but its reaction to E2 at EFEMP1 Enh was unchanged. Strikingly, Pol II at the promoter region of TM4SF1 was drastically lost even under the basal condition. This is possibly because enhancer-promoter dissociation caused by eRNA1 depletion was deleterious to the proper loading of Pol II (Li et al., 2013).
Figure 6. E2-Repressed eRNAs Facilitate the Formation of a Functional ERα-Centered Transcriptional Complex at E2-Downregulated Enhancers.

(A) Expression of indicated RNA molecules after KDM2A knockdown in MCF-7 cells treated with 100 nM E2 for 3 h.
(B) ChIP of Pol II and KDM2A at specified enhancers at different time points upon 100-nM E2 treatment in MCF-7 cells. ChIP with normal IgG antibody was included as a negative control.
(C and D) ChIP of KDM2A (C) or Pol II (D) upon knockdown of TM4SF1 eRNA1. MCF-7 cells were treated with (+) or without (−) 100 nM E2 for 5 min (C) or 30 min (D).
(E) In vitro association between KDM2A protein and ERα-Fr-2 protein in the presence of increasing amount of TM4SF1 eRNA1.
(F) In vitro competitive binding assays by mixing the in-vitro-transcribed eRNAs and the double-stranded PCR products containing enhancer regions in the presence of purified recombinant proteins. The precipitates from cobalt resin were subject to either western blot or ethidium bromide (EtBr) staining.
Data in (A)–(D) are presented as mean ± SD with three biological and technical replicates. Statistical significance in (A), (C), and (D) was calculated using t test. *p < 0.05; **p < 0.001. Also see Figure S8.
The decisive role of E2-repressed eRNAs in the transcriptional repression from a specific set of enhancers made us wonder whether these RNA molecules facilitate the association of ERα with KDM2A. Consistent with what has been reported, the DBD-containing fragment of ERα directly bound to recombinant KDM2A in the in vitro binding assay. Even more interestingly, the interaction between these two purified proteins became more robust in the presence of increasing amounts of TM4SF1 eRNA1 (Figure 6E). These results clearly suggested that E2-repressed eRNAs not only help to recruit ERα and particular cofactors but also strengthen their crosstalk on chromatin.
To explain why consolidation of the interaction between ERα and KDM2A is specifically ascribed to E2-repressed eRNAs but not E2-activated ones, we carried out the in vitro competitive binding assays (Figure 6F). We picked FOXC1 eRNA as the paradigm of E2-activated eRNAs (Li et al., 2013) and TM4SF1 eRNA1 as the E2-repressed one. Equal amounts of FOXC1 eRNA and TM4SF1 eRNA1 were in vitro transcribed and then mixed with or without the double-stranded PCR products comprising sequences of their corresponding enhancers (Figure S8B). The purified recombinant proteins KDM2A and ERα-DBD were added into each reaction. In the case of FOXC1, cobalt resin could pull down the His-tagged ERα DBD fragment together with either KDM2A or eRNA when the ERα protein was incubated with them individually. The eRNA itself had a very moderate impact on the interaction between ERα and KDM2A. However, the double-stranded DNA pieces containing FOXC1 enhancer outcompetes both KDM2A and eRNA for the DBD-containing ERα protein. In the case of TM4SF1, the DNA oligos harboring TM4SF1 Enh1 do not interact with ERα at all, suggesting that in case of E2-downregulated enhancers with no ERE sequence, the intrinsic chromatin environment is required for DNA binding of ERα in cells. In contrast, eRNA1 now directly binds to ERα, which is no longer influenced by the presence of DNA oligos. Not only that, eRNA1 remarkably enhances the binding affinity of ERα with KDM2A. These results delineate strikingly different modes of action between E2-activated and -repressed eRNAs as well as between E2-upregulated and -downregulated enhancers. E2-upregulated enhancers, which usually encompass ERE motifs, dominate the association with the DBD of ERα, and thus KDM2A and E2-activated eRNAs are excluded from accessing the same domain for interaction. On the other hand, since E2-downregulated enhancers do not contain the ERE sequences, eRNA molecules are able to approach ERα, which helps tether and dock the nuclear receptor as well as its essential cofactors onto the chromatin.
DISCUSSION
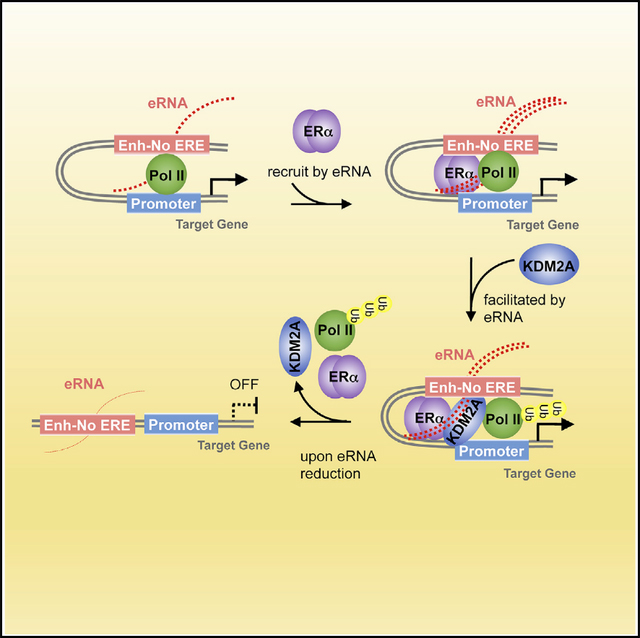
Factors that ensure proper actions of enhancers include transcription factors binding at these DNA elements and high-ordered chromatin landscape (Heinz et al., 2015). Here, we demonstrated that the nascent transcripts that are transcribed from these enhancers are also important contributors to the enhancers’ specificity (Figure 7). In ERα-positive breast cancer cells, we identified a group of eRNAs that are highly expressed before estrogen stimulation. They play a very critical role in linking the functional enhancer and cognate promoter (step 1). They recruit liganded ERα to particular chromatin loci that contain no EREs (step 2). Abundances of the eRNAs lead to fast recruitment of ERα and a rapid increase of themselves. Afterward, eRNAs help associate selective cofactor KDM2A with ERα and reinforce their interaction (step 3). Subsequently, Pol II is ubiquitylated and discharged from the bound chromatin so that specific transcriptional repression is induced at these enhancer regions. Finally, continuously declined eRNAs can no longer hold the ERα-centered transcriptional complex in place nor maintain the chromatin architecture. Thus, transcription of the associated coding gene is turned off (step 4). Overall, our study provided some insights on the mechanism by which activities and specificities of enhancers are dictated by eRNAs.
