


Abstract
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 relies on sRNAs to coordinate expression of metabolic and virulence factors to colonize the host. Here, we focus on the sRNA, named MavR (metabolism and virulence regulator), that is conserved among pathogenic Enterobacteriaceae. MavR is constitutively expressed under in vitro conditions that promote EHEC virulence gene expression. Using MS2-affinity purification coupled with RNA sequencing, the eutR transcript was identified as a putative target of MavR. EutR is a transcription factor that promotes expression of genes required for ethanolamine metabolism as well as virulence factors important for host colonization. MavR binds to the eutR coding sequence to protect the eutR transcript from RNase E-mediated degradation. Ultimately, MavR promotes EutR expression and in turn ethanolamine utilization and ethanolamine-dependent growth. RNAseq analyses revealed that MavR also affected expression of genes important for other metabolic pathways, motility, oxidative stress and attaching and effacing lesion formation, which contribute to EHEC colonization of the gastrointestinal tract. In support of the idea that MavR-dependent gene expression affects fitness during infection, deletion of mavR resulted in significant (∼10- to 100-fold) attenuation in colonization of the mammalian intestine. Altogether, these studies reveal an important, extensive, and robust phenotype for a bacterial sRNA in host-pathogen interactions.
INTRODUCTION
To colonize a host, enteric pathogens must overcome a variety of hurdles, including competing for nutrients with the microbiota, coordinating expression of virulence traits, and evading host defenses. Enterohemorrhagic Escherichia coli O157:H7 (EHEC) is a preeminent example of a pathogen that precisely adapts to its target environment of the colon, with as few as 10–100 bacteria able to cause infection (1). To do this, EHEC exploits a variety of metabolites to sidestep nutritional competition (2) and then traverses the mucus layer to establish a niche at the relatively sterile epithelial border. At the epithelium, EHEC expresses a type three secretion system (T3SS) and effectors which results in intimate adherence to colonocytes and the formation of attaching and effacing (AE) lesions (3,4). AE lesions are characterized by the effacement of the microvilli and rearrangement of underlying host cytoskeleton resulting in the production of a pedestal-like structure beneath the bacterium (5). The locus of enterocyte effacement (LEE) pathogenicity island encodes the T3SS and most of the effectors required for AE lesion formation and is required for host colonization and overall pathogenesis (6–12).
EHEC coordinates expression of traits important for host colonization by sensing signals within the gastrointestinal (GI) tract and precisely controlling gene expression (13). To date, transcriptional regulation of EHEC gene expression is well-recognized to play a key role in niche adaptation (e.g. (14–17)). However, it is becoming increasingly appreciated that post-transcriptional regulation is a critical mechanism for EHEC to control expression of virulence traits (18,19). sRNAs are robust regulators that mediate post-transcriptional gene expression typically by base pairing to target mRNAs and repressing or promoting gene expression (20–22). In EHEC, sRNAs directly affect expression of transcripts encoding T3SS components (e.g., (23)). Moreover, sRNAs may be integrated into transcriptional networks by targeting transcription factors (24,25). To date, most knowledge of sRNAs in EHEC is derived from studies that were performed using nonpathogenic E. coli as the model organism. These studies have provided invaluable insights in understanding mechanisms of post-transcriptional regulation in the Enterobacteriaceae. Notably, during its evolution, EHEC acquired ∼1.34 Mb of unique DNA (26,27). Besides encoding canonical virulence traits, these pathogenicity islands harbor regulatory sRNAs that influence expression of core genes common to nonpathogenic E. coli and EHEC as well as EHEC-specific genes (19,24,28–30). However, the regulatory mechanisms and physiological importance of the majority of EHEC-specific sRNAs remains elusive.
The sRNA sRNA56 was originally identified by RNAseq as present in EHEC but absent in E. coli K-12 (28). Another study confirmed sRNA56 expression and location in an EHEC specific pathogenicity-island (19). Overexpression of sRNA56 influences expression of a T3SS apparatus protein (28), indicating this sRNA affects EHEC virulence. Here, we undertook a comprehensive analysis to characterize the sRNA56 regulon as well as the physiological importance to EHEC fitness and virulence. Based on our findings, we propose that sRNA56 be renamed MavR (metabolism and virulence regulator) and use this nomenclature throughout this paper. Using MS2-affinity purification coupled to RNA sequencing (MAPS) (31,32), we identified transcripts encoding several transcriptional regulators as putative MavR targets. Specifically, the eutR transcript was significantly enriched. EutR is the transcription factor that activates expression of genes required for ethanolamine (EA) metabolism in the Enterobacteriaceae. We provide a detailed mechanism in which MavR promotes EutR expression and EA utilization by stabilizing the eutR transcript. Notably, our findings indicate that MavR interacts with the coding sequence (CDS) of the eutR transcript to antagonize RNase E-mediated degradation. Most characterized sRNAs target the 5′ untranslated region (5′ UTR) of the target mRNA to regulate gene expression (33), whereas only a few sRNAs have been reported to bind the CDS, and the majority of these repress gene expression (24,34–36). Thus, these data reveal a comparatively less well-characterized mechanism of sRNA-based regulation. Moreover, to gain a global understanding of the functional implications of MavR-dependent gene expression, we performed RNAseq using in vitro conditions that recapitulate EHEC gene expression in vivo (15). These data revealed an extensive role for MavR in influencing expression of genes important for growth and virulence during infection. Specifically, deletion of mavR affected expression of genes encoding nutrient acquisition, motility (flagella), oxidative stress responses, and AE lesion formation. In agreement with these findings, MavR was required for robust colonization of the mammalian gastrointestinal tract.
MATERIALS AND METHODS
Bacterial growth conditions and strain construction
Strains, plasmids and oligonucleotides used in this study are listed in Supplementary Tables S1, S2 and S3, respectively. Bacteria were grown overnight in Lura-Bertani (LB) broth with antibiotics when appropriate (ampicillin [100 μg/ml], streptomycin [100 μg/ml], chloramphenicol [20 μg/ml] and kanamycin [50 μg/ml]). The mavR::cat, ΔmavR, ΔmavRΔeutR, ΔphoB, ΔmavRΔphoB, rneΔCTD and ΔmavR rneΔCTD strains were generated using Lamda-red recombination (37). To generate non-polar deletion strains the chloramphenicol resistance cassette was resolved with resolvase plasmid pCP20 (all deletion strains except mavR::cat). As indicated in the text, bacteria were grown in DMEM (Gibco) under aerobic (shaking, atmospheric oxygen) or microaerobic (static, in a 5% CO2 incubator) growth conditions.
Arabinose and IPTG inducible expression vectors were generated by using KpnI and HindIII to insert PCR products (eutR, phoB, flhD, flhC or mavR) into the pBAD/mycHis A or pUCP24 vector. pGEN-mavR was generated using HindIII and NheI to insert the PCR product into pGEN-MCS. pmavR-lux was generated using PmeI and SnaBI to insert the PCR product into pGEN-luxCDABE. Point mutants, pMS2 and pMS2-mavR were generated using the Q5 mutagenesis Kit (NEB). All deletions and plasmids were confirmed by Sanger sequencing. Strains transformed with pBAD/mycHis A vector were grown in the presence of 0.2% arabinose and strains transformed with pUCP24 vector were grown in the presence of 10 μM IPTG.
Growth curves
Overnight cultures were washed once with PBS, then diluted 1:100 into fresh low-glucose DMEM (Gibco) or M9 minimal medium (50 mM Na2HPO4·7H2O, 20 mM KH2PO4, 10 mM NaCl, 2 mM MgSO4, 100 μM CaCl2, 0.4% glycerol, 1 mg/ml thiamine and either 10 mM EA and 150 nM adenosylcobalamin [AdoCbl] or 10 mM NH4) and grown at 37°C aerobically. For in vitro co-culture experiments, overnight cultures were washed and then diluted 1:200 into the indicated medium. Cultures were plated on LB/streptomycin and LB/chloramphenicol when WT was determined to be at mid-exponential phase in single culture.
5′ RACE
5′ RACE was performed as described (38). Briefly, a mavR-specific reverse primer (mavR3′_qRT_R) was used to reverse transcribe to 5′ end of mavR from DNAse-treated RNA. The 5′ end of the transcript was polyadenylated, and the transcript was amplified with a standard primer containing an adapter sequence and a poly-T tract (QT and Qo) and a mavR -specific reverse primer (mavR5′_qRT_R). This product was further amplified using nested primers and sequenced by Sanger sequencing.
Northern blotting
Probes were generated using the T7 in vitro transcription kit (NEB) incorporating Bio-11-UTP (Fisher) and purified with NucAway spin columns (Invitrogen). Cultures were grown aerobically in M9 with ammonium and 0.2% arabinose to O.D.600 of 0.3. For bicyclomycin (BCM) assays, 50 μg/ml BCM or vehicle (ethanol) was added for 20 min. For stability assays, an aliquot was removed at time 0, rifampicin was added to a final concentration of 50 μg/ml and additional aliquots were removed at the indicated times. RNA was extracted using the PureLink RNA Mini Kit (Invitrogen). Total RNA concentration was normalized, and samples were mixed with 2× RNA loading buffer (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol, 0.5 mM EDTA). After heating to 65°C for 10 min, samples were electrophoresed through a 1.5% MOPS/agarose gel containing 1% formaldehyde. Bands were transferred to Zeta-probe membranes (BioRad) overnight by capillary transfer in 20× SSC. After UV crosslinking the RNA to the membrane, methylene blue was used to visualize the 23S and 16S rRNA bands. Membranes were probed overnight in NorthernMax Prehybridization/Hybridization Buffer (Invitrogen) at 68°C. The membranes were washed twice for 5 min with low stringency wash buffer (0.1% SDS, 2× SSC) and once for 15 min with medium stringency wash buffer (0.1% SDS, 1× SSC) and subjected to the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Scientific). Northern blots were visualized using a Gel Doc XR + Gel Documentation System (Bio-Rad).
Luminescence reporter assay
After strains were grown under the indicated condition, a 100 μl aliquot was transferred to a 96-well plate with opaque walls and a transparent bottom. Luminescence readings were accumulated for 10 s by a Wallac Victor 2 plate reader (Perkin Elmer) and normalized to the O.D.600.
