

Abstract
The COVID-19 pandemic is caused by the coronavirus SARS-CoV-2 (SC2). A variety of anti-SC2 vaccines have been approved for human applications, including those using messenger RNA (mRNA), adenoviruses expressing SC2 spike (S) protein, and inactivated virus. The protective periods of immunization afforded by these intramuscularly administered vaccines are currently unknown. An alternative self-administrable vaccine capable of mounting long-lasting immunity via sterilizing neutralizing antibodies would be hugely advantageous in tackling emerging mutant SC2 variants. This could also diminish the possibility of vaccinated individuals acting as passive carriers of COVID-19. Here, we investigate the potential of an intranasal (IN)-delivered DNA vaccine encoding the S protein of SC2 in BALB/c and C57BL/6J immunocompetent mouse models. The immune response to IN delivery of this SC2-spike DNA vaccine transported on a modified gold-chitosan nanocarrier shows a strong and consistent surge in antibodies (IgG, IgA and IgM), and effective neutralization of pseudoviruses expressing S proteins of different SC2 variants (Wuhan, Beta, and D614G). Immunophenotyping and histological analyses reveal chronological events involved in the recognition of SC2 S antigen by resident dendritic cells and alveolar macrophages, which prime the draining lymph nodes and spleen for peak SC2-specific cellular and humoral immune responses. The attainable high levels of anti-SC2 IgA in lung mucosa and tissue-resident memory T cells can efficiently inhibit SC2 and its variants at the site of entry, and also provide long-lasting immunity.
Keywords: SARS-CoV-2, COVID-19, DNA vaccine, mRNA vaccine, intranasal delivery, gold nanoparticles, chitosan
Graphical Abstract.
Schematic illustration of intranasal delivery of spike gene DNA vaccine against SARS-CoV-2 inducing humoral and cell mediated immunity.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic has affected nearly 210 million people around the world as of August 2021 (https://covid19.who.int/).1 The causative pathogen, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, or SC2), belongs to a family of beta coronaviruses.2 Coronaviruses are single-stranded positive-sense RNA viruses that infect humans and mammals. Since this virus shows high transmission and fatality rates specifically in vulnerable populations, a rapid, high-priority approach has been adopted during vaccine development and approval under emergency use authorization.3 There are several strategies currently considered for SC2 vaccine development, including mRNA, DNA, inactivated virus, adenovirus expressing SC2 spike (S) protein, and peptide vaccine varieties.4 Most vaccines are currently targeted against the S protein of SC2 as the primary antigen, such as mRNA-1273 by Moderna, or mRNA-BNT162b2 by Pfizer.5 Inactivated virus (Covaxin) and adenovirus expressing SC2 S protein (Covishield and Sputnik) vaccines are also administered in humans.6
mRNA vaccines are developed from synthetic in vitro transcribed RNA sequences coding for viral protein, but the variations between vaccines are based on the stability of these synthetic mRNAs and that of their nanoformulation vehicles.7 Although intramuscular (IM) vaccination induces systemic humoral and cell-mediated immune responses that protect the lungs against infection and pathology, it does not confer sterilizing immunity.8 Moreover, inadvertent injection of a vaccine into the subcutaneous fat layer with poor vascularity can result in slow mobilization and processing of antigen leading to vaccine failure.9–12 Hence, whether adequate long-lasting active immunity develops or not after IM vaccination is still unknown.8, 12–16 An optimal vaccination strategy would aim for sterilizing vaccines to result in long-lasting immunity. An advantage of both DNA and mRNA constructs coding for antigens is that they are simpler and faster to produce than developing inactivated viruses or making recombinant proteins, and the risks of working with live virus/pathogen can be avoided.17
In contrast to IM injection, intranasal delivery (IN) of vaccine is preferred for respiratory infections to achieve both humoral and innate immune responses, while also producing sterilizing immunity in the respiratory tract and lungs.8, 18, 19 However, IN delivery requires a nanocarrier that can transport the loaded nucleic acid vaccine across the nasal cavity and down into lungs. An efficient nanoparticle (NP) as a delivery system can mount an effective immune response to DNA/RNA vaccines. Similarly, an ideal delivery system should possess high cargo loading capacity, stability, and biocompatibility. In that respect, apart from liposomes, a cationic polysaccharide and natural biopolymer, such as chitosan, has been used as an adjuvant in vaccine delivery systems.20
Recent studies have shown that the nasal cavity may become a reservoir for SC2 in the absence of mucosal immunity, placing patients at risk for reinfection or spread of disease to others.21 IN vaccination can overcome this drawback as it can serve to stimulate broad immune responses via neutralizing IgG, mucosal IgA, and T cells, which can instigate a local mucosal immunity in the nasal cavity that in turn can block both infection and spread from this reservoir. The lungs share many features with other mucosal sites, but preservation of its delicate histomorphological integrity requires a fine interplay between pro- and anti-inflammatory responses in the face of external insults. Well-timed, appropriately located, and tightly regulated T and B cell responses are essential to protect from infection, whereas dysregulated inflammation contributes to disease development and tissue damage.22,23 There are many other advantages to IN delivery, including the avoidance of injections, and likely high tolerance and compliance in clinical practice. Furthermore, respiratory tract immunization via the IN route can target a large surface area for immune response induction, through antigen presenting cells (especially dendritic cells) present in abundance throughout the upper and lower respiratory tract from the nasal mucosa to the lung periphery.24–26 IN vaccination triggers upper and lower respiratory tract mucosal and sub-mucosal surfaces for protective humoral and cellular pathogen-specific immune responses that also remain at high levels at the port of entry for these pathogens. Previous studies have demonstrated that IN vaccination provides a better protection compared to subcutaneous and IM immunizations in the context of respiratory pathogens, capitalizing on the role of mucosal immunity at the site of initial infection.27–30
Alveolar macrophages (AMs), dendritic cells (DCs), epithelial M cells, intraepithelial lymphocytes, as well as lymph nodes and lymphoid tissues of the upper respiratory tract and the bronchial tree all help in mediating a strong immune response to vaccines.31 Tissue resident and circulatory leukocyte migration through the lungs plays a vital role in IN vaccination. To track this dynamic interaction, we evaluate this vaccination approach in C57BL/6J transgenic mice with Ccr2RFPCx3cr1GFP dual-reporter (C57BL/6J-DR), as well as BALB/c mice. The CX3CR1+ receptor is predominantly expressed in leukocytes such as CD4+, CD8+, and γδ T lymphocytes, as well as natural killer (NK) cells, DCs, and monocytes/macrophages. On the other hand, engineered CCR2-RFP enables the tracking of resident monocytes and AMs.32
Chitosan is a nontoxic, biodegradable, bioadhesive, and biocompatible polymer that can penetrate across epithelial cells lining the mucosal surfaces and their tight intercellular junctions for vaccine delivery.33, 34 While chitosan provides effective loading and delivery of nucleic acids across cell membranes as an effective transfection agent, it requires coating onto the surface of a biocompatible solid nanocarrier to provide mobilization across the nasal cavity for transfection into lungs. Here, we develop and evaluate a gold-nanostar-chitosan (AuNS-chitosan) nanoformulation for IN delivery of a DNA vector expressing S protein of SC2, plus mRNA coding for Firefly luciferase reporter protein. In addition to its inherent advantages for IN delivery of nuclei acids, chitosan serves as an immuno-potentiating agent to augment vaccine immunogenicity and effectiveness, as reported by Saenz et al.35 Antigens formulated onto/into NPs can reach the respiratory mucosa in the airways and lungs for uptake by relevant immune cells.36 With growing concerns over IN administration of live-attenuated viral vaccines (especially in immunocompromised patients), NP-based carriers are a promising alternative to generate safer mucosal immunity.37 We thus investigate in mice the potential of AuNS-chitosan for IN delivery of a SC2 vaccine delivered to the upper and lower respiratory tract mucosa. Gold NPs have recently been used as antigen carriers as well as activators of immune cells for different vaccination approches.38 These NPs are non-toxic and have been used in various applications.39 Gold NPs formulated for IN administration have been shown to trigger robust antigen-specific cytotoxic T cell immune responses in the lymph nodes.40 With this in mind, we test DNA (expressing the S protein of SC2) vaccine-mediated antibody production using AuNS-chitosan as a carrier for IN delivery in mice. Additionally, we test the feasibility of IN delivery of AuNS-chitosan carrying mRNA coding for luciferase reporters to target the respiratory airways and as a proof-of-concept and model platform for future adaptation of our strategy to delivery of a SC2 mRNA vaccine. The eventual clinical translation of this approach should be a seamless extension of current mRNA vaccines.
Unlike pDNA (plasmid DNA) vaccines that rely on cell and nuclear membrane poration to reach the nucleus for transcription and further translation into proteins, it is sufficient for an mRNA strand to gain access to the cytosol for translation. On the other hand, stability of DNA may yield a robust vaccine with a longer shelf life suitable for worldwide distribution. However, DNA expression cassettes carry the theoretical risks of genome integration, insertional mutagenesis, long-term expression, and the induction of anti-DNA antibodies. Given the many pros and cons of these two nucleic acid vaccines, we here initially develop and pre-clinically evaluate an IN administered anti-SC2 DNA vaccine using our AuNS-chitosan delivery vehicle. Conceptually, we aim to establish proof-of-principle for our IN delivery platform using this DNA vaccine by firstly validating its stability and successful organ specific expression (using in vivo imaging of simultaneously delivered luciferase reporter mRNA), and to establish the presence of an ensuing robust vaccine-mediated immune effect in mice. However, a critical pre-requisite applicable to both proposed vaccines is whether sufficient SC2 nucleic acids can be transferred across cell membranes using our AuNS-chitosan NPs. Since electroporation is normally required for DNA uptake across cell and nuclear membranes, we reasoned that initial testing of an anti-SC2 DNA vaccine would be useful to establish the ability of this NP vehicle to replace electroporation.
It is well established that S protein mediates viral transduction via interaction with angiotensin-converting enzyme 2 (ACE2) receptors followed by endocytosis. Thus, vaccines based on the S protein are expected to induce antibodies that can block virus binding and fusion with respiratory airway columnar ciliated cells (and their progenitor cells) expressing ACE2 receptors, or neutralize the virus infection.41 Moreover, compared to all structural proteins of SC2, the S protein appears to be the prominent immunogenic protein to induce both humoral and cellular immunity against virus infection.
As compared to the short half-life of injected protein antigens, DNA vaccines can provide tissue specific expressions of antigens over much longer periods, thereby better priming the immune system.42 We therefore designed AuNS-chitosan to IN deliver a SC2 DNA vaccine to stimulate a wide spectrum immune response, including both local immunity (mucosal IgA, and lung resident T cells) in the nasal cavity and respiratory tract, as well as systemic (neutralizing IgG) immunity. We find that this IN-vaccination strategy also achieves prominent levels of anti-SC2 IgA in the lung mucosa and tissue-resident memory (TRM) T cells that efficiently neutralize SC2 pseudovirus and its variants, thus providing long-lasting immunity.
Results and Discussion
In vitro characterization of gold nanostar synthesis, physicochemical properties, stability, chitosan coating, and DNA loading efficiency at different molar ratios.
We prepared AuNS-chitosan using a modified procedure that we outlined previously.39 We optimized the reaction conditions to generate gold nano-octopods that provide ample surface area for incorporating the payload. The surfaces of as-prepared AuNSs were modified using cationic biopolymer chitosan to improve their biocompatibility, colloidal stability, and to achieve sufficient surface potential for loading anionic nucleic acids. The uniform monolayer of chitosan on the modified AuNS was evident on high resolution transmission electron micrographs (TEM), which also correlated with evident changes in NP surface potential measurements. TEM revealed a narrow size distribution of NPs, with an average size of the NP core as 20 nm and protruding spikes of ~20–30 nm (Figure 1a–c). The pristine AuNS had a surface potential of −5.6 mV (±2.89 mV) that shifted to a cationic surface potential of +35.8 mV (±3.59 mV) upon capping with cationic chitosan polymer. We estimated the pcDNA-SC2 (the coding sequence of SC2 S protein) loading efficiency for AuNS-chitosan using a gel retardation assay. We complexed the SC2 plasmid (2 μg) with increasing amounts of AuNS-chitosan and the resultant polyplexes demonstrated a consistent increase in encapsulated pcDNA. The polyplex amount of 2.5 μL AuNS-chitosan encapsulated 2 μg of pcDNA-SC2 plasmid in the NPs, resulting in pDNA being completely retained in the well during electrophoresis in the gel retardation assay (Figure 1d). We also evaluated the polyplexes for hydrodynamic size using dynamic light scattering (DLS) and surface zeta potential measurement, which also agreed with the gel electrophoresis findings. With increasing amounts of SC2 plasmid in polyplexes with AuNS-chitosan, pDNA was increasingly trapped on the surface of NPs by electrostatic interactions, and as a result of which the zeta potential declined to a nearly net neutral surface potential of +2.12 mV (±3.4 mV) at a polyplex ratio of 1 μL NPs with 1 μg of SC2 plasmid, indicating the maximum loading efficiency of AuNS-chitosan. Likewise, at polyplex ratios with more than 1 μg pDNA, the surface zeta potential diminished further to a negative surface potential indicating an excess of loosely bound pDNA on the surface of NPs, which was also clearly evident in the gel retardation assay. Although each ratio of polyplexes displayed different surface potentials, the size of the pDNA loaded AuNS-chitosan was constantly in the range of 35–48 nm (Figure 1e–f). In order to determine the optimum polyplex ratio for plasmid delivery, we loaded AuNS-chitosan with a pcDNA-FLuc-eGFP plasmid and evaluated it for transfection efficiency in A549 (non-small cell lung carcinoma) cells using bioluminescence imaging (BLI). In agreement with the gel retardation assay and zeta potential measurements, we observed maximum transfection efficiency with a combination of 1 μg of plasmid and 1 μL of AuNS. To evaluate S protein expression using pcDNA-SC2 plasmid (the DNA vaccine), we transfected HEK293T cells with different variants of SC2 plasmid (Wuhan, Beta, and D614G) using AuNS with the optimal ratio, and the cell lysates were probed for expression of S protein using rabbit anti-SC2-spike antibody. We also assayed the CoV-2 and CoV-1 proteins using an anti SC2 antibody to validate their implications in subsequent dot blot and ELISA immunoassays (Figure 1h). Overall, the proposed DNA vaccine formulation comprised of three components: AuNS, chitosan polymer, and plasmid DNA.
Figure 1.
