


Abstract
In ribosomal translation, the accommodation of aminoacyl-tRNAs into the ribosome is mediated by elongation factor thermo unstable (EF-Tu). The structures of proteinogenic aminoacyl-tRNAs (pAA-tRNAs) are fine-tuned to have uniform binding affinities to EF-Tu in order that all proteinogenic amino acids can be incorporated into the nascent peptide chain with similar efficiencies. Although genetic code reprogramming has enabled the incorporation of non-proteinogenic amino acids (npAAs) into the nascent peptide chain, the incorporation of some npAAs, such as N-methyl-amino acids (MeAAs), is less efficient, especially when MeAAs frequently and/or consecutively appear in a peptide sequence. Such poor incorporation efficiencies can be attributed to inadequate affinities of MeAA-tRNAs to EF-Tu. Taking advantage of flexizymes, here we have experimentally verified that the affinities of MeAA-tRNAs to EF-Tu are indeed weaker than those of pAA-tRNAs. Since the T-stem of tRNA plays a major role in interacting with EF-Tu, we have engineered the T-stem sequence to tune the affinity of MeAA-tRNAs to EF-Tu. The uniform affinity-tuning of the individual pairs has successfully enhanced the incorporation of MeAAs, achieving the incorporation of nine distinct MeAAs into both linear and thioether-macrocyclic peptide scaffolds.
INTRODUCTION
In prokaryotic translation machinery, all 20 proteinogenic amino acids (pAAs) are charged onto their corresponding tRNAs, and the resulting aminoacyl-tRNAs (pAA-tRNAs) are subsequently accommodated into the ribosomal A site by elongation factor thermo unstable, EF-Tu (Figure 1A) (1,2). EF-Tu recognizes two distinct regions of pAA-tRNAs: the amino acid moiety and the T-stem region of the tRNA (3–5). The binding affinities between EF-Tu and pAA-tRNAs are determined by the sum of these two interactions. Uhlenbeck and colleagues have demonstrated that twenty distinct pAA-tRNAs bind to EF-Tu with near-uniform affinities from −9.5 to −10.5 kcal/mol (6). This is achieved by a compensatory relationship between the two binding regions (6–11). In the case of Glu-tRNAGlu, for example, the intrinsic weak affinity of Glu to EF-Tu is compensated by the strong affinity of the T-stem region of tRNAGlu. Such a narrow range of uniform affinities serves as a threshold for excluding mischarged pAA-tRNAs, for example, Glu-tRNAGln has an insufficient EF-Tu affinity, and is thus poorly accommodated into the ribosome A site, resulting in failure of efficient peptide elongation (12).
Figure 1.
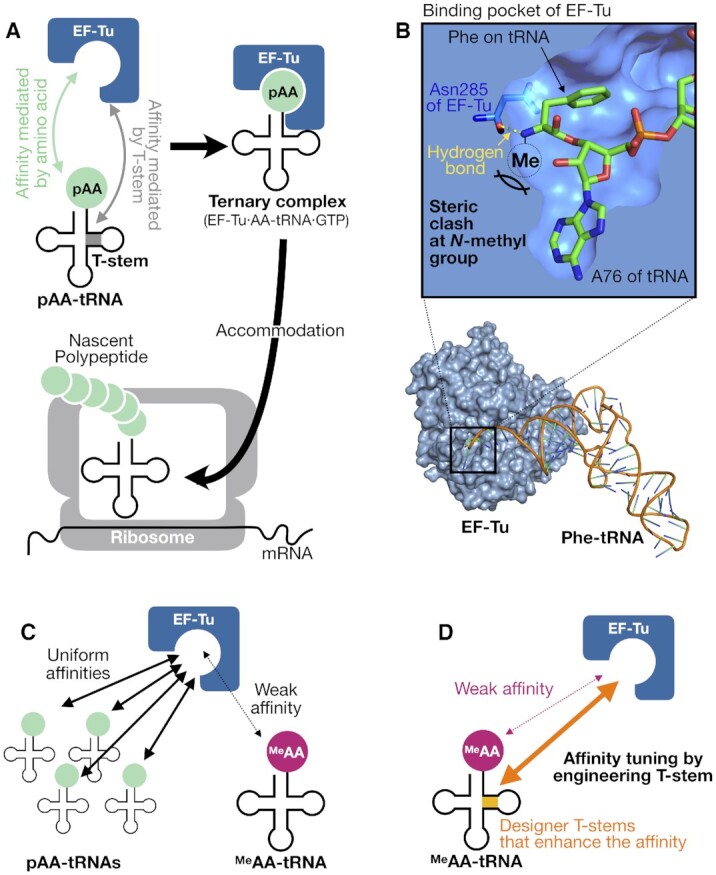
Schematic illustration of the interaction between EF-Tu and aminoacyl-tRNAs. (A) EF-Tu-mediated delivery of pAA-tRNA into the ribosome. (B) A potential steric clash of the N-methyl group of MeAA-tRNAs to EF-Tu. This image was made from the cocrystal structure of Thermus aquaticus EF-Tu and Escherichia coli Phe-tRNAPhe (PDB ID: 1TTT). The cross section around the binding pocket is shown in the upper panel. The hydrogen bond between EF-Tu Asn285 (blue) and Phe-tRNAPhe (green) is indicated in yellow dotted line. (C) Uniform affinities of pAA-tRNAs to EF-Tu versus weak affinities of MeAA-tRNAs to EF-Tu. (D) Schematic representation of the affinity-tuning strategy of MeAAs and tRNAs demonstrated in this study.
Diverse non-proteinogenic amino acids (npAAs) can be also ribosomally incorporated into nascent peptides by means of genetic code manipulation methodologies (13–16). However, ribosomal incorporation of npAAs often suffers from poorer efficiency than the pAA incorporation. The Schultz group has conducted in vitro evolution of amber suppressor tRNAs from semi-random sequences based on Methanocaldococcus jannaschii tRNATyr and obtained those having improved efficiencies for the incorporation of bulky phenylalanine (Phe) analogs into proteins in Escherichia coli (17). Although the fidelity of their incorporation was not clearly defined in their work, the expression level was improved by 2–20-fold likely owing to an affinity enhancement of the npAA-tRNA to EF-Tu. Alternatively, EF-Tu mutants were also engineered to accept certain Phe analogs or O-phosphoserine when they were charged onto cognate tRNAs, where the expression of designated protein containing such npAAs was enhanced (18–20).
The above experiments aimed at expanding the genetic code using the nonsense amber codon, in which npAAs utilized were analogs of pAAs (mostly Phe analogs) having noncanonical sidechains. However, by means of in vitro translation, more aggressive genetic code reprogramming with exotic npAAs, such as N-methyl-amino acids (MeAAs) and d-amino acids, has been attempted. Forster et al. reported that such npAAs are intrinsically poorer substrates for elongation (21,22), and showed that a swapping npAA-tRNAPhe for npAA-tRNAAla could increase the affinity of npAA-tRNA to EF-Tu up to 10-fold, resulting in modestly enhancing the rate of dipeptide formation of fMet-npAA, including N-methylphenylalanine (23,24). The Achenbach group used the flexizyme technology (25) to charge d-amino acids onto derivatives of tRNAGly and tRNATyr that exhibited higher affinities to EF-Tu than other tRNAs, and then the incorporation of d-amino acids was tested (26). The data showed marginal improvements for some d-amino acids but suffered competing incorporations of the natural counter parts. In their work, some mutants of EF-Tu were also tested, giving no major improvement was observed. Our group logically devised a novel chimeric tRNA based on D-arm of tRNAPro1 and T-stem of tRNAGlu, referred to as tRNAPro1E2, which were designed to effectively recruit EF-P and EF-Tu, respectively (27). The use of such designer tRNAs has made remarkable improvements of npAA incorporation by the assist of EF-P in not only single but also multiple and consecutive manners. When certain d-amino acids, β-amino acids, or cyclic γ-amino acids were charged on to this series of tRNAs, their incorporation into nascent peptide chain was improved by up to 13-fold that is a comparable level to their natural counterparts; and in some cases improved from an undetectable expression level to a detectable level (27–32). These results indicate that appropriately designed tRNAs could improve the overall translation efficiency of exotic peptides containing various npAAs with wildtype protein factors, EF-P and EF-Tu.