RT-qPCR
RNA was extracted using the PureLink RNA Mini Kit (Invitrogen) and treated with DNase I (Sigma). RT-qPCR was performed as previously described (24). Briefly, 10 μl reactions containing Power SYBR green master mix (1×, Applied Biosystems), MultiScribe reverse transcriptase (2.5 units, Invitrogen), RNase inhibitor (2 units, Invitrogen), primer mix (0.05 μM each primer) and RNA (50 ng). Reactions were run using the one-step RT-qPCR program on the ABI 7500-FAST sequence detection system and software (Applied Biosystems). Primer sensitivity and specificity were verified by standard curve and melt curve analyses. cDNA generation and amplification were performed as follows: 1 cycle at 48°C for 30 min, 1 cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min. Two technical replicates were averaged for analysis by the relative quantification method in which CT values were normalized to the reference gene rpoA before calculating the ΔΔCT value.
RNAseq
WT and ΔmavR were grown in low glucose DMEM microaerobically for 6 h or aerobically to O.D.600 0.5. RNA was extracted using the PureLink RNA Mini Kit (Invitrogen) and DNase treated (Sigma). For the microaerobic cultures, triplicate RNA samples were pooled and sequenced by Novogene. For the aerobic cultures, triplicate (unpooled) RNA samples were barcoded and sequenced by the University of Maryland Genomic Research Core. After rRNA depletion, sequencing libraries were generated and sequenced on an Illumina Novaseq platform. Reads were trimmed and mapped to the EHEC Sakai (NCBI accession NC_002695.2) or EDL933 (NCBI accession NZ_CP008957.1) genome, respectively. HTSeq and the DEGSeq R package were used to determine the FPKM (Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced) and differential gene expression. Differential expression (FC > 2, P < 0.05) of selected genes from microaerobic growth conditions was confirmed by RT-qPCR.
MAPS
To purify the MS2 coat protein, a 1 L culture of BL21 (DE3) cells transformed with pHMM (39) was grown to O.D.600 0.7 and induced with 0.5 mM IPTG for 3 h. Cells were pelleted at 5000 RPM for 10 min at 4°C and resuspended in 25 ml of sodium phosphate buffer (50 mM sodium phosphate [pH 7.4], 300 mM NaCl, 10 mM imidazole) containing 250 μl of protease inhibitor cocktail (Sigma) and 100 μg of DNase I (Sigma). The cells were lysed by Emulsiflex C3 (Avestin) and the lysate was clarified by centrifugation at 15 000 RPM for 30 min. The lysate was incubated with Ni-NTA agarose (Qiagen) for 2 h with gentle rocking. The lysate/Ni-NTA agarose mixture was applied to a polypropylene column (Qiagen) and the flow through discarded. After washing the column thrice with 4 ml wash buffer (50 mM sodium phosphate pH 7.4, 300 mM NaCl, 25 mM imidazole), the protein was eluted in 5 ml elution buffer (50 mM sodium phosphate pH 7.4, 300 mM NaCl, 250 mM imidazole) and concentrated using Amicon centrifugal filter units (Millipore). The purified concentrated protein was diluted in 5 ml buffer A (20 mM Tris-HCl pH 8.0, 150 mM KCl, 1 mM MgCl2, 10% glycerol) and concentrated again.
MAPS was performed as previously described with a few modifications (31,32). Briefly, cultures were washed and resuspended in buffer A. The cells were lysed by Emulsiflex C3 (Avestin) and the lysate was clarified by centrifugation. MS2-MavR and MS2 RNAs and interacting partners were immunoprecipitated using MS2 coat protein fused to MBP. Eluted RNAs were purified by phenol-chloroform extraction, DNase treated and ethanol precipitated. Sequencing and analysis were completed by the Maryland University Genomic Research Center. Briefly, libraries were prepared using the NEBNext Ultra Directional RNA Library Prep Kit (NEB). Libraries were sequenced using Illumina HiSeq4000 75PE. Reads were mapped to the EHEC EDL933 (NCBI accession NZ_CP008957.1) genome using Bowtie v0.12.7. HTseq and DEseq were used to determine read counts for each gene and calculated fold-enrichment. Target genes were considered to be enriched if > 100 reads mapped to the gene in the MS2-MavR sample and the fold-enrichment was at least 5-fold for the aerobic dataset and at least 2-fold for the microaerobic dataset with a P value < 0.1.
Motility assay
A 1 μl aliquot of WT and ΔmavR grown to mid-exponential phase in DMEM was stab inoculated into motility plates (LB with 0.3% agar). Plates were incubated for 6 h at 37°C and halo diameter was measured.
H2O2 survival assay
Cultures were grown aerobically in DMEM to mid-exponential phase. Samples were collected immediately prior to and then at 0.5, 1 and 1.5 h after the addition of 5 mM H2O2. All samples were diluted and plated on LB containing ampicillin immediately after collection. Plates were incubated overnight at 37°C and CFUs were enumerated.
Mouse colonization
All experiments were approved by the Institutional Animal Care and Use Committee at the University of Virginia School of Medicine. Female 5- to 6-week-old CD-1 mice (Envigo) were infected by oral gavage with a total of 4 × 108 CFUs of bacteria (2 × 108 CFUs of each strain) resuspended in sterile PBS. Fecal samples were collected daily and mice were euthanized 8 days post infection to harvest the ceca and colons. Fecal and tissue samples were homogenized in PBS and CFUs were enumerated on MacConkey agar supplemented with streptomycin or chloramphenicol. The competitive index was calculated as the ratio of mavR::cat to WT normalized to the inoculum.
Fluorescent actin staining (FAS) assay
HeLa cells were seeded at a cell density of 5 × 105 cells/well on coverslips in a 12-well dish. The following day, HeLa cells were washed and infected with the indicated EHEC strains at a multiplicity of infection (M.O.I.) of 100 in low-glucose DMEM. Infected HeLa cells were incubated at 37°C with 5% CO2 for 6 h, replacing the media at 3 h. Coverslips were washed thrice with PBS and cells were fixed with 0.75% formaldehyde for 20 min. Cells were permeabilized with 0.2% Triton-X for 6 min and stained with 1 μg/ml fluorescein isothiocyanate (FITC)-phalloidin for 20 min at 37°C to visualize actin. After RNase A treatment (1 mg/ml for 10 min), coverslips were stained with 4 μg/ml DAPI to visualize DNA. Pedestals were enumerated for at least 98 HeLa cells per condition.
Western blotting
Bacterial overnight cultures were diluted 1:100 into M9 minimal medium and grown aerobically to an O.D.600 of 0.4 (Supplementary Figure S5) or diluted 1:100 into DMEM and grown microaerobically for 6 h. Cultures were pelleted by centrifugation and resuspended in sterile PBS. After the addition of SDS sample buffer, samples were boiled for 10 min and electrophoresed through SDS-PAGE gels and transferred to PVDF membranes (BioRad). Membranes were blocked with 5% dry milk in TBST, washed with TBST thrice and incubated with primary anti-His (1:1000, Cell Signaling), anti-DnaK (1:10 000, AbCam), anti-GAPDH (Invitrogen) or anti-EspA (1:7500, Vanessa Sperandio) antibodies for 1 h at room temperature. After three TBST washes, membranes were incubated with secondary anti-mouse or anti-rabbit antibodies conjugated to horse radish peroxidase for 30 min at room temperature. After TBST washes, ECL (PerkinElmer) was added to the membranes and bands were visualized on a Gel Doc XR + Gel Documentation System.
Electrophoretic mobility shift assays (EMSAs)
T7 in vitro transcription (NEB) was performed for eutR and mavR incorporating Bio-11-UTP (Fisher) where indicated. Transcripts were purified using NucAway Spin Columns (Invitrogen). Purified transcripts were incubated at the indicated concentrations in 1× structure buffer (Ambion RNase T1 kit), 2 ng/µl yeast RNA (Ambion) and brought to a total volume of 20 μl with RNase-free water. Samples were incubated at 85°C for 3 min followed by 20 min at 37°C. Following the addition of 5× RNA loading dye (50% glycerol, 0.1% bromophenol blue), samples were subjected to electrophoresis on 5% native TBE gels using 1× TBE as running buffer. Electrophoresed samples were transferred by capillary action to Zeta-probe membranes (BioRad) in 20× SSC. After UV crosslinking the RNA to the membrane, the membranes were subjected to the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Scientific). EMSAs were visualized using a Gel Doc XR + Gel Documentation System.
RNase E cleavage assay
The N-terminus of RNase E (AAs 1–527, N-Rne) was cloned into pBAD/mycHis A for protein purification as described above (see MAPS methods) with the modification that expression was induced with 0.2% arabinose. In vitro transcribed eutR was biotinylated on the 3′ end using the Pierce RNA 3' End Biotinylation Kit (Thermo). Transcripts were incubated at the indicated concentrations in 1× structure buffer (Ambion RNase T1 kit), 2 ng/μl yeast RNA (Ambion), and brought to a total volume of 10 μl with RNase-free water. Samples were incubated at 85°C for 3 min followed by 20 min at 37°C. RNase E was added to a final concentration of 7.5 μM and samples were incubated at 37°C for an additional 30 or 60 min. Then, 2× denaturing loading buffer (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol, 0.5 mM EDTA) was added and samples were subjected to electrophoresis on 10% TBE-urea gels at 150 V. After electrophoretic transfer (50 V, 2 h) to Zetaprobe membrane, the membranes were crosslinked. Membranes were developed and visualized as previously described.
Bioinformatic analyses
All programs used for bioinformatic analyses are listed and referenced in Supplementary Table S4.
RESULTS
MavR is a pathogen-specific sRNA
MavR is an Hfq-dependent sRNA that is produced under conditions that promote EHEC virulence gene expression (19,28) (Figure 1A). MavR is encoded within O-island 48 in the reverse orientation of genes encoding tellurite resistance (ter) (Figure 1B). O-islands are DNA regions present in EHEC but absent in E. coli K-12 (27). Further analyses revealed that MavR is conserved in diarrheagenic E. coli as well as in other pathogenic Enterobacteriaceae (Figure 1C) but absent from nonpathogenic bacteria. Notably, the genomic organization of mavR within the ter locus is maintained in all bacteria that encode this sRNA.
Figure 1.

MavR is a pathogen-specific sRNA. (A) Northern blot of MavR in WT, Δhfq, and ΔmavR grown under microaerobic conditions. 23S and 16S rRNA are shown as loading controls; N = 2. (B) Schematic of the genomic context of mavR. (C) Genomic alignment of MavR from the indicated bacteria. Each vertical bar represents a base mismatch with respect to the E. coli O157:H7 sequence. Therefore, areas without vertical bars represent more highly conserved sequences to E. coli O157:H7 compared to regions with vertical bars. Asterisks indicate genomes in which mavR is plasmid- encoded. Percent identity is also indicated on the right side.
