

ABSTRACT
Bemarituzumab (FPA144) is a first-in-class, humanized, afucosylated immunoglobulin G1 monoclonal antibody (mAb) directed against fibroblast growth factor receptor 2b (FGFR2b) with two mechanisms of action against FGFR2b-overexpressing tumors: inhibition of FGFR2b signaling and enhanced antibody-dependent cell-mediated cytotoxicity (ADCC). Bemarituzumab is being developed as a cancer therapeutic, and we summarize here the key nonclinical data that supported moving it into clinical trials. Bemarituzumab displayed sub-nanomolar cross-species affinity for FGFR2b receptors, with >20-fold enhanced binding affinity to human Fc gamma receptor IIIa compared with the fucosylated version. In vitro, bemarituzumab induced potent ADCC against FGFR2b-expressing tumor cells, and inhibited FGFR2 phosphorylation and proliferation of SNU-16 gastric cancer cells in a concentration-dependent manner. In vivo, bemarituzumab inhibited tumor growth through inhibition of the FGFR2b pathway and/or ADCC in mouse models. Bemarituzumab demonstrated enhanced anti-tumor activity in combination with chemotherapy, and due to bemarituzumab-induced natural killer cell-dependent increase in programmed death-ligand 1, also resulted in enhanced anti-tumor activity when combined with an anti-programmed death-1 antibody. Repeat-dose toxicity studies established the highest non-severely-toxic dose at 1 and 100 mg/kg in rats and cynomolgus monkeys, respectively. In pharmacokinetic (PK) studies, bemarituzumab exposure increase was greater than dose-proportional, with the linear clearance in the expected dose range for a mAb. The PK data in cynomolgus monkeys were used to project bemarituzumab linear PK in humans, which were consistent with the observed human Phase 1 data. These key nonclinical studies facilitated the successful advancement of bemarituzumab into the clinic.
KEYWORDS: Bemarituzumab, anti-FGFR2b antibody, fibroblast growth factor receptor, pharmacokinetics, toxicology, afucosylated antibody; antibody-dependent cell-mediated cytoxicity; phosphorylation in vitro, cell proliferation in vitro, anti-tumor efficacy
Introduction
The role of fibroblast growth factors (FGFs) in cancer is well known: FGFs can stimulate the transformation and proliferation of tumor cells and angiogenesis.1 The FGF family consists of 22 ligands and 4 fibroblast growth factor receptors (FGFRs).1 Splice variations lead to several receptor variants, including 2 predominant isoforms each of FGFR1–3, termed b and c. The expression of individual FGFs and FGFRs is generally restricted to specific tissues, cell types, and/or developmental stages. In particular, tissues of epithelial origin such as the stomach and skin express the fibroblast growth factor receptor 2 IIIb isoform (FGFR2b), whose 3 major ligands are FGF7, FGF10, and FGF22.2,3
FGFR2b can be highly expressed in tumors through amplification of the FGFR2 gene or transcriptional upregulation of the FGFR2b isoform. As early as 1990, subsets of patients with gastric cancer were noted to have amplification of the FGFR2 gene.4 More recently, either overexpression of the FGFR2b receptor or amplification of FGFR2 have been identified as having prognostic importance in patients with gastric cancer.5–8 Overexpression of FGFR2b is significantly more common in the absence of amplification in advanced stage gastric cancer,9 and recent prospective evaluation in front line advanced and metastatic gastric cancer estimates the prevalence of FGFR2b overexpression at approximately 32%.10 Furthermore, alterations in the FGF/FGFR2 signaling pathway have been observed in other cancers as well, including breast, ovarian, endometrial, lung, and bile duct cancers.11–14 Thus, inhibition of FGFR2 signaling may be an effective mechanism of action for multiple cancer indications.5,6
Bemarituzumab, also referred to as FPA144 or AMG 552, is a first-in-class, recombinant, humanized, afucosylated immunoglobulin (Ig) G1 kappa monoclonal antibody (mAb) directed against FGFR2b. Bemarituzumab has 2 demonstrated mechanisms of action: blocking FGFR2b signaling by competitive binding inhibition of FGFs and eliciting enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) against FGFR2b-overexpressing tumor cells. Here, we demonstrate in nonclinical studies that bemarituzumab can suppress FGFR2b signaling in a time- and concentration-dependent manner, and requires Fc gamma receptor (FcγR) engagement to significantly inhibit tumor growth in vivo. Moreover, the anti-tumor activity of bemarituzumab can be enhanced upon combination either with immune checkpoint blockade via anti-programmed death-1 (PD-1) or chemotherapy. Finally, when administered to rats and cynomolgus monkeys, bemarituzumab demonstrated a greater than dose-proportional increase in exposure at lower doses with linear clearance and an acceptable toxicology profile at exposures expected to be efficacious in cancer patients. Together, these data were used to project the pharmacokinetic (PK) profile in humans and supported the advance of bemarituzumab into a first-in-human Phase 1 dose escalation and expansion study.
Results
Bemarituzumab binding affinity to FGFR2b and FcγRIIIa
To confirm rat and cynomolgus monkey were appropriate species for toxicology studies, the binding affinity of bemarituzumab to the extracellular domain (ECD) of FGFR2b from rat, monkey, and human was measured by surface plasmon resonance and found to be similar across all three species (Table 1). Bemarituzumab demonstrated sub-nanomolar equilibrium dissociation constants (KD) for rat, cynomolgus monkey, and human FGFR2b ECD with comparable associate and dissociate constants (Kon and Koff). Based on these data, rat and cynomolgus monkey were deemed appropriate species in which to perform toxicology studies with bemarituzumab.
Table 1.
Binding affinities of bemarituzumab to FGFR2b ECDs from 3 species
| Source of FGFR2b | Kon (1/Ms) | Koff (1/s) | KD (Koff/Kon) (nM) |
|---|---|---|---|
| Rat | 4.34E+04 | 2.65E-05 | 0.611 |
| Cynomolgus monkey | 4.39E+04 | 3.38E-05 | 0.770 |
| Human | 8.42E+04 | 4.88E-05 | 0.579 |
Kon = associate constant; Koff = dissociate constant; KD = equilibrium dissociation constant.
Surface plasmon resonance was used to determine binding affinities; 6 concentrations (0–500 nM) of FGFR2b ECD IgG fusion proteins were tested. Kon, Koff, and KD were calculated using the Biacore T100 Evaluation Software 1:1 binding model.
After antibodies of the human IgG1 isotype bind to their target on tumor cells, immune cells that express FcγRIIIa, especially natural killer (NK) cells and macrophages, are recruited and activated to promote cancer cell death in a process known as ADCC. Bemarituzumab was specifically engineered to elicit enhanced ADCC against FGFR2b-expressing tumor cells. Bemarituzumab was produced in a Chinese hamster ovary (CHO) cell line that lacks the FUT8 gene (α1,6-Fucosyltransferase) and therefore lacks a core fucose in the polysaccharide portion of the Fc domain of the antibody. The lack of this fucose resulted in higher affinity (>20-fold) of bemarituzumab to human FcγRIIIa compared to the fucosylated molecule (FPA144-F, the fucosylated version of the mAb) (Table 2).
Table 2.
Binding affinities of bemarituzumab and FPA144-F to human FcγRIIIa (V158)
| Antibody | KD (Koff/Kon) (nM) |
|---|---|
| FPA144-A (bemarituzumab) | 9.2 |
| FPA144-F (the fucosylated version) | 207 |
Kon = associate constant; Koff = dissociate constant; KD = equilibrium dissociation constant.
Surface plasmon resonance was used to determine binding affinities; 5 concentrations (0–1000 nM) of FcγRIIIa (V158) were tested. Kon, Koff, and KD were calculated using the Biacore T100 Evaluation Software 1:1 binding model.
Bemarituzumab in vitro activities
Multiple studies were performed to evaluate the in vitro activity of bemarituzumab. FGF-mediated phosphorylation of FGFR2b activates a signal transduction cascade that ultimately results in cell proliferation.15 Therefore, the effect of bemarituzumab on FGFR2 phosphorylation and tumor cell proliferation was assessed in the SNU-16 human gastric cancer cell line. In the presence of increasing concentrations of bemarituzumab, FGF7-induced FGFR2 phosphorylation and cell proliferation were inhibited in a concentration-dependent manner in SNU-16 cells (Figure 1(a,b)). The inhibition reached maximum at bemarituzumab concentrations ≥6.25 µg/mL, which achieved >95% receptor occupancy based on binding affinity of bemarituzumab to FGFR2b.16 Furthermore, compared to the fucosylated anti-FGFR2b antibody (FPA144-F) (FPA144-F/FGFR2b, Figure 2), bemarituzumab displayed enhanced concentration-dependent immune cell-mediated tumor cell killing for tumor cells with FGFR2b overexpression, as indicated by higher specific lysis (bema/FGFR2b, Figure 2). No ADCC was induced on Ba/F3 expressing FGFR2c (bema/FGFR2c, Figure 2), confirming bemarituzumab specificity for the FGFR2b isoform. Therefore, bemarituzumab demonstrated multiple mechanisms of action in vitro, including blockade of the FGFR2b signaling pathway and increased ADCC due to enhanced affinity to FcγRIIIa.
Figure 1.
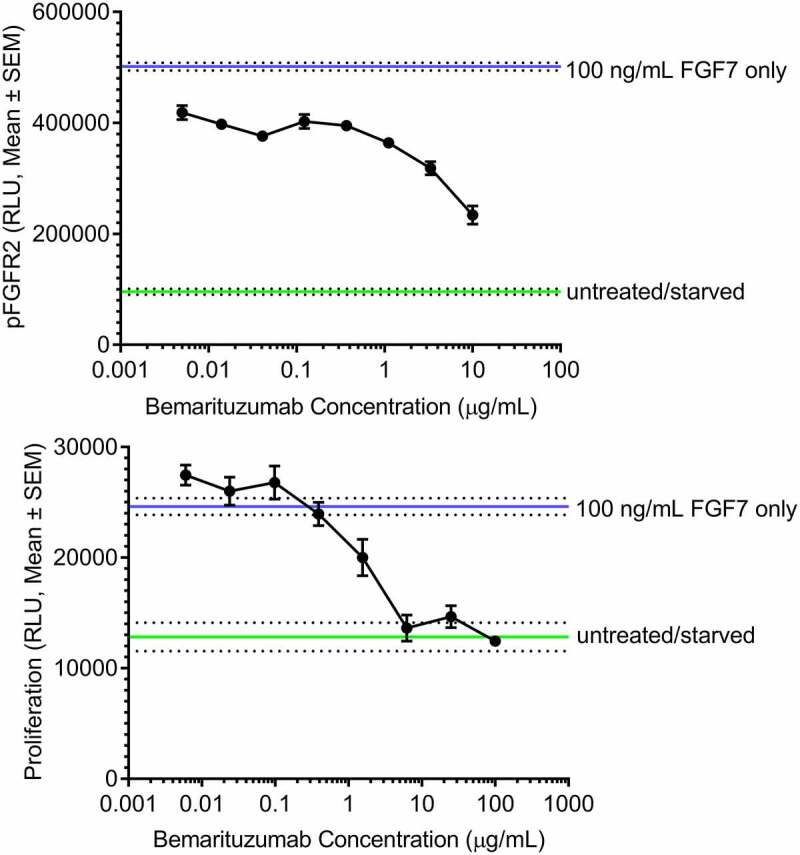
Bemarituzumab inhibits FGFR2 phosphorylation and FGFR2b-mediated proliferation of SNU-16 cells. a: FGFR2 phosphorylation (pFGFR2) in SNU-16 cells treated with varying concentrations of bemarituzumab followed by 100 ng/mL FGF7 is shown. FGFR2 phosphorylation was measured via ELISA. b: Proliferation of SNU-16 cells treated with varying concentrations of bemarituzumab followed by 100 ng/mL FGF7 is shown. Proliferation was measured via cell viability assay. The experiments were performed in triplicate; data are presented as mean ±standard error of mean (SEM) relative light units (RLU)
Figure 2.