MeAAs are invaluable building blocks to install druglike properties to the peptides, for example, peptidase resistance and membrane-permeability (33–38). Our previous study reported that when MeAAs were charged onto tRNAAsnE2 (our standard suppressor tRNA for various npAA incorporations), only five MeAAs (MeG, MeA, MeS, MeF and MeY) showed good incorporation efficiencies (>80% relative to a pAA control); six MeAAs (MeT, MeC, MeQ, MeM, MeH and MeW) were moderate (10–80%); and the other eight MeAAs (MeV, MeL, MeI, MeN, MeD, MeE, MeR and MeK) were poor or undetectable (<10%) (39). It should be noted that the above observation was made by the study of a single incorporation of MeAA into nascent peptide chain (39,40). However, when the frequency of incorporating MeAA residues in nascent peptide chain increases, the expression level as well as fidelity detrimentally decrease (39). Moreover, the consecutive incorporation of MeAAs or even alternating incorporation of MeAA and pAA (e.g. MeAA-pAA-MeAA) is far less efficient when compared to pAA elongation in such peptide sequences. Thus, there remains an important challenge to incorporate not only a greater number of distinct MeAAs but also consecutively and/or alternately into the nascent peptide chain.
Here, we have aimed at improving the incorporation efficiencies of MeAAs by tuning the affinity between MeAA-tRNA and EF-Tu. According to the crystal structure of the ternary complex of EF-Tu·Phe-tRNAPhe·GTP, the N-methyl modification on the amino acid would hamper the interaction of EF-Tu with MeAA-tRNA (Figure 1B) (3), which could weaken their affinity, thereby resulting in reduced incorporation efficiency of MeAA (Figure 1C). Since the T-stem of tRNA plays a critical role in tuning the affinity between AA-tRNA and EF-Tu, we have hypothesized that an appropriate engineering of tRNA T-stem would allow us to uniformly tune the affinity between the corresponding MeAA-tRNA and EF-Tu similar to that of pAA-tRNAs and EF-Tu, and thereby the MeAA incorporation efficiencies into nascent peptide chain could be improved (Figure 1D).
MATERIALS AND METHODS
Preparation of tRNAs, flexizymes, mDNAs and mRNAs
All oligonucleotides listed in Supplementary Table S1 were purchased from Eurofins, Japan. In the nucleotide sequences, N(Me) indicates a 2′-O-methylated nucleotide, which aims at suppressing undesired addition of extra nucleotides at 3′ end of RNA in transcription (41). The sequences of tRNA transcripts prepared in this study are listed in Supplementary Table S2. Note that tRNA bearing GAA anticodon (decoding Phe codon) has a U20A substitution in order to avoid undesired Phe-charging catalyzed by native PheRS (42). Double-stranded DNA templates that encode tRNAs were prepared by primer extension followed by PCR as follows: Appropriate forward and reverse primers (1 μM each, see Supplementary Table S3 for the primers used) were mixed in 100 μl PCR mixture [10 mM Tris–HCl (pH 9.0), 50 mM KCl, 2.5 mM MgCl2, 0.25 mM each dNTPs, 0.1% (v/v) Triton X-100, and 45 nM Taq DNA polymerase]. Primer extension was conducted by denaturing (95°C for 1 min) followed by 5 cycles of annealing (50°C for 1 min) and extending (72°C for 1 min). 1 μl of the resulting reaction mixture was diluted 200-fold with 199 μl of PCR mixture containing appropriate forward and reverse primers (0.5 μM each, see Supplementary Table S3 for the primers used), and PCR was conducted for 12 cycles of denaturing (95°C for 40 s), annealing (50°C for 40 s), and extending (72°C for 40 s). Amplification of the PCR product was confirmed by 3% agarose gel electrophoresis and ethidium bromide staining. The resulting DNA was purified by phenol/chloroform/isoamyl alcohol extraction, phenol/chloroform extraction, and ethanol precipitation. The DNA pellet was dissolved in 20 μl water.
In vitro transcription reaction for tRNA was conducted by incubating 200 μl transcription mixture [40 mM Tris·HCl (pH 8.0), 1 mM spermidine, 0.01% (v/v) Triton X-100, 10 mM DTT, 22.5 mM MgCl2, 3.75 mM each NTPs, 5 mM GMP, 22.5 mM KOH, 10% (v/v) DNA template (prepared as described above), and 120 nM T7 RNA polymerase] at 37°C overnight. The transcription mixture was mixed with MnCl2 (100 mM, 4 μl) and RQ1 RNase-Free DNase (1 U/μl, 1 μl, Promega), and incubated at 37°C for 30 min. The resultant tRNA transcript was precipitated by isopropanol and dissolved in water. The tRNA transcript was purified by 8% denaturing PAGE and ethanol precipitation, and then dissolved in 10 μl water. The concentrations of tRNA were measured by A260 of a 10-fold diluted solution.
Flexizymes (dFx and eFx) and tRNAIni were prepared by in vitro transcription from their respective DNA templates, as previously described (25).
Double stranded DNA templates that encode mRNAs (mDNAs, see Supplementary Table S4 for the mRNA sequences) were prepared by primer extension and PCR (see Supplementary Table S5 for the primers used). The resulting mDNAs were purified by phenol/chloroform extraction and ethanol precipitation, and then dissolved in 10 μl of water. The concentrations were measured by 8% native PAGE and ethidium bromide staining with 100 bp Quick-Load DNA Ladders (New England BioLabs) as reference.
mRNAs were prepared by in vitro transcription as follows: the 200 μl transcription mixture [40 mM Tris·HCl (pH 8.0), 1 mM spermidine, 0.01% (v/v) Triton X-100, 10 mM DTT, 30 mM MgCl2, 5 mM each NTPs, 30 mM KOH, 10% (v/v) DNA template prepared above and 120 nM T7 RNA polymerase] was incubated at 37°C overnight. The transcription mixture was mixed with MnCl2 (100 mM, 4 μl) and RQ1 RNase-Free DNase (1 U/μl, 1 μl, Promega), and incubated at 37°C for 30 min. The resultant mRNA was precipitated by isopropanol and dissolved in water. The mRNA transcript was purified by 8% denaturing PAGE and ethanol precipitation, and then dissolved in 10 μl water. The concentrations of mRNA were measured by A260 of a 10-fold diluted solution.
Synthesis of aminoacyl-tRNAs by flexizymes
Amino acids activated with an appropriate ester group (Phe-CME, Tyr-CME, Ser-DBE, MeG-DBE, MeS-DBE, MeA-DBE, MeF-CME, MeL-DBE, MeM-DBE, MeT-DBE, MeY-CME, MeD-DBE, MeV-DBE, MeNl-DBE, MeYm-CME, MeNv-DBE, AcK-DBE and ClAcY-CME; CME: cyanomethyl ester; DBE: 3,5-dinitrobenzyl ester) were synthesized as previously reported (39,43,44).
