


Abstract
RNA guanine quadruplexes (rG4) assume important roles in post-transcriptional regulations of gene expression, which are often modulated by rG4-binding proteins. Hence, understanding the biological functions of rG4s requires the identification and functional characterizations of rG4-recognition proteins. By employing a bioinformatic approach based on the analysis of overlap between peaks obtained from rG4-seq analysis and those detected in >230 eCLIP-seq datasets for RNA-binding proteins generated from the ENCODE project, we identified a large number of candidate rG4-binding proteins. We showed that one of these proteins, G3BP1, is able to bind directly to rG4 structures with high affinity and selectivity, where the binding entails its C-terminal RGG domain and is further enhanced by its RRM domain. Additionally, our seCLIP-Seq data revealed that pyridostatin, a small-molecule rG4 ligand, could displace G3BP1 from mRNA in cells, with the most pronounced effects being observed for the 3′-untranslated regions (3′-UTR) of mRNAs. Moreover, luciferase reporter assay results showed that G3BP1 positively regulates mRNA stability through its binding with rG4 structures. Together, we identified a number of candidate rG4-binding proteins and validated that G3BP1 can bind directly with rG4 structures and regulate the stabilities of mRNAs.
INTRODUCTION
The RNA guanine quadruplexes (rG4s) are non-canonical four-stranded RNA structures that form in guanine (G)-rich regions of the transcriptome (1). rG4s comprise two or more stacked layers of G-quartets, in which four guanines are assembled in a planar configuration via Hoogsteen hydrogen bonding and are stabilized by K+ ions (1). In vitro rG4 sequencing (rG4-seq) and in silico rG4 prediction revealed over 13 000 potential rG4-forming sites in the human transcriptome (2–5). Although these rG4s were suggested to be largely unfolded in cells (3), immunofluorescence microscopy analysis using G4 structure-specific antibody, live-cell imaging with rG4-binding fluorescent probes, and live-cell RNA labeling with N3-kethoxal followed by deep sequencing all support the existence of rG4s in cells (6–9). Hence, rG4 structures might be highly dynamic in cells.
Bioinformatic predictions and rG4-seq results revealed that rG4s are enriched within 5′- and 3′-untranslated regions (UTRs) of mRNAs, which may constitute an important mechanism for post-transcriptional regulations of gene expression (2,3,10). Indeed, rG4s are implicated in the control of mRNA targeting, processing, translation and degradation (11–14). The biological functions of rG4s in cells often involve cellular proteins (1,15,16). For instance, DHX36 unwinds rG4 structures located in 3′-UTRs of mRNAs to prevent their accumulation in stress granules (17). Thus, understanding fully the biological functions of rG4s entails the identification and functional assessments of rG4-recognition proteins.
In recent years, various methods have been developed for the identification of rG4-binding proteins. Most methods rely on affinity pull-down followed by mass spectrometric analysis (18–22). Several rG4-binding proteins have been identified so far, including AVEN, CNBP, DDX21, DDX3X, DHX36, FMRP, nucleolin, etc. (18,22–27). There are, however, likely many yet identified rG4-binding proteins.
The Encyclopedia of DNA Elements (ENCODE) project has produced hundreds of enhanced UV crosslinking and immunoprecipitation-sequencing (eCLIP-seq) datasets for RNA-binding proteins (RBPs) (28). We posit that the binding sites for rG4-binding proteins in the transcriptome should overlap extensively with rG4 sites identified from rG4-Seq. Herein, we employed a bioinformatic analysis to identify candidate rG4-binding proteins on the basis of similarity between RBP-binding sites and rG4 structure loci in the transcriptome, which are obtained from publicly available eCLIP-seq (29,30) and rG4-seq (2) datasets, respectively. We demonstrated that, among the many putative rG4-binding proteins identified, G3BP1 (Ras GTPase-activating protein-binding protein 1), a stress granule protein, could bind directly and selectively with rG4. We also revealed that this binding modulates the stabilities of mRNAs bearing rG4 structures in the UTRs.
MATERIALS AND METHODS
Cell culture
HeLa and HEK293T (293T) cells, which were purchased from ATCC (Manassas, VA), were cultured in Dulbecco's modified Eagle's medium (DMEM, life Technologies) containing 10% fetal bovine serum (Invitrogen) and 1% penicillin and streptomycin (Invitrogen), and the cells were maintained at 37°C in an incubator containing 5% CO2.
Bioinformatic analysis
ENCODE data were retrieved from the ENCODE portal under eCLIP assay title and cell line biosample classification. A total of 223 experimental results (Supplementary Table S1) were downloaded and the merged narrowpeak files were employed for overlapping analysis. rG4-seq data of HeLa cells were obtained using GEO accession number GSE77282 (2). IntervalStats (31) was employed for overlapping analysis. hg19 genome, rG4-KPDS-hit peak, and merged narrowpeak files of RBP eCLIP-seq peaks were used as domain, reference, and query, respectively. The resulting output was further filtered with a P-value cutoff of 0.05. Overlapping percentage was calculated as (# of overlapped peaks)/(total # of peaks for the target protein) × 100%. Binding motif and peak annotation were analyzed by using HOMER (v4.11) (32). Signal enrichment was analyzed by using bwtool (33). The mapped reads were visualized using the Integrative Genomics Viewer (IGV_2.6.0) (34).
Purification of recombinant proteins
The plasmid for producing recombinant GST-G3BP1 was constructed by first amplifying the G3BP1 gene from a cDNA library with primers containing BamHI and XhoI restriction recognition sites. The PCR amplicons were restriction digested and ligated into pGEX-4T1 vector, where the successful incorporation of the G3BP1 coding sequence (CDS) was confirmed by sequencing. For truncated proteins, the corresponding CDS was amplified by PCR and inserted into the pGEX-4T1 vector using the same method.
The plasmids were transformed into competent Rosetta (DE3) pLysS Escherichia coli cells, and protein expression was induced by incubating cells with 1 mM isopropyl β-d-1-thio-galactopyranoside (IPTG, Sigma) at 16°C for 20 h. The cells were subsequently harvested by centrifugation and lysed by sonication in a 20-ml ice-cold PBS buffer containing 10% (v/v) glycerol and 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma) for 10 min. The cell lysate was then centrifuged at 10 000 g for 15 min. The GST-tagged proteins were purified from the supernatant by using glutathione agarose beads (Pierce) following the manufacturer's recommended procedures. The full-length GST-G3BP1 protein was further purified using size-exclusion chromatography with a Superdex 200 increase 5/150 GL column and an AKTA Purifier 10 FPLC system (GE Healthcare). Protein purity was verified by SDS-PAGE analysis, quantified by Quick Start Bradford Protein Assay kit (Bio-Rad), and used immediately or stored at −80°C until use.
Fluorescence anisotropy
Fluorescently labeled RNA probes (500 nM, Integrated DNA Technologies, Supplementary Table S2) were dissolved in an RNase-free buffer containing 10 mM Tris–HCl (pH 7.5), 100 mM KCl and 0.1 mM EDTA. The annealing was conducted by heating the solution to 95°C for 5 min, followed by cooling slowly to room temperature over 3 h. The binding assays were performed with 10 nM RNA probes and the indicated concentrations of recombinant G3BP1 protein in a 60-μl binding buffer containing 10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 100 mM KCl, 0.1 mM DTT and 10 μg/ml BSA. After incubation on ice for 30 min, fluorescence anisotropy was recorded on a Horiba QuantaMaster-400 spectrofluorometer (Photon Technology International), with the excitation and emission wavelengths being set at 550 and 580 nm, respectively, as described previously (35). The instrument G factor was determined before anisotropy measurements, and the Kd values were calculated with GraphPad Prism 8 software using non-linear regression for curve fitting with a one site-specific binding model.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed using a previously reported method with some modifications (36). Briefly, various concentrations of recombinant G3BP1 protein were incubated with 200 fmol of fluorescently labeled RNA probes in a binding buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 100 mM KCl, 0.1 mM DTT, 10 μg/ml BSA). The mixtures were incubated on ice for 30 min and the protein-bound RNA substrate was separated from free RNA on a 6% native polyacrylamide gel using 1× TAE (40 mM Tris-acetate, pH 8.0, 2 mM EDTA) by electrophoresis at 4°C. Electrophoresis was performed at 120 V for 22 min and the gel was imaged with an Odyssey Imaging System (LI-COR Biosciences).
