

Abstract
Chronic low-grade retinal inflammation is an essential contributor to the pathogenesis of diabetic retinopathy (DR). It is characterized by increased retinal cell expression and secretion of a variety of inflammatory cytokines; among these, IL-1β has the reputation of being a major driver of cytokine-induced inflammation. IL-1β and other cytokines drive inflammatory changes that cause damage to retinal cells, leading to the hallmark vascular lesions of DR; these include increased leukocyte adherence, vascular permeability, and capillary cell death. Nuclear factor of activated T-cells (NFAT) is a transcriptional regulator of inflammatory cytokines and adhesion molecules and is expressed in retinal cells. Consequently, it may influence multiple pathogenic steps early in DR. We investigated the NFAT-dependency of IL-1β-induced inflammation in human Müller cells (hMC) and human retinal microvascular endothelial cells (hRMEC). Our results show that an NFAT inhibitor, Inhibitor of NFAT-Calcineurin Association-6 (INCA-6), decreased IL-1β-induced expression of IL-1β and TNFα in hMC, while having no effect on VEGF, CCL2, or CCL5 expression. We also demonstrate that INCA-6 attenuated IL-1β-induced increases of IL-1β, TNFα, IL-6, CCL2, and CCL5 (inflammatory cytokines and chemokines), and ICAM-1 and E-selectin (leukocyte adhesion molecules) expression in hRMEC. INCA-6 similarly inhibited IL-1β-induced increases in leukocyte adhesion in both hRMEC monolayers in vitro and an acute model of retinal inflammation in vivo. Finally, INCA-6 rescued IL-1β-induced permeability in both hRMEC monolayers in vitro and an acute model of retinal inflammation in vivo. Taken together, these data demonstrate the potential of NFAT inhibition to mitigate retinal inflammation secondary to diabetes.
1. Introduction
Diabetic retinopathy (DR) is the leading cause of blindness in working-age Americans.1 DR has two clinically defined stages, the non-proliferative (NPDR) and the proliferative (PDR). Once a patient has reached the proliferative stage of DR, the retina has sustained irreparable damage which places complex demands on potential therapeutics. Therefore, the identification and blockade of therapeutic targets relevant to early NPDR could halt disease onset and progression, preempting PDR and its vision-threatening consequences.2-4 Current therapies that target vascular endothelial growth factor (VEGF) are invasive and only effective in about half of DR patients.5 These unsatisfactory outcomes have motivated the exploration of therapeutic alternatives, perhaps targeting other inflammatory mediators in addition to VEGF. Chronic low-grade inflammation is a component of early NPDR and, in recent years, its potential as a therapeutic target has received considerable attention. Elevated retinal or vitreous levels of cytokines, including IL-1β, TNFα, IL-8, IL-6, CCL2, and CCL5, have been observed early in DR in both animal models 6-10 and patients.5-7,11-18 These cytokines not only initiate retinal vascular damage but also activate autocrine and paracrine signaling cascades that promote chronic retinal inflammation, all of which contributes to the development of vision-threatening pathology.3,5 Numerous studies have identified IL-1β as a primary driver of inflammation in a wide-range of pathological conditions, including neuro-inflammatory diseases, diabetes, and DR.3,19-23 Increased levels of IL-1β are observed in the serum and vitreous of diabetics and, unlike VEGF, correlate closely with DR severity and progression.3,24,25 Abundant data suggest that IL-1β plays a causative role in several hallmark DR pathologies, including glial activation,26 cell death,27 vascular permeability,28 leukocyte adhesion,29,30 and vasoregression.27 Furthermore, inhibition of the IL-1β-activating enzyme caspase 1 reduced retinal IL-1β levels and retinal capillary regression in both STZ and galactose-fed mice; deletion of the IL-1β receptor IL-1R1 also protected against capillary loss in both models.31 We became particularly interested in IL-1β as a driver of retinal inflammation after RNA sequencing experiments comparing human Müller cell (hMC) responses to treatment with IL-1β, TNFα, IL-8, and IL-6 demonstrated that IL-1β caused the greatest and longest lasting inflammatory response in hMC (manuscript in preparation). Interestingly, these data also identified IL-1β itself as one of the most highly upregulated genes in IL-1β-treated hMC, supporting existing evidence that increased IL-1β levels via auto-amplification may exacerbate and sustain retinal inflammation in DR.23 In aggregate, this evidence suggests that IL-1β acts as a “master regulator” of inflammation in the pathological cascade of DR, and that preventing its downstream effects in retinal cells may therefore significantly slow inflammation-induced damage.19,23,32
IL-1β mediates downstream inflammatory signaling via the activation of several transcription factors, including nuclear factor of activated T-cells (NFAT).33,34 The NFAT family consists of five proteins, four of which (c1-c4) are regulated by the calcium-dependent phosphatase calcineurin (CN).35-37 In quiescent cells, NFAT exists in the cytosol in a hyperphosphorylated state. Upon challenge with an inflammatory stimulus, CN dephosphorylates NFAT, causing it to shuttle to the nucleus where it increases the transcription of inflammatory genes.38-40 The small molecule inhibitor, Inhibitor of NFAT-Calcineurin Association-6 (INCA-6), blocks CN-NFAT association, thereby preventing the dephosphorylation/activation of the four CN-dependent NFAT isoforms.41,42 NFAT is known to play a role in the regulation of inflammatory mediators, extracellular matrix proteins, vascular permeability, and adhesion molecules,35,38,43-54 and as such may control multiple pathogenic steps early in DR. Additionally, published evidence demonstrates that NFAT inhibition is a successful strategy in preventing pathogenic retinal cell behaviors downstream of both hyperglycemia and TNFα.43,50 Combined, these data demonstrate that NFAT mediates a variety of characteristic pathogenic changes that arise in response to multiple components of the diabetic environment; therefore, we hypothesized that NFAT may likewise influence the response of retinal cells to elevated IL-1β in the diabetic milieu.
The diabetic retina is a complex environment of multiple cell types, including endothelial cells, pericytes, astrocytes, and Müller cells.55 Pathogenic behaviors by each of these cell types do not exist in isolation, rather they can produce both autocrine and paracrine factors that potentiate disease progression in surrounding cells. For instance, VEGF, a major target of DR therapeutics, is secreted predominantly by Müller cells and subsequently causes downstream pathogenic responses in endothelial cells.4,56,57 Since multiple retinal cell types are involved in the DR cascade and cell-specific targeting is difficult in vivo, it would be ideal to identify a therapeutic target with the capacity to inhibit pathological responses in more than one retinal cell type. Therefore, in this study, we sought to investigate the role of NFAT in regulating key cell-specific responses to IL-1β, specifically Müller cell auto-amplification of IL-1β and endothelial cell inflammation, leukocyte adhesion, and permeability. We found that inhibition of NFAT not only attenuates Müller cell amplification of inflammation but also inhibits the downstream pathologic response of human retinal microvascular endothelial cells (hRMEC) to IL-1β challenge. These results suggest that targeting NFAT has significant therapeutic potential for slowing early inflammation and subsequent endothelial cell dysfunction in DR.
2. Methods
2.1. Human Müller cell (hMC) isolation and culture
Primary hMC were isolated from human donor tissue (NDRI) within 24 hours post-mortem, using an adapted protocol from previously developed methods.58 Briefly, the retina was dissected from the eye cup and dissociated in Dulbecco’s Modified Eagle Medium (DMEM; Gibco; Carlsbad, CA) supplemented with trypsin and collagenase. After incubation in dissociation medium, hMC were cultured in DMEM supplemented with 10% FBS (R&D Systems; Minneapolis, MN) and 1x antibiotic/antimycotic solution (Gibco). Passages 4-6 were used for all experiments. Cultures were incubated at 37 °C, 5% CO2, and 20.9% O2 and 95% relative humidity.
