

Abstract
Creating small-molecule antivirals specific for SARS-CoV-2 proteins is crucial to battle COVID-19. SARS-CoV-2 main protease (Mpro) is an established drug target for the design of protease inhibitors. We performed a structure-activity relationship (SAR) study of noncovalent compounds that bind in the enzyme’s substrate binding subsites S1 and S2 revealing structural, electronic and electrostatic determinants of these sites. The study was guided by the X-ray/neutron structure of Mpro complexed with Mcule-5948770040 (compound 1), in which protonation states were directly visualized. Virtual reality-assisted structure analysis and small-molecule building were employed to generate analogs of 1. In vitro enzyme inhibition assays and room-temperature X-ray structures demonstrated the effect of the chemical modifications on Mpro inhibition, showing that 1) maintaining correct geometry of an inhibitor’s P1 group is essential to preserve the hydrogen bond with the protonated His163; 2) a positively charged linker is preferred; and 3) subsite S2 prefers non-bulky modestly electronegative groups.
Graphical Abstract

INTRODUCTION
Since the start of the COronaVIrus Disease 2019 (COVID-19) pandemic in early 2020, several preventative and treatment options have been developed, including several vaccines and antiviral therapies.1–4 The COVID-19 vaccines developed in record time are now potentially saving millions of lives. However, due to vaccine hesitancy, preexisting health conditions, and vaccine escape variants of SARS-CoV-25, a significant proportion of the population will remain at risk, creating an urgent priority to advance existing therapeutics. COVID-19 is caused by a novel coronavirus, SARS-CoV-2, believed to be of zoonotic origin6,7, raising concerns that other easily transmissible respiratory viruses will emerge to cause future pandemics. The arsenal of therapeutic intervention options will undoubtedly be expanded by designing multiple small-molecule drugs that inhibit various viral targets disrupting essential steps in the SARS-CoV-2 replication cycle.8 This bolstered preparedness strategy has the potential to yield broad-spectrum antivirals providing a means of tackling future pathogenic coronaviruses.
3-Chymotrypsin-like protease (3CLpro), also known as the main protease (Mpro), from SARS-CoV-2 is a viral cysteine protease enzyme9 and an important drug target that has attracted considerable attention from structural and computational biologists and drug designers.10–15 SARS-CoV-2 is a single-stranded, positive-sense RNA virus with a genome of ~30k nucleotides resembling mRNA.16 Host cell ribosomes partially translate the genomic mRNA to generate two polyproteins, pp1a and pp1ab, encoded by the viral replicase gene during the initial steps of the virus replication cycle.16,17 The proteolytic cleavage of pp1a and pp1ab is vital for SARS-CoV-2 proliferation and liberates 16 individual viral protein components necessary for the viral genome transcription activity. This essential step in the SARS-CoV-2 replication cycle is accomplished by Mpro, and another cysteine protease, the papain-like protease (PLpro), through hydrolyzing peptide bonds within the two polyproteins at specific locations. Small-molecule inhibitors of the viral protease enzymatic activity have strong clinical precedence for blocking virus replication, hence the fervent interest of the scientific community to develop coronavirus-specific protease inhibitors. The active site of Mpro is distinct from the known human proteases; thus off-target binding of specific Mpro inhibitors can be minimized.10,11 Conversely, conservation of the Mpro active site across various coronaviruses creates an opportunity to design pan-coronavirus antivirals.18
Significant effort in the design of Mpro inhibitors against SARS-CoV-28,19–21 has focused on the reversible and irreversible (suicidal) covalent inhibitors, including compounds initially designed for the inhibition of SARS-CoV Mpro.22–25 Such inhibitors contain chemical groups, or warheads, that are reactive towards the catalytic cysteine of Mpro, Cys145. Hepatitis C virus clinical protease inhibitor boceprevir and the feline peritonitis virus inhibitor GC-376 were initially considered for drug repurposing, leading to the rational design of hybrid inhibitors.26–28 In addition, noncovalent competitive and allosteric inhibitors have captured interest due to the availability of high-throughput virtual and experimental screening of large compound libraries that leverage new advances in supercomputing and fast X-ray crystallographic screening.15,29–32 Compounds can be fed into structure-based drug design pipelines and chemically modified to improve their potency to inhibit SARS-CoV-2 Mpro.33–36 Moreover, noncovalent inhibitors may have higher selectivity for Mpro compared to covalent compounds that can also target host proteases and can possess elevated cytotoxicity due to binding to other human proteins.33,36
The active site of Mpro consists of subsites S1’ through S5, which can accommodate substrate and inhibitor groups at positions P1’ through P5. Subsites S1 and S2 are selective for Gln and a medium-sized hydrophobic residue like Leu or Phe, respectively.11,37–39 A recent study analyzed the effect of chemical modifications in a noncovalent inhibitor ML188 on its binding to Mpro.36 Here, we report a structure-activity relationship (SAR) study performed on a competitive noncovalent inhibitor Mcule-5948770040 (compound 1) of a novel scaffold that we recently discovered through a large-scale virtual screening and validated using in vitro enzyme inhibition assays and X-ray crystallography.40 The aim of our SAR study was to chemically modify compound 1 that binds across the Mpro catalytic site in the substrate binding subsites S1 and S2 in order to reveal structural, electronic and electrostatic determinants of these ligand binding sites. Compound 1 has a general architecture of P1-Linker-P2 (Scheme 1). We initiated the study by obtaining a joint X-ray/neutron (XN) structure of Mpro-1 complex at near-physiological temperature and neutral pH (Figure 1a). The XN structure permitted us to fully map the hydrogen positions (observed as deuterium atoms) in the Mpro active site and compound 1, accurately determining protonation states of the enzyme amino acid residues and the inhibitor. With this information in hand, we systematically derivatized P1, P2 and linker groups producing a series of compounds code-named HL-3 (Scheme 1). Virtual reality-assisted structure analysis and small-molecule building were employed to generate derivatives of 1, considering the geometric constraints of the Mpro subsites S1 and S2 and the feasibility of the syntheses. In vitro enzyme inhibition assays demonstrated the effect of the chemical modification on the ability of the modified compounds to inhibit Mpro. In contrast, subsequent X-ray crystallographic analysis at room temperature identified the structural determinants for P1, P2, and linker binding. Moreover, we designed an improved inhibitor, compound HL-3-68, that showed several-fold better inhibition of Mpro in vitro.
Scheme 1.