Circular dichroism (CD) spectroscopy
The CD spectra for G3BP1 protein, and annealed PITX1 rG4 and NRAS rG4 (at 3 μM each) in an RNase-free buffer (10 mM Tris–HCl, pH 7.5, 100 mM KCl and 1 mM EDTA) were recorded separately in the wavelength range of 200–320 nm on a Jasco-815 spectropolarimeter. Additionally, a 3-μM solution of annealed PITX1 rG4 was incubated with an equal concentration of G3BP1 protein in the same RNase-free buffer at 4°C for 30 min, and the CD spectrum of mixture was subsequently acquired in the same wavelength range. The CD spectrum of G3BP1 protein was subtracted from the composite CD spectrum of the mixture to yield the CD spectrum of rG4 in the G3BP1–rG4 complex.
In vitro pull-down experiment
Biotin-conjugated PITX1 rG4 and rM4 (Supplementary Table S3) were annealed individually in a buffer containing 10 mM Tris–HCl (pH 7.5), 100 mM KCl and 0.1 mM EDTA. The annealed RNA probes were incubated with high-capacity streptavidin agarose beads (Thermo Pierce) in a buffer containing 10 mM Tris–HCl (pH 7.5), 100 mM KCl, 1 mM EDTA and 0.1 mM DTT at 4°C for 1 h. After washing for three times with the binding buffer, the RNA-bound beads were incubated with the lysate of HEK293T cells at 4°C for 2 h in a buffer containing 10 mM Tris–HCl (pH 7.5), 100 mM KCl, 1 mM EDTA, 0.1 mM DTT, protease inhibitor (Sigma) and 0.4 units μl−1 RNase inhibitor (NEB). The beads were then washed thoroughly with the binding buffer supplemented with increasing concentrations of NaCl (100, 150 and 200 mM), followed by eluting the bound proteins from the beads through boiling in 3× SDS-PAGE loading buffer for 5 min. The resulting samples were subjected to Western blot analysis.
CRISPR/Cas9-mediated ablation of G3BP1 gene
G3BP1−/− HEK293T and HeLa cells were generated by genome editing with the CRISPR/Cas9 system following the previously reported protocol (37), where the single guide RNA (sgRNA) was designed according to previously published method (38). The guide sequence for the production of sgRNA targeting G3BP1 gene was inserted into the hSpCas9 plasmid pX330 (Addgene) at the BbsI digestion sites. After transfection and clonal isolation, successful deletion of the G3BP1 gene in single-cell clones was screened by Western blot using anti-G3BP1 antibody and the deleted loci in genomic DNA was further identified by Sanger sequencing. The sgRNA sequence is shown in Supplementary Table S3.
Western blot
Protein samples were separated on a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane (Bio-Rad). After blocking with blotting-grade blocker (Bio-Rad), the membrane was incubated with PBS-T (PBS buffer with 0.05% Tween 20) containing primary antibody and 5% BSA for 2 h, and then incubated with the HRP-conjugated secondary antibody in a 5% blotting-grade blocker. Following thorough washing with PBS-T, the immunoblots were detected using ECL Western blotting detection reagent (Amersham). Primary antibodies used in this study included G3BP1 (MBL International, RN048PW; 1:2000), PITX1 (Proteintech, 10873-1-AP; 1:1000), KHSRP (Proteintech, 55409-1-AP; 1:500), ACTR2 (Proteintech, 10922-1-AP; 1:2000), FLAG-tag (Cell Signaling Technology, D6W5B; 1:2000), and GAPDH (Santa Cruz, sc-32233; 1:10 000).
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted using Omega Total RNA Kit I (Omega) and quantified. Reverse transcription was conducted using M-MLV Reverse Transcriptase (Promega) to obtain the cDNA library. RT-qPCR was performed using Luna® Universal qPCR Master Mix (NEB) on the CFX96 RT-qPCR detection system (Bio-rad). Primers used for RT-qPCR are listed in Supplementary Table S3.
Dual-luciferase reporter assay
The wild-type plasmid (PITX1-WT) for the reporter assay was constructed from PCR amplification of the PITX1 3′-UTR (1045 bp) with primers containing XbaI and FseI restriction recognition sites. The digested PCR product was ligated into pGL3-promoter vector (Promega) and the successful incorporation of the PITX1 3′-UTR was confirmed by sequencing. The corresponding plasmid with the quadruplex sequences being mutated (PITX1-3Qm) was constructed by site-directed mutagenesis (39), and the successful mutation was again verified by sequencing. The Flag-PITX1-WT and Flag-PITX1-3Qm plasmids were constructed in two steps. First, the PITX1 CDS (945 bp) was amplified by PCR using primers containing NotI and EcoRI restriction recognition sites, and the digested PCR product was ligated into a pRK7 plasmid to give the pRK7-PITX1 plasmid. Subsequently, the coding region of Flag-PITX1 was amplified from the pRK7-PITX1 plasmid with primers containing NcoI and XbaI restriction recognition sites, and the digested PCR product was ligated into the above-mentioned PITX1-WT and PITX1-3Qm reporter plasmids by replacing the coding sequence of the luciferase gene. The successful constructions of Flag-PITX1-WT and Flag-PITX1-3Qm plasmids were confirmed by Sanger sequencing.
For the reporter assay, HEK293T cells and the isogenic G3BP1−/− cells were seeded in 12-well plates at a density of 2 × 105 cells per well. After 24 h, the cells (at ∼50% confluency) were co-transfected with the 0.05 μg renilla luciferase plasmid (pRL-CMV, Promega) and 1 μg firefly luciferase plasmid (PITX1-WT or PITX1-3Qm). After another 12 h, the cells were treated with 20 μM pyridostatin (PDS) or mock-treated with water. The cells were harvested for measurements 12 h later. For luciferase reporter assay, the attached cells were lysed in 1× passive lysis buffer and vortexed to obtain a homogeneous cell lysate. The firefly and renilla luciferase activities of the cell lysates were measured, with a 10-s read time, using the dual-luciferase® reporter assay system and a luminometer (Promega), following the manufacturer's instructions. For mRNA expression detection, total RNA was extracted and again quantified using RT-qPCR.
RNA half-life measurements
HEK293T and the isogenic G3BP1−/− cells were seeded in 6-well plates at a density of 5 × 105 cells per well. After 24 h, the cells (at ∼50% confluency) were untreated or treated with 20 μM PDS for 12 h before adding 5 μg/ml of actinomycin D to inhibit transcription. After the addition of actinomycin D, the cells were harvested at 0, 0.5, 1, 1.5, 3, 4.5, 6 and 9 h for total RNA extraction and the levels of transcripts of interest at different time points were quantified using RT-qPCR. The levels of target mRNAs at these time points were normalized to that of GAPDH and further normalized to that at 0 h to obtain the percentages of remaining mRNAs. RNA half-life was calculated with GraphPad Prism 8 software by fitting the percentages of remaining mRNA with single-phase exponential decay kinetics.
Single-end enhanced cross-linking and immunoprecipitation-sequencing (seCLIP-Seq) and data analysis
HEK293T cells were plated in 150 mm dishes at a density of 1 × 107 cells per dish. After 24 h, the cells (at ∼60% confluency) were untreated or treated with 20 μM PDS for 12 h, followed by irradiating with UVC to induce protein-RNA cross-linking, and the subsequent seCLIP experiments (40) were performed with an eCLIP Library Prep Kit (Eclipse BioInnovations) following the manufacturer's instructions. Two biological replicates were conducted for each experiment, and 20 million cells were used for each replicate. seCLIP cDNA libraries were subsequently quantified using an Agilent 2100 Bioanalyzer and multiplexed for sequencing on an Illumina HiSeq 4000 Sequencing System with single-end 100-bp read length. Sequencing data were analyzed following the eCLIP-seq processing pipeline with the default setting (28). For the comparison between ‘Ctrl’ and ‘PDS’ datasets, BEDtools was used with the criteria of at least 1 or 10 bp overlap for defining overlapped peaks (41). Metagene analysis was conducted using MetaPlotR Perl/R pipeline (42). G4 prediction was performed using G4Hunter, with the window size and threshold score being 25 and 1.4, respectively (43).
RESULTS
Bioinformatic discovery of candidate rG4-binding proteins
By employing a previously reported bioinformatic method for evaluating the similarity of chromatin immunoprecipitation-sequencing (ChIP-seq) data (44), we assessed, by calculating the P-values for proximity, the percentages of overlap between rG4 loci, which were uncovered by rG4-Seq (2), and the RNA-binding sites of 150 RBPs, which are based on the 233 eCLIP-seq datasets in the ENCODE database (29,30) (Figure 1 and Supplementary Table S1). To illustrate, we compared two RBPs with markedly different overlapping percentages with rG4-Seq data, i.e., AKAP1 (68.85%, a total of 5878 peaks) and MATR3 (10.32%, a total of 7168 peaks) (Supplementary Figure S1). Our results showed that most AKAP1 eCLIP-seq peaks overlap with rG4-seq peaks (Supplementary Figure S1A, B); MATR3 eCLIP-seq peaks, however, are staggered with rG4-seq peaks (Supplementary Figure S1C-D), suggesting the robustness of the bioinformatic approach in revealing overlaps between eCLIP-seq and rG4-seq peaks.