2.2. hMC qRT-PCR
hMC were cultured in 6-well plates until 70% confluence, then cultured in serum-reduced conditions (2% FBS) for 12 hours before treatment. hMC were treated in serum-reduced media containing 50pg/mL IL-1β (Sino Biological; Wayne, PA) with or without 1 or 2.5μM INCA-6 (Tocris; Minneapolis, MN) for 8 hours. Total RNA was collected using an RNeasy Mini kit (Qiagen; Valencia, CA), according to the manufacturer’s protocol. The High-Capacity cDNA Archive Kit (Applied Biosystems; Waltham, MA) was used to reverse transcribe the total RNA. Quantitative RT-PCR was performed using TaqMan Gene Expression Assays (Applied Biosystems). Using technical duplicates, the co-amplification of human IL1Β, TNFA, VEGF, CCL2, and CCL5 cDNA was compared with TBP (TATA-binding protein) as the normalization control. Data were analyzed using the comparative Ct method. Experiments were performed using a minimum of 3 biologically independent samples as well as technical replicates for each sample.
2.3. Human retinal microvascular endothelial cell (hRMEC) culture
Primary hRMEC (Cell Systems; Kirkland, WA) were cultured in phenol red-free endothelial basal medium (EBM; Cell Systems) supplemented with 10% FBS (R&D Systems) and SingleQuots (Lonza, Inc.; Allendale, NJ) and grown on attachment factor- (Cell Systems) coated culture dishes. Passages 5-8 were used for all experiments.
2.4. hRMEC qRT-PCR
hRMEC were plated in 6-well plates and grown in 10% EBM to 75-85% confluency before treatment. Cells were treated for 2 hours with 1ng/mL IL-1β in the presence or absence of 1, 2.5, or 5μM INCA-6 in serum-reduced media (2% FBS EBM supplemented with antibiotic). Subsequently, cells were lysed and qRT-PCR analysis for human IL1Β, TNFA, IL8, IL6, CCL2, CCL5, VCAM1, ICAM1, or SELE (E-selectin) expression was completed as described above for hMC. Experiments were performed using a minimum of 3 biologically independent samples as well as technical replicates for each sample.
2.5. Parallel plate flow chamber assay (PPFC)
hRMEC were plated onto attachment factor-coated glass slides (Thermo Fisher; Waltham, MA). Once cells formed a complete monolayer, slides were treated with 1ng/mL IL-1β with or without 5μM INCA-6 in serum-reduced media. After 4 hours, slides were used to assemble the PPFC (GlycoTech; Gaithersburg, MD) and leukocyte adhesion was assayed as previously described.43 Briefly, human peripheral blood mononuclear cells (PBMC; Precision for Medicine; Fredrick, MD) were resuspended at a concentration of 5 × 105 cells/mL in Hank’s Buffered Salt Solution (HBSS; Gibco). PBMC were flowed at a shear stress of 1 dyn/cm2 over treated monolayers for 7 minutes. Non-adherent cells were then washed away by flowing HBSS over the monolayers at 2 dyn/cm2 for 2 minutes. Eight fields per slide were randomly selected prior to PBMC flow and images were captured before and after flow. A masked observer compared the two sets of images to count adherent leukocytes in the after images. Each data point represents the average number of adherent leukocytes across the eight captured fields per slide divided by the count area (mm2).
2.6. Retinal leukostasis
All experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee and were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Six- to eight-week-old C57BL/6J mice (Charles River; Wilmington, MA) received intravitreal injections of vehicle, IL-1β (50pg), INCA-6 (150ng), or IL-1β (50pg) + INCA-6 (150ng). Retinal leukostasis was performed as described previously.43,59 Briefly, 12 hours after treatment mice were anesthetized with ketamine/xylazine and then perfused with 0.9% saline for 2.5 minutes followed by FITC-conjugated concanavalin-A (40μg/mL in 5.0mL PBS; Vector Laboratories; Burlingame, CA). Saline was then perfused for 2.5 minutes to remove any non-adherent leukocytes. Retinas were immediately dissected into 4% paraformaldehyde, flat-mounted, and images were captured with a Zeiss LSM710 Confocal Microscope (Zeiss; Pleasanton, CA) at 20x magnification. Using ImageJ (NIH; Bethesda, MD), a masked observer selected 4 regions per retina and adherent leukocytes in the superficial plexus of the retinal vasculature were counted. Each data point represents the average number of adherent leukocytes across the 4 regions divided by the count area (mm2).
2.7. Transendothelial electrical resistance (TEER) measurements
hRMEC were plated at a density of 40,000 cells/well in 96W10idf plates (Applied Biophysics; Troy, NY) in 10% FBS EBM. Per the manufacturer’s instructions, hRMEC were allowed to settle onto plates for 15 minutes at room temperature before insertion into the Electric Cell-substrate Impedance Sensing (ECIS) Z-Theta instrument (Applied Biophysics). After resistance plateaued between 24 and 48 hours, monolayers were treated with 1ng/mL IL-1β with or without 5μM INCA-6. Resistance was monitored for 24 hours after treatment and normalized resistance was calculated using Applied Biophysics software.60,61 Each well was normalized to its resistance 1 hour prior to treatment.
2.8. Quantitative fluorescein angiography (qFA)
Six- to eight-week-old C57BL/6J mice (Charles River) received intravitreal injections of vehicle, IL-1β (50pg), INCA-6 (150ng), or IL-1β (50pg) + INCA-6 (150ng). Eight hours after treatment, vascular permeability was assessed using qFA. Mice were anesthetized with an intraperitoneal injection of ketamine/xylazine and their pupils were dilated with tropicamide and phenylephrine before receiving an intraperitoneal injection of sodium fluorescein (NDC 17478-250-20; Akorn; Lake Forest, IL) at 10μL/g body weight. At 2 and 4 minutes after injection, fluorescent fundus images were captured with the Micron IV retinal imaging system (Phoenix Research Labs; Pleasanton, CA). A masked observer checked all images for quality, ensuring proper visualization of the vasculature was achieved in each. ImageJ software was then used to quantify the fluorescence intensity of the images using the integrated density function, an algorithm that measures the mean gray value in a selected area of the image and then multiplies it by the selected area. The integrated density was measured for both the 2 minute and 4 minute images. The integrated density at 2 minutes (baseline) was then subtracted from the integrated density at 4 minutes, with the difference in integrated density between 2 and 4 minutes employed as a readout of vascular leakage.
2.9. Statistical analysis
All data were analyzed with Prism software (GraphPad; La Jolla, CA). Analysis of variance (ANOVA) with Tukey’s multiple comparisons post hoc analysis was used; values of p < 0.05 were considered statistically significant. Grubbs’ test was utilized to identify outliers. For ECIS, two-way ANOVA with Tukey’s multiple comparisons post hoc analysis was used.
3. Results
3.1. INCA-6 decreases IL-1β-induced inflammatory cytokine expression in hMC
Based on Müller cells’ known role as major propagators of retinal inflammation,55,57,62,63 we first sought to determine if NFAT is involved in the regulation of hMC inflammatory responses in an autocrine and paracrine manner. IL-1β increased the expression of IL-1β (152-fold, p<0.0001), TNFα (86-fold, p<0.0001), VEGF (1.3-fold, p=0.0132), CCL2 (15-fold, p<0.0001), and CCL5 (31-fold, p<0.0001) in hMC. IL-1β and TNFα expression levels were decreased 34% (p=0.0367) and 36% (p=0.0303), respectively, in hMC co-treated with IL-1β and 2.5 μM INCA-6, compared to IL-1β alone (Figure 1A-B). INCA-6 had no effect on the IL-1β-induced expression of VEGF, CCL2, or CCL5 at either of the concentrations tested (Figure 1C-E).