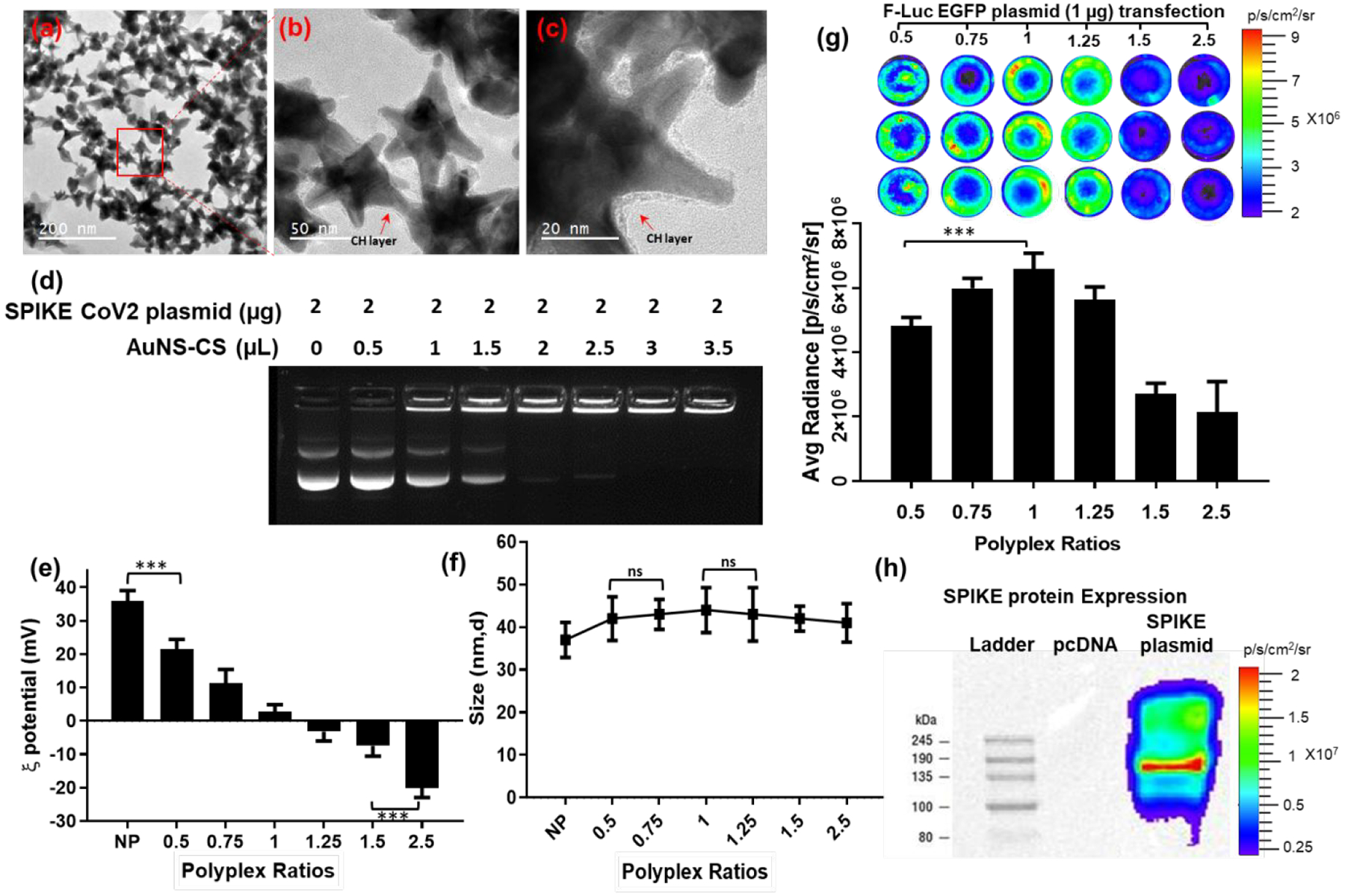
In vitro characterization of SC2 DNA vaccine loaded on AuNS-CS NPs. (a,b&c) FE-SEM micrographs indicate uniform morphology of AuNS-chitosan and SC2 DNA; (d) Evaluation of DNA loading efficiency of AuNS-chitosan by gel retardation assay; (e-f) DLS results measured for zeta potential and particle size (nm) of SC2 vaccine loaded AuNS at different ratios; (g) Transfection efficiency of AuNS-chitosan evaluated by delivery of pcDNA-FLuc-eGFP plasmid by bioluminescence imaging; (h) Immunoblot analysis for expression of SC2 S protein transfected in HEK293 cells by AuNS-chitosan. The data are plotted as mean ± SEM; The significance of comparison was determined by one-way ANOVA with Bonferroni post hoc test. The comparisons were considered statistically significant at adjusted p-values <0.05. The notations used to indicate statistical significance were as follows: * represents p<0.05, ** represents p<0.01, *** represents p<0.001, **** represents p<0.0001, and ns represents no-significant difference.
Intranasal administration of AuNS-chitosan loaded with SC2 DNA vaccine in transgenic C57BL/6J-DR and BALB/c mice.
The growing interest in IN vaccination, and the recent phase I clinical trial of COVI-VAC – an attenuated live viral IN vaccine by Codagenix, developed by introducing hundreds of underrepresented mammalian codons through a deoptimization process – have both prompted us to develop this proof-of-principle IN DNA vaccine platform as the groundwork for a future similar IN mRNA vaccine. A long-term study is required to evaluate the clinical reversion rate of the live attenuated viral vaccine of Codagenix. More recently, it has been shown that a subunit vaccine of SC2 S protein, along with a liposomal STING agonist as an adjuvant, can induce a strong mucosal immunity upon IN delivery in a mouse model.21 However, a subunit vaccine may not elicit neutralizing antibodies sufficient to cover protection against the wide range of variants currently spreading across the globe. Hence, we evaluated our IN-DNA vaccine against SC2 S protein, and this strategy can, in theory, be extended to mRNA vaccines coding for different SC2 structural proteins (S, N, E, and M) to elicit immunity that can protect from all different variants in all viral proteins. We used BALB/c and C57BL/6J-DR transgenic mice to validate the broad immunization capabilities of this IN delivered vaccine, while the C57BL/6J-DR transgenic mice allowed more specifically for evaluation of T cell activation and trafficking using the engineered fluorescence proteins. In the past, there was great interest in using the IN route to deliver influenza A virus vaccines because of their potential to elicit cellular and humoral immune responses along the respiratory tract.43 Indeed, prevention of influenza A virus re-infection requires sterilizing immunity primed by local adaptive immune responses in the lungs. This can only be achieved by IN delivery of antigens, and not through IM inoculation44. To evaluate the efficiency of IN delivery of the SC2 DNA vaccine, we delivered pDNA expressing S protein loaded onto AuNS-chitosan via IN delivery in BALB/c and C57BL/6J-DR mice (N=5, each). Mice were given 20 μg of DNA at the intervals shown in Figure 2a–b.
Figure 2.
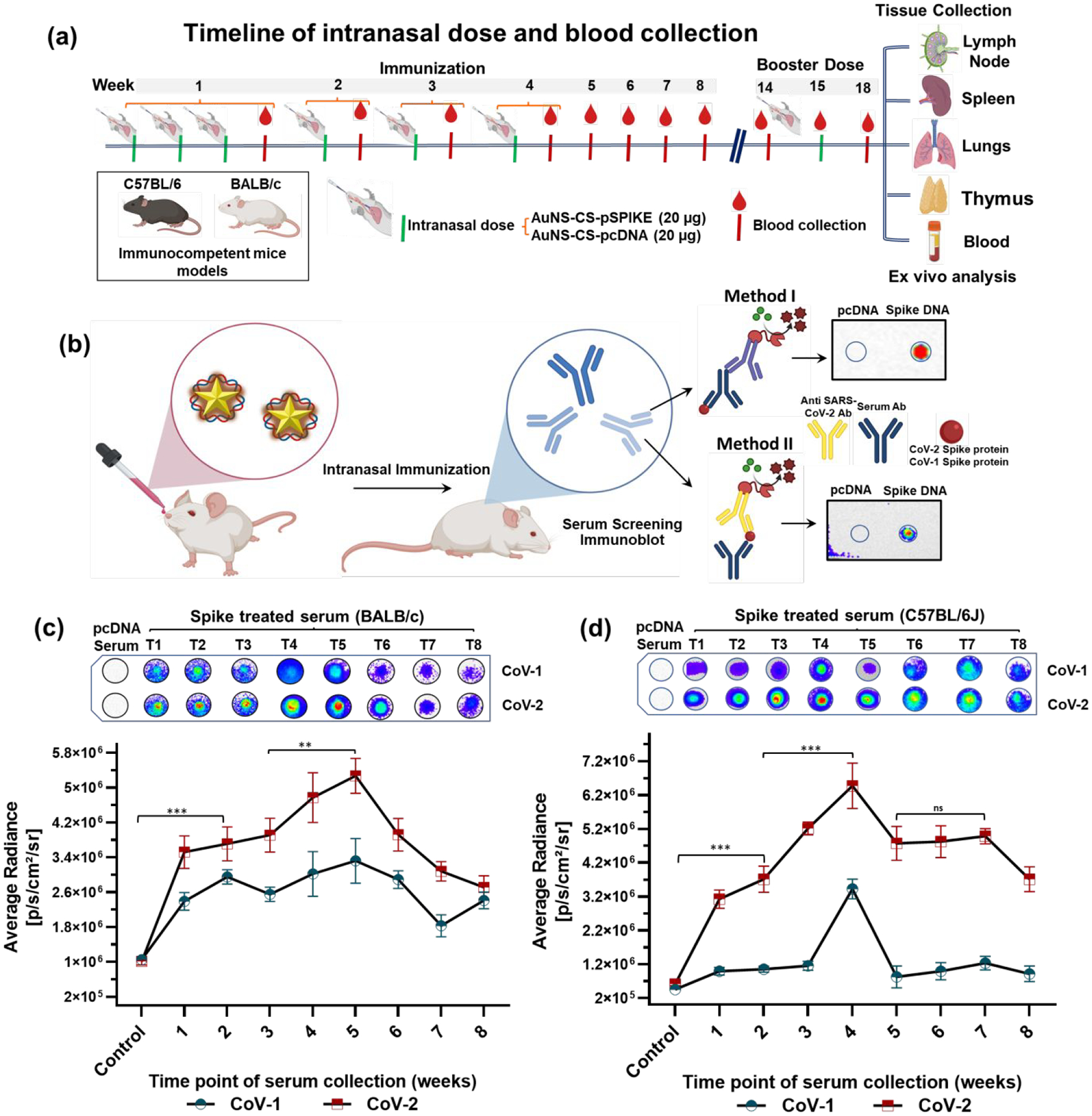
(a) Schematic representation of the experimental design: Five-weeks-old BALB/c mice and C57BL/6J mice were immunized with AuNS-chitosan loaded with control DNA or SC2-S DNA vaccine administered via the IN route, the serum was collected every week and assessed for anti-SC2 antibody against purified proteins of CoV-1 and CoV-2; (b) Chemiluminescence based dot blot immunoassay for screening anti-SC2 antibody levels in serum collected from, (c) BALB/c and (d) C57BL/6J mice at different time points of treatment with their respective quantitative plots (n=5). The data are plotted as mean ± SEM; The significance of comparison was determined by two-way ANOVA with Turkey T test. The comparisons were considered statistically significant at adjusted p-values <0.05. The notations used to indicate statistical significance were as follows: * represents p<0.05, ** represents p<0.01, *** represents p<0.001, **** represents p<0.0001 significance, and ns represents no-significant difference.
The reactivity of sera from SC2-vaccinated mice with the S protein of SARS-CoV-1 and SC2 determined using S protein-based dot blot assay.
With the established in vitro evidence for SC2 delivery and expression by AuNS-chitosan, we investigated the anti-SC2 specific immunoglobulins generated in mice upon IN vaccination. We also investigated the sera cross-reactivity with the S1 subunit of SARS-CoV-1 (CoV-1) from the 2003 SARS outbreak.45 We tested sera collected at different time points from both control and SC2 vaccinated BALB/c and C57BL/6J-DR mice for the anti-S protein antibody against purified S protein from SARS-CoV-1 and SARS-CoV2 (CoV-2) using a chemiluminescence dot blot assay. We clearly observed that the antibodies to the delivered SC2 DNA vaccine were produced in as early as two weeks after vaccination (p<0.001) (Figure 2c). Even though the S protein of CoV-1 shows significant homology with that of CoV-2, the serum of mice induced using the SC2 vaccine showed less sensitivity to the CoV-1 S protein compared to CoV-2. Evaluation of serum collected from both BALB/c and C57BL/6J-DR mice at all time points indicated that the sensitivity of CoV-2 protein detection was much higher compared to CoV-1, and was also consistent in both mouse models (Figure 2c). The CoV-1 detection signal was nearly 40–60% lower (p<0.01, for time point 1 to time point 7) than that of CoV-2, indicating significant cross reactivity, which is consistent with the earlier findings that the vaccination approach outlined here can potentially provide protection against related viruses of the sarbecovirus subgenus, with similar efficiency compared with CoV-2.38, 45–47 This provided the evidence for a B cell-mediated humoral immune response triggered upon DNA vaccination (Figure 2c–d). Overall, the trend in serum detection levels followed the same pattern as observed in the ELISA assay, with the highest serum levels of anti-SC2 S protein antibody peaking at Weeks 4 and 5 of treatment in C57BL/6J-DR and BALB/c mice, respectively. In addition, the peak antibody levels in serum of C57BL/6J-DR was ~25% higher than in BALB/c mice. The observed difference in serum antibody levels and their time to peak values were possibly owing to immunological differences in these two inbred mouse strains.48, 49 However, the vaccination efficacy and pattern in humoral immune response was prominent in both models.
Intranasal vaccination boosts cross-variant humoral immune response against mutant variants of SC2.
With the global surge in SC2 infections, the viral susceptibility to undergo mutations also increases with spread and time, which results in emergence of new mutant variants.50 Emerging SC2 variants have raised concerns because of resistance to neutralizing antibodies generated upon by vaccination or previous infection. Mutations found in emerging S protein variants limit the outcome of neutralization by convalescent plasma, monoclonal antibodies, and sera from vaccinated individuals.51 As a result of such growing concerns, we further evaluated whether IN SC2 (Wuhan) vaccinated mice sera could show cross-reactivity with other emerging variants of SC2. We used an immunoblot assay with total cell lysates of HEK-293 cells transfected with plasmids encoding SC2-Wuhan S protein and SC2-Beta-mutant S protein to probe serum collected from BALB/c and C57BL/6J-DR mice for cross-variant neutralizing antibodies against SC2-Beta-mutant. We found that both SC2-Wuhan and SC2-Beta-mutant S proteins were equally detected by serum collected at all time points (Figure 3a–b). These findings were consistent in both mouse models, and they strongly indicate the efficacy of our vaccination approach against new strains of SC2. On the other hand, these results also point out the importance and need of future vaccination to both previously infected and uninfected subjects to elicit cross-variant neutralizing antibodies when using our proposed strategy.52 We verified the trend in serum levels of SC2 S antigen specific immune response by using different assays. Moreover, in order to establish the consistency of this vaccination approach, we compared serum levels of neutralizing antibody from two independent studies with a batch of five C57BL/6J-DR mice in each group (Figure 3c). Comparative immunoblot analyses of serum collected four weeks after immunization were evaluated against purified SC2 and SC1 proteins, and the results indicated similar serum levels of neutralizing antibodies in both batches, supporting the reproducibility of this IN immunization approach.
Figure 3.
