


Abstract
The Saccharomyces cerevisiae gene deletion collection is widely used for functional gene annotation and genetic interaction analyses. However, the standard G418-resistance cassette used to produce knockout mutants delivers strong regulatory elements into the target genetic loci. To date, its side effects on the expression of neighboring genes have never been systematically assessed. Here, using ribosome profiling data, RT-qPCR, and reporter expression, we investigated perturbations induced by the KanMX module. Our analysis revealed significant alterations in the transcription efficiency of neighboring genes and, more importantly, severe impairment of their mRNA translation, leading to changes in protein abundance. In the ‘head-to-head’ orientation of the deleted and neighboring genes, knockout often led to a shift of the transcription start site of the latter, introducing new uAUG codon(s) into the expanded 5′ untranslated region (5′ UTR). In the ‘tail-to-tail’ arrangement, knockout led to activation of alternative polyadenylation signals in the neighboring gene, thus altering its 3′ UTR. These events may explain the so-called neighboring gene effect (NGE), i.e. false genetic interactions of the deleted genes. We estimate that in as much as ∼1/5 of knockout strains the expression of neighboring genes may be substantially (>2-fold) deregulated at the level of translation.
INTRODUCTION
The haploid genome of the budding yeast Saccharomyces cerevisiae contains approximately 6000 genes (1). Almost two decades ago, the complete gene knockout library was produced by the Saccharomyces Genome Deletion Project (SGDP) (2,3) to assign a function to each ORF through phenotypic analysis of the mutants. For this, a PCR-based gene deletion strategy was used (4) to generate a complete start-to-stop-codon deletion of every ORF in the yeast genome. The ORF was replaced with a KanMX4 cassette (http://www-sequence.stanford.edu/group/yeast_deletion_project/project_desc.html#cassettes, (5)) harboring the G418 resistance gene under control of the strong eEF1A promoter. The eEF1A terminator, adjacent vector-derived sequences, as well as additional artificial barcodes, were inserted along with the gene (Figure 1A).
Figure 1.
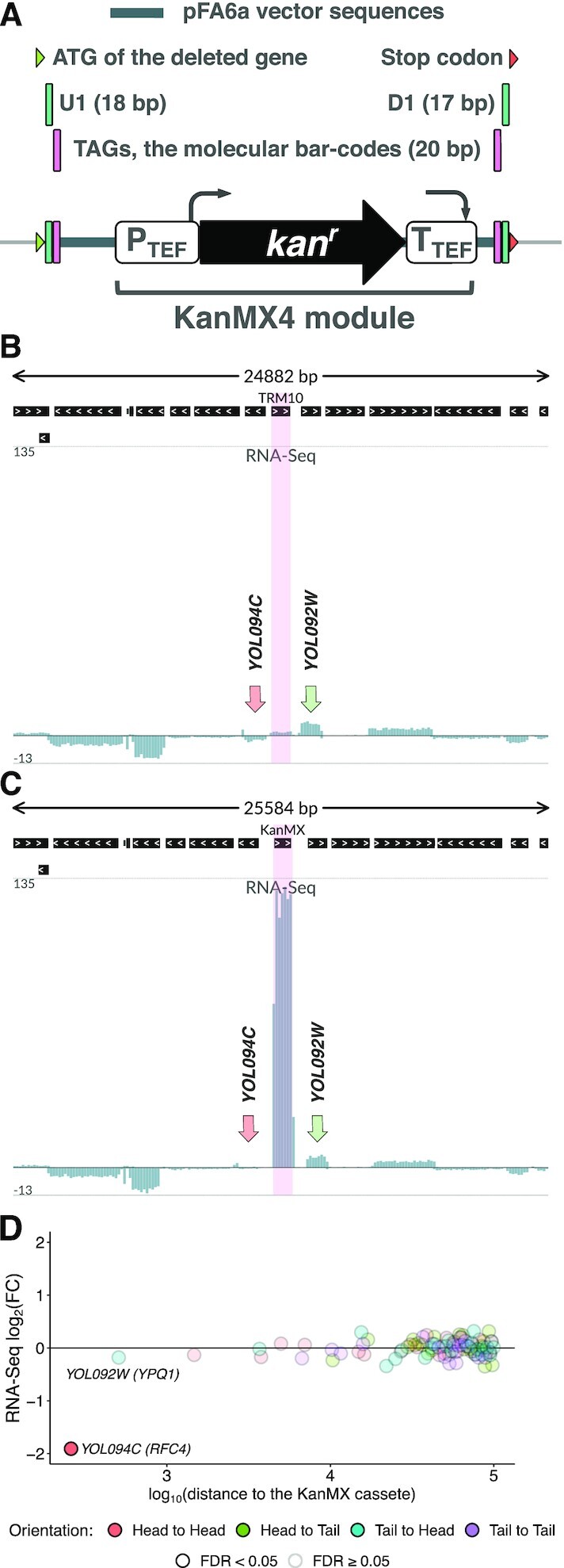
KanMX cassette used to generate gene deletions is excessively expressed within the mutated locus. (A) A schematic representation of the KanMX cassette replacing the knockout target gene. The cassette harbors the G418 resistance gene under the control of the strong A.gossypii eEF1A promoter and eEF1A terminator, as well as adjacent vector-derived sequences and barcodes. (B, C) Excessive kan gene expression in the mutant strain, as compared to other genes in the vicinity. RNA-Seq data for the TRM10 locus (including 15 genes in the vicinity) in wt (B) and trm10Δ (C) strains are shown as normalized total read coverage, positive and negative values correspond to the direct and reverse complementary DNA strands, respectively. Pink highlighting indicates the TRM10 CDS (wt strain) and the KanMX module (trm10Δ strain). RNA-Seq coverage of the neighboring genes, RFC4 (YOL094C) and YPQ1 (YOL092W), is indicated by arrows. (D) Differential expression of genes within the ±100 kb region from the KanMX module at the transcriptional level, trm10Δ compared to wt. FDR: false discovery rate.
The creation of the SGDP yeast deletion collection revolutionized the field of eukaryotic molecular genetics. Since then, a huge amount of information was generated using the knockout strains. In particular, many genes have been functionally characterized according to the analysis of phenotype and genetic interactions of the corresponding deletion strains (6–8).
However, the target genetic loci were substantially modified during the procedure of knockout strain production. As S. cerevisiae has a compact genome with very short intergenic regions, the introduction of a highly expressed gene module could, in principle, significantly alter transcription within the loci, affecting the expression of neighboring genes. There were a few studies of the neighboring gene effects (NGE) in genetic interactions (9–11) but the mechanisms of such effects have never been analyzed.