Figure 1.

Bioinformatic discovery of rG4-binding proteins. (A) A schematic diagram showing the workflow of the bioinformatic method. (B) A scatter plot illustrating the results obtained from bioinformatic analysis. (C) The distribution of G3BP1 eCLIP-seq peaks relative to the center of rG4-seq peaks; (D) the center of the overlapping peaks between G3BP1 eCLIP-seq and rG4-seq datasets. ‘Input’ represents size-match input dataset (ENCSR907EBB), which is a control eCLIP-seq dataset. (E) The most enriched motif of the overlapping peaks.
Importantly, the rG4-seq peaks exhibited high percentages of overlap with eCLIP-seq peaks of some known rG4-binding proteins, including DDX3X, FMR1, GRSF1, SRSF1 and YBX3 (Supplementary Table S1 and Figure 1B) (18,20,45), demonstrating the feasibility of this method in discovering novel rG4-binding proteins. We also evaluated the overlap between our rG4-binding proteins and the published rG4-binding proteins (Supplementary Table S2), and found that ∼50% of known rG4-binding proteins were identified in this study (Supplementary Figure S2). Aside from these known rG4-binding proteins, our bioinformatic analysis led to the discovery of a large number of candidate new rG4-binding proteins (Figure 1B and Supplementary Table S1).
G3BP1 is an rG4-binding protein
Among these candidate rG4-binding proteins, G3BP1, a well-known stress granule protein, exhibits a 49.1% overlap in its eCLIP-seq peaks (3210 out of 6541) with rG4-seq peaks. Signal enrichment analysis also revealed strong overlap between G3BP1 and rG4-seq peaks (Figure 1C). Results from motif analysis of the overlapping peaks revealed that the most enriched motif contains a G-rich sequence satisfying the criteria for rG4 formation (46) (Figure 1D). In addition, two representative overlapping regions contain putative rG4-forming sequences (Supplementary Figure S3), including the previously validated rG4 sequence in the 3′-UTR of LRP5 mRNA (47). Moreover, G3BP1 was recently shown to bind preferentially with mRNAs with highly structured 3′-UTRs (48). Together, these results suggest that G3BP1 is an rG4-binding protein, and we decided to choose this protein for further study.
We recognized that some proteins may bind to rG4 structures indirectly via protein-protein interactions, which may also give rise to high levels of overlap between their eCLIP-seq peaks and rG4-seq peaks. Hence, we next asked whether G3BP1 can bind directly with rG4 structures. To this end, we generated recombinant full-length G3BP1 protein (Supplementary Figure S4) and measured, by using fluorescence anisotropy, its binding affinities toward two previously characterized rG4 probes, one derived from the 5′-untranslated region (5′-UTR) of NRAS mRNA and the other from the 3′-UTR of PITX1 mRNA (13,49), and the corresponding mutated probes (rM4) that are unable to fold into G4 structure. CD measurement results showed that both PITX1 and NRAS rG4 sequences can assemble into parallel G4 topology, as manifested by negative and positive CD peaks at around 240 and 260 nm, respectively (Supplementary Figure S5A-B) (50). The fluorescence anisotropy results showed that G3BP1 binds strongly with both rG4 probes, with the Kd values being 63 ± 10 and 60 ± 5 nM for rG4 structures derived from the mRNAs of PITX1 and NRAS genes, respectively (Figure 2A, B). Moreover, the binding capacities of rG4 probes are much higher than those of the corresponding rM4 probes, with the Kd values being 761 ± 73 and 266 ± 36 nM, respectively (Figure 2A, B).
Figure 2.

G3BP1 binds directly and selectively with rG4 structures. Fluorescence anisotropy for measuring the binding affinities of G3BP1 protein toward rG4 structures derived from PITX1 (A) and NRAS (B) mRNAs and their corresponding mutants (rM4s). Error bars represent S.D. (n = 3). (C) Western blot images and quantitative results obtained from in vitro pull-down of G3BP1 protein from whole-cell protein lysates with the use of biotin-labeled PITX1 rG4 and rM4 probes. Error bars represent S.E.M. (n = 3). (D) A schematic diagram depicting the domain structure of G3BP1 protein. (E–H) EMSA for monitoring the interactions between the four truncated forms of G3BP1 with PITX1 rG4 probe. (I) Fluorescence anisotropy for measuring the binding affinities of G3BP1-RRM-RGG with PITX1 rG4 and rM4. Error bars represent S.D. (n = 3). The P values were calculated by unpaired, two-tailed Student's t-test. ***P < 0.001.
We next assessed the binding capacities of G3BP1 toward rG4 and rM4 in cell lysate. To this end, we performed an in vitro pull-down experiment with biotin-labeled PITX1 rG4 and rM4 probes. The results showed that, with the use of the same amount of lysate, PITX1 rG4 probe was capable of pulling down >10-fold more G3BP1 than the corresponding rM4 probe under the same experimental conditions (Figure 2C). This is consistent with our fluorescence anisotropy results (Figure 2A), suggesting that G3BP1 is able to bind selectively to rG4 over rM4 in complex sample matrix (i.e., whole-cell protein lysate).
We also found that the G3BP1–rG4 interaction could be disrupted by PDS, a small-molecule ligand that can bind to and stabilize G4 structures (Supplementary Figure S6) (51). Additionally, CD measurement results showed that the G3BP1-rG4 interaction did not perturb the quadruplex folding of rG4 (Supplementary Figure S5). Together, these results establish G3BP1 as an rG4-recognition protein.
The C-terminal RGG domain of G3BP1 is involved with rG4 recognition
G3BP1 is an evolutionarily conserved, multi-domain protein harboring an N-terminal NTF2-like domain, an acidic residue-rich region, PXXP motifs, an RNA-recognition motif (RRM) and a C-terminal arginine–glycine–glycine (RGG) domain (Figure 2D) (52,53). The RRM and RGG domains are common RNA-binding modules (54); therefore, we assessed whether these domains of G3BP1 are required for rG4 recognition.
We generated several truncated forms of G3BP1 protein, including those with the RRM and RGG domains being deleted (ΔRRM–RGG, 1–339 aa), or carrying both the RRM and RGG domains (340–466 aa, G3BP1–RRM–RGG) or either domain alone (340–415 aa, G3BP1–RRM and 430–466 aa, G3BP1–RGG) (Supplementary Figure S4). We next examined the binding capacities of these truncated forms of G3BP1 toward PITX1 rG4 using EMSA. We found that G3BP1–RRM and ΔRRM–RGG, both of which lack the RGG domain, did not display appreciable interaction with rG4 (Figure 2E, F); G3BP1–RRM–RGG and G3BP1–RGG can bind with rG4, though the latter exhibited lower binding affinity than the former (Figure 2G, H).
We also measured the binding affinities of G3BP1–RRM–RGG toward rG4 and rM4 by using fluorescence anisotropy. The Kd values for the truncated G3BP1 in binding with PITX1 rG4 and rM4 were 78 ± 9 and 4500 ± 900 nM, respectively (Figure 2I); hence, G3BP1–RRM–RGG exhibits a comparable binding affinity and a much higher binding selectivity toward rG4 structure than the full-length protein. Together, these results demonstrated that the binding capacity of G3BP1 toward rG4 structure arises mainly from the C-terminal RGG domain and is enhanced by the RRM domain.
G3BP1 regulates the stability of PITX1 mRNA in an rG4-dependent manner
Encouraged by the in-vitro binding results, we next examined the biological functions of G3BP1–rG4 interaction. One of the rG4 sequences employed for the aforementioned in-vitro binding assay was derived from the 3′-UTR of PITX1 mRNA, which was previously employed for assessing rG4-mediated regulation of mRNA translation (24,49). Thus, we chose PITX1 as a target to investigate whether G3BP1 can modulate the stability and translational efficiency of PITX1 mRNA through its binding with rG4 structures. To this end, we generated 293T cells with the G3BP1 gene being knocked out with CRISPR-Cas9 (G3BP1−/−). The successful depletion of G3BP1 protein in 293T cells was confirmed by Western blot (Figure 3A), and the deleted loci in genomic DNA were further identified by Sanger sequencing (Supplementary Figure S7A).
Figure 3.