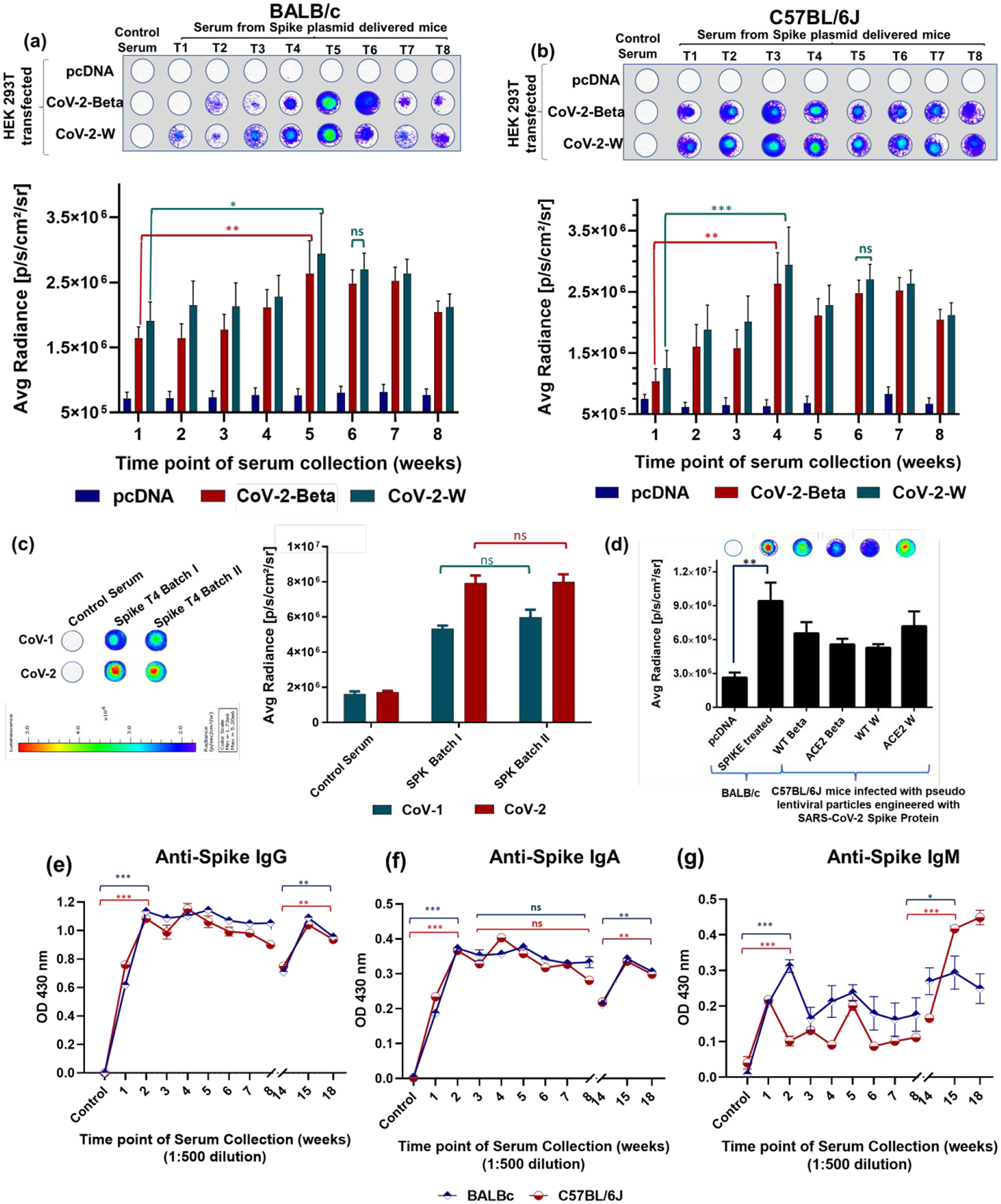
Dot blot immunoassay for screening anti-SC2 antibody levels in (a) BALB/c and (b) C57BL/6J mice at different time points of treatment. The serum was probed against the cell lysate of HEK-293 cells transfected with plasmid encoding S protein of SC2-Beta-mutant and SC2-Wuhan variant to determine the efficacy of vaccination strategy in mounting an immune response against different variants of SC2. (c) Response in anti SC2 antibody levels in serum of two different treatment batches (each with n=5) indicates the consistent antibody response; (d) Comparison of anti-SC2 antibody levels generated in BALB/c mice administered with AuNS-chitosan loaded pcDNA/pcDNA-SC2 DNA vaccine with wild type and ACE-2 engineered C57BL/6J upon challenge with pseudovirus engineered with SC2-W and SC2-Beta S proteins (*p<0.05, ** p<0.01, ** p<0.001, ** p<0.0001, ns: not significant); Antibody mediated immune responses after IN immunization of SC2 S protein DNA vaccine using AuNS-chitosan. Antibody responses in sera of immunized mice at different time points of treatment were evaluated using ELISA. ELISA assay measured against SC2 S protein-specific IgA (e), IgG (f) and IgM (g) levels. Data generated from pooled serum of 3–5 BALB/c and 3–5 C57BL/6J mice. All mice received a booster dose on Week 14 to evaluate mucosal and cell-mediated immune responses. (n=5) The data are plotted as mean ± SEM; The significance of comparison was determined by two-way ANOVA with Turkey T test. The comparisons were considered statistically significant at adjusted p-values <0.05. The notations used to indicate statistical significance were as follows: * represents p<0.05, ** represents p<0.01, *** represents p<0.001, **** represents p<0.0001, and ns represents no-significant difference.
To draw comparisons between DNA vaccine-mediated immunity and actual infection-mediated immunity, we investigated immune responses of transgenic mice expressing human ACE2 receptor along with respective control strains (C57BL/6J and C57BL/6J-ACE2) to SC2 pseudovirus delivered via the IN route. We tested two different variants of pseudoviruses corresponding to SC2-Wuhan and SC2-Beta-mutant in wild type and ACE2 engineered C57BL/6J mice. Evaluation of serum collected from mice five days after pseudovirus infection showed significant (p<0.01) levels of anti-SC2 antibody in both wild type and ACE2 transgenic mice (Figure 3d). We tested immune responses in wt-C57BL/6J and ACE2-C57BL/6J animals IN delivered with lenti-pseudovirus expressing SC2 S protein and compared with antiserum taken from BALB/c animals IN treated using pcDNA (control) and SC2 S DNA loaded on chitosan capped AuNS for comparison. Although both procedures are entirely different in terms of induced immunity, the results indicated that the extent of humoral immune response generated by IN administration of our SC2 DNA vaccine loaded onto AuNS-chitosan NPs was nearly 30% higher than that achieved by pseudovirus-mediated transduction at the similar time point of the study (5 days post treatment of 3 doses of SC2 DNA vaccine or SC2-Wuhan and SC2-Beta pseudoviruses).
Intranasal delivery of DNA vaccine expressing SC2 S protein using AuNS-chitosan showed effective activation of humoral pulmonary immunity.
The entry of SC2 into cells is mediated by the interaction of ACE2 receptors present on the target cell membrane with the receptor-binding domain (RBD) of the viral S glycoprotein.53 Antibodies generated against SC2 S antigen can be screened using S protein-based ELISA. Immunization through the IN route can trigger mucosal immune responses with elevated levels of secretory IgA antibodies to confer protection at or near the site of initial entry of respiratory pathogens.54 To assess the immune response and protective efficacy of AuNS-chitosan loaded with SC2 S DNA vaccine, we used two different mouse strains, BALB/c and C57BL/6J-DR.
We analyzed the S protein antibody responses in serum collected at different time points from SC2-DNA vaccinated mice to determine SC2 S protein specific IgA, IgG and IgM titers in vaccinated mice as compared to control mice. As indicated in the ELISA results, IN immunization of AuNS-chitosan-SC2-spike, but not control DNA loaded AuNS-chitosan, induced high levels of S protein specific IgA, IgG and IgM antibodies in serum (Figure 3e–g). We measured vaccine induced IgG, IgA and IgM in serum serially, up to eight weeks in both BALB/c and C57BL/6J-DR mice using an ELISA assay. S antigen-specific IgG levels rose exponentially in both mice strains as early as in the first week (after three doses) and remained at the peak for eight weeks, independent of further doses of vaccination (Figure 3e). The animals were maintained for 14 weeks without any further doses. In the 14th week, we collected blood samples and given an additional booster dose. Upon administration of the booster dose in Week 14, IgG levels increased further, reaching a maximum on Week 15, and remained elevated for subsequent weeks (Figure 3e). Likewise, AuNS-chitosan-SC2-spike DNA vaccination also elicited IgA specific to S antigen with similar kinetics of induction, as well as time to reach the peak levels (Figure 3f). It also exhibited a sustained peak plateau in serum for two to eight weeks, which is not usually observed after IM vaccination, especially because of its short half-life and seroconversion pattern.8, 55
This distinct pattern of consistently higher levels of IgA generated over the course of treatment can be regarded as a critical advantage, specifically because of the spatial distribution of IgA on mucosal surfaces and the IN route of administration used in this research.55 Multiple studies have found that IgA possesses superior antiviral properties when compared to the IgG for influenza and for SC2. Sterlin et al. recently reported that IgA dominates the early neutralizing response to SC2, and they deduced that serum IgA is 7-fold more potent in viral neutralization compared to serum IgG;56 these advantages could be effectively harnessed by the vaccination strategy adopted in our study. Our finding of the heightened mucosal immunity via IgA could confer an important advantage in preventing SC2 infections, given that the virus attacks respiratory epithelial cells by docking to ACE2 protein on the surface of type-2 alveolar cells.57, 58
On the other hand, existing evidence indicates good correlations between serum and salivary IgG and IgM antibody levels, whereas there is a much weaker correlation between salivary and serum IgA antibodies. This is not unexpected, as salivary IgG and IgM are mainly derived from the circulation, whereas salivary IgA is mostly secreted locally in the salivary glands.59 Thus, it can be inferred with confidence that the levels of S antigen specific IgA in the bronchoalveolar lavage and saliva of vaccinated mice would be much higher than the values determined from serum. SC2 specific IgM and IgA were generated as early antibody responses followed by SC2 specific IgG antibodies.56 The IgG levels are assumed to continue lifelong as protective antibodies against SC2. However, onset of seroconversion also determines respective levels of immunoglobulins, including IgM and IgG synchronous seroconversion; IgM seroconversion later than IgG, and IgM seroconversion earlier than IgG.60 This could plausibly account for the intriguing spike in IgM levels observed after booster dose administration in C57BL/6J mice. IgG and IgA sustained their levels till Week 14 and rose even higher with a single booster dose (Figure 3e–g). The quick surge in levels of IgG, IgA, and IgM following a booster dose in Week 14 also represents clear evidence of long-lasting memory T and B cells that are able to trigger a memory cell mediated rapid recall response. Humoral immune responses are typically characterized by primary IgM antibody responses followed by secondary immune memory antibody responses composed of IgA, IgG and IgE. Here, we observed humoral responses to SC2 in the form of SC2-specific antibodies in the blood pool. Overall, our findings demonstrate that AuNS-chitosan-SC2-spike DNA vaccines effectively induce S-antigen specific IgG, IgA, and IgM responses in immunocompetent mice, with marked differences in their persistence in serum.
Intranasal delivery of DNA vaccine expressing the S-protein if SC2 using AuNS-chitosan showed efficient activation of pulmonary immunity with neutralizing antibodies.
After successful evaluation of antibody induction by the delivered DNA vaccine, we tested the neutralizing effects of antibodies using lenti-pseudoviruses displaying the S protein of SC2 and expressing Firefly luciferase (FLuc)-ZsGreen reporter gene as a pseudovirus for a neutralization assay (BEI resources, NIAID). We first tested the specificity of spike-lenti-pseudoviruses for their infectivity to cells expressing the human ACE2 receptor. We infected viruses of the same titer to control cells and ACE2 expressing HEK293T (HEK293T-ACE2) cells, and assessed for infectivity 72 h post transduction using BLI. We observed selective transduction of pseudovirus into HEK293T-ACE2 cells, which established them as a suitable model to mimic infectivity of SC2 in the presence of neutralizing antibodies in different conditions (Figure 4a). After confirmation of pseudovirus for its selectivity, we used the virus along with serum from mice (BALB/c and C57BL/6J-DR) collected different time points after DNA vaccine delivery to evaluate neutralizing antibody effects. Antibodies from mice immunized with AuNS-chitosan-SC2-spike protein of Wuhan strain neutralized luciferase-expressing SC2 pseudovirus encoding the S protein of the same strain, which was reflected in the decline of FLuc signal. The time dependent variation in the antibody titer was also measured using ZsGreen-based FACS analysis, which correlated well with the trend observed in BLI (Figure 4b–d). The histogram for pseudovirus transduced ZsGreen expression in HEK293T-ACE2 cells in the presence of serum collected from vaccinated mice on Week 3 was fully displaced towards the lower end and overlapped with that of control cells that were not subjected to pseudovirus transduction. Our findings indicated presence of a high titer of neutralizing anti-SC2 S antigen specific antibodies generated in the vaccinated mice, which could completely prevent the infection of SC2 pseudovirus in HEK293T-ACE2 cells.
Figure 4.
Evaluation of the specificity of lentivirus expressing SC2 S protein as a pseudovirus to cells expressing ACE2 receptor. (a) Mechanism of SC2 transduction in cells; lentivirus expressing SC2 S protein and Fluc-ZsGreen reporter gene were engineered, and these pseudoviruses were transduced in control and ACE2 receptor expressing cells, and subsequent infectivity was quantified using bioluminescence imaging. (b&c) DNA vaccine-mediated induction of anti-SC2-S protein specific antibody evaluated for its neutralizing effect using engineered pseudovirus assessed by quantifying pseudovirus-mediated ZsGreen expression in the infected cells in the presence of neutralizing antibody from serum of mice treated with DNA vaccine; and (d) Neutralizing effect of serum collected at different time points after vaccination. Commercial antibody was used as positive (+) control. T1, T2, T3 indicates time point of serum collection, i.e., after 1 week, 2 weeks, and 3 weeks, respectively; The neutralizing antibodies induced by the IN administration of SC2-DNA vaccine measured for viral infectivity inhibition using lenti-pseudoviral particles engineered to display SC2 S protein of different variants and expressing FLuc-ZsGreen reporter gene in HEK-293 cells engineered to express ACE2 receptor. Serum samples from of SC2-DNA vaccinated C57BL/6J mice at Week 18 (pooled serum from n=3 animals) were assayed for neutralizing activity in comparison with commercial SC2-spike antibody. The relative inhibition in infectivity was performed against lentiviral particles engineered with S protein SC2-Wuhan (e), SC2-Beta-mutant (f) and SC2-D614G-mutant (g) variants. Each point represents the mean of serum collected from three mice with three technical replicates. The data are plotted as mean ± SEM (n=5); The significance of comparison was determined by One-way ANOVA with Bonferroni post hoc test as indicated. The comparisons were considered statistically significant at adjusted p-values <0.05. The notations used to indicate statistical significance were as follows: * represents p<0.05, ** represents p<0.01, *** represents p<0.001 and **** represents p<0.0001, and ns represents no significant difference.
Intranasal delivery of DNA vaccine expressing the S-protein of SC2 using AuNS-chitosan induced efficient production of neutralizing antibodies effective against different variants of SC2.
To investigate the efficacy of neutralizing antibodies generated in vaccinated mice against new emerging mutant variants of SC2, we evaluated the infectivity inhibition of vaccinated mice serum against SC2 pseudovirus with the S protein of Wuhan strain, D614G mutant, and the South African variant (SC2-Beta-mutant).61 We observed a dose dependent neutralizing effect by serum collected from mice treated with the DNA vaccine, and the results were represented as relative inhibition of infectivity (Figure 4e–g). The HEK293T-ACE2 cells transduced with pseudovirus in the presence of serum collected from control DNA treated mice served as control, with 100% infectivity. In the absence of SC2 S antigen specific antibodies in the control DNA treated mice sera, the levels of pseudovirus infectivity were similar to those of cells transduced using pseudovirus in the absence of serum. We used vaccinated C57BL/6J mice serum with peak titers of anti-SC2 antibody achieved after booster dose administration for evaluating the neutralization assay, which corresponded to serum collected at Week 18 of the study. We used the serum at different dilutions to study the correlation of antibody titers in serum with infectivity of pseudovirus, and compared with that of commercial anti-SC2 antibody. We observed a dose dependent (serum dilutions) decline in infectivity, and at 1:10 dilution of serum, we found nearly complete inhibition of infectivity, which was almost similar to that achieved by commercial antibody at 4μg/mL concentration (SARS-CoV/SARS-CoV-2 Spike antibody, Chimeric MAb). These trends were consistent across all three strains of pseudoviruses engineered for different variants of SC2 S proteins (i.e., SC2-Wuhan, SC2-Beta-mutant, and SC2-D614G-mutant) with observable difference in inhibition at higher serum dilutions. At 1:10 serum dilution, the infectivity of both SC2-Wuhan pseudovirus and SC2 D614G-mutant variant diminished to 38% ± 22% and 38% ± 5%, respectively, whereas for SC2-Beta-mutant variant, the infectivity dropped to only 67% ± 7%. Despite these variations at higher dilutions of serum, when we used 1:10 serum dilution for the assay, infectivity of all three variants was inhibited completely, indicating the efficacy of this DNA vaccination approach against emerging mutant variants (Figure 4e–g). The IC50 infectivity inhibition concentrations were determined to be 1:83.8, 1:47.5 and 1:150 serum dilutions for SC2-Wuhan, SC2-Beta-mutant, and SC2-D614G-mutant variants, respectively (Figure S1).
