

Summary
Congenital heart defects are a feature of several genetic haploinsufficiency syndromes, often involving transcriptional regulators. One property of haploinsufficient genes is their propensity for network interactions at the gene or protein level. In this article we took advantage of an online dataset of high throughput screening of mutations that are embryonic lethal in mice. Our aim was to identify new genes where the loss of function caused cardiovascular phenotypes resembling the 22q11.2 deletion syndrome models, that is, heterozygous and homozygous loss of Tbx1. One gene with a potentially haploinsufficient phenotype was identified, Setd5, thought to be involved in chromatin modification. We found murine Setd5 haploinsufficiency to be associated with double outlet right ventricle and perimembranous ventricular septal defect, although no genetic interaction with Tbx1 was detected. Conditional mutagenesis revealed that Setd5 was required in cardiopharyngeal mesoderm for progression of the heart tube through the ballooning stage to create a four‐chambered heart.
Keywords: birth defects, early development, heart, mesoderm
1. INTRODUCTION
Syndromic congenital heart disease (CHD) encompasses several haploinsufficiency conditions. Haploinsufficient genes may be present at a “hub” of interacting networks (Huang, Lee, Marcotte, & Hurles, 2010), and phenotypic similarity has been used in human genetics to cluster syndromes based on the premise that these groupings inform biological relationships of the genes involved that is, the underlying genes may function within the same developmental “module” (Oti & Brunner, 2007). The candidate gene for 22q11.2 deletion syndrome (22q11DS) is TBX1, and is haploinsufficient in mice (Jerome & Papaioannou, 2001; Merscher et al., 2001). In mice, Tbx1 heterozygotes present with hypo‐ or aplasia of the fourth PAA at embryonic day (E)10.5 which gives rise to great vessel defects such as interrupted aortic arch type B and aberrant right subclavian artery at later stages. Other cardiovascular defects include double outlet right ventricle (DORV) and ventricular septal defect (VSD) (Lindsay et al., 2001; Merscher et al., 2001).
An initiative entitled Deciphering the Mechanisms of Developmental Disorders (DMDD) aimed to take advantage of embryonic lethal mouse lines containing insertions that initially offer a gene‐trap‐like reporter allele with heterozygous or hypomorphic effect (https://dmdd.org.uk/; Mohun et al., 2013). Upon Flpe recombination, this converts to a conditional allele offering the potential for tissue‐specific studies of heart and brain development. Thus, the DMDD browser provides an online database of mutants where standard phenotype ontology terms can be used to search for genes with phenotypes related to known birth defect syndromes. The database contains high‐resolution episcopic microscopic (HREM) images of embryos at E9.5 and E14.5 allowing local rephenotyping. The database contained 209 genes analysed at E9.5 and E14.5. Querying the online browser with key phenotype ontology terms that define the Tbx1 haploinsufficient phenotype such as “DORV” and “VSD” generated a list of genes that caused these defects when mutated. Out of the 209 total genes, a mutation in only a single gene caused DORV and VSD when heterozygous: Setd5 (MGI allele: 4432631). We concluded Setd5 was a strong candidate for causing haploinsufficient cardiac defects that are similar to those observed due to Tbx1 haploinsufficiency. Moreover, there was a severe loss of function, lethal phenotype in Setd5 nulls (https://dmdd.org.uk/).
Setd5, a ubiquitously expressed gene (Osipovich, Gangula, Vianna, & Magnuson, 2016), is thought to act as a histone modifier. Unlike other SET‐domain proteins, SETD5 appears to lack histone methyltransferase activity (Deliu et al., 2018; Osipovich et al., 2016). Immunoprecipitation and mass spectrometry studies indicate SETD5 associated with HDAC3, a protein complex containing histone‐deacetylating activity; however, there is conflicting evidence on whether SETD5 is required for HDAC3 recruitment (Deliu et al., 2018; Nakagawa et al., 2020; Osipovich et al., 2016). Clinical reports reveal the implications of SETD5 haploinsufficiency in human intellectual disability, with some patients presenting with cardiac abnormalities (Fernandes et al., 2018; Grozeva et al., 2014; Szczałuba et al., 2016). Furthermore, CHD is present in up to 50% of patients with deleterious mutations in chromatin modification proteins underlying neurodevelopmental or psychiatric syndromes (Zaidi & Brueckner, 2017). Despite this, the role of Setd5 in cardiac development has not been extensively investigated.
Osipovich et al showed that mice lacking Setd5 (MGI allele: 5576778) die before E10.5 and were developmentally delayed; embryos were severely underdeveloped with many presenting with hemorrhaging compared to littermate heterozygous controls (Osipovich et al., 2016). Interestingly, unlike the heterozygous embryos presented in the DMDD browser, Osipovich et al reported heterozygous mice as viable and indistinguishable from the wild‐type embryos. Furthermore, the embryonic lethality of Setd5 nulls by E10.5 limits the full assessment of the cardiac phenotype. The purpose of our study was, primarily, to more fully delineate the cardiac phenotype caused by the disruption of Setd5 expression, and secondarily to explore whether there was any genetic interaction between Setd5 and Tbx1. Currently, attempts to understand the Setd5 function are focused on neuronal contexts; here we show the importance of this protein during heart development.
2. MATERIALS AND METHODS
2.1. Mouse strains, breeding, and genotyping
All animal husbandry, maintenance, and procedures were carried out in accordance to the UK Home Office regulations. All mice were maintained on the C57Bl/6 background.
