


Abstract
CRISPR-Cas is a powerful tool for genome editing in bacteria. However, its efficacy is dependent on host factors (such as DNA repair pathways) and/or exogenous expression of recombinases. In this study, we mitigated these constraints by developing a simple and widely applicable genome engineering tool for bacteria which we termed SIBR-Cas (Self-splicing Intron-Based Riboswitch-Cas). SIBR-Cas was generated from a mutant library of the theophylline-dependent self-splicing T4 td intron that allows for tight and inducible control over CRISPR-Cas counter-selection. This control delays CRISPR-Cas counter-selection, granting more time for the editing event (e.g. by homologous recombination) to occur. Without the use of exogenous recombinases, SIBR-Cas was successfully applied to knock-out several genes in three wild-type bacteria species (Escherichia coli MG1655, Pseudomonas putida KT2440 and Flavobacterium IR1) with poor homologous recombination systems. Compared to other genome engineering tools, SIBR-Cas is simple, tightly regulated and widely applicable for most (non-model) bacteria. Furthermore, we propose that SIBR can have a wider application as a simple gene expression and gene regulation control mechanism for any gene or RNA of interest in bacteria.
INTRODUCTION
Homologous recombination (HR) combined with CRISPR-Cas counter-selection is a powerful approach for genome editing in a wide range of bacterial species (1,2). Double stranded DNA (dsDNA) breaks generated by CRISPR-Cas can initiate recombinational repair through RecBCD (3), but its efficiency might be outcompeted by the strong CRISPR-Cas counter-selective pressure (4). We therefore anticipate that gaining control over CRISPR-Cas counter-selection, would be beneficial for genome editing purposes. In other words, to achieve high editing efficiencies, generation of the desired edit through HR should precede CRISPR-Cas counter-selection. Typically, the efficiency of HR in prokaryotes is low, making genome editing through CRISPR-Cas counter-selection cumbersome.
To enhance HR frequencies, the heterologous expression of recombinases has been used. However, this method is often laborious (maintenance of multiple plasmids) and success is not guaranteed as the recombinases may be incompatible with the target organism (5). As an alternative, several regulation systems have been developed to control the expression and activity of the CRISPR-Cas module. Examples include the use of inducible promoters (6–8), inducible intein splicing (9,10), split Cas proteins (11,12), inducible conformation change (13), inducible inhibition through aptamers (14) and inducible translation through riboswitches (15). Other approaches focused on the inducible guide RNA functionality using ribozymes (16), riboswitches (17,18) and photocaging (19–22). The control of the CRISPR-Cas modules gives enough time for HR to occur before CRISPR-Cas counter-selection is induced. Whilst the existing approaches are tailored for the organism, Cas protein or guide RNA (gRNA) of interest, these solutions are typically not widely applicable. Therefore, CRISPR-Cas engineering tools would benefit from widely applicable and tight regulation of their counter-selective properties to provide enough time for HR to take place.
Ribozymes and riboswitches are gene regulation systems found in a wide range of bacterial species. The catalytic and/or regulatory functionality of these RNA molecules relies on their primary, secondary and tertiary structures, making them great candidates for developing universal tools for regulating gene expression, without the use of proteins (23–27). To this end, several studies used ribozymes and riboswitches to control the expression of a gene of interest (GOI), but also for regulating the activity and function of CRISPR-Cas (14–18,24). Nevertheless, these approaches leave room for improvement. For example, the technology developed by Tang et al. (2017) (16), requires base pairing of the CRISPR spacer sequence with the 5′ end of the hammerhead ribozyme; something that requires modification in case the CRISPR spacer needs to be changed. Moreover, the studies by Kundert et al. (2019) (18), Siu et al. (2019) (17) and Zhao et al. (2020) (14) rely on the secondary structure of the Cas9 single guide RNA (sgRNA), which rules out the use of other CRISPR-Cas systems. Lastly, the RiboCas technology developed by Cañadas et al. (2019) (15), regulates the expression of Cas9 by masking the RBS with a theophylline-dependent riboswitch. Whereas this is a smart alternative to previous approaches, it can be cumbersome to use in organisms that either do not use the canonical RBS sequence, or in cases that the secondary structure of the 5′-UTR sequence interferes with the theophylline aptamer (28–30).
A unique type of ribozymes includes the self-splicing Group I introns. Group I introns have been described to control gene expression and RNA processing in bacteria and phages but also in some eukaryotes (protozoa and plants) (31–33). Due to their prevalence and simplistic nature, Group I introns have the potential to be used as universal, synthetic ribozymes to control gene expression. Especially when ribozymes are associated with a specific ligand-binding sequence (RNA aptamer), the presence/absence of such a ligand acts as an ON/OFF switch for splicing (riboswitch), thereby controlling the expression of an associated gene. An example of a natural Group I intron-based riboswitch has been discovered in the bacterium Clostridium difficile, where its sequence resides between the RBS and the ATG start codon of an adjacent gene (34,35). After transcription, this results in a secondary structure in the 5′-UTR that prevents recruitment of the ribosome, hence hampering translation initiation. After induction by intracellular GTP or c-di-GMP, this ribozyme induces its splicing from the precursor transcript, resulting in appropriate re-positioning of the RBS upstream the start codon, thereby allowing for the ribosome to start the translation process (34,35). Although this natural mechanism is a beautiful case of gene expression control, its requirement for specific endogenous inducers (GTP and c-di-GMP) as well as its dependency on specific secondary structures (including both the ribozyme and the coding sequence) complicates its general applicability. A synthetic alternative was provided by Thompson et al. (2002), when they combined the self-splicing Group I intron of the T4 bacteriophage with a theophylline aptamer towards a functional inducible gene expression system (36). Although this system was restricted to controlling the original thymidylate synthase (td) gene, we here describe its repurposing as a generic system to tune gene expression.
In this study, we developed the Self-splicing Intron-Based Riboswitch (SIBR) system. SIBR is based on the bacteriophage T4 td Group I self-splicing intron and has been engineered and repurposed as a modular, tightly regulated system that can control the expression of any GOI in a wide range of bacterial species. To illustrate this, we used SIBR to control the Cas12a nuclease from Francisella novicida (named SIBR-Cas) and demonstrated efficient genome editing in three wild-type (WT) bacterial species (Escherichia coli MG1655, Pseudomonas putida KT2440 and Flavobacterium IR1) without the use of exogenous recombinases nor the use of inducible promoters. SIBR-Cas is an elegant solution for the widespread problem of engineering prokaryotic organisms with poor recombination efficiencies. We also suggest that SIBR can be used as a universal OFF/ON or ON/OFF switch for individual genes or multiple genes in a polycistronic operon.
MATERIALS AND METHODS
Bacterial strains, handling and growth conditions
E. coli DH5a (NEB) was used for general plasmid propagation and standard molecular techniques. E. coli DH10B T1R (Invitrogen) was used for the LacZ assays. E. coli MG1655 (ATCC) was used for targeting and knock-out assays. Unless specified otherwise, E. coli strains were grown at 37°C in LB liquid medium (10 g L–1 tryptone, 5 g l–1 yeast extract, 10 g l–1 NaCl) or on LB agar plates (LB liquid medium, 15 g l–1 bacteriological agarose) containing the appropriate antibiotics: spectinomycin (100 mg l–1), kanamycin (50 mg l–1), ampicillin (100 mg l–1) or chloramphenicol (35 mg l–1). Transformation of electro-competent E. coli cells was performed in 2 mm electroporation cuvettes with an ECM 63 electroporator (BTX) at 2500 V, 200 Ω and 25 μF.
P. putida strain KT2440 was obtained from DSMZ. Cells were grown at 30°C in LB liquid medium or on LB agar plates containing kanamycin (50 mg l–1). Electro-competent P. putida cells were transformed in 2 mm electroporation cuvettes using 2500 V, 200 Ω and 25 μF.
Flavobacterium species Iridescence 1 (sp. IR1) was kindly provided by Hoekmine BV. WT Flavobacterium IR1 was grown at 25°C in ASW medium (5 g l–1 peptone, 1 g l–1 yeast extract, 10 g l–1 sea salt) or plated on ASW agar (ASW medium, 15 g l–1 agar) containing erythromycin (200 mg l–1) where appropriate. Electro-competent Flavobacterium IR1 cells were transformed in 1 mm electroporation cuvettes using 1500 V, 200 Ω and 25 μF.