Aminoacyl-tRNAs were prepared by the following procedure: 12 μl HEPES–KOH buffer (pH 7.5, 83 mM) containing 42 μM tRNA and 42 μM flexizyme (eFx for CME-activated amino acids and dFx for DBE-activated amino acids) was heated at 95°C for 2 min and cooled to 25°C over 5 min. MgCl2 (3 M, 4 μl) was added and the mixture was incubated at 25°C for 5 min. The reaction was initiated by addition of each activated amino acid substrate in DMSO (25 mM, 4 μl) and incubated on ice for acylation (incubation time: 2 h for Phe-CME, Tyr-CME, MeG-DBE, MeA-DBE, AcK-DBE, and ClAcY-CME; 6 h for Ser-DBE, MeS-DBE, MeF-CME, MeL-DBE, MeM-DBE, and MeYm-CME; 10 h for MeY-CME; and 24 h for MeT-DBE, MeD-DBE, MeV-DBE, MeNl-DBE and MeNv-DBE). After acylation, the reaction was quenched by addition of 80 μl of 0.3 M sodium acetate (pH 5.2), and the RNA was precipitated by ethanol. The pellet was rinsed twice with 70% ethanol containing 0.1 M sodium acetate (pH 5.2), and once with sole 70% ethanol. The resulting aminoacyl-tRNA was dissolved in 1 mM sodium acetate (pH 5.2) just before addition to in vitro translation reaction mixture.
For translation of mRNA2 and 3 (Figure 4 and 5), the method to prepare multiple MeAA-tRNAs was changed as follows: after quenching the flexizyme-catalyzed acylation with 0.3M sodium acetate (pH 5.2), all reaction solutions were mixed into one tube and precipitated by ethanol. The pellet was rinsed twice with 70% ethanol containing 0.1 M sodium acetate (pH 5.2), and once with 70% ethanol. The resulting aminoacyl-tRNA was dissolved in 1 mM sodium acetate (pH 5.2) just before addition to in vitro translation reaction mixture. The concentration of aminoacyl-tRNA was adjusted as follows: 0.25 μl of the aminoacyl-tRNA solution was diluted 5000-fold with 1250 μl of 1 mM sodium acetate (pH 5.2) and the concentrations of the total RNA including tRNA and flexizyme were measured by A260 of the diluted solution. The concentration of aminoacyl-tRNA was calculated based on the total RNA concentration and the flexizyme-catalyzed acylation efficiencies (Supplementary Table S6), the concentration of each MeAA-tRNA was adjusted to 10 μM. For the synthesis of a macrocyclic peptide, ClAcY-tRNAIni was prepared separately and its concentration was adjusted to 25 μM without considering the flexizyme-catalyzed acylation efficiency.
Figure 4.

Expression of an N-methyl-peptide containing 9 MeAAs and 15 pAAs. (A) Affinities of MeAA-tRNA#X to EF-Tu using all-#2, all-#4, and uniform sets. The orange bands indicate the range of ΔG values of pAA-tRNAs determined in Figure 2A (−8.0 to −9.4 kcal/mol). (B) Schematic representation of the EF-Tu affinity-tuning strategy. An appropriate combination of MeAA and tRNA#2–4 generates uniform affinities of MeAA-tRNA#Xs to EF-Tu comparable to canonical pAA-tRNAs. (C) The codon table reprogrammed with 9 MeAAs and 15 pAAs used in this study. (D) Sequences of mRNA2 and P2 peptide. (E) MALDI-TOF-MS analysis of P2 expressed using each set of MeAA and tRNA#X pairs. Arrowhead shows the corresponding monoisotopic peak of P2 or P2*. MALDI-TOF mass spectrum of P2-pAA is also shown.
Figure 5.

Expression of a macrocyclic peptide containing 9 MeAAs and 14 pAAs. (A) Sequences of mRNA3 and P3 peptide. (B) Structure of P3 closed via thioether bond. (C) MALDI-TOF MS analysis of P3 expressed with the uniformed set. Arrowhead shows the corresponding monoisotopic peak of P3.
Measurement of the efficiency of flexizyme-catalyzed aminoacylation
The pellet of ethanol-precipitated 50 pmol tRNA pre-charged with pAA or npAA was dissolved in 0.52 μl of 10 mM sodium acetate (pH 5.2) and mixed with 5.0 μl of acid PAGE loading buffer [83% (v/v) formamide, 150 mM sodium acetate (pH 5.2), and 10 mM EDTA]. The solution was loaded on an acid denaturing polyacrylamide gel [12% (w/v) acrylamide/bisacrylamide (19:1), 8M urea, and 50 mM sodium acetate (pH 5.2)] and electrophoresis was conducted at 300 V (approximately 10 V/cm) for 20 h. The gel was stained with ethidium bromide and analyzed using a Typhoon FLA 7000 (GE Healthcare). Aminoacylation efficiency was calculated based on the band intensities of aminoacyl-tRNA (A) and free tRNA (T) and is presented as (A)/[(A) + (T)]. As flexizymes recognize only the 3′-terminal CCA consensus sequence, the observed acylation efficiencies were similar independent of the tRNA species (45). The mean acylation values for each amino acid are listed in Supplementary Table S6.
Purification of EF-Tu
BL21(DE3) pLysS cells were transformed with pET21a-tufA and grown in 6 l of LB medium containing 100 μg/ml ampicillin, 20 μg/ml chloramphenicol, and 5% (w/v) glucose at 37°C until the OD600 reached 0.4. The expression of EF-Tu was induced with 0.5 mM IPTG and the cells were incubated at 37°C for additional 3 h. The cells were harvested and resuspended in 100 ml of buffer A [20 mM Tris–HCl (pH 7.6), 20 mM imidazole, 300 mM NaCl, 10 μM GTP, 1 mM β-mercaptoethanol] with 0.1 mg/ml PMSF. The cells were sonicated on ice for 10 min and the lysate was centrifuged (13 000 rpm, 4°C, for 15 min, CR22GIII equipped with an R15A rotor, Hitachi Koki). The supernatant was filtered thorough Minisart prefilter-GF and Minisart 0.45 μm syringe filters (Sartorius) and then applied to a 5 ml HisTrap HP column at 4°C (GE Healthcare) equipped on an AKTA avant 25 (GE Healthcare). The column was washed with 20 column volumes of buffer A, and the protein was eluted with a linear gradient from buffer A to buffer B [20 mM Tris–HCl (pH 7.6), 250 mM imidazole, 150 mM NaCl, 50 mM KCl, 10 μM GTP, 1 mM β-mercaptoethanol] over 20 column volumes. The fractions containing the protein were combined and dialyzed in 2 l of buffer C [10 mM Tris–HCl (pH 7.6), 50 mM KCl, 10 μM GTP, 1 mM DTT] with 3.5K MWCO membrane. The concentration of EF-Tu was measured by protein assay kit.
Radiolabeling of 3′-terminus of tRNA
The 3′-terminus of a tRNA was radiolabeled as follows: 7.5 μl of 16.7 μM tRNA lacking 3′-terminal adenosine was incubated at 80°C for 3 min, and then put on ice for 10 min. The 25 μl of CCA-adding reaction mixture {120 mM Gly-NaOH (pH 9.0), 75 mM MgCl2, 30 mM DTT, 5 μM tRNA lacking 3′-terminal adenosine prepared above, 10 mM non-radiolabeled ATP, 0.67 μM [α-32P]-ATP and 200 nM CCA-adding enzyme} was incubated at 37°C for 20 min. The tRNA was purified by phenol/chloroform extraction, Micro Bio-Spin 30 column (Bio-Rad), and ethanol precipitation. The tRNA pellet was dissolved in 5 μl water and the concentrations of tRNA were measured by A260 of a 100-fold diluted solution.