RNAseq analyses indicated that MavR is an ∼370 bp sRNA ((28) and data herein). To examine mavR transcriptional control, we performed 5′ RACE and in silico analyses using Bprom, which suggested that mavR contains a housekeeping sigma 70 recognition sequence in the promoter (Supplementary Figure S1A). In support of constitutive MavR expression, transcriptional analyses indicated that mavR is similarly expressed throughout growth as well as under microaerobic and aerobic conditions (Supplementary Figure S1B–D). To study MavR-dependent gene expression, we generated a mavR deletion in EHEC strain 86–24 (Figure 1A) and performed genome sequencing (Illumina and PacBio) to confirm that strain 86–24 encodes mavR only in single copy (EHEC strain EDL933 encodes two copies) as well as to ensure no off-target effects (e.g., spontaneous mutations) occurred. We also confirmed that the mavR deletion did not affect expression of surrounding genes (Supplementary Figure S2).
Global mapping of MavR targets
To identify MavR targets, we performed MS2 affinity purification coupled to RNA sequencing (MAPS) (31,32). For these experiments, we fused the MS2 aptamer to the 5′ end of mavR, and this chimera was cloned under control of an arabinose-inducible (pBAD) promoter, generating pBAD-MS2::MavR. The MS2::MavR construct is functional as it complemented gene expression in the ΔmavR strain (Figure 7D). Affinity purification was performed using ΔmavR carrying the pBAD-MS2::MavR or the empty pBAD-MS2 construct to eliminate competition with native copies of MavR and to control for false positives interacting with the MS2 aptamer. Oxygen is an important signal that influences EHEC gene expression (24,40); therefore, to more comprehensively map the MavR interactome, MAPS was performed under aerobic and microaerobic growth conditions.
Figure 7.

Overview of MavR-dependent gene expression. (A) Comparison of MAPS and RNAseq datasets. (B) Pathway analysis of microaerobic RNAseq data. (C) RT-qPCR of indicated genes identified as differentially expressed in the microaerobic RNAseq data set in WT and ΔmavR. (D) RT-qPCR of indicated genes in WT, ΔmavR, ΔmavR + pBAD-mavR and pBAD-MS2::mavR. WT and ΔmavR carry the empty vector, N = 3. (E) RT-qPCR of indicated genes in WT, ΔmavR and ΔmavR+ pGEN-mavR. WT and ΔmavR carry the empty vector. Bars represent the mean and error bars indicate SEM; * P < 0.05, ** P < 0.01, *** P < 0.001, † P < 0.1 (Student’s two-sample t-test).
We identified 36 transcripts enriched in the microaerobic dataset and 185 transcripts enriched in the aerobic dataset (>100 mapped reads, P < 0.1). Kegg pathway enrichment analyses revealed enrichment of diverse processes, including metabolism, translation, motility/chemotaxis and gene regulation (Figure 2A). Enriched targets also included several genes previously reported to contribute to EHEC virulence (Figure 2B). We repeated the MS2 affinity purification assay and validated targets via RT-qPCR. In agreement with the MAPS data, we measured enrichment of the evgS, cheA, and comR transcripts in the MS2::MavR affinity purification compared to the MS2 affinity purification (Figure 2C). Enrichment of the eutR transcript was more variable (Figure 2C), most likely because this assay was performed under conditions in which eutR is expressed at low levels (see subsequent results sections and the discussion section).
Figure 2.

Overview of MAPS data. (A) Pathway analysis of the MAPS data. (B) Table of transcripts encoding virulence-associated factors enriched in the MAPS data set. (C) RT-qPCR (triangles) of transcripts identified as enriched by MAPS (squares) from MS2::MavR affinity purified RNAs normalized to MS2 affinity purified RNAs.
MavR interacts with the 3′ region of the eutR coding sequence to promote expression
To investigate MavR-dependent gene regulation in more detail, we focused on eutR, which was identified as a putative target by MAPS. EutR was initially characterized nearly 30 years ago as the transcriptional activator of the eut (EA utilization) locus that is required for EA metabolism (41,42). EA metabolism enhances pathogen growth during host infection (43–48). Recent work in our lab established that EutR also regulates virulence genes, including T3SS expression in EHEC, Salmonella enterica serovar Typhimurium and Citrobacter rodentium (16,43,47–50). To examine MavR interaction with the eutR transcript, we performed bioinformatic analysis using IntaRNA (51–54) to identify potential interaction sites. These analyses predicted an interaction between MavR and the 3′ region of the eutR coding sequence (CDS) (Figure 3A, free energy value of -12.16). To test this prediction, we performed RNA electrophoretic mobility shift assays (EMSAs) using in vitro transcribed and biotinylated MavR and increasing amounts of the 3′ region of the eutR transcript (Figure 3B) or 9S (negative control) transcript. Upon addition of the eutR transcript, we measured a shift in the MavR RNA, indicating direct base pairing, whereas no shift occurred upon addition of the 9S transcript (Figure 3C).
Figure 3.

MavR binds the eutR transcript. (A) Predicted MavR-eutR RNA base-pairing. Point mutations to generate the disrupted alleles in the eutRmut transcript are indicated. Numbering on the eutR sequence indicate nucleotides after the translation start site. Numbering on the MavR sequence indicate nucleotides after the transcription start site. (B) Schematic showing the in vitro transcribed RNAs used for the EMSAs. (C) RNA EMSA of labeled MavR and eutR transcripts. 9S precursor RNA is the negative control. (D) Competition RNA EMSA of labeled eutR and unlabeled MavR transcripts competed with indicated unlabeled eutR transcripts. (E) Quantification of competition RNA EMSA in (D). (F) Predicted interaction between eutRmut and MavR transcripts.
We repeated the EMSAs using biotinylated eutR and increasing concentrations of the MavR transcript. These data were consistent with the previous EMSA results, as we measured a shift in the labeled eutR transcript upon addition of MavR to the reactions (Figure 3D–E). Next, we performed competition EMSAs using labeled eutR and increasing amounts of unlabeled wildtype eutR, mutated eutR or truncated eutR (as indicated in Figure 3B). The eutR transcript effectively competed with labeled eutR for MavR binding. To substantiate these data, we generated mutations in the eutR transcript (shown in red in Figure 3A, which were designed to disrupt G-C pairing) and repeated the EMSAs. Surprisingly, the mutated eutR transcript (eutRmut) was able to interact with MavR, as indicated by the decreased intensity of shifted eutR transcript (Figure 3D–E). Subsequent in silico analysis revealed sequence complementarity between MavR and eutRmut transcripts (Figure 3F), suggesting we simply changed the binding site. This conclusion is supported by previous work which showed that altering of nucleotides in an sRNA-RNA binding site can shift target binding and include interactions with sequences that exhibit complementarity to the mutated binding site (55). Therefore, we generated a truncated eutR transcript (eutRtrunc) to remove the entire 3′ end of the eutR transcript. The truncated transcript did not compete for MavR binding as there was virtually no difference in the amount of shifted eutR in the absence or presence of eutRtrunc (Figure 3D–E). Collectively, these data support MavR interaction with the 3′ region of the eutR transcript.
Next, we examined the effect of MavR on EutR expression. For these experiments, eutR was fused to a Myc-His tag and cloned under the control of an arabinose-inducible vector to specifically assay post-transcriptional regulation. The resulting plasmid (pEutR::His) was introduced into WT and ΔmavR. EutR::His expression was decreased in ΔmavR compared to WT (Supplementary Figure S3A,B). To substantiate MavR regulation of EutR expression, we utilized a two plasmid-based assay in which WT and ΔmavR were transformed with pEutR::His or pEutR::His and pMavR. Dual plasmid systems are routinely used to monitor the effects of a sRNA on the target mRNA and enables the specific effects of the sRNA on the target RNA to be assessed (19,24,30,56,57). RT-qPCR and western analysis confirmed MavR positively regulated eutR/EutR expression, as expression was decreased in ΔmavR, and this defect was rescued when MavR was expressed in trans (Figure 4A–C). Collectively, these data indicated that MavR positively regulates EutR expression.
Figure 4.
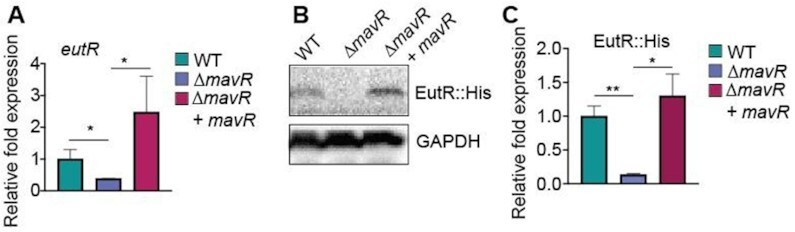
MavR promotes expression of eutR/EutR. (A) RT-qPCR of eutR transcript levels expressed from pBAD-eutR::His in WT, ΔmavR, and ΔmavR+ pUCP24-mavR. WT and ΔmavR carry the empty vector, N = 3. (B) Western blot of EutR::His in WT, ΔmavR and ΔmavR+ pUCP24-mavR. GAPDH is the loading control. (C) Quantification of EutR::His expression in WT, ΔmavR and ΔmavR+ pUCP24-mavR, N = 9. Bars represent the mean and error bars indicate standard error of the mean (SEM). ns P > 0.05, * P < 0.05, ** P < 0.01 (Student’s two-sample t-test).