Figure 1: NFAT inhibition attenuates cytokine expression in hMC.
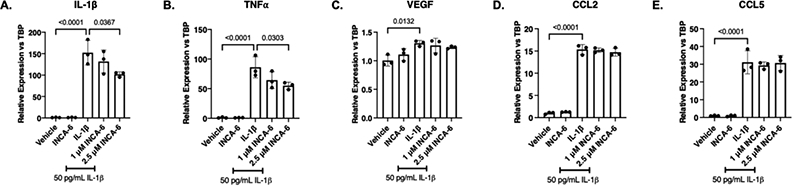
hMC were treated for 8hr with 50pg/mL IL-1β with or without INCA-6. Cells were collected and assayed for expression of (A) IL-1β, (B) TNFα, (C) VEGF, (D) CCL2, or (E) CCL5. Expression data are reported as fold induction over vehicle with bars representing mean +/− SD (n=3).
3.2. INCA-6 decreases IL-1β-induced inflammatory cytokine expression in hRMEC
Similar to hMC, hRMEC respond to cytokine treatment by increasing expression and secretion of inflammatory cytokines, thereby propagating and sustaining local inflammation.5,23 Therefore, we examined the NFAT-dependency of the hRMEC inflammatory response to IL-1β. IL-1β increased the expression of IL-1β (80-fold, p<0.0001), TNFα (3479-fold, p<0.0001), IL-8 (179-fold, p<0.0001), and IL-6 (66-fold, p=0.0001) in hRMEC. IL-1β, TNFα, and IL-6 expression decreased 81% (p<0.0001), 83% (p<0.0001), and 59% (p=0.0051), respectively, in hRMEC co-treated with IL-1β and 5μM INCA-6 compared to IL-1β alone. However, INCA-6 had no significant effect on IL-1β-induced IL-8 expression (Figure 2A-D). Additionally, we examined the chemokines, CCL2 and CCL5. IL-1β increased the expression of CCL2 (53-fold, p<0.0001) and CCL5 (122-fold, p<0.0001) in hRMEC. CCL2 and CCL5 expression decreased 60% (p=0.0003) and 87% (p<0.0001), respectively, in hRMEC co-treated with IL-1β and 5μM INCA-6, compared to IL-1β alone (Figure 2E-F).
Figure 2: INCA-6 inhibits IL-1β-induced cytokine expression in hRMEC.
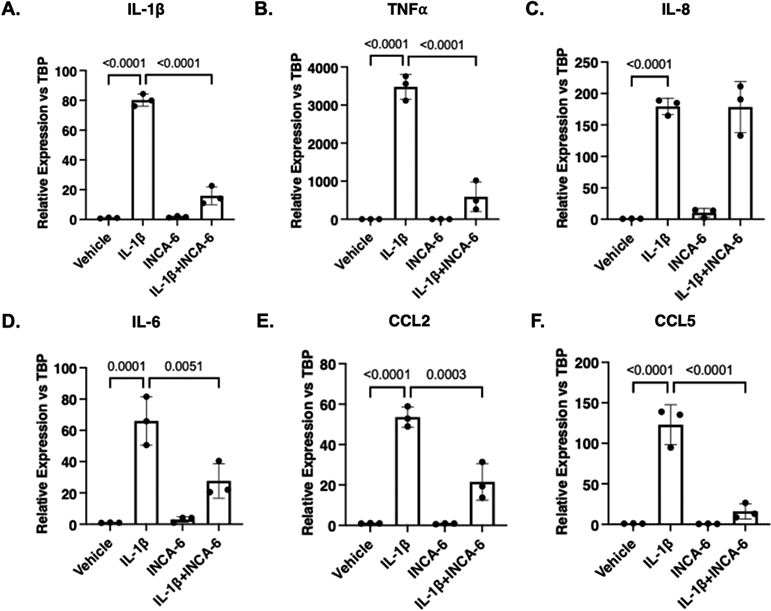
hRMEC were treated for 2hr with 1ng/mL IL-1β with or without 5μM INCA-6. Cells were collected and assayed for expression of (A) IL-1β, (B) TNFα, (C) IL-8, (D) IL-6, (E) CCL2, or (F) CCL5. Expression data are reported as fold induction over vehicle with bars representing mean +/− SD (n=3).
3.3. INCA-6 mitigates IL-1β-induced leukocyte adhesion to hRMEC
We sought to investigate the efficacy of INCA-6 in preventing inflammation-induced leukocyte-endothelial cell adhesion, a characteristic pathogenic event in NPDR.5 We performed a brief dose response experiment to determine the optimal concentration of INCA-6 to mitigate IL-1β-induced adhesion molecule expression in hRMEC. IL-1β increased VCAM-1, ICAM-1, and E-selectin expression 920-(p=0.0003), 229-(p<0.0001), and 1417-fold (p<0.0001), respectively, in hRMEC. Statistically significant decreases in ICAM-1 (51%; p=0.0002) and E-selectin (32%, p=0.0103) expression were achieved in hRMEC co-treated with IL-1β and 5μM INCA-6 (highest dose) compared to IL-1β alone (Figure 3A-C).
Figure 3: INCA-6 inhibits IL-1β-induced expression of leukocyte adhesion molecules in hRMEC.
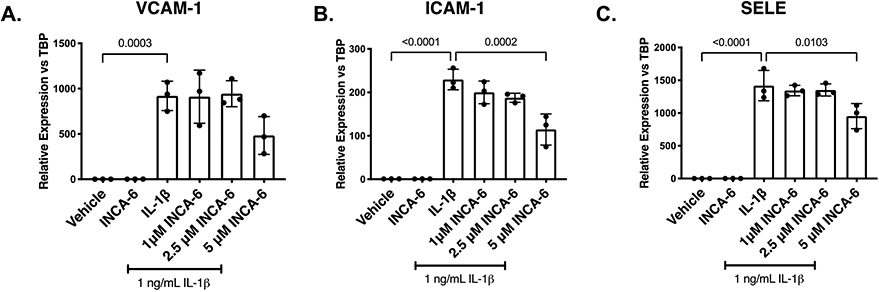
hRMEC were treated for 2hr with 1ng/mL IL-1β with or without INCA-6. Cells were collected and assayed for expression of (A) VCAM-1, (B) ICAM-1, or (C) E-selectin (gene name: SELE). Expression data are reported as fold induction over vehicle with bars representing mean +/− SD (n=3).
Next, we sought to determine if these changes in adhesion molecule expression translated into similar trends in PBMC adhesion to hRMEC monolayers using the parallel plate flow chamber (PPFC) assay. IL-1β caused an 11-fold (p<0.0001) increase in PBMC adhesion to hRMEC monolayers. When monolayers were co-treated with IL-1β and 5μM INCA-6, the optimal dose identified in Figure 3, PBMC adhesion to hRMEC monolayers decreased 41% (p=0.0485) compared to monolayers treated with IL-1β alone (Figure 4).
Figure 4: INCA-6 prevents IL-1β-induced PBMC adhesion to hRMEC.