Chemical diagrams of Mcule-5948770040 (compound 1) and its derivatives. For the generalized structure of the HL-3 series not all possible combinations of X, Y and Z substituents were examined. The full list of HL-3 series of compounds, including their chemical structures, is given in Table S2.
Figure 1. Compound 1 binds to the S1 and S2 subsites of Mpro.
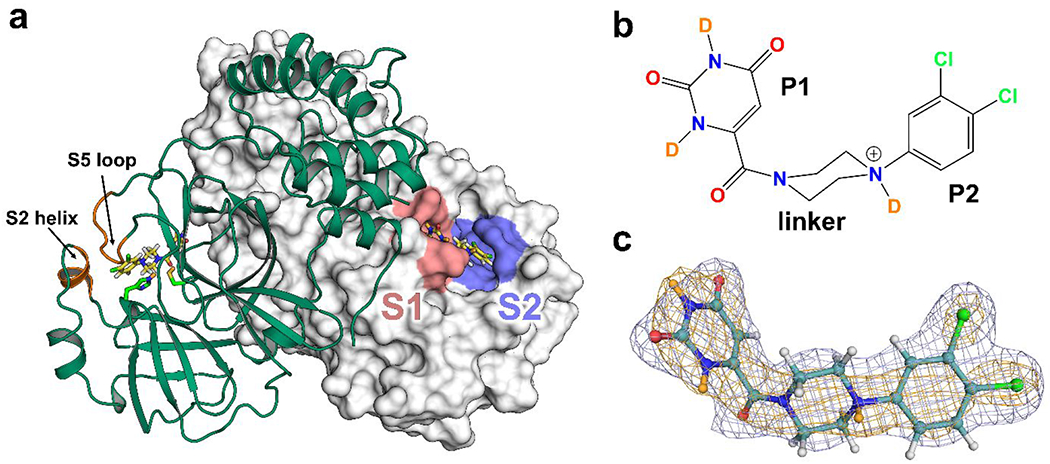
(a) Dimer of Mpro-1 is represented with one protomer as cartoon (green) and the other as surface (white). The locations of subsite S1 (pink) and S2 (purple) are highlighted. The S2 helix and S5 loop are labeled in orange. The Cys145 and His41 catalytic dyad are shown as sticks. PDB ID 7N8C.
(b) Chemical structure of compound 1 as observed in Mpro-1.
(c) Compound 1 from Mpro-1 shown in a ball-and-stick representation (teal carbons). H/D exchanged D atoms are colored in orange. Electron density 2Fo-Fc map (blue mesh) and nuclear density 2Fo-Fc map (orange mesh) contoured to 1σ.
RESULTS
Protonation states in Mpro-1 complex
Neutron crystallography of the Mpro-1 complex was employed to accurately resolve the position of hydrogen atoms in the active site of Mpro and compound 1. Neutron and X-ray diffraction datasets to 2.5 and 2.2 Å resolutions, respectively, were collected at room temperature and neutral pH from a large deuterated protein crystal and then jointly refined to produce accurate positions of both deuterium and heavy atoms (Table S1). The henceforth analysis includes comparisons to previously determined neutron structures of ligand-free Mpro 41 and Mpro bound to the covalent α-ketoamide inhibitor telaprevir.42. Compound 1 (Figure 1b) was modeled into the electron and nuclear density maps with high confidence (Figure 1c). For hydrogen bonds, distances between a deuterium (D) atom and the heavy atom are reported henceforth.
Direct interactions between Mpro and the P1 and P2 groups of 1 are shown in Figure 2a with 2FO-FC and D-omit FO-FC nuclear density maps. The uracil-like P1 group of 1 contains a carbonyl that forms a short 1.7 Å hydrogen bond with a doubly protonated His163. This carbonyl’s second lone pair of electrons makes a weaker unconventional C-H…O hydrogen bond with Cδ2 of His172. The other carbonyl of the P1 group forms a D2O-mediated interaction with Ser1’ of the second Mpro protomer. Another D2O-mediated interaction to Asn142 arises from the amide ND group at the P1 group’s 2 position, whereas the amide ND at the 4 position forms a 2.0 Å hydrogen bond with the carboxylate sidechain of Glu166. To facilitate this interaction, the Glu166 carboxylate rotates from its position observed in the ligand-free Mpro structure towards 1, and His163 gains a D atom on Nε2 to become positively charged in Mpro-1 (Figure S1a). Interestingly, the same conformational change of the Glu166 side chain was observed in the telaprevir-bound neutron structure, where His163 was also found in the doubly protonated state, relative to the ligand-free Mpro (Figure S1b). However, telaprevir possesses a short hydrophobic norvaline P1 group that prevents a direct polar interaction with the enzyme; instead, a water molecule is recruited to this position to hydrogen bond with His163. The aromatic di-chlorobenzene P2 group orchestrates itself into the hydrophobic S2 subsite by displacing Met49 and S2 helix (residues 46-52) and rearranging His41 and Gln189 to create π-π stacking interactions.40 P1 and P2 groups of 1 are connected by a saturated heterocyclic piperazine-amide linker that includes a carbonyl aimed toward the oxyanion hole and a potentially ionizable tertiary amine preceding P2. Analysis of the nuclear density demonstrated that the latter amine nitrogen is protonated with the D atom directed away from His41 and into the bulk solvent. As a result, compound 1 is a cation with a +1 positive charge.
Figure 2. Atomic details of SARS-CoV-2 Mpro complex with compound 1 determined by X-ray/neutron crystallography.
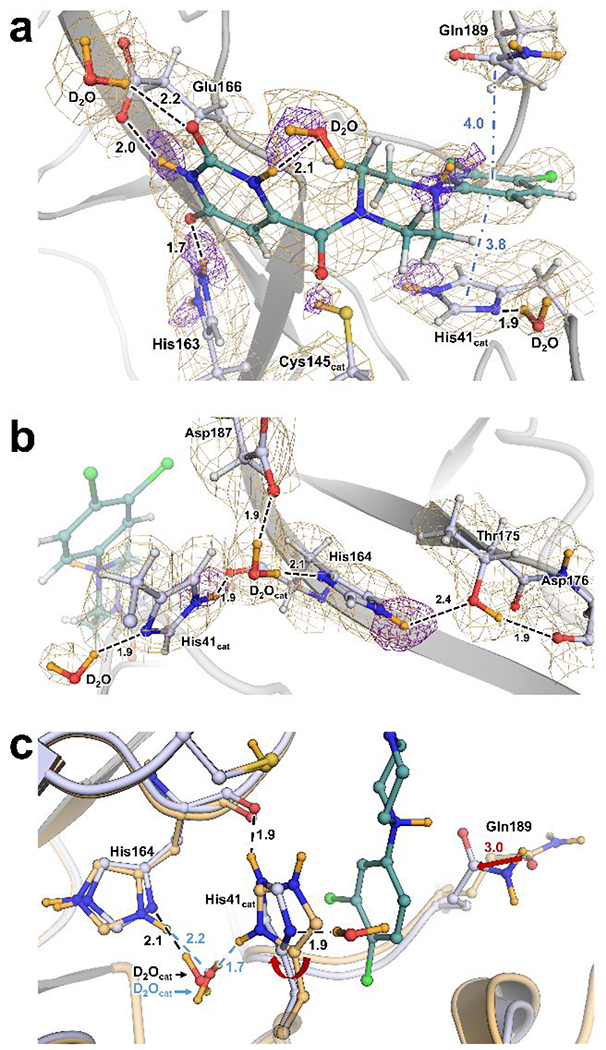
(a) Mpro protonation states and molecular interactions with 1. Protein structures are presented as cartoon with side chains and ligand in ball-and-stick representation. H-bonds represented as dashes while π-interactions are represented as blue dash-dots. Distances in Å. 2Fo-Fc nuclear density map as orange mesh contoured at 1σ. Omit maps for D atoms are shown as purple mesh and contour levels are adjusted for clarity as follows: linker amine D is 2.0σ, P1 amine Ds are 3.0σ, His41 Nε2 D is 2.5σ, His163 Nδ1 D is 4.0σ and Nε2 D is 3.0σ.
(b) H-bond network of the catalytic D2O and His41 side chain. 2Fo-Fc nuclear density map as orange mesh contoured at 1.5σ. Omit map for His164 Nδ1 D contoured at 3.0σ.
(c) Superposition of Mpro-1 (blue carbons) and Mpro ligand-free (light orange carbons, PDB code 7JUN) showing His41 flip and Gln189 shift in the complex. Red arrows indicate conformational shifts from apo to compound 1 complex. Ligand-free is labeled in blue where different. Superposition calculated by least-squares-fitting on Cα atoms.
The catalytic site of Mpro is composed of a non-canonical catalytic dyad, Cys145 and His41, thought to be assisted by a highly coordinated conserved water molecule (D2Ocat). The catalytic dyad exists in the zwitterionic state in ligand-free Mpro as discrete anionic thiolate and cationic imidazolium sidechains.41 The hydrogen bonding landscape of His41 and D2Ocat in Mpro-1 are presented in Figure 2b. In this complex, the catalytic dyad is neutralized with Cys145 observed as a thiol (-SD), and His41 singly protonated on Nε2 but not on Nδ1. The Cys145 thiol deuterium was refined to ~85% D occupancy. Relative to the ligand-free Mpro and Mpro-Telaprevir neutron structures, the His41 sidechain is flipped about 180°, its position in Mpro-1 being stabilized by a new hydrogen bond formed between Nε2-D and the His164 main chain carbonyl (Figure 2c, S1b). Consequently, a conserved hydrogen bond with D2Ocat observed in the other two neutron structures is eliminated to create a new hydrogen bond made by the Nδ1 with a D2O molecule recruited from the bulk solvent.
D2Ocat in Mpro-1 is oriented by donating its D atoms in hydrogen bonds with the His164 and Asp187 side chains in a rotation pose not seen in either the ligand-free or telaprevir-bound neutron structures (Figure S1a, S1b). In contrast to ligand-free Mpro in which His164 is doubly protonated, His164 is neutrally charged in Mpro-1, possessing strong nuclear density for Nε1-D that is hydrogen bonded to Thr175 (Figure 2b). Moreover, this neutral protonation state configuration of His164 is different from that observed in the Mpro-Telaprevir neutron structure where Nε2 is protonated, and the hydrogen bond to Thr175 is absent. Thus, in Mpro-1 D2Ocat rotates to donate its D atoms in hydrogen bonds with His164 Nε2 and Asp187 carboxylate.
Design of compound 1 derivatives – HL-3 series
The architecture of compound 1 can be divided into three parts: P1 and P2 groups and the linker (Scheme 1, Figure 1b). P1 is a uracil-like, 6-pyrimidine-2,4(1H,3H)-dione substituent capable of direct hydrogen bonding with Mpro residues His163 and Glu166 in the S1 subsite. The linker is a piperazine-1-carbonyl moiety whose carbonyl group is anchored by the oxyanion hole of the catalytic site. P2, a substituted benzene substituent, inserts into the mostly hydrophobic S2 subsite bordered by Met49 from the S2 helix, mainchain atoms of Arg188 and Gln189, and side chains of His41, Cys44, Tyr54 and Gln189. The Tyr54 phenolic hydroxyl is the only group that potentially can play a role of a hydrogen bonding partner with a P2 group of an inhibitor. To improve inhibitor affinity and probe the chemical determinants for ligand binding to S1 and S2, we designed an array of compound 1 derivatives, which we call HL-3 series, to methodically assess how the P1, linker, and P2 groups contribute to Mpro inhibition.
Our design strategy of compound 1 derivatives, i.e. HL-3 compounds, (Scheme 1 and Table 1) included chemical modifications to each of its three parts, taking into consideration specific geometric and hydrogen bonding constraints of S1 and S2 subsites and the feasibility of syntheses for designed compounds. We employed virtual reality software to immerse into the Mpro structure, to modify the scaffold of compound 1, and to perform structural analysis of the modeled complexes. First, we examined the effect of saturating the olefinic portion of the uracil-like (pyrimidine-2,4(1H,3H)-dione) P1 group that creates a non-planar sp3-hybridized endocyclic carbon. Both enantiomers were made to mimic the γ-lactam ring commonly used as inhibitor’s P1 (HL-3-51, HL-3-53). Such modification should not eliminate the P1 group’s ability to hydrogen bond with His163, which is a prerequisite for Mpro inhibitors. Next, we examined the effect of removing a positive charge from the linker, where the aniline nitrogen is observed in the protonated quaternary ammonium state in our neutron structure. To achieve this, the aniline nitrogen was replaced with a saturated carbon by substitution of a piperidine moiety for the piperazine in the linker (HL-3-69). The most extensive modifications were made to the P2 group by varying substituents at positions 3, 4, and 5 of the phenyl ring. We examined the effect of removing one Cl from either position 3 or 4 and synthesized singly substituted derivatives at position 4 containing groups such as F, I, CN, CF3, CHO (aldehyde), and CH2OH. In addition, we retained Cl in position 3 and varied substituents in position 4 to include Br, CF3, CHO, and CH2OH. In another compound, Cl in position 4 remained, but position 3 contained a CF3 group. Lastly, we synthesized derivatives with three substituents in positions 3, 4, and 5. In this series, the 3,5-meta positions had Cl groups, whereas para position 4 consisted of Cl, CF3 or CH3.
Table 1.
IC50 values for the inhibition of SARS-CoV-2 Mpro by the series of HL-3 compounds. X-ray crystallographic statistics for the obtained structures is given in Table S2.
| Compound | Chemical Structure | IC50, μM | PDB ID |
|---|---|---|---|
| Compound 1 (Mcule-5948770040) |