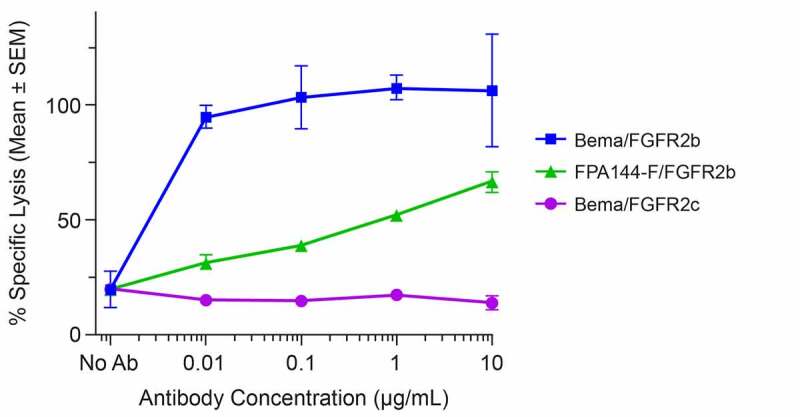
Bemarituzumab elicits potent induction of ADCC activity against FGFR2b-expressing cells. Specific lysis of target cells as a percentage of maximal lysis in the presence of varying concentrations of bemarituzumab or FPA144-F is shown: Ba/F3 cells expressing full-length human FGFR2b treated with bemarituzumab (Bema/FGFR2b); Ba/F3 cells expressing full-length human FGFR2b treated with FPA144-F (FPA144-F/GFGFR2b); Ba/F3 cells expressing full-length human FGFR2c treated with bemarituzumab (Bema/FGFR2c). Data are presented as mean ±standard error of mean (SEM); n = 3 per concentration/group
Bemarituzumab in vivo activity as monotherapy and in combination with anti-PD-1 or chemotherapy
In vivo nonclinical efficacy of bemarituzumab was assessed using xenograft models in mice. First, the efficacy of bemarituzumab monotherapy to inhibit growth of FGFR2-amplified tumors was measured. For these experiments, OCUM-2M human gastric cancer cells were implanted in CB17/SCID mice. Bemarituzumab displayed therapeutic efficacy against OCUM-2M gastric tumors at all dose levels tested compared to the negative control (albumin) group. The inhibition of tumor growth was dose-dependent, with the 1 mg/kg dose having the least inhibitory effect and the highest dose of 5 mg/kg resulting in the greatest inhibition (Figure 3). Specifically, tumor growth inhibition and tumor regression were seen starting at the 1 mg/kg dose, with complete tumor regression observed in 1/15 mice at the 1.5 mg/kg and 2 mg/kg doses, 4/15 mice at the 3 mg/kg dose, and 6/15 mice at the 5 mg/kg dose by Day 42.
Figure 3.
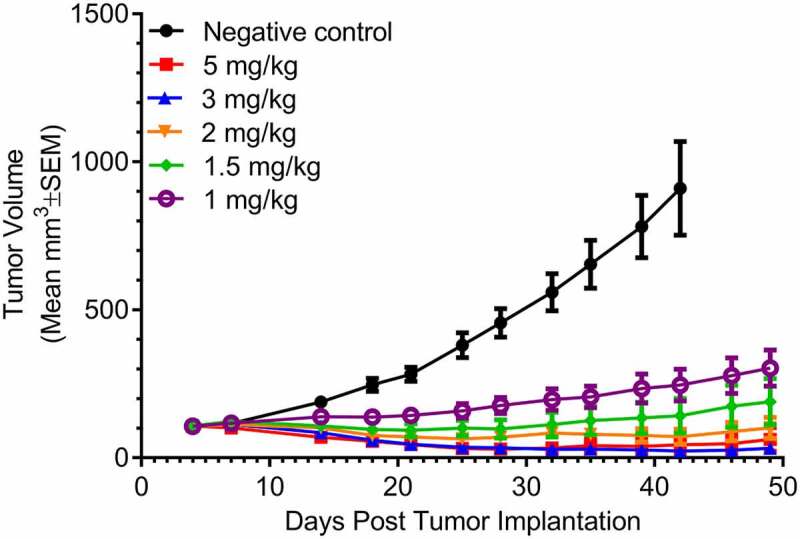
Bemarituzumab inhibits tumor growth in OCUM-2M tumor-bearing mice. Tumor volume (mm3) over time in OCUM-2M tumor-bearing mice treated with twice per week IV bemarituzumab at varying dose levels is shown. Albumin (5 mg/kg, IV) was used as the negative control. The first day of the treatment was day 4 post tumor implantation. Data are presented as mean ±standard error of mean (SEM); n = 15 per group
Next, the ability of bemarituzumab to block in vivo FGFR tumor signaling was examined. Specifically, CB17/SCID mice bearing SNU-16 xenografts were treated with a single intravenous (IV) dose of bemarituzumab at 10 mg/kg. Mice were sacrificed at 2, 6, 24, and 72 hours post treatment, tumors were isolated, and western blotting was performed. The ability of bemarituzumab to decrease FGFR2b levels, decrease FGFR phosphorylation, and decrease phosphorylation of an immediate downstream signaling molecule (FGFR substrate 2; FRS2) is illustrated in Figure 4(A). All results are shown compared to a common housekeeping enzyme, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Sustained decreases in receptor phosphorylation and receptor protein levels were obtained in CB17/SCID mice bearing SNU-16 tumors that were treated over 82 days with twice weekly intraperitoneal (IP) doses of bemarituzumab at 10 mg/kg (Figure 4(B)).
Figure 4.
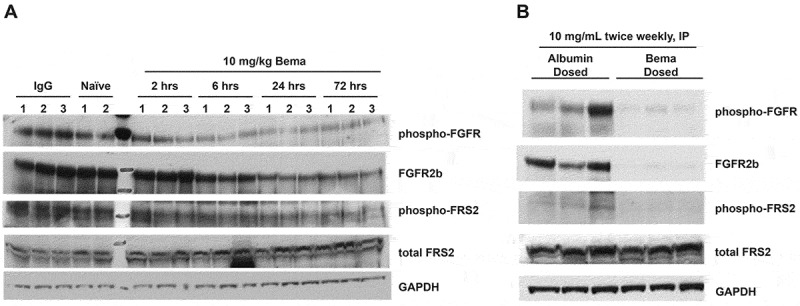
Bemarituzumab decreases FGFR2 signaling in SNU-16 tumor-bearing mice. Western blot analyses of pan-phosphorylated FGFR (phospho-FGFR), total FGFR2b, phosphorylated FRS2 (phospho-FRS2), and total FRS2 in tumors from SNU-16 tumor-bearing mice are shown. All results were compared to a common housekeeping enzyme (GAPDH). a: Tumors from mice treated with a single IV dose of bemarituzumab (10 mg/kg) were collected at 2, 6, 24, and 72 hours post treatment (n = 3 per time point). Control groups included tumors collected at 2 hours from naive animals (n = 2) and at 24 hours post IgG treatment (10 mg/kg; n = 3). Bemarituzumab decreased phospho-FGFR and phospho-FRS2 within 2 hours, and decreased total FGFR2b levels within 6 hours. b: Tumors from mice treated with twice weekly IP doses of 10 mg/kg bemarituzumab or albumin were collected at 82 days post first bemarituzumab/albumin treatment (n = 3 per group). Compared with the albumin control group, bemarituzumab decreased phospho-FGFR, phospho-FRS2, and total FGFR2b levels
To understand the contribution of ADCC in bemarituzumab’s anti-tumor efficacy, an antibody engineered to abrogate binding to FcγR (bemarituzumab-N297Q) was compared to bemarituzumab in the syngeneic 4T1 model that expresses FGFR2b but to a lesser degree than FGFR2 gene-amplified or FGFR2b-overexpressed xenografts, such as OCUM-2M or SNU-16. In the syngeneic 4T1 model, twice weekly bemarituzumab treatment (10 mg/kg, IP) significantly decreased tumor burden compared to the human Fc-IgG1 control, while bemarituzumab-N297Q showed no discernible effect on tumor growth (Figure 5). These data support a role for ADCC activity through FcγR binding as a required mechanism for bemarituzumab efficacy.
Figure 5.
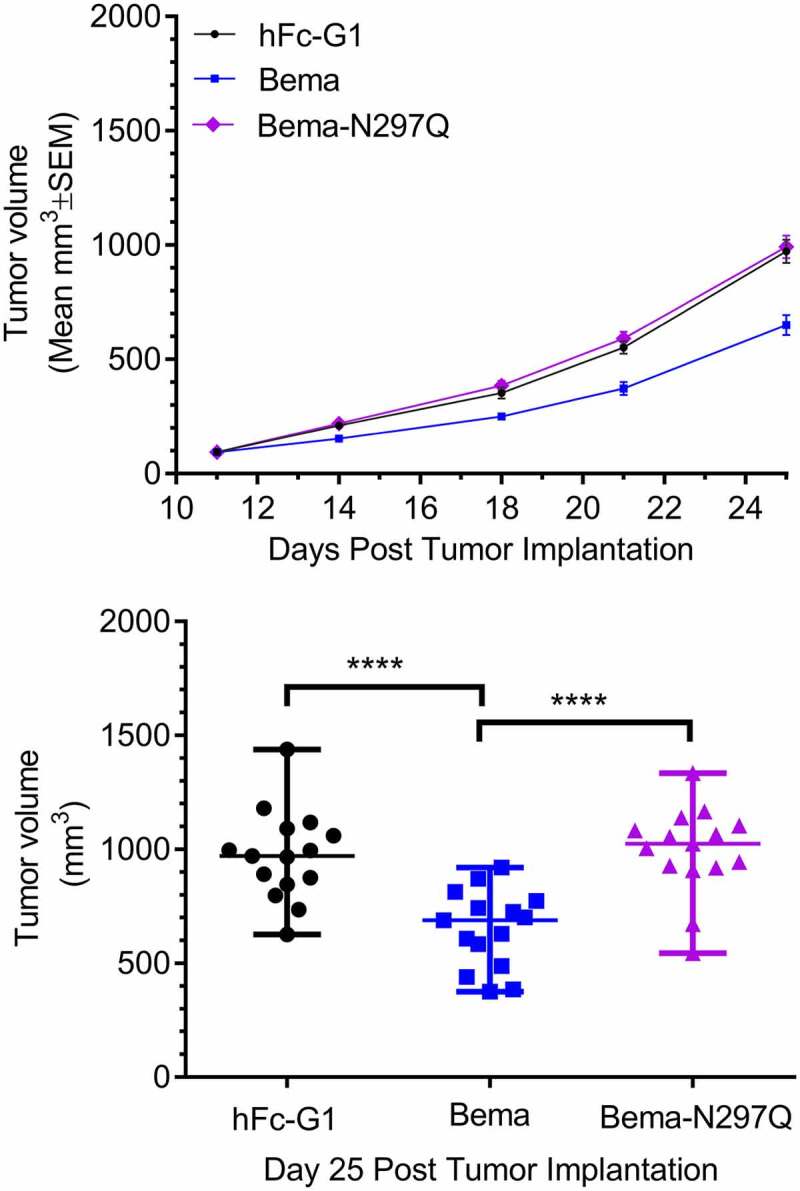
Bemarituzumab, but not an ADCC-deficient FGFR2b antibody, leads to tumor suppression in a syngeneic tumor model with modest FGFR2b expression. a: Tumor volume (mm3) over time in mice bearing 4T1 mammary orthotopic tumors treated with twice per week bemarituzumab (10 mg/kg, IP) or bemarituzumab-N297Q (10 mg/kg, IP) are shown. Human Fc-IgG1 (hFc-G1,10 mg/kg, IP) was used as the negative control. The first day of treatment was Day 11 post tumor implantation. Data are presented as mean ±standard error of mean (SEM); n = 15 per group. b: at day 25 post tumor implantation, mean tumor volumes from mice in panel a were significantly different (****p < .0001). Statistical significance was determined by 1 way ANOVA followed by Tukey’s multiple comparisons test. Each symbol represents an individual animal
To assess the effects of bemarituzumab on the tumor microenvironment, changes in infiltrating leukocytes post-bemarituzumab exposure were analyzed in 4T1 tumors. Mice harboring 4T1 tumors were treated with 1 or 2 doses of bemarituzumab (20 mg/kg, IP) on Day 0 or Day 0 and Day 3, respectively, and euthanized 24 hours after 1 or 2 doses for immunohistochemistry analyses. Twenty-four hours after the first dose, bemarituzumab treatment let to a recruitment of NK cells and programmed death-ligand 1 (PD-L1) expressing cells were increased within the tumor (data not shown). As shown in Figure 6, bemarituzumab treatment led to a persistent recruitment of NK cells 24 hours after the second dose. In addition, PD-L1-expressing cells were persistently increased within the tumor after bemarituzumab treatment compared with either human Fc-IgG1 control or bemarituzumab-N297Q treatment. Furthermore, an increase in cluster of differentiation 3 (CD3)-positive T cells was observed within tumors from mice treated with bemarituzumab that was not evident in mice treated with the bemarituzumab post first dose, or human Fc-IgG1 control or bemarituzumab-N297Q, suggesting that these changes in the tumor microenvironment were dependent on both FGFR2b and FcγR engagement.