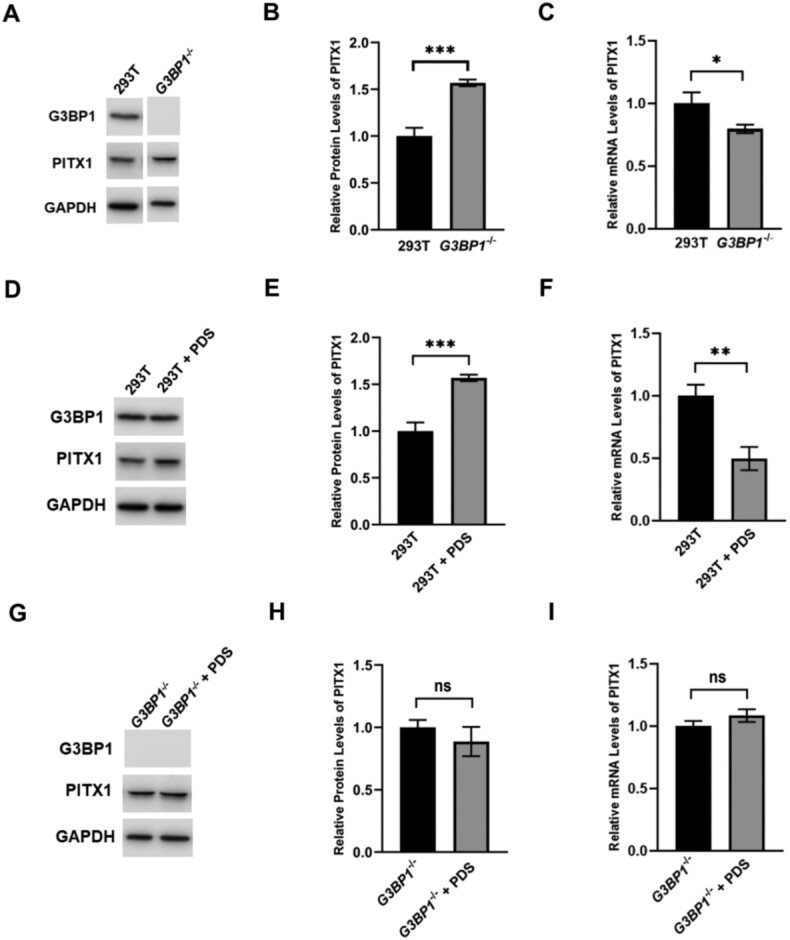
Genetic depletion of G3BP1 and PDS treatment confer similar effects on up-regulating the protein level and down-regulating the mRNA level of PITX1. Western blot and RT-qPCR analyses for monitoring the protein and mRNA levels of PITX1 in 293T cells and the isogenic G3BP1−/− cells (A–C), in 293T cells with or without PDS treatment (D–F), and in G3BP1−/− cells with or without PDS treatment (G–I). Error bars represent S.D. (n = 3). The P values were calculated by unpaired, two-tailed Student's t-test. *, 0.01 ≤ P < 0.05; **, 0.001 ≤ P < 0.01; ***, P < 0.001.
Results from Western blot analysis showed that the level of PITX1 protein was significantly increased after genetic depletion of G3BP1 in 293T cells (Figure 3B). This result suggests that G3BP1 may regulate the decay of PITX1 mRNA by binding to rG4 in its 3′-UTR or negatively modulate the translation of PITX1 mRNA. To distinguish these two scenarios, we monitored the mRNA level of PITX1 by RT-qPCR. The results showed that genetic depletion of G3BP1 in 293T cells led to a decrease in PITX1 mRNA level (Figure 3C), indicating that complete removal of G3BP1 may affect the stability of PITX1 mRNA. Together, G3BP1 positively modulates the stability of PITX1 mRNA, but it negatively regulates the translation of PITX1 mRNA.
We next investigated whether this regulation depends on G3BP1-rG4 interaction. Because PDS can disrupt the interactions between G3BP1 and rG4 structures in vitro (Supplementary Figure S6), we examined how PDS treatment modulates the translation of PITX1 mRNA and whether this depends on G3BP1. Our results showed that PDS treatment led to an increased level of PITX1 protein in 293T cells (Figure 3D-F); this increase, however, was abolished in the isogenic G3BP1−/− cells (Figure 3G-I). Additionally, PDS treatment elicits a decrease in the mRNA level of PITX1 in 293T cells, but not in G3BP1−/− cells (Figure 3F, I).
To further investigate the roles of G3BP1 and PDS in modulating mRNA stability, we measured the half-lives of PITX1 mRNA in 293T cells and G3BP1−/− cells with or without PDS treatment. As expected, genetic depletion of G3BP1 and PDS treatment both result in significant diminutions in the half-life of PITX1 mRNA in 293T cells (Figure 4A, B), with more pronounced effect being observed for PDS-treated cells. Additionally, PDS treatment can also diminish the half-life of PITX1 mRNA in G3BP1−/− cells (Figure 4A, B), indicating the potential role of PDS in regulating the stability of PITX1 mRNA through G3BP1-independent mechanism(s) (e.g., via displacing other proteins from rG4 structures in PITX1 mRNA). Together, these results underscore that PDS disrupts the interactions of G3BP1 with rG4 structures located in the 3′-UTR of PITX1 mRNA, thereby abrogating the regulatory effects of G3BP1 on the stability and translational efficiency of PITX1 mRNA.
Figure 4.
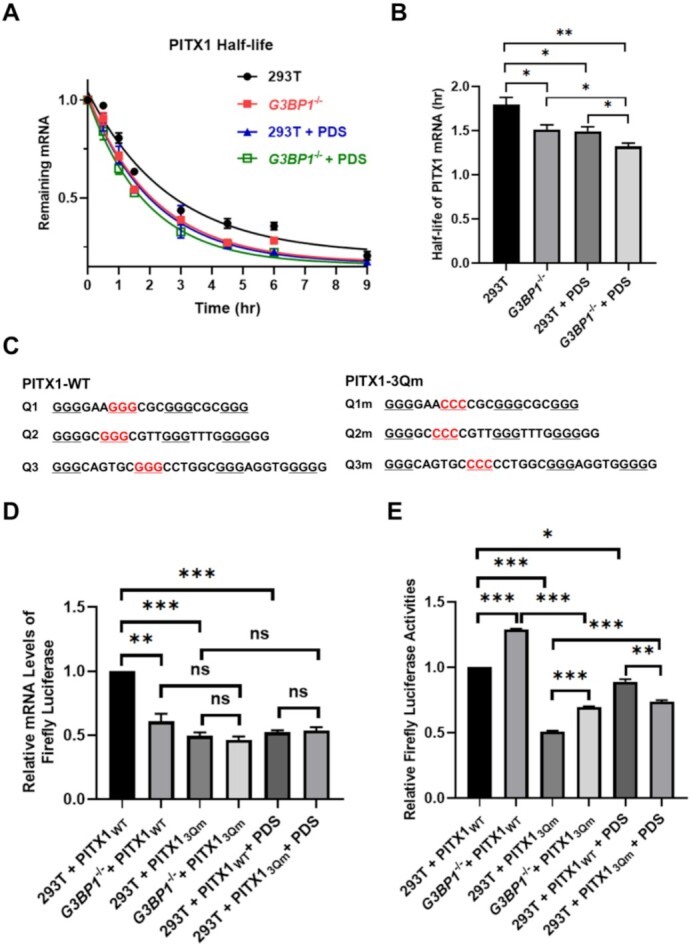
G3BP1 regulates the stability of PITX1 mRNA through its interaction with rG4 structures. (A) RT-qPCR results showing the half-lives of PITX1 mRNA in 293T cells, G3BP1−/− cells, PDS-treated 293T cells, and PDS-treated G3BP1−/− cells. (B) Bar chart showing the half-lives calculated from the above RT-qPCR results. (C) rG4-forming sequences in the 3′-UTR of PITX1 mRNA and their corresponding mutants. (D) Relative firefly luciferase mRNA levels (normalized to the level of renilla luciferase mRNA) and (E) relative firefly luciferase activities (normalized to renilla luciferase activity) in 293T cells, G3BP1−/− cells and PDS-treated 293T cells expressed from PITX1-WT or PITX1-3Qm plasmid. Error bars represent S.E.M. (n = 3). The P values were calculated by using unpaired, two-tailed Student's t-test. ns, P > 0.05; **, 0.001 ≤ P < 0.01; ***P < 0.001.