MavR is required for robust expression of the eut locus and maximal EA-dependent growth
The eut locus is comprised of 17 genes (Figure 5A) that encode proteins that function in the transport and catabolism of EA as well as a protein microcompartment that contains toxic breakdown products of EA metabolism (41,42,58–61). EutR is encoded by the last gene in the eut locus and is required for transcriptional activation of the entire locus (41,42). eutR is constitutively expressed at low levels from an internal P2 promoter. EutR binds to the primary P1 promoter upstream of eutS, and in the presence of EA and adenosylcobalamin (AdoCbl), EutR activates transcription. This results in robust expression of the entire operon, including readthrough of the P2 promoter and positive autoregulation (42,62). Because MavR promotes EutR expression, we reasoned that ΔmavR would be impaired for eut expression. To test this idea, we examined native expression of eutS, eutB, eutL and eutR (as representative genes in the beginning, middle and end of the locus) (Figure 5A) after growth without or with EA and AdoCbl. No differences in eut gene expression were measured when EHEC was grown without EA and AdoCbl supplementation; however, under eut-inducing conditions, we measured a 2- to 3-fold decrease in expression of all genes in ΔmavR compared to WT (Figure 5B–E). Consistent with MavR being a Hfq-dependent sRNA, there was a similar 3-fold decreases in eut gene expression in Δhfq compared to WT when these strains were grown in DMEM supplemented with EA and AdoCbl (Supplementary Figure S4A–D). To determine whether EA or AdoCbl influenced post-transcriptional eut expression (as has been reported for Firmicutes (63,64)), we measured EutR::His expression after growth in medium without supplementation or with EA, AdoCbl, or EA and AdoCbl. In all conditions, EutR::His was detected at lower levels in ΔmavR compared to WT (Supplementary Figure S5). Moreover, no differences in EutR::His expression were detected in the WT strain regardless of EA and/or AdoCbl, and similarly, these molecules did not affect EutR::His levels in ΔmavR (Supplementary Figure S5), indicating that MavR regulates EutR expression independently of EA and AdoCbl.
Figure 5.

MavR is required for robust expression of the ethanolamine utilization (eut) locus and EA utilization. (A) Schematic of the eut locus. (B) RT-qPCR of eutS expression in WT and ΔmavR grown without or with ethanolamine (EA) and adenosylcobalamin (AdoCbl) supplementation. (C) RT-qPCR of eutB expression in WT and ΔmavR grown without or with EA and AdoCbl supplementation. (D) RT-qPCR of eutL expression in WT and ΔmavR grown without or with EA and AdoCbl supplementation. (E) RT-qPCR of eutR expression in WT and ΔmavR grown without or with EA and AdoCbl supplementation. (F) Growth curves of WT and ΔmavR in minimal medium containing EA as the sole nitrogen source. (G) Growth curves of ΔeutR + pBAD-eutR, ΔeutRΔmavR + pBAD-eutR and ΔeutRΔmavR+ pBAD-eutR and pUCP24-mavR in minimal medium containing EA as the sole nitrogen source. (H) Growth curves of WT and ΔmavR in minimal medium containing NH4 as the sole nitrogen source. (I) Competition assay between WT and ΔmavR::cat grown in minimal medium containing EA or NH4 as the sole nitrogen source and in DMEM. Bars represent the mean and error bars indicate SEM. N = 4, ** P < 0.01, *** P < 0.001, **** P < 0.0001 (Student’s two-sample t-test).
To functionally test the consequence of MavR-dependent eut expression, we measured growth of WT and ΔmavR in minimal medium containing EA as the sole nitrogen source. We measured a longer lag phase of ΔmavR compared to WT (Figure 5F). To confirm the role of MavR on EA utilization, we utilized the dual plasmid-based assay (described above) in which ΔeutR or ΔeutRΔmavR was transformed with pEutR::His or pEutR::His and pMavR. MavR expression rescued growth of the ΔeutRΔmavR strain to near WT (ΔeutR + pEutR::His) levels (Figure 5G). Importantly, this growth defect was specific to EA utilization, as WT and ΔmavR grew similarly in minimal medium containing ammonium as the sole nitrogen source (Figure 5H) as well as in DMEM (Supplementary Figure S6A,B). In agreement with single strain growth assays, during growth in co-culture, ΔmavR::cat (unresolved deletion strain) was significantly outcompeted by WT in minimal medium supplemented with EA, but was recovered at similar levels to WT during growth in minimal medium supplemented with ammonium or in DMEM (Figure 5I). Collectively, these data demonstrate that MavR is necessary for maximal eut expression and EA utilization.
MavR stabilizes the eutR transcript
sRNAs regulate gene expression typically by enhancing or preventing translation initiation (21). However, sRNAs may also antagonize Rho-dependent transcription termination or affect stability of the target transcript (65,66). Because of the proximity of the MavR-eutR binding site to the 3′ end of the transcript as well as that MavR influenced eutR transcript levels, we excluded translation initiation as a potential mechanism. To test whether MavR affected Rho-dependent transcription termination, we added the Rho inhibitor bicyclomycin (BCM) (67) to cultures of WT and ΔmavR and assessed eutR levels by northern analysis. The eutR transcript levels were decreased in ΔmavR compared to WT regardless of the addition of BCM to the culture media (Supplementary Figure S7A). We repeated these experiments to measure endogenous eut expression following growth of WT and ΔmavR under eut-inducing conditions. Native eut expression was ∼2-fold reduced in ΔmavR compared to WT without or with BCM treatment (Supplementary Figure S7B). These data indicated that MavR does not antagonize Rho to promote eutR/ eut expression.
To examine the effect of MavR on eutR stability, we grew WT and ΔmavR transformed with pEutR::His. After growth, cultures were treated with rifampicin to inhibit transcription initiation, and RNA was extracted from sample aliquots immediately prior to and at indicated timepoints following rifampicin treatment. eutR transcript abundance and stability was then determined by northern blot analysis. At time 0, eutR transcript levels were decreased nearly 4-fold in ΔmavR compared to WT (Figure 6A,B), which was consistent with data shown in Figures 4A and 5E. Furthermore, the eutR transcript was ∼3–4 fold less stable in ΔmavR (half-life ∼0.25 min) as compared to WT (half-life ∼1 min) (Figure 6A, C and D). Because the His tag could influence the stability and/or processing of the eutR transcript, we repeated these experiments using WT and ΔmavR transformed with pEutR (expressed from pBAD24). These data revealed only a modest decrease in eutR transcript stability in ΔmavR compared to WT (Supplementary Figure S8A,B). We were unable to demonstrate that MavR affects the half-life of the native eutR transcript (data not shown), possibly because eutR is the last gene in the long eut operon (68). Because the data shown in Figure 6A–D suggested that MavR influences eutR stability, we further interrogated this hypothesis, as described below.
Figure 6.

MavR stabilizes the eutR transcript. (A) Northern blot of eutR transcripts expressed from pBAD-eutR::His in WT and ΔmavR at indicated time points before or after addition of rifampicin. 16S rRNA is the loading control. (B) Relative quantification of eutR expression in WT and ΔmavR prior to addition of rifampicin (time 0, in panel (A)), N = 3. (C) Decay curves of eutR RNA in WT and ΔmavR. The signal obtained at 0 min. was set to 1 for each strain, and the amount of RNA remaining at each timepoint was plotted on the y-axis versus time on the x-axis. The time point at which 50% of the eutR mRNA had been decayed (dashed line) was calculated to determine the half-life (t1/2), N = 3. (D) Half-life of eutR expressed from pBAD-eutR::His. N = 3. (E) Northern blot of eutR transcripts expressed from pBAD-eutR::His in WT, ΔmavR, rneΔCTD and ΔmavR rneΔCTD. (F) RT-qPCR of eut gene expression in WT and ΔmavR, N = 3. (G) RT-qPCR of eut gene expression in rneΔCTD and ΔmavR rneΔCTD, N = 3. For (B), (D), (F) and (G), bars represent the mean and error bars indicate SEM. ns P > 0.05, * P < 0.05, ** P < 0.01 (Student's two-sample t-test). (H) In vitro cleavage of eutR by N-Rne in the absence or presence of MavR. (I) Model: MavR interaction with the eutR transcript protects eutR from RNase E-mediated degradation. Stabilized eutR transcripts result in maximal EutR expression and EutR-dependent activation of eut expression.
RNase E initiates degradation of most transcripts in the Enterobacteriaceae (69–73). A previous, unbiased analysis reported that the eutR transcript co-immunoprecipitated with RNase E in EHEC strain Sakai (74). Therefore, we investigated the hypothesis that MavR protects the eutR transcript from RNase E-dependent degradation. Because RNase E is encoded by the essential rne gene, we first generated a truncated RNase E protein that lacks the C-terminal domain (CTD) (75,76) in WT and ΔmavR (generating rneΔCTD and ΔmavR rneΔCTD). The CTD functions as a scaffold in assembly of the degradosome. Bacteria that lack this domain are viable but accumulate RNA processing intermediates (76–78). We performed northern analysis to compare eutR expression in WT, ΔmavR, rneΔCTD and ΔmavR rneΔCTD strains (as an experimental control, eutR expression was also measured in WT and ΔmavR, Figure 6E, left side). Although total eutR levels were decreased in the rne background compared to WT, this truncation ablated differences in eutR expression between rneΔCTD and ΔmavR rneΔCTD strains at time 0 (Figure 6E). Moreover, the RNase E truncation stabilized the eutR transcript as no measurable degradation of the eutR RNA occurred during the course of the experiment in either rneΔCTD or ΔmavR rneΔCTD (Figure 6E). As a complementary approach, we examined whether limiting RNase E activity would rescue endogenous eut expression in ΔmavR compared to WT. In agreement with the plasmid-based data, there was no significant difference in eut gene expression between rneΔCTD and ΔmavR rneΔCTD (Figure 6F and G).
Next, we performed in vitro RNase E cleavage assays. The rationale for this experiment was two-fold. First, this experiment would indicate whether the lack of degradosome in the rneΔCTD strains vs RNase E catalytic activity in the previous assays was responsible for rescue of eutR/eut expression. Second, the in vitro cleavage assay experiment also addresses the role of MavR per se in antagonizing RNase E-dependent degradation of the eutR transcript. For these experiments, we expressed and purified the N-terminal region of RNase E (N-Rne) that contains the catalytic domain and possesses full cleavage activity (79,80). Upon addition of N-Rne, the eutR transcript was partially degraded within 30 min and nearly undetectable after 60 min. However, the addition of MavR to the reactions prevented RNase E hydrolysis (Figure 6H). These data demonstrate that MavR is sufficient to protect the eutR transcript from RNase E-mediated degradation independent of additional regulatory factors. Collectively, these data support a model in which MavR protects eutR from RNase E mediated cleavage to promote eut expression (Figure 6I).