Figure 6.
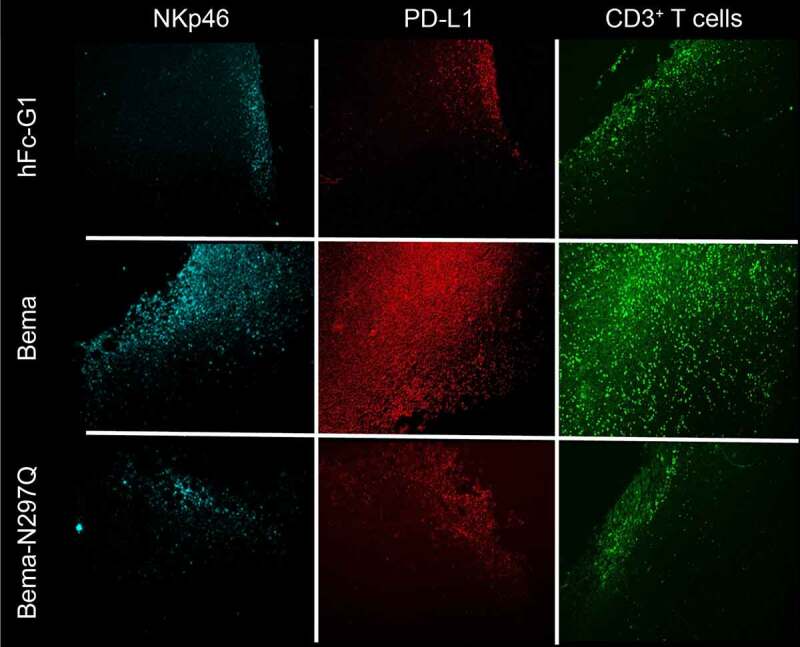
Bemarituzumab induces tumor-specific innate and adaptive immune profile changes. Mice bearing 4T1 tumors were treated with 20 mg/kg IP bemarituzumab (Bema), bemarituzumab-N297Q (Bema-N297Q), or human Fc-IgG1 (hFc-G1) on day 0 (when tumors reached approximately 150 mm3) and day 3, and then euthanized 24 hours later on day 4 for immunohistochemistry analysis. Representative fluorescence of NKp46, PD-L1, and CD3-positive T cells (CD3 T cells) 24 hours post second dose of treatment are shown
To confirm that tumor CD3-positive T-cell infiltration was reliant on bemarituzumab NK cell FcγR engagement, NK cells were depleted with an anti-asialo GM1 antibody (anti-ASGM1) in combination with 2 doses of bemarituzumab as described above in mice harboring 4T1 tumors. After treatment with bemarituzumab alone, NK cells were more numerous. However, this increase was not observed when bemarituzumab was combined with anti-ASGM1, confirming that a majority of NK cells were successfully depleted with a single dose of anti-ASGM1 (Figure 7). Treatment with anti-ASGM1 alone had no effect on CD3-positive T cells within the tumor compared to human Fc-IgG1 control. However, anti-ASGM1 treatment in combination with bemarituzumab lead to a decrease in CD3-positive T cells within the tumor compared to bemarituzumab treatment alone. As demonstrated in Figure 6, treatment with bemarituzumab alone increased the number of PD-L1-positive cells, but when bemarituzumab was given in combination with anti-ASGM1, PD-L1 staining was reduced to the level of human Fc-IgG1 control (Figure 7). Together, these data suggest that depleting NK cells in mice harboring 4T1 tumors leads to an inability to recruit CD3-positive T cells and PD-L1-expressing cells to the tumor in response to bemarituzumab administration.
Figure 7.
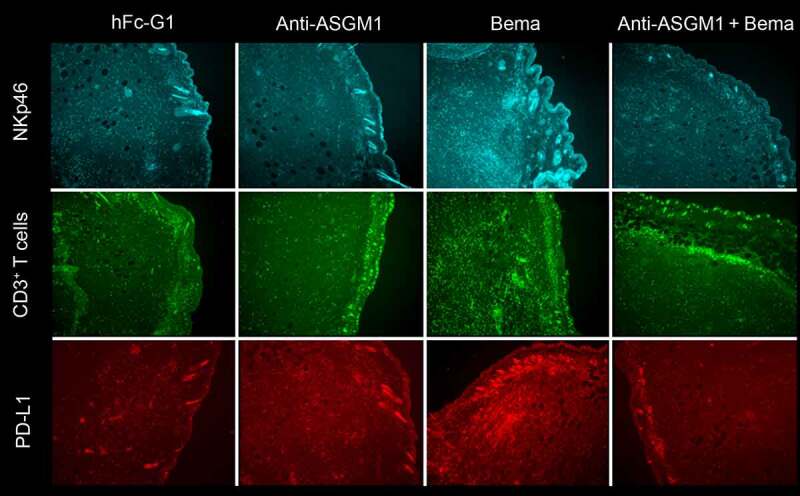
Bemarituzumab-induced increases in PD-L1 and CD3-positive T cells depends on NK cell recruitment. Mice bearing 4T1 tumors were treated with either: human Fc-IgG1 negative control (10 mg/kg, IP) on day 0 (when tumors reached approximately 125 mm3) and day 3 (hFc-G1), anti-ASGM1 (50 mg/kg, IV) once on day 0, bemarituzumab (10 mg/kg, IP) on day 0 and day 3 (Bema), or anti-ASGM1 (50 mg/kg, IV) on day 0 and bemarituzumab (10 mg/kg, IP) on day 0 and day 3 (anti-ASGM1 + Bema). Mice were euthanized on day 4 for immunohistochemistry analysis. Representative fluorescence of NKp46, PD-L1, and CD3-positive T cells (CD3+ T cells) are shown
The observation that bemarituzumab treatment led to NK cell-dependent increases in PD-L1 at the tumor suggested that combining bemarituzumab with PD-1/PD-L1 blockade, such as anti-PD-1 therapy, could result in increased anti-tumor efficacy. To assess this, bemarituzumab in combination with anti-PD-1 (RMP1.14) was tested in mice harboring 4T1 tumors. Anti-PD-1 alone had minimal effect on tumor growth compared to albumin control (Figure 8(a,b)). In contrast, twice weekly bemarituzumab treatment (10 mg/kg, IP) significantly inhibited mean tumor burden as a single agent by approximately 30%. The combination of bemarituzumab and anti-PD-1 led to an additional approximately 20% mean tumor growth inhibition over bemarituzumab alone, suggesting that modulating the 2 pathways together may lead to enhanced and sustained efficacy in tumors with moderate FGFR2b expression, although the difference was not statistically significant.
Figure 8.
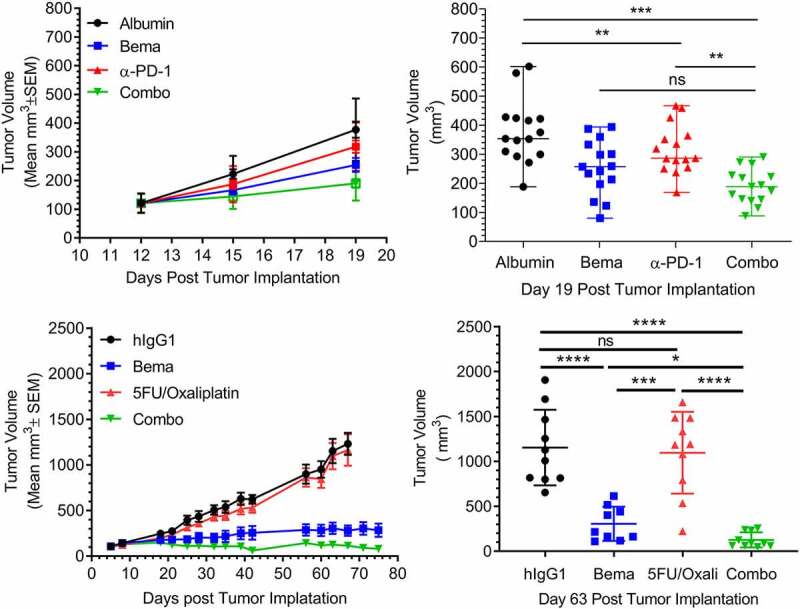
Bemarituzumab displays enhanced anti-tumor activity when combined with anti-PD-1 or chemotherapeutics. a: Tumor volume (mm3) over time in mice bearing 4T1 mammary orthotopic tumors treated with twice per week bemarituzumab (10 mg/kg, IP), anti-PD-1 (5 mg/kg, IP), or bemarituzumab (10 mg/kg, IP) and anti-PD-1 (5 mg/kg, IP) in combination (Combo) are shown. Albumin (10 mg/kg, IP) was used as the negative control. The first day of the treatment was day 12 post tumor implantation. Data are presented as mean ±standard error of mean (SEM); n = 15 per group. b: at day 19 post tumor implantation, mean tumor volumes from mice in panel a were either statistically significant (**p < .01, ***p < .001) or not statistically significant (ns). Each symbol represents an individual animal. c: Tumor volume (mm3) over time in OCUM-2 M tumor-bearing mice treated with twice per week bemarituzumab (5 mg/kg, IP), weekly 5-FU (30 mg/kg, IP) and oxaliplatin (5 mg/kg, IP), or twice per week bemarituzumab (5 mg/kg, IP) and weekly 5-FU (30 mg/kg, IP)/oxaliplatin (5 mg/kg, IP) in combination (Combo) are shown. Human IgG1(5 mg/kg, IP) was used as the negative control (hIgG1). The first day of the treatment was post tumor implantation day 5 for 5-FU/oxaliplatin and day 11 for bemarituzumab. Data are presented as mean ±SEM; n = 9–10 per group. d: at day 63 post tumor implantation, mean tumor volumes from mice in Panel c were either statistically significant (*p < .05, ***p < .001, ****p < .0001) or not statistically significant (ns). Each symbol represents an individual animal. Statistical significance was determined by 1 way ANOVA followed by Tukey’s multiple comparisons
The combination of bemarituzumab with chemotherapeutic regimens commonly used for treatment of patients with gastric cancer was also evaluated in the OCUM-2M xenograft model. Twice weekly bemarituzumab (5 mg/kg, IP) combined with weekly 5-fluorouracil (5-FU) (30 mg/kg, IP) and oxaliplatin (5 mg/kg, IP) demonstrated greater inhibition of tumor growth in the OCUM-2M xenograft model of gastric cancer compared to monotherapy bemarituzumab or 5-FU/oxaliplatin at the doses tested (Figure 8(c,d)). Importantly, the addition of bemarituzumab to chemotherapeutics did not increase toxicity associated with chemotherapy as measured by weight loss in mice up to the last measurement of body weight on Day 32. In other animal models, such as HSC-39 xenografts, the tumors were too sensitive to chemotherapy alone for combination experiments to be informative.