To further determine whether the G3BP1-mediated regulation of PITX1 mRNA is rG4-dependent, we performed dual-luciferase reporter assay using the wild-type 3′-UTR of PITX1 mRNA (PITX1-WT) as well as its variant with all three rG4-forming sequences being mutated (PITX1-3Qm) (Figure 4C). We monitored both the mRNA and protein expression levels of firefly luciferase by using the corresponding signals from renilla luciferase as internal standard. Our results showed that genetic depletion of G3BP1 led to markedly diminished level of luciferase mRNA arising from transcription of PITX1-WT plasmid (Figure 4D). No appreciable difference in mRNA levels was, however, observed between two cell lines when transfected with PITX1-3Qm. These results support that G3BP1 positively regulates the stability of PITX1 mRNA and this regulation depends on rG4 structures located in the 3′-UTR of PITX1 mRNA. Moreover, the mRNA level of PITX1-3Qm was much lower than that of PITX1-WT in 293T cells, which again supports the role of the 3′-UTR rG4 structures in stabilizing of PITX1 mRNA.
In line with what was observed for endogenous PITX1 protein, we found that the firefly luciferase activity of PITX1-WT exhibited a pronounced increase upon genetic ablation of G3BP1 in 293T cells (Figure 4E). A similar increase was also observed for PITX1-3Qm (Figure 4E), indicating that G3BP1 also regulated the translation of PITX1 mRNA in an rG4-independent manner. A combination of mRNA and luciferase activity results showed that G3BP1 exerts a greater effect on the translational efficiency of PITX1-WT over PITX1-3Qm mRNA (Figure 4D-E). Additionally, the luciferase activity of PITX1-3Qm was also lower than PITX1-WT in G3BP1−/− cells (Figure 4E), underscoring that rG4s can also enhance the translation of PITX1 mRNA through a G3BP1-independent mechanism.
We also examined the expression levels of PITX1-WT and PITX1-3Qm in 293T cells with or without PDS treatment. We observed a significant decrease in the mRNA level of PITX1-WT upon PDS treatment (Figure 4D), which is in agreement with the aforementioned result of the endogenous PITX1 mRNA (Figure 3I). PDS treatment, nevertheless, did not alter the mRNA level of PITX1-3Qm, and the treatment also abolished the difference in mRNA levels of PITX1-WT and PITX1-3Qm in 293T cells (Figure 4D). Moreover, while PDS treatment led to a slight decrease in the luciferase activity of PITX1-WT, the decrease is not as pronounced as that observed at the mRNA level (Figure 4D-E). This result suggests an elevated translational efficiency elicited by PDS treatment and parallels what we observed for endogenous PITX1 mRNA.
To explore further how G3BP1-rG4 interaction modulates the expression of PITX1 at mRNA and protein levels, we constructed Flag-PITX1-WT and Flag-PITX1-3Qm plasmids by replacing the coding sequence of the luciferase gene in the aforementioned PITX1-WT and PITX1-3Qm reporter plasmids with that of an N-terminally Flag-tagged PITX1. Our RT-qPCR results revealed that, after transfection with the same amounts of plasmids, the mRNA expression level of Flag-PITX1-WT was significantly higher than that of Flag-PITX1-3Qm in HEK293T cells (Supplementary Figure S8A); genetic depletion of G3BP1, however, abolishes the difference observed for the Flag-PITX1-WT and Flag-PITX1-3Qm mRNAs (Supplementary Figure S8D).
Our Western blot data showed that, upon transfection of HEK293T cells with the same amounts of plasmids, Flag-PITX1-3Qm protein was expressed at 45% level relative to that of Flag-PITX1-WT (Supplementary Figure S8B, C). At first glance, this appears to be incongruent with what we may predict from our observations made for the endogenous PITX1 gene. Nevertheless, the Flag-PITX1 mRNA transcribed from the transfected plasmid does not carry the 5′-UTR of endogenous PTIX1 mRNA, and it additionally contains the coding sequence for the Flag epitope tag. These differences may modulate differential mRNA recognitions by other RNA-binding proteins and/or ribosomes, thereby exerting different effects on the translation between endogenous PITX1 mRNA and that transcribed from the ectopically introduced plasmid. Nevertheless, genetic ablation of G3BP1 led to a markedly higher level (by ∼60-fold) of expression of Flag-PITX1-WT than Flag-PITX1-3Qm protein (Supplementary Figure S8E, F). Together, comparison of the above results obtained for Flag-PITX1-WT and Flag-PITX1-3Qm in HEK293T and the isogenic G3BP1-deficient background allowed us to conclude that G3BP1–rG4 interaction increases the stability of PITX1 mRNA and attenuates its translation efficiency, which mirrors what we observed for the endogenous PITX1 mRNA.
Together, these results support that G3BP1 positively regulates mRNA stability through its interaction with rG4 structures and negatively modulates the translation of mRNAs with rG4 structures in the 3′-UTRs. Nevertheless, our results also suggest the contributions of some mechanisms that are independent of rG4 and/or G3BP1 in these processes.
G3BP1 binds with rG4 structures in cells to regulate the stabilities of other mRNAs
To further explore the G3BP1-rG4 interactions in cells, we conducted seCLIP-seq experiments in HEK293T cells with (‘PDS’) or without (‘Ctrl’) PDS treatment. These experiments resulted in the identification of ∼1400 and ∼2900 mRNA binding sites for G3BP1 in ‘Ctrl’ and ‘PDS’ datasets, respectively (Figure 5A), and the overall signal intensities of G3BP1 peaks were higher in the ‘PDS’ dataset than the ‘Ctrl’ dataset (Figure 5B). The comparison between ‘Ctrl’ and ‘PDS’ seCLIP-seq datasets allowed for the identification of ∼800 peaks with at least 1 bp of overlap (Figure 5A); among these overlapping peaks, ∼100 contain putative rG4 sequences. Strikingly, the signal ratios of PDS/Ctrl of these rG4-containing peaks were significantly lower than the overall ratios of all overlapping peaks (Figure 5C), indicating the ability of PDS to displace G3BP1 from rG4 loci in the transcriptome. This finding corroborates the aforementioned in-vitro binding result and suggests that G3BP1 interacts with rG4 structures in cells.
Figure 5.

seCLIP-seq analyses of G3BP1 in 293T cells without (‘Ctrl’) or with (‘PDS’) PDS treatment reveal the G3BP1-rG4 interaction in cells. (A) A Venn diagram showing the overlap between ‘Ctrl’ and ‘PDS’ datasets. (B) The comparison between ‘Ctrl’ and ‘PDS’ peak intensities in log2(FoldChange) of IP sample and Input sample. (C) Signal ratios of PDS/Ctrl in all overlapping peaks and rG4-containing overlapping peaks. (D) Metagene analyses for profiling the transcriptomic distributions of ‘Ctrl’ and ‘PDS’ datasets. (E) Metagene analyses for profiling the transcriptomic distributions of decreased peaks, increased peaks and Ctrl-only peaks in ‘Ctrl’ dataset relative to the ‘PDS’ dataset. (F) IGV plots showing the comparison of ‘Ctrl’ and ‘PDS’ peaks around the G4-forming sequences located in the 5′- and 3′-UTRs of KHSRP mRNA. ‘Input’ represents size-match input sample. (G, H) RT-qPCR results showing the relative mRNA levels of KHSRP (G) and ACTR2 (H) genes in 293T cells and G3BP1−/− cells without or with PDS treatment. (I–K) Western blot analysis for monitoring the protein levels of KHSRP (J) and ACTR2 (K) genes in 293T cells and G3BP1−/− cells, and PDS-treated 293T cells. Error bars represent S.D. (n = 3). The P values were calculated by using unpaired, two-tailed Student's t-test. ns, P > 0.05; **, 0.001 ≤ P < 0.01; ***P < 0.001.
The density plots obtained from metagene analysis showed that the binding sites of G3BP1 are enriched in the 5′-UTR, followed by 3′-UTR and CDS regions (Figure 5D). However, after PDS treatment, the peak in the 3′-UTR completely shifted to the CDS regions, which is accompanied with a slight drop in signal in the 5′-UTR (Figure 5D), indicating that PDS can specifically disrupt the mRNA binding sites of G3BP1 at UTRs, especially the 3′-UTRs. Additionally, the peak annotation results showed an increased occupancy of G3BP1 in CDS regions, yet a decrease at the UTRs after PDS treatment (Supplementary Figure S9).