Electro-competent cell preparation
Pre-cultures of E. coli or P. putida were grown overnight at 37°C in fresh 10 ml LB broth. 5 ml of the overnight culture was inoculated in 500 ml of pre-warmed 2× YP medium (16 g l–1 peptone, 10 g l–1 yeast extract) and incubated at 37°C shaking at 200 rpm until an OD600 of 0.4 was reached. The culture was then cooled down to 4°C. Next, the culture was aliquoted into two sterile 450 ml centrifuge tubes and centrifuged at 3000 g for 10 min at 4°C. The supernatant was decanted and the pellet was washed with 250 ml ice-cold sterile miliQ water followed by centrifugation at 3000 g for 10 min. The supernatant was decanted and the pellet was resuspended using 5 ml of ice cold 10% glycerol. The two resuspensions were combined in one tube and ice-cold 10% glycerol was added to reach a final volume of 250 ml, followed by centrifugation at 3000 g for 10 min. The supernatant was decanted and the pellet was washed with 250 ml of ice-cold 10% glycerol and centrifuged at 3000 g for 10 min. The supernatant was decanted and the pellet was resuspended with 2 ml of ice-cold 10% glycerol and aliquoted into tubes of 40 μl. All the electrocompetent cells were stored at −80°C prior to transformation. To prepare electro-competent cells, Flavobacterium IR1 was grown overnight in 10 ml ASW at 25°C, shaking at 200 rpm. The overnight culture was used to inoculate 500 ml ASW in 2 l baffled flask to a starting OD600 of 0.05 and incubated at 25°C and shaking at 200 rpm until the cell density reached an OD600 equal to 0.3–0.4. The cells were cooled down at 4°C and kept cold on ice for the rest of the procedure. The culture was divided into two sterile 450 ml centrifuge tubes and centrifuged at 3000 g for 10 min at 4°C. The supernatant was decanted, and the cell pellet was washed twice with 250 ml ice cold washing buffer (10 mM MgCl2 and 5 mM CaCl2) followed by centrifugation at 3000 g for 10 min at 4°C. The supernatant was removed, and the pellet was resuspended using 5 ml of the washing buffer. All the resuspensions were combined in one tube and washed by adding 250 ml of the washing buffer. It was then washed once with 10% glycerol followed by centrifugation at 3000 g for 10 min. The supernatant was decanted, and the resulting cell pellet was resuspended with 5 ml of ice cold 10% (v/v) glycerol and 100 μl aliquots were stored at −80°C until use.
Plasmid construction
The LacZ reporter plasmid series were constructed from pEA001 [PWW]. The LacZ reporter plasmid series contain the E. coli LacZ gene under the control of the constitutive lacUV5 promoter. Ten amino acids flanking the T4 td intron (five from each side) were introduced between D6 and S7 of LacZ, omitting the intron itself. For cloning purposes, the ten amino acids were in turn flanked by a BspTI and PstI restriction sites. Generating the complete mutant series was performed by PCR, digestion with BspTI and PstI (Thermo Fisher Scientific) and ligation into pEA001 [PWW].
The main components (origin of replication, antibiotic resistance gene and promoters) of the SIBR-Cas plasmids for E. coli and P. putida were designed to be functional in both organisms. The constitutive lacUV5 promoter was used to drive the expression of the FnCas12a variants (WT and Int1−4) and an additional lacUV5 promoter was used to drive the expression of the crRNA. The empty vectors pSIBR001-005 were designed to allow convenient insertion of new spacers through Golden Gate Assembly using the BbsI-HF® enzyme and the T4 DNA ligase (NEB), following the protocol as previously described by Batianis et al. (37). Homology arms (500 bp) were introduced to the SIBR-Cas plasmids at a multiple cloning site (MCS). Briefly, homology arms were amplified from genomic DNA and introduced to the MCS of the linearized SIBR-Cas plasmid using the NEBuilder® HiFi DNA Assembly Master Mix (NEB). The plasmid was linearized using Esp3I (NEB). The DNA sequence of all plasmids was verified through Sanger sequencing (Macrogen Europe B.V.). To construct the SIBR-Cas plasmids for Flavobacterium IR1, the backbone of pSpyCas9Fb_NT (38) was used but the Cas9 and the sgRNA were replaced with the FnCas12a variants (WT and Int1−4) and the crRNA, respectively. Spacers and homology arms were introduced through Golden Gate using BsaI-HF® enzyme and NEBuilder® HiFi DNA Assembly, respectively, as described above. Other plasmids and oligonucleotides used in this study are listed in Supplementary Tables S1 and S2 respectively. The complete plasmid maps including the intron sequences, the Cas12a coding sequences, homology arms and the crRNAs sequences used for each plasmid can be accessed through the Benchling links in Supplementary Table S1. Also, the spacer moieties of the crRNA used in this study are highlighted in Supplementary Table S2.
Chemicals and reagents
Unless otherwise specified, all chemical reagents were purchased from Sigma-Aldrich. Sea salt was purchased from Sel Marine. A 40 mM theophylline (Sigma-Aldrich) stock was prepared by dissolving theophylline in dH2O followed by filter sterilization using a 0.2 μm Whatman® puradisc syringe filter. When necessary, 0–10 mM theophylline was added to the liquid or solid medium. 20 mg ml–1 X-Gal (Sigma-Aldrich) stock was prepared by dissolving X-Gal in N,N-dimethylmethanamide. The final X-Gal concentration for blue/white colony screening was 0.2 mg ml–1.
β-Galactosidase activity assay
LacZ activity was assayed in E. coli DH10B T1R in triplicate. Transformed E. coli cells carrying a single variant of the pEA001 plasmid series (Supplementary Table S1) were grown overnight at 37°C, after which 20 μl of culture was mixed with 80 μL of permeabilization solution (100 mM Na2HPO4, 20 mM KCl, 2 mM MgSO4, 0.8 g l–1 CTAB, 0.4 g l–1 sodium deoxycholate and 5.4 ml l–1 β-mercaptoethanol) and incubated at 30°C for 30 min. 600 μl of pre-warmed substrate solution (60 mM Na2HPO4, 40 mM NaH2PO4, 1 g l–1o-nitrophenyl-β-d-galactopyranoside and 2.7 mL l–1 β-mercaptoethanol) was added and incubated at 30°C until sufficient colour had developed. 700 μl of stop solution (1 M Na2CO3) was added to quench the reaction. The reaction was filtered through a 0.2 μM filter and measured in a spectrophotometer at 420 nm in a 1 cm cuvette. LacZ activity was calculated according to the following equation:
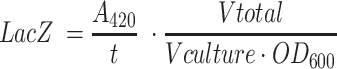 |
The LacZ activities of all clones were divided by the LacZ activity exhibited by the WT intron.
SIBR-Cas targeting and editing assays in E. coli
For both the targeting and the editing assays, E. coli MG1655 electrocompetent cells were transformed with a single variant of the pSIBR-Cas series (Supplementary Table S1). 10 ng of plasmid DNA was used to transform 20 μl electro-competent cells as described above. Transformed cells were recovered in 1 ml LB liquid medium for 1 h at 30°C and shaking at 200 rpm. For the targeting assay, the transformants were ten-fold serially diluted in LB liquid medium and 3 μl were used for spot dilution assays on LB agar plates containing kanamycin (50 mg l–1) in the presence or absence of 2 mM theophylline and incubated for 24 h at 30°C. For the editing assay, transformants were plated on LB agar plates containing kanamycin (50 mg l–1) and 7 mM theophylline and incubated at 30°C for 24 h. Colony PCR was performed on 16 colonies from each transformation to define the editing efficiencies. Triplicate transformations were used for each SIBR-Cas plasmid. Mutant colonies were sequenced through Sanger sequencing (Macrogen BV) to confirm complete deletion of the target gene.
