

Abstract
Lysosomal acid lipase deficiency (LAL-D) is a multi-organ autosomal recessive disease caused by mutations in LIPA. We reviewed data from 681 samples (white blood cells [WBC] n = 625, fibroblasts = 30, liver = 4, amniocytes = 13, chorionic villus = 9) received for analysis of lysosomal acid lipase (LAL) activity over a 15-year period. LIPA sequencing was performed in 49 patients with reduced (n = 26) or deficient (n = 23) LAL activity. The Exome Aggregation Consortium and Genome Aggregation Database dataset were used for LAL-D prevalence calculations. LAL WBC activity was reduced in 67 patients (10.72%) and deficient in 37 (5.92%). The average of LAL activity ± margin of error (CI 95%) was 19.32 ± 0.86 pmol/min/mg for reduced activity patients and 5.90 ± 1.42 pmol/min/mg for deficient patients. The average age at diagnosis for LAL-D was 23.6 years with several patients older than age 30. The correlation between the age at diagnosis and LAL activity showed a significant moderate direct correlation (Pearson’s r = 0.46, P < 0.005). Homozygous or compound heterozygous mutations were identified in 9 out of 23 patients with deficient results (detection rate 39.1%). The average LAL activity in molecularly confirmed patients was 4.02 ± 2.02 pmol/min/mg protein, while in molecularly negative patients was 13.886 ± 1.49 pmol/min/mg (P < 0.0001). Twenty-two different mutations were identified including two novel variants (c.309C>A and c.856G>C). A carrier frequency of approximately 1 in 350 was inferred. LAL activity in WBC is a validated tool for LAL-D diagnosis. Higher residual enzymatic activity might result in a milder phenotype leading to diagnosis delay. A cut-off below 12 pmol/min/mg protein might be useful to discriminate patients with LIPA mutations.
Keywords: acid lipase, cholesterol ester storage disease, lysosomal acid lipase deficiency, LAL activity, LAL-D, LIPA mutations
1 |. INTRODUCTION
Lysosomal acid lipase deficiency (LAL-D; OMIM #278000) is an autosomal recessive disease caused by mutations in LIPA (OMIM 613497) at chromosomal locus 10q23.31, which encodes a hydrolase involved in the degradation of lysosomal cholesterol esters and triglycerides. These molecules accumulate predominantly in the liver, spleen, adrenals, bone marrow, lymph nodes, and in macrophages throughout the body, particularly in the intestinal villi. Clinically, LAL-D has been reported as one of two principal phenotypic presentations: infantile-onset LAL-D (formerly known as Wolman disease [WD]), an early onset fulminant subtype with absent or very low lysosomal acid lipase (LAL) activity1 and childhood/adult-onset LAL-D (formerly known as cholesterol ester storage disease [CES-D]), a late-onset or partial LAL deficiency, with low residual LAL activity.2 The disease onset and severity depend upon the amount of residual LAL activity, with partial enzymatic function leading to a more benign clinical course.2 Infantile-onset LAL-D typically presents in the first 6 months of life and is the most rapidly fatal presentation.3 Childhood/adult-onset LAL-D typically shows a heterogeneous phenotypic presentation, with an age of manifestation ranging from the first to the sixth decade. Later onset patients may be the most under-diagnosed cohort, since they often appear asymptomatic, other than having type IIb hyperlipoproteinemia, until stroke, aneurysm, aorto-coronary disease or premature sudden death from liver failure lead to the diagnosis.2
Clinical diagnostic suspicion can be confirmed by demonstrating markedly deficient LAL activity or by LIPA molecular analysis. For diagnostic purposes, LAL activity can be assessed in peripheral blood leukocytes (WBC), cultured skin fibroblasts, or liver with the use of radiolabeled natural substrates or the synthetic derivatives 4-methylumbelliferone or p-nitrophenol. Prenatal diagnosis of LAL activity can also be reliably determined by testing cultured chorionic villi or fetal cells.
LAL-D is a pan-ethnic disease, but accurate disease incidence is lacking. Genetic testing is useful and cost-effective when the analysis is confined to LIPA exon 8, which enables the identification of the most common donor-splice-site mutation: c.894G>A, p.Ser275_Gln298del. The frequency of this variant is different across geographic locations.4 The allele frequencies of the c.894G>A mutation from the New York metropolitan area (involving unrelated multiethnic subjects) ranged from 0.0005 to 0.0015 which translated to carrier frequencies of 1 in 1000 to 1 in 333, respectively.4 These estimates are in apparent conflict with the small number of LAL-D patients reported in the literature, and this is probably due to grossly under-diagnosed cases or incomplete penetrance of the disease.1
We report the analysis of 681 patient samples over 15 years of LAL enzyme testing in a clinical laboratory, including pediatric and adult diagnostics. LIPA sequencing analysis was performed in a subset of tested patients allowing study of correlation to biochemical data and discovery of novel pathogenic mutations. LAL-D prevalence data has been inferred by Exome Aggregation Consortium (ExAc) and Genome Aggregation database (gnomaAD). We aim at providing biochemical and molecular insights for improving diagnosis of this likely under-reported but treatable condition.