Figure 7.

Schematic Diagram of Our Working Model That Depicts How a Group of E2-Repressed eRNAs Contributes to E2-Mediated, ERα-Dependent Transcriptional Repression
The requirement of intact eRNAs for the production of nearby mRNAs has been observed for E2-activated genes (Li et al., 2013) and in other biological systems (Hsieh et al., 2014; Schaukowitch et al., 2014). In all the previous cases, however, eRNA-regulated genes are transactivated, and our work showed that transcriptional repression can be controlled by eRNAs as well. They do so by recruiting ERα as well as its corepressor KDM2A that was recently shown to bring the major E3 ubiquitin ligase for Pol II degradation, NEDD4 (Anindya et al., 2007), to ERα-bound enhancers of E2-repressed genes (Tan et al., 2018). Although the prior study scrutinized the action of the ERα-KDM2A complex on Pol II dismissal, it did leave multiple questions, for example, how the complex gets in contact with the cis-regulatory elements and how the target enhancers are selected. Findings from our study filled these gaps and pointed out the necessity of eRNAs in inducing transcriptional repression upon E2 treatment.
Current theories of ERα-mediated gene silencing include directly recruiting corepressors (Stossi et al., 2006) and hijacking coactivators from associated enhancers (Guertin et al., 2014). Both models highlight the dynamic displacement of ERα cofactors in mediating distinct gene expression patterns upon E2 stimulation. However, one fundamental question is what factors contribute to the directional movement of these coregulators. In this study, we found eRNAs as such factors, and the flow of ERα cofactors was totally changed when the levels of eRNAs were manipulated. Our data support the idea that eRNAs are decisive in forming a suitable composition of the E2-stimulated, ERα-associated transcriptional complex at target enhancers.
Even though we focused on the roles of eRNAs in the regulation of the ERα-dependent transcriptional program, it is plausible that what we demonstrated here may be applicable to other members of the nuclear receptor superfamily in response to their own ligands. For example, the glucocorticoid receptor (GR) has also been reported to induce not only transcriptional activation but also repression of thousands of genes upon glucocorticoid treatment (Oakley and Cidlowski, 2013). Current knowledge of GR-mediated transcriptional silencing is very much alike to what is known for E2-induced gene repression (Auphan et al., 1995; Langlais et al., 2012). Therefore, our model may also shed some light on the mechanism underlying glucocorticoid-induced transcriptional repression. More interestingly, the DBD of GR has been found to function as a robust structure-specific RNA-binding domain as well because it directly binds to a diverse range of RNA hairpin motifs with high affinity in vitro (Parsonnet et al., 2019). Investigating whether eRNAs orchestrate the differential transcriptional programs from distinct subsets of GR-bound enhancers is warranted, which will corroborate our findings and extend the reach of our work.
It has been suggested that ERα may be tethered to chromatin through other transcription factors (Jakacka et al., 2001; Porter et al., 1997) or even a trans-acting Mega complex (Liu et al., 2014). Unfortunately, we did not identify any transcription-factor-binding motifs that are uniquely enriched at E2-repressed enhancers, suggesting that molecules in addition to protein factors may facilitate ERα chromatin binding. Indeed, eRNAs that are abundantly present before hormone stimulation were proven to grab E2-activated ERα and anchor it to the target enhancers so that downstream transcriptional events were elicited. Although ncRNAs are constantly reported to be estrogen responsive in breast cancer cells (Hah and Kraus, 2014; Klinge, 2009; Niknafs et al., 2016), only a few of them, such as steroid receptor RNA activator (SRA) (Lanz et al., 1999) and HOTAIR (Xue et al., 2016), were found to physically associate with ERα and influence its transcriptional potential (Colley et al., 2008; Norris et al., 2002). Here, we provided some evidence showing a direct interaction between eRNAs and the ERα protein, expanding the pool of ERα-associated ncRNAs. We further pinned down the DBD on the ERα protein to mediate such an interaction. Both a recent report and our work showed that KDM2A competes with the ERE-containing DNA oligos for binding with the DBD domain of ERα (Tan et al., 2018), and here, we found that the same domain may coordinate the association of both ERα cofactor and ncRNAs. How this central part accommodates all the macromolecules (DNAs, RNAs, and proteins) definitely needs further exploration.