|
0.68 [0.48, 0.97]*40 | 7LTJ |
| HL-3-68 |

|
0.29 [0.22, 0.40] | 7RLS |
| Mcule-CSR-494190-S1 |

|
0.29 [0.19, 0.43] | 7RM2 |
| HL-3-78 |

|
0.61 [0.37, 0.96] | 7RMB |
| HL-3-52 |

|
1.4 [0.80, 2.3] | 7RME |
| HL-3-87 |

|
1.4 [0.9, 2.2] | N/D |
| HL-3-70 |

|
6.2 [4.8, 8.0] | 7RMT |
| HL-3-63 |

|
6.4 [4.3, 9.5] | 7RMZ |
| HL-3-69 |

|
8.8 [6.3, 13] | 7RN4 |
| HL-3-45 |

|
> 20 | 7RNH |
| HL-3-71 |

|
> 20 | 7RNK |
| HL-3-46 |

|
> 20 | N/D |
| HL-3-43 |

|
> 20 | N/D |
| HL-3-44 |

|
> 20 | N/D |
| HL-3-49 |

|
> 20 | N/D |
| HL-3-62 |

|
> 20 | N/D |
| HL-3-50 |

|
> 20 | N/D |
| HL-3-51 HL-3-53 |

|
> 20 | N/D |
| HL-3-65 |

|
> 20 | N/D |
95% Confidence Interval (CI).
N/D – Not Determined.
Various halides (F, Cl, I), nitrile and trifluoromethyl were examined at the para position to modulate the electronic properties of the P2 group. In contrast, aldehyde and hydroxymethyl substituents were investigated to determine whether the Tyr54 side chain can act as a hydrogen bond partner, donor or acceptor. Sterically bulkier substituents were not considered for synthesis due to geometric constraints of the S2 subsite.
Inhibition of Mpro by HL-3 compounds
To determine the effect of the chemical modifications within the HL-3 series of compounds on their ability to inhibit Mpro compared to compound 1, an initial Mpro activity inhibition screen was performed at 20 μM inhibitor concentration. Eight compounds showed at least 50% Mpro activity inhibition at 20 μM (Table 1) and were further characterized by assaying inhibition across a range of concentrations to determine 50% inhibitory concentrations (IC50) values (Equation 1 in Materials and Methods). The rest of the compounds were excluded from further measurements because their IC50 values were well above 20 μM. Compound 1 produced the IC50 of 0.68 μM in the current experiments (Table 1).
Replacing P1 uracil-like group with either of dihydropyrimidine-2,4(1H,3H)-dione enantiomers (HL-3-51 and HL-3-53) had a considerable effect, with the IC50s for the two derivatives above 20 μM. Therefore, the structurally conserved S1 subsite cannot accommodate the bent structures of these P1 groups. Interestingly, replacing the protonated positively charged aniline nitrogen with carbon to give HL-3-69 also negatively affected the compound’s potency (IC50 = 8.8 μM), even though this nitrogen does not make close contact with Mpro residues. Hence, it appears that the more than an order of magnitude increase in IC50 for HL-3-69 compared to compound 1 can be attributed to the altered electrostatics of the inhibitor.
Removal of the -Cl substituent at the 3-position of the P2 group to generate the monosubstituted derivative HL-3-45, and then substituting -Cl at the 4-position with various groups, such as -F, -I, -CHO, -CH2OH, CN, and CF3, leads to dramatic losses of these derivatives’ potencies. A similar result was obtained for compounds HL-3-46 and HL-3-71, in which the chlorine at the 4-position is replaced respectively with -H or -CH2OH. These observations clearly indicated that -Cl at the 3-position of the P2 group is essential for a compound to maintain its potency. Indeed, adding -Cl back to the 3-position for monosubstituted compounds with -CHO (HL-3-44) or -CF3 (HL-3-50) at the 4-position significantly restored potency, with HL-3-70 and HL-3-63 possessing respective IC50s of 6.2 and 6.4 μM. However, the potencies of HL-3-70 and HL-3-63 are an order of magnitude worse than that of compound 1, indicating that the presence of sterically larger and more electronegative substituents than -Cl at the 4-position reduces an inhibitor’s potency. Replacing -Cl at the 4-position with -Br, whose van der Walls radius of 1.97 Å is slightly larger than that of -Cl (1.90 Å) and electronegativity lower than that of -Cl, while maintaining -Cl at the 3-position, resulted in compound HL-3-78 which demonstrated potency as good as compound 1. Unexpectedly, we determined that, when -Cl and -CF3 in HL-3-63 swap positions to give HL-3-52, the IC50 improves several fold to 1.4 μM, which is only about twice as high as that for compound 1. Adding an extra -Cl substituent to HL-3-63 at the 5-position (HL-3-87) saw a similar reduction in IC50. The latter two observations demonstrated that the 3-position of the P2 group could accommodate a bulkier and more electronegative substituent, and addition of -Cl to 5-position on the benzene ring is beneficial for an inhibitor’s potency.
With this in mind, we analyzed the potencies of two molecules, HL-3-68 and Mcule-CSR-494190-S1, with -Cl in positions 3 and 5 and either -Cl or -CH3 at position 4 respectively (Table 2). Both showed improved inhibition potency relative to compound 1, indicating the preference of S2 subsite for less bulky substituents with moderate-to-low electronegativity. We thus determined Ki values for HL-3-68 and Mcule-CSR-494190-S1 from initial rates determined across a range of inhibitor and substrate concentrations (Equation 2 in Materials and Methods). Ki measurements confirmed that HL-3-68 was the highest affinity inhibitor, with a sub-μM Ki of 0.89 μM, followed by Mcule-CSR-494190-S1 with a Ki of 1.4 μM. The significant accuracy of Ki measurements indicates that the potency of HL-3-68 and Mcule-CSR-494190-S1 are 3-fold and 2-fold better than the previously reported compound 1 Ki of 2.9 μM40. Of note, none of the compounds demonstrated antiviral activity against SARS-CoV-2 in cell-based assays (Figure S2).
Table 2.
Binding affinities of compound 1 and selected inhibitors determined by in vitro assays (Ki) and isothermal titration calorimetry (Kd and thermodynamic parameters ΔH, ΔS and ΔG of binding).
| Compound | Chemical Structure | Ki, μM | Kd, μM | Stoichiometry, N | ΔH, kcal/mol | ΔS, cal/mol·K | ΔG, kcal/mol |
|---|---|---|---|---|---|---|---|
| Compound 1 Mcule-5948770040 |

|
2.9 [2.2, 4.0]40 | 1.30±0.18 | 0.960±0.014 | −8.32±0.17 | −0.7 | −8.11 |
| HL-3-68 |

|
0.89 [0.72, 1.1] | 0.69±0.21 | 0.9±0.2 | −7.75±0.27 | 2.4 | −8.5 |
| Mcule-CSR-494190-S1 |

|
1.4 [1.1, 1.7] | 1.32±0.29 | 0.97±0.02 | −9.1±0.28 | −3.16 | −8.15 |
Isothermal titration calorimetry (ITC).
To directly assess the thermodynamic binding properties of compound 1 and the two most potent inhibitors HL-3-68 and Mcule-CSR-494190-S1, we performed isothermal titration calorimetry (ITC, Table 2 and Figure S3). Kd values measured by ITC are in good agreement with the Ki values obtained by enzyme kinetics. HL-3-68 demonstrates sub-μM affinity to Mpro, binding ~2-fold better to the enzyme than the other two compounds. Binding of all three compounds to Mpro is driven primarily by enthalpy. Compound 1 binds to the enzyme with essentially no change in entropy, whereas ΔS of binding for Mcule-CSR-494190-S1, although small (−3.16 cal/mol·K), is negative and therefore contributes unfavorably to the binding with -TΔS of +0.95 kcal/mol. Conversely, substitution of the methyl at P2 group 4-position with -Cl reverts ΔS to a small positive value of 2.4 cal/mol·K. Interestingly, ΔH of binding is the most favorable for Mcule-CSR-494190-S1, although its ΔS is the most unfavorable. The interplay of the enthalpy and entropy components results in HL-3-68 possessing the highest affinity for Mpro (Kd = 0.69 μM).
Room-temperature X-ray structures of Mpro in complex with HL-3 compounds
To shed light on the SAR between Mpro and HL-3 compounds, we obtained 9 room temperature X-ray structures of Mpro co-crystallized with selected HL-3 compounds at 1.85-2.10 A resolution (Table S2, Figure S4). We did not obtain crystal structures of Mpro complexes with other compounds because they were either significantly less potent than compound 1 or their complexes did not crystallize. Comparisons of these structures with Mpro-1 provide insights into how substitutions at the aromatic P2 group positions 3, 4 and 5 alter binding of HL-3 compounds and correlate with their inhibition potencies. Inhibitors of all determined structures bind in identical fashion (Figure 3a) anchored to Mpro by invariant hydrogen bonds (2.6 ±0.1 Å between the heavy atoms) of the uracil P1 group with His163 and the linker’s carbonyl with main chain amide nitrogen of Gly143. Some shifts in the positions of the aromatic P2 group up to 0.5 Å are observed due to the various substituents in positions 3, 4 and 5.
Figure 3. Mpro inhibition by HL-3 compounds.