The Setd5 Fl/Fl line was derived from the targeted mouse line (Setd5tm1a[EUCOMM]Wtsi MGI:4432631) (Wellcome Trust Sanger Institute) and kindly donated by the Basson laboratory (KCL). Setd5 heterozygotes were generated by crossing the floxed line with mice expressing Actin‐Cre (Lewandoski, Meyers, & Martin, 1997). F1 mice harboring the Actin‐Cre were backcrossed with wild‐type mice in order to generate mice without the Actin‐Cre for use in downstream experiments. Generation of double heterozygotes involved breeding Setd5 +/− with Tbx1 lacZ/+ heterozygotes, described previously (Lindsay et al., 2001). To generate Tbx1‐cKOs, Setd5 Fl/Fl mice were crossed with Tbx1 Cre/+ heterozygous mice, described previously (Huynh, Chen, Terrell, & Baldini, 2007). To delete Setd5 in the cardiopharyngeal mesoderm, Setd5 Fl/Fl mice were bred with mice expressing Mesp1‐Cre, described previously (Saga et al., 1999). Timed matings for embryo collection involved pairing sexually mature mice overnight and checking the presence of a copulation plug the following morning, which indicated 0.5 days post copulation. The PCR strategies for genotyping are available upon request.
2.2. Tissue processing, whole‐mount analysis, and histological analysis
Whole embryos were fixed in 4% paraformaldehyde overnight at 4°C and then stored in PBS until required. For histological analyzes, whole embryos or hearts were dehydrated in ethanol, and then processed in Histoclear and several washes in wax. Samples were embedded in paraffin and cut into 10 μm sections, air‐dried overnight and then rehydrated through a decreasing gradient of ethanol and stained with hemotoxylin and eosin. Whole‐mount embryo pictures were taken using the Zeiss Lumar V12 Stereoscope. Histological sections were imaged using the Zeiss Axioplan or the slide scanning facility at UCL IQPath. Stage‐matching was performed by assessment of the limb buds and pharyngeal arches (Boehm et al., 2011; Musy et al., 2018).
2.3. Calculation of double heterozygote expected frequencies
In the absence of a synergistic genetic interaction, the expected frequency of defects in a double heterozygote in which two genes of interest are haploinsufficient is calculated as follows:
(A + B[1 – A])*n.
A = percentage of defects observed in one single heterozygote. B = percentage of defects observed in alternate single heterozygote. n = total number of double heterozygotes collected.
Taking the example of VSDs presented in Table 1, A = 75% = 0.75; B = 31% = 0.31; n = 14.
TABLE 1.
Summary of heart defects observed in WT, Setd5 heterozygotes (Setd5 +/−), Tbx1 heterozygotes (Tbx1 lacZ/+) and double heterozygotes (Setd5 +/−; Tbx1 lacZ/+) at E14.5
| H&E analysis | |||||
|---|---|---|---|---|---|
| WT(n = 8) | Setd5 +/− (n = 12) | Tbx1 lacZ/+ (n = 13) | Setd5 +/−; Tbx1 lacZ/+ (n = 14) | ||
| Observed | Expected n number | ||||
| CAT | 0 | 0 | 0 | 0 | 0 |
| Rotational defects | 0 | 6 (50%)* | 1 (8%) | 8 (57%) | 8 |
| VSD | 1 (13%) | 9 (75%)* | 4 (31%) | 12 (86%) | 12 |
| Great vessel defects | |||||
| AbRS | 0 | 0 | 1 (8%) | 3 (21%) | 1 |
Note: A Fisher's exact test was performed to determine whether the defects observed in Setd5 +/− were statistically significant compared to wild‐type control embryos (*p < .05 compared with WT). The expected frequency of defects in double heterozygotes (Setd5 +/−; Tbx1 lacZ/+) was calculated as described in the methods. Rotational defects encompass both double outlet right ventricle and overriding aorta.
Abbreviations: AbRS, aberrant right subclavian artery; CAT, common arterial trunk; VSD, ventricular septal defect.
(0.75 + 0.31[1–0.75])* 14 = 12.
2.4. Statistical analysis
To determine whether genetically modified mice or embryos presented at Mendelian ratios, Chi‐squared analysis was performed. To determine the association of genotype with the frequency of defects observed, Fisher's exact test was performed. A p value less than .05 was taken to be significant.
3. RESULTS
3.1. Setd5 haploinsufficiency leads to outflow tract rotational defects and VSDs
Setd5 Fl/Fl mice were derived from the targeted mouse line (Setd5 tm1a[EUCOMM]Wtsi). The floxed line was then bred with Tmem163 Tg(ACTB‐cre)2Mrt (Actin‐Cre) mice in order to generate Setd5 heterozygotes (Setd5 +/−) for use in downstream embryo experiments. At E14.5, normal heart development results in the septation of the outflow tract (OFT) into the pulmonary trunk and aorta, with an intact interventricular septum dividing the left and right ventricles. A rotational defect, such as DORV or overriding aorta, occurs when the OFT fails to align with the two future ventricles at the looping stage. Correct looping of the heart tube is necessary to align the two definitive outflow vessels with their corresponding ventricular chambers (Christoffels et al., 2000; Moorman & Christoffels, 2003). If the OFT is misaligned such that an abnormal amount of aortic blood originates from the right ventricle, then this is known as an overriding aorta. If more than 50% of the aortic blood arises from the right ventricle, the result is DORV (Creazzo, Godt, Leatherbury, Conway, & Kirby, 1998).
We found that wild‐type hearts displayed correctly OFT septation with the outflow vessels originating from separate ventricles (Figure 1a‑a‴). In contrast, despite correct OFT septation, 50% of Setd5 heterozygotes presented with rotational defects which included DORV or overriding aorta (Figure 1b‑b‴) (p < .05, Fisher's exact test, Table 1). DORV is always accompanied by a VSD, since the fusion of the interventricular septum with the OFT cushions is necessary to complete ventricular septation. If the outflow vessels of the OFT are misaligned with the ventricular chambers, as in DORV, then this fusion cannot take place which leads to a perimembranous VSD (Anderson, 2003; Lin, Lin, Chen, Zhou, & Chang, 2012). Figure 1b″″ shows a perimembranous VSD, which was observed in 75% of Setd5 heterozygotes (p < .05, Fisher's exact test, Table 1).
FIGURE 1.