SIBR-Cas targeting and editing assays in P. putida
40 μL electro-competent P. putida cells were transformed with 200 ng of a single variant of the pSIBR-Cas series (Supplementary Table S1) and recovered in 1 ml LB liquid medium for 2 h at 30°C, shaking at 200 rpm. Targeting was assayed by spot dilution assays on LB agar plates containing kanamycin (50 mg l–1) in the presence or absence of 2 mM theophylline, followed by overnight incubation at 30°C. P. putida cells bearing the editing plasmid were plated on LB agar plates containing kanamycin (50 mg l–1) and 2 mM theophylline and incubated at 30°C for 24 h. Grown colonies were screened through colony PCR to define the editing efficiency. For each SIBR-Cas plasmid, transformations were performed in triplicate. Mutant colonies were sequenced through Sanger sequencing (Macrogen BV) to confirm complete deletion of the target gene.
SIBR-Cas editing assays in Flavobacterium IR1
Electro-competent Flavobacterium IR1 cells (100 μl) were transformed with 2 μg plasmid of a single variant of the pSIBR-Cas series (Supplementary Table S1). Transformed cells were recovered in 1 ml ASW and incubated for 4 h at 25°C, shaking at 200 rpm. Due to very low transformation efficiency, the recovered cells were transferred in 10 ml ASW liquid medium containing erythromycin (200 mg l–1) and incubated for 96 h at 25°C, shaking at 200 rpm. 10–6 or 10–7 cells were then plated on ASW agar containing erythromycin (200 mg l–1) and 2 mM theophylline. Plates were incubated at 25°C for 2–3 days and grown colonies were screened for editing through colony PCR. Each editing assay was performed in triplicate. Mutant colonies were sequenced through Sanger sequencing (Macrogen BV) to confirm complete deletion of the target gene.
RESULTS AND DISCUSSION
The flanking regions of the T4 td intron are amenable to modifications
To create a versatile gene control system, we focused on promoter-, sequence- and organism-independent mechanisms. We chose the T4 bacteriophage Group I self-splicing intron that resides in the thymidylate synthase (td) gene as the appropriate mechanism to control the expression of the GOI (Figure 1A). The self-splicing ribozyme activity of the T4 td intron requires only ubiquitous cofactors such as GTP and Mg2+, making its use widely applicable in bacterial species (39,40). Moreover, similar to other introns, the T4 td intron terminates the translation of the unspliced precursor mRNA due to the presence of in-frame stop codons (Figure 1B). Therefore, the presence of the intron in the precursor mRNA will result in a truncated non-functional protein, whereas the spliced mRNA allows for the full translation of the protein of interest.
Figure 1.

Schematics and function of the T4 td intron. (A) Schematic representation of the predicted secondary and tertiary structure of the wild-type (WT; right) and the theophylline-dependent T4 td intron (left). The structure follows the format of Cech et al. (69). P1 to P10 represent the pairing domains of the intron. Exon sequences are indicated as blue boxes. Orange triangles indicate the splicing sites. Base pairs are indicated by ‘-’ and wobble pairs by ‘•’. The grey boxes at P6 highlight the difference between the WT and the theophylline-dependent ribozyme. Grey lines show interactions within the intron. (B) Schematic representation of the transcription and translation of a gene containing the T4 td intron in its open reading frame. At the top, the intron in-frame stop codon (TAA) is depicted in red, the 5′ flanking region is highlighted with a blue box and a part of the intron sequence is highlighted with a light grey box. 1 depicts transcription, 2 depicts self-splicing (left path) or no self-splicing of the intron (right path) and 3 depicts translation of the full protein (left path) or the translation of a truncated protein (right path) when the intron is spliced or retained, respectively.
The naturally occurring T4 td intron is specific for the td gene because the exonic flanking regions are necessary to preserve the secondary structure of the P1 and P10 stems of the T4 td intron (41) (Figures 1A and 2A). Hence, transferring the intron along with the flanking exonic regions to another gene will disrupt the coding sequence of the target gene. On the other hand, changing the exonic flanking regions of the intron to preserve the coding sequence of the target gene may affect (or completely inhibit) the splicing activity of the intron. Since moving the T4 td intron without changing the flanking exons is not a viable option, we opted to create several variants of the T4 td intron by mutating the flanking exons. Some of the boundaries that determine the effect of the flanking exons on the splicing efficiency have been elucidated previously (41). However, to predict the effect of the altered flanking regions on splicing, we studied them in more detail.
Figure 2.

T4 td intron mutant library generation and LacZ assays. (A) Detailed illustration of the WT and mutant 5′ and 3′ flanking regions of the T4 td intron. Exons are indicated with blue boxes. Orange triangles show the splicing sites. Base pairs are indicated by ‘-’ and wobble pairs by ‘•’. Mutant nucleotides are highlighted with yellow circles and follow the IUPAC nucleotide nomenclature (B = C/G/T, D = A/G/T, H = A/C/T and Y = C/T). (B) LacZ transcription-translation cascade controlled by the T4 td intron library. (C) LacZ activity of position −7 and + 296 mutants. ‘*’ indicates the WT intron and its relative LacZ activity is set to 1. All other LacZ activities are a fraction of the WT intron LacZ activity. (D) LacZ activity of all possible combinations for pair, wobble pair and mismatch at positions −6 to −4. ‘*’ indicates the WT intron and its relative LacZ activity is set to 1. All other LacZ activities are a fraction of the WT intron LacZ activity. ‘**’ indicates the stop codon UGA. The numbers above the bars refer to the intron variants that were selected for the subsequent experiments (1:Int1, 2:Int2, 3:Int3 and 4:Int4). Bars represent the means and error bars represent the standard deviation of three independent experiments.
To assess whether alterations at the flanking exons can affect the splicing efficiency, the T4 td intron variants were placed in between the coding sequence encoding amino acids D6 and S7 of the LacZ gene (Figure 2A and B). We hypothesize that by placing the intron at the beginning of the LacZ gene, potential interference by exon-intron interactions or by other disturbances related to translation will be avoided (42). Splicing was assessed through the well-established β-galactosidase assay in E. coli DH10B.
Single base modifications at the −7 (C to T or G) or +296 (A to T or C) positions decreased the splicing activity of the intron compared to the WT T4 td intron sequence (Figure 2C). Position −7 (C) preferably pairs with position +15 (G) in the WT intron since a weaker interaction in the form of a wobble base pair (T) or no interaction in the form of a mismatch (G), impeded the splicing of the intron by 20% and 35%, respectively. The opposite was observed for position +296 where a mismatch (A) allows for the highest intron splicing activity. The weak wobble base pair (T) impeded splicing by 8%, while the stronger pair (C) decreased the splicing by 29%.
Regardless of the impeded self-splicing of the mutant introns at position −7 and +296, self-splicing was still observed indicating that the flanking regions of the T4 td intron are amenable to modifications. To this end, we created pair, wobble or mismatch base substitutions at the −4, −5 and −6 positions and characterized the self-splicing activity of the resulting T4 td intron variants (Figure 2D). For simplicity reasons, the rest of the flanking regions of the T4 td intron (including the −7 and +296 positions) were kept the same as the WT intron sequence.
Surprisingly, several intron variants showed better LacZ activity compared to the WT intron (−4G, −5T, −6T; Figure 2D). A mismatch at position −4 (T) is preferred in almost all variants, except for those in which both −5 (G) and −6 (C) positions are mismatched too. Compared to the WT intron, a 40% increase in LacZ activity was observed when position −4 was mismatched (T) accompanied by a paired (C) or a wobble paired (T) −5 position and a paired (T) −6 position. A wobble base pair at position −5 (T) negates to a large extent the effect that −4 and −6 have on the splicing. In contrast, a pair (C) or a mismatch (G) at position −5 and depending on −4 and −6 positions, can alter the splicing efficiency from very high (136%) to very low (20%). Position −6 in general appears in favour of being paired (T). However, the complete stabilisation of the secondary structure of P1 (−4A, −5C, −6T) is inhibiting splicing almost completely as it shows 20% relative LacZ activity. Completely mismatching positions −4 to −6 (−4T, −5G, −6C) impedes splicing to around 71%. Complete stabilization or complete destabilization of the −4, −5 and −6 positions of the P1 stem was previously observed by Pichler et al. (41). However, the authors report contradicting results to our study as both their stabilized and destabilized variants show increased splicing efficiency. The observed differences in splicing efficiency by stabilizing or destabilizing the P1 stem may be attributed to the different experimental setup as we investigated splicing efficiency based on enzymatic activities whereas Pichler et al. performed cis splicing assays by isolating total RNA from E. coli cells carrying the different intron variants. Lastly, the −6T, −5G, −4A acts as a negative control as this combination forms a stop codon (UGA), as reflected by the absence of relative LacZ activity using this combination.