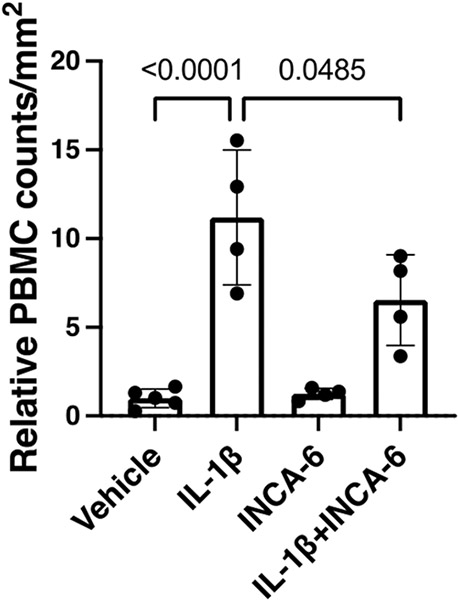
hRMEC were grown on slides until a confluent monolayer formed and then treated for 4hr with 1ng/mL IL-1β with or without 5μM INCA-6. Slides were placed in a PPFC, PBMC were flowed over the monolayers, and non-adherent cells were washed away. Adherent PBMC were counted in 8 fields per slide and averaged. Average counts were then divided by field area. Data are reported as fold induction over vehicle with bars representing mean +/− SD (vehicle: n=5; IL-1β: n=4; INCA-6: n=4; IL-1βINCA-6: n=4).
3.4. INCA-6 attenuates IL-1β-induced leukostasis in mice
Next, we investigated whether the efficacy of INCA-6 for inhibiting leukocyte adhesion in vitro could be translated in vivo. We used a mouse model of acute cytokine-induced retinal inflammation to test the efficacy of INCA-6 against retinal leukostasis. Retinal leukostasis increased 3.72-fold (p<0.0001) in C57BL/6J mice receiving intravitreal injections of IL-1β compared to vehicle. When mice received intravitreal injections of an IL-1β/INCA-6 cocktail, retinal leukostasis decreased to near vehicle levels (p<0.0001) (Figure 5).
Figure 5: INCA-6 prevents IL-1β-induced retinal leukostasis in an acute model of retinal inflammation.
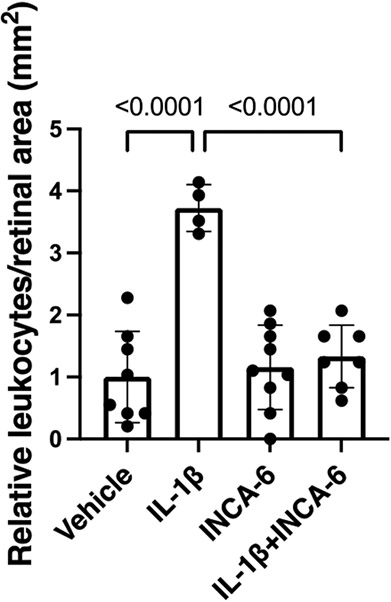
Mice received intravitreal injections of IL-1β +/− INCA-6. 12hr-post injection, leukostasis analysis was performed. Adherent leukocytes were counted in 4 fields per retina and averaged. Average counts were then divided by field area. Data are reported as fold induction over vehicle with bars representing mean +/− SD (vehicle: n=8; IL-1β: n=4; INCA-6: n=9; IL-1β+INCA-6: n=7)
3.5. INCA-6 rescues IL-1β-induced permeability
Finally, we were interested in determining whether INCA-6 was similarly efficacious in attenuating inflammation-induced vascular hyperpermeability, another characteristic pathology of NPDR.5 Transendothelial electrical resistance (TEER) measurements were performed to assess whether INCA-6 has the capacity to correct IL-1β-induced defects in hRMEC barrier function. IL-1β decreased the normalized resistance of hRMEC monolayers 40% (t=2-23 hours, p<0.0001) compared to vehicle. This decreased barrier function was essentially reversed in hRMEC monolayers co-treated with IL-1β and INCA-6 (t=2-23 hours, p<0.0001) (Figure 6). This is clearly demonstrated during the last 6 hours of monitoring, during which there were no significant differences between the normalized resistance measures of hRMEC monolayers treated with vehicle compared to those co-treated with IL-1β and INCA-6; control phenotype was completely restored. Interestingly, INCA-6 alone caused an increase in resistance over vehicle (t=2-23 hours, p<0.0001).
Figure 6: NFAT inhibition prevents IL-1β-induced hRMEC monolayer hyperpermeability.

hRMEC were allowed to settle and form a monolayer on 96-well ECIS plates. Once resistance plateaued, monolayers were treated with 1ng/mL IL-1β with or without 5μM INCA-6 and changes in resistance were monitored over 24hr. Resistance was normalized to plateaued resistance directly prior to treatment (timepoint 0). Data represent mean +/− SD (vehicle, IL-1β, and IL-1β+INCA-6: n=12; INCA-6: n=6).
Finally, we tested the capacity of INCA-6 to rescue retinal vascular hyperpermeability in vivo, assessed by fluorescein angiography, in our acute model of retinal inflammation. The fluorescein intensity in retinal fundus images nearly doubled (p=0.0296) in mice receiving intravitreal injections of IL-1β, indicating pronounced leakage from retinal capillaries. The fluorescein intensity decreased 84% (p=0.0351) in mice receiving intravitreal injections of an IL-1β/INCA-6 cocktail (Figure 7A-B).
Figure 7: INCA-6 prevents IL-1β-induced vascular hyperpermeability in an acute model of retinal inflammation.

Mice received intravitreal injections of IL-1β +/− INCA-6. 8hr post-injection, fluorescein was injected and fluorescent fundus images were captured at 2 and 4min post-fluorescein injection. (A) The difference in integrated density between images was calculated and recorded as a readout of vascular leakage. Bars represent mean +/− SD (vehicle: n=5; IL-1β: n=9; INCA-6: n=5; IL-1β+INCA-6: n=8). (B) Representative images of fluorescent fundus images obtained 2min (top row) and 4min (bottom row) after fluorescein injection.
4. Discussion
Our findings demonstrate for the first time that NFAT controls multiple IL-1β-induced inflammatory responses in both Müller cells and retinal microvascular endothelial cells. We had previously demonstrated that NFAT inhibition prevents TNFα-induced retinal leukostasis.43 However, since the diabetic retinal environment contains a complex mix of inflammatory cytokines, it is desirable to identify a therapeutic target that can universally slow pathogenic responses to multiple stimuli. Additionally, mounting evidence indicates that IL-1β acts as a master regulator of the retinal inflammation consequent to diabetes. Therefore, we tested the potential of INCA-6 to inhibit IL-1β-induced Müller cell and endothelial cell inflammation, leukocyte-endothelial cell adhesion, and vascular hyperpermeability in order to investigate the importance of CN/NFAT signaling in multiple pathogenic events of relevance to DR.