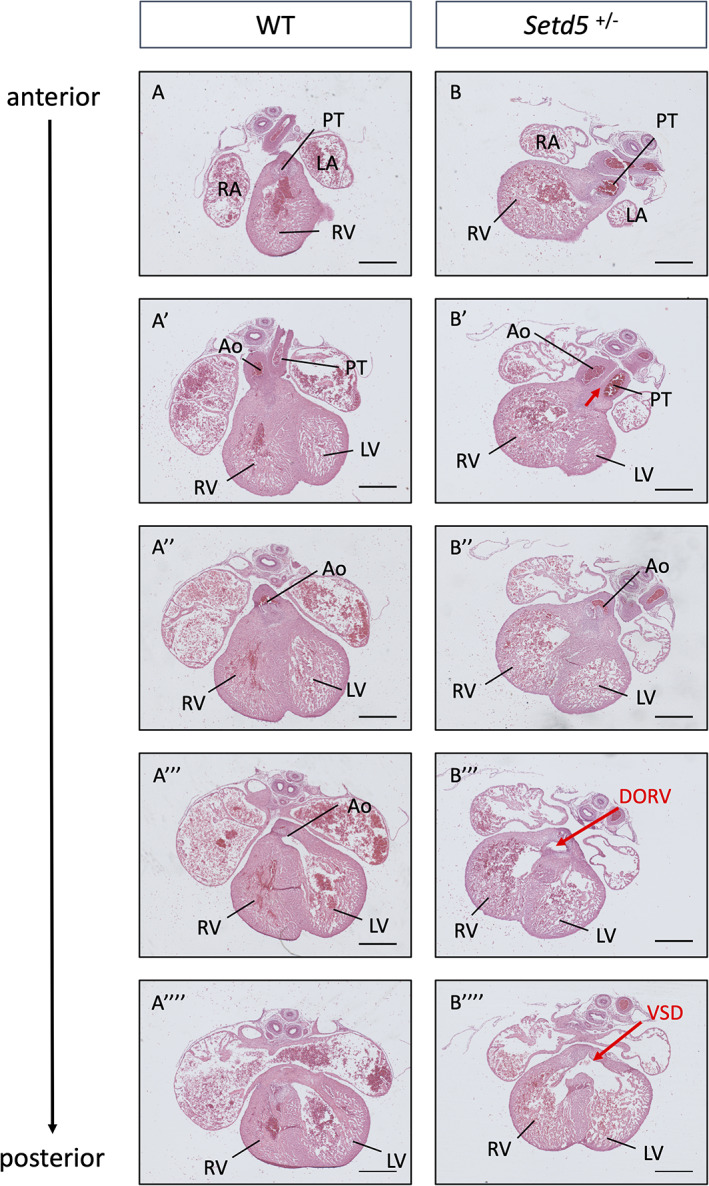
Rotational defects, including DORV was observed in 50% of Setd5 heterozygotes (Setd5 +/−) and VSD were observed in 75% of Setd5 +/− embryos at E14.5. In the WT heart, the pulmonary trunk and aorta open into separate ventricles: the right and left ventricle, respectively (a‑a‴). In Setd5 +/− hearts, DORV was observed. In panel B′ the aorta and pulmonary trunk are juxtaposed, and in posterior sections b‴, the aorta eventually opens into the right ventricle; both the pulmonary trunk and aorta open into the right ventricle. A perimembranous VSD is observed in Setd5 +/− (panel b″″). Ao, aorta; DORV, double outlet right ventricle; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle; VSD, ventricular septal defects; WT, wild‐type. Scale bars = 400 μm
Therefore, in alignment with the DMDD browser, our results show that Setd5 haploinsufficiency causes OFT rotation defects and VSDs.
3.2. No detectable genetic interaction between Setd5 and Tbx1, a well‐established haploinsufficient gene
Because some of the phenotypes observed in Setd5 heterozygotes overlap with those observed in Tbx1 heterozygotes, the genetic interaction between Setd5 and Tbx1 was investigated by generating double heterozygotes, in which both genes of interest are haploinsufficient. Setd5 heterozygotes were mated with Tbx1 lacZ/+ heterozygotes (Tbx1 tm1Bld) and embryos were harvested at E14.5. An exacerbation of defects, that is, an increased frequency of defects over that expected by additive effects, or an increase in severity of defects, would be observed in the double heterozygotes in the case of synergistic genetic interaction.
There was no difference in the number of embryos at E14.5 based on the genotypes (Table STable 1, Chi‐squared, p > .05). Gross morphological analysis during embryo dissection and internal assessment by histological methods revealed no exacerbation or increased frequency of defects in double heterozygotes, suggesting that there was no detectable synergistic genetic interaction between Setd5 and Tbx1 and that these two genes are not functionally interdependent (Table 1).
3.3. Conditional mutagenesis of Setd5 reveals a role in cardiogenic mesoderm
Our results show that the haploinsufficiency of Setd5 led to rotational defects and VSD. These defects are reminiscent of the second heart field (SHF)‐specific defects seen in Tbx1 mutants. To investigate whether Setd5 has a specific role in the SHF, and/or pharyngeal epithelia, Setd5 was deleted using a Tbx1‐Cre driver known to be active in these tissues (Huynh et al., 2007). Figure 2 presents the gross morphology of control and several Tbx1 conditional knockout (Tbx1‐cKO) hearts at E15.5. Minor abnormalities such as retroesophageal right subclavian artery were observed (one in both Tbx1 conditional heterozygote and knockout); this was likely due to the effect of the Cre knock‐in to the Tbx1 locus. Similarly, assessment of internal heart morphology revealed no obvious differences between the genotypes (Figure SFigure 1). Therefore, we concluded Setd5 likely exerts its role earlier during cardiac development, or in other lineages, or both.
FIGURE 2.