2 |. MATERIALS AND METHODS
2.1 |. Patient samples
Samples included in this study were referred to our laboratory for LAL-D testing, including 681 samples for enzymatic analysis and 49 samples for LIPA sequencing, over a 15-year period from 2002 to 2017. The age range for all subjects enrolled was 15 days to 89 years of age (average 25.1 years, median 15 years) for WBC enzyme testing, and 6 to 90 years (average 46.36 years, median 53 years) for molecular analysis. This retrospective analysis of our biochemical and molecular data was obtained as part of routine clinical management through our clinical lab toward better correlation of molecular and enzymatic findings and improvement of diagnostic assessment. This study was approved with a waiver of consent by the Baylor College of Medicine Institutional Review Board based upon the United States code of federal regulations governing Human Subjects Research.
2.2 |. Enzymatic analysis
The assays herein were performed by Baylor College of Medicine Medical Genetics Laboratories/Baylor Genetics, as previously described.5 In brief, WBC pellets were prepared from whole blood collected in heparin or ACD tubes. Cultured skin fibroblasts and amniocytes were harvested from 7-day post confluent T75 flasks. WBC pellets were sonicated in a buffer containing 0.3% NaCl, 0.25% sodium cholic acid, and 0.1% Triton. Lysates were diluted to a protein concentration of 2 mg/mL. A natural substrate, 14C-glycerol trioleate (Perkin Elmer, PerkinElmer Genetics, Pittsburgh, PA 15275 USA) was added to the reaction cocktail and incubated for 60 minutes at 37°C. Reactions were stopped by adding 0.05 M sodium acetate, pH 4.5. Extraction of enzyme product was achieved using 0.3 M NaOH and an organic mixture of oleic acid, benzene, chloroform and methanol. Extracted radioactive products were quantified using the Beckman LS 6000 Scintillation Counter, and LAL activity (pmol/min/mg protein) calculated.
2.3 |. Molecular testing
Molecular analysis of LIPA exons and intron/exon boundaries was performed with Sanger sequencing at Baylor Genetics. Primers for all exons flank the respective intron/exon junctions and are available upon request. Direct sequence analysis of PCR products was performed in both forward and reverse directions using automated fluorescence dideoxy sequencing methods. Reference sequences NM_000235.2 and NT_030059.12 were used for comparison. The nomenclature is based on the convention recommended by the Human Genome Variation Society (http://www.hgvs.org/mutnomen/). Chromosomal microarray was not performed.
2.4 |. Variant frequency
The ExAC Cohort (http://exac.broadinstitute.org/gene/ENSG00000107798) and gnomAD Cohort (http://gnomad.broadinstitute.org/gene/ENSG00000107798) were used to obtain allele frequencies of known pathogenic LIPA variants, as well as pathogenic variants segregating in populations through bioinformatic tools (CADD, SIFT, PolyPhen2) which predict damaging effects (databases accessed May 2018). Data were assumed to be in Hardy-Weinberg equilibrium, and standard Hardy-Weinberg calculations were used to determine carrier frequency and disease prevalence.
2.5 |. Statistical analysis
Two-tailed and one-tailed Student t tests, anova were used as statistical tests for mean comparisons and margins of errors. Statistical analyses were performed by using the R package for statistical computing version 3.3.1, Prism7, and Excel. Correlation and scatter plots were designed with GraphPad Prism7. Analyses with values below 0.05 were considered as statistically significant.
3 |. RESULTS
Diagnostic samples were referred to our clinical laboratory from 2002 to 2017 for LAL enzyme analysis under the suspicion of lysosomal acid lipase deficiency. Sample types included WBC (n = 625), fibroblasts (n = 30), liver (n = 4), amniocytes (n = 13), and chorionic villus tissue (n = 9) (Table 1, and Supporting Information Table S1). Limited clinical data were received for each sample. Mostly, indications for referral are elevated LDL and cholesterol, elevated liver enzymes and hepatomegaly.
TABLE 1.
LAL activity in white blood cells of 625 samples from 2002 to 2017
| Normal LAL activity (>25 pmol/min/mg protein) | Reduced LAL activity (12–25 pmol/min/mg protein) | Deficient LAL activity (0–12 pmol/min/mg protein) | |
|---|---|---|---|
| Results from WBC enzyme assays (n. total = 625) | 521 | 67 | 37 |
| LAL activity (pmol/min/mg), average ± margin of error (CI 95%) | 62.92 ± 3.98 | 19.32 ± 0.86 | 5.90 ± 1.42 |
| Average age at testing (range, years) | 23.71 (0.04–89) | 36.75 (0.1–85) | 23.6 (0.06–82) |
| Females | 201 | 32 | 16 |
| Males | 300 | 35 | 18 |
| Gender unknown | 20 | 0 | 3 |
LAL activities average ± margin of error, age distribution and gender are shown.