We also compared the transcriptomic distributions of decreased peaks (ratios of PDS/Ctrl <0.67, with at least 10 bp overlap) and increased peaks (ratios of PDS/Ctrl >1.5, with at least 10 bp overlap) in ‘Ctrl’ dataset after PDS treatment, as well as Ctrl-only peaks (no overlap with PDS). Here, we assumed that Ctrl-only peaks contain G3BP1 binding loci which are completely competed off by PDS. The density plots showed that Ctrl-only peaks and peaks that are decreased upon PDS treatment are predominately located in the UTRs, whereas those that are increased upon PDS treatment are mainly distributed in the CDS regions (Figure 5E). These results support that PDS displaces selectively G3BP1 from UTRs in mRNAs. Given that rG4 structures are highly enriched in UTRs (2), these results indicate the ability of PDS in disrupting specifically the interactions between G3BP1 and rG4 structure sites in cells. In this vein, it is worth noting that G3BP1 binding peaks around rG4 loci of PITX1 and NRAS mRNAs were very weak (Supplementary Figures S10-S11), which might be due to the low abundance of these transcripts, dynamic nature of rG4 structures in these mRNAs, their dynamic interactions with G3BP1, and/or insufficient sequencing depth.
We next asked whether G3BP1 depletion and PDS treatment exert similar effects on the stabilities and translational efficiencies of target transcripts. We monitored the mRNA and protein levels of two representative genes (i.e., KHSRP and ACTR2), which were chosen on the basis of decreased G3BP1 occupancy at rG4 sites in UTRs after PDS treatment (Figure 5F and Supplementary Figures S12 and S13). The results showed that the mRNA levels of the KHSRP and ACTR2 genes were markedly attenuated in G3BP1-depleted and PDS-treated cells, with the decreases being much more pronounced in PDS-treated cells than G3BP1-depleted cells (Figure 5G, H). We also monitored the stabilities of KHSRP and ACTR2 mRNAs, and found that the half-lives of these two mRNAs were substantially decreased in G3BP1−/− and PDS-treated 293T cells relative to parental 293T cells without any treatment (Supplementary Figure S14). Western blot results showed a significant increase in protein level of KHSRP in G3BP1-depleted cells, and an elevated translational efficiency of KHSRP upon PDS treatment (Figure 5I, J). Similarly, we observed a substantial increase in translational efficiency of ACTR2 mRNA in PDS-treated cells. In this vein, the ratio in the expression levels of ACTR2 mRNA was 0.29 ± 0.06 in PDS-treated over untreated HEK293T cells, and the corresponding ratio of ACTR2 protein was 0.73 ± 0.14 (Figure 5I, K). Loss of G3BP1, on the other hand, led to a slight, though statistically insignificant increase in translational efficiency of ACTR2 mRNA, where the ratios of mRNA and protein expression levels were 0.68 ± 0.13 and 0.81 ± 0.21, respectively, in G3BP1−/− over parental HEK293T cells (Figure 5I&J). The more pronounced effect exerted by PDS treatment over genetic ablation of G3BP1 may emanate from translational regulation of ACTR2 mRNA by other protein(s) that recognize rG4 structures in its 3′-UTR. Cumulatively, these results again support a role of G3BP1–rG4 interaction in modulating the stabilities and translational efficiencies of mRNAs.
We next examined whether the above findings made for 293T cells are general. To this end, we first investigated the effects of G3BP1 ablation and PDS treatment on the mRNA and protein levels of PITX1, KHSRP and ACTR2 genes in HeLa cells. RT-qPCR and Western blot results showed that the mRNA levels of the three genes dropped pronouncedly after PDS treatment, which is accompanied with slight increases in translation efficiencies (Supplementary Figure S15A–C). In addition, CRISPR-mediated genetic ablation of G3BP1 in HeLa cells (Supplementary Figure S7B) led to diminished mRNA levels and elevated translation efficiencies of these three genes (Supplementary Figure S15). Moreover, by analyzing the publicly available RNA-seq data from ENCODE, we found decreased mRNA levels of the three genes in G3BP1-depleted HepG2 and K562 cells (Supplementary Figure S16). These results suggest that the effects of G3BP1 depletion on rG4-bearing transcripts are general in mammalian cells.
DISCUSSION
G-rich sequences in RNAs can fold into rG4 structures, which modulate the stabilities and translational efficiencies of mRNAs (55). Some RBPs, which can bind to rG4s in their folded and unfolded forms, are highly correlated with the rG4-mediated regulations of mRNAs. For instance, RNA helicases in cells can bind to and unwind rG4 structures into single-stranded RNAs which are subsequently recognized by G-rich element-binding proteins (e.g., hnRNP H/F and CNBP) and prevent their refolding into rG4 structures, thereby increasing the translational efficiencies of mRNAs (26,56). In addition, GRSF1 regulates the degradation of rG4-containing mRNAs through unwinding rG4 structures to facilitate degradosome-mediated decay (57). Therefore, there is a growing interest in identifying rG4-binding proteins and characterizing their functions.
We employed a bioinformatic approach, relying on the analysis of overlapping peaks between publicly available rG4-seq dataset (2) and eCLIP-seq datasets for RBPs (28), to uncover putative novel rG4-binding proteins. Our analysis captured some previously reported rG4-binding proteins, which display high levels of occupancy at rG4 loci in the transcriptome (Figure 1 and Supplementary Table S1), validating the ability of the method in identifying candidate rG4-binding proteins. Importantly, our analysis also led to the identification of a very large number of putative rG4-binding proteins whose capabilities in binding to rG4 structures have not been previously documented. This provides an important list of proteins for the research community to assess their direct interactions with rG4s and to explore their biological functions. It is worth noting a limitation of our analysis. In particular, the rG4-seq dataset and eCLIP-seq datasets were generated from different cell lines and the distributions in rG4 in mRNAs and RNA-protein interactions can vary with cell lines, which may lead to false-negative discovery of putative rG4-binding proteins.
Among the top-ranked candidate rG4-binding proteins are splicing factors (e.g., FMR1, RMB15, PPIG, PRPF8 and SRSF1) or helicases (e.g., AQR, DDX3X, DDX55, DDX6, G3BP1 and UPF1). These results are in accordance with the fact that rG4s are implicated in control of mRNA processing and translation through splicing factors and RNA helicases, respectively (11,14,49).
G3BP1 plays an essential role in stress granule formation, DNA-triggered cGAS/STING pathway, RIG-I-mediated cellular antiviral response and innate immune response (58–61). It also displays Mg2+- and ATP-dependent helicase activity (62). Here, we identified G3BP1 as a direct rG4-binding protein with low-nM binding affinities, which are much stronger than those for binding with the corresponding rM4 probes (Figure 2A–C). We also found that the C-terminal RGG domain of G3BP1 is indispensable for its binding toward rG4 structures (Figure 2E-H). Moreover, the G3BP1–RRM–RGG truncated protein exhibits a much higher rG4-binding selectivity than the full-length G3BP1 protein (Figure 2I). In this vein, RGG domain is the second most common RNA-binding domain present in the human proteome (63), and several known G4-binding proteins, including CIRBP, FMRP and TLS/FUS, recognize G4 structures through their RGG domains (45,64,65).
We also found that PDS, a small-molecule G4 ligand, could disrupt pronouncedly the G3BP1-rG4 interaction in vitro. In addition, our seCLIP-seq results revealed that, upon PDS treatment, the signal intensities of rG4-containing peaks decreased (Figure 5C) and the transcriptome-wide distribution of G3BP1-binding sites exhibited a drastic shift from 3′-UTRs to CDS regions (Figure 5D, E & Supplementary Figure S8). Given that rG4-forming sequences are highly enriched in UTRs (2), our results provide strong evidence to support that PDS can disrupt G3BP1–rG4 interactions in cells.
Over the last few years, increasing lines of evidence support that rG4 structures assume critical roles in regulating pre-mRNA processing (splicing and polyadenylation), mRNA stability and translation (1,17,24). These regulatory processes often entail rG4-binding proteins to modulate G4 conformation and/or serve as bridges to recruit additional regulatory proteins. Here, we demonstrated that G3BP1 can enhance the stability and suppress the translational efficiency of PITX1 mRNA, which harbors three rG4 structures in its 3′-UTR. Further analysis revealed that treatment with PDS and genetic depletion of G3BP1 (Figure 3), both of which disrupted the interactions between G3BP1 and rG4 structures in PITX1 mRNA, abolished the regulatory effect of G3BP1, underscoring the importance of G3BP1–rG4 interaction in RNA metabolism. This notion finds additional support from dual-luciferase reporter assay results, showing that loss of G3BP1 and PDS treatment led to diminished mRNA levels and elevated translation of luciferase mRNA harboring PITX1 3′-UTR (PITX1-WT). The modulatory effects of G3BP1 depletion and PDS treatment were also observed for two other target transcripts (i.e., KHSRP and ACTR2) chosen based on seCLIP-seq results (Figure 5G, H), further illustrating the rG4-dependent regulatory functions of G3BP1 on the stabilities and translation efficiencies of mRNA. In this vein, the roles of rG4 in modulating the stabilities of mRNAs are also supported by the observation that genetic depletion of some putative rG4-binding proteins led to more pronounced alterations in mRNA expressions of those genes with putative G4 structures in the UTRs than those without (66).