(a) Binding modes of 9 HL-3 compounds co-crystallized with Mpro.
(b) HL-3-68 (cyan) bound in the active site of Mpro (pink). Electron density omit Fo-Fc map (grey mesh) contoured at 3σ. H-bonds as black dashes, Distances in Å. PDB ID 7RLS.
(c) Superposition of Mpro-1 X-ray/neutron structure (PDB ID 7N8C) with HL-3-68 (PDB ID 7RLS) and Mcule-CSR-494190-S1 (PDB ID 7RM2) complex X-ray structures showing rotation of Met49 to accommodate the -C1 at 5-position of the aromatic P2 group. Van der Waals contacts for improved inhibitors represented as dotted lines. Superposition calculated by least-squares-fitting on Cα atoms.
Mpro-HL-3-68 (Figure 3b) and Mpro-Mcule-CSR-494190-S1 superimpose with the Mpro-1 joint X-ray/neutron structure with Cα r.m.s.d. of 0.2-0.23 Å. Substituting -Cl or -methyl at the P2 4-position probes the effect of the chemical group’s hydrophobicity at the S2 site with a small difference in van der Waals radii. Increased hydrophobicity of the CH3 group in Mpro-Mcule-CSR-494190-S1 compared to -Cl in Mpro-HL-3-68 translates structurally only into the nearby Pro52 ring that adopts the exo-conformation instead of the endo geometry, respectively. Pro52 terminates the short S2 α-helix spanning residues 46-52, where the Met49 side chain acts to gate the S2 subsite. The effect on the S2 subsite arising from -Cl at the P2 5-position can be observed here through superpositions of the Mpro-1 neutron structure with the Mpro-HL-3-68 and Mpro-Mcule-CSR-494190-S1 complexes (Figure 3c). In both structures, the Met49 side chain is sterically rotated away from its position in Mpro-1 to accommodate the additional -Cl atom, which enables van der Waals contacts with Cys44 but does not lead to an additional shift in the position of the S2 helix.
Crystal structures of complexes exhibiting similar or worse potency compared to compound 1 were also analyzed to assess the structural determinants of noncovalent ligand binding to Mpro. HL-3-78 substitutes -Br at the P2 4-position probing the effect of a slightly larger van der Waals radius at this position compared to compound 1. Binding of HL-3-78 resulted in no significant changes in protein structure but comparable inhibition properties. However, bulkier and highly electronegative -CF3 groups at the 3- or 4-position (HL-3-52/63) bring about unfavorable distal shifts in the flexible S5 loop (>1.5 Å for Ala191 Cα, Figure S5a). Tyr54’s phenol oxygen faces the S2 subsite and presents an attractive target for direct H-bonding, as tested by substituting -CH2OH (HL-3-71) and -CHO (HL-3-70) at the P2 4-position. Unfortunately, the Mpro-HL-3-70 and Mpro-HL-3-71 crystal structures show the primary alcohol and the aldehyde oxygens are rotated away from the Tyr54 phenol oxygen, which keeps its conserved hydrogen bond with the main chain carbonyl of Asp187 (Figure S5b). Eliminating the cationic potential of the ligand by changing the linker from piperazine to piperidine (HL-3-69) produced no significant structural changes, except for a 0.4 Å shift in the position of the linker and P2 groups away from S2 helix reducing van der Waals contacts.
Molecular dynamics (MD) simulations
MD simulations of Mpro (ligand-free), the Mpro-HL-3-68 and Mpro-1 complexes reveal a consistent picture of how the HL-3-68 ligand is more stable within the primary binding site of Mpro compared to compound 1 reported in our previous study.40 We quantified the conformational changes using the root-mean square deviations (RMSD) analysis across each trajectory (Figures 4 and S6). The distribution of the RMSDs determined from at least three replicates of the simulations (shown in Figure 4a as a histogram of all conformers from MD trajectories) further reveals that HL-3-68 stabilizes the binding pocket of Mpro more than compound 1. For each system, we did observe slightly different fluctuations in chain A and chain B, which agrees with the previous simulation results.13,40 Across the three simulation systems, the Mpro-HL-3-68 complex had the lowest average RMSD from both chains when compared with Mpro, and Mpro-1 system (Figure S6). Per-residue fluctuations were characterized by calculating the root mean square fluctuations (RMSF) of the Cα-atoms using the average conformation of each trajectory as the reference structure. Despite the fluctuation at the C-termini, the fluctuation patterns are largely in agreement, except that the Mpro-HL-3-68 system depicts suppressed fluctuations across the entire protein (Figure 4). Lower RMSFs were observed for the primary ligand binding site of the Mpro-HL-3-68 system, whereas other regions remain largely unaffected by the binding of the ligand(s). In our previous simulations40 and as is demonstrated here, compound 1 can potentially move away from the primary binding site to occupy various novel sites on the surface of Mpro; however, in crystallographic studies compound 1 has not been observed to bind to other sites. Given that both crystallographic studies and biochemical assays indicate that the HL-3-68 is more stabilizing, our simulations also confirm that over the course of the timescales of our simulations, it appears that the HL-3-68 stabilizes the primary interactions in S1 and S2 subsites by “locking” in the site, thus considerably reducing the flexibility of the loops surrounding the primary binding site. Thus, our analyses support the observation that HL-3-68 ligand binding stabilizes the Mpro structure, forming stronger interactions than compound 1.
Figure 4. Root mean square analysis of MD trajectories.

(A) The distribution of the root-mean square deviations (RMSD) captured as a histogram over the course of MD simulations from the three replicas showing that chain A exhibits higher fluctuations than chain B in ligand-free state (top panel). In the ligand-bound states, the asymmetric nature of fluctuations are still present however, Mpro-HL-3-68 (middle panel) exhibits considerably lower degree of fluctuations compared to Mpro compound-1 (bottom panel) as a consequence of the increased stability of the interactions within the primary binding site. Starting model was PDB ID 7JUN for apo-Mpro simulation, 7RLS for HL-3-68 complex, and 7N8C for complex with compound 1.
(B) The root-mean square fluctuations (RMSF) of the individual chains captured across the three sets of replicas further reinforce the observation that both ligands stabilize the Mpro binding site. The shaded regions highlight the variation across three independent replicas of the simulations (for each system).
(C) Putty-like representation of the RMSF of the ligand-free Mpro dimer simulations depicts large-scale fluctuations along the primary binding site (red tubes).
(D, F, H) A depiction highlighting RMSF in one monomer of the protein, ligand-free protein in (D), Mpro-HL-3-68 complex in (F), and Mpro-1 complex in (H).
(E) Putty representation of the RMSF of the Mpro dimer complexed with HL-3-68.
(G) Putty representations of the RMSF of the Mpro dimer complexed with compound 1. The ligands in each case are highlighted using a ball-and-stick representation.
DISCUSSION
Design and development of small-molecule therapeutics is a crucial component of the ongoing efforts to battle COVID-19 and prepare for future pandemics.43 SARS-CoV-2 Mpro is an attractive target for specific protease inhibitors that can be further developed into clinical drugs. Studying the structure, function and inhibition of the enzyme in detail is important for accelerating this process. Similarly, understanding the SAR profile of the designed compounds is crucial to determine how structural, electronic and electrostatic properties of certain chemical groups affect inhibitor binding to the Mpro active site. Therefore, our SAR study was guided by the XN structure of Mpro-1 complex, where hydrogen atom positions, protonation states and electric charges of Mpro residues and compound 1 were directly determined, providing the most detailed information to date for an Mpro in complex with a noncovalent inhibitor.
We observed in the XN structure of Mpro-1 that Mpro adapts protonation states of the active site residues to maintain a net +1 charge within the binding site found in ligand-free41 and telaprevir-bound42 Mpro (Figure 5 and Table 3). Protonation state modulations occur through His163 in the S1 subsite, Cys145, and the His41-D2Ocatt-His164 network. As predicted computationally,44 His163’s Nε1 becomes protonated upon ligand binding. The active site electrostatics can thus be tuned to allow the Cys145 side chain to exist as a thiol in Mpro-1, or as a thiolate primed for catalysis as observed in the ligand-free form. The imidazole ring of His41 is not only flipped 180° relative to its conformation in the ligand-free and telaprevir complexes, but also neutral. Hence, while the catalytic dyad is zwitterionic in the ligand-free structure, it is neutral in Mpro-1. Whether the protonation states of His-Cys dyad are interdependent is currently an open question. Each protonation state combination of His41 and His164 has now been captured individually in the three neutron structures, suggesting that D2Ocat-mediated H-bonding between these two sidechains is not required for inhibition or a stable active site. Taken together, the protonation states determined from these three neutron structures suggest a mechanism where charges are shuffled between His163, the catalytic dyad, and His164 maintaining an overall +1 charge by active site residues when binding to inhibitors.
Figure 5. Comparison of protonation states of ionizable residues in the active site of SARS-CoV-2 Mpro determined from neutron crystallographic structures.