3.1 |. Biochemical analysis
Standard normal ranges of LAL activity were for WBC pellets approximately 25 to 70 pmol/min/mg protein, for fibroblasts 100 to 290 pmol/min/mg, for liver 20 to 200 pmol/min/mg protein, and for fetal cells 40 to 160 pmol/min/mg protein (Table S1) as supported by mutation analysis discussed in the “Molecular testing and population screening” paragraph. In detail, WBC LAL activity is considered reduced at 12 to 25 pmol/min/mg protein, while values <12 pmol/min/mg protein are considered deficient consistent with LAL-D. Mean plus margins of error of LAL activity in WBC for 625 individuals is shown in Table 1 along with patients’ gender and age at diagnosis. The patients are grouped into: normal, reduced, and deficient categories (Table 1 and in Figure 1). Values for all WBC LAL activity cohort samples showed a correct stratification of the normal, reduced and deficient categories (P < 0.001, t test and anova) (Figure 1). Normal values were detected in 521 subjects with suspected LAL-D based on clinical findings, with average LAL activity values of 62.92 ± 3.98 pmol/min/mg protein (CI 95%), 67 patients with reduced activity showed average values of 19.32 ± 0.86 pmol/min/mg protein (CI 95%), and 37 patients diagnosed with deficient LAL activity had mean values of 5.9 ± 1.42 pmol/min/mg protein (CI 95%) (Figure 1). Out of 37 patients diagnosed biochemically with LAL-D, only six patients showed null LAL activity. Detection rate of LAL deficient results in patients referred for enzymatic testing was 5.92% (37 out of 625 individuals).
FIGURE 1.
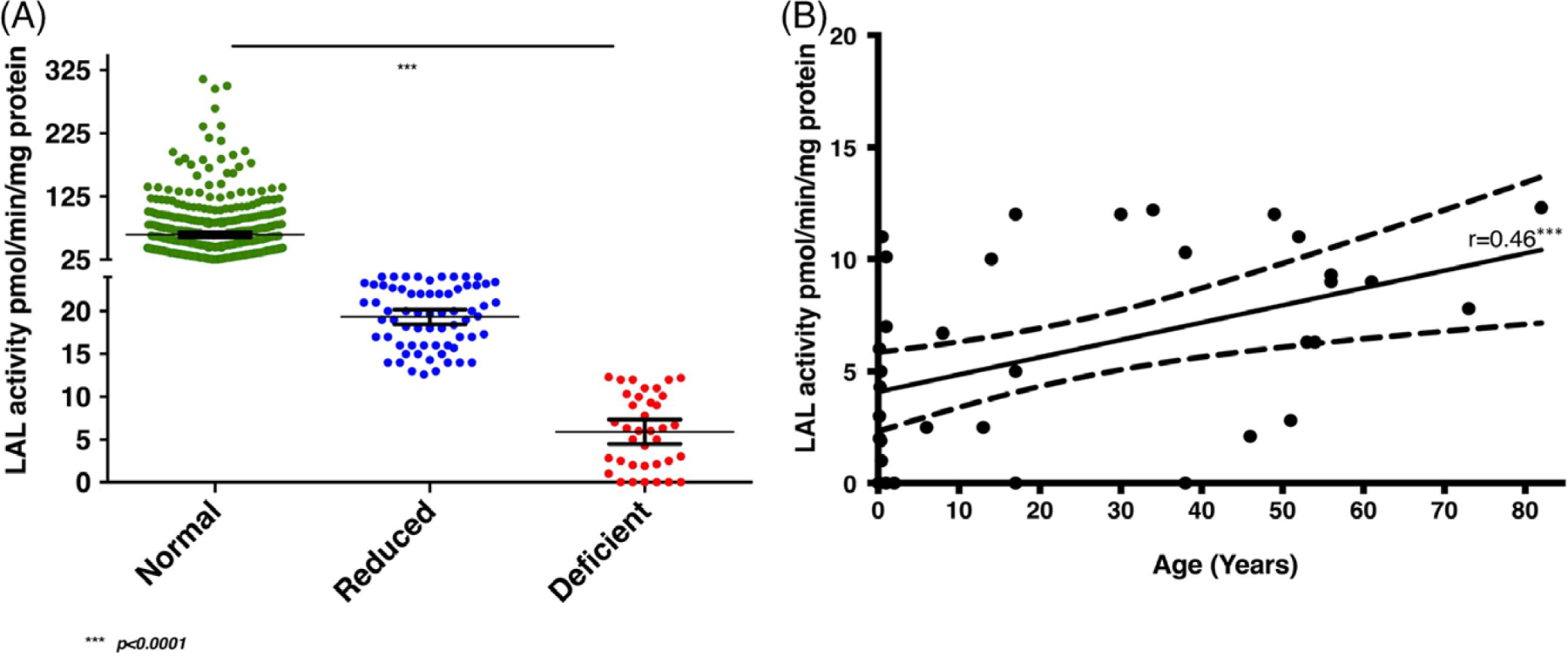
A, Results from 625 WBC analyses: Normal LAL activity (cut-off >25 pmol/min/mg protein) (n = 521, average = 62.92 ± 3.98 pmol/min/mg protein, 95% confidence interval), reduced LAL activity (cut-off >12 and < 25 pmol/min/mg protein) (n = 67, average = 19.32 ± 0.86 pmol/min/mg protein, 95% confidence interval), and deficient LAL activity (cut-off <12 pmol/min/mg protein) (n = 37, average = 5.9 ± 1.42 pmol/min/mg protein, 95% confidence interval). A significant distribution of normal and affected individuals emerged. t test comparing two by two categories and anova showed the three groups different with high statistical significance P < 0.0001. B, Correlation between the age at diagnosis and LAL activity in the cohort. The Pearson’s r resulted 0.46 (95% confidence interval 0.1591 to 0.6819) and P < 0.005 (one-tailed). A moderate linear relationship emerged suggesting higher LAL enzyme activity does correlate with moderate power with a later age of diagnosis. Dots represent deficient LAL activities (0–12 pmol/min/mg protein) of 37 patients and dotted lines enclose the 95% confidence interval values
3.2 |. Patient epidemiological parameters
Of the 37 patients diagnosed biochemically with LAL-D, the average age of diagnosis was 23.6 years of age (range 21 days-82 years), and the male:female ratio was 1.125 (18:16), while in three patients gender was unknown (Table 1). These data include 351 males tested for WBC LAL activity (18/351, 5.1%) and 251 females (16/251, 6.3%). Pearson’s correlation between age at diagnosis and deficient LAL activity (values <12 pmol/min/mg protein) was 0.46 (P < 0.005) suggesting a moderate direct correlation of these two variables. Higher LAL enzyme activity is moderately correlated with a later age at diagnosis (Figure 1B).