Here, using RNA-Seq and ribosome profiling (Ribo-Seq) data available for a number of knockout strains, we revealed drastic changes of gene expression at many loci in which the target gene and its neighbor are in close proximity. Among the observed side-effects, there were major alterations in transcription start sites (TSS) and polyadenylation site (PAS) usage of neighboring genes. These changes led to dramatic deregulation of the neighboring gene expression mainly at the level of mRNA translation that was further confirmed by analysis of protein abundance. Based on several dozen cases analyzed in this study, we roughly estimate that in as much as 10–20% of knockout strains in the yeast deletion library, the presence of the KanMX cassette substantially (>2-fold) deregulates expression of neighboring genes at the translational level.
MATERIALS AND METHODS
Data sources
Ribosome profiling (Ribo-Seq) and RNA-Seq data for knockout and wild-type S. cerevisiae strains (12,13) were downloaded from GEO [GSE100626, GSE122039]. Genome reference annotation and sequence for the BY4741 wt strain (version R64-1-1) were downloaded from Ensembl (14). Coordinates of alternative polyadenylation sites were taken from (15).
Data processing
Knockout strain genome reference annotations and sequences were obtained with reform (https://github.com/gencorefacility/reform, (16)) through modification of the Ensembl (R64-1-1) annotation and sequences. SRA data were downloaded and transformed into fastq with SRA toolkit v2.9.4 (17). The adapters were trimmed with cutadapt v1.18 (18).
Read counting and mapping to the original and modified genome annotations were performed with STAR v2.7.0 (19). BedGraph profiles were produced from SAM data with samtools (20) and bedtools v2.27.1 (21). Coverage profiles were normalized using normalization factor and library size estimates from differential expression analysis (see below) separately for each bedGraph profile. Genomic signal tracks were visualized with svist4get (22).
Ribo-Seq and RNA-Seq differential expression analysis
Raw gene counts processing and statistical analysis were performed in R using edgeR Bioconductor package (23). Genes not reaching 2 read counts per million (CPM) in at least 3 RNA-Seq and 4 Ribo-Seq libraries were excluded from the analysis. To detect differentially expressed genes (for RNA-Seq, Ribo-Seq, and ribosome occupancy), we used the generalized linear model (glmQLFit, glmQLFTest of the edgeR package) with the strain as a categorical variable.
Yeast strains
Yeast strains producing proteins tagged with C- and N-terminal Green Fluorescent Protein (GFP) were taken from systematic collections (24,25) and modified in order to delete genes adjacent to the gene of interest using the same strategy as in the original SGDP, except that the deletion cassette was obtained via one round of PCR with long primers. A list of the strains and primers used for the gene deletions is presented in Supplementary Tables S1 and S2, respectively. The gene deletion was verified by PCR of yeast genomic DNA using the primers listed in Supplementary Table S2.
qPCR-based quantification of normal and extended transcripts
Yeast strains with deletions of particular genes were grown in 100 ml of YPD to OD600 0.5 at 30°С, collected, and frozen in liquid nitrogen. Total RNA was extracted according to Collart et al. (26) using the hot acid phenol method. RNA samples were treated with RQ1 DNase (Promega). 1 mcg of total RNA was used for reverse transcription with MMLV-RT kit (Evrogen) according to the recommended protocol with oligo(dT)15 and random (dN)10 primers at a ratio of 1:3. Real-time PCR for detection of normal length and extended transcripts was performed with Eva Green I, kit №R-441 (Syntol), with primers listed in Supplementary Table S2. PCR protocol: 95°C – 5 min [95°C – 15 s, 64°C – 15 s, 72°C – 30 s, signal detection] × 43 cycles. Expression levels were calculated using the comparative Ct method.
Flow cytometric analysis of the GFP-fusion protein abundance
Cytometric analysis was performed with 12 sets of three yeast strains (encoding a GFP-tagged protein (WT-GFP), the same strain containing a deletion of the adjacent gene (KO-GFP), and a strain harboring only the adjacent gene deletion (KO)), plus a wild-type strain as a control (WT). The strains were grown in complete synthetic medium (0.67% (w/v) Yeast Nitrogen Base (Difco), 2% (w/v) glucose, complete amino acid supplementation) overnight in 96-well flat bottom plates (Eppendorf), diluted into fresh medium with a dilution factor of 200, and grown for another 4 hours. After that they were analyzed using a Beckman Cytoflex S (488 nm laser, 525/40 bandpass emission filter) or a MACS Quant Analyzer, Miltenyi Biotech (488 nm laser, FITC emission filter), both equipped with an autosampler. Data on GFP fluorescence (FITC-A) were collected for ∼30 000 cells in six independent cultures of each sample, which were analyzed on two distinct days (three in 1 day, three during another).
The raw data were extracted from *.fcs files with the FlowJo v.10 software. The FITC-A replicates of each strain were merged, the resulting distributions were compared between WT-GFP and KO-GFP strains with the Mann-Whitney U test. Additionally, we estimated the difference in medians Δmed_KO and Δmed_WT (by separately comparing wt and knockout fluorescence data against the matched autofluorescence data). Next, we computed the Δmed_KO/Δmed_WT ratio as the effect size estimate. To visualize distribution with the kernel density estimates we used the ggplot2 R package.
RESULTS
KanMX cassette expression is excessive in comparison to neighboring genes
Ribosome profiling combined with RNA-Seq provides expression data at the transcriptional and translational levels on a transcriptome-wide scale (27). A number of ribosome profiling studies were performed on yeast deletion strains. For our analysis, we took the unique dataset produced by Chou et al. (12), where Ribo-Seq and RNA-Seq were performed for 57 yeast knockout strains, and our own ribosome profiling data (13). Both datasets were obtained from BY4741-based S. cerevisiae strains using the classical ribosome profiling protocol with cycloheximide (27). Although this drug may affect ribosome footprint distribution at the beginning of CDS (28) and at particular codons (29), it is nevertheless applicable for the analysis of overall ribosome coverage and differential expression (28).
First, we examined the transcriptional changes within a modified genomic locus, in which a target gene is replaced with the KanMX module (Figure 1A). To this end, we reconstructed the complete genome annotation for the knockout strains and mapped RNA-Seq reads to these genomes (KO datasets) and the original genome of the wt BY4741 strain (wt dataset), respectively. Figure 1B-C illustrate the results of such mapping for trm10Δ and wt strains. We observed disproportionally high kan gene expression in the mutant strain, as compared to the transcription efficiency of other genes within the same locus (including at least a dozen upstream and downstream genes). Considering the whole transcriptome, kan is among the top 1% most highly expressed genes in the trm10Δ strain (Supplementary Figure S1A). The same effect is exhibited in Ribo-Seq data (Supplementary Figures S1A, B), illustrating a high load of the kan transcript on the cellular translational machinery.