(a) Mpro bound to compound 1 (b) Ligand-free Mpro (PDB code 7JUN) (c) Mpro complexed with covalent α-ketoamide HCV protease inhibitor telaprevir (PDB code 7LB7). Charges are represented with transparent surface colored grey for neutral, red for positively charged, and blue for negatively charged. Non-polar deuterium atoms shown in orange. Mpro-1 determined at pD=7.4 and 7JUN & 7LB7 are pH=7.0.
Table 3.
Summary of protonation states and corresponding electric charges of the ionizable residues in the SARS-CoV-2 Mpro active site observed in the neutron structures of the ligand-free enzyme and in complex with compound 1 and telaprevir.
| Residue | Mpro ligand-free (PDB ID 7JUN) | Mpro-Telaprevir (PDB ID 7LB7) | Mpro-1 (PDB ID 7N8C) | |||
|---|---|---|---|---|---|---|
|
|
||||||
| Charge | Species | Charge | Species | Charge | Species | |
| Cys145cat | −1 | Thiolate (-S−) | 0 | S-C-OD (hemithioketal) | 0 | Thiol (-SD) |
| His41cat | +1 | Nδ1-D, Nε2-D | 0 | Nδ1-D | 0 | Nε2-D |
| His163 | 0 | Nδ1-D | +1 | Nδ1-D, Nε2-D | +1 | Nδ1-D, Nε2-D |
| His164 | +1 | Nδ1-D, Nε2-D | 0 | Nδ1-D | 0 | Nε2-D |
| His172 | 0 | Nε2-D | 0 | Nε2-D | 0 | Nε2-D |
|
| ||||||
| Net charge | +1 | +1 | +1 | |||
The hydrogen bond between the P1 group of the HL-3 compounds and the protonated positively charged His163 is essential for binding. Distorting the P1 group planarity by introducing a partial saturation as in compounds HL-3-51/53 appears to disrupt the hydrogen bond leading to a dramatic loss of affinity. The cationic nature of the linker positioned above the neutral catalytic dyad is beneficial for the compound’s potency. However, antiviral activity of some cationic drugs may be attributed to induced phospholipidosis rather than their specific function;45 thus, novel Mpro inhibitors should be designed with this knowledge in mind. We determined that the substituents on the aromatic P2 group should have both moderate steric size and electronegativity as the binding is sensitive to small changes in atomic properties. In addition, compounds with only one substituent on P2 group are poor inhibitors. Highly electronegative substituents such as -F or -CF3 are disadvantageous, as are less electronegative but sterically larger -CHO and -CH2OH, that push against S5 loop and Tyr54, respectively. Adding a third -Cl to position 5 of the P2 group in compound 1 to give HL-3-68 improved inhibition 2-3 fold based on Ki and Kd values, indicating that its proximity to Cys44 and 3.5-4.3 Å contacts are favorable.
It is interesting that our ITC measurements of compound 1, Mcule-CSR-494190-S1 and HL-3-68 binding to Mpro demonstrated that these noncovalent inhibitors bind with a limited hydrophobic effect, i.e. the entropy ((ΔS) of binding is small. A combination of several opposing factors may result in the measured values of ΔS of binding. First, ligand-free Mpro has few water molecules in the active site41 whereas several waters are recruited from the bulk solvent when the inhibitors bind. Second, the P2 groups access the S2 subsite by carving out the pocket blocked by Met49 and the S2 helix, limiting the conformational space for favorable binding. Third, a compound would lose some conformational freedom once bound to Mpro. These three factors would contribute unfavorably to the ΔS of binding, while the loss of the compound’s hydration shell when it binds to the enzyme would increase the entropy, contributing favorably to the ΔS of binding. In this way, substituting -Cl with -CH3 at P2 4-position produces enough difference in conformational entropy and hydration entropy to elicit significant differences in the ΔS of binding. Changes in the protein dynamics upon inhibitor binding, and specifically in the vibrational dynamics,46–48 would also contribute to the ΔS (and ΔH) of binding, although the effect of these changes is not known.
CONCLUSION
In summary, the current SAR study of Mpro combines neutron and X-ray crystallography, chemical synthesis, in vitro measurements, and molecular dynamics simulations to profile the binding of a noncovalent ligand discovered through a new high-throughput screening approach.40 Protonation states of critical sidechains in the Mpro active site are intrinsically variable, thus hard to predict, a feature that presents challenges for in silico modeling and inhibitor design. The active site and especially the hydrophobic S2 pocket are sensitive to small changes in ligand properties. We show one atom differences in the studied noncovalent ligand’s P2 group were enough to significantly alter binding entropy, potency, and complex dynamics. Taken together, these characterizations elucidate new details of Mpro as a drug target.
Experimental
General Information
Virtual reality-assisted analysis and model building was used to design and visualize derivatives of compound 1 (Mcule-5948770040). Modifications to the ligand scaffold starting from PDB code 7LTJ40 were modeled and assessed using the MedChem tool in Nanome.49,50 Ni-NTA columns were purchased from Cytiva (Piscataway, New Jersey, USA). His-tagged Human Rhinovirus (HRV) 3C protease was purchased from Sigma (MilliporeSigma, St. Louis, MO). Crystallization reagents and supplies were purchased from Hampton Research (Aliso Viejo, California, USA). Crystallographic supplies for crystal mounting and X-ray and neutron diffraction data collection at room temperature were purchased from MiTeGen (Ithaca, New York, USA) and Vitrocom (Mountain Lakes, New Jersey, USA). The FRET substrate DABCYLKTSAVLQSGFRKM-E(EDANS) trifluoroacetate salt was purchased from Bachem (PN 4045664). NMR spectra were obtained at the Center for Nanophase Materials Sciences on a Bruker Avance NEO NMR console coupled to an 11.74 T actively shielded magnet (Magnex Scientific/Varian) operating at 499.717 MHz for proton. Mcule-5948770040 and 3,5-dichloro, 4-methylphenyl derivative (Mcule-CSR-494190-S1) were purchased from Mcule, Inc (Palo Alto, CA, USA). All HL-3 compounds were synthesized at the Center for Nanophase Materials Sciences (Oak Ridge National Laboratory). Full details of the syntheses and NMR and LDI-TOF structural data are provided in the Supplementary Information section.
General synthetic procedure of HL-3 compounds
The HL-3 compounds were synthesized using techniques described previously.51 In a typical procedure, a 20-mL vial was charged with a stir bar, the appropriate aryl piperazine derivative (1 eq.), orotic acid (1 eq.), HOBt·H2O (0.07 eq.) and anhydrous DMF (5 mL) at ambient temperature. The solution was cooled to 0 °C, and EDC·HCl (1 eq.) was added. The reaction mixture was stirred at room temperature overnight, and DMF removed under reduced pressure. The residue was purified by silica gel chromatography (DCM to DCM:CH3OH = 10:1). Removal of the solvents yielded the products as solids in yields > 90%. Purity is >95% for all compounds as measured by NMR and mass spectrometry analyses (see Supporting Information).