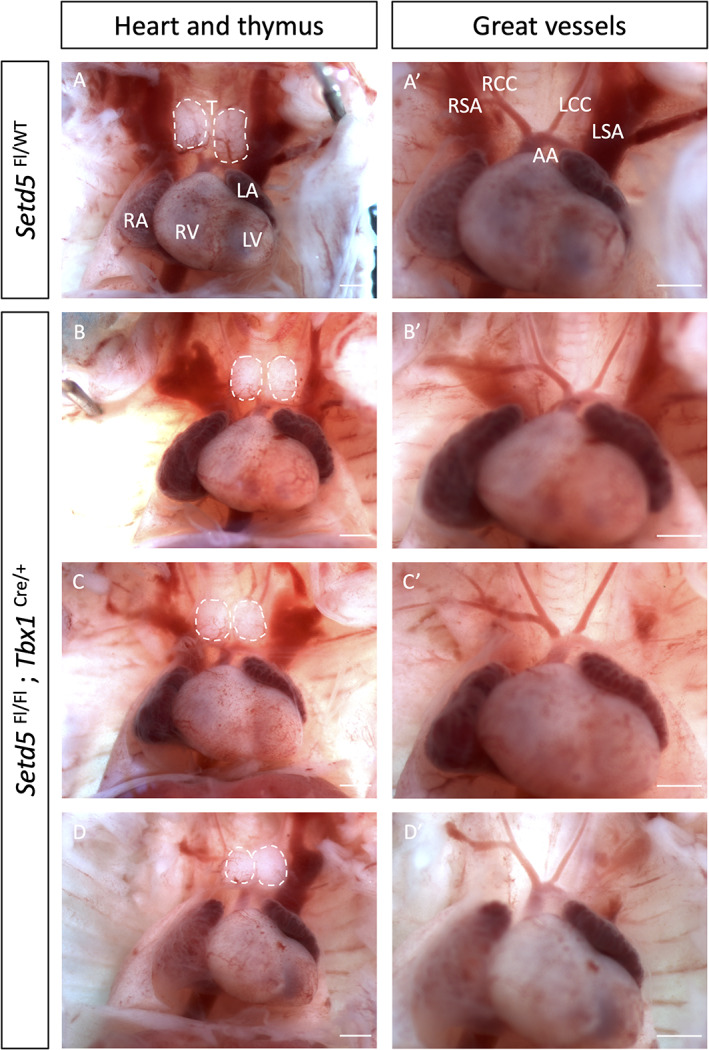
No obvious heart malformations were observed when Setd5 was homozygously deleted using the Tbx1‐Cre at E15.5. No great vessel defects were observed across the genotypes (n = 19 conditional mutants analysed). AA, aortic arch; LA, left atria; LCC, left common carotid; LSA, left subclavian artery; LV, left ventricle; RA, right atria; RCC, right common carotid; RSA, right subclavian artery; RV, right ventricle; T, thymus. Scale bars = 1 mm
Indeed, the DMDD browser revealed an embryonic lethality of homozygous Setd5 mutation (no surviving embryos at E14.5), and Osipovich et al., showed that mice constitutively null for Setd5 die before E10.5, and are severely underdeveloped. Hearts at E9.5 were swollen with a single ventricular chamber (Osipovich et al., 2016). The embryonic lethality of Setd5 constitutive null embryos by E10.5 limits analysis of the importance of Setd5 in cardiac development. Lineage tracing experiments show that the majority of cardiac cells are derived from Mesp1‐expressing progenitor cells (Saga et al., 1999). Therefore, we used the Mesp1‐Cre driver to delete Setd5 in mesoderm encompassing both the first heart field (FHF) and SHF, as well as the pharyngeal mesoderm through which the (non‐Mesp1‐expressing) neural crest migrates. Homozygous deletion in this cardiopharyngeal mesoderm lineage led to embryonic lethality before E12.5. At E10.5, Mesp1‐cKO embryos were harvested at the expected Mendelian ratio (Table S2) but exhibited varying degrees of abnormal cardiac morphogenesis compared to stage‐matched control embryos (p < .05, Table 2). 90% of Mesp1‐cKOs displayed abnormal cardiac chamber ballooning, with 25% of these exhibiting a dumbbell‐shaped heart (Figure 3b), while others displayed right ventricular hypoplasia and abnormal atrial ballooning (p < .05, Figure 3d). We assessed the internal heart morphology by histological techniques and found that 100% of Mesp1‐cKO embryos presented with a short OFT, indistinct ventricular chambers, and poorly developed atria, revealing the importance of Setd5 in OFT elongation and cardiac chamber ballooning (Figure 4). These defects were accompanied by a statistically significant growth delay, determined by the crown‐to‐rump length (p < .05, Figure SFigure 2). The OFT length was further assessed by quantifying the number of paraffin sections containing the OFT and expressing this as a ratio to the crown‐to‐rump length, confirming the shorter OFT in Mesp1‐cKO compared to control embryos (Figure SFigure 3). Morphological analysis of Mesp1‐Cre conditional heterozygotes (n = 6) did not reveal any OFT phenotype, and histological analysis revealed only a single embryo as having poorly developed cardiac chambers.
TABLE 2.
Summary of defects observed in Setd5 Fl/Fl, Setd5 Fl/WT, Setd5 Fl/WT; Mesp1 Cre/+, and Setd5 Fl/Fl; Mesp1 Cre/+ embryos at E10.5
| Abnormal external morphology | ||||
|---|---|---|---|---|
| Setd5 Fl/Fl(n = 17) | Setd5 Fl/WT (n = 23) | Setd5 Fl/WT ; Mesp1 Cre/+ (n = 17) | Setd5 Fl/Fl ; Mesp1 Cre/+ (n = 19) | |
| Hemorrhage | 0 | 0 | 0 | 3 (16%) |
| Pericardial effusion* | 0 | 3 (13%) | 0 | 14 (74%) |
| Abnormal cardiac chamber ballooning* | 0 | 0 | 0 | 18 (95%) |
| H&E analysis | ||||
| Setd5 Fl/Fl (n = 2) | Setd5 Fl/WT (n = 4) | Setd5 Fl/WT ; Mesp1 Cre/+ (n = 6) | Setd5 Fl/Fl ; Mesp1 Cre/+ (n = 6) | |
| Short OFT* | 0 | 0 | 0 | 6 (100%) |
| Abnormal ventricular ballooning* | 0 | 0 | 1 (17%) | 6 (100%) |
| Abnormal atrial ballooning* | 0 | 0 | 0 | 6 (100%) |
Note: The upper part of the table describes the defects observed during the dissection stage. The abnormal external cardiac morphology was further delineated by sectioning and H&E analysis. Fisher's exact test was used to determine the association between the genotype and the frequency of any defects observed.
p < .05.