In summary, we identified a group of eRNAs that are actively involved in gene repression. In addition to bridging enhancers and associated promoters, these eRNAs carry distinct biological functions: they help recruit ERα to target enhancers, facilitate the joining of a selective cofactor, and team up with all the components to induce gene silencing in response to E2 stimulation. Our work further confirms the highly divergent and context-specific functions of eRNAs and suggests that one should investigate the roles of eRNAs in transcriptional regulation on a caseby-case basis.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kexin Xu (xuk3@uthscsa.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Original data have been deposited to GEO: GSE135341. For the information regarding the computational pipelines used in this study, please see the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-ERα | Bethyl | A300-495A; RRID: AB_451030 |
| Rabbit polyclonal anti-RNA Polymerase II | Santa Cruz Biotechnology | SC-9001; RRID: AB_2268548 |
| Mouse monoclonal anti-RNA Polymerase II | Santa Cruz Biotechnology | SC-56767; RRID: AB_785522 |
| Mouse monoclonal anti-RNA Polymerase II | Abcam | ab817; RRID: AB_306327 |
| Rabbit polyclonal anti-KDM2A | Proteintech | 24311-1-AP |
| Rabbit monoclonal anti-NEDD4 | Cell Signaling Technology | 3607; RRID: AB_2149311 |
| Mouse monoclonal anti-GST tag | Proteintech | 66001-2 |
| Mouse monoclonal anti-His tag | Proteintech | 66005-1; RRID: AB_11232599 |
| Mouse monoclonal anti-Lamin A/C | Abcam | ab8984; RRID: AB_306913 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | SC-365062; RRID: AB_10847862 |
| Rabbit polyclonal anti-RAD21 | Abcam | ab992; RRID: AB_2176601 |
| Mouse monoclonal anti-FLAG | Sigma | F1804; RRID: AB_262044 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Rosetta 2(DE3) Singles ™ Competent Cells | Emdmillipore | 71400-3 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| 17β-estradiol | Sigma | E2758 |
| fulvestrant | Medchem Express | HY13636 |
| 4-Thiouridine | Sigma | T4509 |
| RNase A/T | Thermo Fisher | EN0551 |
| RNase Cocktail™ Enzyme Mix | Thermo Fisher | AM2286 |
| SYBR Green Supermix | BioRad | 1725124 |
| GST-KDM2A | Abcam | ab151923 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Dual-Luciferase reporter assay kit | Promega | E1910 |
| ThruPLEX DNA-seq kit | Rubicon | R400427 |
| Q5 Site-Directed Mutagenesis kit | NEB | E0554S |
|
| ||
| Deposited Data | ||
|
| ||
| Raw and processed sequencing data | This paper | GEO: GSE135341 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MCF-7 | ATCC | HTB-22 |
| ZR-75-1 | ATCC | CRL-1500 |
| MCF-7 with BLRP-ERα-wild type | Liu et al., 2014 | GSE135341 |
| MCF-7 with BLRP-P-Box mutant | Liu et al., 2014 | GSE135341 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siRNAs targeting eRNAs: see Table S1 | This paper | N/A |
| LNAs targeting eRNAs: see Table S1 | This paper | N/A |
| Primers for detecting mRNAs by RT-qPCR: see Table S2 | This paper | N/A |
| Primers for detecting eRNAs by RT-qPCR: see Table S2 | This paper | N/A |
| Primers for targeted ChIP-qPCR: see Table S4 | This paper | N/A |
| Primers for 3C: see Table S3 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pGEX-ERα-fragments | Zhang et al., 2013 | N/A |
| pCDH-EF1-MCS-T2A | Systembioscience | CD527A-1 |
| pGL3 | Li et al., 2013 | N/A |
| pET28a(+) | EMD Biosciences | 69864-3 |
|
| ||
| Software and Algorithms | ||
|
| ||
| STAR v2.5.2b | Dobin et al., 2013; Theodorou et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.1 | Liao et al., 2014 | http://subread.sourceforge.net/ |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie v1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| MACS2 v2.1.1.20160309 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| HOMER v4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html |
| edgeR v3.16.5 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
|
| ||
| Other | ||
|
| ||
| H3K27ac ChIP-Seq data | Li et al., 2013 | GEO: GSE45822 |
| H3K4me1 ChIP-Seq data | Theodorou et al., 2013 | GEO: GSE40129 |
| DHS-Seq data | He et al., 2012 | GEO: GSE33216 |
| FOXA1 ChIP-Seq data | Kong et al., 2011 | GEO: GSE26831 |
| RNA Pol II ChIP-Seq data | Li et al., 2013 | GEO: GSE45822 |
| ERα ChIP-Seq data 1 | Schmidt et al., 2010 | ArrayExpress: E-TABM-828 |
| ERα ChIP-Seq data 2 | Tsai et al., 2010b | GEO: GSE24166 |
| CTCF, STAG1 and RAD21 ChIP-Seq data | Schmidt et al., 2010 | ArrayExpress: E-TABM-828 |
| Time course ERα ChIP-Seq data 1 | GEO: GSE94023 | |
| Time course ERα ChIP-Seq data 2 | Honkela et al., 2015 | GEO: GSE62789 |
| GRO-Seq data 1 | Li et al., 2013 | GEO: GSE45822 |
| GRO-Seq data 2 | Franco et al., 2015 | GEO: GSE59532 |
| RNA Pol II ChIA-PET data | Li et al., 2012; Natoli and Andrau, 2012 | GEO: GSE33664 |
| CTCF ChIA-PET data | ENCODE Project Consortium, 2012 | GEO: GSE39495 |
All the genome-wide datasets generated in this study, including ERα ChIP-Seq in MCF-7 cells with or without RNase treatment, anti-HA-targeted ChIP-Seq in MCF-7 expressing the wild-type ERα, DDBD or DAF2 truncation mutant, biotin-ChIP-Seq in MCF-7 expressing the wild-type ERα and P-Box mutant and RNA-Seq in MCF-7 replacement system upon E2 stimulation were deposited at the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with an accession number GSE135341. The H3K27ac and H3K4me1 ChIP-Seq data were downloaded from GSE45822 and GSE40129; the DHS-Seq, FOXA1 and RNA Pol II ChIP-Seq data were downloaded from GSE33216, GSE26831 and GSE45822, respectively. The ERα ChIP-Seq data were downloaded from ArrayExpress: E-TABM-828 and GEO: GSE24166. The CTCF, STAG1 and RAD21 ChIP-Seq data were downloaded from ArrayExpress: E-TABM-828. The two time-course ERα ChIP-Seq data were downloaded from GSE94023 and GSE62789. The two GRO-Seq datasets were downloaded from GSE45822 and GSE59532. The RNA Pol II and CTCF ChIA-PET data were downloaded from GSE33664 and GSE39495 respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MCF-7 and ZR-75–1 cells were originally obtained from ATCC. They are maintained in DMEM and RPMI 1640 (Corning), respectively, both of which are supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% penicillin-streptomycin (Thermo Fisher). Before hormone stimulation, cells were incubated in phenol red-free medium with 5% charcoal-stripped FBS for at least 72 hr. MCF-7 stable clones expressing FLAG- and biotin-tagged wild-type ERα or P-Box mutant were established by Dr. Zhijie Liu (Liu et al., 2014), and kept in DMEM supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% penicillin-streptomycin (Thermo Fisher), 0.3 μg/ml puromycin, 200 μg/ml G418, and 150 μg/ml Hygromycin B. Stable clones of MCF-7 cells expressing ERα mutants were established using lentiviral transduction and selected in 2 μg/mL of puromycin (Sigma) (Schaukowitch et al., 2014).