Gene construction, expression and purification of hydrogenated and partially deuterated SARS-CoV-2 Mpro
A codon optimized gene sequence of Mpro (NSP5) from SARS-CoV-2 was cloned into a plasmid harboring the kanamycin resistance cassette (pD451-SR, Atum, Newark, CA). The Mpro construct is flanked upstream by a gene for maltose binding protein (MBP) and downstream by a His6 tag.52 Native N-terminus is achieved during expression through an Mpro autoprocessing site SAVLQ↓SGFRK where ↓ denotes the cleavage site) corresponding to the cleavage between NSP4 and NSP5 in the viral polyprotein. Native C-terminus is produced through an HRV-3C protease cleavage site (SGVTFQ↓GP). This strategy enables a two-step Ni-affinity chromatography purification. Hydrogenated Mpro was expressed in Escherichia coli and purified according to the established procedures.52 Partially deuterated Mpro was expressed using a bioreactor and purified as described recently.42 Final protein yields for hydrogenated Mpro preparations averaged ~4 mg per 1 g cells (~17 mg per L of cell culture), whereas partially deuterated preparations yielded ~0.8 mg per 1 g of cell paste (−40 mg per L of cell culture).
Similar in strategy to the above, a second construct was also used to express and purify the wild-type Mpro. This construct differs from the first construct by having an additional 36 residue spacer sequence corresponding to the immunoglobulin binding domain B1 of GB1 inserted between MBP and a 6 amino acid flanking nsp4 sequence TSAVLQ. Expression and purification were carried out as described above and in ref. 39. Peak fractions were concentrated and stored in aliquots at −20°C. This second source of Mpro was used to determine the binding constants by ITC independent of the measurement carried out through enzyme kinetic measurements using the first enzyme source.
Crystallization of the Mpro-inhibitor complexes
Detailed instructions for crystallizing high-quality Mpro crystals starting from hydrogenated and partially deuterated enzyme are accessible.42,53 Crystallization conditions for flower-shaped crystal aggregates of Mpro were initially discovered by automated high-throughput screening at the Hauptman-Woodward Medical Research Institute (HWI).54 Crystal aggregates of apo-Mpro were reproduced locally and converted into microseeds for seeding subsequent crystallization experiments. Protein for co-crystallization was concentrated to 5 mg/mL in 20 mM Tris, 150 mM NaCl, and 1 mM TCEP (tris(2-carboxyethyl)phosphine) pH 8.0 and used fresh or stored at −30°C for no longer than 2 weeks. 50 mM stocks of compound 1 derivatives were prepared in 100% dimethyl sulfoxide (DMSO) for crystallization purposes and stored at −30°C. Mpro was mixed with the ligands at 1:5 molar ratio and allowed to incubate at room temperature for a minimum of 30 minutes prior to setting up crystal trays. All crystals grown for room-temperature X-ray diffraction used sitting drop vapor diffusion methodology with 18-21% PEG3350, 0.1 M Bis-Tris pH 6.5 or pH 7.0 as the precipitant solution. Crystallization drop volumes of 20-30 μL at 1:1 ratio were seeded with 0.2 μL apo-Mpro microseeds (1:200 dilution). Crystals appeared after 3 days of incubation at 14°C and continued to grow in volume for additional 7 days. Typical Mpro-ligand complex crystals grew to ~ 0.1 mm3 before mounting in MiTeGen (Ithaca, NY) room-temperature capillary setups (Figure S7).
The crystal used for joint XN crystallography started from partially deuterated Mpro at 10 mg/mL mixed with compound 1 at a 1:5 molar ratio, incubated at room temperature for 30 minutes, and filtered through a 0.2 μm centrifugal filter. A Hampton 9-well sandwich box was set up with 220 μL drops at a 1:1 ratio of protein to 18% PEG3350, 0.1 M Bis-Tris pH 7.0 reservoir solution and 0.2 μL microseeds at 1:200 dilution. After 11 days of incubation at 14°C, the temperature was reduced to 12°C, and crystals were allowed to grow for 30 more days. This process afforded 3 protein crystals of > 2 mm3 in volume, with the final crystal used for neutron data collection measuring ~2x1.5x0.7 mm (2.1 mm3) (Figure S8). The crystal was mounted in a fused quartz capillary accompanied by 19% PEG3350 prepared with 100% D2O to allow labile hydrogens to exchange at 18°C for 2 weeks before starting neutron data collection. The final pH of the crystallization drop at the time of crystal mounting was 7.0 as measured by a microelectrode, corresponding to a final pD of 7.4 (pD = pH + 0.4).
Room temperature X-ray diffraction data collection and structure refinement
All room temperature X-ray crystallographic data was collected with a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics, and a DECTRIS Eiger R 4M hybrid photon counting detector. Diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX) and then reduced and scaled using Aimless55 from the CCP4 suite56. Structures were solved by molecular replacement using PDB code 7LTJ40 with Phaser57 from CCP4. Each model was iteratively refined using Phenix.refine from PHENIX suite58 and COOT59,60 graphics program aided by Molprobity61 for geometry validation. All ligand restraints were generated with eLBOW62 using geometry optimized by quantum mechanical calculations in Gaussian 16 at B3LYP/6-31g(d,p) level of theory.63 Final data collection and refinement statistics are organized in Supplementary Table S3.
Neutron diffraction data collection
Room temperature neutron diffraction data were collected using the Macromolecular Neutron Diffractometer Instrument (MaNDi) at the Spallation Neutron Source of Oak Ridge National Laboratory.64–66 The crystal was held stationary at room temperature while diffraction data were collected for 20 h using all neutrons between 2 and 4.16 Å. Following this, the crystal was rotated by Δφ = 10°, and a subsequent data frame was collected again for 20 h. A total of 21 data frames were collected in the final neutron data set. Diffraction data were reduced using the Mantid package, with integration carried out using three-dimensional TOF profile fitting.67 Wavelength normalization of the Laue data was performed using the Lauenorm program from the Lauegen suite.68,69 Neutron data collection statistics are shown in Supplementary Table S1.
Joint X-ray/neutron (XN) refinement
Joint XN refinement of deuterated Mpro-1 complex was performed using nCNS,69,70 and the structure was manipulated in COOT.59,60 After initial rigid-body refinement, several cycles of positional, atomic displacement parameter, and occupancy refinement were run. Correctness of sidechain conformations, hydrogen bonding, and orientations of D2O water molecules in the structure were based on the mFO-DFC difference neutron scattering length density maps. The 2mFO-DFC and mFO-DFC neutron scattering length density maps were then examined to determine the correct orientations of hydroxyl (Ser, Thr, Tyr), thiol (Cys), and ammonium (Lys) groups, and protonation states of the enzyme residues and compound 1. The protonation states of some disordered side chains on the protein surface could not be obtained directly and remained ambiguous. Water molecules were refined as D2O where water oxygen atoms were centered on their electron density peaks and each molecule was rotated in accordance with the neutron scattering length density maps. Hydrogen positions in the protein were modeled as deuterium atoms because the protein was partially deuterated. Compound 1 is ionizable at the piperazine amine and was modeled as the protonated species with a D atom. Occupancies of D atoms were refined individually within the range of −0.56 (pure H) to 1.00 (pure D) because the neutron scattering length of H is −0.56 times that of D. Before depositing the neutron structure to the PDB, coordinates of a D atom were split into two records corresponding to a H and a D partially occupying the same site, both with positive partial occupancies that add up to unity. The percent D at a specific site is calculated according to the following formula: .
Enzyme inhibition assay
Compounds were dissolved in DMSO at 10 mM concentration and stored at −20°C. Mpro initial rates were measured, and the data were analyzed using a previously established fluorescence resonance energy transfer (FRET) peptide substrate assay method.71,72 For the initial inhibition screen, performed in duplicate, final assay concentrations were 250 nM enzyme, 20 μM inhibitor, and 40 μM peptide substrate. Inhibitors with 50% residual activity or less in the initial screen were further characterized across 7 inhibitor concentrations in the range of 0.03-100 μM in at least duplicate, and the resulting initial rates were normalized with 0 as 0% residual activity and the average of positive control rates without inhibitor as 100% residual activity. The [Inhibitor] vs. normalized response – Variable slope equation in GraphPad Prism 9 was fit to the normalized data to determine IC50s:
| Eq. 1 |
where A is the residual activity, IC50 is the inhibitor concentration at which 50% inhibition is observed, [I] is the inhibitor concentration, and b is the Hill slope.
The two inhibitors with the lowest IC50 values were further characterized to determine their Ki values as previously described for compound 1.40 Initial rates were measured in triplicate without inhibitor and with inhibitor at final concentrations of 2.5, 7.5, and 25 μM, with 150 nM enzyme, and substrate final concentrations of 20-500 μM. The competitive inhibition equation below in GraphPad Prism 9 was fit to the resulting initial rates, , to determine the Michaelis-Menten enzyme parameters and and the affinities of the inhibitors.
| Eq. 2 |
Isothermal Titration Calorimetry
Purified wild-type Mpro was diluted from a stock solution to 60 μM and dialyzed overnight at 4 °C against 25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP (ITC buffer). Concentration of the enzyme was estimated based on its 280 nm absorbance. Stock solutions of inhibitors were diluted in ITC buffer to 0.3 mM and contained a final concentration of 0.5% DMSO. The protein solution was also adjusted to contain the same concentration of DMSO. Titrations were performed at 28 °C on an iTC200 microcalorimeter (Malvern Instruments Inc., Westborough, MA). A control titration of buffer with inhibitor showed negligible response. Data were processed and plots were generated using the Origin software provided with the instrument. For competitive inhibitors that bind at only one site, dissociation constant (Kd = 1/Ka) is equivalent to the inhibition constant measured by enzyme kinetics (Ki).
MD simulations and analysis
MD simulations were performed for three different systems (ligand-free, or apo-, Mpro; Mpro in complex with compound 1 and Mpro with HL-3-68) to study the protein stability upon binding with different ligands. The simulation runs were carried out with OpenMM package on Nvidia V100 GPUs. The protein atomic interactions were described with Amberff14SB force field and tip3p water model. The ligands, compound 1 and HL-3-68, were modeled using the antechamber package with GAFF force field. Each system was neutralized with counterions. The nonbonded interactions were cut off at 10 Å and long-range interactions were calculated with Particle Mesh Ewald method. The simulations were run at 310 K and 2 fs time step with Langevin integrator. Chemical bonds with hydrogen atoms were fixed. System pressure was fixed at 1 bar with Monte Carlo barostat. Each of the three systems were equilibrated using a procedure described in previous work40 and three replicas (each with a 250-ns production run) were generated; snapshots from the simulation were saved every 50 ps. For all the trajectories, we calculated the root mean square deviations (RMSD) for the overall protein structure and root mean square fluctuation (RMSF) to quantify per-residue fluctuations.