MavR influences expression of diverse genes important for EHEC fitness and virulence
To globally assess the biological impact of MavR on EHEC gene expression, we performed RNAseq using RNA purified from WT and ΔmavR grown under aerobic and microaerobic conditions. There were no putative targets/ differentially expressed genes common to all of the MAPS and RNAseq data sets (Figure 7A). However, we identified a handful of shared genes between the aerobic MAPS and RNAseq data, the microaerobic MAPS and RNAseq data sets, as well as transcripts that were enriched in both the aerobic and microaerobic MAPS. Under aerobic growth, MavR affected expression of 87 genes (fold change ≥ 2-fold and P ≤ 0.05). Of the differentially expressed genes, 11 genes were increased in ΔmavR compared to WT. These genes encode metabolic regulators and enzymes (tcdR, tcdB, wcaK, ccmE, dcuC), a putative diguanylate synthase (cdgI), as well as hypothetical/ uncharacterized proteins (Z2249, Z0326, Z2717/ydiL, Z2395 and Z1866). Genes that were decreased in ΔmavR compared to WT primarily encode flagella and motility (detailed in the next section).
Under microaerobic conditions, MavR impacted expression of 52 genes. Although only three overlapping transcripts were identified in the MAPS and RNAseq data sets, Kegg pathway enrichment analyses were consistent between the data in revealing enrichment of metabolic- and ribosome-associated pathways among the differentially regulated genes (Figure 7B). We performed RT-qPCR using RNA harvested from a distinct set of biological replicates to confirm MavR-dependent expression of genes encoding metabolic enzymes involved in glycolysis and amino acid metabolism as well as elaB that plays a role in stress responses (Figure 7C,D).
The transcriptomic data also revealed that genes that encode the T6SS and the T3SS, which contribute to or are required for EHEC virulence, respectively (1,81,82) were decreased in ΔmavR compared to WT (Supplementary Figure S9 and detailed in a subsequent section) and that genes encoding oxidative stress responses, including exposure to H2O2, were upregulated in ΔmavR compared to WT (Supplementary Figure S10A). RT-qPCR data were consistent with the RNAseq data as expression of dps (involved in oxidative stress and nutrient deprivation) and poxB (oxidative stress, metabolism) was increased in ΔmavR compared to WT, and these differences in expression were partially rescued by mavR complementation (Figure 7E). To test the biological significance of MavR in influencing EHEC survival following oxidative stress, we treated mid-logarithmic cultures of WT, ΔmavR and mavR complemented strains with 10 mM H2O2 and enumerated CFUs over time after exposure. At 1 h post-treatment, viable WT cells were at the limit of detection, whereas the ΔmavR strain was only slightly affected by the addition H2O2, which is consistent with the gene expression data. Furthermore, we were able to partially complement survival under H2O2 stress with pmavR (Supplementary Figure S10B). Collectively, these data suggest an extensive role for MavR in EHEC gene expression.
MavR promotes expression of genes encoding flagella under aerobic conditions
Notably, expression of nearly every gene that encodes flagellar biosynthesis or chemotaxis was decreased in ΔmavR compared to WT during aerobic growth (Figure 8A). We performed RT-qPCR to confirm differences in expression of flhD, fliK, fliC, and cheA (Figure 8B,C), which encode genes required for flagellar biosynthesis, motility, and chemotaxis. Trans-complementation with mavR expressed from the native promoter resulted in partial to nearly full complementation (Figure 8C). To determine whether MavR affected flagellar expression through EutR, we examined flhD and fliK expression in WT and ΔeutR grown without or with EA and AdoCbl supplementation. RT-qPCR analyses did not reveal a role for EutR in controlling expression of these genes. No significant differences in flhD or fliK expression were measured among any of the strains or growth conditions (Supplementary Figure S11A,B); however, EutR was required for EA/AdoCbl-dependent eutS expression (Supplementary Figure S11C). These data suggest that MavR influences expression of genes encoding flagella/ motility independent of EutR.
Figure 8.

MavR promotes expression of genes encoding flagella. (A) RNAseq data showing flagella and motility gene expression in ΔmavR compared to WT grown aerobically. The dotted line indicates 2-fold change, N = 3. (B) RT-qPCR of flhD in WT and ΔmavR. (C) RT-qPCR of indicated genes identified as differentially expressed in the RNAseq data set in WT, ΔmavR and ΔmavR + pGEN-mavR. WT and ΔmavR carry the empty vector, N = 3. (D) Western blot of FlhD::His and FlhC::His in WT and ΔmavR. DnaK is the loading control. (E) Quantification of FlhD::His and FlhC::His expression in WT and ΔmavR,N = 3. (F) RT-qPCR of flhD/flhD::his and fliK in WT, ΔmavR and ΔmavR + pUCP24-mavR. WT and ΔmavR carry the empty vector, N = 3. (G) Quantification of WT, ΔmavR, and ΔmavR+ pGEN-mavR motility assays, N = 3. Bars represent the mean and error bars indicate SEM; ** P < 0.01, *** P < 0.001, † P < 0.1 (Student's two-sample t-test).
The heteromeric master regulator FlhDC controls transcription of genes encoding flagella and chemotaxis (83–85). Therefore, we examined whether MavR might affect flagella/chemotaxis gene expression via post-transcriptional control of FlhD and/or FlhC. For these experiments, we cloned each gene under the control of an arabinose-inducible promoter to remove native transcriptional regulation. FlhD expression was significantly decreased in ΔmavR compared to WT, whereas MavR did not influence FlhC expression (Figure 8D,E). To assess MavR modulation of flhD expression and the impact on FlhD target gene expression, we utilized the dual plasmid-based assay previously described. We measured a ∼2-fold decrease in flhD and fliK expression in ΔmavR compared to WT that was complemented by pmavR (Figure 8F). These data suggest that MavR promotes flagellar/chemotaxis gene expression by post-transcriptionally influencing expression of FlhD. Consistent with the gene expression data, ΔmavR was slightly less motile compared to WT, and this difference could be rescued upon complementation (Figure 8G). These results indicate that MavR post-transcriptionally promotes FlhD expression, which affects downstream genes.
MavR promotes LEE expression and AE lesion formation
The LEE pathogenicity island carries 41 genes that are mostly organized into five major operons (Figure 9A). The RNAseq data indicated that 16 LEE-encoded genes were at least 1.5-fold downregulated in ΔmavR compared to WT (Figure 9B). Importantly, expression of ler, that encodes Ler the master regulator of the LEE (11), was decreased 1.7-fold in ΔmavR (P = 0.079) (Figure 9B). We further analyzed LEE transcript levels by RT-qPCR. These data revealed at least 2-fold decreased LEE expression in ΔmavR vs WT, including decreased ler expression (Figure 9C). Moreover, western blot analysis confirmed that levels of EspA, which encodes the T3SS filament (encoded in LEE4) (86), were decreased in ΔmavR strain compared with WT EHEC (Figure 9D,E). Although overexpression of MavR (sRNA56) was previously reported to result in increased expression of espA (28), MavR overexpression did not complement the ΔmavR strain in these assays (data not shown). The LEE is required for the formation of attaching and effacing (AE) lesions on epithelial cells, and consistent with the gene expression data, ΔmavR formed significantly fewer AE lesions on HeLa cells compared to WT (Figure 9F,G). Together, these data reveal that MavR is required for robust LEE expression and demonstrate a role for MavR in EHEC virulence.
Figure 9.

MavR is required for maximal LEE gene expression and AE lesion formation. (A) Schematic of the LEE. (B) RNAseq data comparing LEE gene expression in ΔmavR compared to WT under microaerobic conditions; N = 3. Columns are color-coded according to operon (shown in (A)). The red dotted line indicates 2-fold change, and the blue dotted line indicates 1.5-fold change. (C) RT-qPCR of LEE genes in WT and ΔmavR; N = 3. (D) Western blot of EspA expression in the WT and ΔmavR. DnaK is the loading control. (E) Quantification of EspA expression in the WT and ΔmavR. (F) FAS assay showing AE lesions on HeLa cells infected with WT or ΔmavR. AE lesions are indicated by arrows. (G) Quantification of AE lesions on HeLa cells infected with WT or ΔmavR. N = 98–120 HeLa cells. Bars represent the mean and error bars indicate SEM. ** P < 0.01, *** P < 0.001 (Student’s two-sample t-test).
Bioinformatic queries did not predict MavR interaction with LEE transcripts and LEE transcripts were not enriched by MAPS, suggesting that MavR affects LEE expression indirectly. To date, over 40 transcription factors have been reported to control ler transcription (87–89). EutR directly activates ler expression (48,50). Additionally, the MAPS data indicated phoB, which encodes another transcriptional regulator of LEE expression (90), was a potential MavR target. We confirmed that MavR post-transcriptionally promoted PhoB expression (Supplementary Figure S12A,B) as well as PhoB-dependent gene expression (Supplementary Figure S12C). To test whether MavR and EutR- or PhoB-dependent LEE expression were functionally linked, we generated an eutR or phoB deletion in the ΔmavR background (generating ΔmavRΔeutR and ΔmavRΔphoB strains) and assessed EspA expression. Lower levels of EspA were detected in ΔeutR and ΔphoB compared to WT as previously reported. Notably, EspA levels were further decreased in ΔmavR and ΔmavRΔeutR, as well as in ΔmavRΔphoB (Supplementary Figure S12D-E). These results raise the possibility that MavR affects LEE expression by pleiotropic mechanisms.
MavR is important for EHEC colonization of the mammalian GI tract
The extensive role for MavR in EHEC gene expression suggests MavR plays a critical role in EHEC adaptation to and fitness within the GI tract. Therefore, we performed competition experiments using streptomycin-treated mice. This model does not recapitulate LEE-dependent adherence to epithelial cells or AE lesion formation, but rather is used to evaluate the relative colonization capacity of an E. coli strain, including EHEC (91,92). Mice were orally infected with a 1:1 mixture of WT and ΔmavR::cat strains. At 2 days post infection and throughout the duration of the experiment, ΔmavR::cat was outcompeted by WT (∼10–100 fold) as reflected by CFUs in fecal samples (Figure 10A). These data were consistent with numbers of ΔmavR::cat and WT recovered from the cecum and colon (Figure 10B). These findings demonstrate that MavR is required for robust intestinal colonization.
Figure 10.