The excessive transcription could skew the overall activity of the entire locus at the epigenetic level. Analysis of differential expression of the genes adjacent to TRM10/YOL093W showed that mRNA abundance of RFC4/YOL094C was reduced almost 4-fold, while the level of other mRNAs encoded in this locus did not experience observable changes (Figure 1D, Supplementary Figure S1B).
Similar analysis of RNA-Seq data for the other 57 strains in the dataset showed pervasive changes in the transcription levels of neighboring genes (Supplementary Figure S2). Such alterations were unexpected and could potentially affect the abundance of encoded proteins and phenotype of the mutant strains.
KanMX cassette shifts TSSs of the neighboring genes generating new uORFs and inducing translational silencing
To further characterize unintended changes within mutated loci, we examined particular cases of the immediately adjacent genes in more detail. In the case of the deg1Δ strain, we observed a ∼3-fold decrease of the neighboring SPB4 gene expression at the translational level. Ribo-Seq tracks of wt and deg1Δ strains are presented in Figure 2A. Quantifications of Ribo-Seq and ribosome occupancy (RO) are shown in Figure 2B. Hereafter, we use RO as Ribo-Seq coverage of a CDS normalized to its RNA-Seq coverage that serves as a proxy for translation efficiency of the transcript. The two genes were located in the 5′-to-5′ orientation (hereafter called ‘head-to-head’), with only 196 bp between their ATGs. Although overall SPB4 mRNA level changed only slightly (Figure 2A, cf. RNA-Seq tracks, quantification is shown in Figure 2B), we observed a clear shift of the SPB4 TSS toward the KanMX cassette in the case of the deg1Δ strain. Due to the shift, an upstream AUG codon was included in the new 5′ UTR, thus forming an inhibitory uORF (Figure 2C). This inhibited translation of the SPB4 main coding region (Figure 2B, right panel), as uORFs usually dramatically decrease mRNA translation in yeast and are relatively rare in S. cerevisiae mRNAs (30–32). In the case of the altered SPB4 transcript, the acquired uAUG was in the optimal nucleotide context (AggAUGA) and thus should be effectively recognized by the ribosome (33). Indeed, we observed ribosome coverage of a region corresponding to the uORF (Figure 2A). In this regard, the slightly decreased abundance of the SPB4 mRNA (Figure 2A and B, RNA-Seq tracks) could be related to its reduced stability (34), as uORF-containing transcripts are subject to nonsense-mediated decay in yeast (35,36).
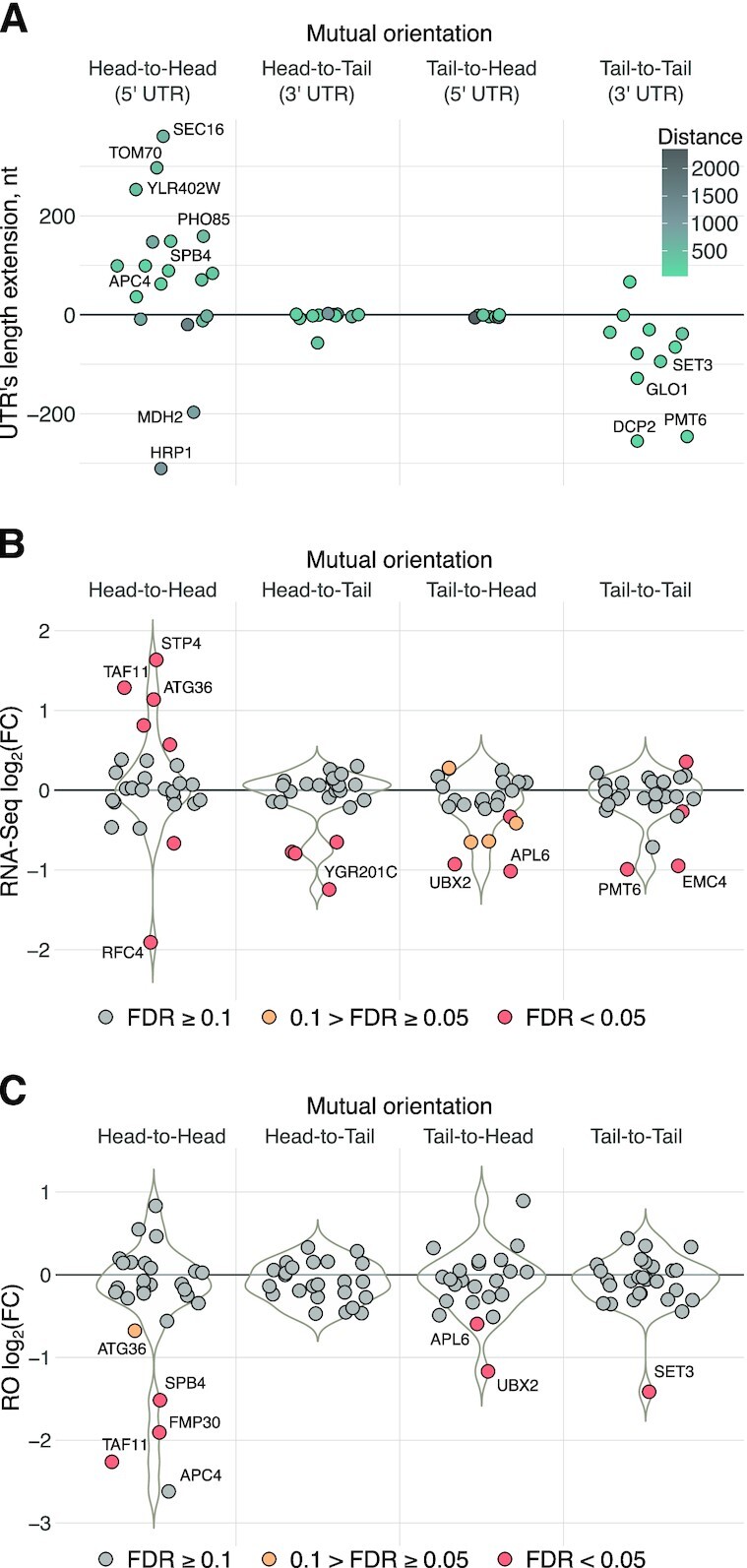
Figure 2.