Intranasal delivery of DNA vaccine expressing SC2 S protein using AuNS-chitosan effectively induced cell-mediated immunity in C57BL/6J-DR mice.
A cell-mediated immune response plays a critical role in combating viral infections.62 It is comprised of T cell responses that fundamentally differ from humoral responses in that they establish receptor mediated cell-to-cell interaction to eliminate the infection. Cell-mediated immunity is primarily driven by mature T cells, macrophages, DCs, NK cells, and the released cytokines, in response to antigen delivery.63 In order to deduce the role of cell-mediated immunity after IN DNA vaccination, we performed immunophenotyping of leukocytes collected from lungs, spleen, thymus, and lymph nodes of mice delivered using control DNA and DNA coding for S protein of SC2 using AuNS-chitosan NPs. The major immune cell subsets that confer protection to the pulmonary immune system include T helper (TH) lymphocytes, AMs, DCs, cytotoxic T lymphocytes, NK cells, memory lymphocytes, and B cells.64, 65 We succeeded in isolating CD45+ positive populations with greater than 90% purity in all four sample sources (Figure S2) and phenotyped them for different immune cell populations.
SC2-spike DNA vaccine mediated antigen processing and immune cell activation in the lungs and spleen.
The important cellular mediators of pulmonary immunity consist of NK cells and phagocytic cells (AMs, eosinophils, neutrophils).66 They have the capacity to recognize and neutralize SC2-spike antigen expressing cells. Although prominent cell mediated immunity is manifested by circulating naïve T lymphocytes, their ability to leave the blood stream and migrate into peripheral tissues is limited. Thus, adaptive immunity of SC2 requires initial transport of S antigen from the site of initial exposure to the T cells of the draining lymph nodes.67 Such a transportation of antigen via afferent lymphatics is a specialized function of alveolar DCs.68 We observed these initial events of interaction in our immunohistochemistry analysis of lungs from SC2-spike vaccinated mice. Figure S3 shows the expression of transfected S protein in the endothelial cells lining the bronchi and alveoli, which are selectively recognized by the DCs. Some of these spike-DNA NPs are also directly internalized into DCs and antigen presenting cells (APCs), which possess cellular extensions through epithelial junctions of alveoli.69 These S antigen primed DCs are expected to process the antigen and drain into lymph nodes to prime other components of the cell-mediated immune response to home to the lungs. The presence of DCs in high density in the proximal airways and their inherent high phagocytic ability places them in a perfect position to capture S antigens expressed by the endothelial cells. Their strategic distribution, and their ability to capture and process antigen, and present them to T cells in the lymph nodes, all make DCs the key APCs in the lungs and in other mucosal surfaces.70 Our FACS analysis results of lymphocytes in spleens of S treated mice revealed an increase in CD11c+ DCs (7.5%), also accompanied by a surge in CD8+ T cells (5.5%). This explains the arrival of CTLs (MHC class I-restricted T cells) from lungs to spleen (Figure S2a). The CD11c+ DCs represent the major DC subset required for cross-presenting antigens to CD8+ T cells, directing T helper type 2 (Th2) responses to S antigen and promoting viral clearance.71 On the other hand, CD4+ T cells respond to the antigen processed and presented with MHC-II complex on the surface of APCs.
A plausible exposure to S antigen can induce naïve CD4+ T cell expansion and differentiation into effector cells, TH1, TH2, TH17, or Treg-phenotypes, which eliminate the infection. On the other hand, a small fraction of these activated CD4+ T cells undergo further differentiation into memory cells that reactivate rapidly upon antigen re-exposure.72 Thus, we evaluated the expansion of CD4+ T cells in lungs of S vaccinated mice, not only for effector and memory functions, but also for their role as T helper cells in the germinal centers of spleen and lymph nodes.73 We observed the levels of CD4+ T helper cells in S treated mice to be increased marginally by 4.2%, which plausibly accounts for the presence of surviving T cells that remain in the alveoli as resident effector memory cells (n=3). Once activated, T helper cells activate B cells in the lymph node (B cell zone) and redirect them into lungs via the systemic circulation.74 In agreement with this pathway, SPK treated mice manifested a 6.1% increase in CD19+ B cell populations (n=3) (Figure S2). As these lung resident B cells represent a major component of adaptive immunity and account for antigen specific immunoglobulins against SC2 vaccine, it can be deduced with certainty that increase in B cells in the lungs correlate with generation of systemic SC2 specific IgM, IgA and IgG immunoglobulin responses (Figure 3e–g).
In addition to DCs and T cells, >90% of the cells in the alveolar lumen are comprised of AMs, which are indirectly in contact with DCs in the alveolar wall that prompts a response for S antigen presentation by DCs.75 We also observed a prominent increase (10.3%) in CD11b+ macrophage levels in lungs of S vaccinated mice to indicate their role in arming the cell-mediated immune response (n=3). As these resident AMs reside in close proximity with endothelial cells, they also come into contact with conventional DCs that extend dendritic snorkels into the alveolar lumen (Figures 5, S3&S4). From the circulation, these DCs can enter the spleen in the marginal zone (MZ) sinus that separates the white and red pulp (RP) to mount a response. We observed such events of DC migration from marginal zones into GCs on spleen histology of S vaccinated mice (Figure 6 &S5). This stood in stark contrast to spleens from pcDNA treated mice. Increased presence of DCs in the white pulp (WP) triggers adaptive immune responses from the spleen against simultaneous pancreas kidney (SPK) antigen.76 Therefore, IN immunization induces mass DC migration into the WP enriched with T cells (Figures S2 and 6). In addition, such differential intrasplenic migration of DC subsets tailor adaptive immunity. Based on function, the structure of the spleen is divided into WP and RP, which are demarcated by the marginal zone (MZ). The cDCs arriving from spike vaccinated lungs modulate the dynamic distribution of immune cells in the spleen to mount appropriate T and B cell responses. Upon activation by an innate immune response to S antigen, the cDCs migrate from RP and MZ of the spleen to the WP, i.e., to the splenic T cell zone. DC migration from marginal zones into germinal centers leads to selective induction of T cell responses (either CD4+ or CD8+) (Figure 6). Likewise, FACS analysis of splenocytes from SC2-spike DNA vaccinated BALB/c mice also indicated 7.4% increase in CD11c positive dendritic cells, which was accompanied by 6.31% increase in B cell population (n=3). These results correlated well with histological evidence of cDC (circulatory DC) migration from marginal zones into GCs (Figure S2a & Figure 6). The synchronous surge in DCs and B lymphocytes in the spleen of vaccinated mice suggested that DCs were involved in transport and transfer of SC2 antigen to naive B lymphocytes, and upon this adoptive antigen transfer, B cells triggered SC2 specific antibody responses (Figure 3e–g).77, 78 On the other hand, SC2 activated cDC interaction with B lymphocytes also primes subsequent T cell dependent response, and in agreement with this postulation, vaccinated mice manifested ~7.9% increase in CD4-CD8 double positive T cells (n=3) (Figure S2a).
Figure 5.
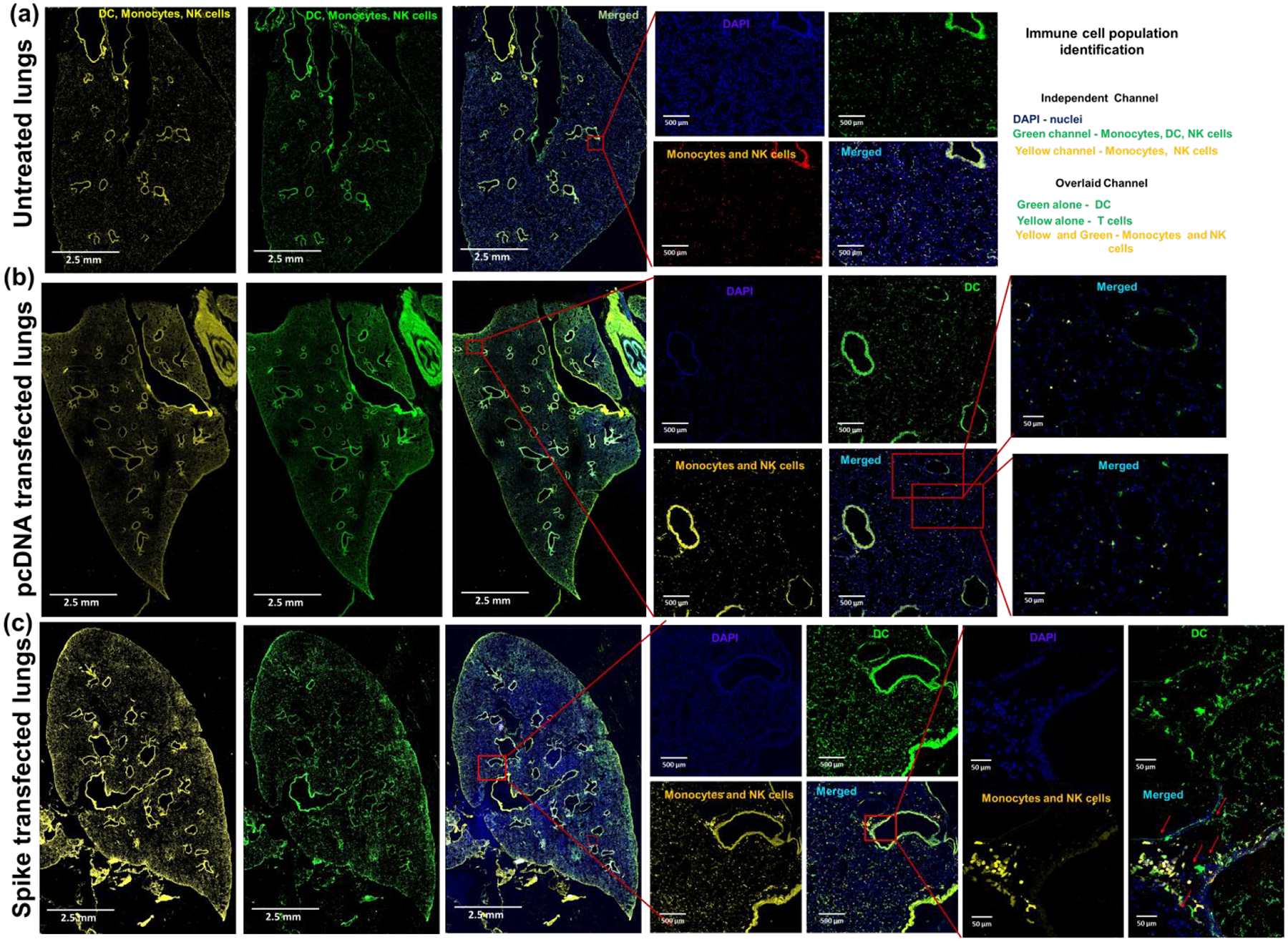
Distribution of monocytes, NK cells, and DCs in lungs of (a) untreated mice, (b) pcDNA treated mice, and (c) SPK DNA vaccine transfected mice. The increased presence of DCs (GFP cells in merged image) in the alveoli and bronchi indicate the arrival of circulatory DCs to complement the role of tissue resident alveolar DCs in recognition and processing of SPK antigen. The monocytes and alveolar macrophages are the other class of resident lung phagocytes that are recruited to the alveoli as well as closely associated to the bronchial epithelium expressing S protein to mediate recruitment of additional leukocyte subsets to the lungs (n=5).
Figure 6.
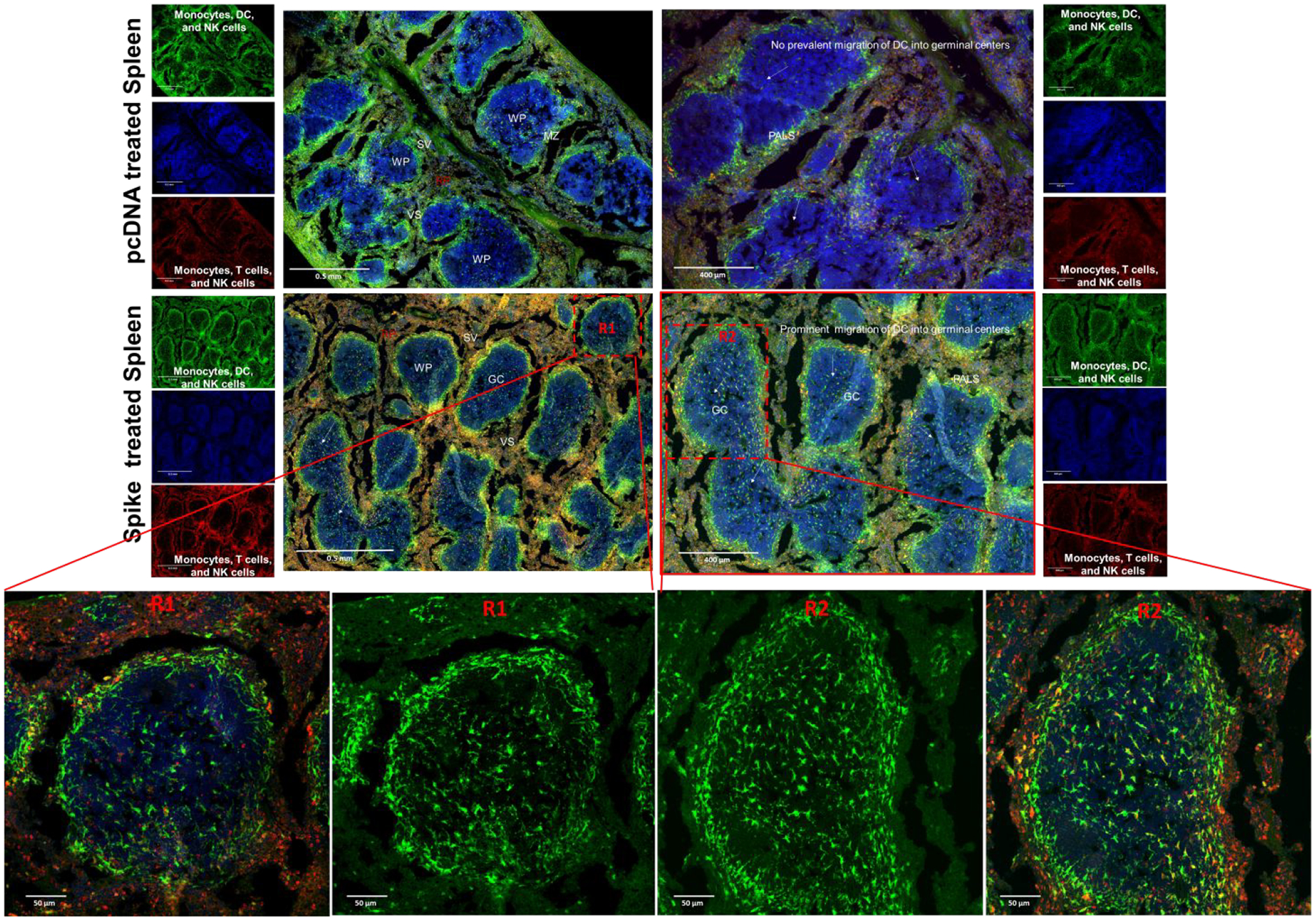
Histological section of germinal centers depicting intrasplenic migration of DC subsets (GFP positive) in white pulp (WP), red pulp (RP), and marginal zone (MZ) of spleen harvested from C57BL/6J-DR mice treated with SC2 vaccine and pcDNA.