Taken together, these results demonstrate that T4 td intron variants were generated with a range of splicing efficiencies, allowing for tuneable control over the LacZ protein expression based solely on the intron variant. In addition, the transfer of the T4 td intron variants from the td gene to the start of the LacZ gene demonstrates the flexibility of the intron and its potential use as a tunable gene expression control mechanism.
SIBR-Cas targeting efficiency is tuneable and inducible
To translate our setup to a CRISPR-Cas engineering context, we tested the ability of the T4 td intron variants to control the expression of a Cas nuclease. We selected Cas12a from Francisella novicida (FnCas12a), due to our prior expertise on this particular Cas nuclease (43). We selected four intron variants with distinct splicing efficiency (Int1: −4A, −5C, −6T; Int2: −4G, −5C, −6T; Int3: −4G, −5T, −6T; Int4: −4T, −5C, −6T; Figure 2D) and inserted them directly after the start codon of the FnCas12a gene (Figure 3A). The intron variants are numbered in order of increasing splicing efficiency with Int1 having the worst and Int4 the highest splicing efficiency. Moreover, to develop a tool compatible for most non-model organisms, where inducible promoters are either not known or not characterised, we used the constitutive lacUV5 promoter as a representative constitutive promoter to drive the expression of the Intron-Cas12a variants in E. coli.
Figure 3.

SIBR-Cas targeting assays in E. coli MG1655. (A) Schematic diagram of SIBR-Cas in the presence or absence of the theophylline inducer. In the presence of theophylline, the intron is self-spliced leading to the translation of FnCas12a with an additional four amino acids at its N-terminus (left path). In the presence of a targeting CRISPR-RNA (crRNA), FnCas12a targets the genome and cleaves it, causing cell death. The LacZ target site is shown as an example. The bold 5′-CAACGTCGTGACTGGGAAAA-3′ sequence indicates the target LacZ protospacer. The red 5′-TTA-3′ nucleotides indicate the PAM sequence. The repeat sequence of the crRNA is indicated with grey nucleotides. In the absence of theophylline, the intron cannot splice itself out of the pre-mRNA, leading to the translation of a short, five amino acid long, peptide (right path). (B) Targeting and induction efficiency of SIBR-Cas in E. coli MG1655. The genome of E. coli MG1655 was targeted at the LacZ locus with a targeting or a non-targeting spacer. Four intron variants (Int1−4; bad splicer to good splicer) were used to control the translation of FnCas12a and a WT-FnCas12a was used as a control. Transformed E. coli cells were serially plated on LB solid medium with the appropriate antibiotic in the absence (left panel) or the presence (right panel) of the theophylline inducer.
According to our design, unspliced precursor mRNAs will result short (5 amino acid) peptides due to the TAA stop codon present at the start of the intron (position + 1, +2, +3). In contrast, excision of the intron will result in the full FnCas12a protein fused to a short 4 amino acid tag (SSGL for Int1,2 and 4 or SLGL for Int3) at its N-terminus. Furthermore, to make splicing inducible, we added a theophylline aptamer at the P6 stem loop of the T4 td intron as previously described (36), resulting in a new tightly-controlled CRISPR-Cas system, which we named SIBR-Cas (Figure 3A). Wild-type FnCas12a (WT-FnCas12a, without intron) was used as a reference for comparison to the SIBR-Cas variants. The efficiency of targeting for the SIBR-Cas and WT-FnCas12a variants was assessed by transforming E. coli MG1655 cells with plasmids expressing either of the different Cas12a variants with either a LacZ targeting (T) or a non-targeting (NT) crRNA. After transformation, the cells were serially diluted and plated on media with or without the presence of the theophylline inducer (Figure 3B and Supplementary Figure S1).
The NT crRNA controls showed colonies up to the 10–5 dilution, both in the presence or absence of theophylline for all the SIBR-Cas variants and WT-FnCas12a (Figure 3B). No colonies were observed when the T crRNA and the WT-FnCas12a combination was used, regardless of induction with theophylline, demonstrating the strong Cas12a-mediated counter-selection. In contrast, transformants targeting LacZ and expressing either of the four SIBR-Cas variants (Int1−4), showed a notable reduction in colony number formation only when theophylline was present in the medium. Intriguingly, the targeting efficiency directly reflected the splicing efficiency of the intron variants tested for LacZ (Figure 2D), with Int1 (-4A, -5C, -6T) showing the least targeting efficiency (10-fold reduction) and Int4 (−4T, −5C, −6T) showing the highest targeting efficiency (103-fold reduction) upon induction.
Our results demonstrate consistency in the splicing activity of the intron variants regardless of its genomic context (LacZ or FnCas12a). We therefore hypothesized that the minimalistic nature of the ‘tag’ sequence that SIBR leaves behind after splicing (i.e. 12 nucleotides at the 5′ of the mRNA, four amino acid residues at the N-terminus of the resulting protein) will be of minor influence on the stability, translation and folding efficiency, although this might be different with other genes. The difference between the intron variants Int1, Int2 and Int4 is only a single nucleotide (Int1: UCCUCAGGU; Int2: UCCUCGGGU; Int4: UCCUCUGGU). Therefore, it is unlikely to be of major importance for the stability and folding of the mRNA, strongly suggesting that the difference in enzyme activity can mostly be attributed to the splicing efficiency of the intron. Moreover, the varied protein activity (LacZ and FnCas12a) is unlikely to be caused by the small N-terminal tag as Int1, Int2 and Int4 have the exact (SSGL for Int1, 2 and 4) or very similar (SLGL for Int3) N-terminal amino acid sequence.
SIBR-Cas is an efficient genome engineering tool for bacteria
For efficient genome editing in bacteria, HR should precede CRISPR-Cas counter-selection (Figure 4A). To assess whether tight control over CRISPR-Cas targeting could bolster the efficiency of CRISPR-Cas mediated genome editing by allowing more time for HR to occur, we used SIBR-Cas and targeted the LacZ gene of E. coli MG1655 for knock-out through HR and CRISPR-Cas counter-selection using a blue/white screening colony assay. To facilitate HR, we added 500 bp up- and down-stream homology arms to the plasmids expressing the four SIBR-Cas (Int1−4) and WT-FnCas12a variants that target the LacZ gene. After 1 h recovery, we induced the expression of the SIBR-Cas variants to counter-select the WT from the mutant colonies.
Figure 4.

SIBR-Cas genome editing assays in E. coli MG1655, P. putida KT2440 and Flavobacterium IR1. (A) Schematic for SIBR-Cas editing procedure. The time required for recovery and HR differs amongst the different bacterial species as described in the materials and methods section. (B) Editing efficiency of the LacZ gene in E. coli MG1655. Blue/white screening was performed to distinguish the edited (white) from the unedited (blue) colonies when using either of the four different SIBR-Cas variants Int1 (0%), Int2 (1% ± 0.35%), Int3 (49% ± 8.75%) and Int4 (80% ± 5.57%) or the WT-FnCas12a (0%, n.d.). The percentage on top of each variant indicates the percentage of white colonies from the total number of colony forming units mL–1 (CFUs ml–1). (C) Representative plates of edited E. coli MG1655 cells at the LacZ locus using the four different SIBR-Cas variants (Int1-4; Int1 is the worst and Int4 is the best splicer). (D) Unbiased (omitting the presence of X-Gal in the medium) editing efficiency of the LacZ gene using the four different SIBR-Cas variants Int1 (0%), Int2 (0%), Int3 (29% ± 18.04%), Int4 (38% ± 6.41%) or the WT-FnCas12a (0%, n.d.). All of the NT controls showed 0% targeting efficiency. (E) Editing efficiency of the EndA [Int1 (21% ± 36.08), Int2 (29% ± 49.49%), Int3 (19% ± 17.35%), Int4 (63% ± 27.24%) or the WT-FnCas12a (0%, n.d.)] and FlgM [Int1 (0%), Int2 (41% ± 13.84%), Int3 (40% ± 6.41%), Int4 (70% ± 9.85%) or the WT-FnCas12a (0%, n.d.)] genes in P. putida KT2440. All of the NT controls showed 0% targeting efficiency. (F) Editing efficiency of the SprF [Int1 (75% ± 38.29), Int2 (100% ± 0%), Int3 (100% ± 0%), Int4 (0%, n.d.) or the WT-FnCas12a (0%, n.d.)] and GldJ [Int1 (68% ± 29.03%), Int2 (72% ± 41.26%), Int3 (96% ± 7.22%), Int4 (0%, n.d.) or the WT-FnCas12a (0%, n.d.)] genes in Flavobacterium IR1. Individual bars represent the mean of triplicate experiments and ‘•’ represents the value of each replicate. N.d., not determined. All of the NT controls showed 0% targeting efficiency. (G) Comparison between WT Flavobacterium IR1 and ΔSprF and ΔGldJ strains generated with SIBR-Cas. Images were taken after incubation at room temperature for 2 days by inoculating 3 μl spot on ASWBC solid medium (ASW supplemented with black ink and carrageenan). The WT strain (left) is 18 mm across.