The current clinical approach to targeting the CN/NFAT signaling axis utilizes drugs (cyclosporin A and Tacrolimus) that block CN’s ability to phosphorylate any substrate.39,40,42 Therefore, they indiscriminately prevent downstream signaling, including important signaling hubs like MAP-K, contributing to severe side effects. Recent efforts have focused on identifying small molecule inhibitors that can specifically inhibit NFAT activation with minimal effects on CN’s other substrates.40 INCA-6 prevents the protein-protein interaction between CN and NFAT at the NFAT docking site and was identified based on its ability to compete out the high affinity VIVIT peptide. INCA-6 not only inhibited NFAT activation – as measured by phospho-NFAT Western blot, NFAT nuclear translocation staining, and expression levels of known NFAT target mRNA – but also showed no inhibition of CN phosphatase activity, no changes in MAP-K activation, and no changes in non-NFAT controlled mRNA expression.42 Based on this favorable specificity profile, we chose to utilize INCA-6 for our studies, selecting dose ranges in the lowest concentrations conventionally used in vitro.43,64-68
Müller cells are retina-specific macroglia that act as sensors of the local environment, and it is well established that Müller cells become activated in the diabetic milieu.63,69 We have previously demonstrated that, among non-neuronal retinal cells, Müller cells have the most potent inflammatory response to a variety of diabetes-relevant stimuli.57 Furthermore, we have also observed that IL-1β potently induces its own expression in hMC, supporting published findings that suggest IL-1β auto-amplification propagates and sustains retinal inflammation.23 Although NFAT has been previously implicated in the control of inflammatory responses in other glial cell types,53,70-72 a potential role in Müller cell inflammation has not been established. To determine whether NFAT inhibition could attenuate Müller cell inflammatory responses, we examined hMC expression of key representative inflammatory mediators: IL-1β, TNFα, VEGF, CCL2 (also MCP-1), and CCL5 (also RANTES). Analogous to findings in other investigations of glial cell responses,53,70-72 our data demonstrated that INCA-6 effectively inhibits the potent IL-1β-induced expression of IL-1β and TNFα in hMC. Recent evidence, in microglia, has indicated that NFAT can signal not only via calcium-dependent nuclear translocation but also via a calcium-independent mechanism involving mitochondrial translocation.73 However, since INCA-6 acts by blocking the binding of CN and NFAT, we believe that, here, NFAT is acting in hMC via its canonical calcium-dependent mechanism, not this novel calcium-independent mechanism. INCA-6 was not efficacious in preventing the modest IL-1β-induced increases in VEGF, CCL2, or CCL5 expression in hMC. We believe it is likely an advantage that this therapeutic approach acts independently of VEGF regulation. Despite diabetic macular edema (DME) being the leading cause of vision loss in DR patients, a recent meta-analysis comparing the efficacies of aflibercept, ranibizumab, and bevacizumab predicted that only 3-4 out of every 10 DME patients will experience improvement after one year of anti-VEGF therapy.74 Consequently, numerous combination therapies in which anti-VEGF therapy is combined with an additional target are currently in development with the goal of achieving greater efficacy in a wider patient population than anti-VEGF alone.75,76 Therefore, our data suggest that NFAT inhibition may be a potent and effective complement to anti-VEGF, though further study is certainly needed. Overall, these data suggest that NFAT inhibition can block the effects of both autocrine and paracrine sources of inflammation in hMC and represent the first report of the role of NFAT in regulating Müller cell expression of inflammatory genes.
Retinal microvascular endothelial cells demonstrate a wide range of molecular responses to the diabetic environment that contribute to the development of characteristic DR lesions.77 Therefore, the identification of therapeutic targets whose inhibition could halt these endothelial responses would be highly beneficial in slowing DR pathogenesis. Although endothelial cell inflammatory responses to primary metabolic stimuli are lower in magnitude compared to those seen in Müller cells,57 endothelial cells still become activated in response to cytokines and contribute to autocrine and paracrine inflammatory signaling.77 Indeed, it is likely that the potent response of Müller cells to primary metabolic stimuli in the diabetic environment contributes heavily to the initiation and subsequent propagation of inflammation by endothelial cells. NFAT inhibition has been shown to mitigate vascular inflammation in a variety of disease contexts.47,49,51 Importantly, previous findings demonstrated that NFAT inhibition attenuated TNFα-induced chemokine expression in hRMEC43,44 and prevented diabetes-induced IL-10 reduction in STZ mice.50 Therefore, we investigated whether INCA-6 could inhibit hRMEC expression of an array of inflammatory cytokines and chemokines, in a manner similar to the results we saw in hMC. When hRMEC were treated with IL-1β in the presence of INCA-6, NFAT inhibition caused a significant decrease in the expression of IL-1β, TNFα, IL-6, CCL2, and CCL5. Interestingly, NFAT showed differential regulation of inflammatory genes in hRMEC compared to hMC. Given the capacity of each of these cytokines to induce downstream pathological behaviors in endothelial cells, including leukostasis and vascular hyperpermeability,5,10,18,31,78 we find it highly encouraging that INCA-6 attenuates cytokine expression across a range of genes in hRMEC. Particularly in the case of CCL2 and CCL5, whose primary function is chemotaxis of leukocytes, inhibition in hRMEC is significant owing to the immediate proximity of the endothelium to the circulation. Overall, these results demonstrate that NFAT regulates IL-1β-dependent inflammatory responses in both Müller cells and microvascular endothelial cells, suggesting that targeting NFAT might significantly inhibit retinal inflammation at multiple points along the pathogenic cascade of DR.
Increased leukocyte adhesion, or leukostasis, is considered a landmark event of NPDR and has been observed in both diabetic animals79-82 and patients. 83-86 Adherent leukocytes contribute to DR pathogenesis in multiple ways, including eliciting and propagating local inflammation as well as occluding capillaries, leading to focal retinal ischemia.4,5 The diabetic milieu induces the expression of several endothelial cell adhesion proteins leading to increased leukocyte adherence. Specifically, E-selectin is responsible for the initial tethering and rolling of leukocytes on the endothelium, while VCAM-1 and ICAM-1 are responsible for the arrest and firm anchoring of leukocytes.87,88 Our previous studies demonstrated that targeting NFAT using INCA-6 or NFAT isoform-specific siRNA inhibited TNFα-induced expression and presentation of adhesion proteins in hRMEC and decreased leukocyte adherence to both hRMEC monolayers in vitro and blood vessel walls in vivo.43 Consistent with these data, NFAT inhibition has also proven efficacious in preventing inflammation-induced increases in adhesion molecule levels in a variety of disease contexts.46,47,51 Therefore, we advanced the hypothesis that INCA-6 would mitigate IL-1β-induced leukocyte adherence owing to decreased NFAT-dependent leukocyte adhesion molecule expression; our results suggest that this is indeed the case. Furthermore, we confirmed that these changes had beneficial functional outcomes by demonstrating the efficacy of INCA-6 against leukocyte adhesion in vitro using the PPFC. Finally, we chose to utilize an acute inflammation model to confirm the therapeutic potential of our NFAT inhibitory strategy in vivo. Using intraocular injections of IL-1β and an intraocular dose of INCA-6 that had proven efficacious in our hands,43,68 we modeled the increased leukostasis characteristic of early DR in a high throughput approach with a short experimental timeline and demonstrated that INCA-6 significantly attenuates this IL-1β-induced increase in retinal leukostasis. It is important to note that all our in vitro data are exclusively focused on endothelial cells’ role in endothelial cell-leukocyte adhesion. However, NFAT was initially characterized in T-lymphocytes and extensive evidence exists supporting its role in leukocyte biology.89-91 Therefore, it is possible that in the in vivo setting INCA-6 is acting not only on endothelial cells, as characterized by our PPFC experiments, but also on circulating immune cells. Further characterization of these effects will be sought in future studies.
The barrier function of the retinal endothelium depends on highly-organized junctional complexes that occlude the paracellular cleft between adjacent endothelial cells.92 Metabolic dysfunction and increased retinal levels of inflammatory cytokines occurring in DR activate molecular signals that lead to junctional complex disorganization and the associated deficit of barrier function. The changes result in vascular hyperpermeability leading to the development of characteristic fundus lesions of NPDR and/or DME, the most common cause of vision loss in DR patients.4,5 It is well established that IL-1β promotes retinal vascular hyperpermeability,5,28,93 and evidence in other disease models suggests that NFAT is involved in control of vascular permeability. For instance, NFAT inhibition attenuated lung vascular permeability in an animal model of sepsis48 and completely abrogated the retinal vascular hyperpermeability observed in Akita mice.50 Therefore, we utilized ECIS to measure TEER across hRMEC monolayers and demonstrated that INCA-6 had significant efficacy in attenuating the decreased resistance seen in IL-1β-treated monolayers. We further supported these findings by using qFA to demonstrate that INCA-6 similarly rescued IL-1β-induced hyperpermeability in our model of acute retinal inflammation. Interestingly, in our ECIS experiments, treatment with INCA-6 alone caused an increase in resistance over vehicle, suggesting that INCA-6 can improve barrier function of even healthy hRMEC monolayers. Taken together, our findings demonstrate that NFAT inhibition mitigates multiple inflammatory responses in hRMEC.