METHOD DETAILS
Plasmids and RNA knockdown
Complementary DNA encoding full-length human ERα was subcloned into pCDH-EF1-MCS-T2A vector that was a generous gift from Dr. Chi Zhang at UTHSCSA, and all the ERα truncations were constructed based on this plasmid by mutagenesis PCR using Q5 Site-Directed Mutagenesis kit (NEB). Vector pET28a (+) was kindly shared by Dr. Qing Zhang at UT Southwestern Medical Center. Control vectors and backbones in RNA-tethering luciferase assay were generously provided by Dr. Wenbo Li at UTHealth Houston, which was originally a kind gift from Dr. Howard Chang at Stanford University. TM4SF1 promoter was cloned in pGL3-basic vector downstream of 4 3 UAS, and the 1.2 kb enhancer region of TM4SF1 containing ERα-binding area or the sense-strand sequence deletion was inserted right before 4 × UAS site. Templates for in vitro transcription of the sense and antisense strands of HOTAIR were generously given by Dr. Yin-Yuan Mo at University of Mississippi Medical Center. Small hairpin RNA targeting 3′-UTR (untranslated region) of ERα was purchased from Sigma-Aldrich (TRCN0000010774). The small interfering RNAs used in this study include: siGENOME Non-Targeting siRNA Pool #2 (Dharmacon, D-001206–14) and KDM2A siRNA kit (QIAGEN FlexiTube GeneSolution, GS22992). eRNA-specific siRNAs were designed and purchased from Dharmacon, and the locked nucleic acid (LNA) probes from QIAGEN. Their sequences were listed in Table S1.
siRNAs or LNA-RNAs targeting individual eRNAs were transfected using Lipofectamine 2000 transfection reagent (Invitrogen) following the manufacturer’s instruction.
Antibodies and reagents
Antibodies used in this study include: αERα (Bethyl #A300–495A) for ChIP-qPCR, ChIP-Seq, RIP-qPCR, PAR-CLIP and immunoblotting; αKDM2A (ProteinTech #24311–1-AP) for ChIP-qPCR; αPol II (Santa Cruz Biotechnology #SC-9001, #SC-56767 and Abcam #ab817) for ChIP-qPCR; αRAD21 (Abcam #ab992) for ChIP-qPCR. Other antibodies that were applied in western blot include: αGST (ProteinTech #66001–2); αHis (ProteinTech #66005–1); αHA (BioLegend, #901501); αNEDD4 (CST #3607); αPol II (Santa Cruz Biotechnology #SC-56767), αLaminA/C (Abcam, # ab8984), αGAPDH (Santa Cruz Biotechnology #sc-365062) and αFLAG (Sigma #F1804). Reagents that were utilized for immunoprecipitation purposes are: anti-HA magnetic beads (Thermo Fisher #88837); streptavidin beads (NEB #S1420S); HisPurTM cobalt Superflow agarose beads (Thermo Fisher #25228); Dynabeads Protein A (Thermo Fisher #10006D) or Protein G (Thermo Fisher #10007D). Normal anti-rabbit IgG antibody was purchased from Invitrogen (#02–6102), 17β-estradiol (E2) from Sigma (#E2758), fulvestrant from Medchem Express (#HY13636), RNase A/T from Thermo Fisher (#EN0551), RNase/T1 cocktail from Thermo Fisher (#AM2286) and iTaq Universal SYBR Green Supermix from BioRad (1725124). Antibodies information can also be found in Key Resources Table.
Real-time RT PCR
Total RNA was extracted using Trizol (Invitrogen) and reverse transcribed using SuperScript III First-Strand Synthesis System (Invitrogen) with random primers. Primers of RT-qPCR are listed in Table S2.
RNA tethering luciferase assay
The BoxB-lN tethering luciferase assay was performed as previously described (Li et al., 2013). Basically, the reporter plasmid was transfected either alone or together with the tethering plasmids into MCF-7 cells that have been hormone-stripped for 3 days. Renilla control plasmid was co-transfected into each sample as well. Culturing medium was changed 6 hr post transfection and 100 nM E2 was then applied for 5 min. Afterward, cells were lysed using Dual-Luciferase reporter assay kit (Promega) and was read by Cytation 5 under the luciferase program (BioTek).
Chromosome Conformation Capture (3C) assay
3C assay was performed according to the published protocol with some optimization (Hagège et al., 2007). Briefly, 10 × 106 cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched in 0.125 M glycine for 5 min. Nuclei were extracted in lysis buffer [10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 1 3 complete protease inhibitor (Roche)] for 10 min on ice and then pelleted. BglII digestion was carried out at 37°C for overnight. T4 ligase was added into the diluted nuclei, and the inter-molecular ligation was executed at 16°C for 4 hr and then at room temperature for 30 min. DNA was purified using phenol-chloroform after de-crosslinking. Chromosome interaction was quantified by real-time PCR (qPCR), and the primer sequences were listed in Table S3. qPCR products were examined on 2% agarose gel and sent for Sanger sequencing to confirm the specific ligation.
Chromatin immunoprecipitation (ChIP)-qPCR and ChIP-Seq
ChIP was performed as previously described (Xu et al., 2012). Briefly, cell pellets were collected after crosslinking in 1% formaldehyde for 10 min at room temperature. In cases of KDM2A and RAD21 ChIPs, cells were crosslinked in 2 mM DSG (CovaChem) for 30 min at room temperature first. Nuclei were extracted using buffers LB1 [50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 10% (v/v) glycerol, 0.5% NP-40, 0.25% Triton X-100 and 1 × complete protease inhibitor] and LB2 [10 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0) and 1 × complete protease inhibitor]. Afterward, cell nuclei were suspended in buffer LB3 [10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.1% Na-deoxycholate, 0.5% N-lauroyl sarcosine and 1 × complete protease inhibitor], and fragmented using Q800R sonicator (QSONICA). The sheared chromatin was incubated with antibody-conjugated Protein A/G Dynabeads at 4°C for overnight. For ChIP with RNase treatment, the lysates were pretreated with RNase A/T for 30 min at room temperature before incubated with Dynabeads at 4°C overnight. Biotin-ChIP was performed using streptavidin magnetic beads (NEB) to pull down protein-DNA (Liu et al., 2014). ChIP’d DNA was subject to either real-time PCR or library construction using ThruPLEX DNA-seq kit (Rubicon). Primers for ChIP-qPCR are listed in Table S4.