The WT-FnCas12a variant targeting the LacZ gene produced no colonies, demonstrating the targeting efficiency of WT-FnCas12a but also the inefficient HR system of the WT E. coli MG1655 strain (Figure 4B). In contrast, SIBR-Cas variants produced multiple colonies of which 80% of the total CFUs ml–1 were white when Int4 was used, followed by Int3 (49%), Int2 (1%) and Int1 (0%) variants (Figure 4B and C). Similar to the previous results, the high editing efficiencies obtained with Int4 suggest that its high splicing efficiency translates into a stronger counter-selective pressure. No white colonies were observed for the non-targeting controls, demonstrating that the efficiency of editing without CRISPR-Cas counter-selection is negligible (Supplementary Figure S2).
Since disruption of LacZ can also be achieved through non-HR mediated approaches (spontaneous mutations or occasional error-prone DNA repair following DNA cleavage by Cas12a), not all gene deletions can be screened phenotypically. Therefore, we repeated our experiment, but X-gal was omitted from the medium to eliminate the possibility of false-positives. Randomly selected colonies that were obtained were screened by PCR for LacZ deletion showing a 0%, 0%, 29% and 38% editing efficiency for Int1, Int2, Int3 and Int4 SIBR-Cas variants, respectively (Figure 4D and Supplementary Figure S3). The WT-FnCas12a variant targeting LacZ did not yield any colonies and all the colonies obtained from the NT controls had the intact, wild-type LacZ locus. The observed decrease in editing efficiency (compared to the blue/white screening) might be attributed to spontaneous LacZ mutations that escape CRISPR-Cas counter-selection. Nevertheless, an editing efficiency of 38% was observed when SIBR-Cas Int4 was used without the use of recombinases or any other complex systems.
Following the successful use of SIBR-Cas in E. coli, we continued to demonstrate the efficiency of SIBR-Cas by testing it in other bacteria. For this purpose, we selected Pseudomonas putida KT2440, an organism with rather complex engineering tools and low HR efficiencies (44). After establishing the successful induction and targeting of SIBR-Cas in P. putida (Supplementary Figure S4), genome editing experiments were conducted to knock-out the EndA and FlgM genes. High knock-out efficiencies were obtained when Int4 was used, with editing efficiencies of 63% and 70% for EndA and FlgM, respectively (Figure 4E, Supplementary Figures S5 and S6). Lower editing efficiencies (<40%) were observed for the other introns, whereas no transformants were obtained with the WT-FnCas12a variant when used with a targeting spacer. Control transformants with the NT crRNA had a WT genotype (Supplementary Figures S6 and S7).
Lastly, we focused on the non-model organism Flavobacterium IR1, which is a recent isolate best known for its iridescent, structural colour (45–47). The lack of genomic tools, low transformation efficiency and the low HR efficiency of IR1 are currently the main bottlenecks holding back the fundamental characterization and commercial exploitation of this phenomenon (i.e. development of photonic paints). In addition, Flavobacterium species do not have a canonical RBS (TAAAA rather than GGAGG) (28–30), which render other widely applicable gene control systems, such as Ribo-Cas (15), inadequate for this type of bacterial species. To this end, we transformed Flavobacterium IR1 with a series of pSIBR plasmids (Supplementary Table S1) and assessed the editing efficiency after 96 h of growth by plating the transformants on plates that contained the theophylline inducer. By using SIBR-Cas, 100% editing efficiency was achieved for both Int2 and Int3 variants when SprF was targeted for knock-out (Figure 4F and Supplementary Figure S8). In accordance, the phenotype of the SprF mutants displayed similar characteristics when compared to previous studies (45,46) (Figure 4G). 100% editing efficiency was also achieved for some of the replicates of Int2 (1 out of 3) and Int3 (2 out of 3) when the GldJ gene was targeted for knock-out (Figure 4F and Supplementary Figure S9). Furthermore, SIBR-Cas was successful in creating a clean GldJ mutant, that could not be achieved by previous endeavours using transposon mutagenesis (45,46). Similar to the SprF mutant, the GldJ mutant could not develop the iridescence phenotype displayed by WT Flavobacterium IR1 (Figure 4G). The WT-FnCas12a variants did not yield any colonies when the SprF or GldJ genes were targeted for knock-out and the non-targeting controls were all confirmed to be unedited (Figure 4F, Supplementary Figures S8 and S9). To achieve high editing efficiency in Flavobacterium IR1, a 96 h incubation (without Cas induction) was required as shorter incubation times (24 and 48 h) did not yield any viable colonies (Supplementary Figure S10). This is in agreement with our assumption that more time is required for HR to occur before the induction of CRISPR-Cas counter-selection and that dsDNA-break stimulated repair by HR was of minor importance. These results also demonstrate that, in this context, the prolonged incubation time did not enrich for escapees (crRNA, protospacer, PAM or Cas12a mutations). Surprisingly and in contrast to E. coli and P. putida, Int4 failed to sustain growth in the recovery stage (data not shown) and hence was not plated. We expect this to be caused by leakiness of the Int4 variant during the recovery phase.
Highly efficient CRISPR-Cas tools have previously been developed for E. coli MG1655 (1,48–51). The study by Jiang et al. (50) for example, demonstrated editing efficiencies close to 100%. This study uses a double vector system with inducible promoters to induce Cas counter-selection and bolstered homologous recombination with the use of the λ red recombinase. Another study used a more simplistic one-plasmid system (52), where the control of Cas9 was under the inducible L-arabinose promoter, allowing for inducible counter-selection. Without the use of any recombinases, 20.8% knock-out efficiency was observed when the PoxB gene was targeted for editing. In contrast, when the λ red recombinase was included, 100% editing efficiency of PoxB was observed, indicating again the importance of exogenously expressed recombinases for high editing efficiency. In general, even for the model organism E. coli, the use of recombinases and inducible promoters is required for efficient genome engineering. SIBR-Cas was specifically developed to alleviate the need for these requirements, as both recombinases and inducible promoter might not be available for (or incompatible with) the host, especially in non-model organisms.
Similar to E. coli, various CRISPR-Cas tools have been developed for P. putida KT2440 (44,53–56). In the study of Sun et al. (2018), for example, a double vector system was used to express the λ red recombinase genes, the Cas nuclease (either Cas9 or Cas12) and the gRNA scaffold. Here, the requirement for exogenously expressed recombinases was even more evident, showing 93% editing with and 0% without the use of the λ red recombination system respectively (44). Nevertheless, successful genome editing in P. putida KT2440 has been achieved without the use of exogenous recombinases (56). Here, a plasmid was constructed expressing a thermostable variant of Cas9 (ThermoCas9) under the control of a 3-methylbenzoate-inducible Pm-promoter, a sgRNA targeting the PyrF gene and contained a homologous recombination template. Transformants were first screened for plasmid integration by PCR, after which a single colony was used for overnight growth in selective media, followed by inoculation in fresh medium. After another 6 h of growth in inducing media, dilutions were plated, ultimately resulting in a 50% editing efficiency rate (56). Lastly, ssDNA recombineering and CRISPR-Cas counter-selection approaches for editing P. putida KT2440 have been developed by Aparicio et al. (53) and Wu et al. (54). Both studies used inducible recombinases (Ssr or Redβ) expressed from an additional plasmid. Of note is the editing efficiency of EndA (54.2%) and FlgM (93.2%) as reported by Aparicio et al. (2018) (53) compared to our results (63% for EndA and 70% for FlgM). Whilst high editing efficiencies are reported by other CRISPR tools for P. putida KT2440, SIBR-Cas is a more widely applicable and simplistic approach (single plasmid, no need for integrant verification, no recombinase or inducible promoters needed) achieving similar or, in some instances, higher editing efficiencies.