Antiviral assays
Evaluation of antiviral activity of compound 1 (Mcule-59487700), HL-3-68, and Mcule-CSR-494190-S1 was carried out in Vero E6 TMPRSSS cells as described in Bocci et al.73 using the USA-WA1/2020 (deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH, NR-52281). Compounds were evaluated in a dose response format starting at 33 micromolar and 6 additional 2-fold dilutions in duplicate. These compounds demonstrated no antiviral activity.
Supplementary Material
Acknowledgments
This research was supported by the DOE Office of Science through the National Virtual Biotechnology Laboratory (NVBL), a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act. This research used resources at the Center for Nanophase Materials Sciences, the Spallation Neutron Source and the High Flux Isotope Reactor, which are DOE Office of Science User Facilities operated by the Oak Ridge National Laboratory. The Office of Biological and Environmental Research supported research at ORNL’s Center for Structural Molecular Biology (CSMB), a DOE Office of Science User Facility. This research used resources of the Spallation Neutron Source Second Target Station Project at Oak Ridge National Laboratory (ORNL). ORNL is managed by UT-Battelle LLC for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. We thank Dr. Hugh M. O’Neill from ORNL for assistance during expression of the partially deuterated protein. L.C. acknowledges support by the NIH (R01-GM071939). We thank Annie Aniana from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) for excellent technical assistance. This work was also supported by the Intramural Research Program of NIDDK, NIH.
Abbreviations Used:
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- 3CL Mpro
chymotrypsin-like main protease
- Mpro
wild-type main protease of SARS-CoV-2
Footnotes
Ancillary Information
Supporting Information contains Tables S1 and S2, Figures S1–S8, Supporting Material and Methods, 1H and 13C NMR spectra and mass spectra. The coordinates and structure factors for the joint X-ray/neutron structure of SARS-CoV-2 Mpro-Compound 1 complex has been deposited in the PDB with the accession code 7N8C. The coordinates and structure factors for the room-temperature X-ray structures of other reported complexes have been deposited in the PDB with the accession codes as follows: 7RLS for Mpro-HL-3-68, 7RM2 for Mpro-Mcule-CSR-494190-S1, 7RMB for Mpro-HL-3-78, 7RME for Mpro-HL-3-52, 7RMT for Mpro-HL-3-70, 7RMZ for Mpro-HL-3-63, 7RN4 for Mpro-HL-3-69, 7RNH for Mpro-HL-3-45, and 7RNK for Mpro-HL-3-71. Authors will release the atomic coordinates upon article publication. Any other relevant data are available from the corresponding authors upon reasonable request.
Conflict of interest
The authors declare no conflicts of interest.
REFERENCES
- (1).Gavor E; Choong YK; Er SY; Sivaraman H; Sivaraman J Structural Basis of SARS-CoV-2 and SARS-CoV Antibody Interactions. Trends Immunol. 2020, 41 (11), 1006–1022. 10.1016/j.it.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Meo SA; Bukhari IA; Akram J; Meo AS; Klonoff DC COVID-19 Vaccines: Comparison of Biological, Pharmacological Characteristics and Adverse Effects of Pfizer/BioNTech and Moderna Vaccines. Eur. Rev. Med. Pharmacol. Sci 2021, 25 (3), 1663–1679. 10.26355/eurrev_202102_24877. [DOI] [PubMed] [Google Scholar]
- (3).Doroftei B; Ciobica A; Ilie O-D; Maftei R; Ilea C Mini-Review Discussing the Reliability and Efficiency of COVID-19 Vaccines. Diagnostics 2021, 11 (4), 579. 10.3390/diagnostics11040579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bidram E; Esmaeili Y; Amini A; Sartorius R; Tay FR; Shariati L; Makvandi P Nanobased Platforms for Diagnosis and Treatment of COVID-19: From Benchtop to Bedside. ACS Biomater. Sci. Eng 2021, 7 (6), 2150–2176. 10.1021/acsbiomaterials.1c00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang P; Nair MS; Liu L; Iketani S; Luo Y; Guo Y; Wang M; Yu J; Zhang B; Kwong PD; Graham BS; Mascola JR; Chang JY; Yin MT; Sobieszczyk M; Kyratsous CA; Shapiro L; Sheng Z; Huang Y; Ho DD Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593 (7857), 130–135. 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- (6).Vilcek S SARS-CoV-2: Zoonotic Origin of Pandemic Coronavirus. Acta Virol. 2020, 64 (3), 281–287. 10.4149/av_2020_302. [DOI] [PubMed] [Google Scholar]
- (7).Singh D; Yi SV On the Origin and Evolution of SARS-CoV-2. Exp. Mol. Med 2021, 53 (4), 537–547. 10.1038/s12276-021-00604-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15 (11), 907–932. 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tong L Viral Proteases. Chem. Rev 2002, 102 (12), 4609–4626. 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- (10).Liu Y; Liang C; Xin L; Ren X; Tian L; Ju X; Li H; Wang Y; Zhao Q; Liu H; Cao W; Xie X; Zhang D; Wang Y; Jian Y The Development of Coronavirus 3C-Like Protease (3CLpro) Inhibitors from 2010 to 2020. Eur. J. Med. Chem 2020, 206, 112711. 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582 (7811), 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- (12).Ton AT; Gentile F; Hsing M; Ban F; Cherkasov A Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform 2020, 39 (8). 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Suárez D; Díaz N SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model 2020, 60(12), 5815–5831. 10.1021/acs.jcim.0c00575. [DOI] [PubMed] [Google Scholar]
- (14).Acharya A; Agarwal R; Baker MB; Baudry J; Bhowmik D; Boehm S; Byler KG; Chen SY; Coates L; Cooper CJ; Demerdash O; Daidone I; Eblen JD; Ellingson S; Forli S; Glaser J; Gumbart JC; Gunnels J; Hernandez O; Irle S; Kneller DW; Kovalevsky A; Larkin J; Lawrence TJ; LeGrand S; Liu S-H; Mitchell JC; Park G; Parks JM; Pavlova A; Petridis L; Poole D; Pouchard L; Ramanathan A; Rogers DM; Santos-Martins D; Scheinberg A; Sedova A; Shen Y; Smith JC; Smith MD; Soto C; Tsaris A; Thavappiragasam M; Tillack AF; Vermaas JV; Vuong VQ; Yin J; Yoo S; Zahran M; Zanetti-Polzi L Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Model 2020, 60 (12), 5832–5852. 10.1021/acs.jcim.0c01010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pathak N; Chen YT; Hsu YC; Hsu NY; Kuo CJ; Tsai HP; Kang JJ; Huang CH; Chang SY; Chang YH; Liang PH; Yang JM Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing. ACS Nano 2021, 15 (1), 857–872. 10.1021/acsnano.0c07383. [DOI] [PubMed] [Google Scholar]
- (16).Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y; Yuan M-L; Zhang Y-L; Dai F-H; Liu Y; Wang Q-M; Zheng J-J; Xu L; Holmes EC; Zhang Y-Z A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Xu J; Zhao S; Teng T; Abdalla AE; Zhu W; Xie L; Wang Y; Guo X Systematic Comparison of Two Animal-to-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12 (2), 244. 10.3390/vl2020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ullrich S; Nitsche C The SARS-CoV-2 Main Protease as Drug Target. Bioorganic Med. Chem. Lett 2020, 30(17), 127377. 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yang KS; Ma XR; Ma Y; Alugubelli YR; Scott DA; Vatansever EC; Drelich AK; Sankaran B; Geng ZZ; Blankenship LR; Ward HE; Sheng YJ; Hsu JC; Kratch KC; Zhao B; Hayatshahi HS; Liu J; Li P; Fierke CA; Tseng CK; Xu S; Liu WR A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem 2020, 16 (6), 942–948. 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hattori S. ichiro; Higashi-Kuwata N; Hayashi H; Allu SR; Raghavaiah J; Bulut H; Das D; Anson BJ; Lendy EK; Takamatsu Y; Takamune N; Kishimoto N; Murayama K; Hasegawa K; Li M; Davis DA; Kodama EN; Yarchoan R; Wlodawer A; Misumi S; Mesecar AD; Ghosh AK; Mitsuya H A Small Molecule Compound with an Indole Moiety Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun 2021, 12 (1), 668. 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bai B; Belovodskiy A; Hena M; Kandadai AS; Joyce MA; Saffran HA; Shields JA; Khan MB; Arutyunova E; Lu J; Bajwa SK; Hockman D; Fischer C; Lamer T; Vuong W; Belkum M. J. van; Gu Z; Lin F; Du Y; Xu J; Rahim M; Young HS; Vederas JC; Tyrrell DL; Lemieux MJ; Nieman JA Peptidomimetic α-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in Vitro Biological Stability. J. Med. Chem 2021. 10.1021/ACS.JMEDCHEM.lC00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pillaiyar T; Manickam M; Namasivayam V; Hayashi Y; Jung SH An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem 2016, 59 (14), 6595–6628. 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pillaiyar T; Meenakshisundaram S; Manickam M Recent Discovery and Development of Inhibitors Targeting Coronaviruses. Drag Discov. Today 2020, 25 (4), 668–688. 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ; Solowiej JE; Steppan CM; Taggart B Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem 2020, 63 (21), 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Konno S; Kobayashi K; Senda M; Funai Y; Seki Y; Tamai I; Schäkel L; Sakata K; Pillaiyar T; Taguchi A; Taniguchi A; Gütschow M; Müller CE; Takeuchi K; Hirohama M; Kawaguchi A; Kojima M; Senda T; Shirasaka Y; Kamitani W; Hayashi Y 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem 2021. 10.1021/ACS.JMEDCHEM.lC00665. [DOI] [PubMed] [Google Scholar]
- (26).Fu L; Ye F; Feng Y; Yu F; Wang Q; Wu Y; Zhao C; Sun H; Huang B; Niu P; Song H; Shi Y; Li X; Tan W; Qi J; Gao GF Both Boceprevir and GC376 Efficaciously Inhibit SARS-CoV-2 by Targeting Its Main Protease. Nat. Commun 2020, 11 (1). 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30 (8), 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Xia Z; Sacco M; Hu Y; Ma C; Meng X; Zhang F; Szeto T; Xiang Y; Chen Y; Wang J Rational Design of Hybrid SARS-CoV-2 Main Protease Inhibitors Guided by the Superimposed Cocrystal Structures with the Peptidomimetic Inhibitors GC-376, Telaprevir, and Boceprevir. ACS Pharmacol. Transl. Sci 2021, 4 (4), 1408–1421. 10.1021/ACSPTSCI.lC00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ghahremanpour MM; Tirado-Rives J; Deshmukh M; Ippolito JA; Zhang CH; Cabeza De Vaca I; Liosi ME; Anderson KS; Jorgensen WL Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett 2020, 11 (12), 2526–2533. 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li Z; Li X; Huang YY; Wu Y; Liu R; Zhou L; Lin Y; Wu D; Zhang L; Liu H; Xu X; Yu K; Zhang Y; Cui J; Zhan CG; Wang X; Luo H. Bin. Identify Potent SARS-CoV-2 Main Protease Inhibitors via Accelerated Free Energy Perturbation-Based Virtual Screening of Existing Drugs. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (44), 27381–27387. 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Douangamath A; Fearon D; Gehrtz P; Krojer T; Lukacik P; Owen CD; Resnick E; Strain-Damerell C; Aimon A; Ábrányi-Balogh P; Brandão-Neto J; Carbery A; Davison G; Dias A; Downes TD; Dunnett L; Fairhead M; Firth JD; Jones SP; Keeley A; Keserü GM; Klein HF; Martin MP; Noble MEM; O’Brien P; Powell A; Reddi RN; Skyner R; Snee M; Waring MJ; Wild C; London N; von Delft F; Walsh MA Crystallographic and Electrophilic Fragment Screening of the SARS-CoV-2 Main Protease. Nat. Commun 2020, 11 (1), 5047. 