FIGURE 3.
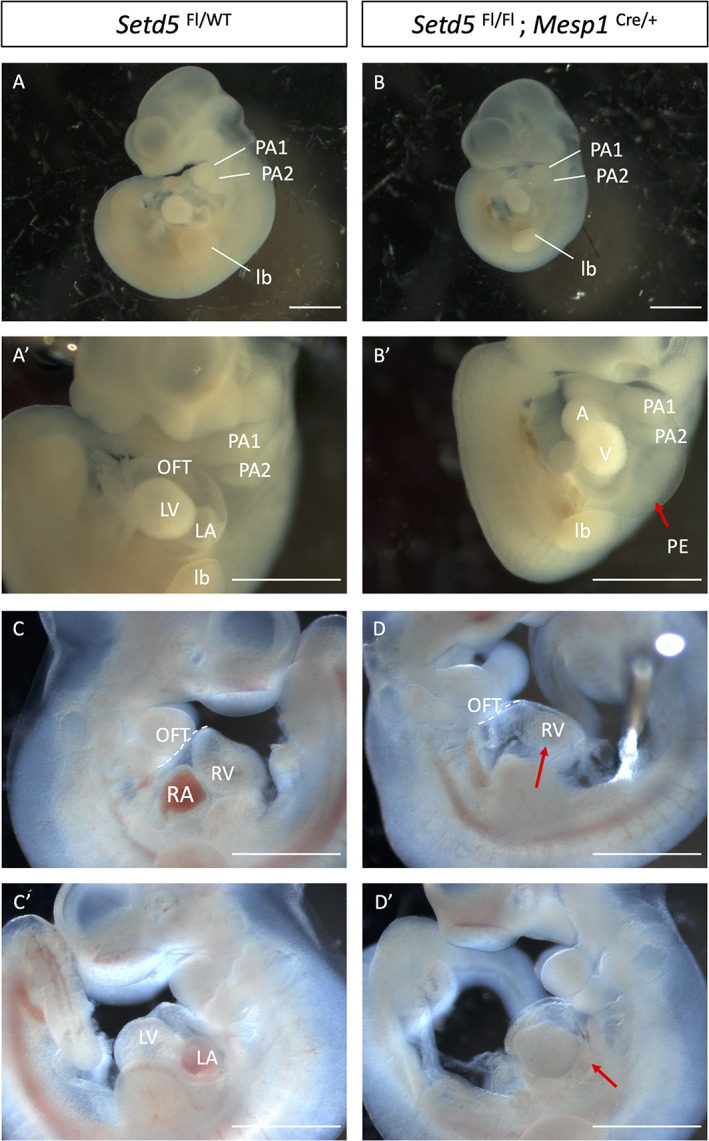
Mesp1‐cKO (Setd5 Fl/Fl; Mesp1 Cre/+) embryos displayed abnormal cardiac chamber ballooning and pericardial effusion at E10.5. At E10.5, Mesp1‐cKO embryos presented with abnormal cardiac morphogenesis in 90% of the cases with 25% of these exhibiting a dumbbell shaped heart: a single atria and single ventricle (b,b′) compared with control embryos (Setd5 Fl/Fl or Setd5 Fl/WT). Pericardial effusion was observed in 70% of Mesp1‐cKO embryos (red arrow, b′). (d,d′) show a Mesp1‐cKO embryo with a short outflow tract, right ventricular hypoplasia, and abnormal atrial ballooning (red arrow). A, atrium; LA, left atrium; LV, left ventricle; lb, limb bud; OFT, outflow tract; PE, pericardial effusion; PAA, pharyngeal arch artery. Scale bars = 1 mm
FIGURE 4.
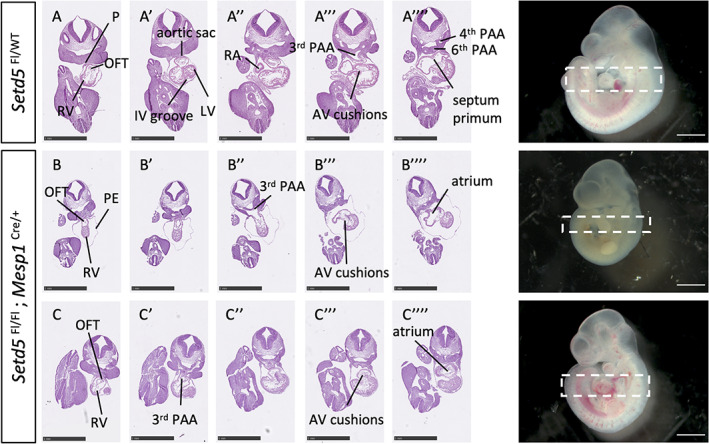
Short OFT and abnormal chamber morphology in Setd5 Fl/Fl; Mesp1 Cre/+ embryos at E10.5. (A‑A″″) Transverse sections of whole E10.5 Setd5 Fl/WT control embryo stained with H&E. The IV groove is observed in panel A'. The emergence of the septum primum (A″″) occurs during atrial septation. (B‑B″″) Transverse sections of whole E10.5 Setd5 Fl/Fl; Mesp1 Cre/+ (cKO) embryo with dumbbell‐shaped heart. Short OFT is depicted by the fewer number of sections between the point at which the OFT emerged and when it fed into the 3rd PAA. The atria are poorly developed with a thickened wall (B””). (C‑C””) Transverse sections of whole E10.5 Setd5 Fl/Fl; Mesp1 Cre/+ (cKO) embryo with a slightly milder phenotype than in B. Embryo presents with a short OFT and no evidence of the interventricular groove, and thickened atrial wall. Sections presented are from top to bottom of the dashed box in whole embryo photograph. AV, atrioventricular; IV, interventricular; LV, left ventricle; OFT, outflow tract; PE, pericardial effusion; P, pericardium; PAA, pharyngeal arch artery; RA, right atrium; RV, right ventricle. Scale bars = 1 mm
4. DISCUSSION
Our results indicate that haploinsufficiency of Setd5, a gene implicated in human intellectual disability (Fernandes et al., 2018; Grozeva et al., 2014; Szczałuba et al., 2016) with reported histone‐modifying activity (Deliu et al., 2018; Nakagawa et al., 2020; Osipovich et al., 2016), leads to cardiac defects in mice. It is worth noting that only one Mesp1‐Cre conditional heterozygote presented with poorly developed cardiac chambers. The remaining embryos exhibited normal cardiac morphology, highlighting the fact that deleting one allele of Setd5 in the Mesp1 lineage does not fully recapitulate the global heterozygotes and that haploinsufficiency of Setd5 requires constitutive heterozygosity. Since haploinsufficient genes exist in hubs or networks and are more likely to interact with other haploinsufficient genes (Huang et al., 2010), and because haploinsufficiency of Setd5, and of Tbx1 lead to similar cardiac defects, one of our initial aims were to investigate their genetic interaction. However, our findings did not reveal a detectable synergistic genetic interaction, suggesting that Setd5 and Tbx1 are not functionally interdependent. Nevertheless, homozygous deletion of Setd5 in the cardiopharyngeal mesoderm revealed a novel role of Setd5 in cardiac development: its importance in OFT elongation and cardiac chamber ballooning.