A number of important questions remain before NFAT inhibition can be further developed as a therapeutic strategy. For instance, since INCA-6 prevents the binding of NFAT to the activating phosphatase CN, INCA-6 inhibits the action of all four calcium-dependent NFAT isoforms. However, it has been demonstrated that distinct NFAT isoforms can oppose each other in their control of inflammatory gene expression. Indeed, we have previously demonstrated that NFATc2 knockdown prevented TNFα-induced inflammatory responses in hRMEC while NFATc3 increased them.43 Therefore, it is possible that greater efficacy could be achieved by identifying the isoform(s) responsible for the behaviors observed here, which would then compel the development of isoform-specific therapies. Our intention in this study was not to investigate specific mechanisms of action of NFAT regulation of each of these events but rather to investigate if NFAT inhibition had broad efficacy across multiple pathogenic events characteristic of early DR. Therefore, isoform-specific studies were outside the scope of this investigation; however, future studies will involve investigation of the isoform-specific roles in controlling each pathogenic behavior with the hopes of developing an NFAT-directed therapeutic strategy with enhanced efficacy. Although NFAT inhibitors have only been tested in pre-clinical settings, CN inhibitors are approved for clinical use and are used in post-kidney transplant and chemotherapy regimens,39,94 suggesting that therapeutic targeting of this signaling axis is tolerated in humans.
Our studies utilized an acute model of retinal inflammation to allow for the rapid assessment of efficacy of our NFAT inhibitory strategy, and it will be important to expand investigations of NFAT inhibition to diabetic animals with retinopathy. Furthermore, because a number of characteristic NPDR events, such as pericyte dropout or acellular capillaries, are not recapitulated by our acute inflammation model, we did not directly investigate the role of NFAT in their regulation. However, since chronic retinal inflammation is ultimately responsible for driving all types of NPDR lesions, we believe that by attenuating the propagation of early inflammation in DR, NFAT inhibition will likewise attenuate downstream events. Moreover, though our primary focus here was prevention of damage early in DR pathology, significant evidence from our lab and others suggests that NFAT inhibition could also be efficacious against the angiogenesis and neovascularization characteristic of late stage DR.47,68,95-100 of particular relevance, our previous work demonstrated that INCA-6 inhibits VEGF-induced hRMEC proliferation and tube formation and that both INCA-6 and the CN inhibitor, FK-506, significantly reduced pathologic neovascularization in a rat model of oxygen-induced retinopathy.68 These findings further support the therapeutic potential of NFAT inhibition to slow or prevent DR progression at any stage.
There is an urgent need for therapies targeting early DR that prevent disease progression to the irreparable damage caused by PDR. Since early DR pathology involves a variety of altered behaviors in several retinal cells, identifying a therapy that can target multiple pathogenic events in multiple cell types is ideal. To this end, we have demonstrated that NFAT inhibition can attenuate multiple IL-1β-induced pathogenic cell behaviors in both hMC and hRMEC. Combined with our previous findings that NFAT inhibition can similarly prevent TNFα-induced retinal leukostasis,43 we have significant evidence that NFAT is a target with substantial therapeutic potential for slowing retinal inflammation under DR-relevant conditions. Furthermore, previous studies have demonstrated that NFAT inhibition is similarly efficacious against hyperglycemia-induced retinal changes.50 When considered in combination with our previous TNFα and current IL-1β findings, it appears that NFAT inhibition can block retinal pathogenic responses elicited by a range of diabetes-relevant stimuli, suggesting NFAT is an excellent target to globally slow the downstream inflammatory effects of the complex diabetic milieu. Overall, we have identified a multi-faceted therapeutic target to inhibit not only Müller cell amplification of diabetic inflammation but also endothelial pathogenic responses, including increased inflammation, leukocyte adhesion, and vascular hyperpermeability.
Highlights.
In DR, retinal inflammation initiates, propagates, and sustains disease progression
Targeting NFAT attenuates IL-1β-induced Müller cell inflammation
NFAT inhibition reduces IL-1β-induced microvascular endothelial cell inflammation
NFAT inhibition rescues vascular hyperpermeability and leukostasis
Targeting NFAT has therapeutic potential for attenuating retinal inflammation
5. Acknowledgements
This research was supported by the following sources: National Institutes of Health grants R01-EY07533 (JP), R01-EY023639 (JP), R01-EY023397 (JP), P30-EY008126, and T32-EY021453; an unrestricted grant from Research to Prevent Blindness, Inc.; the Phyllis G. and William B. Snyder Endowment; and the Carl Marshall Reeves & Mildred Almen Reeves Foundation, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Yau JW et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35, 556–564, doi: 10.2337/dc11-1909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank RN Diabetic retinopathy. N Engl J Med 350, 48–58, doi: 10.1056/NEJMra021678 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Tang J & Kern TS Inflammation in diabetic retinopathy. Prog Retin Eye Res 30, 343–358, doi: 10.1016/j.preteyeres.2011.05.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonetti DA, Klein R & Gardner TW Diabetic retinopathy. N Engl J Med 366, 1227–1239, doi: 10.1056/NEJMra1005073 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Rubsam A, Parikh S & Fort PE Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci 19, doi: 10.3390/ijms19040942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kern TS Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res 2007, 95103, doi: 10.1155/2007/95103 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W, Liu H, Al-Shabrawey M, Caldwell RW & Caldwell RB Inflammation and diabetic retinal microvascular complications. J Cardiovasc Dis Res 2, 96–103, doi: 10.4103/0975-3583.83035 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammad G, Mairaj Siddiquei M, Imtiaz Nawaz M & Abu El-Asrar AM The ERK1/2 Inhibitor U0126 Attenuates Diabetes-Induced Upregulation of MMP-9 and Biomarkers of Inflammation in the Retina. J Diabetes Res 2013, 658548, doi: 10.1155/2013/658548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Z et al. Dendrobium chrysotoxum Lindl. alleviates diabetic retinopathy by preventing retinal inflammation and tight junction protein decrease. J Diabetes Res 2015, 518317, doi: 10.1155/2015/518317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rangasamy S et al. Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One 9, e108508, doi: 10.1371/journal.pone.0108508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzman ML et al. Profile of lipid and protein autacoids in diabetic vitreous correlates with the progression of diabetic retinopathy. Diabetes 59, 1780–1788, doi: 10.2337/db10-0110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki Y, Nakazawa M, Suzuki K, Yamazaki H & Miyagawa Y Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn J Ophthalmol 55, 256–263, doi: 10.1007/s10384-011-0004-8 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Rangasamy S, McGuire PG & Das A Diabetic retinopathy and inflammation: novel therapeutic targets. Middle East Afr J Ophthalmol 19, 52–59, doi: 10.4103/0974-9233.92116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAuley AK et al. Vitreous biomarkers in diabetic retinopathy: a systematic review and meta-analysis. J Diabetes Complications 28, 419–425, doi: 10.1016/j.jdiacomp.2013.09.010 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Vujosevic S et al. Proteome analysis of retinal glia cells-related inflammatory cytokines in the aqueous humour of diabetic patients. Acta Ophthalmol 94, 56–64, doi: 10.1111/aos.12812 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Funatsu H, Noma H, Mimura T, Eguchi S & Hori S Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 116, 73–79, doi: 10.1016/j.ophtha.2008.09.037 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Maier R et al. Multiplex bead analysis of vitreous and serum concentrations of inflammatory and proangiogenic factors in diabetic patients. Mol Vis 14, 637–643 (2008). [PMC free article] [PubMed] [Google Scholar]