G3BP1 is the central node and molecular switch that trigger RNA-protein phase separation (67,68), and rG4 structures can promote RNA phase separation (69,70). Our findings are in agreement with the previous observation that the formation of G3BP1–mRNA ribonucleoprotein particles or stress granules could protect mRNAs from degradation, while concomitantly confer a poor translation efficiency (60). It is of note that this G3BP1-mediated effect was observed in stressed cells, and there is little evidence to support a similar function of G3BP1 in unstressed cells. Another possibility is that, G3BP1 preferentially interacts with G4-containing RNAs, which may attenuate partially the auto-inhibitory effect of G3BP1 under normal conditions (67), thereby increasing the accessibility of G3BP1 to other RNAs and proteins to form large protein-RNA complexes. Like ribonucleoprotein particles and stress granules, these protein–RNA complexes can protect mRNAs from degradation and suppress their translation. Thus, it is also possible that G3BP1–rG4 complex acts as a scaffold to recruit other RNAs and proteins.
G3BP1 contains an NTF2-like domain and multiple intrinsically disordered regions (IDRs), which regulate the dimerization of G3BP1 and G3BP1-RNA interaction, respectively (67). The IDRs of G3BP1 resemble the RNA recognition motifs (RRMs) of other stress granule proteins, including hnRNPA1 and hnRNPA2B1 (Supplementary Figure S17), whose RRMs were shown to be capable of substituting the IDRs of G3BP1 to support the RNA-dependent liquid-liquid phase separation and stress granules assembly (67). Different from other stress granule proteins, G3BP1 exhibits intramolecular interaction between IDR1 and IDR3, and is susceptible to dimerization via the NTF2-like domain, which are essential for liquid-liquid phase separation and the maintenance of stress granules in cells. Hence, these two properties may endow G3BP1 the ability to be the core component of the stress granule network.
It will be important to examine, in the future, other top-ranked candidate rG4-binding proteins identified from our bioinformatics analysis, e.g., UPF1, DDX55, DDX6 and RBM15. In this regard, UPF1, an ATP-dependent RNA helicase, was found to regulate the decay of highly structured RNA in cooperation with G3BP1 (48) and target GC-rich region to trigger RNA decay (71). Notably, the function of G3BP1 in the UPF1-G3BP1-mediated RNA decay is distinct from its protective role proposed here. In the UPF1–G3BP1-mediated RNA decay, UPF1 dominates the regulation by recognizing highly structured RNAs especially double-stranded RNAs, unwinding them, thereby facilitating the enrichment of G3BP1 in proximity to UPF1 (48). Thus, the differences in modes of interactions between G3BP1 and targeted transcripts may contribute to the different functions of G3BP1 in modulating mRNA stabilities.
In summary, we identified multiple candidate rG4-binding proteins with a bioinformatic approach, and we validated that one of these proteins, G3BP1 can bind directly with rG4 with low-nM binding affinity. We also found that the binding of G3BP1 with rG4 structures in the UTRs stabilized mRNAs and suppressed their translation, which revealed a new function of G3BP1. Together, the results from the present study uncovered a number of candidate rG4-binding proteins and expanded the functions of G3BP1.
DATA AVAILABILITY
The seCLIP-seq data were deposited into the NCBI GEO database (GSE168943).
Supplementary Material
Contributor Information
Xiaomei He, Department of Chemistry, University of California, Riverside, CA 92521-0403, USA.
Jun Yuan, Department of Chemistry, University of California, Riverside, CA 92521-0403, USA.
Yinsheng Wang, Department of Chemistry, University of California, Riverside, CA 92521-0403, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35 ES031707]. Funding for open access charge: NIH [R35 ES031707].
Conflict of interest statement. None declared.
REFERENCES
- 1. Millevoi S., Moine H., Vagner S.. G-quadruplexes in RNA biology. Wiley Interdiscip. Rev.: RNA. 2012; 3:495–507. [DOI] [PubMed] [Google Scholar]
- 2. Kwok C.K., Marsico G., Sahakyan A.B., Chambers V.S., Balasubramanian S.. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods. 2016; 13:841. [DOI] [PubMed] [Google Scholar]
- 3. Guo J.U., Bartel D.P.. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science (New York, N.Y.). 2016; 353:aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Puig Lombardi E., Londoño-Vallejo A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2019; 48:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang S.Y., Lejault P., Chevrier S., Boidot R., Robertson A.G., Wong J.M.Y., Monchaud D. Transcriptome-wide identification of transient RNA G-quadruplexes in human cells. Nat. Commun. 2018; 9:4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biffi G., Di Antonio M., Tannahill D., Balasubramanian S.. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 2013; 6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laguerre A., Hukezalie K., Winckler P., Katranji F., Chanteloup G., Pirrotta M., Perrier-Cornet J.-M., Wong J.M.Y., Monchaud D. Visualization of RNA-Quadruplexes in Live Cells. J. Am. Chem. Soc. 2015; 137:8521–8525. [DOI] [PubMed] [Google Scholar]
- 8. Chen X.-C., Chen S.-B., Dai J., Yuan J.-H., Ou T.-M., Huang Z.-S., Tan J.-H.. Tracking the dynamic folding and unfolding of RNA G-quadruplexes in live cells. Angew. Chem. Int. Ed. 2018; 57:4702–4706. [DOI] [PubMed] [Google Scholar]
- 9. Weng X., Gong J., Chen Y., Wu T., Wang F., Yang S., Yuan Y., Luo G., Chen K., Hu L.et al.. Keth-seq for transcriptome-wide RNA structure mapping. Nat. Chem. Biol. 2020; 16:489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huppert J.L., Bugaut A., Kumari S., Balasubramanian S.. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008; 36:6260–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolfe A.L., Singh K., Zhong Y., Drewe P., Rajasekhar V.K., Sanghvi V.R., Mavrakis K.J., Jiang M., Roderick J.E., Van der Meulen J.et al.. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014; 513:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darnell J.C., Jensen K.B., Jin P., Brown V., Warren S.T., Darnell R.B.. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001; 107:489–499. [DOI] [PubMed] [Google Scholar]
- 13. Kumari S., Bugaut A., Huppert J.L., Balasubramanian S.. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007; 3:218–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cammas A., Millevoi S.. RNA G-quadruplexes: emerging mechanisms in disease. Nucleic Acids Res. 2017; 45:1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fay M.M., Lyons S.M., Ivanov P.. RNA G-quadruplexes in biology: principles and molecular mechanisms. J. Mol. Biol. 2017; 429:2127–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kwok C.K., Merrick C.J.. G-Quadruplexes: prediction, characterization, and biological application. Trends Biotechnol. 2017; 35:997–1013. [DOI] [PubMed] [Google Scholar]
- 17. Sauer M., Juranek S.A., Marks J., De Magis A., Kazemier H.G., Hilbig D., Benhalevy D., Wang X., Hafner M., Paeschke K.. DHX36 prevents the accumulation of translationally inactive mRNAs with G4-structures in untranslated regions. Nat. Commun. 2019; 10:2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herdy B., Mayer C., Varshney D., Marsico G., Murat P., Taylor C., D'Santos C., Tannahill D., Balasubramanian S. Analysis of NRAS RNA G-quadruplex binding proteins reveals DDX3X as a novel interactor of cellular G-quadruplex containing transcripts. Nucleic Acids Res. 2018; 46:11592–11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serikawa T., Spanos C., von Hacht A., Budisa N., Rappsilber J., Kurreck J.. Comprehensive identification of proteins binding to RNA G-quadruplex motifs in the 5′ UTR of tumor-associated mRNAs. Biochimie. 2018; 144:169–184. [DOI] [PubMed] [Google Scholar]