RNA Immunoprecipitation (RIP)
RIP was carried out according to the published protocol (Fei et al., 2017). Briefly, 10 × 106 cells were crosslinked in 0.3% formaldehyde for 10 min at room temperature and then quenched in 0.125 M glycine for 5 min. Cell pellets were collected and lysed in RIPA lysis buffer [50 mM Tris (pH 7.6), 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1 × complete protease inhibitor and RNase inhibitor] for 10 min on ice. RNAs were fragmented into the range of 300–700 bp by sonication and mixed with antibody-conjugated Protein A/G Dynabeads at 4°C for overnight. After washing twice in low-salt (150 mM NaCl) and three times in high-salt (1 M NaCl) RIPA buffers, RNAs were eluted in elution buffer (0.1 M NaHCO3 and 1% SDS with Proteinase K and RNase inhibitor) for 10 min at room temperature. Meanwhile, one tenth of the beads were kept to check IP efficiency by western blot. After DNase I treatment, equal amounts of RNAs under each treatment condition were used for reverse transcription using SuperScript III kit and then subject to qPCR. Primers for these qPCR reactions are listed in Table S2.
In vitro RNA pull-down assay
In vitro RNA pull-down was performed as described previously (Tsai et al., 2010a). Biotin-labeled RNAs were in vitro transcribed using Biotin RNA Labeling Mix (Roche) and T7 RNA polymerase (Promega), treated with DNase I (NEB) and purified with Agencourt RNAClean XP beads (Sigma) (Schaukowitch et al., 2014). 1 μg of biotinylated RNAs were processed to form proper secondary structure in 2 × RNA structure buffer [10 mM Tris (pH 7.0), 0.1 M KCl, 10 mM MgCl2 and 0.4U RNase inhibitor] by being incubated at 95C for 5 min followed by snap-cooling on ice for 5 min RNAs were allowed to refold by standing at room temperature for 30 min.
Nuclei from 10 × 106 MCF-7 cells were suspended in 1 mL RIP buffer [150 mM NaCl, 25 mM Tris (pH 7.4), 0.5 mM DTT, 0.5% (v/v) NP-40 and 1 × complete protease inhibitor), sheared and precleaned with yeast tRNA before they were mixed with the RNA-conjugated streptavidin beads (NEB) at 4°C for 4 hr. Beads were washed four times in low-salt (RIP buffer with 500 mM NaCl and SuperaseIn) and four times in high-salt washing buffers (RIP buffer with 750 mM NaCl and SuperaseIn), and then examined by immunoblotting.
In Vitro protein-RNA interaction
In vitro protein-RNA interaction was performed according to the literature, with some adaptions (Rudman et al., 2018). Basically, GST-tagged proteins were incubated with 500 ng of properly refolded RNAs in RNA binding buffer [20 mM Tris (pH 7.4), 100 mM KCl, 0.2 mM EDTA, 0.05% (v/v) NP-40, 0.4U RNase inhibitor and 1 × complete protease inhibitor) at 4°C for 1 hr. Protein-RNA complexes were recovered using PierceTM Glutathione Magnetic Agarose beads (Thermo Fisher) at 4°C for 1 hr. Beads were washed three times in RNA washing buffer [20 mM Tris (pH 7.4), 200 mM KCl, 0.2 mM EDTA, 0.05% (v/v) NP-40, 0.4U RNase inhibitor and 1 × complete protease inhibitor], and RNAs were eluted using TRIzol LS reagent (Thermo Fisher). Purified RNA samples were resolved on a 4% denaturing TBE urea gel and stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen) for 30 min before the gel images were scanned by the Chemidoc Imaging system (Bio-Rad).
Interaction between KDM2A and ERα in the presence of eRNA was performed according to previously described method (Tan et al., 2018). Briefly, purified recombinant proteins, 150 ng His-tagged ERα and 1 mg GST-tagged KDM2A, were mixed in 1 × PBS supplemented with 10 mM imidazole in the presence of either 1 μg yeast tRNA only or increasing amounts of in vitro transcribed TM4SF1 eRNA1 at 4°C for 4 hr. Complex was incubated with HisPurTM cobalt Superflow agarose beads in the presence of 10 mM imidazole, and finally washed 4 times with 1 × PBS plus 0.05% (v/v) Triton X-100 and 10 mM imidazole. The precipitated ERα and KDM2A were detected by western blot.
In vitro competitive binding assay
The assay was carried out as previously described with some adjustments (Tan et al., 2018). Briefly, the purified recombinant proteins, 100 ng His-tagged DNA-binding domain (DBD) of ERα and 1 μg GST-tagged KDM2A were incubated with or without enhancer DNA oligos and/or eRNAs in 500 μL binding buffer [150 mM NaCl, 20 mM HEPES-KOH (pH 7.9), 0.1% (v/v) NP-40, 1 mM sodium butyrate and 0.4U RNase inhibitor] for 8 hr at 4°C. RNA-protein complexes were pulled down by HisPur™ cobalt Superflow agarose beads in the presence of 10 mM imidazole, and then washed 5 times with binding buffer plus 10 mM imidazole. Bound proteins were eluted in SDS sample buffer and analyzed by immunoblotting. RNAs and DNAs were treated with Proteinase K, purified using phenol:chloroform and finally precipitated by ethanol. The purified nucleic acids were subject to electrophoresis on a 1.5% agarose gel and stained with ethidium bromide.
The sequences of ERα-bound enhancer region and eRNA of FOXC1 were obtained from previous study (Li et al., 2013). The double-stranded DNA oligos (dsDNA) representing the enhancers of FOXC1 and TM4SF1 were PCR amplified using genomic DNA from MCF-7 cells as the template. For in vitro transcription, the genomic area that produces the eRNAs of FOXC1 or TM4SF1 was PCR amplified with a forward primer containing the T7 promoter sequences, so the PCR products could be used for in vitro RNA synthesis using T7 RNA polymerase (Promega). Synthesized RNAs were treated with DNase I (NEB) and purified with RNA Clean and Concentrator-5 (Zymo research). The newly synthesized RNAs were then subject to the refolding process as described above.