Quantification of the affinity between EF-Tu and aminoacyl-tRNA (RNase A protection assay)
Aminoacyl-tRNA was prepared as described above except that the acylation reaction was conducted using a mixture of 3′-radiolabeled tRNA (10%) and non-radiolabeled tRNA (90%). The resulting pellet of aminoacyl-tRNA was dissolved in 3.2 μl of 10 mM sodium acetate (pH 5.2). The concentration of aminoacyl-tRNA was adjusted as follows: 0.7 μl of the aminoacyl-tRNA solution was diluted 100-fold with 69.3 μl of 10 mM sodium acetate (pH 5.2) and the concentrations of the total RNA including tRNA and flexizyme were measured by A260 of the diluted solution. The concentration of aminoacyl-tRNA was calculated based on the total RNA concentration and the flexizyme-catalyzed acylation efficiency (Supplementary Table S6). The concentration of aminoacyl-tRNA was adjusted to 2.0 μM by adding an appropriate volume of 10 mM sodium acetate (pH 5.2). Just prior to use, the concentration of aminoacyl-tRNA was adjusted to 100 nM by mixing 2.0 μM aminoacyl-tRNA solution with buffer D [50 mM HEPES–KOH (pH 7.6), 100 mM KOAc, 12 mM Mg(OAc)2, 1 mM GTP, 1 mM DTT, 20 mM creatine phosphate, 2 mM spermidine, 3 mM phosphoenol pyruvate and 0.1 μg/μl pyruvate kinase from rabbit muscle (Sigma)].
EF-Tu was incubated in buffer D at 37°C for 30 min in order to be fully converted to a GTP-bound form. 12 μl samples containing different concentrations of EF-Tu, typically ranging from 1.5 nM to 25 μM, were prepared by twofold serial dilution on ice. 9.6 μl of the EF-Tu solution was mixed with 2.4 μl of 100 nM aminoacyl-tRNA and incubated on ice for 20 min. Under equilibrium binding condition, 10 μl of each solution was mixed with 1 μl of 1 mg/ml RNase A (Sigma) on ice to digest the tRNA not bound to EF-Tu. After 20 s, the digestion was quenched by addition of 50 μl of 10% (v/v) trichloroacetic acid (TCA) containing 0.1 mg/ml unfractionated yeast tRNA on ice. The precipitate was filtered using 0.45 μm pore-size nitrocellulose membrane assembled in Bio-Dot microfiltration apparatus (Bio-Rad), and washed by six times of 200 μl each 5% (v/v) TCA. The membrane was soaked in 95% (v/v) ethanol for 5 min and dried. The dried membrane was analyzed by autoradiography using a Typhoon FLA 7000 (GE Healthcare). To correct for the background signal derived from any aminoacyl-tRNA that may remain after the 20 s treatment with RNase A, a no EF-Tu control was analyzed in parallel and its radioactivity was subtracted from the experimental data. The resultant radioactivity was converted to the concentration of ternary complex using the conversion factor determined by the calibration aminoacyl-tRNA samples in buffer D containing 20, 4, 0.8 and 0.16 nM tRNA. Equilibrium dissociation constants were determined by fitting the binding data to the following equation (1) using KaleidaGraph program (Hulinks). As the concentration of aminoacyl-tRNA varied slightly in every experiment due to its multistep preparation, both KD and the aminoacyl-tRNA concentration were set as variables in the fitting analysis.
The KD equation is defined as follows, where aatRNA, EFTu, and aatRNA·EFTu denote aminoacyl-tRNA, EF-Tu, and their complex, respectively.
 |
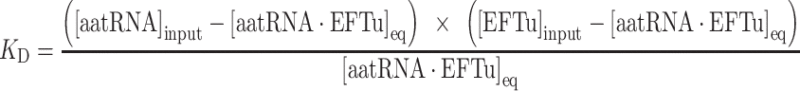 |

The ΔG value was calculated based on determined KD value using the following equation (2).
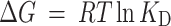 |
(2) |
In vitro translation
The reconstituted cell-free translation system (46) contained all necessary components for translation except for RF1. Concentrations of translation components were optimized in our previous studies as follows (25): 50 mM HEPES–KOH (pH 7.6), 100 mM KOAc, 2 mM GTP, 2 mM ATP, 1 mM CTP, 1 mM UTP, 20 mM creatine phosphate, 12–15 mM Mg(OAc)2, 2 mM spermidine, 1 mM DTT, 100 μM 10-formyl-5,6,7,8-tetrahydrofolic acid, 1.2 μM ribosome, 2.7 μM IF1, 0.4 μM IF2, 1.5 μM IF3, 10 μM EF-Tu, 10 μM EF-Ts, 0.26 μM EF-G, 0.25 μM RF2, 0.17 μM RF3, 0.5 μM RRF, 0.6 μM MTF, 4 μg/ml creatine kinase, 3 μg/ml myokinase, 0.1 μM pyrophosphatase, 0.1 μM nucleotide diphosphate kinase, 0.1 μM T7 RNA polymerase, 0.73 μM AlaRS, 0.03 μM ArgRS, 0.38 μM AsnRS, 0.13 μM AspRS, 0.02 μM CysRS, 0.06 μM GlnRS, 0.23 μM GluRS, 0.09 μM GlyRS, 0.02 μM HisRS, 0.4 μM IleRS, 0.04 μM LeuRS, 0.11 μM LysRS, 0.03 μM MetRS, 0.68 μM PheRS, 0.16 μM ProRS, 0.04 μM SerRS, 0.09 μM ThrRS, 0.03μM TrpRS 0.02 μM TyrRS, 0.02 μM ValRS, 1.5 mg/ml native Escherichia coli tRNA mixture (Roche), 200 μM each 3–20 pAAs (see Supplementary Table S7 for the amino acids added in each experiment), 10–15 μM each tRNAAsnE2s pre-charged with npAAs, and either 0.25 μM mDNA or 6.0 μM mRNA. The details of conditions used in each experiment are described in Supplementary Table S8.
The translation products were analyzed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) and/or autoradiography after tricine-SDS-PAGE. For MALDI-TOF MS analysis, 1.0–5.0 μl of the translation reaction mixture was incubated at 37°C for 3–30 min (see Supplementary Table S8 for the volume and reaction time in each experiment). After the reaction, the mixture was diluted with the same volume of FLAG-purification buffer [100 mM Tris–HCl (pH 7.6), 300 mM NaCl]. The expressed peptide was immobilized on anti-FLAG M2 agarose beads (Sigma) by incubating at 25°C for 1 h. After washing the beads with 25 μl of wash buffer [50 mM Tris–HCl (pH 7.6), 150 mM NaCl], the immobilized peptides were eluted with 15 μl of 0.2% TFA. Following purification, the peptide was desalted with SPE C-tip (Nikkyo Technos) and eluted with 1 μl of 80% acetonitrile, 0.5% acetic acid solution 50% saturated with the matrix (R)-cyano-4-hydroxycinnamic acid (Bruker Daltonics). MALDI-TOF MS measurements were performed using a ultrafleXtreme (Bruker Daltonics) under reflect/positive mode and externally calibrated with peptide calibration standard II (Bruker Daltonics) and/or protein calibration standard I (Bruker Daltonics). For the quantification analysis, the translation reaction was performed in the presence of 50 μM [14C]-Asp instead of 200 μM cold Asp. The translation product was analyzed by 15% tricine-SDS-PAGE and autoradiography using a Typhoon FLA 7000 (GE Healthcare) without FLAG purification. The amount of peptide products was quantified based on the relative band intensity of the peptide product to the sum intensities present in the lane, that is, the sum intensities of unreacted free Asp and peptide products.