3.3 |. Molecular testing and population screening
A subset of samples with reduced and deficient LAL activity (n = 49) was received for both molecular and biochemical testing. Biallelic mutations in LIPA were identified in 9 out of 49 patients with deficient (n = 23) and reduced (n = 26) LAL activity. In patients with biallellic LIPA mutations (n = 9), the mean LAL value was 4.02 ± 2.02 (CI 95%), while in patients with negative molecular results the mean was 13.886 ± 1.49 (CI 95%), and the difference in LAL activities between patients with positive and negative molecular results was significant (P < 0.0001) (Table 2 and Table 3, ID patient 1 to 9). Interestingly, of 49 patients investigated, 4 carriers of LIPA mutations were identified, 3 with deficient biochemical results and one with reduced ones (Table 2 and Table 3, ID patient 10 to 13); in these latter patients, a missed second variant and/or regulatory variant resulting in hypomorphic alleles could not be excluded. LAL enzyme activity below 12 pmol/min/mg protein highly support molecular confirmation since the value of 12 is three standard deviations away from the mean for molecularly negative patients as shown in Table 2. The detection rate of LIPA mutations (homozygous or compound heterozygous) in patients with deficient biochemical result was 39.1% (9/23 patients). In addition, molecular testing was performed in three unrelated samples of fetal cells. Two of these patients had one LIPA mutation and reduced or slightly reduced LAL activity, and one patient had compound heterozygous mutations and deficient LAL activity (Table 3, ID patient 14 to 16). In total, 9 missense, 5 nonsense, plus the c.894G>A splicing variant in 12 alleles were detected in LIPA (Table 2). The most common LIPA pathogenic variant, c.894G>A (at exon 8 splice junction), resulted in low resid- ual LAL activity, in line with previous evidence.4 The variants c.309C>A (p.Ser103Arg) and c.856G>C (p. Ala286Pro) have not been previously reported (Figure 2, highlighted in red and in bold). In our observation, the p. Ser103Arg variant was detected along with the previously known p.Gly266Ter mutation in a patient with deficient LAL activity, while the variant p.Ala286Pro was detected in a patient with reduced LAL activity (Table 2). Bioinformatic tools such as SIFT, Polyphen, CADD, Mutation Taster, LRT, and biochemical data (reduced and deficient LAL activity) strongly support variant pathogenicity (Table S2).
TABLE 2.
LAL activity in patients with LIPA sequencing test results
| LIPA sequencing | LAL activity (pmol/min/mg protein) Average ± margin of error (CI 95%) |
|---|---|
| Patients with biallelic variants in LIPA (n = 9) | 4.02 ± 2.02 |
| Patients with normal LIPA sequence analysis (n = 36) | 13.886 ± 1.49 |
All LIPA coding exons were sequenced in 49 individuals with reduced or deficient LAL activity. Of these cases, seven patients were homozygous or compound heterozygous for pathogenic mutations, four patients were heterozygous for one pathogenic mutation, and 36 showed negative sequencing results. LAL activity from patients with biallelic pathogenic mutations was significant reduced vs patients with negative results (P < 0.001)
TABLE 3.