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Günther S; Reinke PYA; Fernández-Garciá Y; Lieske J; Lane TJ; Ginn HM; Koua FHM; Ehrt C; Ewert W; Oberthuer D; Yefanov O; Meier S; Lorenzen K; Krichel B; Kopicki JD; Gelisio L; Brehm W; Dunkel I; Seychell B; Gieseler H; Norton-Baker B; Escudero-Pérez B; Domaracky M; Saouane S; Tolstikova A; White TA; Hänle A; Groessler M; Fleckenstein H; Trost F; Galchenkova M; Gevorkov Y; Li C; Awel S; Peck A; Barthelmess M; Schlünzen F; Xavier PL; Werner N; Andaleeb H; Ullah N; Falke S; Srinivasan V; Francą BA; Schwinzer M; Brognaro H; Rogers C; Melo D; Zaitseva-Doyle JJ; Knoska J; Penã-Murillo GE; Mashhour AR; Hennicke V; Fischer P; Hakanpää J; Meyer J; Gribbon P; Ellinger B; Kuzikov M; Wolf M; Beccari AR; Bourenkov G; Stetten D. Von; Pompidor G; Bento I; Panneerselvam S; Karpics I; Schneider TR; Garcia-Alai MM; Niebling S; Günther C; Schmidt C; Schubert R; Han H; Boger J; Monteiro DCF; Zhang L; Sun X; Pletzer-Zelgert J; Wollenhaupt J; Feiler CG; Weiss MS; Schulz EC; Mehrabi P; Karnĭcar K; Usenik A; Loboda J; Tidow H; Chari A; Hilgenfeld R; Uetrech C; Cox R; Zaliani A; Beck T; Rarey M; Günther S; Turk D; Hinrichs W; Chapman HN; Pearson AR; Betzel C; Meents A X-Ray Screening Identifies Active Site and Allosteric Inhibitors of SARS-CoV-2 Main Protease. Science 2021, 372 (6542), 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang CH; Stone EA; Deshmukh M; Ippolito JA; Ghahremanpour MM; Tirado-Rives J; Spasov KA; Zhang S; Takeo Y; Kudalkar SN; Liang Z; Isaacs F; Lindenbach B; Miller SJ; Anderson KS; Jorgensen WL Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci 2021, 7, 467–475. 10.1021/acscentsci.lc00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Deshmukh MG; Ippolito JA; Zhang C-H; Stone EA; Reilly RA; Miller SJ; Jorgensen WL; Anderson KS Structure-Guided Design of a Perampanel-Derived Pharmacophore Targeting the SARS-CoV-2 Main Protease. Structure 2021, 29 (8), 823–833. 10.1016/J.STR.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Han SH; Goins CM; Arya T; Shin W-J; Maw J; Hooper A; Sonawane DP; Porter MR; Bannister BE; Crouch RD; Lindsey AA; Lakatos G; Martinez SR; Alvarado J; Akers WS; Wang NS; Jung JU; Macdonald JD; Stauffer SR Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). J. Med. Chem 2021. 10.1021/acs.jmedchem.lc00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kitamura N; Sacco MD; Ma C; Hu Y; Townsend JA; Meng X; Zhang F; Zhang X; Ba M; Szeto T; Kukuljac A; Marty MT; Schultz D; Cherry S; Xiang Y; Chen Y; Wang J Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem 2021, acs.jmedchem.1c00509. 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhu L; George S; Schmidt MF; Al-Gharabli SI; Rademann J; Hilgenfeld R Peptide Aldehyde Inhibitors Challenge the Substrate Specificity of the SARS-Coronavirus Main Protease. Antiviral Res. 2011, 92 (2), 204–212. 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dai W; Zhang B; Jiang X-MM; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang L-KK; Xu Y; Yang H; Liu H Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368 (6497), 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368 (6489), 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Clyde A; Galanie S; Kneller DW; Ma H; Babuji Y; Blaiszik B; Brace A; Brettin T; Chard K; Chard R; Coates L; Foster I; Hauner D; Kertesz V; Kumar N; Lee H; Li Z; Merzky A; Schmidt JG; Tan L; Titov M; Turilli M; Van Dam H; Chennubhotla SC; Jha S; Kovalevsky A; Head M; Stevens R High Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Non-Covalent Inhibitor. bioRxiv 2021. 10.1101/2021.03.27.437323. [DOI] [PubMed] [Google Scholar]
- (41).Kneller DW; Phillips G; Weiss KL; Pant S; Zhang Q; O’Neill HM; Coates L; Kovalevsky A Unusual Zwitterionic Catalytic Site of SARS-CoV-2 Main Protease Revealed by Neutron Crystallography. J. Biol. Chem 2020, 295 (50), 17365–17373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kneller DW; Phillips G; Weiss KL; Zhang Q; Coates L; Kovalevsky A Direct Observation of Protonation State Modulation in SARS-CoV-2 Main Protease upon Inhibitor Binding with Neutron Crystallography. J. Med. Chem 2021, 64 (8), 4991–5000. 10.1021/acs.jmedchem.1c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Adamson CS; Chibale K; Goss RJM; Jaspars M; Newman DJ; Dorrington RA Antiviral Drug Discovery: Preparing for the next Pandemic. Chem. Soc. Rev 2021, 50 (6), 3647–3655. 10.1039/D0CS01118E. [DOI] [PubMed] [Google Scholar]
- (44).Pavlova A; Lynch DL; Daidone I; Zanetti-Polzi L; Smith MD; Chipot C; Kneller DW; Kovalevsky A; Coates L; Golosov AA; Dickson CJ; Velez-Vega C; Duca JS; Vermaas JV; Pang YT; Acharya A; Parks JM; Smith JC; Gumbart J Inhibitor Binding Influences the Protonation States of Histidines in SARS-CoV-2 Main Protease. Chem. Sci 2021, 12 (4), 1513–1527. 10.1039/D0SC04942E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tummino TA; Rezelj VV; Fischer B; Fischer A; O’Meara MJ; Monel B; Vallet T; White KM; Zhang Z; Alon A; Schadt H; O’Donnell HR; Lyu J; Rosales R; McGovern BL; Rathnasinghe R; Jangra S; Schotsaert M; Galarneau J-R; Krogan NJ; Urban L; Shokat KM; Kruse AC; García-Sastre A; Schwartz O; Moretti F; Vignuzzi M; Pognan F; Shoichet BK Drug-Induced Phospholipidosis Confounds Drug Repurposing for SARS-CoV-2. Science 2021, 373 (6554), 541–547. 10.1126/science.abi4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dajnowicz S; Cheng Y; Daemen LL; Weiss KL; Gerlits O; Mueser TC; Kovalevsky A Substrate Binding Stiffens Aspartate Aminotransferaseby Altering the Enzyme Picosecond Vibrational Dynamics. ACS Omega 2020, 5 (30), 18787. 10.1021/ACSOMEGA.0C01900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Balog E; Becker T; Oettl M; Lechner R; Daniel R; Finney J; Smith JC Direct Determination of Vibrational Density of States Change on Ligand Binding to a Protein. 2004. 10.1103/PhysRevLett.93.028103. [DOI] [PubMed] [Google Scholar]
- (48).Balog E; Perahia D; Smith JC; Merzel F Vibrational Softening of a Protein on Ligand Binding. J. Phys. Chem. B 2011, 115 (21), 6811–6817. 10.1021/jp108493g. [DOI] [PubMed] [Google Scholar]
- (49).Kingsley LJ; Brunet V; Lelais G; McCloskey S; Milliken K; Leija E; Fuhs SR; Wang K; Zhou E; Spraggon G Development of a Virtual Reality Platform for Effective Communication of Structural Data in Drug Discovery. J. Mol. Graph. Model 2019, 89, 234–241. 10.1016/jjmgm.2019.03.010. [DOI] [PubMed] [Google Scholar]
- (50).McCloskey S; Leija E; Hessenauer S; Funakawa K Nanome: Creating Powerful, Collaborative and Scientific VR Tools. Available from: https://nanome.ai (2020).
- (51).Bradley DM; Branch CL; Chan WN; Couton S; Dean AW; Doyle PM; Evans B; Gilpin ML; Gough SL; Macritchie JA; Marshall HR; Nash DJ; Porter RA; Ward SE Acylated Piperidines as Glycine Transporter Inhibitors. U.S. Pat. Appl. 2008/0255144 A1 2008.
- (52).Kneller DW; Phillips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A Structural Plasticity of SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-Ray Crystallography. Nat. Commun 2020, 11 (1), 3202. 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kneller DW; Phillips G; Kovalevsky A; Coates L Room-Temperature Neutron and X-Ray Data Collection of 3CL Mpro from SARS-CoV-2. Acta Crystallogr. Sect. F Struct. Biol. Commun 2020, 76 (10), 483–487. 10.1107/S2053230X20011814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Luft JR; Collins RJ; Fehrman NA; Lauricella AM; Veatch CK; DeTitta GT A Deliberate Approach to Screening for Initial Crystallization Conditions of Biological Macromolecules. J. Struct. Biol 2003, 142 (1), 170–179. 10.1016/S1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- (55).Evans PR; Murshudov GN How Good Are My Data and What Is the Resolution? Acta Crystallogr. Sect. D Biol. Crystallogr 2013, 69 (7), 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGWW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40 (Pt 4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Casañal A; Lohkamp B; Emsley P Current Developments in Coot for Macromolecular Model Building of Electron Cryo-Microscopy and Crystallographic Data. Protein Sci. 2020, 29 (4), 1069–1078. 10.1002/pro.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66 (1), 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Moriarty NW; Grosse-Kunstleve RW; Adams PD Electronic Ligand Builder and Optimization Workbench (ELBOW): A Tool for Ligand Coordinate and Restraint Generation. Acta Crystallogr. Sect. D Biol. Crystallogr 2009, 65 (10), 1074–1080. 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JAJ; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Revision A.03. Gaussian Inc.: Wallingford, CT: 2016. [Google Scholar]
- (64).Coates L; Cuneo MJ; Frost MJ; He J; Weiss KL; Tomanicek SJ; McFeeters H; Vandavasi VG; Langan P; Iverson EB The Macromolecular Neutron Diffractometer MaNDi at the Spallation Neutron Source. J. Appl. Crystallogr 2015, 48 (4), 1302–1306. 10.n07/S1600576715011243. [DOI] [Google Scholar]
- (65).Coates L; Cao HB; Chakoumakos BC; Frontzek MD; Hoffmann C; Kovalevsky AY; Liu Y; Meilleur F; dos Santos AM; Myles DAAA; Wang XP; Ye F A Suite-Level Review of the Neutron Single-Crystal Diffraction Instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum 2018, 89 (9), 092802. 10.1063/L5030896. [DOI] [PubMed] [Google Scholar]
- (66).Coates L; Sullivan B The Macromolecular Neutron Diffractometer at the Spallation Neutron Source. In Methods in Enzymology; Academic Press Inc., 2020; Vol. 634, pp 87–99. 10.1016/bs.mie.2019.11.020. [DOI] [PubMed] [Google Scholar]
- (67).Sullivan B; Archibald R; Langan PS; Dobbek H; Bommer M; Mcfeeters RL; Coates L; Wang X; Gallmeier F; Carpenter JM; Lynch V; Langan P Improving the Accuracy and Resolution of Neutron Crystallographic Data by Three-Dimensional Profile Fitting of Bragg Peaks in Reciprocal Space. Res. Pap. Acta Cryst 2018, 74, 1085–1095. 10.1107/S2059798318013347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Campbell JW; Hao Q; Harding MM; Nguti ND; Wilkinson C LAUEGEN Version 6.0 and INTLDM. J. Appl. Crystallogr 1998, 31 (3), 496–502. 10.1107/S0021889897016683. [DOI] [Google Scholar]
- (69).Adams PD; Mustyakimov M; Afonine PV; Langan P Generalized X-Ray and Neutron Crystallographic Analysis: More Accurate and Complete Structures for Biological Macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr 2009, 65 (6), 567–573. 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Mustyakimov M; Langan P NCNS: An Open Source Distribution Patch for CNS for Macromolecular Structure Refinement. Los Alamos National Security: Los Alamos, NM, USA: 2007. [Google Scholar]
- (71).Kuo CJ; Chi YH; Hsu JTA; Liang PH Characterization of SARS Main Protease and Inhibitor Assay Using a Fluorogenic Substrate. Biochem. Biophys. Res. Commun 2004, 318 (4), 862–867. 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kneller DW; Galanie S; Phillips G; O’neill HM; Coates L; Kovalevsky A Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 2020, 28, 1313–1320. 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Bocci G, Bradfute SB, Ye C, Garcia MJ, Parvathareddy J, Reichard W, Surendranathan S, Bansal S, Bologa CG, Perkins DJ, Jonsson CB, Sklar LA, Oprea TI Virtual and in vitro antiviral screening revive therapeutic drugs for COVID-19. ACS. Pharmacol. Transl. Sci 2020, 3, 1278–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.