Bemarituzumab PK in rats and cynomolgus monkeys
The PK of bemarituzumab was first tested in rats. Bemarituzumab PK data following a single IV injection at 1.5 mg/kg (4 rats), 10 mg/kg (4 rats), and 30 mg/kg (3 rats) are shown in Figure 9(a). Bemarituzumab exposure increased greater than dose-proportionally from 1.5 mg/kg to 30 mg/kg, likely due to target-mediated drug disposition (TMDD) that was saturable at higher doses. The area under the concentration-time curve from time 0 extrapolated to infinity was 35.8 ± 2.96 μg×day/mL for the 1.5 mg/kg group, 484 ± 61.8 μg×day/mL for the 10 mg/kg group, and 2620 ± 275 μg×day/mL for the 30 mg/kg group (Table 3). Correspondingly, the clearance rates declined as the dose increased from 42.2 ± 3.67 mL/day/kg in the 1.5 mg/kg group, to 20.9 ± 2.51 mL/day/kg in the 10 mg/kg group, and to 11.5 ± 1.27 mL/day/kg in the 30 mg/kg group. Anti-bemarituzumab antibody was not measured in this study. The clearance of bemarituzumab in rats at high doses suggested that these results were in the range for a typical mAb with linear PK.17
Figure 9.
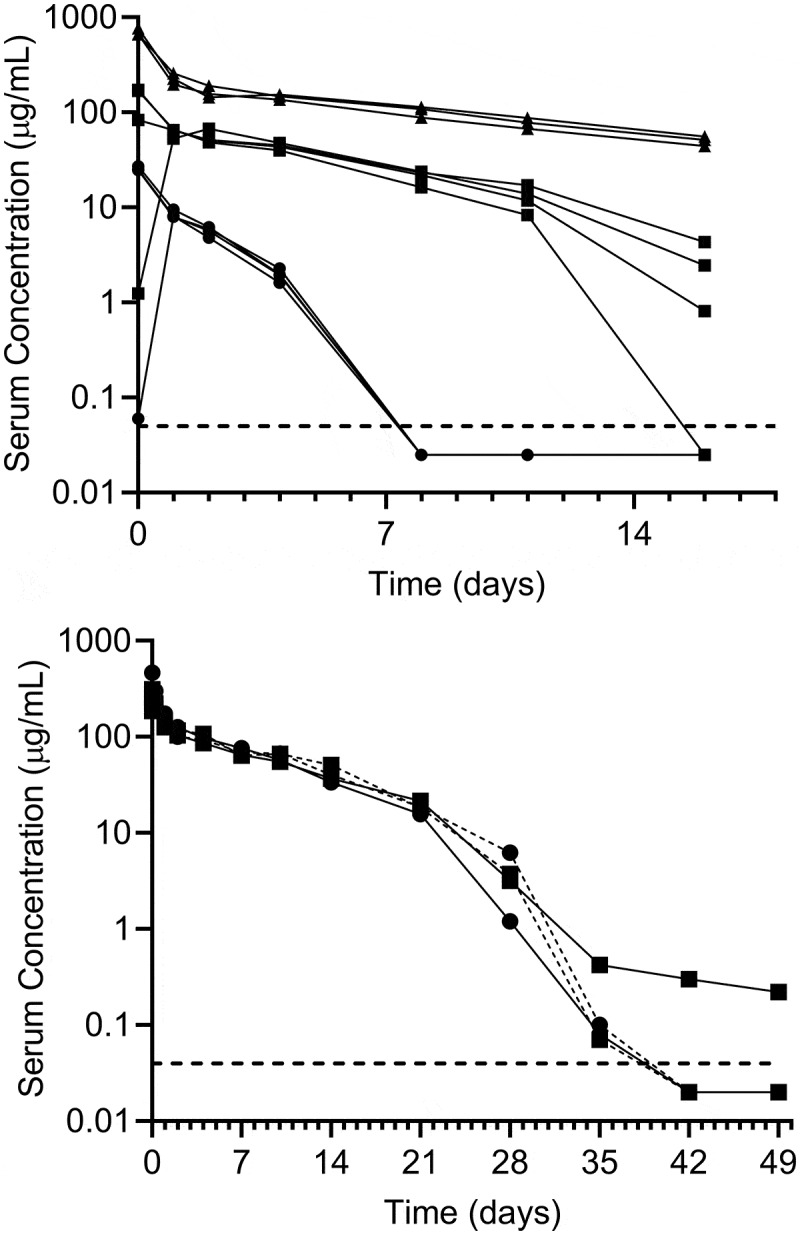
Bemarituzumab demonstrates biphasic disposition in rats and cynomolgus monkeys following a single IV injection. a: Bemarituzumab plasma concentration in rats following a single IV injection at 1.5 mg/kg (●; n = 4), 10 mg/kg (■; n = 4), or 30 mg/kg (▲; n = 3). Based on the concentration profiles, 2 animals (1 in the 1.5 mg/kg group and 1 in the 10 mg/kg group) were suspected of inadvertent subcutaneous dosing. b: Plasma concentrations of bemarituzumab and FPA144-F in cynomolgus monkeys following a single IV injection of bemarituzumab 10 mg/kg (–■̶; n = 1), bemarituzumab 13.5 mg/kg (–●̶; n = 1), or FPA144-F 10 mg/kg (-●-, -■-; n = 2). Each symbol represents a concentration time point for an individual animal. The lower limit of quantification (–) was 50 ng/mL
Table 3.
Summary of rat and cynomolgus monkey PK parameters
| Species | Treatment | Dose (mg/kg) | PK Parameter |
||
|---|---|---|---|---|---|
| Cmax (µg/mL) | AUC0-inf (µg×day/mL) | CL (mL/day/kg) | |||
| Rat | Bemarituzumab | 1.5 (n = 3)a | 25.6 ± 1.30a | 35.8 ± 2.96a | 42.2 ± 3.67a |
| 10 (n = 3)a | 141 ± 50.2a | 484 ± 61.8a | 20.9 ± 2.51a | ||
| 30 (n = 3) | 699 ± 58.7 | 2620 ± 275 | 11.5 ± 1.27 | ||
| Cynomolgus monkey | Bemarituzumab | 10 (n = 1) | 257 | 1380 | 7.20 |
| 13.5 (n = 1) | 466 | 1510 | 8.88 | ||
| FPA144-F (the fucosylated version) | 10 (n = 1) | 297 | 1470 | 6.72 | |
| 10 (n = 1) | 313 | 1570 | 6.48 | ||
AUC0-inf = area under concentration-time curve from time 0 extrapolated to infinity; CL = clearance; Cmax = maximum observed plasma concentration.
Bemarituzumab or FPA144-F was administered as a single IV injection at the indicated doses. Plasma samples were collected at various time points following the dose, and concentrations were determined via ELISA. The PK parameters for rats are displayed as mean ±standard deviation (n = 3 per dose).
aTwo rats (1 of 4 in both the 1.5 and 10 mg/kg groups) were suspected of inadvertent subcutaneous dosing and therefore removed from the PK analysis.
In cynomolgus monkeys, bemarituzumab and FPA144-F had comparable plasma concentration-time profiles following a single IV injection at either 10 mg/kg (3 monkeys) or 13.5 mg/kg (1 monkey) (Figure 9(b)). Dose proportionality could not be assessed since a range of doses were not tested. A rapid drop in plasma concentration was observed after Day 21 following administration of bemarituzumab or the fucosylated version of bemarituzumab (FPA144-F), suggesting the presence of anti-drug antibodies (ADA) and/or TMDD. All 4 monkeys were tested for ADAs at pre-dose and Day 49, and only 1 monkey receiving bemarituzumab showed a weak positive signal for ADAs on Day 49, without any apparent effect on PK. These data suggest the fast clearance in the low concentration range was likely due to TMDD. The clearance was estimated to be approximately 6–9 mL/day/kg for both bemarituzumab and FPA144-F in cynomolgus monkeys (Table 3).
Together, the consistency of nonspecific clearance across species from these PK studies demonstrated that bemarituzumab had PK characteristics consistent with a typical mAb therapeutic, supporting the prospect for a predictable bemarituzumab PK profile in humans.
Toxicology studies in rats and cynomolgus monkeys
Repeat-dose Good Laboratory Practice (GLP) toxicity studies were performed in rats and cynomolgus monkeys. In both species, bemarituzumab was dosed IV once weekly for 13 weeks at doses of 1, 5, and 100 mg/kg, followed by 9-week (rat) or 15-week (cynomolgus monkey) recovery phases.