Molecular and biochemical results are shown for affected individuals
| ID | Gender | Specimen analyzed | Acid lipase activity (pmol/min/mg) | Age at testing (years) | Genotype | Annotation | References | |
|---|---|---|---|---|---|---|---|---|
| Mutation 1 | Mutation 2 | |||||||
| 1 | M | WBC (normal values >25 pmol/min/mg protein) | 2.5 | 13 | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | NM_001127605.2: c.894G>A, p. Ser275_Gln298del | P | 6 |
| 2 | F | WBC (normal values >25 pmol/min/mg protein) | 6.7 | 8 | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P |
7
6 |
| 3 | M | WBC (normal values >25 pmol/min/mg protein) | 0 | 38 | NM_000235.3:c.796G>T, p. Gly266Ter | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P P |
6–8 |
| 4 | M | WBC (normal values >25 pmol/min/mg protein) | 4.3 | 0.25 | NM_000235.3:c.796G>T, p. Gly266Ter | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P P | 6–8 |
| 5 | F | WBC (normal values >25 pmol/min/mg protein) | 6 | 0.16 | NM_000235.3:c.796G>T, p. Gly266Ter | NM_000235.3, c.309C>A, p.SerlO3Arg | P Likely P |
9 |
| 6 | F | WBC (normal values >25 pmol/min/mg protein) | 5 | 17 | NM_001127605.2:c.894G > A, p. Ser275_Gln298del | NM_001127605.2:c.386A>G, p.Hisl29Arg | P P |
10
6 10 |
| 7 | F | WBC (normal values >25 pmol/min/mg protein) | 0 | 1 | NM_000235.3:c.398delC p. Serl33Ter | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P P | 6,7,11 |
| 8 | F | WBC (normal values >25 pmol/min/mg protein) | 6.7 | 6 | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P | 10 |
| 9 | F | WBC (normal values >25 pmol/min/mg protein) | 5 | 17 | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | NM_001127605:c.386A>G, p.Hisl29Arg | P P |
10
10 |
| 10 | F | WBC (normal values >25 pmol/min/mg protein) | 2.5 | 6 | NM_001127605.2:c.894G>A, p. Ser275_Gln298del | P | 10,10 | |
| 11 | M | WBC (normal values >25 pmol/min/mg protein) | 0 | 0.16 | NM_000235.3:c.398de1C p. Serl33Ter | P | 11 | |
| 12 | M | WBC (normal values >25 pmol/min/mg protein) | 16 | 26 | NM_000235.3:c.856G>C, p. Ala286Pro | Likely P | ||
| 13 | M | WBC (normal values >25 pmol/min/mg protein) | 10 | 14 | NM_000235.2:c.253C>A, p. Gln85Lys | P | 12 | |
| 14 | U | Fetal cells from amniocentesis (normal range 40–160 pmol/min/mg protein) | 2.3 | N/A | NM_000235.3:c.894G>C, p. Gln298His | NM_000235.3:c.1033G>A p.Asp345Asn | P Likely P |
13
3 |
| 15 | U | Fetal cells from amniocentesis (normal range 40–160 pmol/min/mg protein) | 23.2 | N/A | NM_000235.3:c.260G>T, p. Gly87Val | P | 14 | |
| 16 | u | Fetal cells from CVS (normal range 40–160 pmol/min/mg protein) | 58 | N/A | NM_000235.3:c.260G>T, p. Gly87Val | P | 14 | |
Abbreviations: F, female; Hmz: homozygous, Htz: heterozygous, M, male; P, pathogenic; likely pathogenic.
LAL activity for WBC (white blood cells), amniocentesis, chorionic villus sampling (CVS), and liver are included. Novel LIPA variants are underscored.
FIGURE 2.
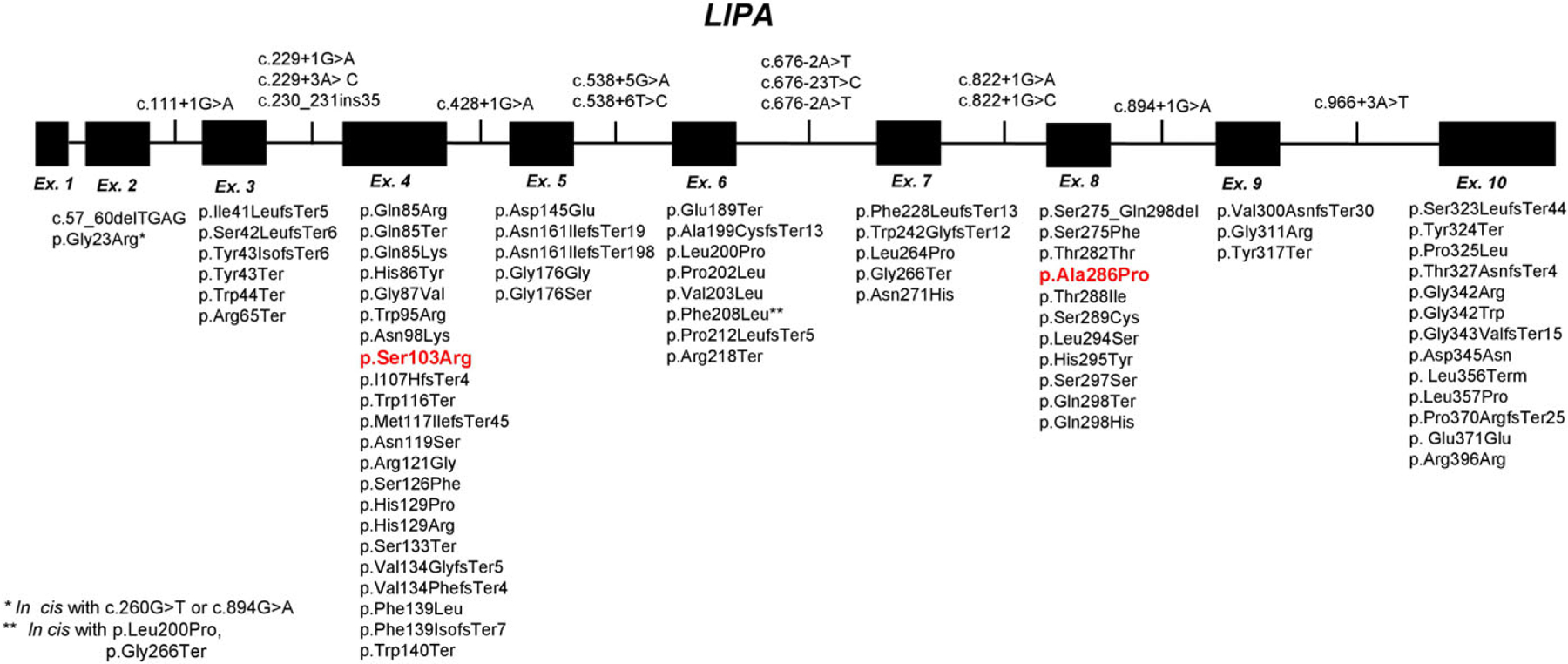
Reported pathogenic variants in LIPA. The 10 exons representing LIPA are shown. Included are all reported mutations in the literature and all loss-of-function mutations in ExAc and ClinVar (91). The two novel mutations identified in our study with associated reduced and deficient LAL activity are red and in bold [c.309C>A (p.Ser103Arg) and c.856G>C (p.Ala286Pro)]