Alongside the professional APCs, such as DCs, other APCs (macrophages) are also actively involved as promoters of germinal center B cell response.79 Splenic macrophages are compartmentalized into white pulp, red pulp and marginal zones, and each of them play distinct roles in immune responses. The arrival of SC2 activated macrophages in the strategic location of the marginal zone and white pulp, places them in close proximity for interactions with both B and T cells to participate in SC2 immune responses (Figure S5). Similarly, we observed increases in prevalence of macrophages and monocytes in the white pulp and the marginal zones of spleens from SC2 vaccinated mice. This correlated well with a synchronous surge in CD11b+ macrophages by 10.3%, accompanied by 6.3% increase in B cells in the spleens of SC2 vaccinated BALBc mice (Figure S2a and Figure 6).
Lastly, we evaluated for the presence of any toxic effects in the lungs of mice treated with multiple doses of the DNA vaccine, and in controls (N=3), using H&E histological staining (Figure S6). We observed that there was no significant tissue damage in the lungs of mice vaccinated with the S DNA vaccine.
SC2-spike DNA vaccine-mediated antigen processing, and B and T cell activation in lymph nodes.
Lymph nodes, house B cells, T cells, and APCs, are the vital command centers of the immune response to orchestrate adaptive immunity.80 The monocytes and DCs are the predominant population of APCs that can internalize the S antigen expressed in the lungs upon IN delivery of DNA vaccine using AuNS-chitosan, and physically transport S antigen from site of expression into lymph nodes. In addition to this role, activated circulating DCs from the lymph nodes mediate antigen cross presentation for priming CD8+ T cells, which are not found in some tissue resident DCs.81 As evident from the interaction of DCs with S protein expressing cells in the lungs (Figures 9, S3&S4), it would be expected that the processing of S antigen into short peptide fragments by DCs can present them onto class I and class II MHC molecules, to enable antigen recognition by CD8+ and CD4+ T cells.82 At the same time, it would be expected that APCs that have encountered SC2-spike antigen to arrive at the nearest lymph nodes and activate naïve T helper cells for S antigen recognition. Upon activation of CD4+ T helper cells, they act in a costimulatory manner to activate B cells that have encountered the same S antigen presented by DCs and macrophages.83 Unlike typical macrophages, the subcapsular sinus macrophages target lymph-borne pathogens by presenting antigens to B cells, and triggering cytokine signaling cascades for recruitment of DCs, neutrophils, NK cells, and in some instance, macrophage mediated antigen presentation to T cells. We observed similar events in SC2 vaccinated mice, which showed the presence of subcapsular sinus macrophages and DCs (Figure 7).84 Activation of B cells is often triggered by the interaction of S antigen captured APCs present in the lymph nodes with the B cells.85 As we could see a consistent dose dependent surge in S specific immunoglobulins (IgG, IgA and IgM) in vaccinated mice, it is certain that B cells in these mice were activated by the delivered S DNA vaccine and orchestrated the antigen specific response. In view of this concept, we further investigated the role of lymph nodes in mounting this B cell response, especially because of their crucial role in maturation and activation of B cells (Figure 7). Draining lymph nodes bring together spike activated cDC, NK cells and macrophages for generating S antigen specific immune response from B cell maturation centers, i.e., germinal centers (GCs). Lymph nodes of vaccinated mice depict characteristics of reactive lymph nodes with multifocal germinal centers with extensive B cell proliferation. The presence of interdigitating follicular dendritic cells (FDCs) in the GCs also validates the S specific B cell assortment in the GC and accounts for the rapid surge in immunoglobulin levels (IgG, IgM and IgA) in vaccinated mice. The events of B cell migration across the marginal zone into the interfollicular space represents short-lived antibody producing plasmablast cells. The interfollicular zone prime appropriate T cell response as well as B cell maturation (Figure 7).
Figure 7.
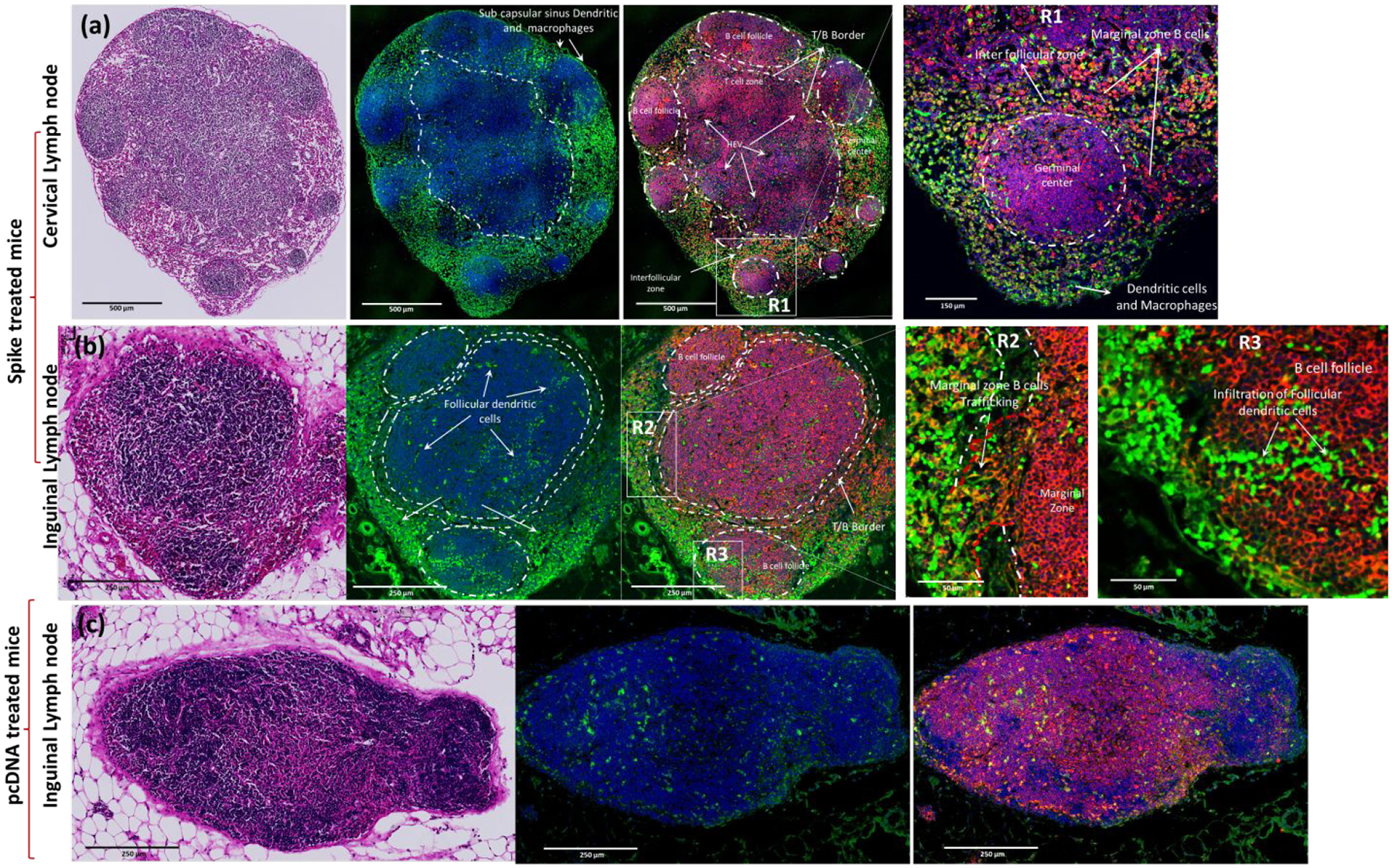
H&E and anti CD19b + stained (red) histological section of lymph nodes harvested from C57BL/6J-DR (GFP positive DCs) treated with SC2 vaccine (a&b) (cervical lymph node &Inguinal lymph node) and pcDNA treated(c) mice.
To investigate whether the SC2 DNA vaccine could trigger such a characteristic S specific immune response in the lymph nodes, we harvested nodes from BALB/c mice treated using SC2 DNA vaccine along with pcDNA treated mice as controls (N=3 animals for each condition) and analyzed using FACS to identify distributions of immune cell populations. The nodes are also where cytotoxic T cells are trained with S antigen presented by professional APCs, especially the DCs.86 Our FACS analysis results indicated this modulation of CD4+ T cells in S vaccinated mice compared to that of pcDNA vaccinated mice (Figure 5, S2). The histology of lungs from vaccinated mice indicated the successful delivery and expression of S antigen and also captured the interaction of DCs with these S expressing cells (Figures 5, S3&S4).
These migratory DCs in the lungs can present antigen to resident DCs in the lymph node.87 These S antigen recognition events direct the activation of innate immune response to the antigen pattern recognition receptors or cytokines, as well as chemokines induced in the lymph nodes in response to S antigen-mediated immunization. FACS analysis of both B and T cells in lymph nodes revealed their S antigen-specific enhancement in treated mice (i.e. 18.6% increase in CD19+ B cells and 6.3% increase in CD4+ T cells). Although preclinical immunization studies in other models indicate that large quantities of antigens are essential for mounting CD8+ T cell responses,87 our approach overcomes this limitation by efficient antigen processing and cross presentation by DCs achieved via the IN route, which also led to a surge in a T cells immune response from the lymph nodes. The presence of high levels of antigen and a greater production of follicular helper T cells can govern lymph node GC responses.
The histology of lymph nodes from C57BL/6J-DR mice vaccinated using SC2-spike indicated a characteristic feature of antigen activation. T and B cells were segregated into distinct locations, with deeper paracortex of the nodes occupied by T cells and follicles populated with B cells (Figures 7). The migrating lymphatic APCs carry the S antigen to the subcapsular sinus of the draining lymph nodes. Recognition of S antigen by B cells drives them into specialized subregions of the follicles called the germinal centers (GCs). Such dynamic mobilization of B cells was evident in the vaccinated mice when compared to control mice (Figure 7).88 The development of GCs in the B cell follicles of lymph nodes is also a clear outcome of T cell-dependent antibody responses.89 The initial events of GCs development and organization triggered by B cells essentially requires activation at the T cell rich MZ by the interdigitating dendritic cells and T helper cells.90 Follicular DCs (FDCs) in the light zones are expected to capture and retain S antigen via complement and Fc receptors and are capable of prolonged presentation to B cells in the GCs.91, 92 B cells acquire antigens from FDCs that are processed and presented on class II MHC molecules within the GC. The activated B cells exit from the GC to become plasmablasts, short-lived antibody producing cells. The results shown in Figure 7 illustrate the migration of B cells from the GC into the light zone as evidence for the movement of antibody producing plasmablasts in response to the SC2 DNA vaccine. The number of T follicular helper cells in lymph nodes directly correlates with the amount of antigen accumulated and GC B cells that develop in immunized lymph nodes.93 Thus, the efficiency of S antigen delivery to follicular DCs will also affect responses to immunization. The FDCs are located along with B cells in the follicles of any secondary lymphoid organs.94 FDCs play a major role in the generation and the selection of high affinity plasmacytes, i.e., memory B lymphocytes, during the adaptive immune response.95 The trapping and display of antigens as immune complexes is the key property of FDCs in a highly stimulatory way to proliferating B cells. FDCs are “antigen retaining reticular cells” that are present within the stromal network of cells in lymph nodes.96 Subsequently, FDCs have been recognized for their selective ability in retaining antigens for prolonged periods. This attribute of FDCs plays a vital role in GC formation and long-term immune memory.97 The histology of lymph nodes from SC2 vaccinated mice indicates the presence of such FDCs within the B cell follicles (BCFs) where GCs develop from the T cell-dependent antibody response. The activated T cells and APCs drain along the nasal lymphatic pathways and eventually access the cervical lymph nodes.98 Hence, we observed a complete remodeling of cervical nodes, with B cells centered on GCs and the presence of DCs in close proximity to B cells in these GCs (Figure 7). Overall, our results confirm that the S antigen expressed by the delivery of DNA vaccine using AuNS-chitosan shows an effective cell-mediated immunity in the lymph nodes.
Considering the fact that IFNγ play a major role in recognizing and eliminating pathogens, it has been identified as a prognostic marker for vaccine response.99 Type I IFNs are pleiotropic antiviral cytokines that control nearly every step of the immune response to SC2 vaccination, ranging from S protein expression, DC activation, to T cell differentiation. Unsurprisingly, type I IFNs have been found to be central mediators of T and B cell responses to SC2 vaccines. We also noticed an increased expression of INFγ by T cells in the lymph nodes and blood (Figure S7).100 The increase in splenic CD8+ T cells expressing IFNγ, which is a signature cytokine of both innate and adaptive immune systems, was evident only in SC2 vaccinated mice but not the pcDNA-control vector treated mice.
The results discussed in the preceding sections establish that AuNS-chitosan can robustly deliver sufficient amount of DNA into the nucleus for S protein expression. This offers other opportunities for a future strategy using mRNA alone, which requires gaining access only to the cytosol for S protein translation, and may therefore improve on the currently reported outcomes using S DNA. To verify if this nanocarrier can be used for IN delivery of a similar mRNA vaccine, we investigated the IN-delivery efficacy using FLuc mRNA as a surrogate for S mRNA.
AuNS-chitosan showed a robust delivery of FLuc mRNA in the lungs of mice upon IN delivery as measured using bioluminescence imaging.
As the IN delivery of DNA vaccine was successful in achieving significant expression and in inducing a pulmonary immune response, we further evaluated the stability and expression of mRNA delivered using AuNS-chitosan in cells, and upon IN delivery in mice using BLI. We used mRNA coding for FLuc reporter gene for this proof-of principle study component. The in vitro results in HEK-293 and A549 cells using the optimal N/P ratio complex transfected with different mRNA concentrations (50, 100, and 200 ng) demonstrated a dose-dependent luciferase expression in both cell types (Figure 8a–b). We used the optimal AuNS-chitosan-FLuc-mRNA complex for IN delivery in mice; 5 μL of NP complex four times in each nostril (a total of 20 μL for each dose; 2 μg of mRNA equivalent). We used 3 mice each for the treatments and controls. We delivered AuNS-chitosan-mRNA, obtained BLI every 24 h after delivery, and continued the dosage every day for three days. Three days after the final dose, the mice were imaged for in vivo bioluminescence signal, and were then sacrificed for ex vivo biodistribution in tissues. The mice revealed strong BLI signals in the lungs (Figure 8c). Ex vivo analysis (lungs, spleen, liver, trachea, and kidneys) showed strong BLI signals in lungs and tracheobronchial junctions. Signal was absent from other organs, including spleen (Figure 8d). These findings clearly supported the efficiency of AuNS-chitosan for in vitro and in vivo delivery of synthetic mRNA, and also the stability and functional efficiency of mRNA for in vivo applications. In the future, we will extend this same strategy to create and evaluate a similar IN administered anti-SC2 mRNA vaccine, and will conduct comparative studies of these two nanotechnologies prior to clinical translation.
Figure 8.