We further showed that SIBR-Cas is compatible for non-model organisms as exemplified by our successful engineering attempt in Flavobacterium IR1. Since Flavobacterium IR1 is a relatively new isolate (45–47), inducible promoters or other gene regulation systems are not described yet. Therefore, SIBR presented a simple and efficient solution to control the expression of CRISPR-Cas and to achieve editing in case the more canonical strategy of using homologous recombination combined with CRISPR-Cas counter-selection fails, as demonstrated here (Figure 4F and Supplementary Figure S10). This is the first report for genome engineering of Flavobacterium IR1 using CRISPR-Cas and can serve as the basis for the easy and efficient engineering of other Flavobacteria species.
Collectively, our results show that SIBR-Cas is a tight and inducible genome engineering tool that can successfully be applied to a wide variety of bacterial species. By delaying CRISPR-Cas counter-selection and thus allowing enough time for HR to occur, we achieved high editing efficiencies in WT model and non-model bacterial species that naturally have very low HR efficiencies. We propose that this tool could be the solution for the difficulties of using CRISPR-Cas for prokaryotic genome engineering, especially in organisms where HR efficiencies are low, the use of recombinases is not possible or inducible promoters are not known or not characterized. We also foresee that SIBR-Cas will significantly decrease the time required for and complexity of CRISPR-Cas mediated genome engineering in prokaryotes.
Lastly, despite the success of SIBR-Cas in the three WT bacterial species chosen for our study, replicative plasmids and transformation protocols existed for all the three species. Therefore, for the application of SIBR-Cas in different (non-model) bacterial species, the existence of replicative plasmids and transformation protocols is a prerequisite. Also, different counterselection schemes may need to be developed for each different bacterium (due to differences in doubling time and/or efficiencies of recombination). For example, 96 h of growth was required for Flavobacterium IR1 to obtain knock-out mutants, whereas only 1 h was required to obtain knock-out mutants in E. coli MG1655. Prolonged incubation time, however, may increase the chances of having escapee mutants. This was likely the case in our study as we observed variable editing efficiencies even amongst biological replicates (Figure 4D−F). This observation may imply that escapees developed early in the recovery phase, replicated further during the recovery phase and then avoided counterselection upon induction. The unpredictable nature of these events demonstrates the need to include replicates for these experiments, as also illustrated by the variable editing obtained in this study. Moreover, we shall not ignore several factors that may affect the splicing efficiency of the T4 td intron and hence the editing efficiency of SIBR-Cas. For example, the splicing efficiency of the T4 td intron may be affected by temperature, the salt and mineral concentration present in the growth medium, the pH of the growth medium and the presence of splicing inhibitors (e.g. certain antibiotics and co-factors) (57–67). Also, the uptake of the theophylline inducer by the bacterial cell is a requirement for splicing of SIBR; although theophylline uptake is common among diverse bacterial species (15,68). To turn SIBR-Cas into a truly universal genome engineering tool for bacteria, future studies should focus on the generation of SIBR systems with a variety of aptamers and a variety of self-splicing introns that overcome the aforementioned limitations.
SIBR-X as a modular, tight and inducible protein expression tool
SIBR was successfully applied to control the expression of the FnCas12a gene in an OFF to ON manner. We suggest that SIBR-X (where X can be any gene/RNA of interest) can be a broader gene regulation tool for virtually any GOI (Figure 5A). This is mainly attributed to the host factor-independent splicing mechanism of the intron variants created during this study. Furthermore, our design of placing the intron directly after the ATG start codon means that it should be compatible with most GOI, leaving only a short four amino acid tag at the N-terminus of the POI, diminishing the risk of interfering with the protein's functionality. Therefore, with the combination of the intron variants and the theophylline inducer concentration, a temporal and tuneable gene expression can be achieved.
Figure 5.
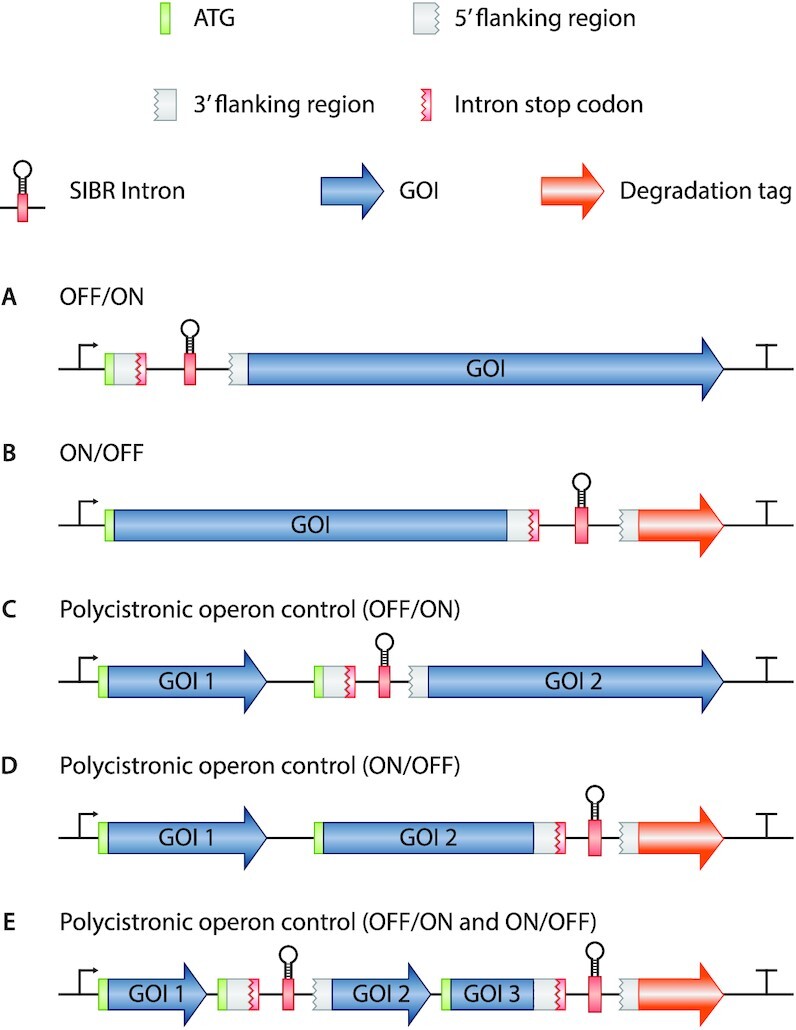
Potential applications of SIBR. (A) OFF to ON switch by interrupting the translation of the GOI. (B) ON to OFF switch by degrading the POI after inducible attachment of a degradation tag. The degradation tag can be replaced by a localization tag as well. (C) Polycistronic operon control allowing constitutive expression of the first gene (Gene of interest 1; GOI 1) and inducible OFF to ON expression of the second gene (GOI 2). (D) Polycistronic operon control by allowing constitutive expression of both GOI 1 and GOI 2 and inducible protein degradation of GOI 2 upon induction. (E) A combination of all the potential SIBR applications in one single polycistronic operon.
SIBR can also be used as an ON to OFF switch (Figure 5B). For example, SIBR can be inserted at the 3′ of the coding sequence with a downstream degradation tag (e.g. SsrA degradation tag). This design will allow for constitutive translation of the POI in the absence of the inducer (terminated at the stop codon of the intron), but will trigger rapid protein degradation after splicing of the intron due to the attached degradation tag. Akin to other down-regulation approaches (such as RNAi and CRISPRi), SIBR-mediated knockdown efficiencies are highly dependent on the turnover speed (and stability) of the protein in question. Other fusions can be envisioned as well, such as a (nuclear) localization tags, signal peptides, etc. Lastly, SIBR can be used as a polycistronic operon control mechanism in different configurations (Figure 5C−E). This approach will be especially useful in organisms where temporal and inducible expression is difficult to achieve by other means (e.g. operons with multiple and/or uncharacterized promoters and terminators).
Conclusively, we foresee various applications within industry and fundamental research, where SIBR-X can be a valuable tool in both model and non-model organisms.