3.4 |. LIPA pathogenic variant frequency
Analysis of population-based exome data from ExAC (http://exac.broadinstitute.org/gene/ENSG00000107798) and gnomAD (http://gnomad.broadinstitute.org/gene/ENSG00000107798) allowed for LIPA pathogenic variant analysis to estimate carrier frequency and disease prevalence of this ultra-rare condition. While most variants identified in individuals with LAL-D were rare private mutations (Figure 2 and Table S3), the most frequent pathogenic LIPA variant was a splicing mutation, c.894G>A, p.Ser275_Gln298del, with an empiric minor allele frequency of 0.0008274 (100/120864 alleles in ExAC) and 0.0008920 (247/276916 alleles in gnomAD). Interestingly, this known pathogenic variant, which is more commonly identified in Caucasian and Latino populations, was not indicated as a loss-of-function variant in the either ExAC or gnomAD. When the ExAC and gnomAD databases were assessed for loss-of-function variants, including nonsense, frameshift, canonical splice variants, and known pathogenic variants which result in enzyme deficiency (Figure 2 and Table S3), an estimated carrier frequency (2pq) and disease prevalence (q2) could be calculated assuming Hardy-Weinberg equilibrium. The carrier rate for LAL-D is estimated to be approximately 1/350, with carrier frequencies of 1/345 and 1/388 calculated from ExAC and gnomAD, respectively. These data estimate the disease prevalence to be approximately 1 in 500,000 or 1/500,000.
4 |. DISCUSSION
LAL-D is a rare, under diagnosed progressive disease related to the metabolism of cholesterol and triglycerides inside the lysosome, with early mortality and significant morbidity mainly due to liver complications. Diagnosis of LAL-D has been mainly based on the enzyme assay of LAL activity in fibroblasts.15 As tissue culture has many disadvantages, especially in terms of invasive sample collection and of the time-consuming and expensive process, potential alternative samples were considered for the evaluation of LAL activity, such as WBC. Colorimetric and fluorescent methods have been used for LAL determination in WBC.16 In our cohort, fibroblast specimens represent only 4.4% (n = 30) and liver 0.64% (n = 4), while WBC are 92% (n = 625) of all specimens. For ease of analysis and quality of data, WBC specimens have been largely preferred. LAL enzyme determination in dried blood spots (DBS) has been quickly applied in recent years for easy sample collection for early diagnosis of LAL deficiency, especially with the availability of LAL enzyme replacement therapy for the disease.17 Using 4-methylumbelliferyl as a substrate, a LAL specific inhibitor, Lalistat 2, is necessary in the DBS assay to exclude other forms of lipase. Our laboratory does not perform DBS analysis for LAL. Several key recommendations have been proposed by an expert committee for an alternative stop solution, excitation wavelength, sample quality and interpretation to improve the DBS assay.18 Our radioactive enzyme assay is performed in acidic pH; thus, interferences of other lipases are restricted, as indicated, with almost zero enzyme activity in some LAL deficient patients. We tested Lalistat 2 in fibroblast assays, and the results were consistent with the original radioactive assay, which showed no Lalistat 2 inhibition is required in radioactive assay.