- 18.Taghavi Y, Hassanshahi G, Kounis NG, Koniari I & Khorramdelazad H Monocyte chemoattractant protein-1 (MCP-1/CCL2) in diabetic retinopathy: latest evidence and clinical considerations. J Cell Commun Signal 13, 451–462, doi: 10.1007/s12079-018-00500-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothwell NJ & Luheshi GN Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci 23, 618–625, doi: 10.1016/s0166-2236(00)01661-1 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Dinarello CA Blocking interleukin-1beta in acute and chronic autoinflammatory diseases. J Intern Med 269, 16–28, doi: 10.1111/j.1365-2796.2010.02313.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masters SL et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 11, 897–904, doi: 10.1038/ni.1935 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandanmagsar B et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17, 179–188, doi: 10.1038/nm.2279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Biarnes Costa M & Gerhardinger C IL-1beta is upregulated in the diabetic retina and retinal vessels: cell-specific effect of high glucose and IL-1beta autostimulation. PLoS One 7, e36949, doi: 10.1371/journal.pone.0036949 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demircan N, Safran BG, Soylu M, Ozcan AA & Sizmaz S Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (Lond) 20, 1366–1369, doi: 10.1038/sj.eye.6702138 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Dong N, Xu B, Wang B & Chu L Study of 27 aqueous humor cytokines in patients with type 2 diabetes with or without retinopathy. Mol Vis 19, 1734–1746 (2013). [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreira R et al. Neuropeptide Y inhibits interleukin-1 beta-induced microglia motility. J Neurochem 120, 93–105, doi: 10.1111/j.1471-4159.2011.07541.x (2012). [DOI] [PubMed] [Google Scholar]
- 27.Kowluru RA & Odenbach S Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol 88, 1343–1347, doi: 10.1136/bjo.2003.038133 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luna JD et al. Blood-retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res 49, 268–280, doi:. (1997). [DOI] [PubMed] [Google Scholar]
- 29.Takeda K et al. Brain-Derived Neurotrophic Factor Inhibits Intercellular Adhesion Molecule-1 Expression in Interleukin-1beta-Treated Endothelial Cells. Cell Biochem Biophys 74, 399–406, doi: 10.1007/s12013-016-0749-2 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Bamforth SD, Lightman SL & Greenwood J Ultrastructural analysis of interleukin-1 beta-induced leukocyte recruitment to the rat retina. Invest Ophthalmol Vis Sci 38, 25–35 (1997). [PubMed] [Google Scholar]
- 31.Vincent JA & Mohr S Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56, 224–230, doi: 10.2337/db06-0427 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Dinarello CA Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732, doi: 10.1182/blood-2010-07-273417 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sama MA et al. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem 283, 21953–21964, doi: 10.1074/jbc.M800148200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaykasli KO et al. ADAMTS9 activation by interleukin 1 beta via NFATc1 in OUMS-27 chondrosarcoma cells and in human chondrocytes. Mol Cell Biochem 323, 69–79, doi: 10.1007/s11010-008-9965-4 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Hogan PG, Chen L, Nardone J & Rao A Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17, 2205–2232, doi: 10.1101/gad.1102703 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Rao A, Luo C & Hogan PG Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15, 707–747, doi: 10.1146/annurev.immunol.15.1.707 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Lopez-Rodriguez C et al. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity 15, 47–58, doi: 10.1016/s1074-7613(01)00165-0 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Pan MG, Xiong Y & Chen F NFAT gene family in inflammation and cancer. Curr Mol Med 13, 543–554, doi: 10.2174/1566524011313040007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin JJ et al. NFAT as cancer target: mission possible? Biochim Biophys Acta 1846, 297–311, doi: 10.1016/j.bbcan.2014.07.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JU, Kim LK & Choi JM Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front Immunol 9, 2747, doi: 10.3389/fimmu.2018.02747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang S, Li H, Rao A & Hogan PG Inhibition of the calcineurin-NFAT interaction by small organic molecules reflects binding at an allosteric site. J Biol Chem 280, 37698–37706, doi: 10.1074/jbc.M502247200 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Roehrl MH et al. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci U S A 101, 7554–7559, doi: 10.1073/pnas.0401835101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bretz CA, Savage SR, Capozzi ME, Suarez S & Penn JS NFAT isoforms play distinct roles in TNFalpha-induced retinal leukostasis. Sci Rep 5, 14963, doi: 10.1038/srep14963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage SR, Bretz CA & Penn JS RNA-Seq reveals a role for NFAT-signaling in human retinal microvascular endothelial cells treated with TNFalpha. PLoS One 10, e0116941, doi: 10.1371/journal.pone.0116941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cobbs SL & Gooch JL NFATc is required for TGFbeta-mediated transcriptional regulation of fibronectin. Biochem Biophys Res Commun 362, 288–294, doi: 10.1016/j.bbrc.2007.07.186 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Funk SD, Finney AC, Yurdagul A Jr., Pattillo CB & Orr AW EphA2 stimulates VCAM-1 expression through calcium-dependent NFAT1 activity. Cell Signal 49, 30–38, doi: 10.1016/j.cellsig.2018.05.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y et al. The role of Ca(2+)/NFAT in Dysfunction and Inflammation of Human Coronary Endothelial Cells induced by Sera from patients with Kawasaki disease. Sci Rep 10, 4706, doi: 10.1038/s41598-020-61667-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karpurapu M et al. Inhibition of nuclear factor of activated T cells (NFAT) c3 activation attenuates acute lung injury and pulmonary edema in murine models of sepsis. Oncotarget 9, 10606–10620, doi: 10.18632/oncotarget.24320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Vaz E et al. Inhibition of NFAT Signaling Restores Microvascular Endothelial Function in Diabetic Mice. Diabetes 69, 424–435, doi: 10.2337/db18-0870 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Zetterqvist AV et al. Nuclear factor of activated T cells is activated in the endothelium of retinal microvessels in diabetic mice. J Diabetes Res 2015, 428473, doi: 10.1155/2015/428473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zetterqvist AV et al. Inhibition of nuclear factor of activated T-cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice. PLoS One 8, e65020, doi: 10.1371/journal.pone.0065020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bendickova K et al. Calcineurin inhibitors reduce NFAT-dependent expression of antifungal pentraxin-3 by human monocytes. J Leukoc Biol 107, 497–508, doi: 10.1002/JLB.4VMA0318-138R (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagamoto-Combs K & Combs CK Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J Neurosci 30, 9641–9646, doi: 10.1523/JNEUROSCI.0828-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang R, Guo L, Gao M, Li J & Xiang S Research Trends and Regulation of CCL5 in Prostate Cancer. Onco Targets Ther 14, 1417–1427, doi: 10.2147/OTT.S279189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abcouwer SF Muller Cell-Microglia Cross Talk Drives Neuroinflammation in Diabetic Retinopathy. Diabetes 66, 261–263, doi: 10.2337/dbi16-0047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Xu X, Elliott MH, Zhu M & Le YZ Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 59, 2297–2305, doi: 10.2337/db09-1420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capozzi ME, McCollum GW, Cousins DB & Penn JS Linoleic Acid is a Diabetes-relevant Stimulator of Retinal Inflammation in Human Retinal Muller Cells and Microvascular Endothelial Cells. J Diabetes Metab 7, doi: 10.4172/2155-6156.1000718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hicks D & Courtois Y The growth and behaviour of rat retinal Muller cells in vitro. 1. An improved method for isolation and culture. Exp Eye Res 51, 119–129, doi: 10.1016/0014-4835(90)90063-z (1990). [DOI] [PubMed] [Google Scholar]