PAR-CLIP qPCR
We performed photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) following the protocol that was established previously (Danan et al., 2016; Hafner et al., 2010). Briefly, 30 × 107 cells were first incubated with 150 μM 4-thiouridine (4-SU) for 16 hr, and then washed with ice-cold 1 × PBS before they were crosslinked using 365nm ultraviolet light (150 mJ/cm2) with a Stratalinker UV crosslinker (Stratagene). Afterward, cells were harvested and lysed in the NP-40 lysis buffer [50 mM HEPES (pH 7.5), 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% (v/v) NP-40, 0.5 mM DTT, complete EDTA-free protease inhibitor cocktail (Roche) and 1U RNase inhibitor] in a volume equal to three times the cell pellets. The cleared cell lysates were incubated with anti-ERα antibody bound to the Protein G Dynabeads for 6 hr at 4°C. Genomic DNA was removed by Turbo DNase I for 30 min at 37°C. Beads were washed three times with wash buffer [20 mM Tris HCl (pH 7.9), 300 mM KCl, 10 mM EDTA, 10 mM EGTA, 0.05% (v/v) NP-40, 2 mM DTT], high-salt wash buffer (wash buffer with 500 mM KCl instead) and TE. Finally, they were resuspended in RNA elution buffer [50 mM HEPES-KOH (pH 7.0), 50 mM NaCl, 1 mM EDTA, 2 mM CaCl2 and 1% (w/v) SDS] for 30 min at 37°C and transferred to a clean microcentrifuge tube for RNA purification. Eluted RNAs were treated with Proteinase K at a final concentration of 4 mg/ml for 30 min at 55°C, and then purified using acidic phenol/chloroform/IAA extraction (25:24:1, pH 4.0) and finally precipitated by ethanol. RNA pellets were dissolved in RNase/DNase free water. The purified RNAs were subject to reverse transcription reactions using random primers with a High Capacity cDNA reverse transcription kit (Applied Biosystems). Real-time qPCR was then carried out to detect the levels of target eRNAs using primers listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq data analysis
The sequencing reads were aligned to human genome (hg19) with STAR 2.5.2b, a spliced-read aligner (Dobin et al., 2013). After removing reads that were mapped to rRNAs, read counts for each gene were conducted by featureCounts package with default parameter (Liao et al., 2014). Genes with less than one read in at least 2 samples were discarded. DESeq2 1.14.1 was used to call differentially expressed genes with fold expression change > = 1.5 and FDR ≤ 0.05 as the cutoff (Love et al., 2014).
ChIP-Seq data analysis
Reads were aligned to human genome (hg19) using bowtie (Langmead et al., 2009) and called for peaks using MACS with a cutoff of q-value at 1E-5 (Zhang et al., 2008). Peaks overlapped with the UCSC blacklist regions were removed. FindMotifs.pl script in HOMER was used to search for potential transcription factor binding motifs against vertebrates motif collector set (Heinz et al., 2010). It scans within a range of 300 bp window size upstream and downstream of the peak summits of interest.
GRO-Seq data analysis
GRO-Seq data was aligned to human genome (hg19) using bowtie (Langmead et al., 2009). Duplicated reads were eliminated from further analysis. To balance between the clonal amplification bias and total useful reads, no more than three reads were allowed for each unique genomic position. When measuring the expression level of genes, mapped reads from the first 30 kb of a gene body were counted, excluding promoter-proximal region [transcription start site (TSS) to 1000 bp downstream of TSS]. If the length of a gene is shorter than 10 kb, the reads within the first 10% of the entire region were excluded. If the length of a gene is shorter than 30 kb, the reads from the whole gene were counted, excluding promoter-proximal region and gene-ending region [500 bp upstream of transcription termination site (TTS) to TTS].
Define E2-upregulated and -downregulated ERα-bound enhancers
All the ERα peaks within 3 kb of TSS of any coding genes were first excluded, and the rest were overlapped with H3K27ac-enriched regions. The resultant list of peaks was regarded as ERα-bound enhancers. To detect differential eRNA responses upon E2 stimulation, we first designated a 2000 bp window that centered at ERα-bound enhancers as the potential eRNA-generating regions, and then used edgeR 3.16.5 to compare the GRO-Seq read counts within that window under E2 treatment condition with vehicle condition (Robinson et al., 2010). Any regions with log2(fold change) > = log2(1.5) and FDR ≤ 0.05 were defined as E2-upregulated ERα-bound enhancers, whereas those with log2(fold change) ≤ −log2(1.5) and FDR ≤ 0.05 as E2-downregulated enhancers.
Associate enhancers with gene promoters
We retrieved the two saturated replicates of RNA Pol II ChIA-PET data from GSE33664 to examine the enhancer-promoter interactions (Natoli and Andrau, 2012). Any ERα-bound enhancer that overlaps with ± 500 bp of the RNA Pol II-linked chromatin regions from any replicate was considered as the enhancer that shows dimensional chromatin interactions. If it happens to associate with ± 1000 bp of the promoter of a coding gene, this is counted as an enhancer-promoter interaction, and the coding gene was selected for further gene expression analysis. When the differential expression between E2 treatment and vehicle condition follows the criteria as log2(fold change) > = log2(1.5) and FDR ≤ 0.05, the gene was defined as E2-activated one, whereas those with log2(fold change) ≤ −log2(1.5) and FDR ≤ 0.05 as E2-repressed one. The rest of the genes were undifferentially expressed upon E2 treatment.
Data collection and visualization
All ChIP-Seq, GRO-Seq and DNase-Seq datasets were visualized in Integrative Genomics Viewer (IGV) (Robinson et al., 2011). For GRO-Seq, the reads were separated by strands and extended to a 100 nt length in the 5′-to-3′ direction. For ChIP-Seq, the reads were extended to 200 nt in the 5′-to-3′ direction. All the genomic datasets were normalized to 10 million mapped reads per sample.
Statistical Analysis
All the experiments were repeated using three biological replicates and three technical replicates per sample. Real-time PCR results were shown as mean ± SD P values were calculated using either Student’s t test or the statistical methods appropriate for the corresponding situations.
Supplementary Material
Highlights.
As active enhancer markers, a group of eRNAs facilitate E2-induced gene repression
These eRNAs help recruit ERα to enhancers with no estrogen response elements
They interact with the DNA-binding domain of the ERα protein
They also consolidate ERα-KDM2A cooperation to decommission RNA polymerase II
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Jindan Yu at Northwestern University for technical support, Dr. Yin-Yuan Mo at University of Mississippi, and Dr. Chi Zhang at UTHCSA for generously sharing their materials. This work was supported by grants from Cancer Prevention and Research Institute of Texas award (RR140072 to K.X.), the Cancer Prevention & Research Institute of Texas (CPRIT)-funded Research Training Award (RP170345 to R.R.the Cancer Prevention & Research Institute of Texas (CPRIT)-funded Research Training Award (RP170345 to R.R.), Voelcker Fund Young Investigator award (to K.X.), V Foundation Translational Award (T2017-010 to K.X.), and Susan Komen Award (CCR19605875 to K.X.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107803.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Varambally et al. (2008).