Analysis of >600 samples for LAL-D diagnosis in WBCs allowed a clear-cut separation of enzyme activities into normal (or unaffected subjects), reduced activity, and deficient (affected patients), which were significantly different from each other (P < 0.001) (Figure 1). In our cohort, 37 patients showed biochemical results supporting LAL-D, including six cases with absent residual activity (average 5.90 ± 1.42 pmol/min/mg protein, margin of error with CI 95%) (Table 1). Of the referred cases, <10% indicated a clinical reason for the analysis; however, elevated cholesterol, elevated triglycerides, and abnormal liver function tests are the most common indications for the analysis, with developmental delay and seizures being the second most common indications. The detection rate of LAL-D by biochemical assay in this population was 5.92%.
In our study, the mean age at diagnosis of LAL-D was 23.6 years ranging from 21 days of age to 82 years. Some patients with LAL-D might remain asymptomatic until adulthood, and older patients with very mild LAL-D phenotype were reported to be diagnosed at 52 and 63 years.10 A review analyzing 131 patients with LAL-D found that the median age at presentation for both genders was 5 years (range: birth-68 years).2 Our data confirm that patients with LAL-D are mainly diagnosed during adolescence or young adulthood. A slightly higher prevalence of LAL-D in females has been described (55% vs 45% in males).2 In our report, the male/female ratio was 1.125; the detection rate in females was 6.3% vs 5.1% in males. We found that residual LAL activity moderately correlated (r = 0.46, P < 0.005, Figure 1B) with age at clinical onset, underscoring that higher residual enzymatic activity might predict later diagnosis.19 Interestingly, we identified a 38-year-old patient (patient number 3, Table 2) with absent LAL activity and two mutations in LIPA (c.796G>T (p.Gly266Ter), c.894G>A (p.Ser275_Gln298del)); these latter mutations were previously described in patients sharing very severe reduction of LAL activity but non-fatal outcomes.12 Conversely, only mild reduction of LAL activity was detected in patients with LAL-D characterized by severe disease progression.2,19 Biochemical data on LAL activity might suggest but not strictly predict clinical course and a more complex disease model might be valid for LAL-D, with yet to be identified genetics and/or environmental modifiers. LAL enzyme activity measured in different clinical laboratories due to the use of different substrates and procedures might also contribute to variability in reported cases.
In our case series, 49 patients with reduced (n = 26) or deficient (n = 23) LAL activity underwent molecular testing: 9 patients with homozygous or compound heterozygous mutations and 4 carriers were found (Table 2 and Table 3). A defined cut-off value for LAL activity has not been established below which is highly probable to find positive molecular results. In patients with positive LIPA molecular results, LAL activity was 4.02 ± 2.02 pmol/min/mg protein [margin of errors with CI 95%] which was statistically different vs LAL activities measured in patients with negative molecular results (13.886 ± 4.41 pmol/min/mg protein, (margin of errors with CI 95%), P < 0.0001). Based upon our findings, we recommend performing LIPA analysis in patients with LAL activity below 12 pmol/min/mg protein for molecular confirmation. To date, 91 mutations and eight gross rearrangements involving the 10 exons of LIPA (exon 4 mainly involved), including those reported in the literature and loss-of-function mutations in ExAC, gnomAD, and ClinVar (Figure 2 and Table S3) have been identified.3,20 Among the detected mutations in our cohort, c.309C>A (p.Ser103Arg) and c.856G>C (p.Ala286Pro) were novel causative mutations (Figure 2 highlighted in red and Table 2), as supported by SIFT, Polyphen, CADD, Mutation taster, LRT and biochemical data (reduced and deficient LAL activity in patients). Asymptomatic carriers of pathogenic mutations in LIPA are reported to share LAL activity that is slightly reduced or low-normal (half-maximal LAL activity),10 which may be responsible for an increased risk of atherosclerosis.10 Also in our cohort, 4 carriers of mutations in LIPA showed reduced or deficient LAL activity level. To date genotype-phenotype correlations in LAL-D remain poorly understood since unrelated affected individuals with the same genotypes share different LAL activity and clinical clues.4 We confirm that LIPA genotypes do not strictly predict residual enzymatic activity. Possibly other factors such as unknown modifier genes or regulating factors could explain these variations.
The exact prevalence of LAL-D in children and adults is not yet established, and ranges from 1 in 150,000 to 1 in 350,000 in Caucasian populations.12,14 However, LAL-D seems to occur at a higher frequency in the Iranian-Jewish community residing in the Los Angeles area, with an overall carrier frequency for the p.Gly87Val mutation of 1:32.4 (3.086%), deducing a much greater prevalence in this population (1 in 4200).14 Population screening for the c.894G>A mutation among healthy West German individuals indicated a heterozygote frequency of 1 in 200 individuals.20,21 According to ExAC and gnomAD databases, we calculated a pan-ethnic carrier frequency of approximately 1 out of 350. LAL-D may be under diagnosed in the face of a high prevalence of LIPA mutations; thus, clinicians and particularly pediatric gastroenterologists should be aware of the continuum spectrum of manifestations of LAL-D to rule out misdiagnosis.