DATA AVAILABILITY
All data included in this study is available upon request by contact with the corresponding author.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Hoekmine B.V. for sharing their IR1 strain and expertise. We thank Rob Joosten and Steven Aalvink for their technical support.
Author contribution: C.P., S.C.A.C., A.Q.A., S.W.M., J.v.d.O. and R.H.J.S. designed the study. C.P., S.C.A.C., A.Q.A., B.A.P. and E.A.G. created all the plasmids. S.C.A.C. and E.A.G. generated and characterized the intron variants. C.P. and A.Q.A. generated and characterized SIBR-Cas. C.J.I. provided lab facilities and expertise for handling Flavobacterium IR1 through Hoekmine B.V.. C.P., S.C.A.C. and R.H.J.S. wrote the manuscript and all authors contributed in reviewing, editing and approving the final manuscript.
Contributor Information
Constantinos Patinios, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Sjoerd C A Creutzburg, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Adini Q Arifah, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Belén Adiego-Pérez, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Evans A Gyimah, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Colin J Ingham, Hoekmine Besloten Vennootschap, Kenniscentrum Technologie en Innovatie, Hogeschool Utrecht, 3584 CS, Utrecht, The Netherlands.
Servé W M Kengen, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
John van der Oost, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
Raymond H J Staals, Laboratory of Microbiology, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Graduate School VLAG, Wageningen University and Research, Netherlands; R.H.J.S. was supported by a VENI grant [016.Veni.171.047 R.H.J.S.], from ‘The Netherlands Organization for Scientific Research’ (NWO); J.v.d.O. was supported by the ‘European Research Council’ (ERC) [ERC-AdG-834279 to J.v.d.O.]. Funding for open access charge: ERC Advanced Grant (European Research Council) [ERC-AdG-834279].
Conflict of interest statement. The authors (C.P., S.C.A.C, J.v.d.O. and R.H.J.S) have filed a patent application based on the results reported in this study.
REFERENCES
- 1. Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A.. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013; 31:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mougiakos I., Bosma E.F., de Vos W.M., van Kranenburg R., van der Oost J.. Next generation prokaryotic engineering: the CRISPR-Cas toolkit. Trends Biotechnol. 2016; 34:575–587. [DOI] [PubMed] [Google Scholar]
- 3. Dillingham M.S., Kowalczykowski S.C.. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008; 72:642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cui L., Bikard D.. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016; 44:4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wannier T.M., Ciaccia P.N., Ellington A.D., Filsinger G.T., Isaacs F.J., Javanmardi K., Jones M.A., Kunjapur A.M., Nyerges A., Pal C.. Recombineering and MAGE. Nat. Rev. Methods Primers. 2021; 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J., Hong W., Zong W., Wang P., Wang Y.. Markerless genome editing in Clostridium beijerinckii using the CRISPR-Cpf1 system. J. Biotechnol. 2018; 284:27–30. [DOI] [PubMed] [Google Scholar]
- 7. Wasels F., Jean-Marie J., Collas F., López-Contreras A.M., Ferreira N.L.. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J. Microbiol. Methods. 2017; 140:5–11. [DOI] [PubMed] [Google Scholar]
- 8. Diallo M., Hocq R., Collas F., Chartier G., Wasels F., Wijaya H.S., Werten M.W., Wolbert E.J., Kengen S.W., van der Oost J.. Adaptation and application of a two-plasmid inducible CRISPR-Cas9 system in Clostridium beijerinckii. Methods. 2020; 172:51–60. [DOI] [PubMed] [Google Scholar]
- 9. Davis K.M., Pattanayak V., Thompson D.B., Zuris J.A., Liu D.R.. Small molecule–triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015; 11:316–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Truong D.-J.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O.. Development of an intein-mediated split–Cas9 system for gene therapy. Nucleic Acids Res. 2015; 43:6450–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zetsche B., Volz S.E., Zhang F.. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015; 33:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nihongaki Y., Kawano F., Nakajima T., Sato M.. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015; 33:755–760. [DOI] [PubMed] [Google Scholar]
- 13. Liu K.I., Ramli M.N.B., Woo C.W.A., Wang Y., Zhao T., Zhang X., Yim G.R.D., Chong B.Y., Gowher A., Chua M.Z.H.. A chemical-inducible CRISPR–Cas9 system for rapid control of genome editing. Nat. Chem. Biol. 2016; 12:980. [DOI] [PubMed] [Google Scholar]
- 14. Zhao J., Inomata R., Kato Y., Miyagishi M.. Development of aptamer-based inhibitors for CRISPR/Cas system. Nucleic Acids Res. 2020; 49:1330–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cañadas I.s.C., Groothuis D., Zygouropoulou M., Rodrigues R., Minton N.P.. RiboCas: a universal CRISPR-based editing tool for Clostridium. ACS Synth. Biol. 2019; 8:1379–1390. [DOI] [PubMed] [Google Scholar]
- 16. Tang W., Hu J.H., Liu D.R.. Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat. Commun. 2017; 8:15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siu K.-H., Chen W.. Riboregulated toehold-gated gRNA for programmable CRISPR–Cas9 function. Nat. Chem. Biol. 2019; 15:217–220. [DOI] [PubMed] [Google Scholar]
- 18. Kundert K., Lucas J.E., Watters K.E., Fellmann C., Ng A.H., Heineike B.M., Fitzsimmons C.M., Oakes B.L., Qu J., Prasad N.. Controlling CRISPR-Cas9 with ligand-activated and ligand-deactivated sgRNAs. Nat. Commun. 2019; 10:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moroz-Omori E.V., Satyapertiwi D., Ramel M.-C., Høgset H.k., Sunyovszki I.K., Liu Z., Wojciechowski J.P., Zhang Y., Grigsby C.L., Brito L.. Photoswitchable gRNAs for spatiotemporally controlled CRISPR-Cas-based genomic regulation. ACS Central Sci. 2020; 6:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jain P.K., Ramanan V., Schepers A.G., Dalvie N.S., Panda A., Fleming H.E., Bhatia S.N.. Development of light-activated CRISPR using guide RNAs with photocleavable protectors. Angew. Chem. Int. Ed. 2016; 55:12440–12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y., Liu Y., Xie F., Lin J., Xu L.. Photocontrol of CRISPR/Cas9 function by site-specific chemical modification of guide RNA. Chem. Sci. 2020; 11:11478–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou W., Brown W., Bardhan A., Delaney M., Ilk A.S., Rauen R.R., Kahn S.I., Tsang M., Deiters A.. Spatiotemporal control of CRISPR/Cas9 function in cells and zebrafish using light-activated guide RNA. Angew. Chem. 2020; 132:9083–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Breaker R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012; 4:a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park S.V., Yang J.-S., Jo H., Kang B., Oh S.S., Jung G.Y.. Catalytic RNA, ribozyme, and its applications in synthetic biology. Biotechnol. Adv. 2019; 37:107452. [DOI] [PubMed] [Google Scholar]
- 25. Serganov A., Nudler E.. A decade of riboswitches. Cell. 2013; 152:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Serganov A., Patel D.J.. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat. Rev. Genet. 2007; 8:776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinberg C.E., Weinberg Z., Hammann C.. Novel ribozymes: discovery, catalytic mechanisms, and the quest to understand biological function. Nucleic Acids Res. 2019; 47:9480–9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen S., Bagdasarian M., Kaufman M., Walker E.. Characterization of strong promoters from an environmental Flavobacterium hibernum strain by using a green fluorescent protein-based reporter system. Appl. Environ. Microbiol. 2007; 73:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gómez E., Álvarez B., Duchaud E., Guijarro J.A.. Development of a markerless deletion system for the fish-pathogenic bacterium Flavobacterium psychrophilum. PLoS One. 2015; 10:e0117969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Accetto T., Avguštin G.. Inability of Prevotella bryantii to form a functional Shine-Dalgarno interaction reflects unique evolution of ribosome binding sites in Bacteroidetes. PLoS One. 2011; 6:e22914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hausner G., Hafez M., Edgell D.R.. Bacterial group I introns: mobile RNA catalysts. Mobile DNA. 2014; 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edgell D.R., Belfort M., Shub D.A.. Barriers to intron promiscuity in bacteria. J. Bacteriol. 2000; 182:5281–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nielsen H., Johansen S.D.. Group I introns: moving in new directions. RNA biology. 2009; 6:375–383. [DOI] [PubMed] [Google Scholar]