REFERENCES
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anindya R, Aygün O, and Svejstrup JQ (2007). Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell 28, 386–397. [DOI] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, and Karin M (1995). Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270, 286–290. [DOI] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149.e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau V, Deschênes J, Laperrière D, Aid M, White JH, and Mader S (2008). Mechanisms of primary and secondary estrogen target gene regulation in breast cancer cells. Nucleic Acids Res. 36, 76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS (2016). Mechanisms of oestrogen receptor (ER) gene regulation in breast cancer. Eur. J. Endocrinol. 175, R41–R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. (2006). Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Colley SM, Iyer KR, and Leedman PJ (2008). The RNA coregulator SRA, its binding proteins and nuclear receptor signaling activity. IUBMB Life 60, 159–164. [DOI] [PubMed] [Google Scholar]
- Danan C, Manickavel S, and Hafner M (2016). PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol. Biol. 1358, 153–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, and Siepel A (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 12, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, Cotter MB, Bowden M, Lis RT, Zhao SG, et al. (2017). Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. USA 114, E5207–E5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, and Kraus WL (2015). TNFa signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol. Cell 58, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Zhang X, Coonrod SA, and Hager GL (2014). Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol. Endocrinol. 28, 1522–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, and Merkenschlager M (2009). Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp AC, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagège H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, and Forné T (2007). Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733. [DOI] [PubMed] [Google Scholar]
- Hah N, and Kraus WL (2014). Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells. Mol. Cell. Endocrinol. 382, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, and Kraus WL (2011). A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, and Kraus WL (2013). Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 23, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, Yokomori K, and Shiekhattar R (2002). A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418, 994–998. [DOI] [PubMed] [Google Scholar]
- He HH, Meyer CA, Chen MW, Jordan VC, Brown M, and Liu XS (2012). Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. 22, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkela A, Peltonen J, Topa H, Charapitsa I, Matarese F, Grote K, Stunnenberg HG, Reid G, Lawrence ND, and Rattray M (2015). Genome-wide modeling of transcription kinetics reveals patterns of RNA production delays. Proc. Natl. Acad. Sci. USA 112, 13115–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X, Sun T, Sweeney CJ, Lee GS, Chen S, et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 111, 7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, and Jameson JL (2001). Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J. Biol. Chem. 276, 13615–13621. [DOI] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM (2009). Estrogen Regulation of MicroRNA Expression. Curr. Genomics 10, 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SL, Li G, Loh SL, Sung WK, and Liu ET (2011). Cellular reprogramming by the conjoint action of ERα, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 7, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Couture C, Balsalobre A, and Drouin J (2012). The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 47, 38–49. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, and O’Malley BW (1999). A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97, 17–27. [DOI] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. (2012). Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. (2014). Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 159, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Yokochi T, Kobayashi R, and Hirano T (2000). Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 150, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G, and Andrau JC (2012). Noncoding transcription at enhancers: general principles and functional models. Annu. Rev. Genet. 46, 1–19. [DOI] [PubMed] [Google Scholar]
- Niknafs YS, Han S, Ma T, Speers C, Zhang C, Wilder-Romans K, Iyer MK, Pitchiaya S, Malik R, Hosono Y, et al. (2016). The lncRNA landscape of breast cancer reveals a role for DSCAM-AS1 in breast cancer progression. Nat. Commun. 7, 12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Fan D, Sherk A, and McDonnell DP (2002). A negative coregulator for the human ER. Mol. Endocrinol. 16, 459–468. [DOI] [PubMed] [Google Scholar]
- Oakley RH, and Cidlowski JA (2013). The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsonnet NV, Lammer NC, Holmes ZE, Batey RT, and Wuttke DS (2019). The glucocorticoid receptor DNA-binding domain recognizes RNA hairpin structures with high affinity. Nucleic Acids Res. 47, 8180–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter W, Saville B, Hoivik D, and Safe S (1997). Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol. Endocrinol. 11, 1569–1580. [DOI] [PubMed] [Google Scholar]
- Rahnamoun H, Lee J, Sun Z, Lu H, Ramsey KM, Komives EA, and Lauberth SM (2018). RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat. Struct. Mol. Biol. 25, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robyr D, Wolffe AP, and Wahli W (2000). Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol. Endocrinol. 14, 329–347. [DOI] [PubMed] [Google Scholar]
- Rudman MD, Choi JS, Lee HE, Tan SK, Ayad NG, and Lee JK (2018). Bromodomain and extraterminal domain-containing protein inhibition attenuates acute inflammation after spinal cord injury. Exp. Neurol. 309, 181–192. [DOI] [PubMed] [Google Scholar]
- Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, and Kim TK (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Ross-Innes CS, Hurtado A, Brown GD, Carroll JS, Flicek P, and Odom DT (2010). A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 20, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, and Brown M (2000). Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, van den Heuvel A, Kolovos P, Jorna R, Leslie K, Grosveld F, and Soler E (2012). Transcription regulation by distal enhancers: who’s in the loop? Transcription 3, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, and Katzenellenbogen BS (2010). Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 30, 3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossi F, Likhite VS, Katzenellenbogen JA, and Katzenellenbogen BS (2006). Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J. Biol. Chem. 281, 16272–16278. [DOI] [PubMed] [Google Scholar]