Bemarituzumab administered to rats resulted in treatment-related findings at all dose levels with a clear dose relationship: most of the effects were more pronounced for animals given doses of 5 mg/kg and 100 mg/kg. The most prominent findings were incisor abnormalities (missing incisors, malocclusion, and discolored white teeth) that were accompanied by body weight loss and lack of weight gain caused by the decreased food intake due to the incisor deformation. Histopathologically, dysgenesis of the incisor was observed and the oral tissue demonstrated increases of ulceration and inflammatory cell infiltrates, most likely caused by the teeth malformation. Dysgenesis of the upper incisor teeth were characterized by primary and secondary changes. Primary changes included the complete loss of the enamel organ (replacement of the enamel organ by a thick zone of dense connective tissue resembling periodontal membrane) and disorganization of the pulp cavity and associated odontoblast and dentin layers. Primary changes were regularly accompanied by changes interpreted to be secondary to incisor malformation. These included remodeling of the tooth alveolus, increased thickness of the periosteum, scalloping of bone at the alveolar margin, formation of new woven bone at the alveolar margin, and the extension of new bone into the alveolar space and/or malformed tooth. The changes in teeth were consistent with the observation that FGFR2b signaling controls regeneration of rodent incisors, which is a rodent-specific phenomenon.18–20
Bemarituzumab-related ophthalmic findings of corneal dystrophy were observed during the dosing phase in the eyes of some rats in the 5 and 10 mg/kg groups. Microscopic findings in the ocular tissues of these animals were corneal epithelial atrophy, corneal neutrophil infiltrates, and anterior chamber neutrophil infiltrates in the eye and Harderian gland (atrophy). Atrophy of the corneal epithelium was characterized by thinning of the corneal epithelium from the typical 5–7 cell layers observed in control animals to 1–3 cell layers in the affected animals. The observed changes in the cornea were consistent with the reported role of FGFR2b in cornea development and maintenance.21,22
Bemarituzumab-related atrophy of the mammary gland was observed in a subset of rats in all dose groups and both genders. The microscopic finding in mammary gland may relate to the importance of FGFR2b signaling for the survival and proliferation of mammary luminal epithelium.23 Additionally, administration of bemarituzumab resulted in exacerbation of background microscopic findings in the prostate gland of male rats in all dose groups, the nonglandular stomach of male and female rats given doses of 5 and 100 mg/kg, and the lung of male and female rats given doses of 100 mg/kg. Apart from the effects on incisors, recovery was evident at the end of the recovery phase for all described findings. The highest non-severely-toxic dose (HNSTD) for bemarituzumab in rats was determined to be 1 mg/kg when given weekly for 13 weeks with a 9-week recovery phase based on the observation that all findings in the 1 mg/kg dose group were minimal, without clinical consequences, and recoverable.
Bemarituzumab was well tolerated in cynomolgus monkeys. There were no bemarituzumab-related effects in clinical observations, ophthalmic exams, safety pharmacology (cardiac, respiratory, or central nervous system) or clinical pathology (hematology, coagulation, clinical chemistry, urinalysis). There were no bemarituzumab effects on the teeth or oral tissues. At terminal necropsy, dose-dependent atrophy of the corneal epithelium was observed in some cynomolgus monkeys given 5 mg/kg and 100 mg/kg, similar to the rat findings. Atrophy was characterized by bilateral thinning of the corneal epithelium from the typical 4–6 cell layers observed in control animals to 2–3 cell layers. The corneal findings were not evident in recovery animals, indicating complete recovery. A dose-dependent atrophy of the mammary gland was also observed in cynomolgus monkey females in all dose groups. This atrophy was not observed at the recovery necropsy, demonstrating complete recovery after the 15-week recovery period. Since the microscopic findings in the cornea and mammary were not associated with clinical sequelae and were recoverable, neither finding was considered adverse. The HNSTD for bemarituzumab in cynomolgus monkeys was considered to be at or above 100 mg/kg when given weekly for 13 weeks with a 15-week recovery phase.
Prediction of human PK of bemarituzumab based on cynomolgus monkey data only
Bemarituzumab PK data from the single IV dose PK study in cynomolgus monkeys up to Day 21 were used to project bemarituzumab linear PK in humans using a species-invariant time method.17 The projected human plasma concentration-time profiles obtained by this method were described well by a linear, 2-compartment model (Figure 10). The following human PK parameter estimates of bemarituzumab were projected: clearance of 5.07 mL/day/kg and volume of distribution phase for the central compartment (V1) of 42.5 mL/kg. Both the projected clearance and V1 were in the range of a typical human IgG1 antibody with linear PK.17 The predicted human linear clearance and V1 were comparable to the population PK estimation of human clearance and V1 (5.43 mL/day/kg and 56.2 mL/kg, respectively) for a typical male patient at 61 kg with albumin at 3.7 g/dL based on PK data from the Phase 1 trial.16
Figure 10.
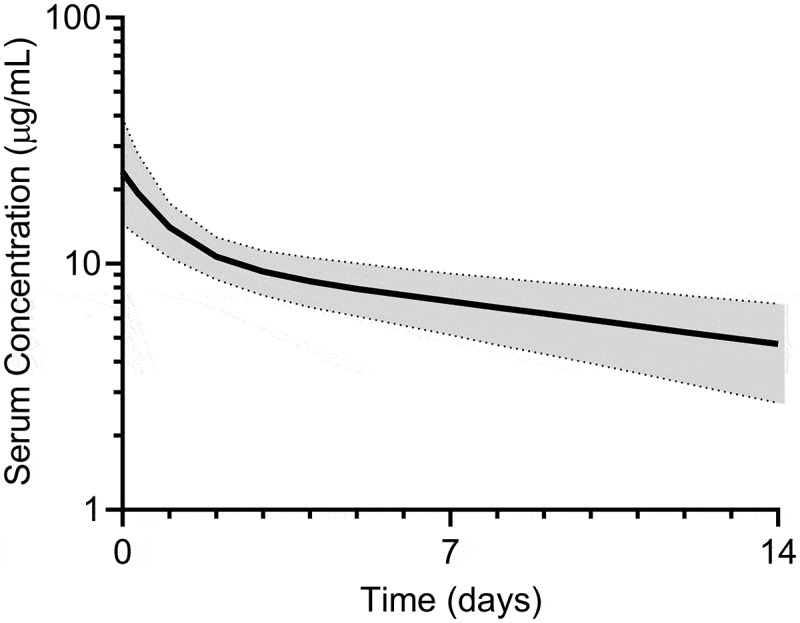
Projected human plasma concentration using PK data from cynomolgus monkeys. Predicted bemarituzumab plasma concentration-time profile (median [solid line], 5% and 95% quantiles [dashed lines], 90% prediction interval [shaded area]) in humans following 1 mg/kg IV administration. Bemarituzumab concentration-time profiles were scaled from cynomolgus monkey PK data using Dedrick approach with exponent of 0.85 and 1 for clearance and V1, respectively
Discussion
Bemarituzumab is a humanized, afucosylated IgG1 kappa mAb that specifically binds to and blocks signaling through FGFR2b, which has been shown to be dysregulated in subsets of patients with gastric,4,5,11,24,25 lung,26 breast,14 ovarian,13 and bile duct cancers,12 among others. Here, we summarize the key nonclinical studies that supported moving bemarituzumab into clinical trials, including assessment of binding affinity to both FGFR2b and FcγRIIIa, in vitro activities through either binding to FGFR2b or FcγRIIIa, anti-tumor activities in xenograft models, PK and toxicology studies in both rats and cynomolgus monkeys, and projection of PK profiles in humans.
Bemarituzumab inhibited FGFR2 signaling and tumor growth in mice bearing FGFR2-amplified tumors (SNU-16 and OCUM-2M human gastric cancer cells, respectively). Bemarituzumab also inhibited tumor growth in mice bearing 4T1 mammary orthotopic tumors that express FGFR2b, but to a lesser degree than FGFR2 gene-amplified or FGFR2b-overexpressed xenografts. However, anti-tumor activity with bemarituzumab was not observed in NUGC-4 and NCI-87 gastric cancer xenografts with low levels of FGFR2b expression (data not shown). These results are consistent with bemarituzumab’s mechanism of action through blockade of FGFR2b signaling.
Bemarituzumab was also specifically engineered to elicit enhanced ADCC against FGFR2b-overexpressing tumors. Bemarituzumab lacks a fucosyl residue in the Fc domain and is produced in FUT8-knockout CHO cells.27 The lack of the fucosyl residue results in enhanced affinity for activating FcγRIIIa and subsequent increased potency in stimulating ADCC activity.
Supporting the contribution of ADCC to mediate the potential efficacy of bemarituzumab, the 4T1 model in immune-competent mice demonstrated both a reduction in tumor burden and a persistent recruitment of NK cells with bemarituzumab treatment. However, a modified antibody lacking Fc effector function neither inhibited tumor growth nor led to the recruitment of NK cells. Together these data support a potential role for ADCC as a mechanism of bemarituzumab tumor growth inhibition. In addition, treatment with bemarituzumab increased PD-L1-expressing cells within the tumor microenvironment, providing a strong rationale that bemarituzumab may combine effectively with PD-1/PD-L1 blockade for additional tumor growth inhibition, which was observed when mice bearing 4T1 mammary orthotopic tumors were treated with bemarituzumab and anti-PD-1.
Bemarituzumab also demonstrated enhanced anti-tumor activity in combination with chemotherapeutic regimens commonly used for treatment of patients with gastric cancer. Advanced-stage gastric cancer is heterogeneous.28 Specifically relevant to bemarituzumab, in the clinic, a single biopsy to test for FGFR2 amplification has a reported sensitivity of only 40% due to genetic variability.29 Therefore, combining orthogonal therapies (such as chemotherapy with an anti-FGFR2b therapy and/or an anti-PD-1 therapy) has the potential to improve patient outcomes.
The mAb clearance pathways appear to determine whether enhancement of binding to FcγR affects the mAb PK. Leabman et al.30 characterized the PK of 3 antibody variants with increased FcγRIIIa binding affinity caused by afucosylation at asparagine residue 297 in the Fc domain (N297) and compared them to their corresponding wild-type antibody in cynomolgus monkeys, and concluded that altered FcγRIIIa binding affinity did not affect PK. Mortensen et al.31 reported that, of 2 high-affinity anti-IgE antibodies (HAE1 and HAE2), HAE2, which contains an Fc region altered to reduce binding to FcγR, has greater exposure relative to HAE1 due to decreased clearance of HAE2:IgE complexes by the Fcγ-mediated clearance pathway. We found that bemarituzumab, an afucosylated IgG1 kappa mAb directed against FGFR2b with >20-fold higher affinity to FcγRIIIa compared with the wild-type mAb, maintained a similar PK profile in cynomolgus monkeys. This suggests that formation of complexes like anti-IgE mAb:IgE might not contribute to bemarituzumab clearance, which is consistent with the low expression of FGFR2b in normal monkeys. Since internalization was observed following bemarituzumab binding to FGFR2b in tumor cell lines (data not shown), bemarituzumab-induced internalization of FGFR2b might contribute to the target-mediated clearance in general.
Bemarituzumab was well tolerated in cynomolgus monkey when administered once per week for 13 weeks up to 100 mg/kg. However, administration to rats once per week for 13 weeks at 1, 5, or 100 mg/kg resulted in treatment-related findings at all dose levels. With the exception of the rodent-specific changes of the incisors, some degree of recovery was evident for all findings at the end of the recovery phase. Since all findings in the 1 mg/kg dose group were minimal, without clinical consequences, and recoverable, the HNSTD was determined to be 1 mg/kg when given weekly for 13 weeks.
The first-in-human starting dose of bemarituzumab in trial FPA144-001 (NCT02318329) was 0.3 mg/kg every 2 weeks.32 The human equivalent dose of the HNSTD of 100 mg/kg in cynomolgus monkey is 32.3 mg/kg, and the safety factor based on a human equivalent dose for 0.3 mg/kg is 108. Although the human equivalent dose of the HNSTD of 1 mg/kg in rat was 0.17 mg/kg, since no lesions in the tongue were observed in monkeys even at the highest dose level of 100 mg/kg, and the mRNA expression pattern of FGFR2b in adult human tissues is very low in the tongue, and if such lesions occurred, they would be monitorable in the clinic by oral examination, the cynomolgus monkey was considered the more relevant species and the starting dose of 0.3 mg/kg in human was deemed to be reasonable.