- 59.Capozzi ME, Hammer SS, McCollum GW & Penn JS Epoxygenated Fatty Acids Inhibit Retinal Vascular Inflammation. Sci Rep 6, 39211, doi: 10.1038/srep39211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lo CM, Keese CR & Giaever I Impedance analysis of MDCK cells measured by electric cell-substrate impedance sensing. Biophys J 69, 2800–2807, doi: 10.1016/S0006-3495(95)80153-0 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robilliard LD et al. The Importance of Multifrequency Impedance Sensing of Endothelial Barrier Formation Using ECIS Technology for the Generation of a Strong and Durable Paracellular Barrier. Biosensors (Basel) 8, doi: 10.3390/bios8030064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bringmann A et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res 25, 397–424, doi: 10.1016/j.preteyeres.2006.05.003 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Coughlin BA, Feenstra DJ & Mohr S Muller cells and diabetic retinopathy. Vision Res 139, 93–100, doi: 10.1016/j.visres.2017.03.013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao L et al. Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res 103, 733–742, doi: 10.1161/CIRCRESAHA.108.171157 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Liu L, Peng Z, Huang H, Xu Z & Wei X Luteolin and apigenin activate the Oct-4/Sox2 signal via NFATc1 in human periodontal ligament cells. Cell Biol Int 40, 1094–1106, doi: 10.1002/cbin.10648 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Liu L, Peng Z, Xu Z, Huang H & Wei X Mouse embryonic fibroblast (MEF)/BMP4-conditioned medium enhanced multipotency of human dental pulp cells. J Mol Histol 49, 17–26, doi: 10.1007/s10735-017-9743-2 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Prasad AM & Inesi G Silencing calcineurin A subunit reduces SERCA2 expression in cardiac myocytes. Am J Physiol Heart Circ Physiol 300, H173–180, doi: 10.1152/ajpheart.00841.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bretz CA, Savage S, Capozzi M & Penn JS The role of the NFAT signaling pathway in retinal neovascularization. Invest Ophthalmol Vis Sci 54, 7020–7027, doi: 10.1167/iovs.13-12183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sorrentino FS, Allkabes M, Salsini G, Bonifazzi C & Perri P The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci 162, 54–59, doi: 10.1016/j.lfs.2016.08.001 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Sompol P & Norris CM Ca(2+), Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front Aging Neurosci 10, 199, doi: 10.3389/fnagi.2018.00199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manocha GD et al. NFATc2 Modulates Microglial Activation in the AbetaPP/PS1 Mouse Model of Alzheimer's Disease. J Alzheimers Dis 58, 775–787, doi: 10.3233/JAD-151203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M & Inoue K P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem 114, 810–819, doi: 10.1111/j.1471-4159.2010.06809.x (2010). [DOI] [PubMed] [Google Scholar]
- 73.Ma B et al. Toll-Like Receptors Promote Mitochondrial Translocation of Nuclear Transcription Factor Nuclear Factor of Activated T-Cells in Prolonged Microglial Activation. J Neurosci 35, 10799–10814, doi: 10.1523/JNEUROSCI.2455-14.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Virgili G, Parravano M, Evans JR, Gordon I & Lucenteforte E Anti-vascular endothelial growth factor for diabetic macular oedema: a network meta-analysis. Cochrane Database Syst Rev 10, CD007419, doi: 10.1002/14651858.CD007419.pub6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sahni J et al. Simultaneous Inhibition of Angiopoietin-2 and Vascular Endothelial Growth Factor-A with Faricimab in Diabetic Macular Edema: BOULEVARD Phase 2 Randomized Trial. Ophthalmology 126, 1155–1170, doi: 10.1016/j.ophtha.2019.03.023 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Beckmann R et al. DutaFabs are engineered therapeutic Fab fragments that can bind two targets simultaneously. Nat Commun 12, 708, doi: 10.1038/s41467-021-20949-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gui F, You Z, Fu S, Wu H & Zhang Y Endothelial Dysfunction in Diabetic Retinopathy. Front Endocrinol (Lausanne) 11, 591, doi: 10.3389/fendo.2020.00591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monickaraj F, Oruganti SR, McGuire P & Das A A potential novel therapeutic target in diabetic retinopathy: a chemokine receptor (CCR2/CCR5) inhibitor reduces retinal vascular leakage in an animal model. Graefes Arch Clin Exp Ophthalmol 259, 93–100, doi: 10.1007/s00417-020-04884-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miyamoto K et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A 96, 10836–10841, doi: 10.1073/pnas.96.19.10836 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim SY et al. Neutrophils are associated with capillary closure in spontaneously diabetic monkey retinas. Diabetes 54, 1534–1542, doi: 10.2337/diabetes.54.5.1534 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Joussen AM et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18, 1450–1452, doi: 10.1096/fj.03-1476fje (2004). [DOI] [PubMed] [Google Scholar]
- 82.Olivares AM et al. Animal Models of Diabetic Retinopathy. Curr Diab Rep 17, 93, doi: 10.1007/s11892-017-0913-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schroder S, Palinski W & Schmid-Schonbein GW Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 139, 81–100 (1991). [PMC free article] [PubMed] [Google Scholar]
- 84.McLeod DS, Lefer DJ, Merges C & Lutty GA Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 147, 642–653 (1995). [PMC free article] [PubMed] [Google Scholar]
- 85.Joussen AM et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158, 147–152, doi: 10.1016/S0002-9440(10)63952-1 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tarr JM, Kaul K, Chopra M, Kohner EM & Chibber R Pathophysiology of diabetic retinopathy. ISRN Ophthalmol 2013, 343560, doi: 10.1155/2013/343560 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Noda K, Nakao S, Ishida S & Ishibashi T Leukocyte adhesion molecules in diabetic retinopathy. J Ophthalmol 2012, 279037, doi: 10.1155/2012/279037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ley K, Laudanna C, Cybulsky MI & Nourshargh S Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7, 678–689, doi: 10.1038/nri2156 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Durand DB et al. Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol Cell Biol 8, 1715–1724, doi: 10.1128/mcb.8.4.1715 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shaw JP et al. Identification of a putative regulator of early T cell activation genes. Science 241, 202–205, doi: 10.1126/science.3260404 (1988). [DOI] [PubMed] [Google Scholar]
- 91.Fric J et al. NFAT control of innate immunity. Blood 120, 1380–1389, doi: 10.1182/blood-2012-02-404475 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Campbell M & Humphries P The blood-retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol 763, 70–84 (2012). [PubMed] [Google Scholar]
- 93.Stahel M, Becker M, Graf N & Michels S SYSTEMIC INTERLEUKIN 1beta INHIBITION IN PROLIFERATIVE DIABETIC RETINOPATHY: A Prospective Open-Label Study Using Canakinumab. Retina 36, 385–391, doi: 10.1097/IAE.0000000000000701 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tanabe K Calcineurin inhibitors in renal transplantation: what is the best option? Drugs 63, 1535–1548, doi: 10.2165/00003495-200363150-00002 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Suehiro J et al. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J Biol Chem 289, 29044–29059, doi: 10.1074/jbc.M114.555235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai Y et al. Role of NFAT in the Progression of Diabetic Atherosclerosis. Front Cardiovasc Med 8, 635172, doi: 10.3389/fcvm.2021.635172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rafiee P et al. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun Signal 2, 3, doi: 10.1186/1478-811X-2-3 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schweighofer B et al. The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb Haemost 102, 544–554, doi: 10.1160/TH08-12-0830 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson EN et al. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem 278, 1686–1692, doi: 10.1074/jbc.M210250200 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Urso K et al. NFATc3 regulates the transcription of genes involved in T-cell activation and angiogenesis. Blood 118, 795–803, doi: 10.1182/blood-2010-12-322701 (2011). [DOI] [PubMed] [Google Scholar]