KanMX cassette shifts transcriptional start sites of neighboring genes generating novel uORFs and inducing translational silencing. (A) RNA-Seq and Ribo-Seq normalized coverage of the DEG1-SPB4 locus in the wt (left subpanel) and deg1Δ (right subpanel) strains. The tracks show normalized coverage, positive and negative values correspond to the direct and reverse complementary DNA strands, respectively. Translation of the novel uORF (highlighted in pink) reduces the SPB4 ribosome occupancy (RO). Primer pairs and the corresponding PCR products used for validation of the TSS shift in (D) are schematically shown below the tracks. (B) Differential expression of genes within the ±100 kb region from the KanMX module at the level of transcription (RNA-Seq, left subpanel), translation (Ribo-Seq, middle subpanel), and translation efficiency (RO, right subpanel), deg1Δ compared to wt. RO (ribosome occupancy) denotes the Ribo-Seq coverage of a CDS normalized to its RNA-Seq coverage. (C) Nucleotide sequence of the SPB4 upstream region and its three-frame translation. The shift of the SPB4 TSS (indicated by arrows) in the deg1Δ strain creates a novel uORF (shadowed). TSSs in both strains are determined as 5′ ends of the longest transcripts present in the RNA-Seq data. (D) RT-qPCR confirms the 5′ UTR extension of the SPB4 mRNA in the deg1Δ strain. Specific primer pairs (shown in (A) by arrows) are used for the extended SPB4 transcript (‘SPB long’), for both shorter and longer isoforms (‘SPB4 both’), or for TAF10 mRNA (control). Product abundances are normalized to those in the wt strain. (E–H) The same as in (A–D) for the TMA64-APC4 locus. Shift of the APC4 TSS in the tma64Δ strain creates a novel uORF, which reduces the APC4 translation efficiency.
The TSS shift of the SPB4 gene in the deg1Δ strain was confirmed experimentally by RT-qPCR with two primer pairs (Supplementary Table S2, schematically shown by arrows in Figure 2A). A forward primer from the first pair corresponded to the extended part of the presumptive extended SPB4 transcript, while that from the other pair annealed to both the extended and original (normal length) mRNAs. In the strain with deleted DEG1, we observed a substantially elevated amount of the extended variant, while the total abundance of both SPB4 transcripts was reduced (Figure 2D).
An even more dramatic effect was observed in the case of TMA64 gene knockout (13), which led to the deregulation of the neighboring essential gene APC4, located in the ‘head-to-head’ orientation (Figure 2E). In tma64Δ we observed a ∼8-fold decrease of the APC4 CDS expression at the translational level (Figure 2F). The region upstream of the original APC4 TSS contained two ATG codons (Figure 2G). Replacing TMA64 with the KanMX module shifted the APC4 TSS upstream, thus forming two uORFs covered by ribosome footprints in the Ribo-Seq track in Figure 2E. The RT-qPCR analysis confirmed that the abundance of the extended APC4 transcript was dramatically increased in the tma64Δ strain (Figure 2H).
APC4 encodes a subunit of the Anaphase-Promoting Complex/Cyclosome (APC/C), involved in the control of cell cycle (37), while the TMA64 gene product is a non-essential translation factor involved in ribosome recycling and reinitiation (38). Curiously, previous analyses of tma64Δ knockout strains revealed strong negative genetic interactions of TMA64 with genes involved in APC/C-mediated anaphase checkpoint (39,40), as well as cell cycle abnormalities (41). Indeed, 7 out of 10 TMA64 genetic interactions listed in SGD (https://www.yeastgenome.org, (42)) and documented in at least 2 data sources, involve genes encoding either APC/C subunits (APC5, CDC20, CDC23, CDC27) or other components of the mitotic checkpoint (Supplementary Figure S3). Our data suggest that these interactions are false positives caused by the defect of the APC4 mRNA translation in the tma64Δ knockout strain that we found in our analysis.
KanMX cassette alters 3′ end processing of the neighboring gene transcripts and deregulates their translation
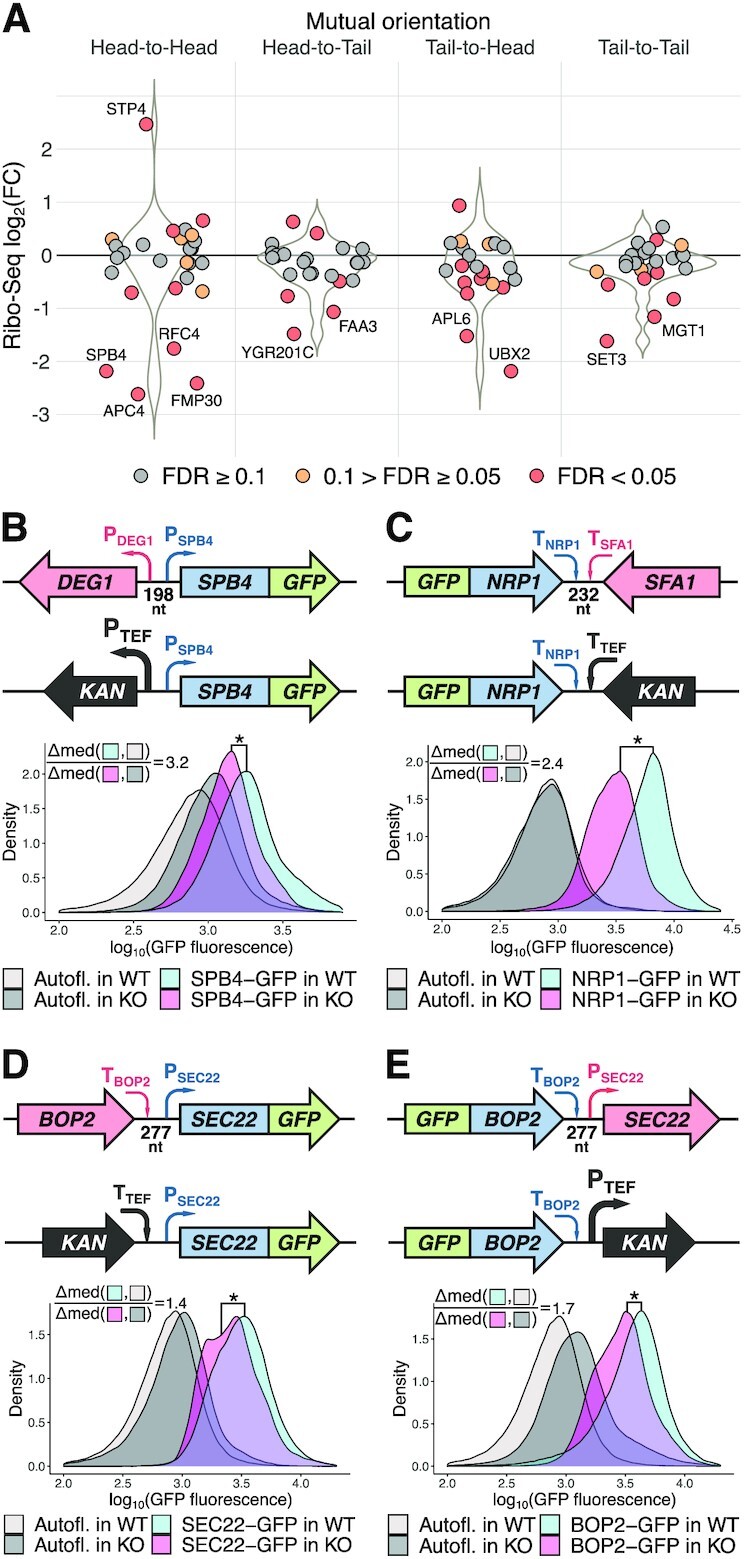
The above cases exemplified the ‘head-to-head’ arrangement of a target gene with its affected neighbor. However, the KanMX module could also disturb proper termination and correct formation of the 3′ end of the transcript in the case of the 3′-to-3′ (‘tail-to-tail’) arrangement. To investigate this, we analyzed several such cases from the same study (12), and indeed found shifts of cleavage and polyadenylation sites (PASs).