The PK of bemarituzumab in cynomolgus monkeys was used to predict the nonspecific PK of bemarituzumab in humans. The cynomolgus monkey was selected to scale PK to humans because it is generally considered the most relevant species for predicting the human PK of mAbs.17 The binding affinity of bemarituzumab in monkeys and humans are comparable (0.770 nM vs 0.579 nM), and the clearance and volume distribution for a mAb that lacks known endogenous host targets in humans can be reasonably predicted based on monkey data, since disposition and elimination pathways are very similar in the 2 species.17,33,34 Cynomolgus monkey data suggested that TMDD and not ADA-mediated clearance or neutralization of bemarituzumab was likely responsible for the faster clearance observed after Day 21, therefore only PK data up to Day 21, which was dominated by nonspecific clearance, was used for the human PK projection. In addition, only linear PK in human is clinically relevant. In monkeys, the estimated clearance was approximately 6–9 mL/day/kg, which is in the expected range of clearance of a typical human IgG1.17 The predicted clearance in humans of 5.07 mL/day/kg and V1 of 42.5 mL/kg are also as expected for a typical human IgG1.17,35,36 The Phase 1 PK data in patients further confirmed the human PK projections: the PK of bemarituzumab was observed to be linear in the dose range of 1–15 mg/kg, with linear clearance as 5.43 mL/day/kg and V1 as 56.2 mL/day/kg.16
In summary, bemarituzumab has sub-nanomolar affinity to FRFR2b with >20-fold enhanced binding affinity to FcγRIIIa compared with the fucosylated wild-type mAb, demonstrated both in vitro and in vivo biological activities, was well tolerated in cynomolgus monkeys with nonspecific clearance similar to typical therapeutic antibodies, and could be reasonably scaled to humans. In addition, the assessment of adequate toxicity in rats, as well as cynomolgus monkey as the most relevant species, enabled the successful progression into clinical trials.
Materials and methods
All research with animals reported here was conducted in compliance with applicable animal welfare acts and with the formal approval of local animal care committees.
Antibody reagents
Bemarituzumab was produced in a CHO cell line that lacks the FUT8 gene (a1, 6-fucosyltransferase) at Five Prime Therapeutics, Inc. (FivePrime; South San Francisco, CA) for the nonclinical studies summarized here. GAL-FR21 was the parent version of bemarituzumab with binding specificity to FGFR2b.37 Bemarituzumab-N297Q was also produced at FivePrime (South San Francisco, CA). The other antibody reagents used are described in the corresponding methods for each study below.
Cell lines
OCUM-2M is a FGFR2 gene-amplified, FGFR2b protein overexpressing gastric cancer cell line that was obtained from Public University Corporation Osaka City University, Japan (Source: Dr. Masakazu Yashiro). SNU-16 gastric cell lines (with FGFR2 amplification) were commercially acquired (ATCC; Catalog #CRL-5974). 4T1 is a derived breast adenocarcinoma cell line and was commercially acquired (ATCC; Catalog #CRL-2539). All cell lines were mycoplasma-negative tested by IDEXX Laboratories (Columbia, MO) using a real-time polymerase chain reaction.
Binding affinity using surface plasmon resonance
The binding affinity of bemarituzumab for FGFR2b ECD IgG fusion protein (FGFR2b-Fc) of rat, cynomolgus monkey, or human was measured using surface plasmon resonance (Biacore T200, GE Healthcare Life Science, Marlborough, MA). Bemarituzumab was immobilized on a dextran chip using the amine coupling kit and 100 mM ethylenediamine in 100 mM sodium borate, pH 8.0, was used as the blocking reagent. Six different concentrations (0–500 nM) of FGFR2b-Fc proteins diluted in HEPES-buffered saline with 0.05% surfactant P20 running buffer were flowed over the immobilized antibody.
To determine binding affinity for FcγRIIIa (V158) by surface plasmon resonance, bemarituzumab or FPA144-F was captured on the chip via Protein A. Protein A was covalently attached to a dextran chip using the same protocol as above. Five concentrations (0–1000 nM) of FcγRIIIa (V158) were diluted in running buffer and flowed over the captured antibody.
The association constant, dissociation constant, and affinity for bemarituzumab binding to human FGFR2b and human FcγRIIIa (V158) were calculated using the Biacore T100 Evaluation Software 1:1 binding model.
In vitro inhibition of SNU-16 cell FGFR2 phosphorylation
The effect of bemarituzumab on tumor cell FGFR2 phosphorylation in vitro was measured in SNU-16 cells. Approximately 10,000 SNU-16 cells were plated onto a 96-well plate in RPMI media (Mediatech; Catalog #MT10-041-CV) with 0.05% bovine serum albumin (BSA) (Bovine Albumin Fraction V Solution [7.5%]; Life Technologies; Catalog #15260037), which were incubated at 37°C with 5% CO2 for 4 hours. Next, SNU-16 cells were treated with varying concentrations of bemarituzumab diluted in RPMI media with 0.05% BSA for 30 minutes. SNU-16 cells were then treated with 100 ng/mL FGF7 (Peprotech; Catalog #100-19) with 1 µg/mL heparin (final concentrations) diluted in RPMI media with 0.05% BSA, and incubated at 37°C with 5% CO2 for 5 minutes. Cold 1X RIPA lysis buffer (EMD Millipore; Catalog #20-188) containing 1X Halt Protease and Phosphatase Inhibitor Cocktail (VWR; Catalog #PI78441) was then added, and the plate was sealed and incubated on ice for 10 minutes. The plate was subsequently shaken for 5 minutes, and stored at −80°C until the enzyme-linked immunosorbent assay (ELISA) procedure.
Phospho-FGFR2 was measured via ELISA using the Phospho-FGF R2 Alpha DuoSet IC (R&D Systems; Catalog #DYC684-2) following the manufacturer’s protocol, except with half of the suggested volumes. After washing out the detection antibody, 50 μL/well of 1:1 premixed SuperSignal ELISA Pico Chemiluminescent Substrate (Thermo Fisher Scientific; Catalog #37069) was added. Plates were protected from light, shaken gently for 30 seconds, and then immediately read on an EnVision Microplate Reader (Perkin Elmer). The experiment was performed in triplicate.
In vitro inhibition of SNU-16 cell proliferation
The effect of bemarituzumab on tumor cell proliferation in vitro was measured in SNU-16 cells. Approximately 5,000 SNU-16 cells were plated onto a 96-well plate in RPMI media (Mediatech; Catalog #MT10-041-CV) with 0% fetal bovine serum (FBS) (Mediatech; Catalog #35-010-CV) to serum starve the cells, which were incubated at 37°C with 5% CO2 for 16 hours/overnight. Next, SNU-16 cells were treated with varying concentrations of bemarituzumab diluted in RPMI media with 0% FBS for 30 minutes. SNU-16 cells were then treated with 100 ng/mL FGF7 (Peprotech; Catalog #100-19) with 1 µg/mL heparin (final concentrations) diluted in RPMI media with 0% FBS, and incubated at 37°C with 5% CO2 for 4 days. To harvest cells for the cell viability assay, the 96-well plate was spun down at 2000 revolutions per minute for 5 minutes. Half of the total volume in the well (approximately 100 μL) was then slowly aspirated without disrupting the cells. CellTiter-Glo reagent (100 μL) from the CellTiter-Glo Luminescent Cell Viability Assay (Promega; Catalog #G7571) was then added, following the manufacturer’s protocol for preparation. Plates were shaken for 2 minutes and then incubated at room temperature for 8 minutes. Plates were protected from light during incubation with the CellTiter-Glo reagent. Cell proliferation was read via luminescence on an EnVision Microplate Reader (Perkin Elmer). The experiment was performed in triplicate.
In vitro ADCC activity measurement
The target cell in this assay was an engineered cell line expressing the full-length human FGFR2b described as Ba/F3 FGFR2b, and the effector cells were peripheral blood mononuclear cells obtained fresh from individual human donors (AllCells; Catalog #PB001). As a negative control, bemarituzumab was also tested using a target cell line that was engineered to express the FGFR2c variant of the receptor (Ba/F3 FGFR2c cells), to which bemarituzumab does not bind. ADCC was measured by combining freshly purified peripheral blood mononuclear cells (effector cells) from a single human donor with Ba/F3 FGFR2b cells (target cells) at a 25:1 ratio for 16 hours for both bemarituzumab and fucosylated anti FGFR2b antibody (FPA144-F).
The CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega; Catalog #G1780) was used to assess ADCC. This assay quantitatively measures lactate dehydrogenase, a stable cytosolic enzyme that is released on cell lysis, or loss of plasma membrane integrity, or necrosis. Released lactate dehydrogenase in culture supernatants was measured with a 30-minute coupled enzymatic assay that resulted in the conversion of a tetrazolium salt (INT) into a red formazan product. The amount of color formed is directly proportional to the number of lysed cells. Where possible, maximal lysis was determined in the presence of 5% Triton X-100, and spontaneous release was determined in the absence of antibody. Percentage of specific lysis was calculated as follows as a percentage of maximal lysis less spontaneous release: (experimental-spontaneous release)/(maximal-spontaneous release)×100. The percentage of specific lysis is positively correlated with ADCC activities.
Efficacy studies in OCUM-2M tumor-bearing mice
Tumors were established by subcutaneously inoculating female CB17 SCID mice (ages 7–8 weeks) from Charles River with 5 × 105 OCUM-2M cells suspended in a solution of 50%/v of Dulbecco’s phosphate-buffered saline (PBS) (Cellgro; Catalog #21-031) and 50%/v of MatriGel (BD Biosciences; Catalog #354234).
In the single-agent tumor growth inhibition study, treatment began when tumors reached volumes of 125–150 mm3, which was 4 days post tumor cell implantation. Dose-responses were assessed using twice per week IV doses of bemarituzumab at 1, 1.5, 2, 3, and 5 mg/kg (15 mice/group). Human albumin U.S.P (Grifols; Catalog #NDC 61953–0002-1) was used as the control (twice weekly doses of 5 mg/kg, IV).
In the combination tumor growth inhibition study with bemarituzumab and chemotherapy, tumor volume was assessed in 4 groups of mice (9–10 mice/group): 1 group received twice weekly doses of bemarituzumab (5 mg/kg, IP); another group received weekly 5-FU (30 mg/kg, IP) and oxaliplatin (5 mg/kg, IP); a third group received both twice weekly doses of bemarituzumab (5 mg/kg, IP) and weekly 5-FU (30 mg/kg, IP) and oxaliplatin (5 mg/kg, IP); and the fourth group received twice weekly doses of human IgG1 (5 mg/kg IP; FivePrime) as the control group. Weekly 5-FU and oxaliplatin treatment began on Day 5 post tumor cell implantation, when tumors reached volumes of 100–150 mm3. Bemarituzumab and human IgG1 treatment began on Day 11 post tumor cell implantation.
For both the single-agent and combination studies, tumor volumes were measured using a digital caliper and the formula (L× W2)/2, where L = tumor length and W = tumor width.
Western blot analyses using tumors collected from SNU-16 xenografic model
Tumors were established by subcutaneously inoculating female CB17 SCID mice (ages 7–8 weeks) from Charles River with 5 × 106 SNU-16 cells suspended in a solution of 50%/v of RPMI (Mediatech; Catalog #MT10-041-CV) and 50%/v of MatriGel (BD Biosciences; Catalog #354234).
To assess the effects of a single dose of bemarituzumab on FGFR signaling in SNU-16 tumor-bearing mice, bemarituzumab (10 mg/kg, IV) was administrated when the average tumor size reached 200 mm3. Tumors from mice treated with bemarituzumab were collected at 2, 6, 24, and 72 hours post treatment (3 mice per time point) for western blot analysis. Tumors were collected at 2 hours from naive animals (2 mice) and at 24 hours post treatment from the IgG (10 mg/kg; Galaxy Biotech) treatment group (3 mice) as controls.