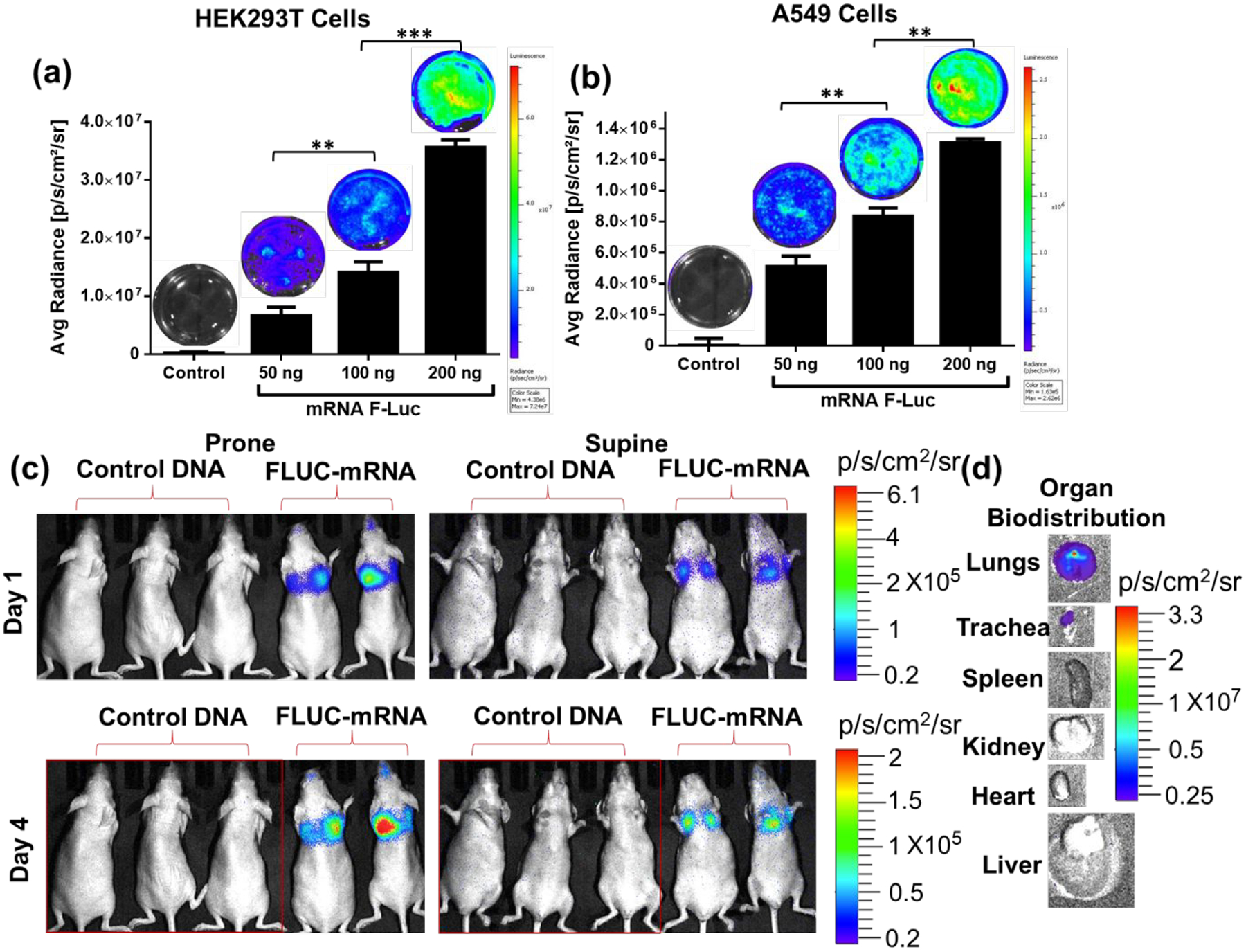
In vitro delivery of Fluc mRNA using AuNS-chitosan in (a) HEK293 and (b) A549 and cells imaged by optical bioluminescence (BLI). (c) In vivo BLI, and (d) ex vivo BLI of tissues after two doses of AuNS-chitosan-FLuc-mRNA delivery. There is significant expression of Firefly luciferase in the lungs and this is supported by the ex vivo tissue imaging findings (n=3 for control group, n=2 for AuNS-chitosan-FLuc-mRNA group-3).
The S protein vaccine used currently for human applications shows strong neutralizing antibody effects to a wide range of SC2 variants. Infection of vaccinated individuals by some of the escape mutants may occur in part owing to the enhanced infectivity of the virus or failure of the individuals to produce sufficient amounts of antibody response to previous vaccination. Recently it has been shown that the murine CTL epitope for SC2-specific CD8+ T cells upon vaccination is different from the human CTL epitopes.101 With the consideration of such differences, it is possible that mutations in the RBD domain of Wuhan, Beta and D614G variant would be expected to induce minimal differences in antibodies, whereas this might not be the case in humans; this difference might account for some variations in immunity induced in humans against the mutant strains.
CONCLUSIONS
We evaluated the potential advantages of IN delivery of a SC2 vaccine using DNA coding for its S protein as antigen. The delivery of DNA vaccine using AuNS-chitosan NPs yields successful expression of this antigen in respiratory mucosa and lungs of mice, which leads to recruitment of antigen presenting DCs to the lungs and an enhanced humoral antibody response. The antibody response develops as early as 1 week after the IN delivery of three doses of DNA vaccine. The antibody levels were consistently elevated for several weeks without a significant decline, as demonstrated in two different mouse models (BALB/c and C57BL/6J-DR). The antibody response results in high levels of IgG and IgA, which show a strong neutralizing effect against pseudoviruses expressing different spike variants of SC2 (Wuhan, D614G and Beta mutant). Additional evaluation using immunostaining-based FACS and confocal microscopy for cell-mediated immune response shows an effective activation of T and B cell responses in the lungs and lymph nodes, which are similar to immune responses normally observed against infectious diseases. Our findings highlight the merits of using AuNS-chitosan as an efficient in vitro and in vivo nanoformulation to deliver DNA and synthetic mRNA, and also its role in stabilizing nucleic acids for functional in vivo transfection for future mRNA vaccine development and applications. This proof-of-principle study highlights the capabilities of this IN SC2 DNA vaccine to yield a strong mucosal immune response, and to also provide a roadmap for the future use of mRNA vaccines coding for different antigens of SC2 (N, E, M and S proteins). This may result in a long-lasting, wide spectrum antibody response to combat the large number of SC2 variants distributed around the world, and which are continuously evolving.
MATERIALS AND METHODS
Materials
We purchased TritonX-100, tetrachloroauric acid, silver nitrate (AgNO3), sodium borohydride (NaBH4), ascorbic acid (AA), L(+)-ascorbic acid (AA), gold(III) chloride trihydrate (HAuCl4·3H2O), trisodium citrate dihydrate, phosphate buffered saline (PBS) 1N hydrochloric acid solution (HCl) and chitosan from Sigma-Aldrich (St Louis, MO). All the chemicals used in this study were of the highest grades. We used double-distilled water in all preparations. Carbon-coated copper TEM grids were obtained from VWR (Radnor, PA). We pretreated all the glassware used in this study for the synthesis of gold NPs using aqua regia for 30 min. We then washed several times using double-distilled water in an ultrasonication (3 min each) condition.
Methods
Synthesis of AuNS.
We synthesized gold nanostar octapods using a modified seed assisted growth of gold nanostars synthesis procedure, as reported previously.39 In brief, gold nanoseeds were generated from AuCl4− by means of NaBH4, in 0.15 M TritonX-100 and the resultant colloidal gold was used as seeds for nanostar growth. 5 μL of seed gold NPs were added to 5 mL of an aqueous solution containing 0.1 M TritonX-100 and 250 μL of 0.004 M AgNO3 in water. To the resulting mixture, 5 mL of HAuCl4 (0.001 M) was supplemented and stirred for 30 min at 70 °C. After solubilization of all the components into a homogenous solution, we lowered the temperature to 37 °C and added 400 μL of 0.0788 M ascorbic acid in an aqueous solution. We stirred the aqueous solution for 30 min and gradually added 0.6 mL of a previously ice-cooled solution of 0.001 M NaBH4 solution dropwise. The reaction mixture was stirred at 1000 rpm, which resulted in a homogenous brownish-red suspension of NPs that subsequently changed to intense green color. After the color change, the NPs growth was stopped by reducing the temperature down to ~4 °C. The resultant gold nanostars were separated by centrifugation at 13,000 rpm for 30 min and washed thrice with distilled water before using them for further in vitro and in vivo studies.
Formulation of SC2 plasmid or pcDNA loaded AuNS.
The chitosan dissolved in 0.2% acetic acid was microfluidized using a LV1-microfluidics system (Microfluidics, Westwood, MA) at 30,000 psi. We extracted the suspension of chitosan at the outlet at a 0.5 mg/mL concentration and used this for coating the AuNS. The AuNS synthesized earlier were dispersed by probe sonication (5 s “On”, and 5s “Off” for 1 min at 40% amplitude) in sterile distilled water at a concentration of 5 mg/mL. The uniformly suspended AuNSs were then incubated with the solubilized chitosan 400 μL (stock - 5 mg/mL) and processed through microfluidic system at 30,000 psi. The collected chitosan coated AuNS-CS were concentrated by centrifuging at 13,000 rpm for 30 min. We pooled the pellets together in 600 μL of sterile double-distilled water and used for further experiments. We used a stock SC2 plasmid (2 μg) diluted to 200 ng/μL in DEPC water and complexed with increasing amount of chitosan capped AuNS, and then incubated at 37 °C for 15 min. The complexes were resolved in 0.7% agarose gel by electrophoresing at 40 V for 45 min. The gel was imaged in BioRad Gel Doc XR+ Gel Documentation system (Bio Rad, Hercules, CA, USA) to further quantify and analyze the extent of DNA encapsulation by checking the gel mobility pattern. The optimized SC2 plasmid or pcDNA loaded AuNS-chitosan was adapted for subsequent in vitro and in vivo studies. The plasmid DNA complexed with the AuNS-chitosan in 20 μL volume was administered at each time point of the study.
Nanoparticle characterization.
The AuNS size before and after surface modification with chitosan (AuNS-CS) was characterized for mean hydrodynamic diameters and zeta potential measurements using a Malvern Zetasizer Nano Z system at 25 °C with a scattering angle of 90°. We determined the ζ-potential (surface charge) of the AuNS based NPs using Smoluchowski approximation. The chitosan capped AuNS prepared in PBS was diluted with deionized water to accurately measure surface charge of NPs low ionic strength. We characterized the particle size and morphology of the AuNS-CS loaded with SC2 plasmid using transmission electron microscopy (TEM, FEI-Tecnai G2 F20 X-TWIN) equipped with an ORIUS CCD camera through digital micrography. For sample preparation, 10 μL of AuNS-CS loaded with SC2 plasmid were drop casted on glow discharged carbon coated copper grids by incubating for 10–15 min. We washed the grids briefly using ultrapure water and used for imaging.
In vitro evaluation of AuNS-chitosan mediated DNA transfection in cells.
We estimated cellular uptake and FLuc plasmid delivery by AuNS-chitosan quantitatively using BLI. The A549 cells were treated with FLuc EGFP (1 μg) plasmid loaded on AuNS-chitosan. At 48 h we imaged the treated cells for bioluminescence signal using an IVIS Lumina-III -In Vivo -Imaging System in the presence of D-Luc (150 μg/mL) substrate. The bioluminescence signal was quantified for all treatment conditions to draw clear correlations.
Mice strains and immunizations.
We purchased 6–8 weeks old BALB/c female mice, from Charles River Laboratories (Wilmington, MA); and C57BL/6J mice, as well as mice carrying the Ccr2RFPCx3cr1GFP dual-reporter from the Jackson Laboratory (Bar Harbor, ME). Mice were maintained under specific pathogen-free conditions. All animal experiments were performed under the guidance of the Administrative Panel on Laboratory Animal Care (APLAC) of Stanford University. We immunized mice IN with 20 μg of SC2 DNA or pcDNA vaccine in solution. We performed the IN delivery of the NP formulation in mice under mild gas anesthesia (0.5% isoflurane in O2), in an induction chamber with oxygen flowmeter set at 0.8–1.5 L O2/min. Once a steady breathing rate was established, 20 μL of the nanoformulation were administered IN by applying 5 μL/drop dose over 2–5 min time frame. We carefully monitored to ensure complete inhalation of the IN dose as well as recovery of steady state breathing. We also monitored the nostrils for any indications of blockage, irritation or bleeding. After completion of the 20 μL dose, each animal was allowed to recover from anesthesia and then transferred to its cage.
Serum neutralization assay with pseudotyped lentivirus.
Virus neutralization assay is a commonly used technique to assess patient antibody levels in many viral diseases. We developed a pseudovirus based neutralization assay to easily and rapidly measure neutralizing antibodies titer in serum and plasma. The SC2 spike D614G pseudotyped lentiviruses were produced using SC2 Spike (Genbank Accession #QHD43416.1; with D614G mutation) as the envelope glycoproteins instead of the commonly used VSV-G. These pseudovirions contain the FLuc gene driven by a CMV promoter; therefore, the spike-mediated cell entry can be conveniently determined via FLuc imaging. The SC2 Spike D614G pseudotyped lentivirus can be used to measure the activity of neutralizing antibody against SC2 in a Biosafety Level 2 facility. A variant called B.1.351 was first identified in the fall of 2020 in the Republic of South Africa. This South African variant, also known as 501Y.V2 or Beta, has many mutations which may lead to higher transmissibility and infectivity. The Spike (B.1.351 Variant) (SC2) pseudotyped lentiviruses were produced using SC2 B.1.351 Variant Spike (Genbank Accession #QHD43416.1 with B.1.351 mutations; as the envelope glycoproteins instead of the commonly used VSV-G.
We seeded HEK293T-ACE2 cells in 96-well plates at 5 × 104 cells per well the day prior to infection. We seeded HEK293FT and HEK293T-ACE2 cells in 96-well plates (ThermoFisher, US) the day before infection. We added pseudotyped viruses to the pre-cultured cells. We cultured cells at 37 °C with 5% CO2 for 2 days. All cells in each well were assayed for luciferase expression levels in the presence of D-Luc substrate. After validating the ACE2 receptor specific transfection of pseudovirus, we evaluated its infectivity in the presence of animal serum. We serially diluted sera in 50 μL using serum free DMEM with P&S as diluent, and pre-incubated with 50 μL of pseudotyped viruses at 37 °C for 1 h. For the positive control infections, virus stocks in similar dilution without any neutralizing serum were used as control. We used a validated neutralizing antibody from a commercial source (SARS-CoV/SARS-CoV-2 Spike antibody, Chimeric Mab [SinoBiological]) as a positive control, while serum collected from control DNA treated mice was used as a negative control. We incubated the serum/Ab in different dilutions with pseudovirus (5×106 viral particles) in 50 μL serum free medium for 1 h and added to cells (0.5 ×104 cells/well in 96-well plates) by mixing with 50 μL of 2X medium. We incubated the cells further for 60 h and used for Luciferase BLI after addition of 100 μg/mL D-Luciferin (D-Luc) using an IVIS Lumina imaging system. We plotted neutralization titers using Prism 8 (GraphPad, US).
Measurement of SPK-specific IgG, IgM and IgA antibodies in peripheral blood.
We drew blood by submandibular vein puncture from each mouse. The time point of blood collection was adopted as indicated in schematic workflow in Figure 2(a). The blood was collected in a Push Cap Micro Tube 1.1 ml Serum-Gel Polypropylene tube, and the serum was separated out to estimate SC2 specific IgA, IgG and IgM levels by enzyme-linked immunosorbent assay (ELISA). Briefly, we coated 96-well plates (Maxisorp, Nunc) with 50 μL of 5 μg/mL SPK peptide in phosphate buffered saline (PBS) overnight at 4 °C. We blocked the plates using 100 μL of 2% bovine serum albumin in PBS for 2 h at room temperature. We diluted serum from individual mice to 1:1600 or 1:400 and added them to wells, and incubated for 1 h at room temperature. After the incubation time, the wells were briefly washed three times with PBS-T (PBS with 0.05% Tween 20), and incubated with secondary antibodies, i.e. rabbit anti-mouse IgG, IgA or IgM (1:2000) conjugated to horseradish peroxidase (Serotec, Oxford, UK) for 1 h at room temperature. At the end of incubation time, the plates were washed three times using 300 μL of PBS-T. The plates were added with 50 μL of substrate TMB (3,3′,5,5′-Tetramethylbenzidine)/H2O2 (BD Biosciences, San Jose, CA, USA) and incubated in the dark for 20 min at room temperature. The reaction was stopped by adding 50 μL of 2N H2SO4. The plates were read at OD450 nm using Tecan Spectrophotometer, and antibody titers were expressed as mean absorbance ± standard deviation (SD).