In humans, loss of function mutations in SETD5 lead to moderate or severe intellectual disability (Fernandes et al., 2018; Grozeva et al., 2014; Powis et al., 2018; Rawlins, Stals, Eason, & Turnpenny, 2017; Szczałuba et al., 2016). It is also one of three genes within the candidate region of 3p25 microdeletion syndrome which is characterized by developmental delay, intellectual disability, low birth weight, microcephaly and craniofacial abnormalities, and some observations of CHD (Grozeva et al., 2014; Kuechler et al., 2015; Szczałuba et al., 2016). Mutations of SETD5 have been reported in other conditions. For instance, one deletion and two mutations were described in patients clinically diagnosed with KBG syndrome without a mutation in ANKRD11, which is usually observed in these cases (OMIM 148050). One such patient presented with mitral stenosis (Crippa et al., 2020). Interestingly, ANKRD11 recruits HDACs and is mutated in autism (Gallagher et al., 2015). Recently, SETD5 has been implicated in moyamoya angiopathy, which is the occlusion of large cerebral arteries leading to childhood stroke (Pinard et al., 2020). Our results reveal that Setd5, a gene implicated in neurodevelopmental disorders, is also important in cardiac development, and may provide some explanation to the clinical observations of CHD when SETD5 is haploinsufficient in humans. Taken together, these data suggest considerable pleiotropy of SETD5 function.
The genetic underpinnings of CHD remain unknown in over 50% of cases, highlighting the importance of identifying novel genes that may be involved in orchestrating cardiac development. Furthermore, genome sequencing CHD studies present an overrepresentation of genes involved in neurodevelopmental disorder (Sigmon, Kelleman, Susi, Nylund, & Oster, 2019; Watkins et al., 2019). As such, screening for mutations in SETD5 during genetic testing could potentially provide some knowledge to families to better understand if there could be recurrent risks of CHD to other family members, or provide information for neurodevelopmental outcome.
Thus, our results provide the basis for further exploration of Setd5 in cardiac development. It will be interesting to investigate how this histone modifier plays a role in orchestrating cardiac development in conjunction with other known cardiac‐relevant genes. Setd5 can now be placed within a growing number of genes encoding chromatin modifiers whose alleles are associated with both neurodevelopmental defects on the one hand, and heart dysmorphogenesis on the other.
Supporting information
Supplementary Figure 1 Transverse sections of hearts of Tbx1‐cKO (Setd5 Fl/Fl ; Tbx1 Cre/+ ) at E15.5.
Panels A – A" show correct outflow tract (OFT) septation across all genotypes. The atrial septum (B – B″) and AV valves (C – C″) were intact across all genotypes
Abbreviations: aorta (Ao), left atrium (LA), left ventricle (LV), pulmonary trunk (PT), right atrium (RA), right ventricle (RV). Scale bars = 200 μm.
Supplementary Figure 2 Box plot showing the crown‐to‐rump length in mm at E10.5.
Setd5 Fl/Fl; Mesp1 Cre/+ cKO embryos are significantly smaller than control embryos (Setd5 Fl/Fl or Setd5 Fl/WT, p < .05, one‐way ANOVA). Error bars present the standard error of mean.
Supplementary Figure 3 Graph showing the OFT length of control embryos (Setd5 Fl/Fl or Setd5 Fl/WT ) and cKO embryos (Setd5 Fl/Fl ; Mesp1 Cre/+ )
The OFT length is presented as a ratio of the number of H&E paraffin sections containing the OFT, to the crown‐to‐rump length. An unpaired student's t test showed that cKO embryos had a shorter OFT than control embryos (*p < .05). Error bars present the standard error of mean.
Supplementary Table 1 Observed and expected number of embryos at E14.5 resulting Setd5 +/− x Tbx1 lacZ/+ cross.
Expected numbers are based on Mendelian ratios, rounded to the nearest whole number. There was no statistical difference in the number of E14.5 embryos across the genotypes, based on Chi‐squared analysis, p > .05.
Supplementary Table 2: Observed and expected number of embryos at E10.5 resulting Setd5 Fl/Fl x Setd5 Fl/WT ; Mesp1 Cre/+ cross.
Expected numbers are based on Mendelian ratios, rounded to the nearest whole number. There was no statistical difference in the number of E10.5 embryos across the genotypes, based on Chi‐squared analysis, p > .05.
ACKNOWLEDGMENTS
The authors thank Professor Albert Basson for kindly providing the Setd5 floxed mice, and the UCL IQPath facility for scanning some H&E slides. This work was supported by the British Heart Foundation (BHF FS/16/61/32740 and RG151431880), and the Fondation Leducq (15CDV01). Research infrastructure within the institute is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. We thank the UCL Biological Services Unit for help with transgenic colonies.