To assess the effects of repeated doses of bemarituzumab on FGFR signaling in SNU-16 tumor-bearing mice, bemarituzumab (10 mg/kg, IP) was administrated twice weekly once the average tumor size reached 100 mm3, which took 7 days post tumor cell implantation in this study. The control group was treated with twice weekly human albumin U.S.P (10 mg/kg, IP; Grifols; Catalog #NDC 61953–0002-1). Tumors from both groups (3 per group) were collected 89 days post tumor cell implantation (82 days post first bemarituzumab/albumin treatment) for western blot analysis.
Each individual tumor collected for western blot analysis was lysed and homogenized with RIPA lysis buffer (EMD Millipore; Catalog #20-188) and an MP Biomedical homogenizer FastPrep-24 Instrument (MP Biomedical; Catalog #116002500), then the Pierce BCA Protein Assay Kit and Reagents (VWR International; Catalog #PI23227) were used to obtain total protein concentrations. Standard western blot procedures were used with the same amount of total protein loaded in each lane. Primary antibodies included anti-phospho-FGFR1-4 (Y653/Y654) (R&D Systems; Catalog #AF3285), anti-phospho-FRS2 Y436 (R&D Systems; Catalog #AF5126), anti-FRS2 (clone 462910) (R&D Systems; Catalog #MAB4069), and anti-GAPDH (GeneTex; Catalog #GTX627408). Secondary detection was performed using peroxidase AffiniPure Goat Anti-Mouse IgG, Fcγ fragment specific (Jackson ImmunoResearch Laboratories; Catalog #115-005-071). For chemiluminescence detection, Pierce ECL Western Blotting Substrate (VWR International; Catalog #PI32106) was used.
Efficacy studies in the 4T1 mouse mammary syngeneic tumor model
For the 4T1 tumor growth inhibition studies, tumors were established in female BALB/c mice (Charles River) by orthotropic injection under the 4th mammary papilla (teat) from the head. Each mouse was inoculated with 5 × 104 4T1 cells (50 µL of the cells suspended in PBS at 1 × 106 4T1 cells/mL concentration). In the first experiment, after tumors reached 125 mm3 ± 25 mm3, mice were sorted according to tumor size and dosed with either human Fc-IgG1 (Bio X Cell; Catalog #BE0096), bemarituzumab, or bemarituzumab-N297Q via biweekly IP injections at 10 mg/kg each (15 mice/group). The first day of treatment was Day 11 post tumor implantation. In the second experiment, after tumors reached 100 mm3 ± 25 mm3, mice were sorted according to tumor size and dosed with either human albumin (Grifols; Catalog #NDC 61953–0002-1) at 5 mg/kg, anti-PD-1 (Bio X Cell; Catalog # BP0146) at 5 mg/kg, bemarituzumab at 10 mg/kg, or anti-PD-1 at 5 mg/kg in combination with bemarituzumab at 10 mg/kg via biweekly IP injections (15 mice/group). The first day of treatment was Day 12 post tumor implantation. For both experiments, tumor volumes were measured using a digital caliper and the formula (L× W2)×3.14/6, where length (L) is a larger value than width (W).
For immunohistochemistry analyses, BALB/c mice (Charles River) were inoculated with 5 × 104 4T1 cells by orthotropic injection as described above and 2 separate experiments were conducted. In the first experiment, once tumors reached approximately 150 mm3 (Day 0), mice were sorted according to tumor size into 3 dosing groups (5 mice per group): bemarituzumab (20 mg/kg, IP), bemarituzumab-N297Q (20 mg/kg, IP), or human Fc-IgG1 (20 mg/kg, IP; Bio X Cell; Catalog #BE0096). Mice received 2 doses of their respective treatment: 1 dose on Day 0, and a second dose on Day 3. Mice were euthanized 24 hours post dose on Day 4 and processed for histology. In the second experiment, once tumors reached approximately 125 mm3 (Day 0), mice were sorted according to tumor size into 4 dosing groups (10 mice per group): Group 1 was dosed with human Fc-IgG1 control (10 mg/kg, IP) on Day 0 and 3; Group 2 was dosed with rabbit anti-asialo GM1 (50 mg/kg, IV; Wako Chemicals), an antibody designed to deplete NK cells, once on Day 0; Group 3 was dosed with bemarituzumab (10 mg/kg, IP) on Day 0 and Day 3; Group 4 was dosed with rabbit anti-asialo GM1 antibody (50 mg/kg, IV) on Day 0 and bemarituzumab (10 mg/kg, IP) on Day 0 and Day 3. Mice were euthanized 24 hours post dose on Day 4 and processed for histology.
For histology, mice were euthanized with CO2 and then perfused with PBS, pH 7.4. Briefly, the mouse chest was opened rapidly, and a syringe with a 20-gauge needle was used to infuse 40 mL of PBS into the aorta via an incision in the left ventricle. Blood and PBS exited through an opening in the right atrium. The 4T1 orthotopic tumors were removed and immersed in 10% neutral buffered formalin at 4°C. After 2 hours the tissues were rinsed 3 times with PBS and then transferred in 30% sucrose in PBS overnight. The next day the tumors were frozen in optimal cutting temperature compound and stored at −80 C.
Serial sections (20 µm thick) of each tumor were cut. Sections were dried on Superfrost Plus slides (VWR) for 1 to 2 hours. Specimens were permeabilized with PBS containing 0.3% Triton X-100 and incubated in 5% goat normal serum in PBS 0.3% Triton X-100 (blocking solution) for 1 hour at room temperature to block nonspecific antibody binding. After 1 hour the blocking solution was removed and the sections were incubated in the primary antibodies overnight. To detect NK cells, sections were incubated with rat anti-NKp46 (CD335; Biolegend; Catalog# 137602) diluted 1:500 in blocking solution. To detect PD-L1, sections were incubated with rat anti-PD-L1 (eBioscience; Catalog #14-5982-82) diluted 1:500 in blocking solution. To detect CD3-positive T cells, sections were incubated with hamster anti-CD3 antibody (BD Biosciences; Catalog #553058) at 1:500 in blocking solution. NK cells and PD-L1 staining were performed in serial sections, as both primary antibodies were generated in rat. Secondary antibody only negative control specimens were incubated in 5% normal serum rather than primary antibodies.
The next day, after rinsing with PBS containing 0.3% Triton X-100, specimens were incubated for 4 hours at room temperature with the following secondary antibodies diluted 1:400 in PBS: Alexa Fluor 594-labeled goat anti-rat (Jackson ImmunoResearch; Catalog #112-585-167) and Alexa Fluor 488-labeled goat anti-hamster (Jackson ImmunoResearch; Catalog #127-545-160). After, specimens were rinsed with PBS containing 0.3% Triton X-100, then were fixed in 1% paraformaldehyde, rinsed again with PBS, and mounted in Vectashield with DAPI (Vector; Catalog #H-1200). DAPI was used to label the cell nuclei. Specimens were examined with a Zeiss Axiophot 2 plus fluorescence microscope equipped with an AxioCam HRc camera.
PK in rats and cynomolgus monkeys
Bemarituzumab PK was tested in female Sprague Dawley rats administered as a single IV injection at 1.5, 10, or 30 mg/kg with 4 animals/dose level. The plasma samples were collected on 0.05, 24, 48, 96, 192, 264, and 384 hours following the dose. The samples were stored at −80°C before assay. In the 30 mg/kg group, data from only 3 animals are available (samples were not received for 1 animal). Two animals (1 in the 1.5 mg/kg group and 1 in the 10 mg/kg group) were suspected of inadvertent subcutaneous dosing and therefore removed from the analysis of PK parameters displayed in Table 3.
Bemarituzumab PK was tested in cynomolgus monkeys administered as a single IV injection at 10 or 13.5 mg/kg per animal (1 monkey/dose). FPA144-F was also tested in cynomolgus monkeys administered as a single IV injection at 10 mg/kg (2 monkeys). The plasma samples were collected at 0.083, 0.5, 1, 2, 6, 24, 48, 96, 168, 240, 336, 504, 672, 840, 1008, and 1176 hours following the dose. Anti-bemarituzumab antibody samples were collected at pre-dose and at Day 49 post-dosing. The samples were stored at −80°C before assay.
Determination of bemarituzumab concentration in rats and cynomolgus monkeys
ELISAs were developed and validated to measure bemarituzumab plasma concentration in rats and cynomolgus monkeys. Briefly, human FGFR2b ECD fused to the Fc domain of IgG1 (FGFR2b-Fc) was used to capture bemarituzumab to a standard microtiter plate. The captured bemarituzumab was detected with horseradish peroxidase-conjugated rabbit anti-human IgG-F(ab’)2 antibody (Jackson ImmunoResearch; Catalog #309-035-006).
The lower limit of quantification (LLOQ) of the method in rat plasma was 50 ng/mL. The overall inter-assay precision calculated as percent coefficient of variation (CV) was 2.3% and the inter-assay accuracy calculated as percent relative error (RE) was 3.1%. The LLOQ of the method in monkey plasma was 50 ng/mL. The overall inter-assay precision calculated as percent CV was 13.8% and the inter-assay accuracy calculated as percent RE was 3.9%.
Measurement of anti-bemarituzumab antibodies in cynomolgus monkey plasma
Immunogenicity was assessed by measuring anti-bemarituzumab antibodies using a bridging assay with electrochemiluminescence detection consisting of screening and confirmation steps. The positive control was an anti-FPA144 antibody (Galaxy Biotech; Lot id-FR21.B.16.1). The test samples were diluted at 1:20 and tested in duplicate.
13-week repeat-dose GLP study in rats
A pivotal 13-week repeat-dose GLP toxicology study with bemarituzumab was conducted in Crl:CD(SD) rats at Covance Laboratories (Madison, WI). Bemarituzumab was administered weekly for 13 weeks by IV infusion. Three dose levels (1, 5, or 100 mg/kg) and a vehicle control (20 mM L-histidine, 270 mM sucrose, and 0.01% polysorbate 20) were evaluated. Main group animals (15 rats/sex/group) were scheduled for terminal necropsy on Day 92, 1 week after the last dose. Recovery group animals (5 rats/sex/group) were maintained for all dose levels for 11 weeks following the end of treatment to assess reversibility, persistence, and delayed occurrence of any observed toxic effects.
13-week repeat-dose GLP study in cynomolgus monkeys
A 13-week repeat-dose GLP toxicology study with bemarituzumab was performed in cynomolgus monkeys (Macaca fascicularis) at Covance Laboratories (Madison, WI). Bemarituzumab was administered weekly for 13 weeks by IV infusion at doses of 1, 5, and 100 mg/kg. A vehicle control (20 mM L-histidine, 270 mM sucrose, and 0.01% polysorbate 20, pH 6) was also evaluated. Main group animals (four monkeys/sex/group) were necropsied on Day 92, 1 week after the final administration. Recovery group animals (2 monkeys/sex/group) were maintained for all dose levels for 16 weeks following the end of treatment to assess reversibility, persistence, or delayed occurrence of any observed toxic effects.