RESULTS AND DISCUSSION
MeAA-tRNAs lack sufficient EF-Tu affinity
We first quantified the affinities of MeAA-tRNAs to EF-Tu (ΔG value) and compared them with those of canonical pAA-tRNAs. The quantification was conducted by means of RNase A protection assay (47) (see methods section for the details). Three canonical pAA-tRNAs (Phe-tRNAPheGAA, Tyr-tRNATyrGUA and Ser-tRNASerCGA) were prepared using flexizymes (25). As expected, all three canonical pAA-tRNAs exhibited similar EF-Tu affinities, ranging from −8.0 to −9.4 kcal/mol (Figure 2A). Although these values were slightly weaker than those previously reported (from −9.5 to −10.5 kcal/mol) (6), this could be attributed to the differences in buffer composition or the presence of the His6 tag at the C-terminus of the EF-Tu used in this assay. For quantification of the affinities of MeAA-tRNAs to EF-Tu, we prepared 13 representative MeAA-tRNAAsnE2GACs [MeG, MeA, MeS, MeT, MeF, MeY, MeV, MeL, MeM, MeD, MeNv (N-methylnorvaline), MeNl (N-methylnorleucine) and MeYm (N-methyl-p-methoxyphenylalanine), Figure 2B] by means of the flexizyme technology (25,38,39). Most of the MeAA-tRNAAsnE2GACs showed undetectably weak affinities to EF-Tu with two exceptions where the affinity of MeG-tRNAAsnE2GAC (−8.4 kcal/mol) and MeS-tRNAAsnE2GAC (−8.4 kcal/mol) are in the affinity range observed for the natural pairs (from −8.0 to −9.4 kcal/mol), while that of MeA-tRNAAsnE2GAC (−7.0 kcal/mol) was measurable but yet out of the range (Figure 2A).
Figure 2.

Weak affinities of npAA-tRNAs to EF-Tu correlating with the inaccurate translation of npAA-containing peptides. (A) The affinities of canonical pAA-tRNAs and npAA-tRNAs to EF-Tu. Asterisk (*) indicates undetectably weak affinity (ΔG > −6 kcal/mol). Error bar indicates the fitting error. (B) MeAAs and ϵ-N-acetyllysine used in this study. MeG, N-methylglycine; MeS, N-methylserine; MeA, N-methylalanine; MeF, N-methylphenylalanine; MeL, N-methylleucine; MeM, N-methylmethionine; MeT, N-methylthreonine; MeY, N-methyltyrosine; MeD, N-methylaspartic acid; MeV, N-methylvaline; MeNl, N-methylnorleucine; MeNv, N-methylnorvaline; MeYm, N-methyl-p-methoxyphenylalanine; and AcK, ϵ-N-acetyllysine. (C) Examination of the translation fidelity of peptides containing each of MeAAs or AcK. In each experiment, the mRNA1 containing GUC codon was translated in the FIT system containing npAA-tRNAAsnE2GAC of interest. Each dagger peak (†) corresponds to a byproduct containing Ile in place of npAA.
In the cocrystal structure of the ternary complex (Thermus aquaticus EF-Tu·E coli Phe-tRNAPhe·GTP), the amino group of Phe esterified on the tRNA forms a hydrogen bond with Asn285 of EF-Tu, defining the orientation of Phe in the binding pocket (Figure 1B) (3). This amino group is tightly surrounded by the pocket allowing no space for additional methylation. Thus, the methylation could cause a steric clash with EF-Tu and disrupt the hydrogen bond formation, resulting in the loss of affinity for most of the MeAA-tRNAAsnE2GACs, since the original T-stem of tRNAAsn has relatively weak binding affinity (4). The three exceptions, MeG-tRNAAsnE2GAC, MeS-tRNAAsnE2GAC, and MeA-tRNAAsnE2GAC might alleviate such a steric clash by virtue of their relatively small sidechain.
To assess the impact of weak binding affinity between MeAA-tRNAs and EF-Tu on the translation efficiency, four kinds of MeAA-tRNAAsnE2GACs (MeG, MeS, MeA and MeL) were used for ribosomal synthesis of a model peptide with a site-specific MeAA incorporation (Figure 2C) using the Flexible In-vitro Translation (FIT) system (25). The FIT system consists of a reconstituted custom-made translation system of E. coli (46) supplemented with synthetic tRNAs pre-charged with npAAs by flexizyme catalysis (43). In the model mRNA template 1 (mRNA1), GUC (Val) codon was reprogrammed to MeAAs by excluding the corresponding Val from the FIT system and instead supplementing one of the MeAA-tRNAAsnE2GAC molecules. The synthesized peptide (P1-npAA) was conventionally purified by anti-FLAG M2 affinity agarose gel through its C-terminal FLAG tag (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) (30) and analyzed by MALDI-TOF MS to determine the incorporation fidelity of the genetic code reprogramming. When MeG-tRNAAsnE2GAC or MeS-tRNAAsnE2GAC with sufficient affinity to EF-Tu was used, the expected peptide containing MeG or MeS (P1-MeG or P1-MeS) was observed as a single major peak in MALDI-TOF MS. On the other hand, in the case of MeA-tRNAAsnE2GAC that has a detectable but weaker affinity to EF-Tu, or MeL-tRNAAsnE2GAC that has an undetectably weak affinity to EF-Tu, a peptide containing Ile in place of MeAA (P1-Ile, see also Supplementary Figure S1 for the identity of the misincorporated amino acid) was observed in MALDI-TOF MS in addition to the desired peptide (P1-MeA or P1-MeL). This misincorporation of Ile (P1-Ile) at GUC codon likely occurred by misreading of endogenous Ile-tRNAIle on GUC codon of the mRNA, indicating that EF-Tu cannot effectively deliver such a MeAA-tRNAAsnE2GAC to the ribosome A site.
The above results demonstrate that the affinities of many MeAA-tRNAAsnE2GAC molecules to EF-Tu were significantly weaker than the mean value of canonical pAA-tRNAs, which causes ineffective reprogramming of the genetic code using MeAA. To demonstrate the universality of such a relationship between EF-Tu affinity and reprogramming fidelity, we tested another npAA, ϵ-N-acetyllysine (AcK) in addition to MeAAs. Indeed, AcK-tRNAAsnE2GAC showed poor affinity to EF-Tu and the reprogrammed translation using AcK-tRNAAsnE2GAC also generated P1-Ile as well as the desired peptide P1-AcK (Figure 2A and C). This indicates that the relationship is applicable to other npAAs in a similar manner.
Reinforcing the affinities of MeAA-tRNAs to EF-Tu
Since poor affinities of MeAA-tRNA to EF-Tu observed above might have caused the drastic decrease in the efficiency of their elongation, we have decided to reinforce their affinities. The Uhlenbeck group has extensively investigated the effects of single base-pair substitutions in the T-stem region on ΔG value, reporting that the T-stem base-pair sequences contribute to the affinity to EF-Tu independently from the kind of amino acids on tRNA. Furthermore, they found that the total ΔG value mediated by the T-stem region can be predicted simply by summing up each contribution of the base-pair substitutions (9,48). Based on their findings, we rationally designed three T-stem variants #1, #3, and #4, while the original T-stem of tRNAAsnE2 was defined as #2 (Figure 3A and Supplementary Figure S2). The tRNAAsnE2 whose T-stem was replaced with #X is denoted as tRNA#X. The predicted ΔΔG value of each T-stem variant compared to the original #2 was calculated as follows: +1.4 kcal/mol for #1, −0.6 kcal/mol for #3, and −1.2 kcal/mol for #4. The EF-Tu affinities of these MeAA-tRNA variants were designed to increase as the T-stem number increases from #1 to #4.
Figure 3.

Reinforcement of EF-Tu affinity of Phe-tRNA and npAA-tRNAs by T-stem engineering. (A) Sequence of the original tRNAAsnE2 (#2) and T-stem variants #1–4. The affinities of tRNA #1–4 were designed to increase as the T-stem number increases from #1 to #4. (B–E) Quantification of the EF-Tu affinities of Phe-tRNAGAC#1–4 (B, C) and MeF-tRNAGAC#2–4 (D, E). Schematic representation of RNase A protection assay, the observed fraction of the ternary complex with the fitting curve to determine the KD value (B, D), and the calculated ΔG value (C, E). Asterisk (*) indicates undetectably weak affinity (ΔG > −6 kcal/mol). Error bar indicates the fitting error. (F–J) The EF-Tu affinities of npAA-tRNA#1–4 and MALDI-TOF MS of P1-npAA examined for MeG (F), MeS (G), MeA (H), MeL (I) and AcK (J). Each dagger peak (†) corresponds to a byproduct containing Ile in place of npAA.