- Tan Y, Jin C, Ma W, Hu Y, Tanasa B, Oh S, Gamliel A, Ma Q, Yao L, Zhang J, et al. (2018). Dismissal of RNA Polymerase II Underlies a Large Ligand-Induced Enhancer Decommissioning Program. Mol. Cell 71, 526–539.e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou V, Stark R, Menon S, and Carroll JS (2013). GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 23, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, and Chang HY (2010a). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, et al. (2010b). TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. (2008). Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. (2012). EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, Li S, Zhao JC, and Yu J (2016). LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 35, 2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Modelbased analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tanaka K, Yan J, Li J, Peng D, Jiang Y, Yang Z, Barton MC, Wen H, and Shi X (2013). Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 110, 17284–17289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang L, Ren S, Wang L, Blackburn PR, McNulty MS, Gao X, Qiao M, Vessella RL, Kohli M, et al. (2016). Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 15, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Dixon JR, van der Reijden MIJA, Ye Z, Kolovos P, Brouwer RWW, van de Corput MPC, van de Werken HJG, Knoch TA, van IJcken WFJ, et al. (2014). Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 111, 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to GEO: GSE135341. For the information regarding the computational pipelines used in this study, please see the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-ERα | Bethyl | A300-495A; RRID: AB_451030 |
| Rabbit polyclonal anti-RNA Polymerase II | Santa Cruz Biotechnology | SC-9001; RRID: AB_2268548 |
| Mouse monoclonal anti-RNA Polymerase II | Santa Cruz Biotechnology | SC-56767; RRID: AB_785522 |
| Mouse monoclonal anti-RNA Polymerase II | Abcam | ab817; RRID: AB_306327 |
| Rabbit polyclonal anti-KDM2A | Proteintech | 24311-1-AP |
| Rabbit monoclonal anti-NEDD4 | Cell Signaling Technology | 3607; RRID: AB_2149311 |
| Mouse monoclonal anti-GST tag | Proteintech | 66001-2 |
| Mouse monoclonal anti-His tag | Proteintech | 66005-1; RRID: AB_11232599 |
| Mouse monoclonal anti-Lamin A/C | Abcam | ab8984; RRID: AB_306913 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | SC-365062; RRID: AB_10847862 |
| Rabbit polyclonal anti-RAD21 | Abcam | ab992; RRID: AB_2176601 |
| Mouse monoclonal anti-FLAG | Sigma | F1804; RRID: AB_262044 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Rosetta 2(DE3) Singles ™ Competent Cells | Emdmillipore | 71400-3 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| 17β-estradiol | Sigma | E2758 |
| fulvestrant | Medchem Express | HY13636 |
| 4-Thiouridine | Sigma | T4509 |
| RNase A/T | Thermo Fisher | EN0551 |
| RNase Cocktail™ Enzyme Mix | Thermo Fisher | AM2286 |
| SYBR Green Supermix | BioRad | 1725124 |
| GST-KDM2A | Abcam | ab151923 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Dual-Luciferase reporter assay kit | Promega | E1910 |
| ThruPLEX DNA-seq kit | Rubicon | R400427 |
| Q5 Site-Directed Mutagenesis kit | NEB | E0554S |
|
| ||
| Deposited Data | ||
|
| ||
| Raw and processed sequencing data | This paper | GEO: GSE135341 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MCF-7 | ATCC | HTB-22 |
| ZR-75-1 | ATCC | CRL-1500 |
| MCF-7 with BLRP-ERα-wild type | Liu et al., 2014 | GSE135341 |
| MCF-7 with BLRP-P-Box mutant | Liu et al., 2014 | GSE135341 |
|
| ||
| Oligonucleotides | ||
|
| ||
| siRNAs targeting eRNAs: see Table S1 | This paper | N/A |
| LNAs targeting eRNAs: see Table S1 | This paper | N/A |
| Primers for detecting mRNAs by RT-qPCR: see Table S2 | This paper | N/A |
| Primers for detecting eRNAs by RT-qPCR: see Table S2 | This paper | N/A |
| Primers for targeted ChIP-qPCR: see Table S4 | This paper | N/A |
| Primers for 3C: see Table S3 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pGEX-ERα-fragments | Zhang et al., 2013 | N/A |
| pCDH-EF1-MCS-T2A | Systembioscience | CD527A-1 |
| pGL3 | Li et al., 2013 | N/A |
| pET28a(+) | EMD Biosciences | 69864-3 |
|
| ||
| Software and Algorithms | ||
|
| ||
| STAR v2.5.2b | Dobin et al., 2013; Theodorou et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.1 | Liao et al., 2014 | http://subread.sourceforge.net/ |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie v1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| MACS2 v2.1.1.20160309 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| HOMER v4.8 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html |
| edgeR v3.16.5 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
|
| ||
| Other | ||
|
| ||
| H3K27ac ChIP-Seq data | Li et al., 2013 | GEO: GSE45822 |
| H3K4me1 ChIP-Seq data | Theodorou et al., 2013 | GEO: GSE40129 |
| DHS-Seq data | He et al., 2012 | GEO: GSE33216 |
| FOXA1 ChIP-Seq data | Kong et al., 2011 | GEO: GSE26831 |
| RNA Pol II ChIP-Seq data | Li et al., 2013 | GEO: GSE45822 |
| ERα ChIP-Seq data 1 | Schmidt et al., 2010 | ArrayExpress: E-TABM-828 |
| ERα ChIP-Seq data 2 | Tsai et al., 2010b | GEO: GSE24166 |
| CTCF, STAG1 and RAD21 ChIP-Seq data | Schmidt et al., 2010 | ArrayExpress: E-TABM-828 |
| Time course ERα ChIP-Seq data 1 | GEO: GSE94023 | |
| Time course ERα ChIP-Seq data 2 | Honkela et al., 2015 | GEO: GSE62789 |
| GRO-Seq data 1 | Li et al., 2013 | GEO: GSE45822 |
| GRO-Seq data 2 | Franco et al., 2015 | GEO: GSE59532 |
| RNA Pol II ChIA-PET data | Li et al., 2012; Natoli and Andrau, 2012 | GEO: GSE33664 |
| CTCF ChIA-PET data | ENCODE Project Consortium, 2012 | GEO: GSE39495 |
All the genome-wide datasets generated in this study, including ERα ChIP-Seq in MCF-7 cells with or without RNase treatment, anti-HA-targeted ChIP-Seq in MCF-7 expressing the wild-type ERα, DDBD or DAF2 truncation mutant, biotin-ChIP-Seq in MCF-7 expressing the wild-type ERα and P-Box mutant and RNA-Seq in MCF-7 replacement system upon E2 stimulation were deposited at the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with an accession number GSE135341. The H3K27ac and H3K4me1 ChIP-Seq data were downloaded from GSE45822 and GSE40129; the DHS-Seq, FOXA1 and RNA Pol II ChIP-Seq data were downloaded from GSE33216, GSE26831 and GSE45822, respectively. The ERα ChIP-Seq data were downloaded from ArrayExpress: E-TABM-828 and GEO: GSE24166. The CTCF, STAG1 and RAD21 ChIP-Seq data were downloaded from ArrayExpress: E-TABM-828. The two time-course ERα ChIP-Seq data were downloaded from GSE94023 and GSE62789. The two GRO-Seq datasets were downloaded from GSE45822 and GSE59532. The RNA Pol II and CTCF ChIA-PET data were downloaded from GSE33664 and GSE39495 respectively.