Cheung, M. Y.‐Q. , Roberts, C. , Scambler, P. , & Stathopoulou, A. (2021). Setd5 is required in cardiopharyngeal mesoderm for heart development and its haploinsufficiency is associated with outflow tract defects in mouse. genesis, 59(7), e23421. 10.1002/dvg.23421
Funding information British Heart Foundation, Grant/Award Numbers: FS/16/61/32740, RG151431880; Fondation Leducq, Grant/Award Number: 15CDV01
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Anderson, R. H. (2003). Development of the heart: (3) formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart, 89(9), 1110–1118. 10.1136/heart.89.9.1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm, B. , Rautschka, M. , Quintana, L. , Raspopovic, J. , Jan, Ž. , & Sharpe, J. (2011). A landmark‐free morphometric staging system for the mouse limb bud. Development, 138(6), 1227–1234. 10.1242/dev.057547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels, V. M. , Habets, P. E. M. H. , Franco, D. , Campione, M. , de Jong, F. , Lamers, W. H. , … Moorman, A. F. M. (2000). Chamber formation and morphogenesis in the developing mammalian heart. Developmental Biology, 223(2), 266–278. 10.1006/dbio.2000.9753 [DOI] [PubMed] [Google Scholar]
- Creazzo, T. L. , Godt, R. E. , Leatherbury, L. , Conway, S. J. , & Kirby, M. L. (1998). Role of cardiac neural crest cells in cardiovascular development. Annual Review of Physiology, 60(1), 267–286. 10.1146/annurev.physiol.60.1.267 [DOI] [PubMed] [Google Scholar]
- Crippa, M. , Bestetti, I. , Maitz, S. , Weiss, K. , Spano, A. , Masciadri, M. , … Finelli, P. (2020). SETD5 gene Haploinsufficiency in three patients with suspected KBG syndrome. Frontiers in Neurology, 11(July), 1–9. 10.3389/fneur.2020.00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliu, E. , Arecco, N. , Morandell, J. , Dotter, C. P. , Contreras, X. , Girardot, C. , … Novarino, G. (2018). Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nature Neuroscience, 21(12), 1717–1727. 10.1038/s41593-018-0266-2 [DOI] [PubMed] [Google Scholar]
- DMDD Deciphering the Mechanisms of Developmental Disorders. (2020). Retrieved from https://dmdd.org.uk/
- Fernandes, I. R. , Cruz, A. C. P. , Ferrasa, A. , Phan, D. , Herai, R. H. , & Muotri, A. R. (2018). Genetic variations on SETD5 underlying autistic conditions. Developmental Neurobiology, 78(5), 500–518. 10.1002/dneu.22584 [DOI] [PubMed] [Google Scholar]
- Gallagher, D. , Voronova, A. , Zander, M. A. , Cancino, G. I. , Bramall, A. , Krause, M. P. , … Miller, F. D. (2015). Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Developmental Cell, 32(1), 31–42. 10.1016/j.devcel.2014.11.031 [DOI] [PubMed] [Google Scholar]
- Grozeva, D. , Carss, K. , Spasic‐Boskovic, O. , Parker, M. J. , Archer, H. , Firth, H. V. , … Raymond, F. L. (2014). De novo loss‐of‐function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. The American Journal of Human Genetics, 94(4), 618–624. 10.1016/j.ajhg.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, N. , Lee, I. , Marcotte, E. M. , & Hurles, M. E. (2010). Characterising and predicting haploinsufficiency in the human genome. PLoS Genetics, 6(10), 1–11. 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, T. , Chen, L. , Terrell, P. , & Baldini, A. (2007). A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis, 45(7), 470–475. 10.1002/dvg.20317 [DOI] [PubMed] [Google Scholar]
- Jerome, L. A. , & Papaioannou, V. E. (2001). DiGeorge syndrome phenotype in mice mutant for the T‐box gene, Tbx1. Nature Genetics, 27(3), 286–291. 10.1038/85845 [DOI] [PubMed] [Google Scholar]
- Kuechler, A. , Zink, A. M. , Wieland, T. , Lüdecke, H. J. , Cremer, K. , Salviati, L. , … Engels, H. (2015). Loss‐of‐function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. European Journal of Human Genetics, 23(6), 753–760. 10.1038/ejhg.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski, M. , Meyers, E. N. , & Martin, G. R. (1997). Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harbor Symposia on Quantitative Biology, 62(1), 159–168. 10.1101/SQB.1997.062.01.021 [DOI] [PubMed] [Google Scholar]
- Lin, C.‐J. , Lin, C.‐Y. , Chen, C.‐H. , Zhou, B. , & Chang, C.‐P. (2012). Partitioning the heart: Mechanisms of cardiac septation and valve development. Development, 139(18), 3277–3299. 10.1242/dev.063495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay, E. A. , Vitelli, F. , Su, H. , Morishima, M. , Huynh, T. , Pramparo, T. , … Baldini, A. (2001). Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature, 410(6824), 97–101. 10.1038/35065105 [DOI] [PubMed] [Google Scholar]
- Merscher, S. , Funke, B. , Epstein, J. A. , Heyer, J. , Puech, A. , Lu, M. M. , … Kucherlapati, R. (2001). TBX1 is responsible for cardiovascular defects in velo‐cardio‐facial/DiGeorge syndrome. Cell, 104(4), 619–629. 10.1016/S0092-8674 [DOI] [PubMed] [Google Scholar]
- Mohun, T. , Adams, D. J. , Baldock, R. , Bhattacharya, S. , Copp, A. J. , Hemberger, M. , … Weninger, W. (2013). Deciphering the mechanisms of developmental disorders (DMDD): A new programme for phenotyping embryonic lethal mice. Disease Models & Mechanisms, 6(3), 562–566. 10.1242/dmm.011957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman, A. F. M. , & Christoffels, V. M. (2003). Cardiac chamber formation: Development, genes, and evolution. Physiological Reviews, 83(4), 1223–1267. 10.1152/physrev.00006.2003 [DOI] [PubMed] [Google Scholar]