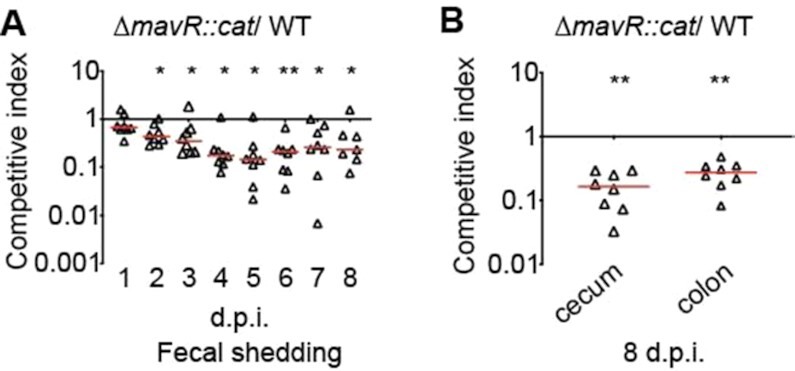
MavR is required for robust colonization of the GI tract. (A) Competition assay between WT and ΔmavR::cat strains harvested from fecal samples at indicated time points. (B) Competition assay between WT and ΔmavR::cat strains harvested from the cecum or colon. Each point represents a competitive index (CI). Horizontal lines represent the median value for each group. dpi, days post infection; N = 8. * P < 0.05, ** P < 0.01 (Wilcoxon log-rank test).
DISCUSSION
MavR was originally discovered via transcriptomic analyses to identify EHEC-specific and Hfq-dependent sRNAs. Initial characterization of MavR revealed that overexpression resulted in increased expression of the LEE-encoded gene espA (28), suggesting a role for MavR in virulence. Here, we employed two unbiased techniques, MAPS and RNAseq, to comprehensively elucidate the MavR regulon in EHEC grown under aerobic and microaerobic conditions. Although there was no overlap of putative targets/ differentially expressed genes among all of the data sets (Figure 7A), some genes were common in the aerobic datasets and microaerobic datasets, as well as transcripts that were enriched in both the aerobic and microaerobic MAPS. By performing these assays under distinct conditions, we determined that MavR affected expression of genes important for all phases of infection. Notably, previous studies have also demonstrated discrete sRNA–RNA interactions and regulatory outcomes depending on growth-phase (30) or growth medium (rich versus minimal/defined) (19,56). Collectively, these findings suggest that the regulon of a sRNA can vary under different growth and/or environmental conditions likely because the transcriptome—and therefore available RNA targets—varies under distinct conditions.
The ability to acquire nutrients is an essential first step in host colonization and may enable a pathogen to overcome nutritional competition (43,46,93–95) (Figure 11). To overcome this challenge, EHEC utilizes diverse metabolic pathways to take advantage of a variety of metabolites (96). Our data reveal that MavR affected expression of genes important for biosynthesis and energy production, including EutR, the DNA-binding transcriptional activator of the eut locus. Subsequently, EHEC uses flagella to traverse the mucus layer and reach the epithelial border (97,98). There, EHEC encounters reactive oxygen species, including H2O2 (99,100). Finally, EHEC forms AE lesions which results in intimate attachment and nutrient acquisition (1,101,102). Our data indicate that MavR promoted expression of genes encoding flagella and the T3SS and repressed expression of genes important for oxidative stress responses (Figure 11). The concentration of H2O2 in the colonic lumen is sublethal (103,104). Therefore, it is possible that repression of oxidative stress response genes by MavR may reduce unnecessary energy expenditure to enhance survival in the GI tract. Finally, the mavR deletion strain was significantly attenuated during colonization of the mammalian GI tract (Figure 10), underscoring the importance of this sRNA to EHEC fitness during infection.
Figure 11.
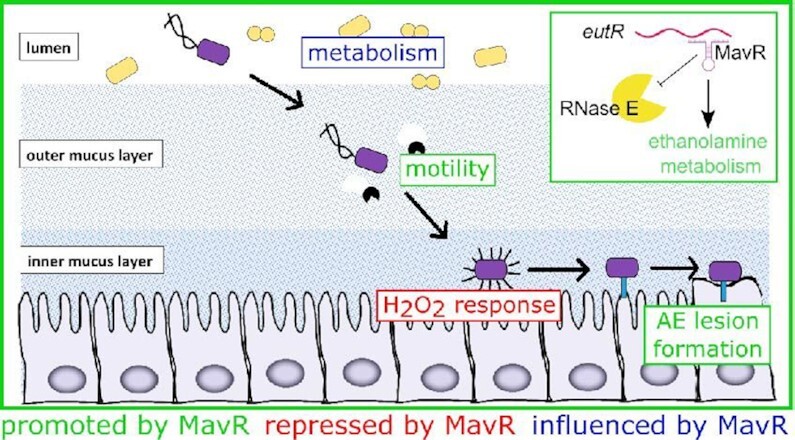
Summary of MavR-dependent gene expression in the context of EHEC host colonization. MavR affects expression of genes important for nutrient acquisition, motility, oxidative stress responses and AE lesion formation. The inset summarizes MavR-dependent activation of EutR expression and the impact on EA utilization.
An important issue is understanding how MavR, and other sRNAs, coordinate expression of various targets. The competing endogenous RNAs (ceRNAs) hypothesis predicts that if a sRNA regulates several targets, an increase in the production of one target will cause sRNA availability for the other targets to decrease below the threshold value and result in deregulation (33,105,106). Maximal expression of EutR requires EA and AdoCbl (42,59). Interestingly, although eutR was identified as a putative MavR target by MAPS, subsequent targeted affinity purification experiments did not reliably reproduce these results (Figure 2). Notably, the affinity purification experiments were performed using medium lacking EA and AdoCbl, and subsequent experiments revealed that MavR affected eutR transcript levels only upon EA and AdoCbl supplementation (Figure 5) (or when native transcriptional control was removed [e.g. using an arabinose-inducible promoter]). Therefore, we hypothesize that MavR may be titrated away from less abundant targets, such as eutR in the absence of EA and AdoCbl, through competitive binding with more abundant transcripts. In addition to eutR, we identified a role for MavR in promoting PhoB expression. Similar to EutR, PhoB expression is responsive to environmental signals and occurs under phosphate starvation (107). A non-conflicting alternative hypothesis as to how MavR coordinates expression of diverse genes in EHEC may be due to MavR-dependent expression of transcription factors. Integration of transcriptional regulators into the regulatory network of an sRNA amplifies the regulon of the sRNA by adding indirect targets (108–110). Besides EutR and PhoB, MavR also post-transcriptionally promoted expression of the transcriptional regulator FlhD. Therefore, ceRNAs and/ or the incorporation of MavR into regulatory circuits may provide a mechanism for MavR to indirectly integrate discrete environmental signals to coordinate EHEC gene expression.
The ability to utilize EA as a carbon, nitrogen, and/or energy source is conserved among diverse bacteria. However, the complexity and organization of the eut genes varies greatly and can include only a few genes or many genes (111). The Enterobacteriaceae and the Firmicutes encode the longest eut operons, containing 17 and 19 genes, respectively. For decades, control of eut expression was thought to occur solely at the level of transcription initiation, via EutR, in the Enterobacteriaceae and the noncanonical two component system EutVW in the Firmicutes (112). More recently, several reports have characterized a complex mechanism in which a riboswitch-containing sRNA controls the activity of EutVW in Enterococcus faecalis and Listeria monocytogenes (Firmicutes) (63,64). Our data support a commonality in which expression of the energetically costly, long eut locus requires multiple layers of regulation and occurs at the level of the transcriptional activator. Expression of these long operons is only advantageous when both the metabolite EA and the required co-factor AdoCbl are present. In both systems, sRNAs positively regulate expression of these long operons only in the presence of both ligands.
The first characterized sRNAs were shown to interact with the 5′ untranslated region (5′ UTR) of the target mRNA. This resulted in sequestration or unmasking of the ribosome binding site (RBS) to inhibit or promote translation, respectively, or modulation of mRNA stability (20,113). Based on these original data, interactions with the 5′ UTR has become the canonical model of sRNA regulation. Notably, unbiased approaches to identify sRNA interacting partners are increasingly detecting interactions between sRNAs and the CDS of detected targets (e.g., (19,55,56); however, only a handful of studies have characterized the outcome of these interactions (including (24,34,36,114)). In the first example, the sRNA MicC was shown to promote target destabilization in S. typhimurium (36). Since then, additional examples have been reported in which a sRNA interacts with the CDS of its target transcript to repress expression (34,35). We previously reported that the sRNA DicF interacts with the CDS of the pchA transcript to promote translation (24). Subsequently, Chen et al. reported that the sRNA GcvB binds the rbn CDS to stabilize the transcript and promote expression (115). Our findings demonstrate that MavR also binds to the CDS to antagonize RNase E-dependent cleavage of the eutR transcript. Although the rifampicin experiments were not consistent, our model is supported by several lines of evidence. First, native eutR transcript levels were consistently decreased in ΔmavR compared to WT (Figure 5E, 6F, Supplementary Figure S7B). Second, the RNase E truncation rescued stability and expression of both tagged and native eutR in the ΔmavR strain compared to WT (Figure 6E–G). Finally, MavR protected the eutR transcript from RNase E mediated cleavage (Figure 6H). Thus, MavR-dependent regulation ensures maximal EutR expression and activation of eut expression and EA utilization (Figure 6I and 11). Thus, the data presented in this study expands upon the model that sRNAs promote gene expression through association with the CDS of a target transcript.
In summary, we provide the initial characterization of a novel sRNA, MavR, describe the global impact of MavR on EHEC gene expression, and present the functional consequences of MavR to fitness, motility, and virulence. We also report mechanistic insights as to how MavR promotes EutR expression. Further investigation is required to verify other potential targets identified in the MAPS experiments as well as to determine how MavR influences expression of these targets. Altogether, the findings presented herein reveal a striking role for a bacterial sRNA in niche adaptation and bacterial-host interactions.
DATA AVAILABILITY
The sequencing data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE166491 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166491).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Beth Melson, Carol Rowley and CJ Anderson for feedback on this project, and the University of Maryland School of Medicine Genomics Resource Center for RNAseq and analysis services. We thank Hallie Rauch for help with RNA purification.