The feasibility of this approach was evaluated using Phe-tRNAGAC#1–4 and MeF-tRNAGAC#2–4. As expected, the affinity of Phe-tRNAGAC#X was enhanced as the T-stem number increased, that is, ΔG = −7.0, −8.7, −9.4 and −9.7 kcal/mol for #1–4, respectively (Figure 3B and C). The affinity of MeF-tRNAGAC#2 to EF-Tu was undetectable but the T-stem replacement to #3 and #4 enhanced the affinity to −7.9 and −9.0 kcal/mol, respectively (Figure 3D and E). A control experiment using uncharged tRNAGAC#1–4 showed that the tRNA itself did not bind to EF-Tu, which is consistent with the notion that EF-Tu selectively binds to aminoacylated tRNAs regardless of the T-stem sequence (Supplementary Figure S3). We also evaluated the EF-Tu affinities for the other twelve distinct MeAAs (MeG, MeS, MeA, and MeL in Figure 3F–I, left panels; MeM, MeT, MeY, MeD, MeV, MeNv, MeNl and MeYm in Supplementary Figure S4) with the series of tRNAs, demonstrating that their reinforcement of EF-Tu affinity can be achieved with all MeAA-tRNAs. Interestingly, in most MeAA cases the affinities measured with T-stem #3 and #4 were stronger than the predicted value based on #2, whereas those of Phe-tRNAs and MeG-tRNAs were close to the predicted value. This suggests that the effect of the affinity reinforcement becomes more pronounced when MeAA-tRNA#2 exhibits undetectably poor affinity. We further evaluated four MeAAs (MeG, MeS, MeA and MeL) and monitored the reprogramming fidelity in expression of P1-MeG, P1-MeS, P1-MeA and P1-MeL using tRNAGAC#1–4 charged with MeG, MeS, MeA, and MeL, respectively (Figure 3F–I, right panels). Consequently, the desired peptide was expressed as the sole product only when the affinity of MeAA-tRNA to EF-Tu was sufficiently reinforced (Figure 3F #2–4, 3G #2–4, 3H #3–4 and 3I #4). In contrast, when the affinity of MeAA-tRNA to EF-Tu was insufficient or undetectable, the expression fidelity of the desired P1-MeAA was poor, giving a peak of P1-Ile (Figure 3F #1, 3G #1, 3H #1–2 and 3I #1–3). It should be noted that MeL was reported as a very poor substrate (39), but P1-MeL was cleanly expressed by using MeL-tRNAGAC#4 (Figure 3I #4). These results clearly show that the reprogramming fidelity can be significantly improved by reinforcing the affinity of MeAA-tRNA to EF-Tu. A similar trend was also observed in expression of P1-AcK, demonstrating the versatility of this strategy in introducing npAAs (Figure 3J).
Expression of N-methyl-peptides by EF-Tu affinity-tuned MeAA-tRNAs
Having the affinity-tuned MeAA-tRNA#1–4 to EF-Tu, we next expressed an N-methyl-peptide containing nine distinct MeAAs using three sets of tRNA: all-#2 set, all-#4 set, and uniform set (Figure 4A). In the all-#2 set, all nine MeAAs were charged onto tRNA#2 with the original tRNAAsnE2 T-stem. In the all-#4 set, all nine MeAAs were charged onto tRNA#4 with the strongest T-stem to simply maximize the EF-Tu affinities. In contrast, the uniform set is a combination of each MeAA and appropriately chosen T-stem with optimal EF-Tu affinities as follows: #2 for MeS and MeG; #3 for MeA, MeNv, MeY and MeF; and #4 for MeYm, MeNl and MeV. In this set, the affinities of MeAA-tRNAs to EF-Tu (ΔG) were tuned in the range of −7.9 to −8.8 kcal/mol, which is comparable to the previously determined ΔG values of pAA-tRNAs (−8.0 to −9.4 kcal/mol, the orange bands in Figure 4A). In this range, EF-Tu can bind to all pAA-tRNAs and MeAA-tRNAs with uniform affinities (Figure 4B). We prepared a FIT system where Phe, Leu, Ile, Val, and Ala were omitted and their vacant codons were reprogrammed with nine MeAAs by the addition of MeG-tRNACAA#X, MeS-tRNAGAG#X, MeA-tRNACGC#X, MeNv-tRNACAC#X, MeY-tRNAGAU#X, MeF-tRNAGGC#X, MeYm-tRNAGAA#X, MeNl-tRNACAG#X and MeV-tRNAGAC#X (Figure 4C) by means of the artificial codon box division methodology (49) to express a linear 32-mer model peptide P2 containing 9 MeAAs (Figure 4D).
The peptide P2 expressed in the FIT system supplemented with a tRNA set (all-#2, all-#4, or uniform set) was analyzed by MALDI-TOF MS (Figure 4E). The exclusive use of the all-#2 set yielded a set of peaks corresponding P2 along with byproducts that have −28 and −14 m/z compared to P2. Interestingly, the exclusive use of the all-#4 set did not show a detectable peak of P2 at all. The calculated m/z value of P2 is 4019.97, but the monoisotopic m/z value of the observed peak in the all-#4 set was 4021.97 (P2*), indicating that the desired peptide P2 was not synthesized by the all-#4 set (Figure 4E). The P2* peak was also accompanied by various byproducts with −14, +14 or +16 m/z difference relative to the P2* peak. These byproducts could be attributed to misincorporations of a certain amino acid(s); for example, a misincorporation of MeNv instead of MeNl could result in −14 m/z difference; a misincorporation of MeNl instead of MeNv could result in +14 m/z difference; or a misincorporation of Ser instead of MeG could result in +16 m/z difference; and P2* itself, with +2 m/z difference from P2 could be derived from the combination of −14 and +16 m/z. It should be noted that a substantial amount of unidentified peptide byproducts was also observed when using the all-#2 or all-#4 set (Supplementary Figure S5B). In contrast, when the uniform set was tested, the desired peptide P2 was correctly synthesized as a nearly sole product, and the degree of byproducts was drastically suppressed, indicating that appropriately tuning to the uniform affinities is effective for the accurate synthesis of the N-methyl-peptide.
The aberrant translation observed when using the all-#2 set can be attributed to the insufficient affinities of the MeAA-tRNAs decreasing the fidelity of the multiple MeAA incorporation, as is the case with the single incorporation (Figure 3F–I). Notably, the inaccurate and inefficient translation observed for the all-#4 set suggests that unnecessary reinforcements of MeAA-tRNA affinity to EF-Tu can cause a disorder in the translation. As predicted, the uniform set has given the best expression in terms of fidelity and efficiency.
We also quantitatively evaluated the expression level of P2 by means of tricine-SDS-PAGE with autoradiographic detection of [14C]-Asp introduced in the C-terminal FLAG tag of the peptide. The use of the uniform set yielded the 0.48 μM P2 peptide, which was approximately 10% of the control peptide (P2-pAA) consisting of only 20 pAAs (Supplementary Figure S5C). However, this expression level is still sufficient for a practical use based on our result using the RaPID (Random nonstandard Peptides Integrated Discovery) selection where de novo macrocyclic peptide binders containing cyclic β-amino acids have been discovered against proteins (30). These results have shown that (i) a major cause of disorder in genetic code reprogramming was attributed to inappropriate affinities of MeAA-tRNAs to EF-Tu, (ii) unnecessary reinforcements of EF-Tu affinity for MeAA-tRNAs do not improve the yield and fidelity of expression of N-methyl-peptides and (iii) this affinity-tuning strategy is able to improve both the synthetic accuracy and translation efficiency of N-methyl-peptides.