Immunoblot analysis.
To determine the expression of SPK protein in transfected cells and screen the specificity of SC2 antibody against mutant variants of SC2 with respect to purified SPK proteins, we performed immunoblot analysis using anti-SC2 SPK antibody. We seeded 3×106 HEK293 cells in 10 cm well plates and transfected with 10 μg pDNA (pcDNA, SC2-Wuhan, SC2-Beta-mutant, and SC2-D614G-mutant) using lipofectamine 3000 reagent (Thermo Fisher Scientific) and after 48 h treatment, we harvested and processed cells further for immunoblot analysis using anti-SPK antibody as outlined in our earlier work.39
Histology of lungs, spleen, and lymph nodes.
We performed cardiac perfusion of animals under deep anesthesia to harvest the organs for histology. Briefly, under anesthesia we dissected each mouse below the diaphragm and cut the rib cage to expose the heart. We made a left ventriculotomy and inserted a needle into the aorta and clamped, and then cut the right atrium to allow flow. Each animal was transcardially perfused using 30 mL PBS for 4–5 min or until the liver was cleared of blood. Next, to preserve tissue morphology and retain the antigenicity of the target molecules, we perfused the animal with 30 mL 4% paraformaldehyde for 4 min. Following aldehyde fixation, we harvested the tissues (spleen, lymph nodes, and lungs), transferred into 30% sucrose in PBS for overnight equilibration, and then processed for OCT embedding. The OCT blocks were sectioned at 5 μm thick tissue sections using a cryostat and thaw-mounted onto gelatin-coated histological slides. We then dried the slides for 30 min at 37 °C and rehydrated in a wash buffer for 10 min. The tissues were blocked using 1% bovine serum albumin in PBS for 30 min at room temperature and then incubated with antigen specific anti-mouse fluorophore tagged antibody (CD4- FITC, CD8-Alexa-700, CD19 Alexa-700) and incubated overnight at 2–8 °C. After the incubation time, we washed the antibodies three times for 15 minutes in wash buffer. We then incubated the slides in 300 μL of the diluted solution of Hoechst 33342 and incubated for 5 min at room temperature. The slide were finally rinsed once with PBS and mounted with an anti-fade mounting media and visualized using a Leica DMi8 confocal microscope under respective filters.
Flow cytometry immunophenotyping:
We performed cell surface marker based immune cell analysis using flow cytometry for lungs, spleen, lymph nodes, thymus, and blood samples. Briefly, we prepared single-cell suspensions from tissues using mechanical dissociation, and red blood cells were removed using ACK lysing buffer. After the final wash, we filtered cells through a 70-μm cell strainer and viability was checked using 0.1% trypan blue. One million cells were labeled with cell surface marker specific anti-mouse antibody labelled with fluorochrome, i.e., CD45–Pac-Blue, CD3/CD4/CD8 PE-CY7/FITC/Alexa-700, CD45/CD11b PacBlue/APC-Cy7, CD45/CD11c PacBlue/PE-Cy7, CD45/CD86 Pac Blue / PE, CD19 Alexa-700, CD22 Alexa-700 (Biolegend). Isotype antibodies were included for gating and compensation. Following addition of antibodies, we kept cells in the dark for 30 min. We washed cells using PBS and suspended in fresh PBS, then analyzed for 20,000 events using a Guava® easyCyte™ Flow Cytometer.
Statistical analyses:
We used GraphPad Prism 8 (version 8.0a; GraphPad Soft-ware, Inc., La Jolla, CA, USA) to plot all graphs and perform statistical analyses. We pooled data from 3–5 mice from independent experiments and presented results as mean ± standard deviation (SD) or standard errors of means (SEM), as indicated in the figure legends, and interquartile range between the first (25th percentile) and third (75th percentile) was adopted for analysis. We compared grouped data using two-tailed Students t-test and calculated multiple comparisons of grouped data. For correlation analysis between ELISA and neutralization assay titers, significance of p-values were calculated using Prism 9.0 (GraphPad). Differences were considered significant when p-values were less than 0.05. If the p-value is not indicated in respective figure legends, the level of significance are as follow: (*denotes 0.01<p<0.05, **denotes 0.001<p<0.01, ***denotes 0.0001<p<0.001, and ****denotes p<0.0001).
Supplementary Material
Acknowledgments
General:
The authors would like to thank Canary Center at Stanford, Department of Radiology for providing the facility and resources to conduct this research. The authors would also like to thank Stanford Animal Histology Services for preparation of histology. We would like to thank Prof. Max Wintermark and Dr. Yanrong Zhang for the help with engineered C57BL-6J-DR animals.
Funding:
This work in part was supported by the Gary Glazer-GE Fund (Department of Radiology, Stanford University). NIH S10OD023518-01A1 Award for the Celigo S Imaging Cytometer (200-BFFL-S) to RP is acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi.
The supporting figures S1–S7 provide the results of serum neutralization IC50 (serum dilution factor) for relative inhibition in infectivity of lentiviral particles, immunophenotypic FACS analysis of spleen, lungs, thymus, and lymph nodes, and H&E and immunohistochemistry of spleen and lung tissues for immune cells (NK cells, DC cells, T cells) by confocal microscopy.
Competing interests: The authors do not have any competing interests to declare.
REFERENCES
- 1.WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int. (March 13, 2021)
- 2.Harapan H; Itoh N; Yufika A; Winardi W; Keam S; Te H; Megawati D; Hayati Z; Wagner AL; Mudatsir M, Coronavirus Disease 2019 (COVID-19): A Literature Review. J Infect Public Health 2020, 13 (5), 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krammer F, SARS-CoV-2 Vaccines in Development. Nature 2020, 586 (7830), 516–527. [DOI] [PubMed] [Google Scholar]
- 4.Arashkia A; Jalilvand S; Mohajel N; Afchangi A; Azadmanesh K; Salehi-Vaziri M; Fazlalipour M; Pouriayevali MH; Jalali T; Mousavi Nasab SD; Roohvand F; Shoja Z; Iran, S. C.-R. R. T. o. P. I. o., Severe Acute Respiratory Syndrome-Coronavirus-2 Spike (S) Protein Based Vaccine Candidates: State of the Art and Future Prospects. Rev Med Virol 2020, e2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettini E; Locci M, SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond. Vaccines (Basel) 2021, 9 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO COVID-19 Vaccine Tracker and Landscape. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines. (March 13, 2021)
- 7.Schlake T; Thess A; Fotin-Mleczek M; Kallen KJ, Developing mRNA-Vaccine Technologies. RNA Biol 2012, 9 (11), 1319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassan AO; Kafai NM; Dmitriev IP; Fox JM; Smith BK; Harvey IB; Chen RE; Winkler ES; Wessel AW; Case JB; Kashentseva E; McCune BT; Bailey AL; Zhao H; VanBlargan LA; Dai YN; Ma M; Adams LJ; Shrihari S; Danis JE, et al. , A Single-Dose Intranasal ChAd Vaccine Protects Upper and Lower Respiratory Tracts against SARS-CoV-2. Cell 2020, 183 (1), 169–184 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groswasser J; Kahn A; Bouche B; Hanquinet S; Perlmuter N; Hessel L, Needle Length and Injection Technique for Efficient Intramuscular Vaccine Delivery in Infants and Children Evaluated through an Ultrasonographic Determination of Subcutaneous and Muscle Layer Thickness. Pediatrics 1997, 100 (3 Pt 1), 400–3. [DOI] [PubMed] [Google Scholar]
- 10.Poland GA; Borrud A; Jacobson RM; McDermott K; Wollan PC; Brakke D; Charboneau JW, Determination of Deltoid Fat Pad Thickness. Implications for Needle Length in Adult Immunization. JAMA 1997, 277 (21), 1709–11. [PubMed] [Google Scholar]
- 11.Shaw FE Jr.; Guess HA; Roets JM; Mohr FE; Coleman PJ; Mandel EJ; Roehm RR Jr.; Talley WS; Hadler SC, Effect of Anatomic Injection Site, Age and Smoking on the Immune Response to Hepatitis B Vaccination. Vaccine 1989, 7 (5), 425–30. [DOI] [PubMed] [Google Scholar]
- 12.Zuckerman JN, The Importance of Injecting Vaccines into Muscle. Different Patients Need Different Needle Sizes. BMJ 2000, 321 (7271), 1237–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baraniuk C, How Long Does Covid-19 Immunity Last? BMJ 2021, 373, n1605. [DOI] [PubMed] [Google Scholar]
- 14.CDC Interim Clinical Considerations for Use of COVID-19 Vaccines Currently Authorized in the United States. https://www.cdc.gov/vaccines/covid-19/clinical-considerations/covid-19-vaccines-us.html. (March 13, 2021)
- 15.Kim DS; Rowland-Jones S; Gea-Mallorqui E, Will SARS-CoV-2 Infection Elicit Long-Lasting Protective or Sterilising Immunity? Implications for Vaccine Strategies (2020). Front Immunol 2020, 11, 571481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward H; Cooke GS; Atchison C; Whitaker M; Elliott J; Moshe M; Brown JC; Flower B; Daunt A; Ainslie K; Ashby D; Donnelly CA; Riley S; Darzi A; Barclay W; Elliott P, Prevalence of Antibody Positivity to SARS-CoV-2 Following the First Peak of Infection in England: Serial Cross-Sectional Studies of 365,000 Adults. Lancet Reg Health Eur 2021, 4, 100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho W; Gao M; Li F; Li Z; Zhang XQ; Xu X, Next-Generation Vaccines: Nanoparticle-Mediated DNA and mRNA Delivery. Adv Healthc Mater 2021, 10 (8), e2001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chavda VP; Vora LK; Pandya AK; Patravale VB, Intranasal Vaccines for SARS-CoV-2: From Challenges to Potential in COVID-19 Management. Drug Discov Today 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King RG; Silva-Sanchez A; Peel JN; Botta D; Meza-Perez S; Allie R; Schultz MD; Liu M; Bradley JE; Qiu S; Yang G; Zhou F; Zumaquero E; Simpler TS; Mousseau B; Killian JT; Dean B; Shang Q; Tipper JL; Risley C, et al. , Single-Dose Intranasal Administration of AdCOVID Elicits Systemic and Mucosal Immunity Against SARS-CoV-2 in Mice. 2020, 331348, bioRxiv, https://www.biorxiv.org/content/10.1101/2020.10.10.331348v1 (March 13, 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suhail M; Rosenholm JM; Minhas MU; Badshah SF; Naeem A; Khan KU; Fahad M, Nanogels as Drug-Delivery Systems: a Comprehensive Overview. Ther Deliv 2019, 10 (11), 697–717. [DOI] [PubMed] [Google Scholar]
- 21.An X; Martinez-Paniagua M; Rezvan A; Fathi M; Singh S; Biswas S; Pourpak M; Yee C; Liu X; Varadarajan N, Single-Dose Intranasal Vaccination Elicits Systemic and Mucosal Immunity Against SARS-CoV-2. 2020, 212357, bioRxiv, https://www.biorxiv.org/content/10.1101/2020.07.23.212357v1 (December 16, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu C; Openshaw PJ, Antiviral B Cell and T Cell Immunity in the Lungs. Nat Immunol 2015, 16 (1), 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tay MZ; Poh CM; Renia L; MacAry PA; Ng LFP, The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat Rev Immunol 2020, 20 (6), 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holt PG, Regulation of Antigen-Presenting Cell function(s) in Lung and Airway Tissues. Eur Respir J 1993, 6 (1), 120–9. [PubMed] [Google Scholar]
- 25.Holt PG, Antigen Presentation in the Lung. Am J Respir Crit Care Med 2000, 162 (4 Pt 2), S151–6. [DOI] [PubMed] [Google Scholar]
- 26.Axel R, Transcription and Chromatin Subunit Structure. Adv Pathobiol 1976, (3), 27–41. [PubMed] [Google Scholar]
- 27.Chen L; Wang J; Zganiacz A; Xing Z, Single Intranasal Mucosal Mycobacterium Bovis BCG Vaccination Confers Improved Protection Compared to Subcutaneous Vaccination Against Pulmonary Tuberculosis. Infect Immun 2004, 72 (1), 238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandtzaeg P, Function of Mucosa-Associated Lymphoid Tissue in Antibody Formation. Immunol Invest 2010, 39 (4–5), 303–55. [DOI] [PubMed] [Google Scholar]
- 29.Neutra MR; Kozlowski PA, Mucosal Vaccines: The Promise and the Challenge. Nat Rev Immunol 2006, 6 (2), 148–58. [DOI] [PubMed] [Google Scholar]
- 30.Xu Y; Yuen PW; Lam JK, Intranasal DNA Vaccine for Protection against Respiratory Infectious Diseases: The Delivery Perspectives. Pharmaceutics 2014, 6 (3), 378–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis SS, Nasal Vaccines. Adv Drug Deliv Rev 2001, 51 (1–3), 21–42. [DOI] [PubMed] [Google Scholar]
- 32.Cain MD; Salimi H; Gong Y; Yang L; Hamilton SL; Heffernan JR; Hou J; Miller MJ; Klein RS, Virus Entry and Replication in the Brain Precedes Blood-Brain Barrier Disruption During Intranasal Alphavirus Infection. J Neuroimmunol 2017, 308, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prego C; Paolicelli P; Diaz B; Vicente S; Sanchez A; Gonzalez-Fernandez A; Alonso MJ, Chitosan-Based Nanoparticles for Improving Immunization against Hepatitis B Infection. Vaccine 2010, 28 (14), 2607–14. [DOI] [PubMed] [Google Scholar]
- 34.Mohammed MA; Syeda JTM; Wasan KM; Wasan EK, An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saenz L; Neira-Carrillo A; Paredes R; Cortes M; Bucarey S; Arias JL, Chitosan Formulations Improve the Immunogenicity of a GnRH-I Peptide-Based Vaccine. Int J Pharm 2009, 369 (1–2), 64–71. [DOI] [PubMed] [Google Scholar]
- 36.Kunda NK; Somavarapu S; Gordon SB; Hutcheon GA; Saleem IY, Nanocarriers Targeting Dendritic Cells for Pulmonary Vaccine Delivery. Pharm Res 2013, 30 (2), 325–41. [DOI] [PubMed] [Google Scholar]
- 37.Mohn KG; Smith I; Sjursen H; Cox RJ, Immune Responses After Live Attenuated Influenza Vaccination. Hum Vaccin Immunother 2018, 14 (3), 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao W; Ziemer KS; Gill HS, Gold Nanoparticle-M2e Conjugate Coformulated with CpG Induces Protective Immunity against Influenza A Virus. Nanomedicine (Lond) 2014, 9 (2), 237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sukumar UK; Bose RJC; Malhotra M; Babikir HA; Afjei R; Robinson E; Zeng Y; Chang E; Habte F; Sinclair R; Gambhir SS; Massoud TF; Paulmurugan R, Intranasal Delivery of Targeted Polyfunctional Gold-Iron Oxide Nanoparticles Loaded with Therapeutic MicroRNAs for Combined Theranostic Multimodality Imaging and Presensitization of Glioblastoma to Temozolomide. Biomaterials 2019, 218, 119342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bastus NG; Sanchez-Tillo E; Pujals S; Farrera C; Kogan MJ; Giralt E; Celada A; Lloberas J; Puntes V, Peptides Conjugated to Gold Nanoparticles Induce Macrophage Activation. Mol Immunol 2009, 46 (4), 743–8. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe K; Watanabe C; Honma T; Tian YS; Kawashima Y; Kawashita N; Takagi T; Fukuzawa K, Intermolecular Interaction Analyses on SARS-CoV-2 Spike Protein Receptor Binding Domain and Human Angiotensin-Converting Enzyme 2 Receptor-Blocking Antibody/Peptide Using Fragment Molecular Orbital Calculation. J Phys Chem Lett 2021, 12 (16), 4059–4066. [DOI] [PubMed] [Google Scholar]