Specifically, the PAS shift happens in the TRM12 knockout, with a dramatic alteration in the PAS position of the neighboring GLO1 gene (Figure 3A). In this particular case, the alteration was probably unavoidable, as the original GLO1 transcript extends into the TRM12 coding region, where its PAS is located normally. In the trm12Δ strain, however, this region is replaced by the KanMX cassette. Mapping of the RNA-Seq reads onto the trm12Δ genome revealed the formation of an alternative 3′ terminus, which probably was the result of activation of the cryptic premature PAS located in this locus (15) (Figure 3A and C). The resulting 3′ UTR is ∼2-fold shorter than the original one. Surprisingly, the dramatic changes in the GLO1 3′ UTR length did not lead to prominent perturbations in either abundance of the transcript or its RO (Figure 3B), at least under the conditions tested. However, in other cases, such alterations could affect gene expression, as 3′ UTRs play important roles in translation regulation and control of mRNA stability (43,44). Indeed, in the case of another ‘tail-to-tail’ oriented gene pair, SET3 and SAP190, we observed a substantial (∼3-fold) reduction in RO of the SET3 coding region in the strain where SAP190 was replaced with the KanMX cassette (Figure 3D and E), as expected from the dramatic shortening of the SET3 3′ UTR (Figure 3D and F).
Figure 3.

KanMX cassette deregulates expression of the neighboring gene transcripts at the translational level by altering 3′ end processing. (A) Normalized RNA-Seq coverage of the GLO1-TRM12 locus in wt (left subpanel) and trm12Δ (right subpanel) strains. The GLO1 3′ UTR (highlighted in pink) is shorter in the mutant strain. (B) Differential expression of genes within the ±100 kb region from the KanMX module at the level of transcription (RNA-Seq, left subpanel), translation (Ribo-Seq, middle subpanel), and translation efficiency (RO, right subpanel), trm12Δ compared to wt. (C) Nucleotide sequence of the GLO1 downstream region. The putative major PASs of GLO1 in both strains are indicated by arrows. Putative PASs from (15) are shown in the additional track, Y-axis in the log2 scale. (D–F) The same for the SET3-SAP190 locus, in (D) the SET3 3′ UTR is highlighted in pink.
Transcriptional and translational deregulation of neighboring genes caused by TSS shifts and alternative PAS activation are pervasive in yeast knockout strains
To systematize TSS shifting and alternative PAS activation, we estimated alterations of 5′ UTR and 3′ UTR lengths of neighboring genes observed in the analyzed datasets, depending on the mutual orientation of the knockout target and the nearest neighboring gene (Figures 4A). The analysis showed that ‘head-to-head’ orientation often leads to TSS shifting: 5′ UTRs were extended by the TSS shifts in 11 of the 12 cases when the distance between the ORFs of the neighboring genes was less than 1 kb. In the case of the ‘tail-to-tail’ orientation, we found a trend towards 3′ UTRs shortening as a result of alternative PAS activation, but this effect was less pronounced in comparison with the TSS shift. In the case of ‘head-to-tail’ and ‘tail-to-head’ orientations, no significant alterations were observed, although particular changes are harder to track in such cases, as RNA sequences of the neighboring and knockout genes are located on the same DNA strand in close proximity.
Figure 4.
KanMX cassette alters 5′ and 3′ UTRs of neighboring genes, affecting transcript abundances and translation efficiencies. (A) Swarmplots illustrating alterations of 5′ UTR and 3′ UTR lengths of neighboring genes induced by the KanMX module depending on their orientation relative to the cassette. Labels indicate particular genes that were discussed in the text and, additionally, those with the UTR’s length extension of more than 150 nt. (B) Swarmplots and underlying violin plots illustrating differential expression of the neighboring genes in the 58 knockout strains analyzed in this study, at the level of transcription (RNA-Seq), as compared to wt. (C) Swarmplots and underlying violin plots illustrating changes in the translation efficiency (RO) of mRNAs encoded by the neighboring genes in knockout strains compared to wt. Labels indicate particular genes that were discussed in the text and, additionally, those with the absolute value of log2 fold change >1 and FDR <0.05.
We then calculated expression changes in the closest neighboring genes in all datasets at both transcriptional and translational levels and grouped the results with respect to the neighboring gene orientation to the deleted genes. Analysis of the RNA-Seq coverage (Figure 4B) showed strong transcriptional deregulation of many ‘head-to-head’-oriented neighboring genes, with either decreased or increased expression; for other mutual orientations, there was a tendency to observe the reduced mRNA level. A similar analysis of RO, which reflects translation efficiency (Figure 4C), revealed a prominent effect of translational downregulation for a substantial proportion of neighboring genes, leading to more than a 2-fold drop (FDR < 0.05) in at least six cases (Figure 4C), four of which can be explained by the above-described effect from novel uORFs (see Supplementary Figure S2).