Finally, we designed a macrocyclic peptide, P3, composed of 9 distinct MeAAs and 14 different kinds of pAAs. For macrocyclization, an N-chloroacetyl-l-tyrosine (ClAcY) at the N-terminus and a Cys residue at a downstream (24th) position were also installed and thereby they underwent spontaneously macrocyclization via a thioether bond (44,50), giving a 24-mer macrocycle (Figure 5A and B). To achieve the desired reprogramming, we used a FIT system that is deficient in Met, Phe, Leu, Ile, Val and Ala, enabling us to install ClAcY (by the addition of ClAcY-tRNAIni) and aforementioned 9 MeAAs by the uniform set of MeAA-tRNAs. As expected, the desired macrocyclic P3 was accurately expressed (Figure 5C) with an expression level of 0.37 μM (1.5 ng/μl).
CONCLUSION
Here, we have reported a new methodology to achieve the ribosomal synthesis of nonstandard peptides containing a rich variety of MeAAs. The inefficient incorporation of MeAAs is attributed to the two major reasons: impaired EF-Tu-mediated accommodation of the MeAA-tRNAs and their slow peptidyl transfer. Indeed, we have quantitatively demonstrated that the binding affinities (ΔG values) of MeAA-tRNAAsnE2s to EF-Tu are generally weaker than those of pAA-tRNAs. In vitro translation experiments using MeAA-tRNAAsnE2s with inadequate affinities indicate that the thermodynamic contributions of the EF-Tu affinities play a major role in the translation. Based on this knowledge, we have engineered the T-stem sequence of tRNA to tune the affinity of MeAA-tRNA to EF-Tu. This affinity-tuning strategy dramatically improves the expression of peptides containing multiple MeAAs with high fidelity and expression level, even though some MeAAs are consecutively and/or alternately placed in the single sequence. Consequently, we succeeded in assigning 24 distinct building blocks in the genetic code, where a set of 15 pAAs and 9 MeAAs, or 14 pAAs, 9 MeAAs and ClAcY, and expressing N-methyl-peptides consisting of these amino acids. The present data have exceeded the previous record of the genetic code reprogramming achieved to place 23 building blocks consisting of 20 pAAs and 3 npAAs (49,51).
Forster et al. kinetically determined the peptidyl-transfer rate where the initiation complex of ribosome and fMet-tRNA was incubated with pre-formed MeF-tRNA·EF-Tu complex, showing that the rate was approximately 8 000-fold slower than the natural pair (Phe-tRNA·EF-Tu) case (21). However, it is hard to generalize this notion to other MeAAs, since the efficiency of MeF incorporation into polypeptides was as high as Phe incorporation in our previous experiment (39). More importantly, when the incorporation of MeAA in a middle of sequence of longer peptides, the observed expression efficiency relies on not only the peptidyl-transfer step but also all steps of elongation. Our work undoubtedly shows that the uniform tuning of MeAA-tRNA affinities to EF-Tu is quite effective to improve their incorporation efficiency, giving the sufficient expression of such exotic peptides in terms of fidelity and yield for the practical application to the RaPID selection. Therefore, the most critical determinant for MeAA incorporation to the nascent peptide chain could be a step of delivery of MeAA-tRNA·EF-Tu complex to the A site, or a step of EF-Tu dissociation and MeAA-tRNA accommodation (10), or both steps.
In addition to MeAAs, we have also demonstrated that the incorporation of AcK was enhanced by engineering the T-stem, indicating that the substrate of this affinity-tuning methodology is applicable to other npAAs. In our most recent work aiming at enhancing the incorporation of d-, β- and γ-amino acids into nascent peptide chain, we logically devise a chimeric tRNAPro1E2 based on the T-stem of tRNA#3 (equal to tRNAGluE2) and D-arm of tRNAPro1 (27). In the case of the consecutive incorporation of such exotic amino acids, the EF-P recruitment by the use of tRNAPro1E2 was essential to enhance the efficiency (27–31). The knowledge established in the tRNA engineering efforts including the present work enables us to further expand the utility of exotic amino acids to the translation chemistry world. Particularly when the knowledge is integrated with the RaPID system, it will allow us to rapid and high-throughput screening of binding peptides to target proteins of interest using a massive library of randomized macrocyclic peptides, yielding therapeutically valuable peptides with improved pharmacological properties (30).
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yuuki Hayashi for providing purified EF-Tu. We also thank Renier Herman Pieter van Neer, Hayden Peacock, Koki Shimbara and Naoya Kawakami for proofreading of the manuscript.
Contributor Information
Yoshihiko Iwane, Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
Hiroyuki Kimura, Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
Takayuki Katoh, Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
Hiroaki Suga, Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for JSPS Fellows [26-9576 to Y.I.]; Grant-in-Aid for Scientific Research (B) [JP18H02080 to T.K.]; Grant-in-Aid for Specially Promoted Research [JP20H05618]; Human Frontier Science Program [RGP0015/2017-302 to H.S.]. Funding for open access charge: Japan Society for the Promotion of Science [JP20H05618].
Conflict of interest statement. None declared.
REFERENCES
- 1. Schmeing T.M., Ramakrishnan V.. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009; 461:1234–1242. [DOI] [PubMed] [Google Scholar]
- 2. Pape T., Wintermeyer W., Rodnina M.. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999; 18:3800–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nissen P., Kjeldgaard M., Thirup S., Polekhina G., Reshetnikova L., Clark B.F.C., Nyborg J.. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP Analog. Science. 1995; 270:1464–1472. [DOI] [PubMed] [Google Scholar]
- 4. Asahara H., Uhlenbeck O.C.. The tRNA specificity of Thermus thermophilus EF-Tu. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3499–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sanderson L.E., Uhlenbeck O.C.. Directed mutagenesis identifies amino acid residues involved in elongation factor Tu binding to yeast Phe-tRNAPhe. J. Mol. Biol. 2007; 368:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. LaRiviere F.J., Wolfson A.D., Uhlenbeck O.C.. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001; 294:165–168. [DOI] [PubMed] [Google Scholar]
- 7. Asahara H., Uhlenbeck O.C.. Predicting the binding affinities of misacylated tRNAs for Thermus thermophilus EF-Tu·GTP. Biochemistry. 2005; 44:11254–11261. [DOI] [PubMed] [Google Scholar]
- 8. Dale T., Sanderson L.E., Uhlenbeck O.C.. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry. 2004; 43:6159–6166. [DOI] [PubMed] [Google Scholar]
- 9. Schrader J.M., Chapman S.J., Uhlenbeck O.C.. Understanding the sequence specificity of tRNA binding to elongation factor Tu using tRNA mutagenesis. J. Mol. Biol. 2009; 386:1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrader J.M., Chapman S.J., Uhlenbeck O.C.. Tuning the affinity of aminoacyl-tRNA to elongation factor Tu for optimal decoding. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:5215–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uhlenbeck O.C., Schrader J.M.. Evolutionary tuning impacts the design of bacterial tRNAs for the incorporation of unnatural amino acids by ribosomes. Curr. Opin. Chem. Biol. 2018; 46:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stanzel M., Schön A., Sprinzl M.. Discrimination against misacylated tRNA by chloroplast elongation factor Tu. Eur. J. Biochem. 1994; 219:435–439. [DOI] [PubMed] [Google Scholar]