Current medical management of LAL-D is limited and includes the use of HMG-CoA reductase inhibitors and other lipid-lowering therapies for disease-associated hypercholesterolemia. Enzyme replacement therapy with sebelipase alfa has been used with good results.22 These results shed light on the prevalence and variability of this underappreciated genetic lipid disorder. Awareness of the disease combined with efficient diagnostic tools will facilitate the correct diagnosis, prognosis, and therapy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tom Lo, Thomas Brown, and Guillermo del Angel for assistance with LIPA variant identification. We are grateful to Carlos Ferreira, Michael Grotewiel, and Katherine Grotewiel for assistance with population data analysis. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Footnotes
CONFLICTS OF INTERESTS
All the authors have no conflicts of interest to report. L.H., Q.S., and S.H.E. are employees of Baylor College of Medicine, which has a partnership with Baylor Genetics and derives revenue from genetic testing. T.R.D. is an employee of PerkinElmer Genetics.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Fasano T, Pisciotta L, Bocchi L, et al. Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab. 2012;105: 450–456. 10.1016/j.ymgme.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013a;58: 1230–1243. 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Pisciotta L, Tozzi G, Travaglini L, et al. Molecular and clinical characterization of a series of patients with childhood-onset lysosomal acid lipase deficiency. Retrospective investigations, follow-up and detection of two novel LIPA pathogenic variants. Atherosclerosis. 2017; 265:124–132. 10.1016/j.atherosclerosis.2017.08.021. [DOI] [PubMed] [Google Scholar]
- 4.Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58:958–965. 10.1002/hep.26327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaudet AL, Lipson MH, Ferry GD, Nichols BL. Acid lipase in cultured fibroblasts: cholesterol ester storage disease. J Lab Clin Med. 1974;84:54–61. [PubMed] [Google Scholar]
- 6.Ameis D, Brockmann G, Knoblich R, et al. A 5′ splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res. 1995;36:241–250. [PubMed] [Google Scholar]
- 7.Klima H, Ullrich K, Aslanidis C, Fehringer P, Lackner KJ, Schmitz G. A splice junction mutation causes deletion of a 72-base exon from the mRNA for lysosomal acid lipase in a patient with cholesteryl ester storage disease. J Clin Invest. 1993; 92:2713–2718. 10.1172/JCI116888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33:85–93. 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- 9.Ries S, Aslanidis C, Fehringer P, Carel JC, Gendrel D, Schmitz G. A new mutation in the gene for lysosomal acid lipase leads to Wolman disease in an African kindred. J Lipid Res. 1996;37:1761–1765. [PubMed] [Google Scholar]
- 10.Ries S, Büchler C, Schindler G, et al. Different missense mutations in histidine-108 of lysosomal acid lipase cause cholesteryl ester storage disease in unrelated compound heterozygous and hemizygous individuals. Hum Mutat. 1998;12:44–51. . [DOI] [PubMed] [Google Scholar]
- 11.Zschenker O, Jung N, Rethmeier J, et al. Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease. J Lipid Res. 2001;42: 1033–1040. [PubMed] [Google Scholar]
- 12.Santillán-Hernández Y, Almanza-Miranda E, Xin WW, et al. Novel LIPA mutations in Mexican siblings with lysosomal acid lipase deficiency. World J Gastroenterol. 2015;21:1001–1008. 10.3748/wjg.v21.i3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Nájera M, Barajas-Medina H, Gallegos-Rivas MC, et al. New diagnostic method for lysosomal acid lipase deficiency and the need to recognize its manifestation in infants (Wolman disease). J Pediatr Gastroenterol Nutr. 2015;60:e22–e24. 10.1097/MPG.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 14.Valles-Ayoub Y, Esfandiarifard S, No D, et al. Wolman disease (LIPA p.G87V) genotype frequency in people of Iranian-Jewish ancestry. Genet Test Mol Biomarkers. 2011;15:395–398. 10.1089/gtmb.2010.0203. [DOI] [PubMed] [Google Scholar]
- 15.Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolman’s disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant. 2009;44:449–450. 10.1038/bmt.2009.57. [DOI] [PubMed] [Google Scholar]
- 16.Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007; 27:1866–1868. 10.1161/ATVBAHA.107.146639. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413:1207–1210. 10.1016/j.cca.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Lukacs Z, Barr M, Hamilton J. Best practice in the measurement and interpretation of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2017;471:201–205. 10.1016/j.cca.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 19.Al-Dirbashi OY, McIntosh N, McRoberts C, et al. Analysis of methylcitrate in dried blood spots by liquid chromatography-tandem mass spectrometry. JIMD Rep. 2014;16:65–73. 10.1007/8904_2014_321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabowski G, Du H, Charnas L (2012) Lysosomal Acid Lipase Deficiencies: The Wolman Disease/Cholesteryl Ester Storage Disease Spectrum. OMMBID doi: 10.1036/ommbid.172 [DOI] [Google Scholar]
- 21.Lohse P, Maas S, Lohse P, et al. Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. J Lipid Res. 2000;41:23–31. [PubMed] [Google Scholar]
- 22.Valayannopoulos V, Malinova V, Honzík T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61:1135–1142. 10.1016/j.jhep.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.