- 34. Lee E.R., Baker J.L., Weinberg Z., Sudarsan N., Breaker R.R.. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010; 329:845–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen A.G., Sudarsan N., Breaker R.R.. Mechanism for gene control by a natural allosteric group I ribozyme. RNA. 2011; 17:1967–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson K.M., Syrett H.A., Knudsen S.M., Ellington A.D.. Group I aptazymes as genetic regulatory switches. BMC Biotech. 2002; 2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Batianis C., Kozaeva E., Damalas S.G., Martín-Pascual M., Volke D.C., Nikel P.I., Martins dos Santos V.A.. An expanded CRISPRi toolbox for tunable control of gene expression in Pseudomonas putida. Microb. Biotechnol. 2020; 13:368–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carrión V.J., Perez-Jaramillo J., Cordovez V., Tracanna V., De Hollander M., Ruiz-Buck D., Mendes L.W., Van Ijcken W.F., Gomez-Exposito R., Elsayed S.S.et al.. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science. 2019; 366:606–612. [DOI] [PubMed] [Google Scholar]
- 39. Chu F.K., Maley G.F., Maley F., Belfort M.. Intervening sequence in the thymidylate synthase gene of bacteriophage T4. Proc. Natl. Acad. Sci. U.S.A>. 1984; 81:3049–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chu F.K., Maley G.F., West D.K., Belfort M., Maley F.. Characterization of the intron in the phage T4 thymidylate synthase gene and evidence for its self-excision from the primary transcript. Cell. 1986; 45:157–166. [DOI] [PubMed] [Google Scholar]
- 41. Pichler A., Schroeder R.. Folding problems of the 5′ splice site containing the P1 stem of the group I thymidylate synthase intron: substrate binding inhibition in vitro and mis-splicing in vivo. J. Biol. Chem. 2002; 277:17987–17993. [DOI] [PubMed] [Google Scholar]
- 42. Sandegren L., Sjöberg B.-M.. Self-splicing of the bacteriophage T4 group I introns requires efficient translation of the pre-mRNA in vivo and correlates with the growth state of the infected bacterium. J. Bacteriol. 2007; 189:980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun J., Wang Q., Jiang Y., Wen Z., Yang L., Wu J., Yang S.. Genome editing and transcriptional repression in Pseudomonas putida KT2440 via the type II CRISPR system. Microb. Cell Fact. 2018; 17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamidjaja R., Capoulade J., Catón L., Ingham C.J.. The cell organization underlying structural colour is involved in Flavobacterium IR1 predation. The ISME Journal. 2020; 14:2890–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johansen V.E., Catón L., Hamidjaja R., Oosterink E., Wilts B.D., Rasmussen T.S., Sherlock M.M., Ingham C.J., Vignolini S.. Genetic manipulation of structural color in bacterial colonies. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2652–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schertel L., van de Kerkhof G.T., Jacucci G., Catón L., Ogawa Y., Wilts B.D., Ingham C.J., Vignolini S., Johansen V.E.. Complex photonic response reveals three-dimensional self-organization of structural coloured bacterial colonies. J. R. Soc., Interface. 2020; 17:20200196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pyne M.E., Moo-Young M., Chung D.A., Chou C.P.. Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl. Environ. Microbiol. 2015; 81:5103–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bassalo M.C., Garst A.D., Halweg-Edwards A.L., Grau W.C., Domaille D.W., Mutalik V.K., Arkin A.P., Gill R.T.. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth. Biol. 2016; 5:561–568. [DOI] [PubMed] [Google Scholar]
- 50. Jiang Y., Chen B., Duan C., Sun B., Yang J., Yang S.. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015; 81:2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ao X., Yao Y., Li T., Yang T.-T., Dong X., Zheng Z.-T., Chen G.-Q., Wu Q., Guo Y.. A multiplex genome editing method for Escherichia coli based on CRISPR-Cas12a. Front. Microbiol. 2018; 9:2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao D., Yuan S., Xiong B., Sun H., Ye L., Li J., Zhang X., Bi C.. Development of a fast and easy method for Escherichia coli genome editing with CRISPR/Cas9. Microb. Cell Fact. 2016; 15:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aparicio T., de Lorenzo V., Martínez-García E.. CRISPR/Cas9-based counterselection boosts recombineering efficiency in Pseudomonas putida. Biotechnol. J. 2018; 13:1700161. [DOI] [PubMed] [Google Scholar]
- 54. Wu Z., Chen Z., Gao X., Li J., Shang G.. Combination of ssDNA recombineering and CRISPR-Cas9 for Pseudomonas putida KT2440 genome editing. Appl. Microbiol. Biotechnol. 2019; 103:2783–2795. [DOI] [PubMed] [Google Scholar]
- 55. Wirth N.T., Kozaeva E., Nikel P.I.. Accelerated genome engineering of Pseudomonas putida by I-SceI-mediated recombination and CRISPR-Cas9 counterselection. Microb. Biotechnol. 2020; 13:233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mougiakos I., Mohanraju P., Bosma E.F., Vrouwe V., Bou M.F., Naduthodi M.I., Gussak A., Brinkman R.B., Van Kranenburg R., Van Der Oost J.. Characterizing a thermostable Cas9 for bacterial genome editing and silencing. Nat. Commun. 2017; 8:1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ahn S.J., Park I.K.. The coenzyme thiamine pyrophosphate inhibits the self-splicing of the group I intron. Int. J. Biochem. Cell Biol. 2003; 35:157–167. [DOI] [PubMed] [Google Scholar]
- 58. Jung C., Shin S., Park I.K.. Pyridoxal phosphate inhibits the group I intron splicing. Mol. Cell. Biochem. 2005; 280:17–23. [DOI] [PubMed] [Google Scholar]
- 59. Jung W.S., Shin S., Park I.K.. Novobiocin inhibits the self-splicing of the primary transcripts of T4 phage thymidylate synthase gene. Mol. Cell. Biochem. 2008; 314:143–149. [DOI] [PubMed] [Google Scholar]
- 60. Kim J.H., Park I.K.. Inhibition of the group I ribozyme splicing by NADP+. Mol. Cell. Biochem. 2003; 252:285–293. [DOI] [PubMed] [Google Scholar]
- 61. LIU Y., TIDWELL R.R., Leibowitz M.J.. Inhibition of in vitro splicing of a group I intron of Pneumocystis carinii. J. Eukaryot. Microbiol. 1994; 41:31–38. [DOI] [PubMed] [Google Scholar]
- 62. Park I.K. Effects of deamido-NAD+ on self-splicing of primary transcripts of phage T4 thymidylate synthase gene. Korean J. Biol. Sci. 2000; 4:141–144. [Google Scholar]
- 63. Park I.K., Kim J.Y., Lim E.H., Shin S.. Spectinomycin inhibits the self-splicing of the group 1 intron RNA. Biochem. Biophys. Res. Commun. 2000; 269:574–579. [DOI] [PubMed] [Google Scholar]
- 64. Park I.K., Kim J.Y.. NAD+ Inhibits the self-splicing of the group I intron. Biochem. Biophys. Res. Commun. 2001; 281:206–211. [DOI] [PubMed] [Google Scholar]
- 65. von Ahsen U., Davies J., Schroeder R.. Antibiotic inhibition of group I ribozyme function. Nature. 1991; 353:368–370. [DOI] [PubMed] [Google Scholar]
- 66. von Ahsen U., Davies J., Schroeder R.. Non-competitive inhibition of group I intron RNA self-splicing by aminoglycoside antibiotics. J. Mol. Biol. 1992; 226:935–941. [DOI] [PubMed] [Google Scholar]
- 67. Waldsich C., Semrad K., Schroeder R.. Neomycin B inhibits splicing of the td intron indirectly by interfering with translation and enhances missplicing in vivo. RNA. 1998; 4:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Groher F., Suess B.. Synthetic riboswitches—a tool comes of age. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014; 1839:964–973. [DOI] [PubMed] [Google Scholar]
- 69. Cech T.R., Damberger S.H., Gutell R.R.. Representation of the secondary and tertiary structure of group I introns. Nat. Struct. Biol. 1994; 1:273–280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in this study is available upon request by contact with the corresponding author.