Contributor Information
Amber B Sauder, Department of Microbiology, Immunology and Cancer Biology, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
Melissa M Kendall, Department of Microbiology, Immunology and Cancer Biology, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health; National Institute of Allergy and Infectious Diseases [AI118732, AI154355, AI163565]; National Institutes of Health, National Institute of Allergy and Infectious Disease Biodefense Training [5 T32 AI055432 to A.B.S.]; UVA SOM Wagner Fellowship. Funding for open access charge: NIH AI163565.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kaper J.B., Nataro J.P., Mobley H.L.. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004; 2:123–140. [DOI] [PubMed] [Google Scholar]
- 2. McKenney E., Kendall M.. Microbiota and pathogen ‘pas de deux’: setting up and breaking down barriers to intestinal infection. Pathogens Disease. 2016; 74:ftw051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hall G.A., Chanter N., Bland A.P.. Comparison in gnotobiotic pigs of lesions caused by verotoxigenic and non-verotoxigenic Escherichia coli. Vet. Pathol. 1988; 25:205–210. [DOI] [PubMed] [Google Scholar]
- 4. Schmidt H., Russmann H., Schwarzkopf A., Aleksic S., Heesemann J., Karch H.. Prevalence of attaching and effacing Escherichia coli in stool samples from patients and controls. Zentralbl. Bakteriol. 1994; 281:201–213. [DOI] [PubMed] [Google Scholar]
- 5. Donnenberg M., Kaper J., Finlay B.. Interactions between enteropathogenic Escherichia coli and host epithelial cells. Trends Microbiol. 1997; 5:109–114. [DOI] [PubMed] [Google Scholar]
- 6. Fitzhenry R.J., Pickard D.J., Hartland E.L., Reece S., Dougan G., Phillips A.D., Frankel G.. Intimin type influences the site of human intestinal mucosal colonisation by enterohaemorrhagic Escherichia coli O157:H7. Gut. 2002; 50:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ritchie J.M., Waldor M.K.. The locus of enterocyte effacement-encoded effector proteins all promote enterohemorrhagic Escherichia coli pathogenicity in infant rabbits. Infect. Immun. 2005; 73:1466–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ritchie J.M., Thorpe C.M., Rogers A.B., Waldor M.K.. Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infect. Immun. 2003; 71:7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abe A., Heczko U., Hegele R.G., Brett Finlay B.. Two enteropathogenic Escherichia coli type III secreted proteins, EspA and EspB, are virulence factors. J. Exp. Med. 1998; 188:1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Posfai G., Koob M.D., Kirkpatrick H.A., Blattner F.R.. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J. Bacteriol. 1997; 179:4426–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong A.R., Pearson J.S., Bright M.D., Munera D., Robinson K.S., Lee S.F., Frankel G., Hartland E.L.. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol. Microbiol. 2011; 80:1420–1438. [DOI] [PubMed] [Google Scholar]
- 12. Hicks S., Frankel G., Kaper J.B., Dougan G., Phillips A.D.. Role of intimin and bundle-forming pili in enteropathogenic Escherichia coli adhesion to pediatric intestinal tissue in vitro. Infect. Immun. 1998; 66:1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kendall M.M., Sperandio V.. What a dinner party! Mechanisms and functions of interkingdom signaling in host-pathogen associations. mBio. 2016; 7:e01748-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Connolly J.P.R., Goldstone R.J., Burgess K., Cogdell R.J., Beatson S.A., Vollmer W., Smith D.G.E., Roe A.J.. The host metabolite D-serine contributes to bacterial niche specificity through gene selection. ISME J. 2015; 9:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Curtis M.M., Hu Z., Klimko C., Narayanan S., Deberardinis R., Sperandio V.. The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe. 2014; 16:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kendall M.M., Gruber C.C., Parker C.T., Sperandio V.. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. mBio. 2012; 3:e00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pacheco A.R., Curtis M.M., Ritchie J.M., Munera D., Waldor M.K., Moreira C.G., Sperandio V.. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012; 492:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kendall M.M., Gruber C.C., Rasko D.A., Hughes D.T., Sperandio V.. Hfq virulence regulation in enterohemorrhagic Escherichia coli O157:H7 strain 86-24. J. Bacteriol. 2011; 193:6843–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tree J.J., Granneman S., McAteer S.P., Tollervey D., Gally D.L.. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014; 55:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gottesman S., Storz G.. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 2011; 3:a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sauder A.B., Kendall M.M.. After the fact(or): post-transcriptional gene regulation in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 2018; 200:e00228-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Storz G., Vogel J., Wassarman K.M.. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011; 43:880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gruber C.C., Sperandio V.. Posttranscriptional control of microbe-induced rearrangement of host cell actin. mBio. 2014; 5:e01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Melson E.M., Kendall M.M.. The sRNA DicF integrates oxygen sensing to enhance enterohemorrhagic Escherichia coli virulence via distinctive RNA control mechanisms. Proc. Natl. Acad. Sci. USA. 2019; 116:14210–14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sy B.M., Lan R., Tree J.J.. Early termination of the Shiga toxin transcript generates a regulatory small RNA. Proc. Natl. Acad. Sci. USA. 2020; 117:25055–25065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayashi T., Makino K., Ohnishi M., Kurokawa K., Ishii K., Yokoyama K., Han C.-G., Ohtsubo E., Nakayama K., Murata T.et al.. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001; 8:11–22. [DOI] [PubMed] [Google Scholar]
- 27. Perna N.T., Plunkett G.r., Burland V., Mau B., Glasner J.D., Rose D.J., Mayhew G.F., Evans P.S., Gregor J., Kirkpatrick H.A.et al.. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001; 409:529–533. [DOI] [PubMed] [Google Scholar]
- 28. Gruber C.C., Sperandio V.. Global analysis of posttranscriptional regulation by GlmY and GlmZ in enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 2015; 83:1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sudo N., Soma A., Muto A., Iyoda S., Suh M., Kurihara N., Abe H., Tobe T., Ogura T., Hayashi H.et al.. A novel small regulatory RNA enhances cell motility in enterohemorrhagic Escherichia coli. J. Gen. Appl. Microbiol. 2014; 60:44–50. [DOI] [PubMed] [Google Scholar]
- 30. Iosub I., van Nues R., McKellar S., Nieken K., Marchioretto M., Sy B., Tree J., Viero G., Granneman S.. Hfq CLASH Uncovers sRNA-target interaction networks linked to nutrient availability adaptation. eLife. 2020; 9:e54655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carrier M.C., Lalaouna D., Massé E.. A game of tag: MAPS catches up on RNA interactomes. RNA Biol. 2016; 13:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carrier M., Morin C., Massé E.. On the prowl: An in vivo method to identify RNA partners of a sRNA. Methods Enzymol. 2018; 612:251–268. [DOI] [PubMed] [Google Scholar]
- 33. Bossi L., Figueroa-Bossi N.. Competing endogenous RNAs: a target-centric view of small RNA regulation in bacteria. Nat. Rev. Microbiol. 2016; 14:775–784. [DOI] [PubMed] [Google Scholar]
- 34. Frohlich K.S., Papenfort K., Berger A.A., Vogel J.. A conserved RpoS-dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acid Res. 2012; 40:3623–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lalaouna D., Morissette A., Carrier M.C., Massé E.. DsrA regulatory RNA represses both hns and rbsD mRNAs through distinct mechanisms in Escherichia coli. Mol. Microbiol. 2015; 98:357–369. [DOI] [PubMed] [Google Scholar]
- 36. Pfeiffer V., Papenfort K., Lucchini S., Hinton J.C.D., Vogel J.. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nature Struct. Mol. Biol. 2009; 16:840–847. [DOI] [PubMed] [Google Scholar]
- 37. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scotto-Lavino E., Du G., Frohman M.A.. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 2006; 1:2555–2562. [DOI] [PubMed] [Google Scholar]
- 39. Batey R., Kieft J.. Improved native affinity purification of RNA. RNA. 2007; 13:1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carlson-Banning K.M., Sperandio V.. mBio. 2016; 7:e01852-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roof D.M., Roth J.R.. Ethanolamine utilization in Salmonella typhimurium. J. Bacteriol. 1988; 170:3855–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roof D.M., Roth J.R.. Autogenous regulation of ethanolamine utilization by a transcriptional activator of the eut operon in Salmonella typhimurium. J. Bacteriol. 1992; 174:6634–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anderson C.J., Clark D.E., Adli M., Kendall M.M.. Ethanolamine signaling promotes Salmonella niche recognition and adaptation during infection. PLoS Pathog. 2015; 11:e1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bertin Y., Girardeau J.P., Chaucheyras-Durand F., Lyan B., Pujos-Guillot E., Harel J., Martin C.. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ. Microbiol. 2011; 13:365. [DOI] [PubMed] [Google Scholar]
- 45. Maadani A., Fox K., Mylonakis E., Garsin D.. Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect. Immun. 2007; 75:2634–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thiennimitr P., Winter S.E., Winter M.G., Xavier M.N., Tolstikov V., Huseby D.L., Sterzenbach T., Tsolis R.M., Roth J.R., Baumler A.J.. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA. 2011; 108:17480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anderson C.J., Satkovich J., Köseoğlu V., Agaisse H., Kendall M.M.. The Ethanolamine Permease EutH Promotes Vacuole Adaptation of Salmonella enterica and Listeria monocytogenes during Macrophage Infection. Infect. Immun. 2018; 86:e00172-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rowley C.A., Sauder A.B., Kendall M.M.. The Ethanolamine-Sensing Transcription Factor EutR Promotes Virulence and Transmission During Citrobacter rodentium Intestinal Infection. Infect. Immun. 2020; 88:e00137-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gonyar L.A., Kendall M.M.. Ethanolamine and choline promote expression of putative and characterized fimbriae in enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 2014; 82:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luzader D.H., Clark D.E., Gonyar L.A., Kendall M.M.. EutR is a direct regulator of genes that contribute to metabolism and virulence in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 2013; 195:4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wright P.R., Georg J., Mann M., Sorescu D.A., Richter A.S., Lott S., Kleinkauf R., Hess W.R., Backofen R.. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014; 42:W119–W123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raden M., Ali S.M., Alkhnbashi O.S., Busch A., Costa F., Davis J.A., Eggenhofer F., Gelhausen R., Georg J., Heyne S.et al.. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018; 46:W25–W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mann M., Wright P.R., Backofen R.. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017; 45:W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Busch A., Richter A.S., Backofen R.. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008; 24:2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faigenbaum-Romm R., Reich A., Gatt Y., Barsheshet M., Argaman L., Margalit H.. Hierarchy in Hfq chaperon occupancy of small RNA targets plays a major role in their regulation. Cell Rep. 2020; 30:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Melamed S., Peer A., Faigenbaum-Romm R., Gatt Y., Reiss N., Bar A., Altuvia Y., Argaman L., Margalit H.. Global mapping of small RNA-Target interactions in bacteria. Mol. Cell. 2016; 63:884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miyakoshi M., Chao Y., Vogel J.. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015; 34:1478–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kofoid E., Rappleye C., Stojiljkovic I., Roth J.. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J. Bacteriol. 1999; 181:5317–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roof D.M., Roth J.R.. Functions required for vitamin B12-dependent ethanolamine utilization in Salmonella typhimurium. J. Bacteriol. 1989; 171:3316–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sheppard D.E., Penrod J.T., Bobik T., Kofoid E., Roth J.R.. Evidence that a B12-adenosyl transferase is encoded within the ethanolamine operon of Salmonella enterica. J. Bacteriol. 2004; 186:7635–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stojiljkovic I., Bäumler A.J., Heffron F.. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J. Bacteriol. 1995; 177:1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheppard D.E., Roth J.R.. A rationale for autoinduction of a transcriptional activator: ethanolamine ammonia-lyase (EutBC) and the operon activator (EutR) compete for adenosyl-cobalamin in Salmonella typhimurium. J. Bacteriol. 1994; 176:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DebRoy S., Gebbie M., Ramesh A., Goodson J.R., Cruz M.R., vanHoof A., Winkler W.C., Garsin D.A.. Riboswitches. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science. 2014; 345:937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mellin J.R., Koutero M., Dar D., Nahori M.A., Sorek R., Cossart P.. Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science. 2014; 345:940–943. [DOI] [PubMed] [Google Scholar]