As much as 1/5 of knockout strains may have substantially (>2-fold) deregulated expression of neighboring genes that can result in changed protein abundance
Since ribosome coverage of a transcript can be used as a proxy for the gene product abundance, we analyzed Ribo-Seq data to calculate a proportion of cases in the analyzed datasets, where the protein synthesis from neighboring gene transcripts was deregulated (Figure 5A). Among the 58 analyzed strains, ribosome footprint coverage of neighboring gene transcripts was altered more than 2-fold (FDR < 0.05) in 11 (19%), >3-fold in 7 (12%) and >4-fold in 5 cases (8%). Weaker effects (from 1.5- to 2-fold) were observed in 20 (34%) of the analyzed strains. This gives a rough estimate of the number of affected strains across the complete yeast deletion library.
Figure 5.
A substantial fraction of analyzed knockout strains has deregulated expression of neighboring genes that can result in corresponding changes of protein abundance. (A) Swarmplot and underlying violin plots illustrating differential expression of the neighboring genes in the 58 knockout strains analyzed in this study at the level of translation (Ribo-Seq) depending on neighboring gene orientation relative to the kan gene (knockout strains compared to wt). Labels indicate genes that were mentioned in the text and, additionally, those with the absolute value of log2 fold change >1 and the FDR <0.05. (B–E) Gene replacement with the KanMX module induces changes in the abundance of the GFP-tagged proteins encoded by neighboring genes. Four cases with different mutual gene orientations (schematically shown in upper subpanels) were analyzed. For each case, four yeast strains were assayed by flow cytometry: two strains with the GFP-tagged neighboring gene (as shown in the schemes) and two corresponding strains without the GFP-tag (to take into account possible autofluorescence changes between the KO strains and wt). The density plots show distribution of the GFP fluorescence (log10 scale, combined results from at least three independent experiments). Effect size estimates (Δmed ratios) are shown on the plots, see Materials and Methods for details. Combined results from at least three independent experiments are shown, *one-sided U-test P-value <10−20.
However, protein abundance is determined not only by synthesis but also degradation. Thus, it was important to experimentally verify the changes in the abundance of proteins encoded by the genes neighboring the KanMX module in knockout strains. We selected several strains expressing GFP-tagged proteins from the GFP-tagged systematic collections (24,25) and replaced the adjacent genes with the standard KanMX cassette. Wt and deletion strains having no GFP tag were used for autofluorescence background correction.
We assayed five strains with ‘head-to-head’ orientation of genes, four strains with ‘tail-to-tail’ orientation and three with ‘head-to-tail’ or ‘tail-to-head’ orientation, in which we could reliably detect GFP fluorescence using flow cytometry. The set included GFP-tagged SPB4 protein corresponding to the DEG1-SPB4 gene analyzed earlier (Figures 2A and 5B) and two complementing pairs of genes: BOP2―SEC22-GFP / GFP-BOP2―SEC22 and RRD1―IMP2′-GFP / IMP2′―RRD1-GFP). Importantly, half of the deleted genes (IMP2′, IRC6, RRD1, SEC22, SFA1, VIP1) were unrelated to translation, thus avoiding a bias of the Ribo-Seq analysis where most of the target genes were encoding tRNA modification enzymes (12).
A statistically significant decrease in the level of GFP fluorescence was observed in 6 out of the tested 12 cases (Figure 5B, C and Supplementary Figure S4), including the GFP-tagged SPB4. For four proteins we were unable to detect any significant changes in the protein level, while in one case the reporter level was increased (Supplementary Figure S4D). To sum up, KanMX-mediated deregulation of the expression of adjacent genes can affect their protein levels in the cell and, consequently, influence the phenotype of the knockout strains.
DISCUSSION
The SGDP yeast deletion library is a powerful and indispensable instrument to study eukaryotic gene function (2,6). However, due to the compact nature of the yeast genome, the replacement of every individual gene with the highly expressed KanMX cassette can potentially perturb the expression of the neighboring genes. Importantly, the undesirable effects of the cassette could extend beyond the transcriptional level, affecting also the translation of mRNAs synthesized from neighboring genes. Such changes may cause inappropriate attribution of the side effects to the absence of the target gene, while in reality they are caused by perturbation of neighboring gene expression. This phenomenon is known as the neighboring gene effect, NGE (10,11). NGE should be taken into account during high-throughput studies of functional gene annotation and genetic interaction analyses. A few systematic studies devoted to this important issue revealed that NGE may be involved in as much as 7–15% of the identified gene-gene interactions (9–11). However, no mechanistic explanations of these effects have been proposed.
In this study, we took advantage of ribosome profiling (45,46) to investigate the molecular mechanisms behind NGE. Although this powerful method was originally developed for S. cerevisiae (27,47), to date only a limited number of Ribo-Seq studies have been performed in knockout yeast strains. We used one of the most comprehensive datasets containing ribosome profiling and RNA-Seq data for 57 mutant strains (12), as well as our own data (13), to characterize the transcriptional and translational changes that occur in genomic regions adjacent to the introduced KanMX cassette. We documented several types of changes, including (i) transcriptional repression of the neighboring genes (Figures 1 and 4B); (ii) shifts of TSSs toward the cassette, causing the appearance of uAUG(s) in the extended 5′ UTRs, reduction of translation efficiency and likely mRNA stability (Figures 2 and 4A) and (iii) triggering alternative pre-mRNA cleavage and polyadenylation events, in some cases also leading to deregulation of mRNA expression at the translational level (Figures 3 and 4A). Finally, using GFP reporters, we showed that such perturbations can result in changes in protein abundance (Figure 5B and C).
Although our study was restrained by the limited amount of published yeast Ribo-Seq datasets, the available examples clearly show that replacement of a gene by the KanMX cassette often significantly affects expression of the neighboring genes via both transcriptional and translational mechanisms.
One could argue that all datasets that we initially analyzed were obtained for strains with mutations of translation machinery components. Indeed, ribosome profiling is usually applied to mutants or conditions where translation is expected to be altered. However, in a large portion of these strains, no phenotype associated with compromised overall translation was observed (12). We also showed that only the adjacent genes were typically exhibiting differential expression at the level of transcription or translation (see Supplementary Figure S2). Moreover, our GFP-based validation assay was also applied to genes not involved in translation. Thus, the observed phenomenon is likely general and not limited to translation-related genes. Extrapolating our results, we estimate that changes in the levels of transcription and translation of neighboring genes of 2-fold and higher may be observed in up to one-fifth of knockout strains in the yeast deletion library.