- 42.Li L; Petrovsky N, Molecular Mechanisms for Enhanced DNA Vaccine Immunogenicity. Expert Rev Vaccines 2016, 15 (3), 313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh M; Briones M; O’Hagan DT, A Novel Bioadhesive Intranasal Delivery System for Inactivated Influenza Vaccines. J Control Release 2001, 70 (3), 267–76. [DOI] [PubMed] [Google Scholar]
- 44.Dutta A; Huang CT; Lin CY; Chen TC; Lin YC; Chang CS; He YC, Sterilizing Immunity to Influenza Virus Infection Requires Local Antigen-Specific T Cell Response in the Lungs. Sci Rep 2016, 6, 32973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y; Yu D; Han Y; Yan H; Chong H; Ren L; Wang J; Li T; He Y, Cross-Reactive Neutralization of SARS-CoV-2 by Serum Antibodies from Recovered SARS Patients and Immunized Animals. Sci Adv 2020, 6 (45). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rappazzo CG; Tse LV; Kaku CI; Wrapp D; Sakharkar M; Huang D; Deveau LM; Yockachonis TJ; Herbert AS; Battles MB; O’Brien CM; Brown ME; Geoghegan JC; Belk J; Peng L; Yang L; Hou Y; Scobey TD; Burton DR; Nemazee D, et al. , Broad and Potent Activity against SARS-like Viruses by an Engineered Human Monoclonal Antibody. Science 2021, 371 (6531), 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan CW; Chia WN; Young BE; Zhu F; Lim BL; Sia WR; Thein TL; Chen MI; Leo YS; Lye DC; Wang LF, Pan-Sarbecovirus Neutralizing Antibodies in BNT162b2-Immunized SARS-CoV-1 Survivors. N Engl J Med 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bleul T; Zhuang X; Hildebrand A; Lange C; Bohringer D; Schlunck G; Reinhard T; Lapp T, Different Innate Immune Responses in BALB/c and C57BL/6 Strains Following Corneal Transplantation. J Innate Immun 2021, 13 (1), 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahler M; Janke C; Wagner S; Hedrich HJ, Differential Susceptibility of Inbred Mouse Strains to Helicobacter Pylori Infection. Scand J Gastroenterol 2002, 37 (3), 267–78. [DOI] [PubMed] [Google Scholar]
- 50.Skums P; Kirpich A; Icer Baykal P; Zelikovsky A; Chowell G, Global Transmission Network of SARS-CoV-2: From Outbreak to Pandemic. medRxiv 2020. [Google Scholar]
- 51.Chen RE; Zhang X; Case JB; Winkler ES; Liu Y; VanBlargan LA; Liu J; Errico JM; Xie X; Suryadevara N; Gilchuk P; Zost SJ; Tahan S; Droit L; Turner JS; Kim W; Schmitz AJ; Thapa M; Wang D; Boon ACM, et al. , Resistance of SARS-CoV-2 Variants to Neutralization by Monoclonal and Serum-Derived Polyclonal Antibodies. Nat Med 2021, 27 (4), 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stamatatos L; Czartoski J; Wan YH; Homad LJ; Rubin V; Glantz H; Neradilek M; Seydoux E; Jennewein MF; MacCamy AJ; Feng J; Mize G; De Rosa SC; Finzi A; Lemos MP; Cohen KW; Moodie Z; McElrath MJ; McGuire AT, mRNA Vaccination Boosts Cross-Variant Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Science 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shang J; Wan Y; Luo C; Ye G; Geng Q; Auerbach A; Li F, Cell Entry Mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020, 117 (21), 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhakal S; Renu S; Ghimire S; Shaan Lakshmanappa Y; Hogshead BT; Feliciano-Ruiz N; Lu F; HogenEsch H; Krakowka S; Lee CW; Renukaradhya GJ, Mucosal Immunity and Protective Efficacy of Intranasal Inactivated Influenza Vaccine Is Improved by Chitosan Nanoparticle Delivery in Pigs. Front Immunol 2018, 9, 934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wisnewski AV; Campillo Luna J; Redlich CA, Human IgG and IgA Responses to COVID-19 mRNA Vaccines. PLoS One 2021, 16 (6), e0249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sterlin D; Mathian A; Miyara M; Mohr A; Anna F; Claer L; Quentric P; Fadlallah J; Devilliers H; Ghillani P; Gunn C; Hockett R; Mudumba S; Guihot A; Luyt CE; Mayaux J; Beurton A; Fourati S; Bruel T; Schwartz O, et al. , IgA Dominates the Early Neutralizing Antibody Response to SARS-CoV-2. Sci Transl Med 2021, 13 (577). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ejemel M; Li Q; Hou S; Schiller ZA; Tree JA; Wallace A; Amcheslavsky A; Kurt Yilmaz N; Buttigieg KR; Elmore MJ; Godwin K; Coombes N; Toomey JR; Schneider R; Ramchetty AS; Close BJ; Chen DY; Conway HL; Saeed M; Ganesa C, et al. , A Cross-Reactive Human IgA Monoclonal Antibody Blocks SARS-CoV-2 Spike-ACE2 Interaction. Nat Commun 2020, 11 (1), 4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ejemel M; Li Q; Hou S; Schiller ZA; Wallace AL; Amcheslavsky A; Yilmaz NK; Toomey JR; Schneider R; Close BJ; Chen DY; Conway HL; Mohsan S; Cavacini LA; Klempner MS; Schiffer CA; Wang Y, IgA MAb Blocks SARS-CoV-2 Spike-ACE2 Interaction Providing Mucosal Immunity. bioRxiv 2020. [Google Scholar]
- 59.Russell MW; Moldoveanu Z; Ogra PL; Mestecky J, Mucosal Immunity in COVID-19: A Neglected but Critical Aspect of SARS-CoV-2 Infection. Front Immunol 2020, 11, 611337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marklund E; Leach S; Axelsson H; Nystrom K; Norder H; Bemark M; Angeletti D; Lundgren A; Nilsson S; Andersson LM; Yilmaz A; Lindh M; Liljeqvist JA; Gisslen M, Serum-IgG Responses to SARS-CoV-2 After Mild and Severe COVID-19 Infection and Analysis of IgG Non-Responders. PLoS One 2020, 15 (10), e0241104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Q; Wu J; Nie J; Zhang L; Hao H; Liu S; Zhao C; Zhang Q; Liu H; Nie L; Qin H; Wang M; Lu Q; Li X; Sun Q; Liu J; Zhang L; Li X; Huang W; Wang Y, The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182 (5), 1284–1294 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shah VK; Firmal P; Alam A; Ganguly D; Chattopadhyay S, Overview of Immune Response during SARS-CoV-2 Infection: Lessons from the Past. Front Immunol 2020, 11, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu WJ; Zhao M; Liu K; Xu K; Wong G; Tan W; Gao GF, T-Cell Immunity of SARS-CoV: Implications for Vaccine Development against MERS-CoV. Antiviral Res 2017, 137, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kopf M; Schneider C; Nobs SP, The Development and Function of Lung-Resident Macrophages and Dendritic Cells. Nat Immunol 2015, 16 (1), 36–44. [DOI] [PubMed] [Google Scholar]
- 65.Martin TR; Frevert CW, Innate Immunity in the Lungs. Proc Am Thorac Soc 2005, 2 (5), 403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar V, Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front Immunol 2020, 11, 1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lambrecht BN; Prins JB; Hoogsteden HC, Lung Dendritic Cells and Host Immunity to Infection. Eur Respir J 2001, 18 (4), 692–704. [PubMed] [Google Scholar]
- 68.Martin-Fontecha A; Lanzavecchia A; Sallusto F, Dendritic Cell Migration to Peripheral Lymph Nodes. Handb Exp Pharmacol 2009, (188), 31–49. [DOI] [PubMed] [Google Scholar]
- 69.Al-Halifa S; Gauthier L; Arpin D; Bourgault S; Archambault D, Nanoparticle-Based Vaccines Against Respiratory Viruses. Front Immunol 2019, 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fries CN; Curvino EJ; Chen JL; Permar SR; Fouda GG; Collier JH, Advances in Nanomaterial Vaccine Strategies to Address Infectious Diseases Impacting Global Health. Nat Nanotechnol 2021, 16 (4), 1–14. [DOI] [PubMed] [Google Scholar]
- 71.Nakano H; Burgents JE; Nakano K; Whitehead GS; Cheong C; Bortner CD; Cook DN, Migratory Properties of Pulmonary Dendritic Cells Are Determined by Their Developmental Lineage. Mucosal Immunol 2013, 6 (4), 678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flaherty S; Reynolds JM, Mouse Naive CD4+ T Cell Isolation and in Vitro Differentiation into T Cell Subsets. J Vis Exp 2015, (98). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Odak I; Barros-Martins J; Bosnjak B; Stahl K; David S; Wiesner O; Busch M; Hoeper MM; Pink I; Welte T; Cornberg M; Stoll M; Goudeva L; Blasczyk R; Ganser A; Prinz I; Forster R; Koenecke C; Schultze-Florey CR, Reappearance of Effector T Cells is Associated with Recovery from COVID-19. EBioMedicine 2020, 57, 102885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bousso P, T-Cell Activation by Dendritic Cells in the Lymph Node: Lessons from the Movies. Nat Rev Immunol 2008, 8 (9), 675–84. [DOI] [PubMed] [Google Scholar]
- 75.Mule JJ, Dendritic Cells: At the Clinical Crossroads. J Clin Invest 2000, 105 (6), 707–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weller S; Reynaud CA; Weill JC, Splenic Marginal Zone B Cells in Humans: Where Do They Mutate Their Ig Receptor? Eur J Immunol 2005, 35 (10), 2789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fayette J; Durand I; Bridon JM; Arpin C; Dubois B; Caux C; Liu YJ; Banchereau J; Briere F, Dendritic Cells Enhance the Differentiation of Naive B Cells into Plasma Cells in Vitro. Scand J Immunol 1998, 48 (6), 563–70. [DOI] [PubMed] [Google Scholar]
- 78.Wykes M; Pombo A; Jenkins C; MacPherson GG, Dendritic Cells Interact Directly with Naive B Lymphocytes to Transfer Antigen and Initiate Class Switching in a Primary T-Dependent Response. J Immunol 1998, 161 (3), 1313–9. [PubMed] [Google Scholar]
- 79.Veninga H; Borg EG; Vreeman K; Taylor PR; Kalay H; van Kooyk Y; Kraal G; Martinez-Pomares L; den Haan JM, Antigen Targeting Reveals Splenic CD169+ Macrophages as Promoters of Germinal Center B-Cell Responses. Eur J Immunol 2015, 45 (3), 747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tozuka M; Oka T; Jounai N; Egawa G; Ishii KJ; Kabashima K; Takeshita F, Efficient Antigen Delivery to the Draining Lymph Nodes is a Key Component in the Immunogenic Pathway of the Intradermal Vaccine. J Dermatol Sci 2016, 82 (1), 38–45. [DOI] [PubMed] [Google Scholar]
- 81.Irvine DJ; Aung A; Silva M, Controlling Timing and Location in Vaccines. Adv Drug Deliv Rev 2020, 158, 91–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou X; Jiang X; Qu M; Aninwene GE 2nd; Jucaud V; Moon JJ; Gu Z; Sun W; Khademhosseini A, Engineering Antiviral Vaccines. ACS Nano 2020, 14 (10), 12370–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Faiq MA, B-Cell Engineering: A Promising Approach towards Vaccine Development for COVID-19. Med Hypotheses 2020, 144, 109948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Louie DAP; Liao S, Lymph Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front Immunol 2019, 10, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kwak K; Akkaya M; Pierce SK, B Cell Signaling in Context. Nat Immunol 2019, 20 (8), 963–969. [DOI] [PubMed] [Google Scholar]
- 86.Buettner M; Bode U, Lymph Node Dissection—Understanding the Immunological Function of Lymph Nodes. Clin Exp Immunol 2012, 169 (3), 205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mueller SN, Spreading the Load: Antigen Transfer between Migratory and Lymph Node-Resident Dendritic Cells Promotes T-Cell Priming. Eur J Immunol 2017, 47 (10), 1798–1801. [DOI] [PubMed] [Google Scholar]
- 88.Pal I; Ramsey JD, The Role of the Lymphatic System in Vaccine Trafficking and Immune Response. Adv Drug Deliv Rev 2011, 63 (10–11), 909–22. [DOI] [PubMed] [Google Scholar]
- 89.van der Poel CE; Bajic G; Macaulay CW; van den Broek T; Ellson CD; Bouma G; Victora GD; Degn SE; Carroll MC, Follicular Dendritic Cells Modulate Germinal Center B Cell Diversity through FcgammaRIIB. Cell Rep 2019, 29 (9), 2745–2755 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tarlinton DM, Immunology: To Affinity and Beyond. Nature 2014, 509 (7502), 573–4. [DOI] [PubMed] [Google Scholar]
- 91.Allen CD; Cyster JG, Follicular Dendritic Cell Networks of Primary Follicles and Germinal Centers: Phenotype and Function. Semin Immunol 2008, 20 (1), 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X; Cho B; Suzuki K; Xu Y; Green JA; An J; Cyster JG, Follicular Dendritic Cells Help Establish Follicle Identity and Promote B Cell Retention in Germinal Centers. J Exp Med 2011, 208 (12), 2497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tam HH; Melo MB; Kang M; Pelet JM; Ruda VM; Foley MH; Hu JK; Kumari S; Crampton J; Baldeon AD; Sanders RW; Moore JP; Crotty S; Langer R; Anderson DG; Chakraborty AK; Irvine DJ, Sustained Antigen Availability during Germinal Center Initiation Enhances Antibody Responses to Vaccination. Proc Natl Acad Sci U S A 2016, 113 (43), E6639–E6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heath WR; Kato Y; Steiner TM; Caminschi I, Antigen Presentation by Dendritic Cells for B Cell Activation. Curr Opin Immunol 2019, 58, 44–52. [DOI] [PubMed] [Google Scholar]
- 95.Kranich J; Krautler NJ, How Follicular Dendritic Cells Shape the B-Cell Antigenome. Front Immunol 2016, 7, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dave RS; Jain P; Byrareddy SN, Follicular Dendritic Cells of Lymph Nodes as Human Immunodeficiency Virus/Simian Immunodeficiency Virus Reservoirs and Insights on Cervical Lymph Node. Front Immunol 2018, 9, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hannum LG; Haberman AM; Anderson SM; Shlomchik MJ, Germinal Center Initiation, Variable Gene Region Hypermutation, and Mutant B Cell Selection without Detectable Immune Complexes on Follicular Dendritic Cells. J Exp Med 2000, 192 (7), 931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pichler WJ; Wyss-Coray T, T Cells as Antigen-Presenting Cells. Immunol Today 1994, 15 (7), 312–5. [DOI] [PubMed] [Google Scholar]
- 99.Takamura S, Persistence in Temporary Lung Niches: A Survival Strategy of Lung-Resident Memory CD8(+) T Cells. Viral Immunol 2017, 30 (6), 438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kak G; Raza M; Tiwari BK, Interferon-Gamma (IFN-Gamma): Exploring Its Implications in Infectious Diseases. Biomol Concepts 2018, 9 (1), 64–79. [DOI] [PubMed] [Google Scholar]
- 101.Yang J; Kim E; Lee JS; Poo H, A Murine CD8(+) T Cell Epitope Identified in the Receptor-Binding Domain of the SARS-CoV-2 Spike Protein. Vaccines (Basel) 2021, 9 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.