Projection of human PK profiles based on cynomolgus monkey data only
The mAb plasma concentration-time profiles in monkeys following 13.5 and 10 mg/kg IV administration of bemarituzumab were transformed to human concentration-time profiles using the species-invariant time method described by the following equations,17 where Timehuman is PK time in human, Timecyno is PK time in cynomolgus monkey, Concentrationhuman is mAb plasma concentration in human, Concentrationcyno is mAb plasma concentration in cynomolgus monkey, Dosehuman is dose in the human PK study (mg/kg), and Dosecyno is dose in the cynomolgus monkey PK study (mg/kg). A scaling exponent of 0.85 was used to estimate human clearance, and a scaling exponent of 1 was used to estimate human V1.
| (1) |
| (2) |
The bi-exponential PK profiles obtained from the species-invariant time method were fit to a 2-compartmental model IV bolus input, first order elimination to estimate the PK parameters using ADAPT 5 User’s Guide,38 which served as input for Monte Carlo simulations of plasma mAb concentrations in humans. The simulations were performed in NONMEM (7.4.3, ICON Development Solutions, Ellicott City, MD) for 1000 subjects. Covariate effects on PK parameters and covariance between clearance and V1 was not included in the model structure. For the simulations, the inter-individual variability on clearance and V1 was assumed to be 30% based on what are generally observed in humans for mAbs.35
Statistical analyses
Statistical significance was determined by 1 way ANOVA followed by Tukey’s multiple comparisons test using GraphPad Prism 7.04. Significance is denoted as *p < .05, **p < .01, ***p < .001, ****p < .0001.
Acknowledgments
The authors acknowledge the entire bemarituzumab team at FivePrime for their contributions through the years of the development of bemarituzumab. The authors thank Bill Buchta, PhD, of Whitsell Innovations, Inc. for providing medical writing and editorial support.
Funding Statement
This work was funded by Five Prime Therapeutics, Inc.
List of Abbreviations
5-FU 5-fluorouracil
ADA anti-drug antibodies
ADCC antibody-dependent cell-mediated cytotoxicity
anti-ASGM1 anti-asialo GM1 antibody
BSA bovine serum albumin
CD3 cluster of differentiation 3
CHO Chinese hamster ovary
CV coefficient of variation
ECD extracellular domain
ELISA enzyme-linked immunosorbent assay
FBS fetal bovine serum
FcγR Fc gamma receptor
FGF fibroblast growth factor
FGFR fibroblast growth factor receptor
FGFR2b fibroblast growth factor receptor 2 IIIb
FivePrime Five Prime Therapeutics, Inc.
FRS2 FGFR substrate 2
GAPDH glyceraldehyde 3-phosphate dehydrogenase
GLP Good Laboratory Practice
HAE high-affinity anti-IgE antibody
HNSTD highest non-severely-toxic dose
Ig immunoglobulin
IP intraperitoneal
IV intravenous(ly)
LLOQ lower limit of quantification
mAb monoclonal antibody
NK natural killer
PBS phosphate-buffered saline
PD-1 programmed death-1
PD-L1 programmed death-ligand 1
PK pharmacokinetic(s)
RE relative error
TMDD target-mediated drug disposition
V1 volume of distribution phase for the central compartment.
Disclosure statement
This research was sponsored by FivePrime. All authors other than Rong Deng were employees of FivePrime and had a business and/or financial interest in the company. Rong Deng declare no potential conflicts of interest. Medical writing services were funded by FivePrime. Five Prime was acquired by Amgen Inc. on April 16th, 2021.
References
- 1.Korc M, Friesel RE.. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. 2009;9(5):639–16. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, Aaronson SA.. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci USA. 1992;89(1):246–50. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev Biol. 1993;158(2):475–86. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 4.Hattori Y, Odagiri H, Nakatani H, Miyagawa K, Naito K, Sakamoto H, Katoh O, Yoshida T, Sugimura T, Terada M. K-sam, an amplified gene in stomach cancer, is a member of the heparin-binding growth factor receptor genes. Proc Natl Acad Sci USA. 1990;87(15):5983–87. doi: 10.1073/pnas.87.15.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung E-J, Jung E-J, Min SY, Kim MA, Kim WH. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum Pathol. 2012;43(10):1559–66. doi: 10.1016/j.humpath.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I, Taniguchi H, Koizumi F, Yanagihara K, Sasaki H, et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer. 2012;106(4):727–32. doi: 10.1038/bjc.2011.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su X, Zhan P, Gavine PR, Morgan S, Womack C, Ni X, Shen D, Bang Y-J, Im S-A, Ho Kim W, et al. FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer. 2014;110(4):967–75. doi: 10.1038/bjc.2013.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seo S, Park SJ, Ryu M-H, Park SR, Ryoo B-Y, Park YS, Na Y-S, Lee C-W, Lee J-K, Kang Y-K. Prognostic impact of fibroblast growth factor receptor 2 gene amplification in patients receiving fluoropyrimidine and platinum chemotherapy for metastatic and locally advanced unresectable gastric cancers. Oncotarget. 2017;8(20):33844–54. doi: 10.18632/oncotarget.12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deshpande AM, Palencia S, Bellovin DI, Gemo AT, Giese T, Stohr B, Pierce KL, Los G. Abstract 2845: expression of FGFR2b in gastric cancer as measured by immunohistochemistry with a highly specific monoclonal antibody. Cancer Res. 2014;74(19Supplement):2845. doi: 10.1158/1538-7445.AM2014-2845. [DOI] [Google Scholar]
- 10.Wainberg ZA, Enzinger PC, Kang Y-K, Yamaguchi K, Qin S, Lee K-W, Oh SC, Li J, Turk HM, Teixeira AC, et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J Clin Oncol. 2021;39(3_suppl):160–160. doi: 10.1200/JCO.2021.39.3_suppl.160. [DOI] [Google Scholar]
- 11.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y-M, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin S-F, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steele IA, Edmondson RJ, Bulmer JN, Bolger BS, Leung HY, Davies BR. Induction of FGF receptor 2-IIIb expression and response to its ligands in epithelial ovarian cancer. Oncogene. 2001;20(41):5878–87. doi: 10.1038/sj.onc.1204755. [DOI] [PubMed] [Google Scholar]
- 14.Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, Geyer FC, Van Kouwenhove M, Kreike B, Mackay A, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29(14):2013–23. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ornitz DM, Nobuyuki I. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4(3):215–66. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang H, Liu L, Gao Y, Ahene A, Macal M, Hsu AW, Dreiling L, Collins H. Population pharmacokinetic analysis of phase 1 bemarituzumab data to support phase 2 gastroesophageal adenocarcinoma FIGHT trial. Cancer Chemother Pharmacol. 2020;86(5):595–606. doi: 10.1007/s00280-020-04139-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng R, Iyer S, Theil F-P, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs. 2011;3(1):61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fletcher AM, Bregman CL, Woicke J, Salcedo TW, Zidell RH, Janke HE, Fang H, Janusz WJ, Schulze GE, Mense MG. Incisor degeneration in rats induced by vascular endothelial growth factor/fibroblast growth factor receptor tyrosine kinase inhibition. Toxicol Pathol. 2010;38(2):267–79. doi: 10.1177/0192623309357950. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Cheng Y-SL, Qin C, Lin C, D’Souza R, Wang F. FGFR2 in the dental epithelium is essential for development and maintenance of the maxillary cervical loop, a stem cell niche in mouse incisors. Dev Dyn. 2009;238(2):324–30. doi: 10.1002/dvdy.21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsa S, Kuremoto K-I, Seidel K, Tabatabai R, Mackenzie B, Yamaza T, Akiyama K, Branch J, Koh CJ, Alam DA, et al. Signaling by FGFR2b controls the regenerative capacity of adult mouse incisors. Development. 2010;137(22):3743–52. doi: 10.1242/dev.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Upadhya D, Lu L, Reneker LW, Connon CJ. Fibroblast growth factor receptor 2 (FGFR2) is required for corneal epithelial cell proliferation and differentiation during embryonic development. PLoS One. 2015;10(1):e0117089. doi: 10.1371/journal.pone.0117089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao WW-Y, Xia Y, Liu C-Y, Saika S. Signaling pathways in morphogenesis of cornea and eyelid. Ocul Surf. 2008;6(1):9–23. doi: 10.1016/s1542-0124(12)70102-7. [DOI] [PubMed] [Google Scholar]
- 23.Parsa S, Ramasamy SK, De Langhe S, Gupte VV, Haigh JJ, Medina D, Bellusci S. Terminal end bud maintenance in mammary gland is dependent upon FGFR2b signaling. Dev Biol. 2008;317(1):121–31. doi: 10.1016/j.ydbio.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Hattori Y, Itoh H, Uchino S, Hosokawa K, Ochiai A, Ino Y, Ishii H, Sakamoto H, Yamaguchi N, Yanagihara K, et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin Cancer Res. 1996;2(8):1373–81. PMID: 9816310. [PubMed] [Google Scholar]
- 25.Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–84. doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamayoshi T, Nagayasu T, Matsumoto K, Abo T, Hishikawa Y, Koji T. Expression of keratinocyte growth factor/fibroblast growth factor-7 and its receptor in human lung cancer: correlation with tumour proliferative activity and patient prognosis. J Pathol. 2004;204(1):110–18. doi: 10.1002/path.1617. [DOI] [PubMed] [Google Scholar]
- 27.Natasha AP, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. 2018;10(5):693–711. doi: 10.1080/19420862.2018.1466767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ec S, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D. ESMO guidelines committee. Gastric cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v38–v49. doi: 10.1093/annonc/mdw350. [DOI] [PubMed] [Google Scholar]
- 29.Ye P, Zhang M, Fan S, Zhang T, Fu H, Su X, Gavine PR, Liu Q, Yin X, Generali D. Intra-tumoral heterogeneity of HER2, FGFR2, cMET and ATM in gastric cancer: optimizing personalized healthcare through innovative pathological and statistical analysis. PLoS One. 2015;10(11):e0143207. doi: 10.1371/journal.pone.0143207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leabman MK, Meng YG, Kelley RF, DeForge LE, Cowan KJ, Iyer S. Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs. 2013;5(6):896–903. doi: 10.4161/mabs.26436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortensen DL, Prabhu S, Stefanich EG, Kadkhodayan-Fischer S, Gelzleichter TR, Baker D, Jiang J, Wallace K, Iyer S, Fielder PJ, et al. Effect of antigen binding affinity and effector function on the pharmacokinetics and pharmacodynamics of anti-IgE monoclonal antibodies. MAbs. 2012;4(6):724–31. doi: 10.4161/mabs.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Catenacci DVT, Rasco D, Lee J, Rha SY, Lee K-W, Bang YJ, Bendell J, Enzinger P, Marina N, Xiang H, et al. Phase I escalation and expansion study of bemarituzumab (FPA144) in patients with advanced solid tumors and FGFR2b-selected gastroesophageal adenocarcinoma. J Clin Oncol. 2020;38(21):2418–26. doi: 10.1200/JCO.19.01834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling J, Zhou H, Jiao Q, Hm D. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J Clin Pharmacol. 2009;49(12):1382–402. doi: 10.1177/0091270009337134. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, Prueksaritanont T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm Drug Dispos. 2010;31(4):253–63. doi: 10.1002/bdd.708. [DOI] [PubMed] [Google Scholar]
- 35.Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 36.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs. 2018;10(5):751–64. doi: 10.1080/19420862.2018.1462429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W, Wang L, Park H, Chhim S, Tanphanich M, Yashiro M, Kim KJ. Monoclonal antibodies to fibroblast growth factor receptor 2 effectively inhibit growth of gastric tumor xenografts. Clin Cancer Res. 2010;16(23):5750–58. doi: 10.1158/1078-0432.CCR-10-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Argenio DZ, Schumitzky A, Wang X. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource , Los Angeles, California, 2009. [Google Scholar]