The revealed TSS shifts (in the case of ‘head-to-head’ gene arrangement) and alternative PAS activation (in ‘tail-to-tail’ orientation of the genes), leading to mRNA translation decline, were the most striking observations of our study (Figure 4A, overviewed in Figure 6). The effects of the TSS shifts and PAS changes in knockout strains may be even more important under different growth conditions, cell stress, or sporulation, when numerous alternative TSSs and PASs are activated (48,49).
Figure 6.
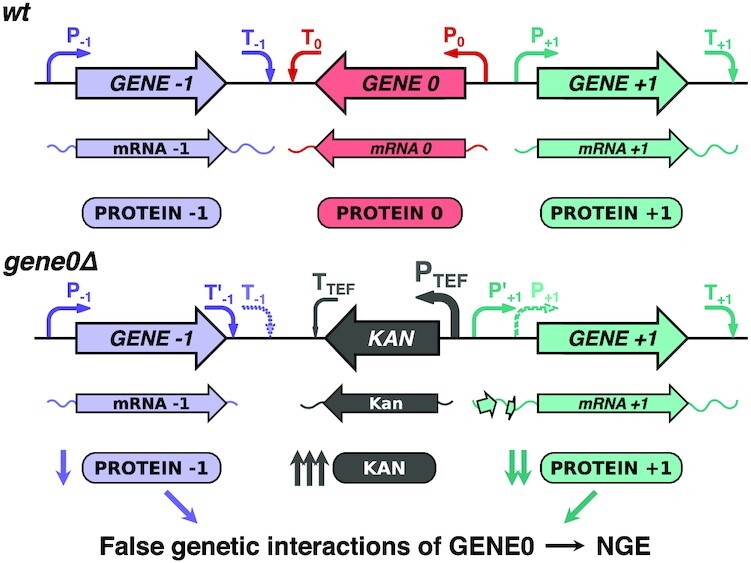
A model explaining NGE in knockout strains arising from perturbation of neighboring gene expression at the translational level. As GENE0 is replaced by the KanMX cassette, its strong regulatory elements (P-TEF and T-TEF) perturb transcription of the neighboring genes, altering PAS and TSS of GENE-1 and GENE + 1, respectively. This decreases their mRNA translation due to the GENE-1 3′ UTR shortening and uORF(s) appearance in the GENE + 1 5′ UTR.
Taking into account the pervasiveness of such effects, we compiled a complete list of potentially affected genes (Supplementary Table S3) where one can find a qualitative estimation of the potential effect that replacement by the KanMX module may induce on the nearest adjacent gene. The estimation is based on a set of empirical rules revealed in this study: mutual gene orientation, the distance between the genes, and, in the case of ‘head-to-head’ orientation, the presence of AUG codon in the spacer located near the TSS of the neighboring gene.
The identified patterns of expression changes of neighboring genes could explain the false genetic interactions of the deleted genes, the so-called NGE phenomenon (Figure 6), both in the cases we examined here and in many other cases. For example, almost half of the neighboring gene pairs with the distance between the genes less than 200 bp and with a genetic interaction profile similarity of PCC >0.1 (according to the TheCellMap.org (9,40)) have at least one ATG codon within 50 bp upstream of a TSS of the neighboring gene, while the same is true for only a quarter of pairs with such arrangement of genes and with PCC < 0.01 (see Supplementary Table S3). This could explain the observed NGE in some of the former gene pairs, with clearer criteria to be formulated when more high-throughput data become available for systematic analysis.
All in all, we propose that in many cases NGE in knockout strains is caused by KanMX-induced defects at the translational level, as revealed in this study. This should be taken into account when using the yeast knockout collection for functional gene annotation and genetic interaction analysis, as well as when interpreting results based on similar methods of targeted gene manipulation in any other model eukaryotic organism.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Pavel Baranov (University College Cork, Ireland) for helpful discussion and the Moscow State University Development Program PNR5 for providing access to the MACS Quant Analyzer flow cytometer, as well as the Shared-Access Equipment Centre ‘Industrial Biotechnology’ of Federal Research Center ‘Fundamentals of Biotechnology’ of Russian Academy of Sciences for providing access to the Beckman Cytoflex S flow сytometer. We also thank Dr Maya Schuldiner for sharing N-terminally GFP-tagged yeast strains. A.A.E., R.O.E., A.S.K. and S.E.D. are members of the Interdisciplinary Scientific and Educational School of Moscow University “Molecular Technologies of the Living Systems and Synthetic Biology”.
Contributor Information
Artyom A Egorov, Phystech School of Biological and Medical Physics, Moscow Institute of Physics and Technology (State University), Dolgoprudny 141700, Russia; Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia.
Alexander I Alexandrov, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; FRC of Biotechnology of the Russian Academy of Sciences, Bach Institute of Biochemistry, Moscow 119071, Russia.
Valery N Urakov, FRC of Biotechnology of the Russian Academy of Sciences, Bach Institute of Biochemistry, Moscow 119071, Russia.
Desislava S Makeeva, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia.
Roman O Edakin, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia; Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow 119234, Russia.
Artem S Kushchenko, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia; Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow 119234, Russia.
Vadim N Gladyshev, Division of Genetics, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115, USA.
Ivan V Kulakovskiy, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia; Vavilov Institute of General Genetics, Russian Academy of Sciences, Moscow 119991, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow 119991, Russia.
Sergey E Dmitriev, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119234, Russia; Sirius University of Science and Technology, 1 Olympic Ave, Sochi 354340, Russia; Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow 119234, Russia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Foundation for Basic Research [19-34-51047 to S.E.D.]; Russian Ministry of Science and Higher Education. Funding for open access charge: Russian Foundation for Basic Research grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M.et al.. Life with 6000 genes. Science. 1996; 274:563–547. [DOI] [PubMed] [Google Scholar]
- 2. Giaever G., Chu A.M., Ni L., Connelly C., Riles L., Veronneau S., Dow S., Lucau-Danila A., Anderson K., Andre B.et al.. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002; 418:387–391. [DOI] [PubMed] [Google Scholar]
- 3. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H.et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 4. Wach A., Brachat A., Pohlmann R., Philippsen P.. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994; 10:1793–1808. [DOI] [PubMed] [Google Scholar]
- 5. Saccharomyces Genome Deletion Project.