- Musy, M. , Flaherty, K. , Raspopovic, J. , Robert‐Moreno, A. , Richtsmeier, J. T. , & Sharpe, J. (2018). A quantitative method for staging mouse embryos based on limb morphometry. Development (Cambridge), 145(7), 1–7. 10.1242/dev.154856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T. , Hattori, S. , Nobuta, R. , Kimura, R. , Nakagawa, M. , Matsumoto, M. , … Nakayama, K. (2020). The autism‐related protein SETD5 controls neural cell proliferation through epigenetic regulation of rDNA expression. IScience, 23(4), 101030. 10.1016/j.isci.2020.101030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovich, A. B. , Gangula, R. , Vianna, P. G. , & Magnuson, M. A. (2016). Setd5 is essential for mammalian development and the co‐transcriptional regulation of histone acetylation. Development, 143(24), 4595–4607. 10.1242/dev.141465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti, M. , & Brunner, H. G. (2007). The modular nature of genetic diseases. Clinical Genetics, 71(1), 1–11. 10.1111/j.1399-0004.2006.00708.x [DOI] [PubMed] [Google Scholar]
- Pinard, A. , Guey, S. , Guo, D. , Cecchi, A. C. , Sharrief, A. Z. , Bergametti, F. , … Tournier‐lasserve, E. (2020). The pleiotropy associated with de novo variants in CHD4, CNOT3, and SETD5 extends to moyamoya angiopathy. Genetics in Medicine, 22(2), 427–431. 10.1038/s41436-019-0639-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis, Z. , Farwell Hagman, K. D. , Mroske, C. , McWalter, K. , Cohen, J. S. , Colombo, R. , … Tang, S. (2018). Expansion and further delineation of the SETD5 phenotype leading to global developmental delay, variable dysmorphic features, and reduced penetrance. Clinical Genetics, 93(4), 752–761. 10.1111/cge.13132 [DOI] [PubMed] [Google Scholar]
- Rawlins, L. E. , Stals, K. L. , Eason, J. D. , & Turnpenny, P. D. (2017). De novo SETD5 nonsense mutation associated with diaphragmatic hernia and severe cerebral cortical dysplasia. Clinical Dysmorphology, 26(2), 95–97. 10.1097/MCD.0000000000000144 [DOI] [PubMed] [Google Scholar]
- Saga, Y. , Miyagawa‐Tomita, S. , Takagi, A. , Kitajima, S. , Miyazaki, J. , & Inoue, T. (1999). MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development, 126(15), 3437–3447.Retrieved from. http://www.ncbi.nlm.nih.gov/pubmed/10393122 [DOI] [PubMed] [Google Scholar]
- Sigmon, E. R. , Kelleman, M. , Susi, A. , Nylund, C. M. , & Oster, M. E. (2019). Congenital heart disease and autism: A case‐control study. Pediatrics, 144(5), 1–8. 10.1542/peds.2018-4114 [DOI] [PubMed] [Google Scholar]
- Szczałuba, K. , Brzezinska, M. , Kot, J. , Rydzanicz, M. , Walczak, A. , Stawiński, P. , … Płoski, R. (2016). SETD5 loss‐of‐function mutation as a likely cause of a familial syndromic intellectual disability with variable phenotypic expression. American Journal of Medical Genetics, Part A, 170(9), 2322–2327. 10.1002/ajmg.a.37832 [DOI] [PubMed] [Google Scholar]
- Watkins, W. S. , Hernandez, E. J. , Wesolowski, S. , Bisgrove, B. W. , Sunderland, R. T. , Lin, E. , … Tristani‐Firouzi, M. (2019). De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nature Communications, 10(1), 1–12. 10.1038/s41467-019-12582-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi, S. , & Brueckner, M. (2017). Genetics and genomics of congenital heart disease. Circulation Research, 120(6), 923–940. 10.1161/CIRCRESAHA.116.309140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Transverse sections of hearts of Tbx1‐cKO (Setd5 Fl/Fl ; Tbx1 Cre/+ ) at E15.5.
Panels A – A" show correct outflow tract (OFT) septation across all genotypes. The atrial septum (B – B″) and AV valves (C – C″) were intact across all genotypes
Abbreviations: aorta (Ao), left atrium (LA), left ventricle (LV), pulmonary trunk (PT), right atrium (RA), right ventricle (RV). Scale bars = 200 μm.
Supplementary Figure 2 Box plot showing the crown‐to‐rump length in mm at E10.5.
Setd5 Fl/Fl; Mesp1 Cre/+ cKO embryos are significantly smaller than control embryos (Setd5 Fl/Fl or Setd5 Fl/WT, p < .05, one‐way ANOVA). Error bars present the standard error of mean.
Supplementary Figure 3 Graph showing the OFT length of control embryos (Setd5 Fl/Fl or Setd5 Fl/WT ) and cKO embryos (Setd5 Fl/Fl ; Mesp1 Cre/+ )
The OFT length is presented as a ratio of the number of H&E paraffin sections containing the OFT, to the crown‐to‐rump length. An unpaired student's t test showed that cKO embryos had a shorter OFT than control embryos (*p < .05). Error bars present the standard error of mean.
Supplementary Table 1 Observed and expected number of embryos at E14.5 resulting Setd5 +/− x Tbx1 lacZ/+ cross.
Expected numbers are based on Mendelian ratios, rounded to the nearest whole number. There was no statistical difference in the number of E14.5 embryos across the genotypes, based on Chi‐squared analysis, p > .05.
Supplementary Table 2: Observed and expected number of embryos at E10.5 resulting Setd5 Fl/Fl x Setd5 Fl/WT ; Mesp1 Cre/+ cross.
Expected numbers are based on Mendelian ratios, rounded to the nearest whole number. There was no statistical difference in the number of E10.5 embryos across the genotypes, based on Chi‐squared analysis, p > .05.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.