- 20. von Hacht A., Seifert O., Menger M., Schütze T., Arora A., Konthur Z., Neubauer P., Wagner A., Weise C., Kurreck J.. Identification and characterization of RNA guanine-quadruplex binding proteins. Nucleic Acids Res. 2014; 42:6630–6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bian W.-X., Xie Y., Wang X.-N., Xu G.-H., Fu B.-S., Li S., Long G., Zhou X., Zhang X.-L.. Binding of cellular nucleolin with the viral core RNA G-quadruplex structure suppresses HCV replication. Nucleic Acids Res. 2018; 47:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lista M.J., Martins R.P., Billant O., Contesse M.-A., Findakly S., Pochard P., Daskalogianni C., Beauvineau C., Guetta C., Jamin C.et al.. Nucleolin directly mediates Epstein-Barr virus immune evasion through binding to G-quadruplexes of EBNA1 mRNA. Nat. Commun. 2017; 8:16043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sexton A.N., Collins K.. The 5′ guanosine tracts of human telomerase RNA are recognized by the G-quadruplex binding domain of the RNA helicase DHX36 and function to increase RNA accumulation. Mol. Cell Biol. 2011; 31:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McRae E.K.S., Booy E.P., Moya-Torres A., Ezzati P., Stetefeld J., McKenna S.A.. Human DDX21 binds and unwinds RNA guanine quadruplexes. Nucleic Acids Res. 2017; 45:6656–6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Gaetano C.M., Williams K.R., Bassell G.J., Mihailescu M.R.. FMRP interacts with G-quadruplex structures in the 3′-UTR of its dendritic target Shank1 mRNA. RNA Biology. 2014; 11:1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benhalevy D., Gupta S.K., Danan C.H., Ghosal S., Sun H.-W., Kazemier H.G., Paeschke K., Hafner M., Juranek S.A.. The human CCHC-type zinc finger nucleic acid-binding protein binds G-rich elements in target mRNA coding sequences and promotes translation. Cell Rep. 2017; 18:2979–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thandapani P., Song J., Gandin V., Cai Y., Rouleau S.G., Garant J.M., Boisvert F.M., Yu Z., Perreault J.P., Topisirovic I.et al.. Aven recognition of RNA G-quadruplexes regulates translation of the mixed lineage leukemia protooncogenes. Elife. 2015; 4:e06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K.et al.. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods. 2016; 13:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chikina M.D., Troyanskaya O.G.. An effective statistical evaluation of ChIPseq dataset similarity. Bioinformatics. 2012; 28:607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pohl A., Beato M.. bwtool: a tool for bigWig files. Bioinformatics. 2014; 30:1618–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinf. 2012; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams P., Li L., Dong X., Wang Y.. Identification of SLIRP as a G quadruplex-binding protein. J. Am. Chem. Soc. 2017; 139:12426–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hellman L.M., Fried M.G.. Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat. Protoc. 2007; 2:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R.et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu H., Naismith J.H.. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008; 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Nostrand E.L., Nguyen T.B., Gelboin-Burkhart C., Wang R., Blue S.M., Pratt G.A., Louie A.L., Yeo G.W.. Robust, cost-effective profiling of RNA binding protein targets with single-end enhanced crosslinking and immunoprecipitation (seCLIP). Methods Mol. Biol. 2017; 1648:177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olarerin-George A.O., Jaffrey S.R.. MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics (Oxford, England). 2017; 33:1563–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bedrat A., Lacroix L., Mergny J.-L.. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chikina M.D., Troyanskaya O.G.. An effective statistical evaluation of ChIPseq dataset similarity. Bioinformatics (Oxford, England). 2012; 28:607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phan A.T., Kuryavyi V., Darnell J.C., Serganov A., Majumdar A., Ilin S., Raslin T., Polonskaia A., Chen C., Clain D.et al.. Structure-function studies of FMRP RGG peptide recognition of an RNA duplex-quadruplex junction. Nat. Struct. Mol. Biol. 2011; 18:796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brázda V., Kolomazník J., Lýsek J., Bartas M., Fojta M., Šťastný J., Mergny J.-L.. G4Hunter web application: a web server for G-quadruplex prediction. Bioinformatics. 2019; 35:3493–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beaudoin J.-D., Jodoin R., Perreault J.-P.. New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res. 2014; 42:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fischer J.W., Busa V.F., Shao Y., Leung A.K.L.. Structure-mediated RNA decay by UPF1 and G3BP1. Mol. Cell. 2020; 78:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Booy E.P., Howard R., Marushchak O., Ariyo E.O., Meier M., Novakowski S.K., Deo S.R., Dzananovic E., Stetefeld J., McKenna S.A.. The RNA helicase RHAU (DHX36) suppresses expression of the transcription factor PITX1. Nucleic Acids Res. 2014; 42:3346–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paramasivan S., Rujan I., Bolton P.H.. Circular dichroism of quadruplex DNAs: applications to structure, cation effects and ligand binding. Methods. 2007; 43:324–331. [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez R., Müller S., Yeoman J.A., Trentesaux C., Riou J.-F., Balasubramanian S.. A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J. Am. Chem. Soc. 2008; 130:15758–15759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J.M., Bertrand E., Tazi J.. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003; 160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53. Parker F., Maurier F., Delumeau I., Duchesne M., Faucher D., Debussche L., Dugue A., Schweighoffer F., Tocque B.. A Ras-GTPase-activating protein SH3-domain-binding protein. Mol. Cell Biol. 1996; 16:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Castello A., Fischer B., Frese Christian K., Horos R., Alleaume A.-M., Foehr S., Curk T., Krijgsveld J., Hentze Matthias W.. Comprehensive identification of RNA-binding domains in human cells. Mol. Cell. 2016; 63:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dumas L., Herviou P., Dassi E., Cammas A., Millevoi S.. G-Quadruplexes in RNA biology: recent advances and future directions. Trends Biochem. Sci. 2020; 46:270–283. [DOI] [PubMed] [Google Scholar]
- 56. Dominguez C., Fisette J.-F., Chabot B., Allain F.H.T.. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat. Struct. Mol. Biol. 2010; 17:853–861. [DOI] [PubMed] [Google Scholar]
- 57. Pietras Z., Wojcik M.A., Borowski L.S., Szewczyk M., Kulinski T.M., Cysewski D., Stepien P.P., Dziembowski A., Szczesny R.J.. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018; 9:2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Markmiller S., Soltanieh S., Server K.L., Mak R., Jin W., Fang M.Y., Luo E.-C., Krach F., Yang D., Sen A.et al.. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018; 172:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Onomoto K., Yoneyama M., Fung G., Kato H., Fujita T.. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014; 35:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khong A., Matheny T., Jain S., Mitchell S.F., Wheeler J.R., Parker R.. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol. Cell. 2017; 68:808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu Z.-S., Cai H., Xue W., Wang M., Xia T., Li W.-J., Xing J.-Q., Zhao M., Huang Y.-J., Chen S.et al.. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019; 20:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Costa M., Ochem A., Staub A., Falaschi A.. Human DNA helicase VIII: a DNA and RNA helicase corresponding to the G3BP protein, an element of the ras transduction pathway. Nucleic Acids Res. 1999; 27:817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thandapani P., O’Connor T.R., Bailey Timothy L., Richard S.. Defining the RGG/RG Motif. Mol. Cell. 2013; 50:613–623. [DOI] [PubMed] [Google Scholar]
- 64. Huang Z.-L., Dai J., Luo W.-H., Wang X.-G., Tan J.-H., Chen S.-B., Huang Z.-S.. Identification of G-quadruplex-binding protein from the exploration of RGG motif/G-quadruplex interactions. J. Am. Chem. Soc. 2018; 140:17945–17955. [DOI] [PubMed] [Google Scholar]
- 65. Takahama K., Takada A., Tada S., Shimizu M., Sayama K., Kurokawa R., Oyoshi T.. Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem. Biol. 2013; 20:341–350. [DOI] [PubMed] [Google Scholar]
- 66. Lee D.S.M., Ghanem L.R., Barash Y.. Integrative analysis reveals RNA G-quadruplexes in UTRs are selectively constrained and enriched for functional associations. Nat. Commun. 2020; 11:527–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang P., Mathieu C., Kolaitis R.-M., Zhang P., Messing J., Yurtsever U., Yang Z., Wu J., Li Y., Pan Q.et al.. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell. 2020; 181:325–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guillén-Boixet J., Kopach A., Holehouse A.S., Wittmann S., Jahnel M., Schlüßler R., Kim K., Trussina I.R.E.A., Wang J., Mateju D.et al.. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell. 2020; 181:346–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jain A., Vale R.D.. RNA phase transitions in repeat expansion disorders. Nature. 2017; 546:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Y., Yang M., Duncan S., Yang X., Abdelhamid M.A.S., Huang L., Zhang H., Benfey P.N., Waller Z.A.E., Ding Y.. G-quadruplex structures trigger RNA phase separation. Nucleic Acids Res. 2019; 47:11746–11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Imamachi N., Salam K.A., Suzuki Y., Akimitsu N.. A GC-rich sequence feature in the 3′ UTR directs UPF1-dependent mRNA decay in mammalian cells. Genome Res. 2017; 27:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The seCLIP-seq data were deposited into the NCBI GEO database (GSE168943).