- 6. Giaever G., Nislow C.. The yeast deletion collection: a decade of functional genomics. Genetics. 2014; 197:451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costanzo M., Kuzmin E., van Leeuwen J., Mair B., Moffat J., Boone C., Andrews B.. Global genetic networks and the genotype-to-phenotype relationship. Cell. 2019; 177:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ooi S.L., Pan X., Peyser B.D., Ye P., Meluh P.B., Yuan D.S., Irizarry R.A., Bader J.S., Spencer F.A., Boeke J.D.. Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2006; 22:56–63. [DOI] [PubMed] [Google Scholar]
- 9. Usaj M., Tan Y., Wang W., VanderSluis B., Zou A., Myers C.L., Costanzo M., Andrews B., Boone C.. TheCellMap.org: a web-accessible database for visualizing and mining the global yeast genetic interaction network. G3. 2017; 7:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atias N., Kupiec M., Sharan R.. Systematic identification and correction of annotation errors in the genetic interaction map of Saccharomyces cerevisiae. Nucleic Acids Res. 2016; 44:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ben-Shitrit T., Yosef N., Shemesh K., Sharan R., Ruppin E., Kupiec M.. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat. Methods. 2012; 9:373–378. [DOI] [PubMed] [Google Scholar]
- 12. Chou H.J., Donnard E., Gustafsson H.T., Garber M., Rando O.J.. Transcriptome-wide analysis of roles for tRNA modifications in translational regulation. Mol. Cell. 2017; 68:978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makeeva D.S., Lando A.S., Anisimova A., Egorov A.A., Logacheva M.D., Penin A.A., Andreev D.E., Sinitcyn P.G., Terenin I.M., Shatsky I.N.et al.. Translatome and transcriptome analysis of TMA20 (MCT-1) and TMA64 (eIF2D) knockout yeast strains. Data in Brief. 2019; 23:103701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G.et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X., Hoque M., Larochelle M., Lemay J.F., Yurko N., Manley J.L., Bachand F., Tian B.. Comparative analysis of alternative polyadenylation in S. cerevisiae and S. pombe. Genome Res. 2017; 27:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khalfan M., Gresham D. Modifying reference sequence and annotation files quickly and easily with reform. OSF. 2021; https://osf.io/5ks7r/. [Google Scholar]
- 17. Leinonen R., Sugawara H., Shumway M.International Nucleotide Sequence Database, C . The sequence read archive. Nucleic Acids Res. 2011; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011; 17:3. [Google Scholar]
- 19. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egorov A.A., Sakharova E.A., Anisimova A.S., Dmitriev S.E., Gladyshev V.N., Kulakovskiy I.V.. svist4get: a simple visualization tool for genomic tracks from sequencing experiments. BMC Bioinformatics. 2019; 20:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003; 425:686–691. [DOI] [PubMed] [Google Scholar]
- 25. Weill U., Yofe I., Sass E., Stynen B., Davidi D., Natarajan J., Ben-Menachem R., Avihou Z., Goldman O., Harpaz N.et al.. Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods. 2018; 15:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collart M.A., Oliviero S.. Preparation of yeast RNA. Curr. Protoc. Mol. Biol. 2001; Chapter 13:Unit13 12. [DOI] [PubMed] [Google Scholar]
- 27. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerashchenko M.V., Gladyshev V.N.. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 2014; 42:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hussmann J.A., Patchett S., Johnson A., Sawyer S., Press W.H.. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLos Genet. 2015; 11:e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Z., Dietrich F.S.. Identification and characterization of upstream open reading frames (uORF) in the 5' untranslated regions (UTR) of genes in Saccharomyces cerevisiae. Curr. Genet. 2005; 48:77–87. [DOI] [PubMed] [Google Scholar]
- 31. Hood H.M., Neafsey D.E., Galagan J., Sachs M.S.. Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annu. Rev. Microbiol. 2009; 63:385–409. [DOI] [PubMed] [Google Scholar]
- 32. Lawless C., Pearson R.D., Selley J.N., Smirnova J.B., Grant C.M., Ashe M.P., Pavitt G.D., Hubbard S.J.. Upstream sequence elements direct post-transcriptional regulation of gene expression under stress conditions in yeast. BMC Genomics. 2009; 10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011; 75:434–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oliveira C.C., McCarthy J.E.. The relationship between eukaryotic translation and mRNA stability. A short upstream open reading frame strongly inhibits translational initiation and greatly accelerates mRNA degradation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1995; 270:8936–8943. [DOI] [PubMed] [Google Scholar]
- 35. Guan Q., Zheng W., Tang S., Liu X., Zinkel R.A., Tsui K.W., Yandell B.S., Culbertson M.R.. Impact of nonsense-mediated mRNA decay on the global expression profile of budding yeast. PLos Genet. 2006; 2:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malabat C., Feuerbach F., Ma L., Saveanu C., Jacquier A.. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. eLife. 2015; 4:e06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zachariae W., Shevchenko A., Andrews P.D., Ciosk R., Galova M., Stark M.J., Mann M., Nasmyth K.. Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science. 1998; 279:1216–1219. [DOI] [PubMed] [Google Scholar]
- 38. Young D.J., Makeeva D.S., Zhang F., Anisimova A.S., Stolboushkina E.A., Ghobakhlou F., Shatsky I.N., Dmitriev S.E., Hinnebusch A.G., Guydosh N.R.. Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR recycle post-termination 40S subunits in vivo. Mol. Cell. 2018; 71:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E.D., Sevier C.S., Ding H., Koh J.L., Toufighi K., Mostafavi S.et al.. The genetic landscape of a cell. Science. 2010; 327:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D.et al.. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016; 353:aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watanabe M., Watanabe D., Nogami S., Morishita S., Ohya Y.. Comprehensive and quantitative analysis of yeast deletion mutants defective in apical and isotropic bud growth. Curr. Genet. 2009; 55:365–380. [DOI] [PubMed] [Google Scholar]
- 42. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R.et al.. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jia J., Yao P., Arif A., Fox P.L.. Regulation and dysregulation of 3'UTR-mediated translational control. Curr. Opin. Genet. Dev. 2013; 23:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matoulkova E., Michalova E., Vojtesek B., Hrstka R.. The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biology. 2012; 9:563–576. [DOI] [PubMed] [Google Scholar]
- 45. Ingolia N.T., Hussmann J.A., Weissman J.S.. Ribosome profiling: global views of translation. Cold Spring Harb. Perspect Biol. 2019; 11:a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Andreev D.E., O’Connor P.B., Loughran G., Dmitriev S.E., Baranov P.V., Shatsky I.N.. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017; 45:513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brar G.A., Yassour M., Friedman N., Regev A., Ingolia N.T., Weissman J.S.. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012; 335:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waern K., Snyder M.. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3. 2013; 3:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng Z., Otto G.M., Powers E.N., Keskin A., Mertins P., Carr S.A., Jovanovic M., Brar G.A.. Pervasive, coordinated protein-level changes driven by transcript isoform switching during meiosis. Cell. 2018; 172:910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.