- 13. Wang L., Schultz P.G.. Expanding the genetic code. Angew. Chem. Int. Ed. 2005; 44:34–66. [DOI] [PubMed] [Google Scholar]
- 14. Chin J.W. Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem. 2014; 83:379–408. [DOI] [PubMed] [Google Scholar]
- 15. Dumas A., Lercher L., Spicer C.D., Davis B.G.. Designing logical codon reassignment – expanding the chemistry in biology. Chem. Sci. 2015; 6:50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirose H., Tsiamantas C., Katoh T., Suga H.. In vitro expression of genetically encoded non-standard peptides consisting of exotic amino acid building blocks. Curr. Opin. Biotechnol. 2019; 58:28–36. [DOI] [PubMed] [Google Scholar]
- 17. Guo J., Melançon C.E. III, Lee H.S., Groff D., Schultz P.G.. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew. Chem. Int. Ed. 2009; 48:9148–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doi Y., Ohtsuki T., Shimizu Y., Ueda T., Sisido M.. Elongation factor Tu mutants expand amino acid tolerance of protein biosynthesis system. J. Am. Chem. Soc. 2007; 129:14458–14462. [DOI] [PubMed] [Google Scholar]
- 19. Gan R., Perez J.G., Carlson E.D., Ntai I., Isaacs F.J., Kelleher N.L., Jewett M.C.. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng. 2017; 114:1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park H.-S., Hohn M.J., Umehara T., Guo L.-T., Osborne E.M., Benner J., Noren C.J., Rinehart J., Söll D.. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011; 333:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pavlov M.Y., Watts R.E., Tan Z., Cornish V.W., Ehrenberg M., Forster A.C.. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liljeruhm J., Wang J., Kwiatkowski M., Sabari S., Forster A.C.. Kinetics of d. -amino acid incorporation in translation. ACS Chem. Biol. 2019; 14:204–213. [DOI] [PubMed] [Google Scholar]
- 23. Zhang B., Tan Z., Dickson L.G., Nalam M.N.L., Cornish V.W., Forster A.C.. Specificity of translation for N-alkyl amino acids. J. Am. Chem. Soc. 2007; 129:11316–11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ieong K.W., Pavlov M.Y., Kwiatkowski M., Ehrenberg M., Forster A.C.. A tRNA body with high affinity for EF-Tu hastens ribosomal incorporation of unnatural amino acids. RNA. 2014; 20:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goto Y., Katoh T., Suga H.. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011; 6:779–790. [DOI] [PubMed] [Google Scholar]
- 26. Achenbach J., Jahnz M., Bethge L., Paal K., Jung M., Schuster M., Albrecht R., Jarosch F., Nierhaus K.H., Klussmann S.. Outwitting EF-Tu and the ribosome: translation with d. -amino acids. Nucleic Acids Res. 2015; 43:5687–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katoh T., Iwane Y., Suga H.. Logical engineering of D-arm and T-stem of tRNA that enhances d. -amino acid incorporation. Nucleic Acids Res. 2017; 45:12601–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoh T., Suga H.. Ribosomal incorporation of consecutive β-amino acids. J. Am. Chem. Soc. 2018; 140:12159–12167. [DOI] [PubMed] [Google Scholar]
- 29. Katoh T., Suga H.. Ribosomal elongation of cyclic γ-amino acids using a reprogrammed genetic code. J. Am. Chem. Soc. 2020; 142:4965–4969. [DOI] [PubMed] [Google Scholar]
- 30. Katoh T., Sengoku T., Hirata K., Ogata K., Suga H.. Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic β-amino acids. Nat. Chem. 2020; 12:1081–1088. [DOI] [PubMed] [Google Scholar]
- 31. Katoh T., Suga H.. Ribosomal elongation of aminobenzoic acid derivatives. J. Am. Chem. Soc. 2020; 142:16518–16522. [DOI] [PubMed] [Google Scholar]
- 32. Katoh T., Iwane Y., Suga H.. tRNA engineering for manipulating genetic code. RNA Biol. 2018; 15:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Conradi R.A., Hilgers A.R., Ho N.F.H., Burton P.S.. The influence of peptide structure on transport across Caco-2 Cells. II. Peptide bond modification which results in improved permeability. Pharm. Res. 1992; 9:435–439. [DOI] [PubMed] [Google Scholar]
- 34. Haviv F., Fitzpatrick T.D., Swenson R.E., Nichols C.J., Mort N.A., Bush E.N., Diaz G., Bammert G., Nguyen A, Rhutasel N.S., Nellans H.N., Hoffman D.J., Johnson E.S., Greer J.. Effect of N-methyl substitution of the peptide bonds in luteinizing hormone-releasing hormone agonists. J. Med. Chem. 1993; 36:363–369. [DOI] [PubMed] [Google Scholar]
- 35. Chikhale E.G., Ng K.Y., Burton P.S., Borchardt R.T.. Hydrogen bonding potential as a determinant of the in vitro and in situ blood–brain barrier permeability of peptides. Pharm. Res. 1994; 11:412–419. [DOI] [PubMed] [Google Scholar]
- 36. Bockus A.T., Schwochert J.A., Pye C.R., Townsend C.E., Sok V., Bednarek M.A., Lokey R.S.. Going out on a limb: delineating the effects of β-branching, N-methylation, and side chain size on the passive permeability, solubility, and flexibility of sanguinamide A analogues. J. Med. Chem. 2015; 58:7409–7418. [DOI] [PubMed] [Google Scholar]
- 37. Nielsen D.S., Shepherd N.E., Xu W., Lucke A.J., Stoermer M.J., Fairlie D.P.. Orally absorbed cyclic peptides. Chem. Rev. 2017; 117:8094–8128. [DOI] [PubMed] [Google Scholar]
- 38. Yamagishi Y., Shoji I., Miyagawa S., Kawakami T., Katoh T., Goto Y., Suga H.. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011; 18:1562–1570. [DOI] [PubMed] [Google Scholar]
- 39. Kawakami T., Murakami H., Suga H.. Messenger RNA-programmed incorporation of multiple N-methyl-amino acids into linear and cyclic peptides. Chem. Biol. 2008; 15:32–42. [DOI] [PubMed] [Google Scholar]
- 40. Subtelny A.O., Hartman M.C.T., Szostak J.W.. Ribosomal synthesis of N-methyl peptides. J. Am. Chem. Soc. 2008; 130:6131–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kao C., Zheng M., Rüdisser S.. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999; 5:1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vasil’eva I.A., Ankilova V.N., Lavrik O.I., Moor N.A.. tRNA discrimination by T. thermophilus phenylalanyl-tRNA synthetase at the binding step. J. Mol. Recognit. 2002; 15:188–196. [DOI] [PubMed] [Google Scholar]
- 43. Murakami H., Ohta A., Ashigai H., Suga H.. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods. 2006; 3:357–359. [DOI] [PubMed] [Google Scholar]
- 44. Goto Y., Ohta A., Sako Y., Yamagishi Y., Murakami H., Suga H.. Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol. 2008; 3:120–129. [DOI] [PubMed] [Google Scholar]
- 45. Xiao H., Murakami H., Suga H., Ferré-D’Amaré A.R.. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature. 2008; 454:358–361. [DOI] [PubMed] [Google Scholar]
- 46. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T.. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001; 19:751–755. [DOI] [PubMed] [Google Scholar]
- 47. Pleiss J.A., Uhlenbeck O.C.. Identification of thermodynamically relevant interactions between EF-Tu and backbone elements of tRNA. J. Mol. Biol. 2001; 308:895–905. [DOI] [PubMed] [Google Scholar]
- 48. Schrader J.M., Uhlenbeck O.C.. Is the sequence-specific binding of aminoacyl-tRNAs by EF-Tu universal among bacteria. Nucleic Acids Res. 2011; 39:9746–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iwane Y., Hitomi A., Murakami H., Katoh T., Goto Y., Suga H.. Expanding the amino acid repertoire of ribosomal polypeptide synthesis via the artificial division of codon boxes. Nat. Chem. 2016; 8:317–325. [DOI] [PubMed] [Google Scholar]
- 50. Vinogradov A.A., Yin Y., Suga H.. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 2019; 141:4167–4181. [DOI] [PubMed] [Google Scholar]
- 51. Ohtsuki T., Manabe T., Sisido M.. Multiple incorporation of non-natural amino acids into a single protein using tRNAs with non-standard structures. FEBS Lett. 2005; 579:6769–6774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.