- 65. Silva I.J., Barahona S., Eyraud A., Lalaouna D., Figueroa-Bossi N., Masse E., Arraiano C.M.. SraL sRNA interaction regulates the terminator by preventing premature transcription termination of rho mRNA. Proc. Natl. Acad. Sci. USA. 2019; 116:3042–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sedlyarova N., Shamovsky I., Bharati B.K., Epshtein V., Chen J., Gottesman S., Schroeder R., Nudler E.. sRNA-Mediated control of transcription termination in E. coli. Cell. 2016; 167:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cardinale C.J., Washburn R.S., Tadigotla V.R., Brown L.M., Gottesman M.E., Nudler E.. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008; 320:935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen H., Shiroguchi K., Ge H., Xie X.S.. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol. Syst. Biol. 2015; 11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 70. Carpousis A.J. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007; 61:71–87. [DOI] [PubMed] [Google Scholar]
- 71. Carpousis A.J., Luisi B.F., McDowall K.J.. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 2009; 85:91–135. [DOI] [PubMed] [Google Scholar]
- 72. Belasco J.G. Ribonuclease E: Chopping knife and sculpting tool. Mol. Cell. 2017; 65:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Belasco J. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell Biol. 2010; 11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Waters S.A., McAteer S.P., Kudla G., Pang I., Deshpande N.P., Amos T.G., Leong K.W., Wilkins M.R., Strugnell R., Gally D.L.et al.. Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. EMBO J. 2017; 36:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lodato P.B., Kaper J.B.. Post-transcriptional processing of the LEE4 operon in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 2009; 71:273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kido M., Yamanaka K., Mitani T., Niki H., Ogura T., Hiraga S.. RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J. Bacteriol. 1996; 178:3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Taraseviciene L., Bjork G.R., Uhlin B.E.. Evidence for an RNA binding region in the Escherichia coli processing endoribonuclease RNase E. J. Biol. Chem. 1995; 270:26391–26398. [DOI] [PubMed] [Google Scholar]
- 78. Khemici V., Carpousis A.J.. The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol. Microbiol. 2004; 51:777–790. [DOI] [PubMed] [Google Scholar]
- 79. Chao Y., Li L., Girodat D., Förstner K.U., Said N., Corcoran C., Śmiga M., Papenfort K., Reinhardt R., Wieden H.J.et al.. In vivo cleavage map illuminates the central role of RNase E in coding and Non-coding RNA pathways. Mol. Cell. 2017; 65:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McDowall K.J., Cohen S.N.. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J. Mol. Biol. 1996; 255:349–355. [DOI] [PubMed] [Google Scholar]
- 81. Collins J.W., Keeney K.M., Crepin V.F., Rathinam V.A.K., Fitzgerald K.A., Finlay B.B., Frankel G.. Citrobacter rodentium: infection, inflammation, and the microbiota. Nat. Rev. Microbiol. 2014; 12:612–623. [DOI] [PubMed] [Google Scholar]
- 82. Wan B., Zhang Q., Ni J., Li S., Wen D., Li J., Xiao H., He P., Ou H.Y., Tao J.et al.. Type VI secretion system contributes to enterohemorrhagic Escherichia coli virulence by secreting catalase against host reactive oxygen species (ROS). PLoS Pathog. 2017; 13:e1006246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Arnosti D. Regulation of Escherichia coli Sigma F RNA Polymerase by flhD and flhC Flagellar Regulatory Genes. J. Bacteriol. 1990; 172:4106–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu X., Matsumura P.. The FlhD/FlhC complex, a transcriptional activator of the Escherichia coli flagellar class II operons. J. Bacteriol. 1994; 176:7345–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu X., Fujita N., Ishihama A., Matsumura P.. The C-terminal region of the alpha subunit of Escherichia coli RNA polymerase is required for transcriptional activation of the flagellar level II operons by the FlhD/FlhC complex. J. Bacteriol. 1995; 177:5186–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Garmendia J., Frankel G., Crepin V.F.. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect. Immun. 2005; 73:2573–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mellies J.L., Barron A.M., Carmona A.M.. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect. Immun. 2007; 75:4199–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mellies J.L., Lorenzen E.. Enterohemorrhagic Escherichia coli virulence gene regulation. Microbiology Spectrum. 2014; 2:EHEC-0004-2013. [DOI] [PubMed] [Google Scholar]
- 89. Furniss R., Clements A.. Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J. Bacteriol. 2017; 200:e00336-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chekabab S., Jubelin G., Dozois C., Harel J.. PhoB activates Escherichia coli O157:H7 virulence factors in response to inorganic phosphate limitation. PLoS One. 2014; 9:e94285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wadolkowski E.A., Burris J.A., O’Brien A.D.. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 1990; 58:2438–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Myhal M.L., Laux D.C., Cohen P.S.. Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1982; 1:186–192. [DOI] [PubMed] [Google Scholar]
- 93. Fabich A.J., Jones S.A., Chowdhury F.Z., Cernosek A., Anderson A., Smalley D., McHargue J.W., Hightower G.A., Smith J.T., Autieri S.M.et al.. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 2008; 76:1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Winter S.E., Thiennimitr P., Winter M.G., Butler B.P., Huseby D.L., Crawford R.W., Russell J.M., Bevins C.L., Adams L.G., Tsolis R.M.et al.. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010; 467:426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Winter S.E., Winter M.G., Xavier M.N., Thiennimitr P., Poon V., Keestra A.M., Laughlin R.C., Gomez G., Wu J., Lawhon S.D.et al.. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013; 339:708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ducarmon Q.R., Zwittink R.D., Hornung B.V.H., van Schaik W., Young V.B., Kuijper E.J.. Gut microbiota and colonization resistance against bacterial enteric infection. Microbiol. Mol. Biol. Rev. 2019; 83:e00007-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Best A., La Ragione R.M., Sayers A.R., Woodward M.J.. Role for flagella but not intimin in the persistent infection of the gastrointestinal tissues of specific-pathogen-free chicks by Shiga toxin-negative Escherichia coli O157:H7. Infect. Immun. 2005; 73:1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rogers T.J., Paton J.C., Wang H., Talbot U.M., Paton A.W.. Reduced virulence of an fliC mutant of Shiga-toxigenic Escherichia coli O113:H21. Infect. Immun. 2006; 74:1962–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Aviello G., Knaus U.G.. ROS in gastrointestinal inflammation: Rescue or sabotage?. Br. J. Pharmacol. 2017; 174:1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jones R.M., Mercante J.W., Neish A.S.. Reactive oxygen production induced by the gut microbiota: pharmacotherapeutic implications. Curr. Med. Chem. 2012; 19:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kamada N., Kim Y., Sham H.P., Vallance B.A., Puente J.L., Martens E.C., Núñez G.. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012; 336:1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lopez C.A., Miller B.M., F. R.-C., Velaquez E.M., Byndloss M.X., Chavez-Arroyo A., Lokken K.L., Tsolis R.M., Winter S.E., Baumler A.J.. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science. 2016; 353:1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Botteaux A., Hoste C., Dumont J.E., Van Sande J., Allaoui A.. Potential role of Noxes in the protection of mucosae: H(2)O(2) as a bacterial repellent. Microbes Infect. 2009; 11:537–544. [DOI] [PubMed] [Google Scholar]
- 104. Corcionivoschi N., Alvarez L.A., Sharp T.H., Strengert M., Alemka A., Mantell J., Verkade P., Knaus U.G., Bourke B.. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe. 2012; 12:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jost D., Nowojewski A., Levine E.. Small RNA biology is systems biology. BMB Rep. 2011; 44:11–21. [DOI] [PubMed] [Google Scholar]
- 106. Levine E., Zhang Z., Kuhlman T., Hwa T.. Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 2007; 5:e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yamada M., Makino K., Amemura M., Shinagawa H., Nakata A.. Regulation of the phosphate regulon of Escherichia coli: analysis of mutant phoB and phoR genes causing different phenotypes. J. Bacteriol. 1989; 171:5601–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sharma C.M., Papenfort K., Pernitzsch S.R., Mollenkopf H.-J., Hinton J.C., Vogel J.. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol. Microbiol. 2011; 81:1144–1165. [DOI] [PubMed] [Google Scholar]
- 109. Lee H.J., Gottesman S.. sRNA roles in regulating transcriptional regulators: Lrp and SoxS regulation by sRNAs. Nucleic Acids Res. 2016; 44:6907–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Coleman S., Smith M., Spicer V., Y L., Mookherjee N., Hancock R.. Overexpression of the Small RNA PA0805.1 in Pseudomonas aeruginosa modulates the expression of a large set of genes and proteins, resulting in altered motility, cytotoxicity, and tobramycin resistance. mSystems. 2020; 5:e00204-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tsoy O., Ravcheev D., Mushegian A.. Comparative genomics of ethanolamine utilization. J. Bacteriol. 2009; 191:7157–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Garsin D.A. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 2010; 8:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Papenfort K., Vanderpool C.K.. Target activation by regulatory RNAs in bacteria. FEMS Microbiol. Rev. 2015; 39:362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lalaouna D., Carrier M., Semsey S., Brouard J., Wang J., Wade J., Massé E.. A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol. Cell. 2015; 58:393–405. [DOI] [PubMed] [Google Scholar]
- 115. Chen H., Previero A., Deutscher M.P.. A novel mechanism of ribonuclease regulation: GcvB and Hfq stabilize the mRNA that encodes RNase BN/Z during exponential phase. J. Biol. Chem. 2019; 294:19997–20008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE166491 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166491).